

Abstract
According to the currently accepted structure–property relationships, aceno-pentalenes with an angular shape (fused to the 1,2-bond of the acene) exhibit higher antiaromaticity than those with a linear shape (fused to the 2,3-bond of the acene). To explore and expand the current view, we designed and synthesized molecules where two isomeric, yet, different, 8π antiaromatic subunits, a benzocyclobutadiene (BCB) and a pentalene, are combined into, respectively, an angular and a linear topology via an unsaturated six-membered ring. The antiaromatic character of the molecules is supported experimentally by 1H NMR, UV–vis, and cyclic voltammetry measurements and X-ray crystallography. The experimental results are further confirmed by theoretical studies including the calculation of several aromaticity indices (NICS, ACID, HOMA, FLU, MCI). In the case of the angular molecule, double bond-localization within the connecting six-membered ring resulted in reduced antiaromaticity of both the BCB and pentalene subunits, while the linear structure provided a competitive situation for the two unequal [4n]π subunits. We found that in the latter case the BCB unit alleviated its unfavorable antiaromaticity more efficiently, leaving the pentalene with strong antiaromaticity. Thus, a reversed structure–antiaromaticity relationship when compared to aceno-pentalenes was achieved.
Introduction
Polycyclic conjugated systems that incorporate [4n]π antiaromatic1−3 subunits are of increasing interest in contemporary organic material design for small-molecule semiconductors. The rationale behind the application of antiaromatic motifs is their ability to efficiently reduce the aromaticity of acene-type systems while maintaining extended π-conjugation, thus ensuring high charge mobilities with reasonable chemical stability.4 In fact, there are recent reports on the successful realization of organic electronic devices (OFETs, solar cells) based on this principle.5−11 Antiaromaticity is also considered a design element in the development of supramolecular systems12−16 and molecular wires.17−21
Among antiaromatic carbocyclic structures, synthetic efforts are focused mainly on indacene,22−31 cyclobutadiene,32−40 and pentalene41−50 derivatives. Pentalene,51,52 as an 8π molecule, is one of the simplest antiaromatic polycyclic conjugated hydrocarbons, which provides several opportunities for modification and hence different degrees of stabilization that is affecting molecular conductivity, crystallization, or thin-film formation properties.
There are distinct approaches to tune the antiaromaticity of the pentalene unit within its π-extended derivatives (Figure 1). These include the fusion of heteroaromatic rings53−57 on the pentalene core, introduction of substituents58−62 with electron-donating or -withdrawing character, and different degrees of π-extension63−68 by the fusion of acene-type ring systems. A particularly important aspect within this latter approach is the topology of the fusion to the antiaromatic subunits.
Figure 1.

General strategies to tune the antiaromaticity of pentalenes. (a) Fusion of aryl/heteroaryl rings; (b) introduction of donor/acceptor substituents either on the pentalene core or on the fused aryl ring; and (c) variation of the fusion pattern around the pentalene core in aceno-pentalene derivatives.
A general pattern that can be recognized from the structure–property studies of these molecules, regardless of if they are monoaryl69,70 or diaryl systems,71−73 is that the more linear the structure (fusion to the 2,3-bond of the acene), the less antiaromatic becomes the pentalene core. This not only is true for pentalenes but also applies to π-extended cyclobutadienes,39 indacenes,74 and pentalene derivatives having N-heteroatoms within the five-membered rings.75 Haley and co-workers rationalized this effect in a combined experimental and theoretical study of these compounds.74 They found a correlation between the bond order of the fused aromatics and the antiaromaticity of the resulting π-extended [4n]π structure. Fusion to the bond with a higher bond order led to increased antiaromaticity and vice versa (Figure 2a).
Figure 2.

Role of bond order in the topology-dependent antiaromaticity of fused pentalenes. (a) Naphtho-pentalenes; (b) biphenyleno-pentalenes.
Based on the above analysis, it was predicted that this correlation may be general to all diareno-fused antiaromatic systems.74 With regard to the geometric structure of the resulting molecules, this implies that—in the case of the fusion of regular acenes to antiaromatic units—the molecules that are less antiaromatic will be more linear (2,3-fusion), while those with higher antiaromaticity will be more angular (1,2-fusion). This pattern leads to a serious limitation among aceno-antiaromatics as it restricts distinct electronic properties to distinct molecular topologies. It is well-known that molecular topology greatly affects the solid-state properties of the molecules, and this strongly influences their efficiency in device applications.4,76,77 With regard to the correlation between the acene bond-order and antiaromaticity, this could mean that the electronically more interesting highly antiaromatic systems could be of limited use in device applications due to their disadvantageous geometric structures. Thus, the question arises whether the correlation between the topology of a molecule and its antiaromaticity can be reversed by a different approach to the molecular design.
As approaches to reverse the relationship between molecular shape and antiaromaticity have not been explored previously, our goal was to deviate from the expected low antiaromaticity in the case of linearly fused acenes and to construct π-systems with a linear shape that maintain high antiaromaticity. A key feature of our design was the identification of a system to be fused with the pentalene unit having reversed bond orders of the 1,2- and 2,3-bonds when compared to the previously used acenes. We turned to biphenylene, a 12π cyclobutadiene derivative, that has been described as antiaromatic, although the description of its aromaticity and the destabilizing effect of its cyclobutadiene subunit are not straightforward.78−81 In this molecule, to decrease the cyclobutadiene character, there is considerable double-bond localization that leads to lower bond order at the 1,2-bond compared to that of the 2,3-bond (Figure 2b).82 This is the opposite trend to what is present in acenes such as naphthalene or anthracene. Hence, we argue that π-extended pentalenes with a linear shape and preserved antiaromaticity could be prepared by the fusion with biphenylene. From a structural point, the proposed molecules could be considered as the linking of isomeric benzocyclobutadiene (BCB) and pentalene, having different levels of antiaromaticity, through an unsaturated six-membered ring in different topologies (Figure 3).
Figure 3.

Combination of benzocyclobutadiene (BCB) and pentalene with different topologies.
In this regard, they are isoelectronic to the recently reported bispentalenes, where two pentalene moieties were fused to a central benzene ring.83 These latter systems were dominated by the two pentalene subunits, which exhibited an antiaromatic character. Thus, the reversed structure–property relationship was expected by the fusion of biphenylene instead of acenes to pentalene. The key question addressed herein is which one of the two unequally antiaromatic subunits alleviates its antiaromaticity to the most extent; in other words, who wins the battle, BCB or pentalene?84
Results and Discussion
We have synthesized two monoannelated biphenyleno-pentalenes 9 and 14 having an angular and a linear topology, respectively. These molecules were studied experimentally by 1H NMR spectroscopy, X-ray crystallography, UV–vis spectroscopy, and cyclic voltammetry, and the computational analysis of their aromaticity was performed (NICS-XY scan, ACID, HOMA, FLU, MCI). Throughout the manuscript, we compared the properties of the newly prepared molecules 9 and 14 to those of the previously reported naphtho-pentalene derivatives 15 and 16 with similar topologies.69
Synthesis
We have synthesized two biphenylene fused monoannelated pentalenes with an angular (9) (Scheme 1) and a linear (14) (Scheme 2) topology. In the angular structure, the pentalene moiety is fused to the 1,2-bond of biphenylene, while in the linear case, it is fused to the 2,3-bond. Both synthesis sequences relied on the modifications of biphenylene (2) earlier explored by McOmie and co-workers.85,86
Scheme 1. Synthesis of Biphenyleno-pentalene 9 with an Angular Topology.

Scheme 2. Synthesis of Biphenyleno-pentalene 14 with a Linear Topology.

Biphenylene itself was prepared from anthranilic acid (1) via a benzyne intermediate with varying yields (27–32%), due to the instability of the diazonium salt generated from 1. Formylation of 2 was performed selectively using dichloromethyl methyl ether in the presence of SnCl4. The reduction of the formyl group of compound 3 to the corresponding alcohol 4 by NaBH4 was necessary for the selective iodination in the following step via the ortho-lithiation/iodination sequence. The benzylic alcohol group of iodinated derivative 5 was oxidized back to the corresponding aldehyde 6 using the Jones reagent. Subsequent Sonogashira coupling with 4-methoxyphenylacetylene, yielding 7, and gem-dibromoolefin formation provided the key intermediate 8 for the pentalene formation. The key step for the synthesis of the pentalene unit was a Pd-catalyzed cascade carbopalladation reaction between alkynes and gem-dibromoolefins pioneered by Diederich and co-workers.43,69 The carbopalladation cascade provided the angular biphenyleno-pentalene 9 in 42% yield.
For the synthesis of linear biphenyleno-pentalene (Scheme 2), compound 2 was first brominated using N-bromosuccinimide (NBS) in N,N-dimethylformamide (DMF), yielding bromobiphenylene 10, which was formylated using the dichloromethyl methyl ether/SnCl4 system to obtain aldehyde 11. The low yield obtained for compound 11 is due to the low selectivity of the reaction. Subsequent Sonogashira coupling with 4-methoxyphenylacetylene, yielding 12, and gem-dibromoolefin formation provided the key intermediate 13 for the pentalene formation. The carbopalladation cascade provided the linear biphenyleno-pentalene 14 in 27% yield.
Based on previous examples,83 the introduction of a methoxy group in the dibromoolefins is advantageous as it increased the yield of the cascade reaction and facilitated the purification of the products. Compound 9 is a deep-purple bench-stable compound, while the orange-brown compound 14 was found to degrade under slightly acidic conditions (during silica column chromatography and to some extent in CDCl3; for measurements, solvents treated with basic alumina were used).
1H NMR Spectroscopy
The ability to sustain diatropic and paratropic ring currents is a characteristic of aromaticity and antiaromaticity, respectively.2,87−89 Such ring current effects are reflected in the proton chemical shifts of aromatic and antiaromatic compounds, which could be used as indicators of the extent of their aromaticity or antiaromaticity. It has been shown recently that [4n]π subunits can sustain paratropicity within larger conjugated frameworks.83 Because the molecules presented here also comprise two different 8π antiaromatic subunits, a BCB and a pentalene, their 1H NMR spectra are a good approximation of whether antiaromaticity is preserved in their structure (Figure 4a).
Figure 4.

(a) Partial 1H NMR spectra of compounds 9 and 14 (CDCl3, 500 MHz, room room temperature (rt)). Protons were assigned by the one-dimensional-nuclear over-Hauser effect spectroscopy (1D-NOESY) technique. (b) Reported monoaryl pentalene structures for comparison of the chemical shifts of the pentalene proton (all measured in CDCl3).
The pentalene protons in both molecules showed the characteristic upfield shift that has been observed for monoaryl-pentalenes earlier.43,69 The absorption of the pentalene proton (H1, Figure 4a) of the angular compound 9 appears at 6.60 ppm, while in the linear compound 14, this signal appears at 6.02 ppm in CDCl3 (protons were assigned by the 1D-NOESY technique; see Section S6, Supporting Information; for spectra recorded in CD2Cl2, see Section S6, Supporting Information). As the potential shielding effect by the proximal pendant phenyl substituents is expected to be the same in both cases, the upfield shifts originate from the interplay of the remaining diatropic ring current in ring c and the paratropic ring current in the pentalene subunit. The results suggest that the antiaromaticity of the pentalene subunit in the linear structure is more preserved.
