

Abstract
Microbial lanthipeptides are formed by a two-step enzymatic introduction of (methyl)lanthionine rings. A dehydratase catalyzes the dehydration of serine and threonine residues, yielding dehydroalanine and dehydrobutyrine, respectively. Cyclase-catalyzed coupling of the formed dehydroresidues to cysteines forms (methyl)lanthionine rings in a peptide. Lanthipeptide biosynthetic systems allow discovery of target-specific, lanthionine-stabilized therapeutic peptides. However, the substrate specificity of existing modification enzymes impose limitations on installing lanthionines in non-natural substrates. The goal of the present study was to obtain a lanthipeptide dehydratase with the capacity to dehydrate substrates that are unsuitable for the nisin dehydratase NisB. We report high-throughput screening for tailored specificity of intracellular, genetically encoded NisB dehydratases. The principle is based on the screening of bacterially displayed lanthionine-constrained streptavidin ligands, which have a much higher affinity for streptavidin than linear ligands. The designed NisC-cyclizable high-affinity ligands can be formed via mutant NisB-catalyzed dehydration but less effectively via wild-type NisB activity. In Lactococcus lactis, a cell surface display precursor was designed comprising DSHPQFC. The Asp residue preceding the serine in this sequence disfavors its dehydration by wild-type NisB. The cell surface display vector was coexpressed with a mutant NisB library and NisTC. Subsequently, mutant NisB-containing bacteria that display cyclized strep ligands on the cell surface were selected via panning rounds with streptavidin-coupled magnetic beads. In this way, a NisB variant with a tailored capacity of dehydration was obtained, which was further evaluated with respect to its capacity to dehydrate nisin mutants. These results demonstrate a powerful method for selecting lanthipeptide modification enzymes with adapted substrate specificity.
Keywords: Lactococcus lactis, NisB, dehydratase, streptavidin, high-throughput screening, bacterial display
Lanthipeptides are (methyl)lanthionine ring-containing ribosomally synthesized and post-translationally modified peptides (RiPPs).1 Many natural lanthipeptides show potent antimicrobial activity against human pathogens and even against antibiotic-resistant human pathogens. Importantly, the lantibiotics duramycin and NVB-302 and the lanthipeptide MOR107 have already been tested in the clinic. The ribosomal synthesis and enzymatic modifications of lanthipeptides provide excellent opportunities for the discovery of lanthionine-stabilized and target-specific therapeutic peptides.2−4
The nisin biosynthetic machinery has been well-studied and is widely used. This system allows us to engineer and heterologously produce novel lanthipeptides. It comprises the NisB dehydratase, which is responsible for serine/threonine dehydration in the core peptide, the NisC cyclase, which couples the dehydroresidues to cysteines yielding (methyl)lanthionine rings, and the transporter NisT, which secretes modified peptides.5 The peptidase NisP is responsible for cleaving off the leader peptide, thus yielding mature lanthipeptides.5 NisB is a fascinating enzyme whose mechanism is glutamyl-tRNAGlu-dependent. The crystal structure of NisB, in complex with its natural substrate peptide NisA, reveals two separate domains, one domain that catalyzes the Ser/Thr glutamylation and another domain that is responsible for glutamate elimination.6 Its Ser/Thr dehydration activity depends in part on the amino acids that directly flank the dehydratable Ser/Thr.7,8 In that light, some substrates appear unsuitable for wild-type NisB-mediated dehydration.7,8 As a consequence, the substrate specificity of wild-type dehydratase NisB can be a limitation for engineering novel lanthipeptides.
Cell surface display is a powerful method for high-throughput screening of genetically encoded peptides with desired properties. Surface display on phage, yeast, and Gram-positive bacteria has been successfully applied in high-throughput screening.9−11 Also lanthipeptide display systems for phage, yeast, and Gram-positive bacteria have been developed and applied.12 A lanthipeptide cell surface display system has been devised in Lactococcus lactis NZ9000.13 This bacterial display system contains a plasmid-encoded linear lanthipeptide precursor fused to the N-terminus of the L. lactis surface protease PrtP C-terminal domain and a second plasmid that encodes the NisBTC enzymes.13
Many cyclic His-Pro-Gln (HPQ) motif-containing peptides showed up to 3 orders of magnitude higher affinities to streptavidin than linear HPQ motif-containing peptides.14,15 In this study, we exploited this high affinity of cyclic streptavidin ligands compared to linear unmodified streptavidin ligands. We employed the NisBC enzymes to introduce a thioether cross-link into a designed strep ligand (SHPQFC), which showed higher affinity for streptavidin than the linear strep ligand. Subsequently, a strep ligand was designed where the Ser to be dehydrated residue is preceded by an Asp residue (DSHPQFC), which is an unsuitable substrate for NisB. By lack of dehydration, this peptide would never be subject to NisC-catalyzed or spontaneous cyclization, thus having lower affinity to streptavidin than the cyclized variants. For high-throughput screening of tailored NisB variants from a genetically encoded NisB library, the unsuitable DSHPQFC substrate was genetically fused to the display scaffold13 and coexpressed with a plasmid encoding NisCT and a mutant NisB library. By use of streptavidin-coupled magnetic beads, cyclized strep ligand displaying bacteria were selected aiming at mutant NisB-catalyzed dehydration of DSHPQFC. The results demonstrate that selection of mutant modification enzymes from genetically encoded libraries can be based on cell surface display of mutant-enzyme-modified products.
