

Abstract
We investigated effects of age, sex and frailty on contractions, calcium transients and myofilament proteins to determine if maladaptive changes associated with aging were sex-specific and modified by frailty. Ventricular myocytes and myofilaments were isolated from middle-aged (~12 mos) and older (~24 mos) mice. Frailty was assessed with a non-invasive frailty index. Calcium transients declined and slowed with age in both sexes, but contractions were largely unaffected. Actomyosin Mg-ATPase activity increased with age in females but not males; this could maintain contractions with smaller calcium transients in females. Phosphorylation of myosin-binding protein C (MyBP-C), desmin, tropomyosin and myosin light chain-1 (MLC-1) increased with age in males, but only MyBP-C and troponin-T increased in females. Enhanced phosphorylation of MyBP-C and MLC-1 could preserve contractions in aging. Interestingly, the age-related decline in Hill coefficients (r = −0.816; p = 0.002) and increase in phosphorylation of desmin (r = 0.735; p = 0.010), tropomyosin (r = 0.779; p = 0.005) and MLC-1 (r = 0.817; p = 0.022) were graded by the level of frailty in males but not females. In these ways, cardiac remodeling at cellular and subcellular levels is graded by overall health in aging males. Such changes may contribute to heart diseases in frail older males, whereas females may be resistant to these effects of frailty.
Subject terms: Cardiovascular biology, Comorbidities
Introduction
Diseases of impaired myocardial contractile function, including heart failure, increase with age in both men and women1. This may not be surprising. Studies suggest that the heart undergoes maladaptive changes during normal aging that may set the stage for the development of heart failure2. However, a key challenge to understanding the milieu in which such diseases develop is that, while older men and women are most likely to develop heart diseases as they age1, current preclinical research studies typically use young, mostly male animals3–5. Although few experimental studies have investigated the influence of age on cardiac contractile function, emerging evidence suggests this may differ between the sexes both in humans and in animal models6–9. To understand the underlying reasons, it is important to identify cellular and subcellular mechanisms that are involved in cardiac aging in both sexes.
Although age modifies the heart, such changes are average responses that may not be present, or present to the same extent, in all individuals of the same age10. For example, on average ventricular contractility declines with age, even though some older men and women perform at the same or even at higher levels when compared to younger adults11. This suggests that aging is heterogenous. The concept of “frailty”, used by demographers in 1979 to describe unmeasured heterogeneity in mortality risk in people of the same age12, is now used to describe unmeasured heterogeneity in the risk of many age-related adverse outcomes in both humans and animals13. While there is no consensus definition of frailty14, it is clinically important as frail individuals are more likely to develop diseases, including heart diseases, than their non-frail counterparts15. It is possible that changes associated with cardiac aging are modified by frailty, but little is known about the impact of age and frailty on the heart, especially in females.
Frailty has been quantified clinically with many different instruments16,17. One common technique is to create a “frailty index”, by dividing the number of health deficits in an individual by the total number of deficits considered to produce a score between 0 and 1, where higher scores denote greater frailty18. We have adapted this approach to quantify the degree of frailty in aging rodents13,19–21. This provides a powerful new tool that can be used to explore the relationship between cardiac aging and overall health (frailty), in mice of both sexes. The goal of this study is to investigate the impact of age, sex and frailty on cardiac contractile function and explore underlying mechanisms that regulate contraction in a mouse model. Studies used isolated ventricular myocytes, Langendorff-perfused hearts and ventricular homogenates from middle-aged (~12 months) and older (~24 months) male and female C57Bl/6 mice. Frailty was evaluated in each animal with a frailty index tool that measures frailty as the accumulation of health deficits across many diverse systems, but not the cardiovascular system per se22.
Results
Calcium transients declined with age, but contractions were largely unaffected in both field-stimulated ventricular myocytes and intact hearts from male and female mice
Initial experiments determined whether ventricular myocyte contractions and the underlying calcium transients were affected by age and whether this differed between the sexes. Figure 1A shows representative contractions recorded from ventricular myocytes (paced at 4 Hz) isolated from the hearts of middle-aged (~12 mos) and older (~24 mos) male and female mice. The mean (± SEM) data show that contraction amplitudes were similar regardless of age or sex (Fig. 1B). The speed of shortening showed a modest increase with age in males, which was significantly different from older females (Fig. 1C). The velocity of lengthening was not affected by age but was slower in older females compared to older males (Fig. 1D). These results indicate that there are few age- or sex-dependent changes in contractions in field-stimulated ventricular myocytes between middle-age and later life.
Figure 1.

Peak calcium transients declined and slowed with age in C57BL/6 mice of both sexes, but contractions were largely unaffected. (A) Representative examples of contractions (cell shortening) recorded from field-stimulated (4 Hz) ventricular myocytes isolated from middle-aged (~12 mos) and older (~24 mos) male and female mice. (B) Mean data show that peak contractions were similar in all four groups. (C) The velocity of shortening increased slightly with age in males and was faster in older male cells compared to female cells. (D) The velocity of lengthening was unaffected by age but was lower in older females than in older males. (E) Representative examples of calcium transients recorded from myocytes from middle-aged and older mice of both sexes. (F) Mean data show that peak calcium transients declined with age in both sexes and were smaller in cells from females than males at both ages. (G) The rates of rise of the calcium transient declined with age and this was significant in females. The rates of rise were slower in females than males at all ages. (H) The rates of decay of the calcium transients declined markedly with age in both sexes. Values represent the mean ± SEM values in each case. Data were analyzed by two-way ANOVA with age and sex as main factors (post-hoc test was Holm-Sidak). The * denotes p < 0.05. For calcium transients n = 15, 18, 22 and 12 myocytes from 6 middle-aged male mice, 4 older males, 9 middle-aged females and 3 older females, respectively. For contractions n = 24, 20, 27 and 12 myocytes from 6 middle-aged male mice, 4 older males, 8 middle-aged females and 3 older females, respectively.
Figure 1E shows examples of calcium transients recorded from ventricular myocytes from the hearts of middle-aged and older mice. Mean data show that calcium transient amplitudes declined with age in both sexes (Fig. 1F). There also was a sex difference where, regardless of age, calcium transients were smaller in cells from females than males. We evaluated the impact of age and sex on calcium transient rates of rise and decay (Fig. 1G,H). The calcium transient rates of rise declined with age and this was significant for females, where rates of rise were slower than for males at both ages (Fig. 1G). Age was also associated with a dramatic slowing of calcium transient decay rate in both sexes (Fig. 1H). These observations show that aging was associated with an overall decrease in the magnitude and speed of calcium transients in both sexes, with few parallel changes in contractions.
To investigate whether cardiac contractile function changed between 12 and 24 months of age, we also compared left ventricular developed pressure (LVDP) in Langendorff-perfused hearts from both sexes. Figure 2A shows representative examples of LVDP recorded from isolated perfused hearts from middle-aged and older male mice. Figure 2B illustrates mean data that show that peak LVDP was similar, regardless of age or sex. Likewise, the rates of pressure development (Fig. 2C) and decay (Fig. 2D) were similar in hearts from middle-aged and older mice of both sexes. Thus, even though aging was associated with smaller, slower calcium transients, contractile function was unaffected in intact hearts and isolated myocytes. To explore potential underlying mechanisms, we next investigated whether changes in the myofilaments occurred during the aging process.