Comparing these chemical shifts to the pentalene protons of the corresponding naphtho-pentalenes 15 and 16 that have been reported earlier,69 the opposite trend was found (Figure 4b). In these latter cases, the pentalene proton of the angular structure 15 appears at 6.11 ppm, while in the linear structure 16, it is at 6.71 ppm, showing lower antiaromaticity of the linear structure in this case. As a further comparison, the chemical shift of the pentalene proton in the related monobenzo-pentalene structure 17 is 6.28 ppm (Figure 4b).43 Note that the effect of the substituent on the peripheral phenyl groups on the chemical shifts is negligible, as described earlier.83 The chemical shifts of the protons on the six-membered rings between the two antiaromatic subunits also appear in the alkene region, which points toward the decreased aromaticity of this ring in both cases (H2 and H3, Figure 4a).
An extensive comparison of the 1H NMR shifts is difficult as only a few examples of diareno-pentalenes with unsubstituted pentalene rings have been reported. Still, the pentalene protons of dibenzo[a,e]pentalene appear at 6.40 ppm,90 while this value for diareno-pentalenes with further π-extension increases up to around 7 ppm, showing diminishing antiaromaticity in these cases.67
X-ray Crystallography
The topology of the aryl-fusion around the pentalene core has been shown to greatly influence the antiaromaticity and hence the optical and electronic properties of the molecules in the case of both monoareno- and diareno-pentalenes.69−73 The general observation for naphthalene- and anthracene-fused systems is that the 1,2-fused (angular) areno-pentalenes are better preserving their antiaromaticity compared to the 2,3-fused (linear) systems. We have examined the synthesized biphenyleno-pentalene molecules in this context, as the behavior of the six-membered ring that is confined between two antiaromatic units (BCB and pentalene) was expected to be particularly interesting and decisive for the extent of antiaromaticity of the subunits.
We could successfully grow single crystals of the angular compound 9 suitable for X-ray crystallographic analysis (Figure 5a) (for further details, see Section S1, Supporting Information). Significant bond-length alteration was found through the periphery of the conjugated core. The calculated bond lengths (Figure 5c) are in agreement with those determined experimentally. It is interesting to note that the polycyclic core of the molecule is not completely planar, and it is slightly out of plane around the four-membered ring (C10–C10A–C10B–C10C dihedral angle is 9.86°). In the calculated structure of compound 9, the same dihedral angle is about 1.5°, while the structure without the phenyl groups is planar. Hence, the observed effect could be attributed partially to intramolecular crowding and partially to forces in the crystal.
Figure 5.

(a) X-ray structure of compound 9 and the corresponding bond lengths; ORTEP representation of 9 is drawn at the 50% probability level; (b) layered structure of compound 9 in the crystalline state presented from the view of the crystallographic b-axis. Blue lines represent intermolecular short contacts (≤ sum of van der Waals radii + 0.1 Å). (c) Calculated bond lengths of compounds 9 and 14.
The crystal structure reveals that double bond-localization occurs within the six-membered ring between the two antiaromatic subunits in such a manner that decreases the bond order at the four- and five-membered ring fusion. By this localization, the antiaromatic characters of both the BCB and pentalene subunits are decreased. Molecules in the crystals are held together by multiple weak secondary interactions that lead to a layered structure (Figure 5b).
Unfortunately, we were not able to crystallize the linear derivative 14, but for comparison, we calculated the bond lengths within the molecule (Figure 5c). Calculations predict bond length alteration in this case as well. Importantly, the predicted bond length at the pentalene/six-membered ring fusion is shorter than that in compound 9 (1.429 and 1.465 nm, respectively), indicating higher bond order at this bond in 14.
Opto-electronic Properties
UV–vis spectra of compounds 9 and 14 were recorded in CHCl3 (Figure 6a) and compared to those of compounds 15 and 16 (Figure 6b).69 Spectra recorded in alternative solvents (CH2Cl2 and tetrahydrofuran (THF)) showed no pronounced differences (see Section S2, Supporting Information). It is immediately apparent that the shape of the absorption bands between 400 and 600 nm is reversed within the two compound pairs. In the case of the more antiaromatic angular naphtho-pentalene 15, a broad absorption band appears with a maximum at 426 nm, while in the case of the less antiaromatic linear compound 16, there are two distinct maxima (at 456 and 486 nm) giving a more structured absorption profile in the same region. In the case of the biphenyleno-pentalenes, however, structure 9 having an angular topology exhibits the absorption profile that resembles that of linear naphtho-pentalene 16. The two distinct absorption maxima in the case of compound 9 are bathochromically shifted to 527 and 568 nm. The spectrum of compound 14 is characterized by a maximum at 391 nm and a broad shoulder between 430 and 550 nm. In the spectra of compounds 15 and 16, low-energy long-wavelength absorptions that are characteristic of the symmetry-forbidden highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) transitions for pentalenes are clearly present (λmax = 660 nm for 15 and 566 nm for 16). These absorptions correspond to HOMO → LUMO transitions of 1.84 and 2.19 eV, respectively. As no clear low-energy absorptions could be identified in the spectra of 9 and 14, we performed time-dependent density functional theory (TD-DFT) calculations (B3LYP/6-311+G(d,p) level of theory) to gain more insight into the electronic properties and transition energies of these compounds.
Figure 6.

(a) UV–vis spectra of compounds 9 and 14 (in CHCl3); (b) UV–vis spectra of naphtho-pentalenes 15 and 16 (in CHCl3).
In the case of angular compound 9, the computed HOMO–LUMO gap was found to be 2.11 eV (λ = 590 nm, f = 0.016), which suggests that the corresponding absorption is overlapping with the shoulder in the region above the 568 nm maximum. In the case of linear compound 14, the transition energy is calculated to 1.61 eV (λ = 770 nm, f = 0.0004). These results could be rationalized by the analysis of the frontier molecular orbitals of the compounds (Figure 7). In the case of compound 14, both the HOMO and the LUMO possess an S-shaped geometry that is responsible for the symmetry-forbidden nature of this transition and explains the lack of the corresponding absorption in its UV–vis spectrum. Compared to the S-shaped geometry of the HOMO in compound 14, the HOMO of compound 9 is distorted; hence, there is a comparably increased absorption in the region where the HOMO → LUMO transition is expected to occur.
Figure 7.

Calculated HOMO and LUMO orbitals of biphenyleno-pentalenes 9 and 14 and naphtho-pentalenes 15 and 16.
Similar to the trend in 1H NMR shifts and UV–vis absorptions, in the case of the orbital features, a reversal of the patterns can be observed when comparing naphtho-pentalenes and biphenyleno-pentalenes. While the S-shape of the frontier orbitals and hence the lower absorption intensity that corresponds to the HOMO → LUMO transition are characteristic of the angular compound 15, in the case of biphenyleno-pentalenes, these features are associated with the linear structure of 14. On the other hand, distorted symmetry and thus higher-intensity UV–vis absorptions that correspond to HOMO → LUMO transitions are characteristic of both the linear naphtho-pentalene 16 and the angular biphenyleno-pentalene 9.
The frontier orbitals were also calculated for the antiaromatic subunits BCB and pentalene and for those units having exo-double bonds (Figure 8). These substructures are expected to have a contribution to the overall structures of molecules 9 and 14.
Figure 8.

Calculated HOMO and LUMO orbitals of BCB, pentalene, exo-BCB, and exo-pentalene.
In line with the experimentally determined bond lengths from the crystal structure of compound 9, the fulvene-like exo-pentalene structure was found to have a strong contribution to the HOMO of this molecule. The exo-double bonds are part of the adjacent six-membered ring that exhibits pronounced double bond-localization. Contrary to compound 9, where this bond localization led to the formation of an exo-pentalene substructure, in the HOMO of compound 14, the contribution of BCB and pentalene is clearly present. Furthermore, the LUMO of both substructures can be recognized in the LUMO of 14. These results suggest that in the linear compound 4, antiaromaticity is more significantly present than in the angular molecule 9.
Although the optical HOMO–LUMO gaps could not be determined experimentally from the UV–vis absorptions, we could obtain experimental support for the calculated energy differences by cyclic voltammetry (CV) measurements (Table 1) (see Section S3, Supporting Information). Both compounds 9 and 14 exhibited first reversible oxidation at 0.55 and 0.25 V and irreversible reduction at −1.47 and −1.32 V, respectively. The electrochemical gap is approximative due to the irreversible nature of the reductions; however, the obtained values of 2.02 V for compound 9 and 1.57 V for compound 14 are in good agreement with the theoretically predicted HOMO–LUMO gaps.
Table 1. Summary of Electrochemical, Optical, and Computational Data for Compounds 9, 14, 15, and 16.
| entry | compound | E1ox [V]a | E1red [V]a | HOMO [eV]b | LUMO [eV]b | ΔEredox [eV]c | ΔEopt [eV] | ΔEcalc [eV]d |
|---|---|---|---|---|---|---|---|---|
| 1 | 9 | 0.55e | –1.47f | –5.35 | –3.33 | 2.02 | 2.11 | |
| 2 | 14 | 0.25e | –1.32f | –5.05 | –3.48 | 1.57 | 1.61 | |
| 3 | 15(69) | 0.44e | –1.44e | –5.24 | –3.36 | 1.88 | 1.84 | 1.85 |
| 4 | 16(69) | 0.60e | –1.53f | –5.46 | –3.27 | 2.13 | 2.19 | 2.15 |
Electrochemical measurements were carried out in 0.1 M Bu4NPF6 in dichloromethane (DCM) at a scan rate of 0.1 V s–1 on a platinum wire working electrode. All potentials are given versus the Fc/Fc+ couple used as the internal standard.
HOMO and LUMO energy levels in electron volt were approximated using the equation HOMO = −(4.80 + E1ox), LUMO = −(4.80 + E1red).91,92
ΔEredox = LUMO - HOMO.
Calculations were performed on the B3LYP/6-311+G(d,p) level of theory.
Reversible first reduction or oxidation wave.
Irreversible first reduction or oxidation wave.
The calculated SOMO features of the radical cation and anion for both biphenyleno-pentalene isomers suggest that the unpaired electrons in the cationic and anionic species are delocalized over the system; however, in the anion, a pronounced pentalene character is present (see Section S4.5, Supporting Information). That may lead to further reactions, resulting in the irreversibility of the reduction process.
Aromaticity Analyses
As described above, substantial differences among compounds 9, 14, 15, and 16 were observed both in their HOMO–LUMO transitions obtained from UV–vis absorption spectroscopy and in their HOMO–LUMO energy gaps provided by electrochemical measurements. Low-intensity absorptions that correspond to the symmetry-forbidden HOMO → LUMO transitions are found in the UV–vis spectra of 14 and 15, while absorptions with increased intensity for the same transitions are detected for 9 and 16 (Figure 6). Below, we show that the associated changes in energies can be linked to aromaticity changes between the singlet (S0) ground state and the first excited states (T1, S1). At this point, it should be noted that the S1 and T1 states for all four compounds are similar in terms of configurations as the excitations according to TD-DFT calculations mainly are described by singly excited HOMO to LUMO (ππ*) configurations.93 As the two states only differ in multiplicity, we explored the T1 state as this state is more straightforward computationally than the S1 state (for comparative MCI and FLU values for the S1 state, see Tables S9 and S11, Supporting Information). Additionally, the trend in HOMO–LUMO gaps matches that of vertical excitations to the T1 state. To explore if the experimentally observed HOMO–LUMO gap variations for 9, 14, 15, and 16 can be linked to (anti)aromaticity changes upon excitation, we first analyzed their (anti)aromatic character in the S0 states and then compared with the T1 states.