Results
Lanthionine-Cyclized HPQF-Containing Peptides Have Enhanced Capacity to Bind Streptavidin Compared to Linear HPQF Peptides
Previous studies demonstrated that thioether cross-linked HPQ-containing cyclic peptides show up to 3 orders of magnitude higher streptavidin affinities than linear peptides.14,15 In this study, a cyclic HPQF-containing strep ligand fused to the C-terminus of nisin fragments was used. To form the cyclic HPQF-containing strep ligand by lanthipeptide synthetases, a Ser and a Cys were added at the N- and C-terminus of HPQF, respectively (SHPQFC). The N-terminus of the designed SHPQFC strep ligand was engineered at the C-terminus of nisin, nisin(1–22), or nisin(1–12) (Supplemental Figure S1). Asn-Lys or Lys was engineered at the C-terminus of the designed SHPQFC strep ligand, since these residues are favorable for the NisC-catalyzed cyclization.8 Five peptides (CS1, CS2, CS3, CS4, and CS5) were designed by following this setup (Supplemental Figure S1). L. lactis NZ9000 with pTLR-BTC was transformed with plasmids encoding the designed peptides, respectively. Following the induction and purification, the mass of the produced peptides was checked by MALDI-TOF MS. Of the designed five peptides, only the construct CS5 was fully dehydrated (Supplemental Figure S2). The formation of the potentially three NisC-formed thioether cross-links, two in nisin(1–12) and one in the designed streptavidin ligand of CS5, was investigated using 1-cyano-4-dimethylaminopyridinium tetrafluoroborate (CDAP), a compound that reacts with unmodified cysteines in peptides. CDAP reaction to cysteine results in an increase of 25 Da in the peptide’s molecular weight.4,16 CDAP treatment was executed under reducing conditions followed by trypsin cleavage and LC-MS/MS analysis. Very little 25 Da adduct was observed for the CS5 main product (Figure 1a), indicating that no unmodified cysteines were present. This implied that most thioether cross-links in CS5 were formed, including the intended thioether cross-link for the strep ligand (Figure 1a). Subsequently, a trypsin-mediated cleavage demonstrated that the cyclic strep ligand was correctly formed (Supplemental Figure S3). Furthermore, LC-MS/MS for CS5(13–20) confirmed the presence of the designed cyclic strep ligand in CS5 (Figure 1c). These results proved the CS5 structure (Figure 1a), a lanthipeptide composed of N-terminal nisin followed by a cyclic strep ligand. Subsequently, CS5 was expressed in the presence of only NisT for production of linear strep ligand. After purification, the streptavidin binding capacity of cyclic and linear CS5 peptides was investigated by using a streptavidin column. After elution, the fractions were analyzed by Tricine-SDS gel (Figure 1b, lanes 3 and 4). The cyclic strep ligand containing CS5 bound to the streptavidin column, since a clear band appeared from the elution fraction of cyclic strep ligand containing CS5 (Figure 1b, lane 3). However, no band was observed from the elution fraction of linear HPQF-containing CS5 (Figure 1b, lane 4), indicating that under the used conditions linear HPQF-containing CS5 had no or too low binding affinity to streptavidin. These data confirm that the cyclic strep ligand containing CS5 peptide has significantly higher affinity to streptavidin than the linear one.
Figure 1.

(a) MALDI-TOF MS data of CS5 before (blue) and after (red) treatment with CDAP and hypothetical structure of CS5. (b) SDS-Tricine gel of CS5 coexpressed with NisBTC or NisT before (&) and after (#) streptavidin column purification. (c) LC-MS/MS data of CS5(13–20). The observed fragments demonstrate that the cyclic strep ligand was formed.
Cyclic HPQF-Containing Peptide Displayed on the Cell Surface by an Improved Display Scaffold
A previous study demonstrated a PrtP protease-based cell surface display system in L. lactis.13 To display the cyclic HPQF strep ligand of CS5 on the cell surface of L. lactis, the CS5 was genetically fused to an extension-improved L. lactis display scaffold described in Materials and Methods (peptides are fused to a cell wall bound part of PrtP protease via an LPXTG motif). A human rhinovirus (HRV)-3C protease cleavage site (sequence LEVLFQGP) was introduced between CS5 and the improved scaffold (stalk and cell wall spacer, LPxTG domain and TMS domain) (BD-CS5, Supplemental Figure S1). To investigate whether the cyclic HPQF strep ligand of CS5 was also formed within the L. lactis displayed BD-CS5, a streptavidin column binding experiment was performed. Briefly, BD-CS5 was expressed in the presence of either NisBTC (modified) or NisT (unmodified). After purification of expressed modified and unmodified BD-CS5 proteins, their streptavidin binding capacity was investigated by using a streptavidin column. After elution, the fractions were investigated by Tricine-SDS gel (Figure 2a, lanes 3 and 4). After the streptavidin column, the fraction corresponding to BD-CS5 coexpressed with the NisBTC enzymes showed a band on the tricine gel (Figure 2a, lane 3). In contrast, the fraction corresponding to BD-CS5 coexpressed with NisT showed no visible band (Figure 2a, lane 4). These results indicated that the thioether cross-link between Ser and Cys in SHPQFC had been installed by NisBC (Figure 2b). Subsequently, a whole cell ELISA assay was performed using horseradish peroxidase (HRP)-conjugated streptavidin to investigate whether the cyclic HPQF strep ligand was displayed on the surface of L. lactis. Figure 2c clearly shows cell surface display for induced L. lactis with plasmids pTLR-BTC and pBD-CS5, whereas only background level was observed in uninduced and NisBC lacking L. lactis. Together, these results clearly demonstrate that cyclic HPQF strep ligand was successfully formed and displayed on the surface of L. lactis.
Figure 2.

(a) SDS-Tricine gel of BD-CS5 coexpressed with NisBTC or NisT before (&) and after (#) streptavidin column purification. (b) Structure of BD-CS5 with a cyclic strep ligand. (c) Whole-cell ELISA on L. lactis NZ9000 cells demonstrates that cyclic HPQF is displayed on the cell surface; each column represents the mean ± SD of three independent experiments; the statistical significance of differences was evaluated by Pearson r2, ns, p > 0.05; *p < 0.05 vs L. lactis NZ9000 cells (without plasmid).
Directly Serine-Flanking Amino Acids Affect NisB-Catalyzed Dehydration of SHPQFC
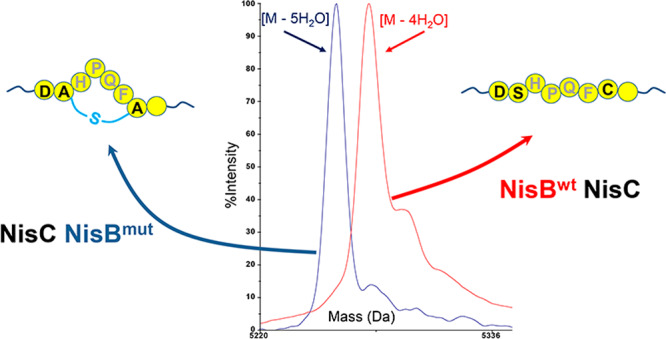
Rink et al.8 demonstrated that some amino acid residues directly flanking Ser or Thr can inhibit or even abolish the dehydration of these Ser and Thr residues by NisB.8 A subsequent study demonstrated that essentially all hydrophobic flanking amino acids favor dehydration of Ser and Thr, whereas hydrophilic amino acids, Asp, Glu, and Arg, and also Gly disfavor hydration.7 Based on these studies,7,8 three NisB-unfavorable (Asp, Glu, Arg) and four favorable (Ala, His, Met, Trp) amino acid codons were genetically inserted in front of SHPQFC of CS5 (CS5D, CS5E, CS5H, CS5M, CS5R, CS5W, and CS5A, Supplemental Figure S1). Plasmids encoding the designed peptides were transformed into L. lactis NZ9000 with pTLR-BTC. After the induction and purification, the mass of the produced peptides was checked by MALDI-TOF MS. Insertion of hydrophobic amino acids, Ala, Met, or Trp, or insertion of His resulting in the sequences ASHPQFC, MSHPQFC, WSHPQFC, or HSHPQFC allowed full dehydration (Figure 3a, Supplemental Figure S4). On the other hand, NisB-unfavorable Glu, Asp, and Arg residues reduced the extent of dehydration (Figure 3a, Supplemental Figure S4). Moreover, insertion of a NisB-unfavorable Asp residue before SHPQFC completely abolished the dehydration of the resulting DSHPQFC (CS5D) (Figure 3a,b, Supplemental Figure S4). Therefore, it would be interesting to find a NisB variant that can fully dehydrate CS5D. Hence, CS5D was selected for further studies (Figure 3b). CS5D was fused to the display scaffold (BD-CS5D, Supplemental Figure S1) and coexpressed with NisBTC enzymes. After purification, BD-CS5D was applied to a streptavidin column for testing its binding capability as described above. BD-CS5D, which contained DSHPQFC, showed no streptavidin column binding capability under the conditions used (data not shown), indicating that the Asp residue also blocked the dehydration of Ser in DSHPQFC within BD-CS5D. After removal of the his6 tag of pBD-CS5D, a pBDD vector (Figure 3c) was constructed for selecting NisB variants that can dehydrate Ser when preceded by an Asp residue in a DSHPQFC sequence.