Figure 2.

Left ventricular contractile function was similar in hearts from middle-aged and older mice, regardless of sex. (A) Examples of pressure recordings from Langendorff-perfused hearts isolated from middle aged and older male mice. Recordings are shown at condensed (left) and expanded (right) time scales. (B–D) Scatterplots demonstrate that LVDP, + dP/dt and −dP/dt were similar in hearts from middle-aged and older mice of both sexes. Data were analyzed by two-way ANOVA with age and sex as main factors (post-hoc test was Holm-Sidak). Samples were hearts from 11 middle-aged male mice, 14 older males, 15 middle-aged females and 11 older females, respectively.
Actomyosin Mg-ATPase activity markedly increased with age in myofilaments from female but not male hearts
We investigated the relationship between activating calcium concentrations and actomyosin Mg-ATPase activity in myofilaments isolated from male and female ventricles at both ages. Results are shown in Fig. 3. When absolute actomyosin Mg-ATPase activity was plotted as a function of calcium in males, there was no difference between middle-aged and older hearts (Fig. 3A). By contrast, actomyosin Mg-ATPase activity increased with age in females, and this increase was statistically significant at physiologically relevant calcium levels above ~500 nM free calcium (Fig. 3B). We also compared maximal actomyosin Mg-ATPase activity between all four groups, as shown in Fig. 4A. The average maximal actomyosin Mg-ATPase activity increased with age in females but not males. Interestingly, activity was lowest in middle-aged females and was significantly lower than age-matched males (Fig. 4A). These data demonstrate that there was an increase in actomyosin Mg-ATPase activity with age across a wide range of activating calcium concentrations, although this effect was sex-specific and occurred only in hearts from females.
Figure 3.
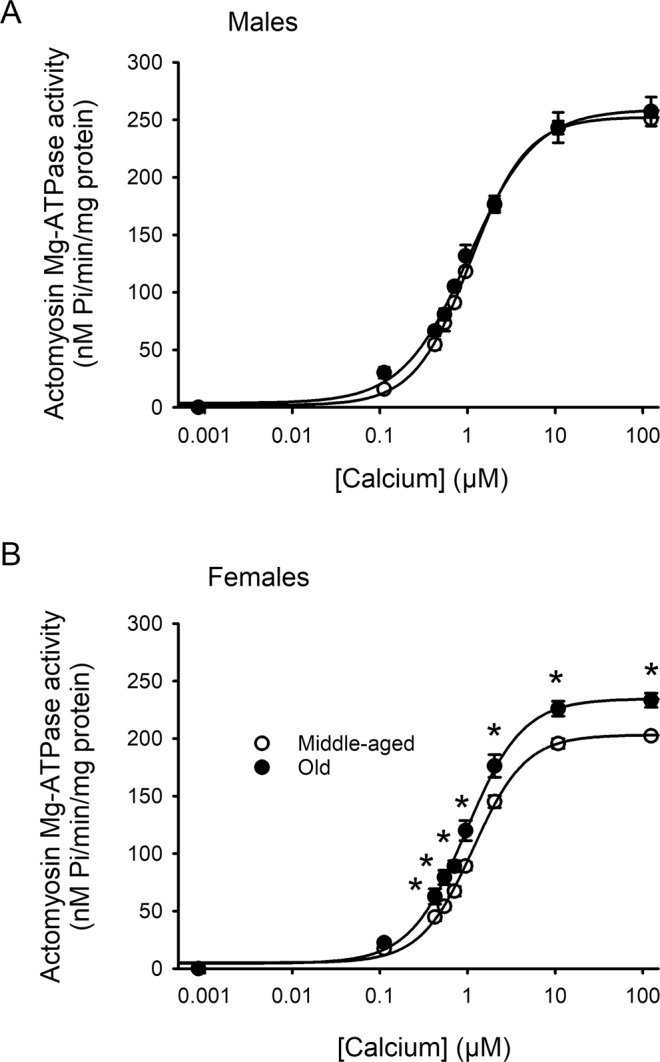
Maximal actomyosin Mg-ATPase activity increased with age in hearts from females but not males. (A) Actomyosin Mg-ATPase activity increased as calcium concentrations increased to the same extent in myofilaments from middle-aged and older male hearts. (B) In females, actomyosin Mg-ATPase activity increased with age at almost all calcium concentrations tested. Values represent the mean ± SEM values. Data were analyzed with a two-way repeated measures ANOVA, with age as the main factor, calcium concentration as the repeated measure and pairwise multiple post-hoc comparisons with a Tukey test. The * symbol indicates a significant effect of age. Values were significant for p < 0.05. Samples were hearts from 5 middle-aged male mice, 6 older males, 5 middle-aged females and 5 older females, respectively.
Figure 4.
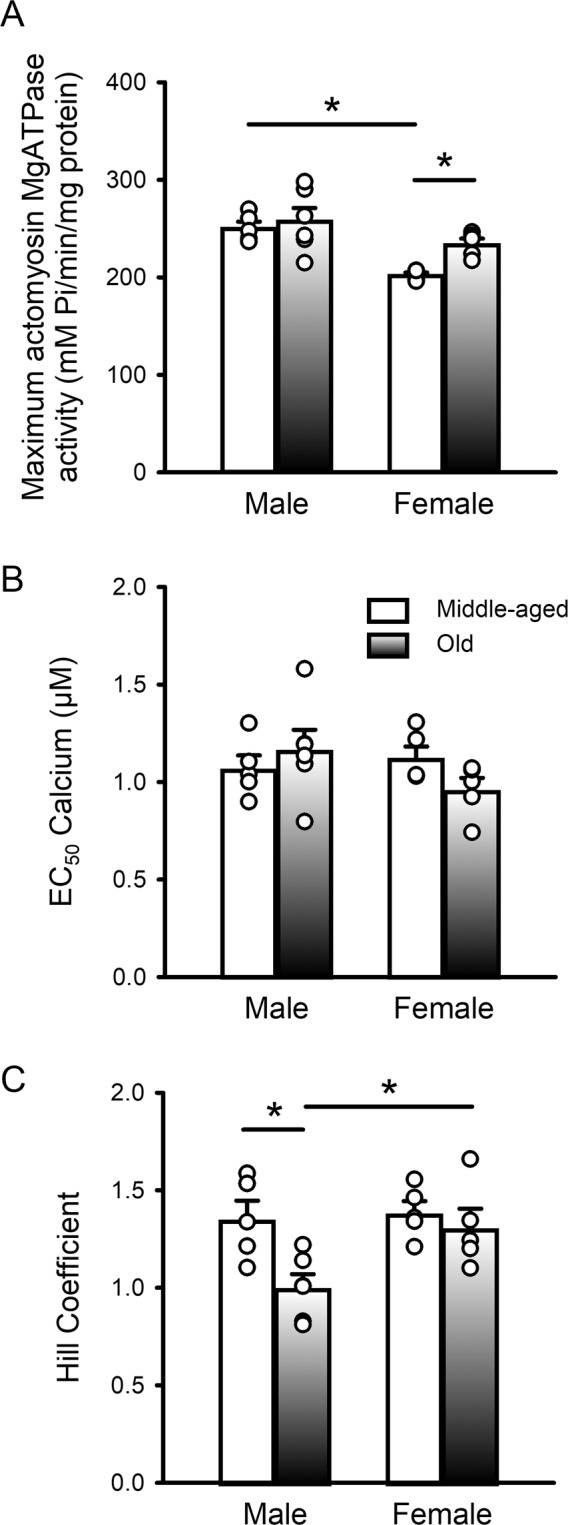
Maximal actomyosin Mg-ATPase activity and Hill coefficients varied with age in a sex-specific fashion, but EC50 values were unaffected. (A) Mean data show that maximal actomyosin ATPase activity increased with age in females but not males. There was also a sex difference at younger ages where activity was higher in middle-aged males than in middle-aged females. (B) EC50 values were similar in all four groups. (C) Average values for the Hill coefficients declined markedly with age in males and were significantly lower in older males compared to older females. Values denote the mean ± SEM in each case. Data were analyzed with a two-way ANOVA with age and sex as main factors (post-hoc test was Holm-Sidak). The * denotes p < 0.05. Samples were hearts from 5 middle-aged male mice, 6 older males, 5 middle-aged females and 5 older females, respectively.