Simplified structures where the Ph substituents of all molecules were replaced by H-atoms (denoted 9′, 14′, 15′, and 16′) were computed. This simplification does not considerably alter the findings (for further details, see Section S4.2, Supporting Information). All geometry optimizations were made with the Gaussian 16 package using the B3LYP hybrid functional and the 6-311+G(d,p) basis set.94−96
The aromatic character has been evaluated by means of magnetic (ACID plots and NICS-XY scans)97−102 and geometric (HOMA)103−105 indices106,107 in their ground (S0) and excited (T1) states, computed at the (U)B3LYP/6-311+G(d,p) level (for further details, see Section S4.1, Supporting Information). Aromatic rings are characterized by (i) clockwise ring currents revealed through ACID plots, (ii) diatropic ring currents detected by negative NICS values, and (iii) HOMA values in the range 0.5–1.0. On the other hand, anticlockwise ring currents, positive NICS, and negative HOMA values are indicative of antiaromaticity.
Here, we briefly point out some critical aspects of the usage of different indices to assess the (anti)aromatic character of a molecule. The usage of NICS alone, without analysis of the ring currents, is not recommendable.108 With regard to HOMA, there is recent criticism regarding the usage of this index for polycyclic aromatic hydrocarbons as it was found that HOMA values of internal six-membered rings in such hydrocarbons are overestimated.109 Still, the broad applicability of the HOMA index also for polycyclic systems is well documented.105 We focus the aromaticity analysis on the BCB and pentalene subunits, realizing that this is a simplification as other circuits in the polycyclic compounds also will contribute to the (anti)aromatic character of the molecules. Yet, the BCB and pentalene substructures play significant roles in determining the electronic character of the molecules as shown in the previous Experimental Section on the properties of 9 and 14. To obtain a qualitative view, we therefore focus on the BCB and pentalene subunits.
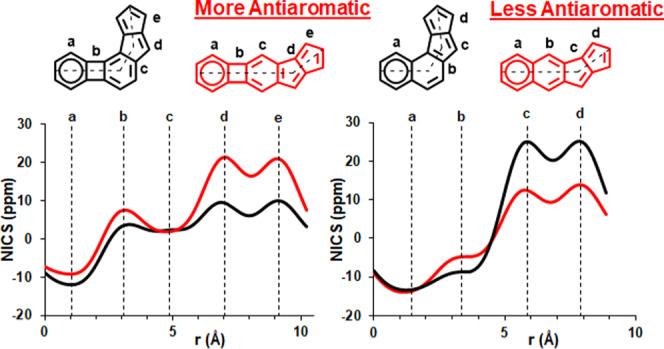
The NICS-XY scans of molecules 9′ and 14′ in their S0 state (Figure 9a) highlight that within these structures, the patterns that are characteristic of BCB and pentalene (see Figure S9, Supporting Information) to a good extent are preserved, even though the antiaromatic characters of the substructures that correspond to BCB (circuit a+b) and pentalene (circuit d+e) are clearly attenuated. Yet, it is important to note that the NICS values of 9′ throughout the scan are lower than those of 14′, which agrees with the lower antiaromaticity of the angular topology determined experimentally by 1H NMR spectroscopy. It is particularly noteworthy that the NICS values in the d+e subunit of 9′ are significantly lower than those within the same substructure of 14′. The ACID plots of 9′ and 14′ in their S0 states (Figure 9c) further support this observation. The opposite trend is obtained for molecules 15′ and 16′ in their NICS values and ACID plots regarding their pentalene (c+d) subunits (Figure 9b). In these cases, structure 15′ having an angular topology exhibits higher antiaromaticity in its c+d subunit compared to that of 16′ with a linear topology. We also calculated the relative energies of BCB and pentalene and found that the latter is more stable by approximately 6 kcal/mol. Complementary to this, as expected, the calculation of the relative energies of the corresponding exo-BCB and exo-pentalene structures shows the opposite, exo-BCB being more stable by about 4.6 kcal/mol. This supports the preferred rearrangement of the BCB unit to the exo-BCB structure in molecule 14. Furthermore, the interaction of BCB and pentalene subunits in 9′ and 14′ is clear from the comparison of the NICS-XY scans of 9′, 14′, and benzopentalene (17′) (see Section S4.4, Supporting Information). The alleviated antiaromaticity of BCB in 14′ enforces a stronger pentalene antiaromaticity in 14′ compared to that in benzopentalene. Furthermore, the remaining aromaticity of the six-membered ring c is higher in benzopentalene as it lacks the BCB subunit, which significantly decreases the aromaticity of ring c in molecules 9′ and 14′. Similarly, the comparison of 9′, 14′, and the corresponding compounds that lack the terminal benzene rings (“cyclobutadieno-benzopentalenes”, structures S4 and S5, Section S4.4, Supporting Information) suggests that the electronic structures of 9′ and 14′ are dominated by the interaction of the BCB and pentalene subunits rather than the cyclobutadiene and pentalene subunits.
Figure 9.

(a) NICS-XY scans of biphenyleno-pentalenes 9′ and 14′ in the S0 (solid line) and T1 (dashed line) states. (b) NICS-XY scans of naphtho-pentalenes 15′ and 16′ in the S0 (solid line) and T1 (dashed line) states. (c) ACID plots of biphenyleno-pentalenes 9′ and 14′ in the S0 and T1 states. (d) ACID plots of naphtho-pentalenes 15′ and 16′ in the S0 and T1 states. (For higher-resolution images, see Section S4.2.1, Supporting Information.) Blue and red arrows correspond to diatropicity and paratropicity, respectively. The width of the arrow denotes the strength of the ring current.
The geometry-based HOMA values for the BCB and pentalene substructures in 9′ and 14′ (Table 2) are in line with the magnetic indices. The HOMA values of both the BCB and the pentalene unit in 9′ reflect its lower antiaromaticity when compared to 14′, and this agrees with the crystallographically as well as the computationally determined bond-length alterations (see Figures S7 and S8, Supporting Information). That the pentalene subunits of 9′ and 14′ are the main antiaromatic subunits, rather than the BCB subunit, becomes clear when analyzing the difference of aromaticity going from the two units as separate molecules to subunits in 9′ and 14′. The perimeter of BCB is slightly more antiaromatic (HOMA = −0.460) than that of pentalene (HOMA = −0.388). When the two molecules are combined to form compounds 14′ and 9′, HOMA reveals a reduction of the antiaromatic character of both subunits, but the alleviation is larger in the BCB than in the pentalene subunit (ΔHOMA = 0.53 and 0.41 versus 0.38 and 0.26, respectively). Moreover, the contribution of BCB in 14′ to LUMO is very small compared to 9′ (Figure 7) as the BCB subunit efficiently alleviates its antiaromaticity contrary to the pentalene subunit (see Tables S14 and S15, Supporting Information). In agreement with the magnetic indices, HOMA values calculated for the c+d subunits of structures 15′ and 16′ (see Figure S6, Supporting Information) show the opposite structure–antiaromaticity relationship, as molecule 15′ with an angular topology maintains a higher antiaromaticity when compared to the linear structure 16′.
Table 2. Geometric (HOMA) Aromaticity Indices Calculated for the BCB and Pentalene Subunits in Their S0 and T1 States.
| compound/subunit | HOMA-S0 | HOMA–T1 |
|---|---|---|
| BCB | –0.460 | 0.568 |
| pentalene | –0.388 | 0.820 |
| a+b(9′) | 0.073 | 0.085 |
| d+e(9′) | 0.004 | 0.339 |
| a+b(14′) | –0.050 | 0.169 |
| d+e(14′) | –0.123 | 0.606 |
| c+d(15′) | –0.150 | 0.673 |
| c+d(16′) | –0.036 | 0.379 |
Despite the fact that the bicyclic a+b and d+e moieties are substructures within molecules 9′ and 14′, it is interesting to compare their HOMA values with those of BCB and pentalene alone (Table 2). In their S0 states, the HOMA values of perimeters of BCB and pentalene are −0.460 and −0.388, respectively, revealing clear antiaromaticity. Upon connecting these units in an angular fashion, as in 9′, these values change to 0.073 (a+b) and 0.004 (d+e), indicating nonaromatic circuits. Such a decrease in the antiaromatic character can be explained as a consequence of the strong double bond localization in ring c as seen in the crystal structure of 9, leading to simultaneous antiaromaticity alleviations in both the BCB and pentalene units. In contrast, when BCB and pentalene are fused in a linear fashion, as in 14′, similar simultaneous antiaromaticity alleviations of the BCB and pentalene units are impossible. According to the HOMA values of 14′, the pentalene unit retains a slightly more antiaromatic character than the BCB unit and the change is larger for the perimeter of the a+b moiety (ΔHOMA = 0.410) than for d+e (ΔHOMA = 0.265). As a consequence, the linear compound 14′ displays an antiaromatic character in the pentalene moiety.
As noted above, 9, 14, 15, and 16 display marked differences in their HOMO–LUMO transitions determined via UV–vis absorption spectroscopy and electrochemical measurements. We now explored if there is a link between the HOMO–LUMO gaps and the (anti)aromaticity changes between the singlet ground state and the first excited state (T1).110−115 We analyzed the T1 state instead of the S1 state for reasons described above. Moreover, we present NICS-XY, ACID, and HOMA results for the T1 states as these are the most readily calculated, while FLU and MCI data for the S1 state are given in the Supporting Information (Tables S9 and S11, respectively). The aromaticity indices corresponding to the BCB and pentalene subunits in 9′ and 14′ in their T1 state (Table 2) can potentially be connected to the ground-state characteristics of these molecules.
Upon excitation of 9′ and 14′ to their T1 states, there is a marked change in the (anti)aromaticity character of the pentalene subunit of 14′ as it clearly becomes Baird-aromatic according to NICS-XY, ACID, as well as HOMA (Figure 9 and Table 2). Thus, the strong antiaromatic character localized to the pentalene subunit in the S0 state of 14′ switches into a considerable aromatic character in the T1 state. In this latter case, both experimental data and computational analyses in the S0 state confirm higher antiaromaticity, especially in the pentalene subunit. Hence, preserved pentalene antiaromaticity is the price for an alleviated BCB antiaromaticity. This ground-state situation is well reflected in the T1 features of 14′. Similar to that in 9′, the nonaromatic BCB subunit has a low contribution to T1 aromaticity of compound 14′.
Overall, compound 9′ exhibits a relatively low antiaromaticity in the S0 state and low aromaticity in the T1 state, while the pentalene unit in compound 14′ has an antiaromatic character in the S0 state and an aromatic character in the T1 state. Hence, the relative (anti)aromaticities in the S0 and T1 states of these compounds, revealed by NICS-XY, ACID, and HOMA, indicate a larger ground-state destabilization and an enhanced aromatic stabilization in the T1 state of 14′, leading to a ΔE(T1–S0) of 0.51 eV for 14′ and 0.85 eV for 9′. This is also in agreement with the experimentally determined as well as the calculated lower HOMO–LUMO gap of 14′ compared to 9′ (Table 1).
Both the previously reported experimental data69 and the calculated aromaticity indices (Figure 9 and Table 2) confirm the stronger antiaromaticity of the pentalene subunit in angular naphtho-pentalene 15′ compared to linear 16′, where the double bond pattern in the fused five-membered rings rather defines a fulvene-like system (just as in 9′). Importantly, in line with the experimental findings, all of the indices confirm a similarly high antiaromaticity and Baird aromaticity of the pentalene subunit within the linear biphenyleno-pentalene 14′ and the angular naphtho-pentalene 15′.
