

SUMMARY
In humans, disruption of nonsense-mediated decay (NMD) has been associated with neurodevelopmental disorders (NDDs) such as autism spectrum disorder and intellectual disability. However, the mechanism by which deficient NMD leads to neurodevelopmental dysfunction remains unknown, preventing development of targeted therapies. Here we identified novel protein-coding UPF2 (UP-Frameshift 2) variants in humans with NDD, including speech and language deficits. In parallel, we found that mice lacking Upf2 in the forebrain (Upf2 fb-KO mice) show impaired NMD, memory deficits, abnormal long-term potentiation (LTP), and social and communication deficits. Surprisingly, Upf2 fb-KO mice exhibit elevated expression of immune genes and brain inflammation. More importantly, treatment with two FDA-approved anti-inflammatory drugs reduced brain inflammation, restored LTP and long-term memory, and reversed social and communication deficits. Collectively, our findings indicate that impaired UPF2-dependent NMD leads to neurodevelopmental dysfunction and suggest that anti-inflammatory agents may prove effective for treatment of disorders with impaired NMD.
Graphical Abstract

In Brief
Johnson et al. discovered that genetic ablation of Upf2-mediated NMD triggers an aberrant immune response and leads to memory, synaptic plasticity, social, and vocal communication deficits. These behavioral and neurophysiological abnormalities were reversed by FDA-approved agents that dampen brain inflammation.
INTRODUCTION
The ability to regulate gene expression is crucial for normal development. Recent genetic studies have shown that variants that alter gene expression are likely risk factors for neurodevelopmental disorders (NDDs) (Sahin and Sur, 2015). Most of the research in the field has focused on proteins and/or signaling pathways that control gene expression via epigenetic, transcriptional, and translational regulation (Ehninger et al., 2008; Sahin and Sur, 2015; Sossin and Costa-Mattioli, 2019). Nonsense-mediated decay (NMD) is an mRNA quality control mechanism that degrades mRNAs containing a premature termination codon (PTC) (Baker and Parker, 2004; Karousis and Muhlemann, 2019; Kervestin and Jacobson, 2012; Popp and Maquat, 2013). By degrading faulty mRNAs, NMD prevents synthesis of C-terminally truncated proteins, which may exert dominant-negative activities and interfere with the normal function of full-length proteins present in the cell. Importantly, nonsense and frameshift variants that generate PTCs cause approximately one-third of all known human monogenic disorders (Mort et al., 2008). In addition to PTC-containing mRNAs, NMD can regulate the expression of up to 20% of cellular transcripts (He and Jacobson, 2015; Karousis and Muhlemann, 2019), indicating that NMD also functions to regulate physiological gene expression. Although NMD is a crucial post-transcriptional gene expression regulator, little is known about its role in brain development and function. The clinical relevance of NMD in brain disorders is highlighted by the identification of copy number variants (CNVs) in UPF2 and other NMD components (Nguyen et al., 2013, 2014) and single-nucleotide pathogenic variants in NMD core component UPF3B (Laumonnier et al., 2010; Tarpey et al., 2007; Xu et al., 2013) in individuals with NDDs, including intellectual disability (ID) and autism spectrum disorder (ASD). However, how dysfunctional NMD leads to impaired brain development and function remains unknown.
To address this, we inhibited NMD in vivo by conditionally removing Upf2 in the murine forebrain. Similar to individuals with deletions encompassing the UPF2 locus, we found that Upf2 forebrain-specific knockout (Upf2 fb-KO) mice exhibit learning and memory impairments and display social deficits and behavioral inflexibility. We also sought to identify the mechanism by which loss of Upf2-dependent NMD leads to these neurodevelopmental impairments in mice. Integrating multiple approaches such as transcriptomics, flow cytometry, immunohistochemistry, pharmacology, and behavior, we provide causal evidence that over-activation of the immune response contributes to the deficits in long-term memory (LTM), synaptic plasticity, and social behaviors caused by loss of Upf2 in mice. In addition, we identified three individuals with novel variants in UPF2 who exhibited speech deficits, implicating Upf2-mediated control of NMD as a new pathway involved in speech and language. Finally, Upf2 fb-KO mice recapitulate the human UPF2 phenotype by showing vocal communication problems that were rescued by Food and Drug Administration (FDA)-approved agents that reduce brain inflammation.