Figure 3.

(a) Dehydration of the designed peptides. Green, full dehydration; red, only four dehydrations. (b) Hypothetical structure of CS5D, which has no cyclic HPQF. (c) BDD construct for selecting NisB variants that can dehydrate Ser when preceded by an Asp residue in a DSHPQFC sequence.
Selected NisBS88P/D234N Dehydrates DSHPQFC
High-throughput selection is a robust approach to improve properties of enzymes or proteins. To find NisB enzyme variants with the ability to dehydrate Ser preceded by Asp in the strep ligand context used here, a full NisB mutant library was constructed by error-prone PCR, which involved about 100 000 NisB variants according to colony counting and gene sequencing. After transformation of the NisB library into a pBDD plasmid-containing (Supplemental Figure S1) L. lactis NZ9000 strain, streptavidin-coupled magnetic beads were used in panning rounds to select cyclic strep ligand displaying L. lactis (Figure 4). After three panning rounds, the cells were plated on GM17 agar with antibiotics. After overnight growth, 20 colonies were randomly picked, followed by sequencing of nisB. Strikingly, the NisBS88P/D234N appeared 18 times out of 20 randomly picked colonies (NisBN292K and NisBT115I appeared in the remaining two colonies). Subsequently, the dehydration capability of the three obtained NisB variants was evaluated by dehydration of CS5D and CS5A. The MALDI-TOF MS showed that fully dehydrated main products of CS5A were obtained in all three NisB variant containing strains, which means that the NisB variants obtained still kept the function of NisBwt (Figure 5b). NisBN292K and NisBT115I showed low-efficiency dehydration of Ser preceded by Asp (CS5D) (data not shown). In contrast, the NisBS88P/D234N variant showed significant improvement in the dehydration of Ser preceded by Asp (CS5D) and produced a fully dehydrated main product (Figure 5a). To investigate whether this improvement also appeared in the displayed proteins, a HRV-3C protease was used to release the N-terminal part of the displayed proteins. NisBS88P/D234N almost completely dehydrated the Ser of SHPQFC preceded by either NisB-favorable Ala or NisB-unfavorable Asp (Figure 5c,d). These results demonstrate that a NisB enzyme with tailored substrate specificity was discovered by selection of a bacterially displayed cyclic HPQF sequence.
Figure 4.

Schematic diagram of selection mutant NisB with improved dehydration capacity. Cyclic strep ligand has higher affinity to streptavidin than linear strep ligand, allowing the use of streptavidin-coupled magnetic beads to fish out the bacteria with NisBmut that can dehydrate Ser preceded by Asp.
Figure 5.

MALDI-TOF MS data of peptides modified by NisBwt (blue) or NisBS88P/D234N (green). (a) CS5D; (b) CS5A; (c) N-terminal part of BDA, which was released by HRV-3C protease from cell surface; (d) N-terminal part of BDD, which was released by HRV-3C protease from cell surface; (e) nisinT2D; (f) nisinT2E; (g) nisin(1–22)K12D; (h) nisin(1–22)K12E; (i) nisin(1–22)G14D; (m) nisin(1–22)G14E.
NisBS88P/D234N Improves the Dehydration of NisBwt Unfavorable Substrates
In order to investigate the dehydration capacity of the selected NisBS88P/D234N, six NisBwt unfavorable substrates were designed and coexpressed with NisBS88P/D234N and NisTC. MALDI-TOF MS results showed that NisBS88P/D234N improved the dehydration of all six designed nisin variants with respect to NisBwt (Figure 5e,f,g,h,i,m, Table 1). Moreover, NisBS88P/D234N showed capacity to dehydrate nisin comparable to NisBwt (Table 1, Supplemental Figure S5a,b). Considering that NisBS88P/D234N is an enzyme with two mutations, single mutant NisBS88P and NisBD234N were constructed and used to modify both NisBwt favorable and unfavorable substrates. Both NisBS88P and NisBD234N improved the dehydration of nisinT2E, nisin(1–22)K12E, nisin(1–22)G14D, and nisin(1–22)G14E with respect to NisBwt (Table 1, Supplemental Figures S6g,h and S7c,d,e,f,g,h). In addition, both NisBS88P and NisBD234N showed dehydration capacity comparable to NisBwt for nisin and CS5A (Table 1, Supplemental Figure S5a,c,d). However, both NisBS88P and NisBD234N showed less dehydration than NisBS88P/D234N for CS5D, nisin(1–22)K12E, and nisinT2D (Figure 5a,e, Table 1, Supplemental Figures S6c,d,e,f and S7a,b). These results demonstrate that, in contrast to NisBwt, NisBS88P/D234N shows significant dehydration of Ser/Thr that are flanked by Asp/Glu. Furthermore, NisBS88P/D234N has better dehydration capacity than the single mutant enzymes NisBS88P or NisBD234N.