Conclusions
We have shown that the generally accepted correlation between fusion pattern and antiaromaticity among π-extended pentalenes (aceno-pentalenes) could be altered by extending the molecular design to unsymmetric bis(antiaromatic) systems. There are two key features in the design of the newly synthesized molecules, which are composed of a biphenylene and a pentalene substructure. On the one hand, the bond-order values of the 1,2- and 2,3-bonds in biphenylene, to which the pentalene unit is fused, are reversed compared to those in acenes. On the other hand, the molecules can be considered as the mergers of two different subunits with an unequal antiaromatic character: a benzocyclobutadiene (BCB), contained in the biphenylene substructure, and a pentalene. When these subunits were fused in an angular fashion (through the 1,2-bond of biphenylene, as in compound 9), bond localization in the confined six-membered ring minimized both the BCB and the pentalene character in the molecule as supported by 1H NMR studies, X-ray crystal structure, and computational aromaticity analyses (NICS, ACID, HOMA). However, when BCB and pentalene were combined in a linear fashion (through the 2,3-bond of biphenylene, as in compound 14), there was no possibility for both subunits to alleviate their aromaticity. In this case, the BCB subunit alleviated its antiaromaticity to a greater extent than the pentalene subunit. Theoretical calculations supported the thermodynamically favored electronic rearrangement of the BCB unit and the strong antiaromaticity of the pentalene structure. The combined experimental (1H NMR, UV–vis, CV, and crystallography) and computational study of the synthesized molecules proved the reversal of the structure–antiaromaticity correlation compared to aceno-pentalenes. It has to be noted that our results do not violate the proposed correlation between bond order and antiaromaticity74 but expand the design to unsymmetric bis(antiaromatic) systems, where the bond orders favor the construction of linear and highly antiaromatic compounds.
The work presented here provides the basis of the syntheses of linear π-systems having strongly antiaromatic subunits and could aid in the design of solid-state structures beneficial for organic electronic device applications. Research on the extension of this design to different antiaromatic subunits and the construction of more extended π-systems is in progress in our laboratories.
Experimental Section
General Information
Commercial reagents, solvents, and catalysts (Aldrich, Fluorochem, VWR) were purchased as reagent-grade and used without further purification. Solvents for extraction or column chromatography were of technical quality. Organic solutions were concentrated by rotary evaporation at 25–40 °C. Thin-layer chromatography was carried out on SiO2-layered aluminum plates (60778-25EA, Fluka). Column chromatography was performed using SiO2–60 (230–400-mesh ASTM, 0.040–0.063 mm from Merck) at 25 °C or using a Teledyne Isco CombiFlash Rf+ automated flash chromatographer with silica gel (25–40 μm, Zeochem). Room temperature refers to 25(±1) °C. NMR spectra were acquired on a Varian 500 NMR spectrometer, running at 500 and 126 MHz for 1H and 13C, respectively. The residual solvent peaks were used as the internal reference. Chemical shifts (δ) are reported in ppm. The following abbreviations are used to indicate the multiplicity in 1H NMR spectra: s, singlet; d, doublet; t, triplet; q, quartet; h, heptet; and m, multiplet. 13C NMR spectra were acquired on a broad-band decoupled mode. Gas chromatography–mass spectrometry (GC–MS) analysis was performed on a Shimadzu GCMS-QP2010 Ultra System operated in the electron impact ionization (EI) mode. Mass spectrometric measurements were performed using a Q-TOF Premier mass spectrometer (Waters Corporation, Milford, MA) in the positive electrospray ionization mode.
General Procedures
General Procedure for the Sonogashira Reactions (GP1)
Orthohalo-formylbiphenylene (1 equiv) and 4-ethynylanisole (1.05 equiv) were dissolved in triethylamine (0.1 M). The solution was added to a vial that contained Pd(PPh3)2Cl2 (0.05 equiv) and CuI (0.05 equiv) under an inert atmosphere (N2). The mixture was heated to 50 °C in an aluminum heating block and stirred for 3–12 h. After the reaction was completed, the mixture was diluted with EtOAc and washed once with 10% HCl and twice with brine. The organic phase was dried over MgSO4. The solvent was evaporated in vacuo, and the crude product was further purified with column chromatography (SiO2, n-hexane/EtOAc).
General Procedure for Gem-dibromoolefination (GP2)
The product of GP1 (1 equiv) and CBr4 (1.5 equiv) was dissolved in 1,2-dichloroethane (DCE, 0.1 M). After the solution was purged with N2 for 10 min, P(OiPr)3 (3 equiv) was added. Following 3 h of stirring, the solution was diluted with DCE and was washed with water. The organic phase was separated and then dried over MgSO4. The solvent was evaporated in vacuo, and the crude product was further purified with column chromatography (SiO2, n-hexane/EtOAc).
General Procedure for the Carbopalladation Cascade Reaction (GP3)
The product of GP2 (1 equiv) and diphenylacetilene (5 equiv) was dissolved in toluene (0.1 M). The solution was added to a vial, which contained Pd(PPh3)2Cl2 (0.1 equiv), Zn (0.1 equiv), and K2CO3 (2 equiv) under an inert atmosphere (N2). The mixture was heated up to 110 °C in an aluminum heating block and stirred for 2 h. Subsequently, the reaction was cooled to rt, hydroquinone (1 equiv) was added to the mixture, and it was purged again with N2. The reaction was heated up to 110 °C and stirred for 16 h at this temperature. This was followed by cooling to rt, and the mixture was diluted with EtOAc and washed twice with water and once with brine. The organic phase was separated and then dried over MgSO4. The solvent was evaporated under reduced pressure, and the crude product was further purified with column chromatography (SiO2, n-hexane/EtOAc).
Synthetic Procedures
Synthesis of Biphenylene (2)116
Isopentyl nitrite (2.4 mL, 18.0 mmol) was added to a solution of anthranilic acid (2 g, 14.6 mmol) in THF (30 mL). This mixture was stirred for 1 h upon which the formation of a red precipitate was observed. Subsequently, a catalytic amount of trichloroacetic acid (∼20 mg) was added to the mixture and stirred until a brown precipitate was observed (approximately 1 h). The brown precipitate was filtered (Caution! Always keep the precipitate wet by solvent! The dried precipitate is highly explosive!) and washed with THF until the solvent was colorless. Subsequently, the residue was washed with 1,2-dichloroethane (3 × 10 mL) and then suspended in 1,2-dichloroethane. This suspension was carefully added to gently boiling 1,2-dichloroethane (60 mL, heated in an oil bath), yielding a dark-brown solution, which was stirred and boiled for another 15 min. The resulting solution was allowed to cool to room temperature and was washed with brine. The organic phase was separated and dried over MgSO4. The solvent was evaporated in vacuo, giving a brown solid as a crude product, which was purified further with column chromatography (SiO2, n-hexane) to give the product as yellowish-white crystals. Yield: 303 mg, 27%. 1H NMR (500 MHz, CDCl3) δ = 6.74 (dd, J = 4.8, 2.9 Hz, 1H), 6.63 (dd, J = 4.8, 2.9 Hz, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 151.5 (4), 128.4 (4), 117.5 (4) ppm.
Synthesis of 2-Formylbiphenylene (3)
SnCl4 (4 mL, 34.2 mmol) was added to a stirred solution of 2 (300 mg, 1.97 mmol) and dichloromethyl methyl ether (1 mL, 11.1 mmol) in 1,2-dichloroethane (30 mL) under a N2 atmosphere. The solution was stirred for 16 h at rt, and then, ice-cold HCl solution (3 M, 50 mL) was added. Following 1 h of vigorous stirring at rt, the mixture was extracted with CH2Cl2. The organic phase was washed with water and brine and then dried over MgSO4. The solvent was removed under reduced pressure, and the crude mixture was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (15%)). Evaporation of the solvent gave the product as yellow crystals. Yield: 225 mg, 64%. 1H NMR (500 MHz, CDCl3) δ = 9.68 (s, 1H), 7.27 (d, J = 7.1 Hz, 1H), 7.07 (s, 1H), 6.86 (dt, J = 17.1, 7.4 Hz, 2H), 6.76 (dd, J = 12.3, 6.4 Hz, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 191.0, 158.3, 152.1, 149.9, 149.5, 137.2, 136.7, 130.2, 129.2, 119.2, 118.7, 116.7, 114.1 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C12H10O+: 181.0653; found 181.0651.
Synthesis of 2-Hydroxymetylbiphenylene (4)
A suspension of NaBH4 (66.1 mg, 1.75 mmol) in ethanol (5.25 mL) was added in one portion to a stirred solution of 3 (210 mg, 1.17 mmol) in THF (585 μL). The mixture was stirred for 1 h at rt, and then water (∼50 mL) was added. The solution was extracted with EtOAc (2 × 20 mL). The organic phase was washed with water and brine and dried over MgSO4. Evaporation of the solvent under reduced pressure gave the product as bright-yellow crystals. Yield: 196 mg, 92%. 1H NMR (500 MHz, CDCl3) δ = 6.78–6.72 (m, 2H), 6.71 (d, J = 7.0 Hz, 1H), 6.67 (s, 1H), 6.64 (dd, J = 5.8, 3.7 Hz, 2H), 6.59 (d, J = 7.0 Hz, 1H), 4.44 (s, 2H), 1.89 (s, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 151.9, 151.0, 150.9, 150.8, 141.2, 128.5, 128.4, 126.6, 117.6, 117.5, 117.2, 116.8, 65.8 ppm; HRMS (ESI): m/z: [M]+, calcd for C12H11O+: 182.0732; found 182.0726.
Synthesis of 2-Hydroxymetylbiphenylene 2-Hydroxymethyl-1-iodobiphenylene (5)
n-BuLi (2.07 mL, 5.2 mmol, 2.5 M solution in hexane) was added slowly to a stirred solution of 4 (445 mg, 2.5 mmol) in diethyl ether (40 mL) at rt. The mixture was heated to reflux in an oil bath and stirred for 1 h. Subsequently, the solution was cooled to room temperature, and 1-chloro-2-iodoethane (340.6 mg, 3.1 mmol) in diethyl ether (10 mL) was added. The resulting solution was refluxed for another 30 min, and then it was cooled to rt and washed with water and brine. The ether solution was dried over MgSO4. The solvent was evaporated under reduced pressure, giving a dark-brown oil. The crude oily product was purified by column chromatography (SiO2, CH2Cl2/MeOH (2%)). The solvent was removed under reduced pressure, yielding the product as bright-yellow crystals. Yield: 335 mg, 44%. 1H NMR (500 MHz, CDCl3) δ = 6.89–6.84 (m, 1H), 6.84–6.78 (m, 2H), 6.75 (d, J = 6.8 Hz, 1H), 6.67–6.61 (m, 1H), 6.54 (d, J = 6.8 Hz, 1H), 4.45 (s, 2H), 1.95 (s, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 157.8, 151.7, 151.0, 149.0, 141.1, 129.4, 128.5, 128.0, 117.8, 116.6, 116.5, 83.8, 68.2 ppm; HRMS (ESI): m/z: [M]+, calcd for C12H9OI+: 307.9701; found 307.9701.
Synthesis of 1-Iodo-2-formylbiphenylene (6)
Jones reagent (80 μL) was added to the solution of 5 (80 mg, 0.26 mmol) in acetone (1 mL). The mixture was stirred for 10 min, and a green solid precipitate was formed. The precipitate was filtered, and acetone was removed in vacuo. The remaining residue was dissolved in EtOAc (10 mL) and washed with water. The organic phase was separated and dried over MgSO4. The solvent was evaporated under reduced pressure, giving an orange oil. The oil was purified by column chromatography (SiO2, n-hexane/EtOAc (10%)). Evaporation of the solvent under reduced pressure gave the product as yellow crystals. Yield: 75 mg, 94%. 1H NMR (500 MHz, CDCl3) δ = 9.80 (s, 1H), 7.31 (d, J = 7.0 Hz, 1H), 6.96 (d, J = 6.4 Hz, 1H), 6.95–6.86 (m, 2H), 6.76 (d, J = 6.4 Hz, 1H), 6.66 (d, J = 7.0 Hz, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 192.7, 158.7, 158.6, 150.6, 148.0, 135.0, 133.6, 130.5, 130.1, 119.4, 117.5, 116.6, 82.2 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C12H8OI+: 306.9620; found 306.9618.