When the actomyosin Mg-ATPase activity data were normalized to the maximum for each group, there were no significant age or sex effects (Supplementary Figure 1A,B). Consistent with this finding, the average EC50 values (concentration of calcium required to produce 50% activation), did not differ between groups (Fig. 4B). The steepness of the actomyosin Mg-ATPase-calcium relationship is indicated by the Hill coefficient such that larger values denote increased cooperativity of calcium activation. To determine if there were age- or sex-related changes in the steepness of the actomyosin Mg-ATPase versus calcium curves, we compared mean Hill coefficients between groups. On average, Hill coefficients declined markedly with age in males, although there was heterogeneity and individual data points for the two age groups overlapped (Fig. 4C). By contrast, there was no age-dependent change in females, but Hill coefficients were significantly higher in older females when compared to older males (Fig. 4C). These findings indicate that cooperativity of the actomyosin Mg-ATPase-calcium relationship declined with age in male hearts.
Overall health, measured with a frailty index, graded age-related changes in myofilament function in males but not females
On average, Hill coefficients declined with age in males (Fig. 4C) whereas maximal actomyosin Mg-ATPase activity increased with age in females (Fig. 4A). However, there was considerable heterogeneity, especially for the Hill coefficients, such that individual values from the two age groups overlapped in many cases. To determine the relationship between parameters derived from the actomyosin Mg-ATPase activity curves and frailty, we plotted maximal actomyosin Mg-ATPase activity, EC50 values and Hill coefficients as a function of frailty index score (Fig. 5). We fitted each curve by linear regression. We found that there was no correlation between maximal actomyosin Mg-ATPase activity and frailty index scores in either males (Fig. 5A) or females (Fig. 5D). Similarly, EC50 values were not correlated with the level of frailty in either sex (Fig. 5B,E). By contrast, Hill coefficients, which declined with age in males only, exhibited a strong negative association with frailty in males (Fig. 5C) but showed no relationship with frailty in females (Fig. 5F). As both age and frailty were related to the decline in Hill coefficients in male hearts, we used multivariable regression to calculate the semi-partial correlations to assess the separate contributions of age and frailty to this relationship. With respect to the semi-partial correlations, we found that both frailty (r = −0.816) and age (r = −0.726) contributed significantly to the decline in Hill coefficients. This indicates that age-dependent changes in actomyosin Mg-ATPase activity in male hearts were graded by the overall health of the animal, as quantified with a frailty index score.
Figure 5.

The Hill coefficients for the actomyosin Mg ATPase activity versus calcium curves were graded by frailty in males only. (A,D) Maximal actomyosin Mg-ATPase activity was not related to frailty scores in either males or females. (B,E) EC50 values also were not correlated with frailty index scores in either sex. (C,F) In contrast, Hill coefficients were graded by the level of frailty, but only in males. Data were fitted with a regression line. Correlation coefficients and p values are indicated on each panel and a line was drawn for statistically significant correlations (p < 0.05). Samples were hearts from 5 middle-aged male mice, 6 older males, 5 middle-aged females and 5 older females, respectively.
Male and female hearts exhibited distinct age-associated changes in myofilament protein phosphorylation
As changes in the phosphorylation of key myofilament proteins could modify contractile function, we compared myofilament phosphorylation patterns in hearts from middle-aged and older mice of both sexes. Figure 6A–D shows gels of myofilament proteins for all samples evaluated in this study. The proteins were stained with Pro-Q (left) to visualize total phosphorylation levels and stained with Coomassie blue (right) to indicate total protein load. Actin was used as a loading control and the uncropped gels are shown in Supplementary Information Files 1–3. The mean data as well as scatterplots of the individual data points are shown in Fig. 7. Results show that, on average, myosin binding protein-C (MyBP-C) phosphorylation increased with age in both sexes (Fig. 7A). There was also a sex difference where MyBP-C phosphorylation was greater in middle-aged males than age-matched females. Mean phosphorylation levels for myosin light chain-1 (MLC-1), desmin and tropomyosin all increased with age in males but there were no age-associated changes in females (Fig. 7B–D). In all cases, there was heterogeneity in data, especially for the older males, such that values from the two different age groups showed considerable overlap. There were also sex differences in the older group, where MLC-1 and desmin phosphorylation were higher in older males than older females (Fig. 7B,C). In contrast, troponin-T phosphorylation increased with age in females only and was higher in older females than in older males (Fig. 7E). Phosphorylation of troponin-I was not affected by age or sex (Fig. 7F). These experiments demonstrate that, on average, phosphorylation of MyBP-C, MLC-1, desmin and tropomyosin increased with age in males, but only MyBP-C and troponin-T phosphorylation increased with age in females.
Figure 6.

Data illustrating the protein phosphorylation of major myofilament proteins in hearts from middle-aged and older males and females of both sexes. Gels illustrate phosphorylation staining (ProQ Diamond) and total protein staining (Coomassie blue) for all samples quantified in this study. Individual samples in each group are indicated numerically at the top of each lane. Protein standards at 25 and 75 kDa are visible in the ProQ images (lane labelled “S”) and the molecular weight ladder is visible in the Coomassie images (lane labelled “L”). Actin (42 kDa) was used as a loading control and is shown with an arrow on each Coomassie image. (A,B) Myofilament proteins from middle-aged and older male ventricles were separated on SDS-PAGE and stained with Pro-Q (left) to compare phosphorylation. They were also stained with Coomassie blue (right) to illustrate protein load. (C,D) Myofilament proteins from middle-aged and older females separated by Pro-Q (left) and stained with Coomassie (right). Major myofilament proteins are indicated on the left side of each panel and molecular weight markers (kDa) are shown on the right. Myosin-binding protein C (MyBP-C), myosin light chain-1 (MLC-1). The gels have been cropped to the edges; the uncropped gels are included as Supplementary Information Files 1–3. Samples were hearts from 5 middle-aged male mice, 6 older males, 5 middle-aged females and 5 older females, respectively.