Table 1. Dehydration of Designed Peptides by Different NisB Mutationsa.
| observed
mass (Da, main product)/dehydrations |
|||||
|---|---|---|---|---|---|
| peptide | predicted mass (Da)/dehydrations | NisBwt | NisBmut(S88P/D234N) | NisBmut(S88P) | NisBmut(D234N) |
| nisin | 5688/8 | 5689/8 | 5687/8 | 5687/8 | 5687/8 |
| CS5D | 5477/5 | 5496/4 | 5477/5 | 5495/4 | 5495/4 |
| CS5A | 5433/5 | 5434/5 | 5434/5 | 5432/5 | 5433/5 |
| nisinT2D | 5720/7 | 5739/6 | 5723/7 | 5736/6 | 5737/6 |
| nisinT2E | 5734/7 | 5747/6 | 5732/7 | 5734/7 | 5732/7 |
| nisin(1–22)K12D | 4460/5 | 4479/4 | 4479/4 | 4477/4 | 4476/4 |
| nisin(1–22)K12E | 4474/5 | 4473/5 | 4473/5 | 4472/5 | 4471/5 |
| nisin(1–22)G14D | 4531/5 | 4548/4 | 4532/5 | 4531/5 | 4530/5 |
| nisin(1–22)G14E | 4545/5 | 4559/4 | 4542/5 | 4543/5 | 4543/5 |
NisinT2D, the second amino acid of nisin was changed from Thr to Asp; nisinT2E, the second amino acid of nisin was changed from Thr to Glu; nisin(1–22)K12D, the 12th amino acid of nisin was changed from Lys to Asp; nisin(1–22)K12E, the 12th amino acid of nisin was changed from Lys to Glu; nisin(1–22)G14D, the 14th amino acid of nisin was changed from Gly to Asp; nisin(1–22)G14E, the 14th amino acid of nisin was changed from Gly to Glu.
The major products with less dehydration than predicted are in italic. The major products with predicted dehydration are in bold.
Discussion
Although L. lactis cells containing NisB have been frequently used for the discovery of lanthionine-stabilized and target-specific therapeutic peptides,13,17−20 unsuitable NisB substrates constitute a limitation in engineering lanthipeptides.8,21,22 The overall structure of the dehydratase NisB was revealed by crystal structure analysis in 2015,6 and some residues of the dehydratase NisB have been proven to be important for the dehydration activity.6,23,24 However, to the best of our knowledge, up to now no LanB variant has been generated with modulated or tailored substrate specificity. Here, we show a valuable approach for de novo selection of tailored intracellular NisB dehydratase by screening of a cell surface displayed, high-affinity strep ligand that was intracellularly modified by mutant NisB.
Directed evolution has arguably become the most effective strategy for improving or altering the activity of enzymes,25−28 but the success of this strategy is dependent on the library size and the efficiency of the selection method. Focused mutagenesis and random mutagenesis are commonly used approaches for gene diversification.25 A focused mutagenesis approach requires phylogenetic information on enzymes, and even if the essential data are available, deducing the determinants of enzyme function at the amino acid level can be challenging.25 Moreover, the small library size of focused mutagenesis may be responsible for the low success rate for selecting enzymes with desired activity. Therefore, in this study, a random mutagenesis approach was used to construct the NisB library, and L. lactis containing this library was subjected to cell surface displayed product screening.
The traditional screening approaches, which are based on agar plate or microplate assays, would require robotics to handle such large libraries. In the absence of robust screening methods, it seems difficult or even impossible to identify enzyme variants with desired properties.29−31 If screening approaches are not amenable to automation, only about 1000 to 2000 of the mutants can be screened for desired properties.26,29 Cell surface display combined with target-displaying magnetic beads for panning rounds has been widely used in antibody32,33 and drug34,35 discovery. In this study, a lanthipeptide L. lactis display system was applied.13 A NisB dehydratase mutation with the capacity to dehydrate substrates that could not be dehydrated by wild-type NisB was successfully discovered (Table 1, Figure 5). These results raise the hope that the here-used principle can be widely applied.
In previous cell surface display studies, protein libraries, which were used for screening protein variants with desired properties, were usually fused to a display scaffold and thereafter displayed on the cell surface.15,36−38 Subsequently, the selections were based on higher target affinity.15,36−38 In contrast, in our present study, the enzyme library itself was not displayed on the cell surface, but the corresponding aimed-for product was (Figure 2c and 4). This constitutes a powerful strategy for high-throughput selecting of intracellular enzyme variants with desired properties.
NisB is a glutamyl-tRNAGlu-dependent lantibiotic dehydratase, and the function of each domain has been revealed in previous studies6 (Figure 6a). The dehydration process of NisB involves glutamyl-tRNAGlu to activate Ser or Thr residues.6 Therefore, glutamyl-tRNAGlu plays a vital role in the NisB-catalyzed dehydration of Ser and Thr residues. In the present study, the NisBS88P/D234N mutation was discovered by the herein developed method. Both of the mutation points of NisBS88P/D234N are located in the glutamyl-tRNAGlu binding site of NisB (Figure 6b). Also the single mutation points of NisBN292K and NisBT115I are located in the glutamyl-tRNAGlu binding site of NisB (Figures S8b and S9b). For further characterization, an amino acid sequence alignment of LanB protein sequences was performed.39 The results show that all four mutation positions are nonconserved positions (Figure S10). Moreover, the mutated amino acid residues of NisBS88P/D234N and NisBN292K increased the probability value of the corresponding positions. These results suggest that the improved substrate tolerance of NisBS88P/D234N is caused by the mutations in NisB that promote a slightly different and more productive positioning of the glutamyl-tRNAGlu, allowing a more efficient transfer of the glutamyl moiety. In addition, a silightly different positioning of the peptide substrate in the NisB mutant may also led to a more efficient glutamyl-tRNAGlu-catalyzed activation of the respective amino acid residues for dehydration. The findings thus open the way for exciting follow-up studies aimed at understanding the exact underlying mechanistic aspects.
Figure 6.

(a) Overall structure of the NisB dehydratase and the functions of each domain.6 (b) Glutamyl-tRNAGlu binding site. Amino acids are shown in green that are known to be important for glutamyl-tRNAGlu binding; mutation points of NisBS88P/D234N are shown in red.
In conclusion, our results demonstrate that a tailored NisB dehydratase was selected from an intrabacterial mutant NisB library by selecting a bacterial cell surface displayed, high-affinity NisB-modified strep ligand in panning rounds with streptavidin-coupled magnetic beads. These results open a new window for high-throughput selection of lanthipeptide synthetases with modulated capacity to modify specific substrate peptides.
Materials and Methods
Microbial Strains Used and Growth Conditions
Strains and plasmids used in this study are listed in Supplemental Table S1. E. coli XL-10 Gold ultracompetent cells were used for construction of the NisB library. E. coli stains were grown in LB medium or LB medium solidified with 1% (w/v) agar at 37 °C. For plasmid selection, 250 μg/mL erythromycin (Sigma) was added.
L. lactis NZ9000 was used for plasmid construction, plasmid maintenance, and protein expression. L. lactis was grown at 30 °C in M17 broth (Oxoid) or M17 broth solidified with 1% (w/v) agar, containing 0.5% (w/v) glucose (GM17) when necessary, supplemented with chloramphenicol (5 μg/mL) or erythromycin (5 μg/mL) for plasmid selection. For protein expression, stationary-phase cultures were inoculated (20-fold diluted) on GM17 broth and induced with nisin (5 ng/mL) at an optical density at 600 nm (OD600) of about 0.4.