RESULTS
Humans Carrying Pathogenic Variants in UPF2 Exhibit Speech and Language Impairments and Impaired NMD
NDDs are often characterized by delays in speech and language development (Morgan and Webster, 2018; Tomblin, 2011). Indeed, variants in genes implicated in ID and ASD, such as the Forkhead box transcription factors (FOXP1/2) and contactin-associated protein-like 2 (CNTNAP2) are also associated with speech- and language-related disorders (Fisher and Scharff, 2009; Hamdan et al., 2015; Morgan and Webster, 2018; Rodenas-Cuadrado et al., 2016). Given that individuals with single allele deletions of the UPF2 locus exhibit NDD (Nguyen et al., 2013), we wondered whether Upf2-mediated NMD might also be implicated in speech and language function. We identified three unrelated individuals who carry novel UPF2 variants and display a range of speech and language deficits. Two unrelated individuals (cases 1 and 2) harbored different de novo frameshift mutations, and case 3 displayed a de novo deletion spanning UPF2 (Figures 1A and 1B; Figure S1). In addition to UPF2, the other deleted genes in case 3 were CELF2, USP6NL, ECHDC3, PROSER2, DHTKD1, SEC61A2, NUDT5, CDC123, AMK1D, CCDC3, OPTN, MCM10, and UMCA. It is noteworthy that only two of these genes are disease causing in Online Mendelian Inheritance in Man (OMIM), and neither has been associated with a pediatric phenotype: a loss-of-function variant in DHTKD1 has been shown (in one family) to be associated with Charcot-Marie-Tooth disease with onset in adolescence, and loss-of-function variants in OPTN have been associated with amyotrophic lateral sclerosis with onset after age 50 years. Cases 1 and 2 had low average intelligence quotient (IQ), low average ability in receptive and expressive language, and a phonological speech sound disorder. Case 3, carrying a UPF2 deletion, had severe ID and ASD and was non-verbal (Figure 1A).
Figure 1. Human UPF2 Variants Are Associated with Language Problems and Impaired NMD.

(A) Summary of clinical phenotypes in affected individuals with UPF2 variants. R, right; AVSD, atrioventricular septal defect; ASD, autism spectrum disorder; N, not present; Y, present; avg, average; ext, extremely; artic, articulation disorder; mod, moderate; NA, not available because the child has severe ID and is non-verbal.
(B) cDNA Sanger sequencing of a lymphoblastoid cell line (LCL) from case 1 (top panel) and a control LCL (bottom panel) across the site of the c.1940 (NM_080599) deletion. The background “noise” in the case 1 sequencing panel is due to the overlapping frameshifted c.1940delA mRNA, which is not 100% degraded by NMD.
(C) Western blot analysis revealed that individuals with UPF2 variants exhibit a reduction in UPF2 protein levels (n = 6 control, n = 1 c.1940delA/case 1, n = 1 UPF2 CNV/case 3, n = 2 UPF2 CNV).
(D) A significant positive correlation was observed when differentially expressed genes (DEGs) were compared between unrelated individuals carrying mutations in different core NMD factor mutations (n = 1 c.1940delA/case 1, n = 1 UPF2 CNV/case 3, n = 2 UPF2 CNV grouped, n = 3 UPF3B mutations).
Error bars represent mean ± SEM.
See also Figures S1 and S2.
UPF2 protein and mRNA levels were decreased in lymphoblastoid cell lines (LCLs) from case 1 (containing a frameshift variant [NM_080599.2: c1940del; Figures 1B and S1B]) and case 3 (carrying a deletion of one allele of the UPF2 gene, HG19 chr10: 10,901,194–13,286,601del) compared with LCLs isolated from healthy control individuals as well as two individuals shown previously by us to have CNV deletions encompassing the UPF2 locus (Nguyen et al., 2013; Figure 1C; Figure S2A). The protein levels of other core NMD factors, UPF1 and UPF3B, were unchanged in LCLs isolated from cases 1 and 3 (Figures S2B and S2C). Unfortunately, we were unable to obtain LCLs from case 2 (containing a frameshift variant [NM_080599.2: c986del; Figure S1C]).