Figure 7.

Total phosphorylation levels for MyBP-C, MLC-1, desmin & tropomyosin increased with age in males, while only MyBP-C and troponin-T increased with age in females. (A) MyBP-C phosphorylation increased moderately with age in males and dramatically with age in females; MyBP-C phosphorylation also was greater in middle-aged males compared to females. (B) MLC-1 phosphorylation increased markedly with age in males but not females; values were higher in older males than age-matched females. (C) Desmin phosphorylation increased with age in males only; values were significantly higher in older males compared to older females. (D) Tropomyosin phosphorylation increased with age in males only. (E) Phosphorylation of troponin-T increased with age in females only and was significantly higher in older females compared to older males. (F) Phosphorylation of troponin-I was not affected by either age or sex. Values denote the mean ± SEM in each case. Data were analyzed with two-way ANOVA (age and sex were main factors; the post-hoc test was Holm-Sidak). The * denotes p < 0.05. Samples were hearts from 5 middle-aged male mice, 6 older males, 5 middle-aged females and 5 older females, respectively. Actin was used as a loading control; it was not affected by either age or sex (not shown). In all cases data were normalized to actin. Myosin-binding protein C (MyBP-C), myosin light chain-1 (MLC-1).
Age-related changes in myofilament phosphorylation were graded by frailty index scores in males but not females
We found that phosphorylation of several key myofilament proteins increased with age, especially in hearts from males (Fig. 7). Still, there was considerable heterogeneity, especially in data from the older males where values from the two age groups exhibited substantial overlap. To determine the relationship between myofilament protein phosphorylation and frailty, we plotted phosphorylation levels as a function of frailty and fitted the curves by linear regression as shown in Fig. 8. Results showed that phosphorylation levels for MLC-1, desmin and tropomyosin exhibited strong positive correlations with frailty in hearts from males (Fig. 8A–C). When we examined the semi-partial correlations, we found that both frailty (r = 0.817) and age (r = 0.828) contributed significantly to the higher phosphorylation levels for MLC-1 in males (Fig. 8A). Frailty and age also contributed significantly to higher phosphorylation levels for desmin (r = 0.735 for frailty and r = 0.724 for age) and tropomyosin (r = 0.779 for frailty and r = 0.645 for age; Fig. 8B,C). Unlike males, there were no correlations between phosphorylation of MLC-1, desmin or tropomyosin and frailty index scores in hearts from females (Fig. 8D–F). Phosphorylation levels for MyBP-C, troponin-T and troponin-I were not related to frailty scores in either sex (Supplementary Figure 2A–F). Together these results show that phosphorylation of key myofilament proteins increased as frailty increased in hearts from males but not females. This indicates that age-dependent increases in the phosphorylation of key myofilament proteins were graded by overall health, as quantified in a frailty index, but only in hearts from male animals.
Figure 8.

Phosphorylation of the key myofilament proteins MLC-1, desmin and tropomyosin were highly correlated with overall health, as indicated by frailty index scores, in hearts from males only. (A–C) The phosphorylation of desmin, MLC-1 and tropomyosin exhibited strong positive correlations with frailty in hearts from males. (D–F) Although phosphorylation levels for many myofilament proteins increased as frailty scores increased in males, there were no correlations between myofilament protein phosphorylation and frailty in hearts from females. The data were fit with a regression line. Correlation coefficients plus p values are indicated on each panel; lines were drawn for all statistically significant correlations (p < 0.05). Samples were hearts from 5 middle-aged male mice, 6 older males, 5 middle-aged females and 5 older females, respectively. Myosin light chain-1 (MLC-1).
Discussion
This study evaluated the impact of age, sex and frailty on cardiac contractile function and explored underlying mechanisms that regulate contraction in the murine heart. Studies in isolated ventricular myocytes showed that calcium transients declined and slowed between middle age and later life in both sexes. By contrast, contractions in myocytes and in intact hearts were relatively unaffected by age. Myofilament analysis showed that actomyosin Mg-ATPase activity increased with age at physiological calcium concentrations in hearts from females, whereas myofilament cooperativity as represented by Hill coefficients declined in males. Age was accompanied by changes in the phosphorylation levels of several major myofilament proteins. However, the patterns of change differed between the sexes, and age effects were much more prominent in males. These age-associated changes in cooperativity and myofilament phosphorylation were correlated with and graded by the level of frailty in males. By contrast, no relationship between frailty and the myofilament parameters measured in this study was seen in females. Our work highlights the substantial heterogeneity in the impact of age on myofilament proteins, especially in male hearts, and show that both frailty and chronological age contribute significantly to this variance. These observations suggest that differences in overall health status contribute importantly to the impact of age on the heart. Even so, these modifications are sex-specific and are most apparent at high levels of frailty in males only.
The results of the present study showed that age had no effect on peak contractions recorded from field-simulated ventricular myocytes from mice of both sexes. This agrees with results of previous studies in field-stimulated ventricular myocytes from aged rodents when compared to young adult males23–25 and females26,27. Here, we have extended these observations to show that contractions did not change between middle age and later life, even though calcium transients declined and slowed in field-stimulated cells from mice of both sexes. We also confirmed that baseline contractile function in isolated intact hearts did not change between middle-age and later life regardless of sex28. Given that cardiac contraction is proportional to the magnitude of intracellular calcium release29, our finding that calcium transients decline with age and contractions do not is unexpected.
To explore mechanisms that might maintain contractile performance in aging in the face of reduced calcium availability, we examined myofilament calcium sensitivity. Previous work showed that myofilament calcium sensitivity declined when 2–4-month-old mice were compared to 2-year-old animals, although this study used only males25. Prior studies of sex differences in myofilament calcium sensitivity used young animals only and found either no sex difference30 or lower calcium sensitivity in hearts from males when compared to females31. To our knowledge, the present study is the first to investigate sex-dependent changes in myofilament calcium sensitivity in the setting of aging. We found that submaximal actomyosin Mg-ATPase activity increased markedly with age, but this was seen in female hearts only. Critically, higher actomyosin Mg-ATPase activity occurred in females at calcium concentrations within the normal physiological range32. Our observation that the influence of age on myofilaments is sex-specific is a key finding. The increase in myofilament calcium sensitivity in the aging female heart is likely to be compensatory and may help preserve contractile function in the face of lower intracellular calcium availability in aging.
How enhanced myofilament calcium sensitivity arises in older females is not yet clear. One possibility is that the low circulating estradiol levels seen in older female mice and rats33 could be involved. In support of this, studies in young, ovariectomized females show that short-term exposure to low circulating estradiol increases myofilament calcium sensitivity in the heart34,35. In addition, higher actomyosin Mg-ATPase activity occurs early in a murine model of menopause in which ovarian function is gradually reduced by 4‐vinylcyclohexene diepoxide injections36. On the other hand, both long-term ovariectomy and longer-term exposure to menopause appear to reduce cardiac myofilament calcium sensitivity in the mouse model36,37. Thus, whether low circulating estradiol levels can explain enhanced myofilament calcium sensitivity in naturally aging animals is not clear and may be dependent on the duration of the estradiol deficiency; additional experiments that explore this question would be of interest.