Molecular Biology Techniques
Lists of oligonucleotide primers used for PCR, cloning, and sequencing in this study are provided in Supplemental Tables S2 and S3, and all the oligonucleotide primers were purchased from Biolegio B.V. (Nijmegen, The Netherlands). Constructs coding for mutants of peptides or display proteins were made by amplifying template plasmid using a phosphorylated downstream sense (or upstream antisense) primer and an upstream antisense (or downstream sense) primer with a peptide-encoding tail. The DNA amplification was carried out using phusion DNA polymerase (Thermo Fisher Scientific, Waltham, MA). Self-ligation of the resulting plasmid encoding linear leader peptide was carried out with T4 DNA ligase (Thermo Fisher Scientific, Waltham, MA). The electrotransformation of L. lactis was carried out as previously described using a Bio-Rad gene pulser (Bio-Rad, Richmond, CA).40 Correctness of all genetic constructs was verified by sequencing. The peptide mutations were verified by sequencing using the forward primer PS1 or the reverse primer PS2, and the NisB variants were verified by sequencing using primers PS3–PS8.
Plasmid Cloning for Lanthipeptide Surface Display
PrtP protease C-terminal domain (1586–1962 aa), here designated stalk and anchor, was amplified from L. lactis SK11 cells using primers Pbd1 and Pbd2. A DNA fragment encoding the NisA promoter, NisA leader peptide, and CS5 core peptide was amplified from the pCS5 plasmid with primers Pbd3 and Pbd4. Part of the structure gene of pCS5 encoding an NdeI cleavage site was amplified from pCS5 with primers Pbd5 and Pbd6. The different amplification products were spin column-purified, and joined by PCR fusion. The full-length amplification product was amplified using oligomers Pbd3 and Pbd6. After spin column purification, the full-length amplification product, and plasmid pCS5 were digested with BglII and NdeI. After collection of the digested fragments, T4 ligase was used to ligate the two fragments and thereafter electroporated to L. lactis NZ9000. The resulting plasmid was designated pBD-CS5 and encoded the NisA promoter, NisA leader peptide, pCS5 core peptide, stalk, and cell wall anchor domain.
NisB Library Synthesis
To construct a NisB library, an error-prone PCR was performed by using the GeneMorph II Random Mutagenesis Kit (Agilent). Plasmid backbone of pTLR-BTC41 was amplified by using high fidelity PCR with primers Ptlr1 and Ptlr2. The nisB gene was amplified by error-prone PCR with primers Ptlr3 and Ptlr4. The error-prone PCR of the nisB gene was performed using 50, 250, and 750 ng of template to obtain a low, medium, and high rate of mutation. In order to remove the template plasmids, a DpnI (FastDigest, Thermo) digestion was performed. After that, the error-prone PCR products were mixed at the same concentration (50 ng) that was used as template for a second generation of PCR amplification by using the enzyme pfu×7.42 For the ligation, various concentrations of insert ranging from 1 to 6 with respect to the backbone (200 ng) were performed. After dialysis, the ligation mixes were transformed into E. coli XL-10 Gold chemo-competent cells. After growth overnight at 37 °C, the efficiency of the transformation was calculated. Cells were harvested from the plates, and NisB library plasmids were isolated by using the NucleoSpin Plasmid EasyPure kit (Macherey-Nagel, Düren, Germany). Gene sequencing verified that the NisB library was obtained as expected.
Expression and Purification of His6-Tagged Peptides and Proteins
L. lactis NZ9000 cells containing pTLR-BTC for expression of the NisBTC enzymes were electroporated with His6-protein plasmids (50 ng), plated on M17 agar plates containing 0.5% (w/v) glucose supplemented with chloramphenicol (5 μg/mL) and erythromycin (5 μg/mL), and grown at 30 °C for 20–24 h. A single colony was used to inoculate 50 mL of GM17 supplemented with chloramphenicol (5 μg/mL) and erythromycin (5 μg/mL) (GM17EmCm), and the culture was grown for 15–18 h at 30 °C. After that, the culture was used to inoculate 1 L (20-fold dilution) of GM17EmCm. Cultures were grown at 30 °C to an OD600 of 0.4. Protein expression was induced by the addition of nisin to a final concentration of 5 ng/mL, and cultures were grown overnight at 30 °C.
Purification of Secreted Peptides
After centrifugation at 15 000 × g for 15 min, supernatants were collected. The pH of supernatants was adjusted to 7.4, and then supernatants were filtered through a 0.45 μm membrane. Supernatants were applied to a HisTrap Excel column (GE Healthcare) equilibrated with 50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole, pH 8.0. The flow-through was discarded, and the column was subsequently washed with 16 column volumes (CV) of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). The peptide was eluted with 15 CV of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). The eluted peptide was then applied to a SIGMA-Aldrich C18 silica gel spherically equilibrated first with 8 CV of MeCN containing 0.1% trifluoroacetic acid followed by 8 CV of 5% aq. MeCN containing 0.1% trifluoroacetic acid. The column was washed with 8 CV of 5% aq. MeCN containing 0.1% trifluoroacetic acid to remove the salts. Peptide was eluted from the column using up to 8 CV of 50% aq. MeCN containing 0.1% trifluoroacetic acid. Fractions containing the eluted peptide were freeze-dried, and the peptide was subsequently dissolved in PBS containing an appropriate amount of NisP and incubated at 37 °C for 2 h to cleave off the leader peptide. The insoluble material was removed by filtration through a 0.2 μm filter, and the supernatant was purified on an Agilent 1260 Infinity HPLC system with a Phenomenex Aeris C18 column (250 mm × 4.6 mm, 3.6 μm particle size, 100 Å pore size). Acetonitrile was used as the mobile phase, and a gradient of 20–30% aq. MeCN over 35 min at 1 mL/min was used for separation. Peptides eluted at 27–29% MeCN.
Purification of Displayed Bacterial Proteins
After centrifugation at 15 000 × g for 15 min, cells were collected. After treatment with lysis buffer (50 mM Tris-HCl, 2 mM EDTA, 100 mM NaCl, 0.5% Triton X-100, pH 8.5) containing 10 mg/mL lysozyme at 37 °C for 1 h, cell homogenates were sonicated for 12 min in total. The insoluble material was subsequently removed by centrifugation at 10 000 × g for 20 min, and supernatants were filtered through a 0.45 μm membrane. The supernatants were applied to a HisTrap Fast Flow column (GE Healthcare) equilibrated with 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0. The flow-through was discarded, and the column was subsequently washed with 16 CV of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). The proteins were eluted with 15 CV of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). After filtration through a 0.2 μm membrane, the eluted proteins were purified on an Agilent 1260 Infinity HPLC system with a Phenomenex Aeris C18 column (250 mm × 4.6 mm, 3.6 μm particle size, 100 Å pore size). Acetonitrile was used as the mobile phase, and a gradient of 20–70% aq. MeCN over 60 min at 1 mL/min was used for separation. Proteins were eluted at 59–61% MeCN.