Synthesis of 1-((4-,Ethoxyphenyl)ethynyl)biphenylene-2-carbaldehyde (7)
This compound was prepared from compound 6 according to GP1. The crude product was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (10%)), giving the product as yellow crystals. Yield: 34 mg, 57%. 1H NMR (500 MHz, CDCl3) δ = 10.25 (s, 1H), 7.47 (dd, J = 13.1, 7.9 Hz, 3H), 6.96–6.81 (m, 5H), 6.75 (d, J = 6.7 Hz, 1H), 6.67 (d, J = 7.1 Hz, 1H), 3.84 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 190.2, 160.4, 157.4, 154.3, 149.6, 149.1, 135.1, 133.4 (2), 132.1, 130.4, 129.6, 119.2, 118.8, 116.4, 114.7, 114.3 (2), 114.1, 98.1, 81.2, 55.5 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C22H15O2+: 311.1072; found 311.1072.
Synthesis of 2-(2,2-Dibromovinyl)-1-((4-methoxyphenyl)ethynyl)biphenylene (8)
This compound was prepared according to GP2. The crude product was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (10%)), giving the product as orange-yellow crystals. Yield: 28 mg, 54%. 1H NMR (500 MHz, CDCl3) δ = 7.63 (s, 1H), 7.47 (d, J = 8.9 Hz, 2H), 7.20 (dd, J = 7.3, 0.8 Hz, 1H), 6.90 (d, J = 8.9 Hz, 2H), 6.84–6.78 (m, 3H), 6.70–6.66 (m, 1H), 6.59 (d, J = 7.2 Hz, 1H), 3.84 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 160.2, 153.3, 151.2, 150.1, 150.1, 136.0, 135.7, 133.3 (2), 129.3, 129.1, 128.6, 118.3, 118.1, 116.2, 115.1, 114.3 (2), 112.8, 97.4, 90.7, 82.7, 55.5 ppm; HRMS (ESI): m/z: [M]+, calcd for C23H14OBr2+: 463.9411; found 463.9416.
Synthesis of 1-(4-Methoxyphenyl)-2,3-diphenylpentaleno[1,2-a]biphenylene (9)
This compound was prepared according to GP3. The crude product was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (5%)), giving the product as purple crystals. Yield: 12 mg, 42%.1H NMR (500 MHz, CDCl3) δ = 7.22–7.11 (m, 8H), 7.01 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 7.2 Hz, 2H), 6.88–6.84 (m, 1H), 6.84–6.78 (m, 3H), 6.73 (t, J = 7.5 Hz, 1H), 6.60 (s, 1H), 6.43 (d, J = 6.7 Hz, 1H), 6.18 (d, J = 6.7 Hz, 1H), 5.76 (d, J = 7.0 Hz, 1H), 3.82 (s, 3H) ppm; 1H NMR (500 MHz, CD2Cl2) δ = 7.25–7.10 (m, 8H), 7.01 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 7.3 Hz, 2H), 6.88 (t, J = 7.5 Hz, 1H), 6.82 (d, J = 8.6 Hz, 3H), 6.75 (t, J = 7.5 Hz, 1H), 6.62 (s, 1H), 6.47 (d, J = 6.7 Hz, 1H), 6.21 (d, J = 6.7 Hz, 1H), 5.76 (d, J = 7.1 Hz, 1H), 3.82 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CD2Cl2) δ = 159.9, 154.0, 151.1, 150.8, 150.3, 149.4, 141.1, 136.2, 135.3, 134.0, 132.3 (2), 131.9, 130.4 (2), 129.2 (2), 129.1, 129.1 128.7 (2), 128.4 (2), 128.2, 127.7, 127.3, 125.3, 120.9, 118.2, 113.9, 113.9 (2), 55.9 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C37H25O+: 485.1905; found 485.1902.
Synthesis of 2-Bromobiphenylene (10)
Compound 2 (700 mg, 4.6 mmol) and NBS (930 mg, 5.2 mmol) were dissolved in dried DMF (12 mL). The mixture was stirred for 2–3 h while the reaction was followed with GC–MS and TLC (SiO2, n-hexane). After the reaction was completed, the solution was diluted with DCM (40 mL) and washed with water (3 × 20 mL). The organic phase was separated and dried over MgSO4, and the solvents were evaporated in vacuo, giving an orange residue. The crude product was further purified with column chromatography (SiO2, n-hexane). Evaporation of the eluent under reduced pressure gave the product as bright-yellow crystals. Yield: 828 mg, 78%. 1H NMR (500 MHz, CDCl3) δ 6.90 (d, J = 7.3 Hz, 1H), 6.79–6.76 (m, 2H), 6.75 (s, 1H), 6.68–6.63 (m, 2H), 6.48 (d, J = 7.3 Hz, 1H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 152.8, 150.4, 150.0, 149.9, 130.6, 129.1, 128.7, 121.6, 121.3, 118.7, 118.2, 118.0; HRMS (ESI): m/z: [M + H]+, calcd for C12H7Br+: 229.9731; found 229.9731.
Synthesis of 3-Bromo-2-formylbiphenylene (11)
SnCl4 (6 mL, 51.3 mmol) was added to a stirred solution of 10 (708 mg, 3.06 mmol) and dichloromethyl methyl ether (1.5 mL, 16.6 mmol) in 1,2-dichloroethane (45 mL) under a N2 atmosphere. The solution was stirred for 16 h at rt, and then, ice-cold HCl solution (3 M, 75 mL) was added. Following 1 h of vigorous stirring at room temperature, the mixture was extracted with CH2Cl2. The organic phase was washed with water and brine and dried over MgSO4. The solvent was removed under reduced pressure, and the crude mixture was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (10%)). Evaporation of the solvent gave the product as yellow crystals. Yield: 149 mg, 19%. 1H NMR (500 MHz, CDCl3) δ = 10.16 (s, 1H), 7.09 (s, 1H), 6.96–6.84 (m, 3H), 6.81 (d, J = 7.3 Hz, 2H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 191.2, 158.6, 150.7, 149.7, 148.6, 133.5, 131.3, 131.0, 129.6, 122.5, 119.9, 119.3, 116.4 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C13H8OBr+: 258.9759; found 258.9759.
Synthesis of 3-((4-Methoxyphenyl)ethynyl)biphenylene-2-carbaldehyde (12)
This compound was prepared according to GP1. The crude product was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (10%)), giving the product as yellow crystals. Yield: 191.5 mg, 77%. 1H NMR (500 MHz, CDCl3) δ = 10.44 (s, 3H), 7.46 (d, J = 8.7 Hz, 7H), 7.13 (s, 3H), 6.94–6.87 (m, 13H), 6.81 (dd, J = 8.3, 4.7 Hz, 10H), 3.84 (s, 11H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 190.9, 160.5, 156.9, 150.8, 150.0, 149.2, 137.2, 133.4 (2), 131.3, 130.6, 129.6, 120.3, 119.6, 119.2, 114.5, 114.4 (2), 113.8, 98.0, 85.1, 55.5 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C22H15O2+: 311.1072; found 311.1076.
Synthesis of 2-(2,2-Dibromovinyl)-3-((4-methoxyphenyl)ethynyl)biphenylene (13)
This compound was prepared according to GP2. The crude product was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (10%)), giving the product as orange-yellow crystals. Yield: 112 mg, 69%. 1H NMR (500 MHz, CDCl3) δ = 7.77 (s, 1H), 7.45 (d, J = 8.8 Hz, 2H), 7.11 (s, 1H), 6.89 (d, J = 8.8 Hz, 2H), 6.86–6.79 (m, 2H), 6.75 (s, 1H), 6.72 (ddd, J = 6.7, 4.4, 1.5 Hz, 2H), 3.84 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CDCl3) δ = 160.1, 150.9, 150.4, 150.2, 137.8, 136.5, 133.2 (2), 129.3, 129.2, 123.3, 119.9, 118.4, 118.4, 116.6, 115.2, 114.3 (2), 96.8, 90.6, 87.2, 55.5 ppm; HRMS (ESI): m/z: [M]+, calcd for C23H14OBr2+: 463.9411; found 463.9409.
Synthesis of 1-(4-Methoxyphenyl)-2,3-diphenylpentaleno[1,2-b]biphenylene (14)
This compound was prepared according to GP3. The crude product was purified by flash column chromatography (SiO2, n-hexane → n-hexane/EtOAc (5%)), giving the product as a brown solid. Yield: 15 mg, 27%. 1H NMR (500 MHz, CDCl3) δ = 7.24–7.09 (m, 6H), 7.05 (dd, J = 6.6, 3.1 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 6.9 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H), 6.69–6.65 (m, 2H), 6.49–6.44 (m, 2H), 6.32 (s, 1H), 6.19 (s, 1H), 6.02 (s, 1H), 3.80 (s, 3H) ppm; 1H NMR (500 MHz, CD2Cl2) δ = 7.22–7.12 (m, 6H), 7.05 (dd, J = 6.6, 3.0 Hz, 2H), 6.97 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 7.0 Hz, 2H), 6.77 (d, J = 8.7 Hz, 2H), 6.70–6.66 (m, 2H), 6.49 (dd, J = 12.9, 5.2 Hz, 2H), 6.32 (s, 1H), 6.23 (s, 1H), 6.04 (s, 1H), 3.79 (s, 3H) ppm; 13C{1H} NMR (126 MHz, CD2Cl2) δ = 160.8, 153.5, 152.2, 150.4, 150.2, 150.1, 149.3, 147.9, 147.6, 140.3, 137.8, 135.5, 134.9, 134.8, 133.6, 131.1 (2), 130.1 (2), 128.8 (2), 128.6 (2), 128.4 (2), 128.3, 128.1, 127.8, 127.7, 126.7, 116.5, 116.3, 115.5, 113.8 (2), 112.6, 55.8 ppm; HRMS (ESI): m/z: [M + H]+, calcd for C37H25O+: 485.1905; found 485.1902.
Acknowledgments
Financial support from the Hungarian Academy of Sciences through the Lendület Program (LENDULET_2018_355 (G.L.)) is gratefully acknowledged. T.H. acknowledges the National Research, Development and Innovation Office, Hungary (NKFIH) (OTKA PD 128504), and the Hungarian Academy of Sciences (János Bolyai Research Scholarship) for funding. The Wenner-Gren Foundations is greatly acknowledged for a postdoctoral fellowship to O.E.B. (grant UPD2017-0243). H.O. is grateful for financial support from the Swedish Research Council (grant 2015-04538). The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and UPPMAX. P.J.M. acknowledges the Campus Mundi Programme (short-term study visit) by the Tempus Foundation for support. Péter Pál Kalapos, Tamás Gazdag, and Dr. István Jablonkai (MTA TTK “Lendület” Functional Organic Materials Research Group) are acknowledged for technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b03119.
The authors declare no competing financial interest.
Dedication
† This article is dedicated to Professor François Diederich.