We also found that age had very little effect on actomyosin Mg-ATPase activity in males, so other regulatory mechanisms that could maintain contraction in the face of lower calcium availability were explored. As age reduces circulating testosterone levels in older mice33, the present results suggest that low testosterone may have little effect on myofilament calcium sensitivity. This agrees with our earlier work in male mice where we showed that gonadectomy did not influence myofilament calcium sensitivity at physiological calcium levels38. However, the present study did show that Hill coefficients declined with age in males. The Hill coefficient represents the steepness of the actomyosin Mg-ATPase-calcium relationship, where a larger value indicates positive cooperativity of calcium activation39. Cooperativity has been attributed to a variety of mechanisms, all of which involve tropomyosin either directly or indirectly39. Interestingly, we found that phosphorylation of tropomyosin increased with age in males but not females. This may influence the degree of cooperativity observed in aging male hearts. In support of this mechanism, there is evidence that cooperativity declines when tropomyosin is phosphorylated in reconstituted cardiac muscle fibres40. Thus, it is possible that enhanced phosphorylation of tropomyosin reduces the cooperativity of calcium activation. However, this change is likely to disrupt rather than preserve contractile function in the aging male heart.
We observed sex-specific changes in phosphorylation of myofilament proteins with age. For example, hearts from older males exhibited an increase in the phosphorylation of both desmin and MLC-1. Desmin is a myofilament protein that provides a scaffold to preserve cardiac myocyte structure and protect the heart from stressors such as mechanical stress41. Phosphorylation of desmin is thought to facilitate desmin misfolding, which may generate toxic protein aggregates that have been implicated in the pathogenesis of diseases such as heart failure42. The dysfunctional role of desmin phosphorylation may be mediated through the accumulation of these amyloid-like oligomers, which increase cytoskeletal cell stiffness and decrease cytoskeletal viscosity thereby presenting a physical impediment to contractility42. Thus, elevated desmin phosphorylation may be maladaptive in the aging male heart. We also observed increased MLC-1 phosphorylation in aging male hearts. Although relatively little is known about its role in cardiac contraction, hypophosphorylation of MLC-1 in a zebra fish model disrupts myocardial force generation and increases the heart’s susceptibility to stress43. Thus, this increase in MLC-1 phosphorylation may represent a beneficial effect to compensate for deleterious effects of desmin hyperphosphorylation in aging male hearts. We also found a significant increase in troponin-T phosphorylation in aging females but the significance of this is unclear. Although troponin-T can be phosphorylated at several different sites, this appears to have little impact on myofilament function44,45.
Our work showed that MyBP-C phosphorylation increased with age in both sexes. MyBP-C regulates cardiac contractile function and is controlled by phosphorylation through multiple signaling pathways, although its contributions to heart function are not fully understood46. Interestingly, dephosphorylation of cardiac MyBP-C is a common finding in diseases of contractile dysfunction, such as heart failure, in both humans and animal models47,48. In addition, transgenic mice with non-phosphorylatable MyBP-C exhibit impaired systolic and diastolic function49. Together these findings indicate that chronic dephosphorylation of MyBP-C disrupts myocardial contractile function. As phosphorylation of cardiac MyBP-C induces a conformational change in myosin that promotes cross-bridge formation50, an increase in MyBP-C phosphorylation may be a compensatory mechanism that helps preserve cardiac contractile function in aging. While MyBP-C phosphorylation increased with age and maybe be a compensatory mechanism in both sexes, opposing phosphorylation changes that impair myofilament function (e.g. desmin phosphorylation) in males may off-set beneficial alterations in the contractile apparatus.
Frailty is a key determinant of overall health status and mortality in mice of similar chronological ages, as it is in humans13. We have previously shown that age-associated adverse remodeling in the atria and ventricles is graded by the level of frailty in male mice10,51,52. This suggests that cardiac aging and overall health are closely linked, at least in males. A novel and important finding in the present study is our observation that, while most age-associated changes seen in male hearts were graded by frailty, none of the age-dependent changes in females exhibited any clear relationship with frailty. As identified by Maric-Bilkan et al.53, it is critically important to conduct basic research in cardiovascular biology in animals of both sexes to address the knowledge gap in how sex can affect research outcomes. Our results highlight the importance of using both male and female animals in such studies, as very different conclusions would have been reached had only one sex been used here. Taken together, our data suggest that poor overall health, quantified in a frailty index, predicts adverse changes and post-translational modifications in myofilaments in the aging male heart but not in the aging female heart. Some of these changes, such as the increased phosphorylation of MLC-1 at high frailty levels, may be compensatory and help preserve contractile function in hearts from frail older men. Still, enhanced phosphorylation of desmin and tropomyosin, as well as smaller Hill coefficients, would be expected to negatively affect heart function and may ultimately promote the development of diseases of impaired contractility in frail older men. Our findings also suggest that females may be more resilient than males to the effects of poor overall health on the heart.
We have demonstrated that intracellular calcium availability declined between middle age and later life in myocytes from male and females, but contractions were preserved. Contractile function was maintained in aging females by an increase in myofilament calcium sensitivity and enhanced phosphorylation of MyBP-C. By contrast, there was no compensatory increase in myofilament calcium sensitivity in males, although enhanced phosphorylation of MLC-1 and MyBP-C in males may help preserve contractile function in aging. Still, the elevated phosphorylation of tropomyosin and desmin, as well as the decrease in positive cooperativity, would be expected to ultimately impair contractile function in older males. We found that the impact of age on myofilament proteins was heterogenous in male hearts and our results demonstrate that both frailty and chronological age contribute significantly to this variance. This suggests that there is a link between cardiac aging and overall health in aging males, while older females may be resistant to the adverse effects of frailty on the heart. These findings may help explain the so-called morbidity-mortality paradox, where older women have higher levels of frailty than men at any age, but live longer54. Further exploration of the mechanistic basis for sex-specific changes in aging and frailty is motivating additional inquires by our group.
Methods
Animals
C57BL/6 mice of both sexes were purchased from Charles River (St. Constant, QC, Canada) and aged in the Carlton Animal Care Facility at Dalhousie University until use. Mice were housed in groups in individually ventilated caging systems (Allentown Inc; 21 °C; 35% humidity). They were kept on a 12-hour light/dark cycle with free access to water and food (ProLab RMH 3000, Purina LabDiet, Aberfoyle, Ontario, Canada). Animal protocols were approved by the Dalhousie University Committee on Laboratory Animals and studies were performed in accordance with the guidelines of the Canadian Council on Animal Care (CCAC, Ottawa, ON: Vol 1, 2nd edition, 1993; revised March 2017).