Whole-Cell ELISA for Detection of Surface-Displayed Cyclic HPQF
L. lactis NZ9000 cells containing plasmids pBD-CS5 and pTLR-BTC/pTLR-T and L. lactis NZ9000 cells were grown with or without nisin (5 ng/mL) for induction of modification enzymes and protein expression. After production, cells were collected by centrifugation, washed three times with MES buffer (0.1 M, pH 7.4) plus 20 mM Cacl2 (MES-Ca), and then resuspended to an OD600 of 20. Aliquots of OD600 = 3.0 from these cell suspensions were incubated with a 4000-fold diluted horseradish peroxidase (HRP)-conjugated streptavidin solution (Thermo Fisher Scientific, Waltham, MA) in a final volume of 1 mL of MES-Ca plus 0.2% Tween 20 and 1% BSA at room temperature for 1 h under rotation. After three washes with MES-Ca-T-BSA, the displayed cyclic HPQF was visualized by the addition of 1 mL of 1-Step ABTS horseradish peroxidase substrate solution (Thermo Fisher Scientific, Waltham, MA). The absorbance was determined at 410 nm after a suitable time period, which is a measure for the displayed cyclic HPQF.
Mass Spectrometry
One microliter of sample was spotted and dried on the target. Subsequently, 0.7 μL of matrix solution (5 mg/mL α-cyano-4-hydroxycinnamic acid from Sigma-Aldrich dissolved in 50% acetonitrile containing 0.1% trifluoroacetic acid) was spotted on top of the sample. Matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) mass spectrometer analysis was performed using a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems) in the linear-positive mode.
Evaluation of (Methyl)lanthionine Formation
After the freeze-dried samples were dissolved in 18 μL of 0.5 M HCL (pH = 3), the samples were treated with 2 μL of 100 mg/mL tris[2-carboxyethyl]phosphine in 0.5 M HCL (pH = 3) for 30 min at room temperature. Subsequently, 4 μL of 100 mg/mL 1-cyano-4-dimethylaminopyridinium tetrafluoroborate (CDAP) in 0.5 M HCL (pH = 3) was added to the samples. After incubation at room temperature for 2 h, the samples were desalted with a C-18 ZipTip (Millipore) and analyzed by MALDI-TOF MS.43
LC-MS/MS
To gain insight into the lanthionine bridging pattern, we performed MS/MS. LC-MS was performed using a Q-Exactive mass spectrometer fitted with an Ultimate 3000 UPLC, an ACQUITY BEH C18 column (2.1 mm × 50 mm, 1.7 μm particle size, 200 Å; Waters), a HESI ion source, and an Orbitrap detector. A gradient of 5–90% MeCN with 0.1% formic acid (v/v) at a flow rate of 0.35 mL/min over 60 min was used. MS/MS was performed in a separate run in PRM mode selecting the doubly and triply charged ion of the compound of interest.
Library Screening by Streptavidin-Coupled Magnetic Bead Assisted Cell Sorting
An aliquot of the NisB library transformed cells (∼5 × 1010 cells) was inoculated in 50 mL of GM17EmCm. The culture was grown at 30 °C until an OD600 of about 0.4. At this point, protein and enzyme production was induced by adding nisin at a final concentration of 5 ng/mL. Incubation was continued overnight. Library-containing cells were collected by centrifugation, washed three times with 0.1 M MES-Ca, and resuspended in 2 mL of MES-Ca-T-BSA. To reduce the number of nonspecifically binding peptides from the initial library, a 1 mL of library cell suspension was incubated for 30 min at room temperature with 200 μL of biotin-saturated streptavidin-coupled magnetic beads (Dynabeads and MyOne Streptavidin T1; Invitrogen). The unbound cells in the supernatant were collected and resuspended in 1 mL of MES-Ca-T-BSA. For the first round of selection, 200 μL of streptavidin-coupled magnetic beads was added and incubated under rotation at room temperature for 1 h. Streptavidin-binding cells were collected by magnetic-bead assisted cell sorting (MACS), washed 3 times with MES-Ca-T-BSA, resuspended in 50 mL of GM17EmCm, grown overnight at 30 °C, and used to produce cells for the next round of selection. Two additional selection rounds were performed. After three rounds of MACS, the cells were plated, and individual clones were picked for sequencing analysis.
Protease Induced Removal of Displayed Peptides from the Cell Surface and Peptide Purification
L. lactis surface display vectors pBDA and pBDD were used to compare the dehydration ability of selected NisB mutation and wild-type NisB. The produced proteins containing the HRV 3C protease recognition sequence (PreScission protease; GE Healthcare) to release the N-terminal part of proteins from the L. lactis cell surface. L. lactis NZ9000 cells displaying BDA or BDD was subjected to 16% trichloroacetic acid (TCA) for 1 h on ice. Cells were washed three times with 10 mL of acetone, dried in a Speed Vac, and washed three times with PBS. The cells were resuspended in 5 mL of PreScission protease cleavage buffer (GE Healthcare) containing 200 U of PreScission protease (GE Healthcare) and incubated at 4 °C for 48 h under rotation. After protease digestion, the supernatant was collected, and the N-terminal parts of the BDA and BDD peptides were purified by HPLC, and subsequently mass spectrometry was performed.
Acknowledgments
We thank Marcel P. de Vries for support with mass spectrometry. X.Z. was financially supported by the Chinese Scholarship Council (CSC, Project No. 201706910062) and The Netherlands Organization for Scientific Research (NWO), research program TTW (Project No. 17241). Y.F. was financially supported by the Chinese Scholarship Council (CSC, Project No. 201806200107). R.C. was financially supported by the Ramon Areces Foundation (Spanish) and The Netherlands Organization for Scientific Research (NWO), research program NACTAR (Project No. 16433).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00130.
Amino acid sequence of designed peptides, MALDI-TOF MS data of CS5 with or without leader peptide, MALDI-TOF MS data of trypsin treated CS5 fragments, MALDI-TOF MS data of CS5A, CS5D, CS5E, CS5H, CS5M, CS5R, and CS5W, MALDI-TOF MS data of nisin modified by different NisB mutants, MALDI-TOF MS data of peptides modified by different NisB mutants, strains and plasmids used in this study, and primers used for PCR in this study (PDF)
Author Contributions
O.P.K. conceived and supervised the project and strategy and corrected the manuscript. G.N.M. helped to design part of experiments in more detail, provided supervision, and corrected the manuscript. R.C. performed experimental work on the NisB library synthesis and did work on finding mutations in the structure of NisB. T.B. discovered the larger cell wall-spanning display scaffold. R.R. provided the basic idea of coupling lanthipeptide-modification-enzyme engineering to screening by bacterially displayed product. Y.F. did experimental work on characterization of the NisB variants. X.Z. designed and carried out the experiments, analyzed data, and wrote the manuscript. All authors contributed to and commented on the manuscript text and approved its final version.