To determine whether NMD was impaired in individuals carrying different UPF2 loss-of-function variants, we performed transcriptomic profiling (RNA sequencing) on LCLs from these two cases (1 and 3), two previously described cases with UPF2 CNV deletions (Nguyen et al., 2013), three previously described cases with loss-of-function mutations in UPF3B (Nguyen et al., 2012; Tarpey et al., 2007), and 7 control individuals (Figures 1D and S2D; Tables S1, S2, and S3; see also Figure S2E for an illustration of how the RNA sequencing [RNA-seq] comparisons were performed). First, it is important to note that, although we previously performed RNA sequencing from individuals carrying the two UPF2 CNV deletions and the UPF3B mutations (Nguyen et al., 2013; Tarpey et al., 2007), the previously generated data were not used here because, at the time, we were only able to collect approximately 15–20 million mappable RNA-seq reads per individual. In view of the current standard in the field, we considered the number of RNA-seq reads to be insufficient. Thus, we generated new data with 50–60 million mappable RNA-seq reads/per LCL line. Second, UPF3B is on the X chromo-some; consequently, males with a deletion in UPF3B are de facto null for this NMD factor. Thus, differentially expressed genes (DEGs) from UPF3B carrying loss-of-function mutations represent an excellent set of “NMD genes” in the human transcriptome. Third, we combined the transcriptional profiles of all three individuals carrying UPF2 CNV deletions (case 3 and the other two previously described cases; Nguyen et al., 2013) as a group (herein defined as “UPF2 CNV grouped”) because the transcriptional profiles did not significantly differ from each other (data not shown). Fourth, we found highly significant positive correlations between the DEGs of case 1 (UPF2 frameshift) and UPF3B mutations (Figure 1D, left panel) and UPF2 CNV grouped and UPF3B mutations (Figure 1D, center panel), consistent with the idea that the frameshifting variant of case 1 and the deletions of a single allele of UPF2 (UPF2 CNV grouped) impair NMD. Finally, DEGs from case 1 and those from UPF2 CNV grouped were also significantly correlated (Figure 1D, right panel), indicating that NMD is similarly impaired in individuals carrying different UPF2 CNVs. Thus, we concluded that loss-of-function UPF2 variants, as identified in cases 1 and 3, lead to impaired NMD.
Inhibition of Upf2-Dependent NMD Impairs LTM and Related Changes in Synaptic Strength
We sought to further investigate the effect of compromised Upf2-dependent NMD in brain development and function using mice. Efficient or specific pharmacological inhibitors of NMD are not available. In addition, although the disorder is constructed to be a disorder of hemizygosity of UPF2, mice lacking only one copy of Upf2 (germline Upf2+/−) show no behavioral abnormalities (unpublished data). Moreover, genetic deletion of core NMD gene components, such as Upf1 or Upf2, is embryonic lethal (Medghalchi et al., 2001; Thoren et al., 2010; Weischenfeldt et al., 2008). To circumvent this problem, we conditionally deleted Upf2 specifically in the forebrain by crossing mice in which exons 2 and 3 of Upf2 are flanked by loxP sites (Upf2loxP/loxP) with mice expressing Cre recombinase under control of the α subunit of the Ca2+/calmodulin-dependent protein kinase II (Camk2α) promoter (Camk2α-Cre), generating Upf2 fb-KO mice (STAR Methods). As expected, in the hippocampus (Figure 2A) and cortex (Figure S3A) of Upf2 fb-KO mice, Upf2 mRNA levels were significantly reduced compared with control mice. However, in the cerebellum, where Cre is not expressed, Upf2 mRNA levels remained unchanged (Figure S3B). To investigate the effect of conditionally deleting Upf2 on NMD function in the forebrain, we measured the levels of several canonical NMD targets by qRT-PCR. As expected, in Upf2 fb-KO mice, the NMD mRNA substrates Atf4 (Mendell et al., 2004), Gadd45b (Viegas et al., 2007), Pdrg1 (Karam et al., 2015), Cars (Mendell et al., 2004), Ddit3 (Weischenfeldt et al., 2008), and Rassf1 (Weischenfeldt et al., 2008) were all significantly upregulated in the hippocampus (Figure 2B) and cortex (Figure S3C), but not in the cerebellum (Figure S3D). In contrast, the expression of non-NMD targets (Gapdh, Vsp4a, and Srp72) was similar in the cortex and hippocampus of control and Upf2 fb-KO mice (Figures S3E and S3F). Thus, conditional deletion of Upf2 inhibits NMD selectively in the forebrain.
Figure 2. Upf2 fb-KO Mice Exhibit Impaired NMD and LTM and Synaptic Plasticity Deficits.