Mouse clinical frailty assessment
Overall health was assessed with a mouse clinical frailty index tool. This instrument is an index of 31 potential deficits in health that can accumulate with age in C57BL/6 mice20. This tool evaluates deficits in overall health (e.g. the integument, musculoskeletal system, vestibulocochlear/auditory systems, ocular/nasal systems, digestive system, urogenital system, respiratory system, signs of discomfort, body weight and body surface temperature) and none of the potential deficits is a measure of cardiovascular health per se. This non-invasive instrument is validated and reliable, as described previously55,56. Mice were assessed in a quiet room after they had acclimatized for approximately 10 minutes. They were individually evaluated for the presence of 31 potential deficits. For each item, mice without the deficit received a score of 0, those with a mild deficit received 0.5, and those with a severe deficit scored a 1. Values for each deficit were then added and divided by the total number of deficits assessed to produce a frailty index score that could theoretically be between 0 and 1.
Field stimulation experiments
Ventricular myocytes were enzymatically dissociated with our established techniques26. Mice were weighed and anesthetized (200 mg/kg sodium pentobarbital IP, 100 U heparin IP). The heart was removed and perfused through the aorta at 2 mL/min (10 mins) with calcium-free buffer (mM): 105 NaCl, 5 KCl, 25 HEPES, 0.33 NaH2PO4, 1.0 MgCl2, 20 glucose, 3.0 Na-pyruvate, and 1.0 lactic acid (pH 7.4; 37 °C; 100% O2). Then the heart was perfused with this buffer plus calcium (50 μM), collagenase (8 mg/30 ml, Worthington Type I, 250 U/mg), dispase II (3.5 mg/30 ml, Roche) and trypsin (0.5 mg/30 ml, Sigma). After 8–10 minutes, the ventricles were removed, minced and stored in high potassium buffer (mM): 50 L-glutamic acid, 30 KCl, 30 KH2PO4, 20 taurine, 10 HEPES, 10 glucose, 3 MgSO4, and 0.5 EGTA (pH 7.4; 21 °C). Cell suspensions were filtered with a 225 μm polyethylene mesh filter (Spectra/Mesh).
Cells were loaded with calcium-sensitive dye (fura-2 AM, 2.5 μM; Invitrogen, Burlington, ON) for 20 minutes in the dark on the stage of an inverted microscope (Nikon Eclipse TE200, Nikon Canada, Mississauga, ON). Cells were superfused (3 mL/min) with buffer (mM): 145 NaCl, 10 glucose, 10 HEPES, 4 KCl, 1 CaCl2, and 1 MgCl2 (pH 7.4) at 37 °C. Cell shortening and calcium transients were simultaneously recorded by splitting the microscope light between the camera (model TM-640, Pulnix America) and photomultiplier tube (PTI, Brunswick NJ, USA) with a dichroic mirror (Chroma Tech. Corp. Rockingham, VT). Cells were viewed on a closed-circuit television monitor linked to a video edge detector (Crescent Electronics, Sandy, UT) to measure cell length (120 samples/sec). Calcium transients were measured with a DeltaRam fluorescence system and Felix software (Photon Technologies International, Birmingham, NJ). Cells were alternately excited at 340 and 380 nm and fluorescence emission at 510 nm was recorded for both wavelengths (200 samples/sec). Recordings were background corrected and emission ratios were converted to calcium concentrations with an in vitro calibration curve as in our earlier studies57,58. Cells were field-stimulated at 4 Hz with bipolar pulses delivered through platinum electrodes via a stimulus isolation unit (Model # SIU-102; Warner Instruments, Hamden, CT) controlled by pClamp 8.1 software (Molecular Devices, Sunnyvale, CA).
Langendorff-perfused heart studies
Langendorff-perfused heart studies were conduced as we have previously described33. In brief, mice were weighed and anesthetized as described above. Hearts were excised, cannulated on a Langendorff apparatus (Radnoti LLC, Monrovia, Ca, USA) and perfused at constant pressure (80 ± 0.5 mmHg; 37 °C) with the following buffer (mM): 126 NaCl, 0.9 NaH2PO4, 4 KCl, 20 NaHCO3, 0.5 MgSO4, 5.5 glucose, and 1.8 CaCl2 (95% O2, 5% CO2; pH 7.4). A fluid filled balloon was inserted into the left ventricle via the left atrium and inflated to 5–10 mmHg. Pressure was recorded with a pressure transducer and PowerLab 8/35 data acquisition system (ADInstruments, Colorado Springs, CO, USA). Data were analyzed with LabChart 7 software (ADInstruments). Left ventricular pressure was measured to quantify LVDP and the maximum rates of pressure development (+dP/dt) and decay (−dP/dt). Hearts were allowed to stabilize for 20–30 minutes before recordings were made; responses over a 10-minute period were averaged.
Myofilament studies
Myofilaments were isolated with our established techniques59. Briefly, mice were anesthetized with sodium pentobarbital (described above) and their hearts were removed. The ventricles were weighed, flash frozen in liquid nitrogen and stored at -80 °C. Tissue was homogenized in ice-cold buffer (mM): 60 KCl, 30 imidazole (pH 7.0), 2 MgCl2, 0.01 leupeptin, 0.1 PMSF, 0.2 benzamidine, and phosphatase inhibitors (P0044, Sigma-Aldrich) and centrifuged (14,000 g; 15 min; 4 °C). The pellet was re-suspended in the homogenizing buffer supplemented with 1% Triton X-100 (45 min, on ice). This solution was then centrifuged (1,100 g; 15 min; 4 °C) and the myofilament pellet was washed three times in ice-cold buffer and re-suspended in homogenizing buffer. Myofilaments were either flash frozen (for subsequent myofilament protein phosphorylation assays) or kept on ice and used immediately to assess actomyosin MgATPase activity. Myofilaments (25 µg) were incubated in ATPase buffers supplemented with increasing concentrations of free calcium (10 min; 32 °C) to quantify actomyosin MgATPase activity, as we have previously described60. Reactions were quenched with 10% trichloroacetic acid and then equal volumes of FeSO4 (0.5%) and ammonium molybdate (0.5%) in 0.5 M H2SO4 were added. The production of inorganic phosphate was measured as the absorbance at 630 nm.
Myofilament protein phosphorylation was assessed with our established techniques59. Briefly, myofilament proteins (10 µg) were separated with SDS-PAGE (12%) and fixed in 50% methanol-10% acetic acid (23 °C) overnight. ProQ Diamond staining was used to assess myofilament protein phosphorylation (Molecular Probes, Eugene, OR). Gels were imaged with a Bio-Rad ChemiDoc MP Imaging System (Bio-Rad Laboratories Ltd., Mississauga, ON) and they were analyzed with ImageJ (NIH, Bethesda, MD, USA). The protein load of each gel was determined by Coomassie staining, after the ProQ Diamond staining and imaging. Actin was selected to represent protein load as we have done previously38. To permit comparisons across gels, an equal amount of protein standard was loaded in multiple lanes of each gel. The protein standards (Bio-Rad 161-0374) at 25 and 75 kDa are visible during ProQ Diamond imaging, allowing for standardization across all gels. These standards showed equal fluorescence across all gels (<3% variation at most).