Supplementary Material
References
- Breslow R. Antiaromaticity. Acc. Chem. Res. 1973, 6, 393–398. 10.1021/ar50072a001. [DOI] [Google Scholar]
- Gomes J. A. N. F.; Mallion R. B. Aromaticity and Ring Currents. Chem. Rev. 2001, 101, 1349–1383. 10.1021/cr990323h. [DOI] [PubMed] [Google Scholar]
- Minkin V. I.; Glukhovtsev M. N.; Simkin B. Y.. Aromaticity and Antiaromaticity; John Wiley and Sons: New York, 1994. [Google Scholar]
- Mei J.; Diao Y.; Appleton A. L.; Fang L.; Bao Z. Integrated Materials Design of Organic Semiconductors for Field-Effect Transistors. J. Am. Chem. Soc. 2013, 135, 6724–6746. 10.1021/ja400881n. [DOI] [PubMed] [Google Scholar]
- Nishida J.-i.; Tsukaguchi S.; Yamashita Y. Synthesis, Crystal Structures, and Properties of 6,12-Diaryl-Substituted Indeno[1,2-b]fluorenes. Chem. – Eur. J. 2012, 18, 8964–8970. 10.1002/chem.201200591. [DOI] [PubMed] [Google Scholar]
- Li C.; Liu C.; Li Y.; Zhua X.; Wang Z. Facile Synthesis of a Pyrrole-Fused Dibenzo[a,e]pentalene and its Application as a New Extended, Ladder-Type Fused Aromatic System. Chem. Commun. 2015, 51, 693–696. 10.1039/C4CC07721K. [DOI] [PubMed] [Google Scholar]
- Dai G.; Chang J.; Zhang W.; Bai S.; Huang K.-W.; Xub J.; Chi C. Dianthraceno[a,e]pentalenes: Synthesis, Crystallographic Structures and Applications in Organic Field-Effect Transistors. Chem. Commun. 2015, 51, 503–506. 10.1039/C4CC07630C. [DOI] [PubMed] [Google Scholar]
- Liu C.; Xu S.; Zhu W.; Zhu X.; Hu W.; Li Z.; Wang Z. Diaceno[a,e]pentalenes: an Excellent Molecular Platform for High-Performance Organic Semiconductors. Chem. – Eur. J. 2015, 21, 17016–17022. 10.1002/chem.201502184. [DOI] [PubMed] [Google Scholar]
- Dai G.; Chang J.; Jing L.; Chi C. Diacenopentalene Dicarboximides as New n-Type Organic Semiconductors for Field-Effect Transistors. J. Mater. Chem. C 2016, 4, 8758–8764. 10.1039/C6TC02601J. [DOI] [Google Scholar]
- Yuan B.; Zhuang J.; Kirmess K. M.; Bridgmohan C. N.; Whalley A. C.; Wang L.; Plunkett K. N. Pentaleno[1,2-a:4,5′]diacenaphthylenes: Uniquely Stabilized Pentalene Derivatives. J. Org. Chem. 2016, 81, 8312–8318. 10.1021/acs.joc.6b01480. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Fana H.; Zhu X. Fast Construction of Dianthraceno[a,e]pentalenes for OPV Applications. Org. Chem. Front. 2017, 4, 711–716. 10.1039/C6QO00867D. [DOI] [Google Scholar]
- Corminboeuf C.; Schleyer P. vR.; Warner P. Are Antiaromatic Rings Stacked Face-to-Face Aromatic?. Org. Lett. 2007, 9, 3263–3266. 10.1021/ol071183y. [DOI] [PubMed] [Google Scholar]
- Bean D. E.; Fowler P. W. Stacked-Ring Aromaticity: an Orbital Model. Org. Lett. 2008, 10, 5573–5576. 10.1021/ol802417n. [DOI] [PubMed] [Google Scholar]
- Aihara J.-i. J. Origin of Stacked-Ring Aromaticity. J. Phys. Chem. A 2009, 113, 7945–7952. 10.1021/jp900258a. [DOI] [PubMed] [Google Scholar]
- Nozawa R.; Tanaka H.; Cha W.-Y.; Hong Y.; Hisaki I.; Shimizu S.; Shin J.-Y.; Kowalczyk T.; Irle S.; Kim D.; Shinokubo H. Stacked Antiaromatic Porphyrins. Nat. Commun. 2016, 7, 13620 10.1038/ncomms13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej B.; Wang J.; Oruganti B. Molecular Photoswitching Aided by Excited-State Aromaticity. ChemPlusChem 2018, 83, 958–967. 10.1002/cplu.201800307. [DOI] [PubMed] [Google Scholar]
- Breslow R.; Foss F. W. Jr. Charge Transport in Nanoscale Aromatic and Antiaromatic Systems. J. Phys.: Condens. Matter 2008, 20, 374104 10.1088/0953-8984/20/37/374104. [DOI] [Google Scholar]
- Schneebeli S.; Kamenetska M.; Foss F.; Vazquez H.; Skouta R.; Hybertsen M.; Venkataraman L.; Breslow R. The Electrical Properties of Biphenylenes. Org. Lett. 2010, 12, 4114–4117. 10.1021/ol1017036. [DOI] [PubMed] [Google Scholar]
- Chen W.; Li H.; Widawsky J. R.; Appayee C.; Venkataraman L.; Breslow R. Aromaticity Decreases Single-Molecule Junction Conductance. J. Am. Chem. Soc. 2014, 136, 918–920. 10.1021/ja411143s. [DOI] [PubMed] [Google Scholar]
- Mahendran A.; Gopinath P.; Breslow R. Single Molecule Conductance of Aromatic, Nonaromatic, and Partially Antiaromatic Systems. Tetrahedron Lett. 2015, 56, 4833–4835. 10.1016/j.tetlet.2015.06.076. [DOI] [Google Scholar]
- Fujii S.; Marqués-González S.; Shin J.-Y.; Shinokubo H.; Masuda T.; Nishino T.; Arasu N. P.; Vázquez H.; Kiguchi M. Highly-Conducting Molecular Circuits Based on Antiaromaticity. Nat. Commun. 2017, 8, 15984 10.1038/ncomms15984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe Y. Non-Alternant Non-Benzenoid Aromatic Compounds: Past, Present, and Future. Chem. Rec. 2015, 15, 86–96. 10.1002/tcr.201402077. [DOI] [PubMed] [Google Scholar]
- Frederickson C. K.; Rose B. D.; Haley M. M. Explorations of the Indenofluorenes and Expanded Quinoidal Analogues. Acc. Chem. Res. 2017, 50, 977–987. 10.1021/acs.accounts.7b00004. [DOI] [PubMed] [Google Scholar]
- Tobe Y. Quinodimethanes Incorporated in Non-Benzenoid Aromatic or Antiaromatic Frameworks. Top. Curr. Chem. 2018, 376, 12 10.1007/s41061-018-0189-0. [DOI] [PubMed] [Google Scholar]
- Fix A. G.; Deal P. E.; Vonnegut C. L.; Rose B. D.; Zakharov L. N.; Haley M. M. Indeno[2,1-c]fluorene: a New Electron-Accepting Scaffold for Organic Electronics. Org. Lett. 2013, 15, 1362–1365. 10.1021/ol400318z. [DOI] [PubMed] [Google Scholar]
- Nobusue S.; Miyoshi H.; Shimizu A.; Hisaki I.; Fukuda K.; Nakano M.; Tobe Y. Tetracyclopenta[def,jkl,pqr,vwx]tetraphenylene: a Potential Tetraradicaloid Hydrocarbon. Angew. Chem., Int. Ed. 2015, 54, 2090–2094. 10.1002/anie.201410791. [DOI] [PubMed] [Google Scholar]
- Marshall J. L.; Uchida K.; Frederickson C. K.; Schutt C.; Zeidell A. M.; Goetz K. P.; Finn T. W.; Jarolimek K.; Zakharov L. N.; Risko C.; Herges R.; Jurchescud O. D.; Haley M. M. Indacenodibenzothiophenes: Synthesis, Optoelectronic Properties and Materials Applications of Molecules with Strong Antiaromatic Character. Chem. Sci. 2016, 7, 5547–5558. 10.1039/C6SC00950F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ie Y.; Sato C.; Yamamoto K.; Nitani M.; Aso Y. A Thiazole-Fused Antiaromatic Compound Containing an s-Indacene Chromophore with a High Electron Affinity. Chem. Lett. 2018, 47, 1534–1537. 10.1246/cl.180818. [DOI] [Google Scholar]
- Frederickson C. K.; Barker J. E.; Dressler J. J.; Zhou Z.; Hanks E. R.; Bard J. P.; Zakharov L. N.; Petrukhina M. A.; Haley M. M. Synthesis and Characterization of a Fluorescent Dianthracenoindacene. Synlett 2018, 29, 2562–2566. 10.1055/s-0037-1610280. [DOI] [Google Scholar]
- Melidonie J.; Liu J. Z.; Fu Y. B.; Weigand J. J.; Berger R.; Feng X. L. Pyrene-Fused s-Indacene. J. Org. Chem. 2018, 83, 6633–6639. 10.1021/acs.joc.8b00925. [DOI] [PubMed] [Google Scholar]
- Li G. W.; Gopalakrishna T. Y.; Phan H.; Herng T. S.; Ding J.; Wu J. S. From Open-Shell Singlet Diradicaloid to Closed-Shell Global Antiaromatic Macrocycles. Angew. Chem., Int. Ed. 2018, 57, 7166–7170. 10.1002/anie.201803949. [DOI] [PubMed] [Google Scholar]
- Iglesias B.; Cobas A.; Perez D.; Guitian E.; Vollhardt K. P. C. Tris(benzocyclobutadieno)triphenylene and its Lower Biphenylene Homologues by Palladium-Catalyzed Cyclizations of 2,3-Didehydrobiphenylene. Org. Lett. 2004, 6, 3557–3560. 10.1021/ol048548o. [DOI] [PubMed] [Google Scholar]
- Engelhardt V.; Garcia J. G.; Hubaud A. A.; Lyssenko K. A.; Spyroudis S.; Timofeeva T. V.; Tongwa P.; Vollhardt K. P. C. The Cobalt-Way to Heterophenylenes: Syntheses of 2-Thianorbiphenylenes, Monoazabiphenylenes, and Linear 1-Aza[3]phenylene {biphenyleno[2,3-a]cyclobuta[1,2-b]pyridine}. Synlett 2011, 2, 280–284. 10.1055/s-0030-1259087. [DOI] [Google Scholar]
- Gu Z.; Boursalian G. B.; Gandon V.; Padilla R.; Shen H.; Timofeeva T. V.; Tongwa P.; Vollhardt K. P. C.; Yakovenko A. A. Activated Phenacenes from Phenylenes by Nickel-Catalyzed Alkyne Cycloadditions. Angew. Chem., Int. Ed. 2011, 50, 9413–9417. 10.1002/anie.201103428. [DOI] [PubMed] [Google Scholar]
- Schlutter F.; Nishiuchi T.; Enkelmann V.; Mullen K. Octafunctionalized Biphenylenes: Molecular Precursors for Isomeric Graphene Nanostructures. Angew. Chem., Int. Ed. 2014, 53, 1538–1542. 10.1002/anie.201309324. [DOI] [PubMed] [Google Scholar]
- Biegger P.; Schaffroth M.; Tverskoy O.; Rominger F.; Bunz U. H. F. A Stable Bis(benzocyclobutadiene)-Annelated Tetraazapentacene Derivative. Chem. – Eur. J. 2016, 22, 15896–15901. 10.1002/chem.201602675. [DOI] [PubMed] [Google Scholar]
- Luppino S. P.; Swager T. M. Differentially Substituted Phenylene-Containing Oligoacene Derivatives. Synlett 2017, 28, 323–326. 10.1055/s-0036-1588650. [DOI] [Google Scholar]
- Dengiz C.; Luppino S. P.; Gutierrez G. D.; Swager T. M. Naphthazarin-Polycyclic Conjugated Hydrocarbons and Iptycenes. J. Org. Chem. 2017, 82, 7470–7480. 10.1021/acs.joc.7b01170. [DOI] [PubMed] [Google Scholar]