(A) mRNA expression of Upf2, determined by qRTPCR analysis, was significantly decreased in the hippocampus (n = 9 control, n = 7 Upf2 fb-KO, unpaired two-tailed t test, t = 5.67) of 3-month-old Upf2 fb-KO mice compared with age-matched control littermates.
(B) mRNA expression of canonical NMD target genes was significantly increased in the hippocampus (n = 8–9/group, unpaired two-tailed t test; Atf4 t = 9.90, Gadd45b t = 3.24, Pdrg1 t = 8.64, Cars t = 4.02, Ddit3 t = 10.78, Rassf1 t = 7.16) of 3-month-old Upf2 fb-KO mice compared with control mice.
(C) Schematic of the contextual fear conditioning paradigm.
(D) Freezing times were recorded before (naive) and 24 h post-training. Reduced freezing 24 h after training in 3-month-old Upf2 fb-KO mice indicates impaired LTM (n = 20control, n = 10 Upf2fb-KO, two-way repeated-measures ANOVA, F1,28 = 84.02, p < 0.0001 [between-group comparison: control versus Upf2 fb-KO naive t = 0.32, p > 0.99, control versus Upf2 fb-KO 24 h t = 11.11]).
(E) Schematic of the Morris water maze test.
(F) In the hidden-platform version of the Morris water maze, escape latencies on days 4–7 were significantly longer in 3-month-old Upf2 fb-KO mice compared with control mice (n = 13 control, n = 11 Upf2 fb-KO, two-way repeated-measures ANOVA, F1,22 = 24.64, p < 0.0001 [between-group comparison: control versus Upf2 fb-KO day 4 t =2.98, day 5 t = 4.04, day 6 t = 4.05, day 7 t = 3.73]).
(G) During the probe trial on day 8, only control mice showed preference for the target quadrant (n = 13 control, n = 11 Upf2 fb-KO, two-way repeated-measures ANOVA, F3,88 = 9.56, p < 0.0001 [between-group comparison: control versus Upf2 fb-KO target t = 4.08]), indicating that spatial LTM was impaired in Upf2 fb-KO mice.
(H) Late LTP (L-LTP) elicited by four trains of high-frequency stimulation (HFS; 4 × 100 Hz) was impaired in 3-month-old Upf2 fb-KO slices versus control slices (n = 11 control, n = 9 Upf2 fb-KO; at 200 min, one-way ANOVA, F1,18 = 12.98). Superimposed single traces were recorded before and 220 min after tetani.
(I) Schematic of the Drosophila learning and memory paradigm.
(J and K) Compared with control flies, knockdown of Upf2 in fly brains (elav-GAL4 > Upf2RNAi/+) caused a significant deficit in LTM (J; Upf2RNAi/+, n = 7/group, unpaired two-tailed t test, t = 2.37) but not in 3-min memory (K; n = 5–6/group, t = 1.80, p = 0.11).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SEM.
See also Figures S3 and S4.
Individuals with pathogenic variants in core NMD components exhibit NDD, including learning and memory problems (Laumonnier et al., 2010; Nguyen et al., 2013, 2014; Tarpey et al., 2007). Because the hippocampus is required for memory formation (Milner and Klein, 2016), we first studied a form of hippocampus-dependent memory called contextual fear conditioning. Mice were trained by pairing a context (conditioned stimulus [CS]) with a foot shock (unconditioned stimulus [US]). Twenty-four hours after training, mice were exposed to the context (CS) and fear responses (i.e., the mouse stops moving [“freezing behavior”]) were measured as an index of the strength of their LTM (Figure 2C). LTM was significantly impaired in 3-month-old Upf2 fb-KO mice (Figure 2D) but not in 1-month-old control and Upf2 fb-KO mice (Figure S4A). These observations are consistent with the temporal expression of the Camk2α promoter (Tsien et al., 1996) and the finding that Upf2 mRNA levels are significantly decreased at 3 months (Figure 2A), but not at 1 month (Figure S4B), in the hippocampus of Upf2 fb-KO mice.
To test whether Upf2 fb-KO mice are also impaired in another hippocampus-dependent memory task, we assessed spatial LTM in the Morris water maze. In this task, mice use visual cues to find a hidden platform in a circular pool of opaque water (Figure 2E). Compared with controls, Upf2 fb-KO mice took significantly longer to find the hidden (submerged) platform (Figure 2F) and failed to remember the platform location during the probe test, performed in the absence of the platform (Figure 2G). It is noteworthy that no significant differences were observed in either swimming speed (Figure S4C) or the visible platform version (Figure S4D) of the task. Hence, inhibition of Upf2-dependent NMD impairs both contextual and spatial LTM storage.