Statistics
Data are expressed as mean ± SEM unless otherwise indicated. The effects of age and sex on each outcome were compared with a 2-way ANOVA followed by Holm-Sidak post-hoc tests. When actomyosin Mg-ATPase activity was plotted as a function of calcium concentration, male and female groups were analyzed separately with a two-way repeated measures ANOVA, with age as the main factor and calcium concentration as the repeated measure and pairwise multiple post-hoc comparisons with a Tukey test. We evaluated relationships between various parameters and frailty with linear regression analysis. When both age and frailty were significantly related to a parameter under study, we used multivariable regression and calculated semi-partial correlations to assess their separate contributions. Sigmaplot software (v15.0, Systat Software Inc.) and SPSS software (v21.0) were used for all statistical analyses and Sigmaplot was used to construct graphs. P values of less than 0.05 were considered significant.
Supplementary information
Acknowledgements
The authors are grateful to Peter Nicholl for expert technical assistance. This work was supported by the Canadian Institutes of Health Research (SEH, grant numbers PGT 162462, MOP 97973); the Heart and Stroke Foundation of Canada (S.E.H., grant number G-19-0026260); the Natural Sciences and Engineering Research Council of Canada (W.G.P., RGPIN-2018-04732); and a Reynolds post-doctoral fellowship from the Department of Pharmacology (AEK).
Author contributions
S.E.H., A.E.K. and W.G.P. contributed to the conception or design of the work; S.E.H., A.E.K., E.S.B., K.M.K. A.G. and W.G.P. contributed to the acquisition, analysis, or interpretation of data for the work; S.E.H. and A.E.K. drafted the work; S.E.H., A.E.K., E.S.B., K.M.K., A.G. and W.G.P. revised it critically for important intellectual content; S.E.H., A.E.K., E.S.B., K.M.K., A.G. and W.G.P. approved the final submission and are accountable for the work.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-66903-z.
References
- 1.Mozaffarian D, et al. American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2016 Update: A Report from the American Heart Association. Circ. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 2012;8:143–164. doi: 10.1016/j.hfc.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlett SE, Rockwood K. Ageing: Develop models of frailty. Letter. Nature. 2014;512:253. doi: 10.1038/512253d. [DOI] [PubMed] [Google Scholar]
- 4.Jackson SJ, et al. Does age matter? The impact of rodent age on study outcomes. Lab. Anim. 2017;51:160–169. doi: 10.1177/0023677216653984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beery AK. Inclusion of females does not increase variability in rodent research studies. Curr. Opin. Behav. Sci. 2018;23:143–149. doi: 10.1016/j.cobeha.2018.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feridooni HA, Dibb KM, Howlett SE. How cardiomyocyte excitation, calcium release and contraction become altered with age. J. Mol. Cell. Cardiol. 2015;83:62–72. doi: 10.1016/j.yjmcc.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Kane AE, Howlett SE. Differences in cardiovascular aging in men and women. Adv. Exp. Med. Biol. 2018;1065:389–411. doi: 10.1007/978-3-319-77932-4_25. [DOI] [PubMed] [Google Scholar]
- 8.Keller KM, Howlett SE. Sex differences in the biology and pathology of the aging heart. Can. J. Cardiol. 2016;32:1065–1073. doi: 10.1016/j.cjca.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 9.Ghimire, A., Kane, A.E., Howlett, S.E. Sex differences in the physiology and pathology of the aging heart in Encyclopedia of cardiovascular research and medicine (eds. Sawyer, D. and Vasan, R) 368-376 (Elsevier, 2018).
- 10.Feridooni HA, et al. The impact of age and frailty on ventricular structure and function in C57BL/6J mice. J. Physiol. 2017;595:3721–3742. doi: 10.1113/JP274134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circ. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- 12.Vaupel JW, Manton KG, Stallard E. The impact of heterogeneity in individual frailty on the dynamics of mortality. Demography. 1979;16:439–454. [PubMed] [Google Scholar]
- 13.Rockwood K, et al. A frailty index based on deficit accumulation quantifies mortality risk in humans and in mice. Sci. Rep. 2017;7:43068. doi: 10.1038/srep43068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodríguez-Mañas L, et al. FOD-CC group (Appendix 1). Searching for an operational definition of frailty: a Delphi method based consensus statement: the frailty operative definition-consensus conference project. J. Gerontol. A. Biol. Sci. Med. Sci. 2013;68:62–67. doi: 10.1093/gerona/gls119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rockwood K, Howlett SE. Age-related deficit accumulation and the diseases of ageing. Mec. Ageing Dev. 2019;180:107–116. doi: 10.1016/j.mad.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 16.de Vries NM, Staal JB, van Ravensberg CD, Hobbelen JS, Olde Rikkert MG. Nijhuis-van der Sanden, M.W. Outcome instruments to measure frailty: a systematic review. Ageing Res. Rev. 2011;10:104–114. doi: 10.1016/j.arr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Bouillon K, et al. Measures of frailty in population-based studies: an overview. BMC Geriatr. 2013;13:64. doi: 10.1186/1471-2318-13-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Searle SD, Mitnitski A, Gahbauer EA, Gill TM, Rockwood K. A standard procedure for creating a frailty index. BMC Geriatr. 2008;8:24. doi: 10.1186/1471-2318-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parks RJ, et al. A procedure for creating a frailty index based on deficit accumulation in aging mice. J. Gerontol. A Biol. Sci. Med. Sci. 2012;67:217–227. doi: 10.1093/gerona/glr193. [DOI] [PubMed] [Google Scholar]
- 20.Whitehead JC, et al. A clinical frailty index in aging mice: comparisons with frailty index data in humans. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:621–632. doi: 10.1093/gerona/glt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yorke A, Kane AE, Hancock Friesen CL, Howlett SE, O’Blenes S. Development of a rat clinical frailty index. J. Gerontol. A Biol. Sci. Med. Sci. 2017;72:897–903. doi: 10.1093/gerona/glw339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bisset ES, Howlett SE. The biology of frailty in humans and animals: understanding frailty and promoting translation. Aging Med. 2019;2:27–34. doi: 10.1002/agm2.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien JD, Howlett SE. Simulated ischemia-induced preconditioning of isolated ventricular myocytes from young adult and aged Fischer-344 rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H768–777. doi: 10.1152/ajpheart.00432.2008. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien JD, Ferguson JH, Howlett SE. Effects of ischemia and reperfusion on isolated ventricular myocytes from young adult and aged Fischer 344 rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2174–2183. doi: 10.1152/ajpheart.00058.2008. [DOI] [PubMed] [Google Scholar]
- 25.Rueckschloss U, Villmow M, Klöckner U. NADPH oxidase-derived superoxide impairs calcium transients and contraction in aged murine ventricular myocytes. Exp. Gerontol. 2010;45:788–796. doi: 10.1016/j.exger.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Ross JL, Howlett SE. Age and ovariectomy abolish beneficial effects of female sex on rat ventricular myocytes exposed to simulated ischemia and reperfusion. PLoS One. 2012;7:e38425. doi: 10.1371/journal.pone.0038425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellor, K. M. et al. Ageing-related cardiomyocyte functional decline is sex and angiotensin II dependent. Age.36, 9630 (2014). [DOI] [PMC free article] [PubMed]