The authors declare no competing financial interest.
Supplementary Material
References
- Fuchs S. W.; Lackner G.; Morinaka B. I.; Morishita Y.; Asai T.; Riniker S.; Piel J. (2016) A Lanthipeptide-like N-Terminal Leader Region Guides Peptide Epimerization by Radical SAM Epimerases: Implications for RiPP Evolution. Angew. Chem., Int. Ed. 55 (40), 12330–12333. 10.1002/anie.201602863. [DOI] [PubMed] [Google Scholar]
- Montalbán-López M.; van Heel A. J.; Kuipers O. P. (2017) Employing the Promiscuity of Lantibiotic Biosynthetic Machineries to Produce Novel Antimicrobials. FEMS Microbiol. Rev. 41 (1), 5–18. 10.1093/femsre/fuw034. [DOI] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; et al. (2013) Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 30 (1), 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Yin Z.; Breukink E.; Moll G. N.; Kuipers O. P. (2020) An Engineered Double Lipid II Binding Motifs-Containing Lantibiotic Displays Potent and Selective Antimicrobial Activity against E. Faecium. Antimicrob. Agents Chemother. 10.1128/AAC.02050-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelski J.; Rink R.; Khusainov R.; Moll G. N.; Kuipers O. P. (2008) Biosynthesis, Immunity, Regulation, Mode of Action and Engineering of the Model Lantibiotic Nisin. Cell. Mol. Life Sci. 65 (3), 455–476. 10.1007/s00018-007-7171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M. A.; Hao Y.; Zhang Q.; Walker M. C.; van der Donk W. A.; Nair S. K. (2015) Structure and Mechanism of the TRNA-Dependent Lantibiotic Dehydratase NisB. Nature 517 (7535), 509. 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink R.; Wierenga J.; Kuipers A.; Kluskens L. D.; Driessen A. J. M.; Kuipers O. P.; Moll G. N. (2007) Production of Dehydroamino Acid-Containing Peptides by Lactococcus Lactis. Appl. Environ. Microbiol. 73 (6), 1792–1796. 10.1128/AEM.02350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink R.; Kuipers A.; de Boef E.; Leenhouts K. J.; Driessen A. J. M.; Moll G. N.; Kuipers O. P. (2005) Lantibiotic Structures as Guidelines for the Design of Peptides That Can Be Modified by Lantibiotic Enzymes. Biochemistry 44 (24), 8873–8882. 10.1021/bi050081h. [DOI] [PubMed] [Google Scholar]
- Schmitz U.; Versmold A.; Kaufmann P.; Frank H.-G. (2000) Phage Display: A Molecular Tool for the Generation of Antibodies—a Review. Placenta 21, S106–S112. 10.1053/plac.1999.0511. [DOI] [PubMed] [Google Scholar]
- Kondo A.; Ueda M. (2004) Yeast Cell-Surface Display—Applications of Molecular Display. Appl. Microbiol. Biotechnol. 64 (1), 28–40. 10.1007/s00253-003-1492-3. [DOI] [PubMed] [Google Scholar]
- Ståhl S.; Uhlén M. (1997) Bacterial Surface Display: Trends and Progress. Trends Biotechnol. 15 (5), 185–192. 10.1016/S0167-7799(97)01034-2. [DOI] [PubMed] [Google Scholar]
- Bosma T.; Rink R.; Moosmeier M. A.; Moll G. N. (2019) Genetically Encoded Libraries of Constrained Peptides. ChemBioChem 20, 1754. 10.1002/cbic.201900031. [DOI] [PubMed] [Google Scholar]
- Bosma T.; Kuipers A.; Bulten E.; de Vries L.; Rink R.; Moll G. N. (2011) Bacterial Display and Screening of Posttranslationally Thioether-Stabilized Peptides. Appl. Environ. Microbiol. 77 (19), 6794–6801. 10.1128/AEM.05550-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel L. B.; Cass R.; Milligan D. L.; Young D.; Arze R.; Johnson C. (1995) Screening of Cyclic Peptide Phage Libraries Identifies Ligands That Bind Streptavidin with High Affinities. Biochemistry 34 (47), 15430–15435. 10.1021/bi00047a006. [DOI] [PubMed] [Google Scholar]
- Katz B. A.; Johnson C.; Cass R. T. (1995) Structure-Based Design of High Affinity Streptavidin Binding Cyclic Peptide Ligands Containing Thioether Crosslinks. J. Am. Chem. Soc. 117 (33), 8541–8547. 10.1021/ja00138a008. [DOI] [Google Scholar]
- Plat A.; Kuipers A.; Lange J. G. d.; Moll G. N.; Rink R. (2011) Activity and Export of Engineered Nisin-(1–22) Analogs. Polymers (Basel, Switz.) 3 (3), 1282–1296. 10.3390/polym3031282. [DOI] [Google Scholar]
- Schmitt S.; Montalbán-López M.; Peterhoff D.; Deng J.; Wagner R.; Held M.; Kuipers O. P.; Panke S. (2019) Analysis of Modular Bioengineered Antimicrobial Lanthipeptides at Nanoliter Scale. Nat. Chem. Biol. 15 (5), 437. 10.1038/s41589-019-0250-5. [DOI] [PubMed] [Google Scholar]
- Van Heel A. J.; Kloosterman T. G.; Montalban-Lopez M.; Deng J.; Plat A.; Baudu B.; Hendriks D.; Moll G. N.; Kuipers O. P. (2016) Discovery, Production and Modification of Five Novel Lantibiotics Using the Promiscuous Nisin Modification Machinery. ACS Synth. Biol. 5 (10), 1146–1154. 10.1021/acssynbio.6b00033. [DOI] [PubMed] [Google Scholar]
- Majchrzykiewicz J. A.; Lubelski J.; Moll G. N.; Kuipers A.; Bijlsma J. J. E.; Kuipers O. P.; Rink R. (2010) Production of a Class II Two-Component Lantibiotic of Streptococcus Pneumoniae Using the Class I Nisin Synthetic Machinery and Leader Sequence. Antimicrob. Agents Chemother. 54 (4), 1498–1505. 10.1128/AAC.00883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll G. N.; Kuipers A.; Rink R. (2010) Microbial Engineering of Dehydro-Amino Acids and Lanthionines in Non-Lantibiotic Peptides. Antonie van Leeuwenhoek 97 (4), 319–333. 10.1007/s10482-010-9418-4. [DOI] [PubMed] [Google Scholar]
- van Heel A. J.; Kloosterman T. G.; Montalban-Lopez M.; Deng J.; Plat A.; Baudu B.; Hendriks D.; Moll G. N.; Kuipers O. P. (2016) Discovery, Production and Modification of Five Novel Lantibiotics Using the Promiscuous Nisin Modification Machinery. ACS Synth. Biol. 5 (10), 1146–1154. 10.1021/acssynbio.6b00033. [DOI] [PubMed] [Google Scholar]