- Jin Z.; Teo Y. C.; Teat S. J.; Xia Y. Regioselective Synthesis of [3]Naphthylenes and Tuning of their Antiaromaticity. J. Am. Chem. Soc. 2017, 139, 15933–15939. 10.1021/jacs.7b09222. [DOI] [PubMed] [Google Scholar]
- Jin Z.; Teo Y. C.; Teat S. J.; Xia Y. Iterative Synthesis of Edge-Bent [3]Naphthylene. Synlett 2018, 29, 2547–2551. 10.1055/s-0037-1610261. [DOI] [Google Scholar]
- Levi Z. U.; Tilley T. D. Versatile Synthesis of Pentalene Derivatives via the Pd-Catalyzed Homocoupling of Haloenynes. J. Am. Chem. Soc. 2009, 131, 2796–2797. 10.1021/ja809930f. [DOI] [PubMed] [Google Scholar]
- Kawase T.; Konishi A.; Hirao Y.; Matsumoto K.; Kurata H.; Kubo T. An Extremely Simple Dibenzopentalene Synthesis from 2-Bromo-1-Ethynylbenzenes Using Nickel(0) Complexes: Construction of its Derivatives with Various Functionalities. Chem. – Eur. J. 2009, 15, 2653–2661. 10.1002/chem.200802471. [DOI] [PubMed] [Google Scholar]
- Rivera-Fuentes P.; von Wantoch Rekowski M.; Schweizer W. B.; Gisselbrecht J.-P.; Boudon C.; Diederich F. Cascade Carbopalladation Reaction Between Alkynes and Gem-Dibromoolefins: Facile Access to Monoannelated Pentalenes. Org. Lett. 2012, 14, 4066–4069. 10.1021/ol301670d. [DOI] [PubMed] [Google Scholar]
- Xu F.; Peng L.; Orita A.; Otera J. Dihalo-Substituted Dibenzopentalenes: their Practical Synthesis and Transformation to Dibenzopentalene Derivatives. Org. Lett. 2012, 14, 3970–3973. 10.1021/ol3017353. [DOI] [PubMed] [Google Scholar]
- Hashmi A. S. K.; Wieteck M.; Braun I.; Nösel P.; Jongbloed L.; Rudolph M.; Rominger F. Gold-Catalyzed Synthesis of Dibenzopentalenes – Evidence For Gold Vinylidenes. Adv. Synth. Catal. 2012, 354, 555–562. 10.1002/adsc.201200086. [DOI] [Google Scholar]
- Maekawa T.; Segawa Y.; Itami K. C–H Activation Route to Dibenzo[a,e]pentalenes: Annulation of Arylacetylenes Promoted by PdCl2–AgOTf–o-Chloranil. Chem. Sci. 2013, 4, 2369–2373. 10.1039/c3sc50585e. [DOI] [Google Scholar]
- Zhao J.; Oniwa K.; Asao N.; Yamamoto Y.; Jin T. Pd-Catalyzed Cascade Crossover Annulation of O-Alkynylarylhalides and Diarylacetylenes Leading to Dibenzo[a,e]pentalenes. J. Am. Chem. Soc. 2013, 135, 10222–10225. 10.1021/ja403382d. [DOI] [PubMed] [Google Scholar]
- Chen C.; Harhausen M.; Liedtke R.; Bussmann K.; Fukazawa A.; Yamaguchi S.; Petersen J. L.; Daniliuc C. G.; Fröhlich R.; Kehr G.; Erker G. Dibenzopentalenes from B(C6F5)3-Induced Cyclization Reactions of 1,2-Bis(phenylethynyl)benzenes. Angew. Chem., Int. Ed. 2013, 52, 5992–5996. 10.1002/anie.201300871. [DOI] [PubMed] [Google Scholar]
- Li H.; Wang X.-Y.; Wei B.; Xu L.; Zhang W.-X.; Pei J.; Xi Z. Intramolecular C–F and C–H Bond Cleavage Promoted by Butadienyl Heavy Grignard Reagents. Nat. Commun. 2014, 5, 4508 10.1038/ncomms5508. [DOI] [PubMed] [Google Scholar]
- Takahashi K.; Ito S.; Shintani R.; Nozaki K. Selective Synthesis of Unsymmetric Dibenzo[a,e]pentalenes by a Rhodium-Catalysed Stitching Reaction. Chem. Sci. 2017, 8, 101–107. 10.1039/C6SC04560J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally T.; Chai S.; Neuenschwander M.; Zhu Z. Pentalene: Formation, Electronic, and Vibrational Structure. J. Am. Chem. Soc. 1997, 119, 1869–1875. 10.1021/ja963439t. [DOI] [Google Scholar]
- Hopf H. Pentalenes – from Highly Reactive Antiaromatics to Substrates for Material Science. Angew. Chem., Int. Ed. 2013, 52, 12224–12226. 10.1002/anie.201307162. [DOI] [PubMed] [Google Scholar]
- Konishi A.; Fujiwara T.; Ogawa N.; Hirao Y.; Matsumoto K.; Kurata H.; Kubo T.; Kitamura C.; Kawase T. Pentaleno[1,2-c:4,5-c′]dithiophene Derivatives: First Synthesis, Properties, and a Molecular Structure. Chem. Lett. 2010, 39, 300–301. 10.1246/cl.2010.300. [DOI] [Google Scholar]
- Yin X.; Li Y.; Zhu Y.; Kan Y.; Li Y.; Zhu D. Synthesis of a Pentalene Centered Polycyclic Fused System. Org. Lett. 2011, 13, 1520–1523. 10.1021/ol200213h. [DOI] [PubMed] [Google Scholar]
- Dai G.; Chang J.; Shi X.; Zhang W.; Zheng B.; Huang K.-W.; Chi C. Thienoacene-Fused Pentalenes: Syntheses, Structures, Physical Properties and Applications for Organic Field-Effect Transistors. Chem. – Eur. J. 2015, 21, 2019–2028. 10.1002/chem.201405652. [DOI] [PubMed] [Google Scholar]
- Oshima H.; Fukazawa A.; Yamaguchi S. Facile Synthesis of Polycyclic Pentalenes with Enhanced Hückel Antiaromaticity. Angew. Chem., Int. Ed. 2017, 56, 3270–3274. 10.1002/anie.201611344. [DOI] [PubMed] [Google Scholar]
- Sekine K.; Stuck F.; Schulmeister J.; Wurm T.; Zetschok D.; Rominger F.; Rudolph M.; Hashmi A. S. K. N-Heterocycle-Fused Pentalenes by a Gold-Catalyzed Annulation of Diethynyl-Quinoxalines and -Phenazines. Chem. – Eur. J. 2018, 24, 12515–12518. 10.1002/chem.201803096. [DOI] [PubMed] [Google Scholar]
- Nakano M.; Osaka I.; Takimiya K. Dibenzo[a,e]pentalene-Embedded Dicyanomethylene-Substituted Thienoquinoidals for n-Channel Organic Semiconductors: Synthesis, Properties, and Device Characteristics. J. Mater. Chem. C 2015, 3, 283–290. 10.1039/C4TC02164A. [DOI] [Google Scholar]
- Xu F.; Peng L.; Shinohara K.; Nishida T.; Wakamatsu K.; Uejima M.; Sato T.; Tanaka K.; Machida N.; Akashi H.; Orita A.; Otera J. One-Shot Double Amination of Sondheimer–Wong Diynes: Synthesis of Photoluminescent Dinaphthopentalenes. Org. Lett. 2015, 17, 3014–3017. 10.1021/acs.orglett.5b01293. [DOI] [PubMed] [Google Scholar]
- Shen J.-J.; Shao J.-Y.; Zhu X.; Zhong Y.-W. Amine–Amine Electronic Coupling Through a Dibenzo[a,e]pentalene Bridge. Org. Lett. 2016, 18, 256–259. 10.1021/acs.orglett.5b03408. [DOI] [PubMed] [Google Scholar]
- Wilbuer J.; Grenz D. C.; Schnakenburg G.; Esser B. Donor- and Acceptor-Functionalized Dibenzo[a,e]pentalenes: Modulation of the Electronic Band Gap. Org. Chem. Front. 2017, 4, 658–663. 10.1039/C6QO00487C. [DOI] [Google Scholar]
- Grenz D. C.; Schmidt M.; Kratzert D.; Esser B. Dibenzo[a,e]pentalenes with Low-Lying LUMO Energy Levels as Potential n-Type Materials. J. Org. Chem. 2018, 83, 656–663. 10.1021/acs.joc.7b02250. [DOI] [PubMed] [Google Scholar]
- Saito M. Synthesis and Reactions of Dibenzo[a,e]pentalenes. Symmetry 2010, 2, 950–969. 10.3390/sym2020950. [DOI] [Google Scholar]
- Shen J.; Yuan D.; Qiao Y.; Shen X.; Zhang Z.; Zhong Y.; Yi Y.; Zhu X. Diaceno[a,e]pentalenes from Homoannulations of o-Alkynylaryliodides Utilizing a Unique Pd(OAc)2/n-Bu4NOAc Catalytic Combination. Org. Lett. 2014, 16, 4924–4927. 10.1021/ol502440d. [DOI] [PubMed] [Google Scholar]
- Kawase T.; Nishida J.-i. π-Extended Pentalenes: the Revival of the Old Compound from New Standpoints. Chem. Rec. 2015, 15, 1045–1059. 10.1002/tcr.201402093. [DOI] [PubMed] [Google Scholar]
- Dai G.; Chang J.; Luo J.; Dong S.; Aratani N.; Zheng B.; Huang K.-W.; Yamada H.; Chi C. Z-Shaped Pentaleno-Acene Dimers with High Stability and Small Band Gap. Angew. Chem., Int. Ed. 2016, 55, 2693–2696. 10.1002/anie.201508919. [DOI] [PubMed] [Google Scholar]
- Wurm T.; Rediger E. C.; Schulmeister J.; Koser S.; Rudolph M.; Rominger F.; Bunz U. H. F.; Hashmi A. S. K. A Golden Access to Acenopentalenes. Chem. – Eur. J. 2018, 24, 2735–2740. 10.1002/chem.201705456. [DOI] [PubMed] [Google Scholar]
- Sekine K.; Schulmeister J.; Paulus F.; Goetz K. P.; Rominger F.; Rudolph M.; Zaumseil J.; Hashmi A. S. K. Gold-Catalyzed Facile Synthesis and Crystal Structures of Benzene-/Naphthalene-Based Bispentalenes as Organic Semiconductors. Chem. – Eur. J. 2019, 25, 216–220. 10.1002/chem.201805637. [DOI] [PubMed] [Google Scholar]
- London G.; von Wantoch Rekowski M.; Dumele O.; Schweizer W. B.; Gisselbrecht J.-P.; Boudon C.; Diederich F. Pentalenes with Novel Topologies: Exploiting the Cascade Carbopalladation Reaction Between Alkynes And gem-Dibromoolefins. Chem. Sci. 2014, 5, 965–972. 10.1039/C3SC52623B. [DOI] [Google Scholar]
- Kato S.-i.; Kuwako S.; Takahashi N.; Kijima T.; Nakamura Y. Benzo- and Naphthopentalenes: Syntheses, Structures, and Properties. J. Org. Chem. 2016, 81, 7700–7710. 10.1021/acs.joc.6b01409. [DOI] [PubMed] [Google Scholar]
- Kawase T.; Fujiwara T.; Kitamura C.; Konishi A.; Hirao Y.; Matsumoto K.; Kurata H.; Kubo T.; Shinamura S.; Mori H.; Miyazaki E.; Takimiya K. Dinaphthopentalenes: Pentalene Derivatives for Organic Thin-Film Transistors. Angew. Chem., Int. Ed. 2010, 49, 7728–7732. 10.1002/anie.201003609. [DOI] [PubMed] [Google Scholar]
- Konishi A.; Okada Y.; Nakano M.; Sugisaki K.; Sato K.; Takui T.; Yasuda M. Synthesis and Characterization of Dibenzo[a,f]pentalene: Harmonization of the Antiaromatic and Singlet Biradical Character. J. Am. Chem. Soc. 2017, 139, 15284–15287. 10.1021/jacs.7b05709. [DOI] [PubMed] [Google Scholar]