Sustained changes in synaptic efficacy are believed to constitute the cellular basis of learning and memory. The most studied manifestation of synaptic plasticity is long-term potentiation (LTP), which refers to long-lasting increases in synaptic strength (Malenka, 1994; Neves et al., 2008). Thus, we next examined LTP, induced by applying four trains of high-frequency stimulation (HFS; 4 × 100 Hz) at Schaffer collateral-CA1 synapses in the hippocampus. Consistent with deficits in hippocampal LTM, long-lasting LTP was significantly impaired in Upf2 fb-KO hippocampal slices (Figure 2H). The impaired LTP in Upf2 fb-KO slices was not due to changes in basal synaptic transmission because input-output curves (Figures S4E and S4F) and paired-pulse facilitation (Figure S4G) did not significantly differ between slices from control and Upf2 fb-KO mice. Thus, deficient Upf2-dependent NMD impairs both hippocampal LTM and long-lasting LTP.
Given that Upf2 is evolutionarily conserved (Karousis and Muhlemann, 2019; Kervestin and Jacobson, 2012; Popp and Maquat, 2013), we next investigated whether its function is required for LTM formation in Drosophila. As in mice, deletion of Upf2 is lethal in flies (Avery et al., 2011; Metzstein and Krasnow, 2006). Thus, we reduced Upf2 levels specifically in the fly brain using the GAL4-upstream activating sequence (UAS) system and a conditional Upf2 RNAi construct (UAS-Upf2RNAi; STAR Methods). As expected, in the brain of Upf2RNAi flies, Upf2 levels were significantly decreased (Figure S4H), and NMD was inhibited, as determined by increased expression of canonical Drosophila NMD substrates (Chapin et al., 2014; Rehwinkel et al., 2005; Figure S4I). Interestingly, when tested in classical olfactory conditioning (Figure 2I)—a well-studied form of associative learning where flies are trained to associate an odor with an aversive electric shock (Tully et al., 1994)—LTM was impaired in Upf2RNAi flies (Figure 2J). This impaired LTM is unlikely to be a consequence of defects in sensorimotor processing because short-term memory (STM) in Upf2RNAi flies did not significantly differ from that of control flies (Figure 2K). Taken together, our data indicate that deficient Upf2-dependent NMD impairs LTM storage in both flies and mice, highlighting the evolutionarily conserved role of Upf2 in learning and memory.
Inhibition of Upf2-Dependent NMD Results in Deficits in Social Behavior and Behavioral Inflexibility
In humans, pathogenic variants in NMD genes are associated with ASD (Laumonnier et al., 2010; Nguyen et al., 2013, 2014; Tarpey et al., 2007). Because deficits in social interaction are salient features of ASD individuals (Mefford et al., 2012), we first studied social behavior in both control and Upf2 fb-KO mice. To this end, we used the three-chamber test to measure (1) sociability by comparing the time mice spent interacting with an empty wire cage (empty) versus one containing a mouse (mouse 1) and (2) preference for social novelty by measuring the time mice spent interacting with a familiar (mouse 1) versus an unfamiliar (stranger) mouse (mouse 2; Figure 3A). Although both control and Upf2 fb-KO mice had normal sociability (Figure 3B), Upf2 fb-KO mice had no preference for social novelty, as indicated by the same amount of time spent with a novel and a familiar mouse (Figure 3C). Consistent with an impairment in social novelty, Upf2 fb-KO mice were also significantly impaired in reciprocal social interaction (Figures 3D and 3E).
Figure 3. Upf2 fb-KO Mice Have Deficits in Social Behavior and Impaired Behavioral Flexibility.