- Majchrzykiewicz J. A.; Lubelski J.; Moll G. N.; Kuipers A.; Bijlsma J. J. E.; Kuipers O. P.; Rink R. (2010) Production of a Class II Two-Component Lantibiotic of Streptococcus Pneumoniae Using the Class I Nisin Synthetic Machinery and Leader Sequence. Antimicrob. Agents Chemother. 54 (4), 1498–1505. 10.1128/AAC.00883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusainov R.; van Heel A. J.; Lubelski J.; Moll G. N.; Kuipers O. P. (2015) Identification of Essential Amino Acid Residues in the Nisin Dehydratase NisB. Front. Microbiol. 6 (FEB), 1–8. 10.3389/fmicb.2015.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusainov R.; Heils R.; Lubelski J.; Moll G. N.; Kuipers O. P. (2011) Determining Sites of Interaction between Prenisin and Its Modification Enzymes NisB and NisC. Mol. Microbiol. 82 (3), 706–718. 10.1111/j.1365-2958.2011.07846.x. [DOI] [PubMed] [Google Scholar]
- Packer M. S.; Liu D. R. (2015) Methods for the Directed Evolution of Proteins. Nat. Rev. Genet. 16 (7), 379–394. 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Zhu L.; Schwaneberg U. (2015) Directed Evolution 2.0: Improving and Deciphering Enzyme Properties. Chem. Commun. 51 (48), 9760–9772. 10.1039/C5CC01594D. [DOI] [PubMed] [Google Scholar]
- Gielen F.; Hours R.; Emond S.; Fischlechner M.; Schell U.; Hollfelder F. (2016) Ultrahigh-Throughput-Directed Enzyme Evolution by Absorbance-Activated Droplet Sorting (AADS). Proc. Natl. Acad. Sci. U. S. A. 113 (47), E7383–E7389. 10.1073/pnas.1606927113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchner O.; Arnold F. H. (1997) Directed Evolution of Enzyme Catalysts. Trends Biotechnol. 15 (12), 523–530. 10.1016/S0167-7799(97)01138-4. [DOI] [PubMed] [Google Scholar]
- Reetz M. T. (2003) Select Protocols of High-Throughput ee-Screening Systems for Assaying Enantioselective Enzymes, Directed Enzyme Evolution, pp 283–290, Springer. [DOI] [PubMed] [Google Scholar]
- Dalby P. A. (2003) Optimising Enzyme Function by Directed Evolution. Curr. Opin. Struct. Biol. 13 (4), 500–505. 10.1016/S0959-440X(03)00101-5. [DOI] [PubMed] [Google Scholar]
- Glieder A.; Farinas E. T.; Arnold F. H. (2002) Laboratory Evolution of a Soluble, Self-Sufficient, Highly Active Alkane Hydroxylase. Nat. Biotechnol. 20 (11), 1135. 10.1038/nbt744. [DOI] [PubMed] [Google Scholar]
- Kretzschmar T.; Von Rüden T. (2002) Antibody Discovery: Phage Display. Curr. Opin. Biotechnol. 13 (6), 598–602. 10.1016/S0958-1669(02)00380-4. [DOI] [PubMed] [Google Scholar]
- Georgiou G.; Ippolito G. C.; Beausang J.; Busse C. E.; Wardemann H.; Quake S. R. (2014) The Promise and Challenge of High-Throughput Sequencing of the Antibody Repertoire. Nat. Biotechnol. 32 (2), 158. 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soendergaard M.; Newton-Northup J. R.; Palmier M. O.; Deutscher S. L. (2012) Peptide Phage Display for Discovery of Novel Biomarkers for Imaging and Therapy of Cell Subpopulations in Ovarian Cancer. J. Mol. Biomarkers Diagn. 1, S2. 10.4172/2155-9929.S2-004. [DOI] [Google Scholar]
- Jose J.; Betscheider D.; Zangen D. (2005) Bacterial Surface Display Library Screening by Target Enzyme Labeling: Identification of New Human Cathepsin G Inhibitors. Anal. Biochem. 346 (2), 258–267. 10.1016/j.ab.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Fellouse F. A.; Esaki K.; Birtalan S.; Raptis D.; Cancasci V. J.; Koide A.; Jhurani P.; Vasser M.; Wiesmann C.; Kossiakoff A. A.; et al. (2007) High-Throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-Displayed Libraries. J. Mol. Biol. 373 (4), 924–940. 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Schofield D. J.; Pope A. R.; Clementel V.; Buckell J.; Chapple S. D. J.; Clarke K. F.; Conquer J. S.; Crofts A. M.; Crowther S. R. E.; Dyson M. R.; et al. (2007) Application of Phage Display to High Throughput Antibody Generation and Characterization. Genome Biol. 8 (11), R254. 10.1186/gb-2007-8-11-r254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai S. A.; Wittrup K. D. (2007) Yeast Surface Display for Protein Engineering and Characterization. Curr. Opin. Struct. Biol. 17 (4), 467–473. 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Böckler B.; Schultz J.; Rahmann S. (2004) HMM Logos for Visualization of Protein Families. BMC Bioinf. 5 (1), 7. 10.1186/1471-2105-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holo H., and Nes I. F. (1995) Transformation of Lactococcus by Electroporation, Electroporation protocols for microorganisms, pp 195–199, Springer. [DOI] [PubMed] [Google Scholar]
- Cebrian R.; Macia-Valero A.; Jati A. P.; Kuipers O. P. (2019) Design and Expression of Specific Hybrid Lantibiotics Active against Pathogenic Clostridium Spp. Front. Microbiol. 10, 2154. 10.3389/fmicb.2019.02154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørholm M. H. H. (2010) A Mutant Pfu DNA Polymerase Designed for Advanced Uracil-Excision DNA Engineering. BMC Biotechnol. 10 (1), 21. 10.1186/1472-6750-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipes G. D.; Kosky A. A.; Abel J.; Zhang Y.; Treuheit M. J.; Kleemann G. R. (2005) Optimization and Applications of CDAP Labeling for the Assignment of Cysteines. Pharm. Res. 22 (7), 1059–1068. 10.1007/s11095-005-5643-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.