- Konishi A.; Okada Y.; Kishi R.; Nakano M.; Yasuda M. Enhancement of Antiaromatic Character via Additional Benzoannulation into Dibenzo[a,f]pentalene: Syntheses and Properties of Benzo[a]naphtho[2,1-f]pentalene and Dinaphtho[2,1-a,f]pentalene. J. Am. Chem. Soc. 2019, 141, 560–571. 10.1021/jacs.8b11530. [DOI] [PubMed] [Google Scholar]
- Frederickson C. K.; Zakharov L. N.; Haley M. M. Modulating Paratropicity Strength in Diareno-Fused Antiaromatics. J. Am. Chem. Soc. 2016, 138, 16827–16838. 10.1021/jacs.6b11397. [DOI] [PubMed] [Google Scholar]
- Qiu L.; Zhuang X.; Zhao N.; Wang X.; An Z.; Lan Z.; Wan X. Benzo[f]benzo[5,6]indolo[3,2-b]indole: a Stable Unsubstituted 4nπ-Electron Acene with an Antiaromatic 1,4-Diazapentalene Core. Chem. Commun. 2014, 50, 3324–3327. 10.1039/C3CC49418G. [DOI] [PubMed] [Google Scholar]
- Wang C.; Dong H.; Hu W.; Liu Y.; Zhu D. Semiconducting π-Conjugated Systems in Field-Effect Transistors: a Material Odyssey of Organic Electronics. Chem. Rev. 2012, 112, 2208–2267. 10.1021/cr100380z. [DOI] [PubMed] [Google Scholar]
- Henson Z. B.; Müllen K.; Bazan G. C. Design Strategies for Organic Semiconductors Beyond the Molecular Formula. Nat. Chem. 2012, 4, 699–704. 10.1038/nchem.1422. [DOI] [PubMed] [Google Scholar]
- Mak T. C. W.; Trotter J. The Crystal and Molecular Structure of Biphenylene. J. Chem. Soc. 1962, 1–8. 10.1039/jr9620000001. [DOI] [Google Scholar]
- Zimmermann R. A Theoretical Study on the Molecular Structure of Biphenylene in its First Excited Singlet and Triplet States: Quantum Chemical Calculations on the Structural Changes of an Antiaromatic Molecule Upon Excitation. J. Mol. Struct. 1996, 377, 35–46. 10.1016/0022-2860(96)88398-0. [DOI] [Google Scholar]
- Wu J. I.-C.; Mo Y.; Evangelista F. A.; von Ragué Schleyer P. Is Cyclobutadiene Really Highly Destabilized by Antiaromaticity?. Chem. Commun. 2012, 48, 8437–8439. 10.1039/c2cc33521b. [DOI] [PubMed] [Google Scholar]
- Randić M. Aromaticity of Polycyclic Conjugated Hydrocarbons. Chem. Rev. 2003, 103, 3449–3606. 10.1021/cr9903656. [DOI] [PubMed] [Google Scholar]
- Glidewell C.; Lloyd D. Bond Orders in 1,5-Benzodiazepinium Cations Compared to those in Biphenylenes. Another Example of Bonds of Low Bond Order which Isolate Π-Electron Systems. Tetrahedron Lett. 1982, 23, 4379–4380. 10.1016/S0040-4039(00)85606-3. [DOI] [Google Scholar]
- Cao J.; London G.; Dumele O.; von Wantoch Rekowski M.; Trapp N.; Ruhlmann L.; Boudon C.; Stanger A.; Diederich F. The Impact of Antiaromatic Subunits in [4n+2] π-Systems: Bispentalenes with [4n+2] π-Electron Perimeters and Antiaromatic Character. J. Am. Chem. Soc. 2015, 137, 7178–7188. 10.1021/jacs.5b03074. [DOI] [PubMed] [Google Scholar]
- An initial version of this work was deposited in ChemRxiv on 21.10.2019, Reference:, https://doi.org/10.26434/chemrxiv.9981806.v1.
- Buckland P. R.; McOmie J. F. W. Biphenylenes–XXIX: Synthesis of Cyclobuta[b]biphenylene-1-Carboxylic Acid and of Cyclopenteno[b]biphenylene-1,2,3-Trione. Tetrahedron 1977, 33, 1797–1801. 10.1016/0040-4020(77)84061-1. [DOI] [Google Scholar]
- Cracknell M. E.; Kabli R. A.; McOmie J. F. W.; Perry D. H. Biphenylenes. Part 33. Synthesis of Bisbenzo[3,4]cyclobuta[1,2-b; 1′,2′-h]- and Bisbenzo[3,4]cyclobuta[1,2-c; 1′,2′-g]-Phenanthrene, and Attempts to Prepare Planar Derivatives of Cyclo-Octatetraene. J. Chem. Soc., Perkin Trans. 1 1985, 115–120. 10.1039/P19850000115. [DOI] [Google Scholar]
- von Schleyer P. R.; Jiao H. What is Aromaticity?. Pure Appl. Chem. 1996, 68, 209–218. 10.1351/pac199668020209. [DOI] [Google Scholar]
- Fowler P. W.; Steiner E.; Havenith R. W. A.; Jenneskens L. W. Current Density, Chemical Shifts and Aromaticity. Magn. Reson. Chem. 2004, 42, S68–S78. 10.1002/mrc.1445. [DOI] [PubMed] [Google Scholar]
- Gershoni-Poranne R.; Stanger A. Magnetic Criteria of Aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. 10.1039/C5CS00114E. [DOI] [PubMed] [Google Scholar]
- Eliasson B.; Edlund U. A Carbon-13 NMR Study of the Ion Pair Structure of the Dibenzo[b,f]pentalene Dianion. Org. Magn. Reson. 1983, 21, 322–327. 10.1002/omr.1270210508. [DOI] [Google Scholar]
- Pommerehne J.; Vestweber H.; Guss W.; Mahrt R. F.; Bässler H.; Porsch M.; Daub J. Efficient Two Layer LEDs on a Polymer Blend Basis. Adv. Mater. 1995, 7, 551–554. 10.1002/adma.19950070608. [DOI] [Google Scholar]
- D’Andrade B. W.; Datta S.; Forrest S. R.; Djurovich P.; Polikarpov E.; Thompson M. E. Relationship between the Ionization and Oxidation Potentials of Molecular Organic Semiconductors. Org. Electron. 2005, 6, 11–20. 10.1016/j.orgel.2005.01.002. [DOI] [Google Scholar]
- Toldo J.; El Bakouri O.; Solà M.; Norrby P.-O.; Ottosson H. Is Excited-State Aromaticity a Driving Force for Planarization of Dibenzannelated 8π-Electron Heterocycles?. ChemPlusChem 2019, 84, 712–721. 10.1002/cplu.201900066. [DOI] [PubMed] [Google Scholar]
- Gaussian 16; revision B.01 (for full reference, see Supporting Information).
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. A 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Krishnan R.; Binkley K. S.; Seeger R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Herges R.; Geuenich D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. 10.1021/jp0034426. [DOI] [Google Scholar]
- Geuenich D.; Hess K.; Köhler F.; Herges R. Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. 10.1021/cr0300901. [DOI] [PubMed] [Google Scholar]
- Gershoni-Poranne R.; Stanger A. The NICS-XY-Scan: Identification of Local and Global Ring Currents in Multi-Ring Systems. Chem. – Eur. J. 2014, 20, 5673–5688. 10.1002/chem.201304307. [DOI] [PubMed] [Google Scholar]
- Stanger A. Obtaining Relative Induced Ring Currents Quantitatively from NICS. J. Org. Chem. 2010, 75, 2281–2288. 10.1021/jo1000753. [DOI] [PubMed] [Google Scholar]
- Stanger A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. 10.1021/jo051746o. [DOI] [PubMed] [Google Scholar]
- Stanger A.; Rahalkar A. Aroma, http://schulich.technion.ac.il/Amnon_Stanger.htm.
- Kruszewski J.; Krygowski T. M. Definition of Aromaticity Basing on the Harmonic Oscillator Model. Tetrahedron Lett. 1972, 13, 3839–3842. 10.1016/S0040-4039(01)94175-9. [DOI] [Google Scholar]
- Krygowski T. M. Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of Pi-Electron Systems. J. Chem. Inf. Model. 1993, 33, 70–78. 10.1021/ci00011a011. [DOI] [Google Scholar]
- Krygowski T. M.; Cyrański M. K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1419. 10.1021/cr990326u. [DOI] [PubMed] [Google Scholar]
- Feixas F.; Matito E.; Poater J.; Solà M. On the Performance of Some Aromaticity Indices: A Critical Assessment Using a Test Set. J. Comput. Chem. 2008, 29, 1543–1554. 10.1002/jcc.20914. [DOI] [PubMed] [Google Scholar]
- Feixas F.; Matito E.; Poater J.; Solà M. Quantifying Aromaticity with Electron Delocalisation Measures. Chem. Soc. Rev. 2015, 44, 6434–6451. 10.1039/C5CS00066A. [DOI] [PubMed] [Google Scholar]
- Van Damme S.; Acke G.; Havenith R. W. A.; Bultinck P. Can the Current Density Map Topology Be Extracted from the Nucleus Independent Chemical Shifts?. Phys. Chem. Chem. Phys. 2016, 18, 11746–11755. 10.1039/C5CP07170D. [DOI] [PubMed] [Google Scholar]
- Makino M.; Nishina N.; Aihara J.-i. Critical Evaluation of HOMA and MBL as Local Aromaticity Indices. J. Phys. Org. Chem. 2018, 31, e3783 10.1002/poc.3783. [DOI] [Google Scholar]
- Baird N. C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. 10.1021/ja00769a025. [DOI] [Google Scholar]
- Ottosson H. Exciting Excited-State Aromaticity. Nat. Chem. 2012, 4, 969–971. 10.1038/nchem.1518. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.; Dahlstrand C.; Kilså K.; Ottosson H. Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. 10.1021/cr300471v. [DOI] [PubMed] [Google Scholar]
- Papadakis R.; Ottosson H. The Excited State Antiaromatic Benzene Ring: A Molecular Mr. Hyde?. Chem. Soc. Rev. 2015, 44, 6472–6493. 10.1039/C5CS00057B. [DOI] [PubMed] [Google Scholar]
- Ayub R.; El Bakouri O.; Jorner K.; Solà M.; Ottosson H. Can Baird’s and Clar’s Rules Combined Explain Triplet State Energies of Polycyclic Conjugated Hydrocarbons with Fused 4nπ- and (4n + 2)π-Rings?. J. Org. Chem. 2017, 82, 6327–6340. 10.1021/acs.joc.7b00906. [DOI] [PubMed] [Google Scholar]
- Oh J.; Sung Y. M.; Hong Y.; Kim D. Spectroscopic Diagnosis of Excited-State Aromaticity: Capturing Electronic Structures and Conformations Upon Aromaticity Reversal. Acc. Chem. Res. 2018, 51, 1349–1358. 10.1021/acs.accounts.7b00629. [DOI] [PubMed] [Google Scholar]
- Logullo F. M.; Seitz A. H.; Friedman L. Benzenediazonium-2-Carboxylate and Biphenylene. Org. Synth. 1968, 48, 12. 10.15227/orgsyn.048.0012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.