- 28.Willems L, Zatta A, Holmgren K, Ashton KJ, Headrick JP. Age-related changes in ischemic tolerance in male and female mouse hearts. J. Mol. Cell Cardiol. 2005;38:245–256. doi: 10.1016/j.yjmcc.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014;76:1071–27. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- 30.Petre RE, et al. Sex-based differences in myocardial contractile reserve. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R810–818. doi: 10.1152/ajpregu.00377.2006. [DOI] [PubMed] [Google Scholar]
- 31.McKee LA, et al. Sexually dimorphic myofilament function and cardiac troponin I phosphospecies distribution in hypertrophic cardiomyopathy mice. Arch. Biochem. Biophys. 2013;535:39–48. doi: 10.1016/j.abb.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb. Perspect. Biol. 2011;3:a004242. doi: 10.1101/cshperspect.a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghimire A, Bisset ES, Howlett SE. Ischemia and reperfusion injury following cardioplegic arrest is attenuated by age and testosterone deficiency in male but not female mice. Biol. Sex. Differ. 2019;10:42. doi: 10.1186/s13293-019-0256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wattanapermpool J. Increase in calcium responsiveness of cardiac myofilament activation in ovariectomized rats. Life Sci. 1998;63:955–964. doi: 10.1016/s0024-3205(98)00353-1. [DOI] [PubMed] [Google Scholar]
- 35.Bupha-Intr T, Oo YW, Wattanapermpool J. Increased myocardial stiffness with maintenance of length-dependent calcium activation by female sex hormones in diabetic rats. Am. J. Physiol. Heart. Circ. Physiol. 2011;300:H1661–H1668. doi: 10.1152/ajpheart.00411.2010. [DOI] [PubMed] [Google Scholar]
- 36.Fernandes RD, et al. Cardiac changes during the peri-menopausal period in a VCD-induced murine model of ovarian failure. Acta. Physiol. 2019;227:e13290. doi: 10.1111/apha.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fares E, et al. The impact of ovariectomy on calcium homeostasis and myofilament calcium sensitivity in the aging mouse heart. PLoS One. 2013;8:e74719. doi: 10.1371/journal.pone.0074719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayaz O, et al. Long-term testosterone deficiency modifies myofilament and calcium-handling proteins and promotes diastolic dysfunction in the aging mouse heart. Am. J. Physiol. Heart. Circ. Physiol. 2019;316:H768–H780. doi: 10.1152/ajpheart.00471.2018. [DOI] [PubMed] [Google Scholar]
- 39.Loong CK, Badr MA, Chase PB. Tropomyosin flexural rigidity and single Ca2+ regulatory unit dynamics: implications for cooperative regulation of cardiac muscle contraction and cardiomyocyte hypertrophy. Front. Physiol. 2012;3:80. doi: 10.3389/fphys.2012.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu X, Heeley DH, Smillie LB, Kawai M. The role of tropomyosin isoforms and phosphorylation in force generation in thin-filament reconstituted bovine cardiac muscle fibres. J. Muscle Res. Cell. Motil. 2010;31:93–109. doi: 10.1007/s10974-010-9213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hnia K, Ramspacher C, Vermot J, Laporte J. Desmin in muscle and associated diseases: beyond the structural function. Cell. Tissue Res. 2015;360:591–608. doi: 10.1007/s00441-014-2016-4. [DOI] [PubMed] [Google Scholar]
- 42.Maloyan A, et al. Biochemical and mechanical dysfunction in a mouse model of desmin-related myopathy. Circ. Res. 2009;104:1021–1028. doi: 10.1161/CIRCRESAHA.108.193516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheid LM, et al. Essential light chain S195 phosphorylation is required for cardiac adaptation under physical stress. Cardiovasc. Res. 2016;111:44–55. doi: 10.1093/cvr/cvw066. [DOI] [PubMed] [Google Scholar]
- 44.Katrukha IA, Gusev NB. Enigmas of cardiac troponin T phosphorylation. J. Mol. Cell. Cardiol. 2013;65:156–158. doi: 10.1016/j.yjmcc.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 45.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J. Biol. Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 46.Gilda JE, Gomes AV. How phosphorylated can it get? Cardiac myosin binding protein C phosphorylation in heart failure. J. Mol. Cell. Cardiol. 2013;62:108–110. doi: 10.1016/j.yjmcc.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 47.Han YS, Arroyo J, Ogut O. Human heart failure is accompanied by altered protein kinase A subunit expression and post-translational state. Arch. Biochem. Biophys. 2013;538:25–33. doi: 10.1016/j.abb.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Armouche A, et al. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J. Mol. Cell. Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Gresham KS, Stelzer JE. The contributions of cardiac myosin binding protein C and troponin I phosphorylation to β-adrenergic enhancement of in vivo cardiac function. J. Physiol. 2016;594:669–686. doi: 10.1113/JP270959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisberg A, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc. Natl. Acad. Sci. USA. 1996;93:8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moghtadaei M, et al. The impacts of age and frailty on heart rate and sinoatrial node function. J. Physiol. 2016;594:7105–7126. doi: 10.1113/JP272979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jansen HJ, et al. Atrial structure, function and arrhythmogenesis in aged and frail mice. Sci. Rep. 2017;7:44336. doi: 10.1038/srep44336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maric-Bilkan C, et al. Report of the National Heart, Lung, and Blood Institute Working Group on sex differences research in cardiovascular disease: Scientific questions and challenges. Hypertension. 2016;67:802–807. doi: 10.1161/HYPERTENSIONAHA.115.06967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gordon EH, Hubbard RE. Do sex differences in chronic disease underpin the sex-frailty paradox? Mech. Ageing Dev. 2019;179:44–50. doi: 10.1016/j.mad.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 55.Kane AE, Ayaz O, Ghimire A, Feridooni HA, Howlett SE. Implementation of the mouse frailty index. Can. J. Physiol. Pharmacol. 2017;95:1149–1155. doi: 10.1139/cjpp-2017-0025. [DOI] [PubMed] [Google Scholar]
- 56.Feridooni HA, Sun MH, Rockwood K, Howlett SE. Reliability of a frailty index based on the clinical assessment of health deficits in C57BL/6J mice. J. Gerontol. A Biol. Sci. Med. Sci. 2015;70:686–693. doi: 10.1093/gerona/glu161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shutt RH, Howlett SE. Hypothermia increases the gain of excitation-contraction coupling in guinea pig ventricular myocytes. Am. J. Physiol. Cell. Physiol. 2008;295:C692–700. doi: 10.1152/ajpcell.00287.2008. [DOI] [PubMed] [Google Scholar]
- 58.Fares E, Parks RJ, Macdonald JK, Egar JM, Howlett SE. Ovariectomy enhances SR Ca2+ release and increases Ca2+ spark amplitudes in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 2012;52:32–42. doi: 10.1016/j.yjmcc.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 59.Feridooni HA, MacDonald JK, Ghimire A, Pyle WG, Howlett SE. Acute exposure to progesterone attenuates cardiac contraction by modifying myofilament calcium sensitivity in the female mouse heart. Am. J. Physiol. Heart. Circ. Physiol. 2017;312:H46–H59. doi: 10.1152/ajpheart.00073.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang FH, Pyle WG. Reduced cardiac CapZ protein protects hearts against acute ischemia-reperfusion injury and enhances preconditioning. J. Mol. Cell. Cardiol. 2012;52:761–772. doi: 10.1016/j.yjmcc.2011.11.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.