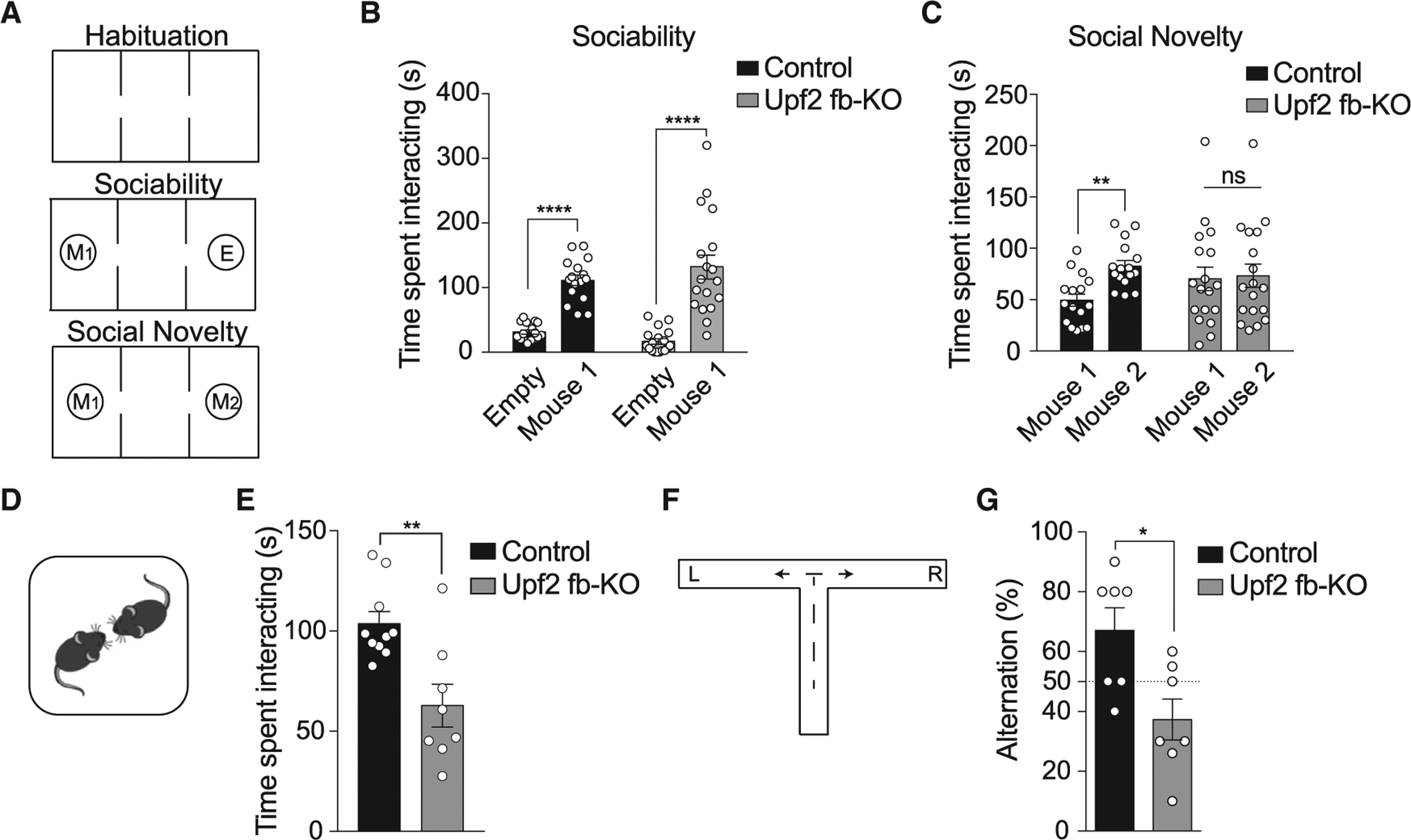
(A) Schematic of the three-chamber social interaction task.
(B) In the sociability test, both 3-month-old control and Upf2 fb-KO mice spent more time interacting with a mouse than with an empty cup (n = 16 control, n = 18 Upf2 fb-KO, two-way ANOVA, F1,64 = 80.21, p < 0.0001 [between-group comparison: control empty versus mouse 1 t = 5.05; Upf2 fb-KO empty versus mouse 1 t = 7.70]).
(C) In the social novelty test, unlike controls, Upf2 fb-KO mice showed no preference for interacting with a novel versus a familiar mouse (n = 16 control, n = 18 Upf2 fb-KO, two-way repeated-measures ANOVA, F1,32 = 5.70, p < 0.05 [between-group comparison: control mouse 1 versus mouse 2 t = 3.02; Upf2 fb-KO mouse 1 versus mouse 2 t = 0.28, p > 0.99]).
(D) Schematic of the reciprocal social interaction test.
(E) Compared with control mice, 3-month-old Upf2 fb-KO mice had significantly reduced reciprocal social interaction (n = 10 control, n = 8 Upf2 fb-KO, unpaired two-tailed t test, t = 3.36).
(F) Schematic of the T-maze spontaneous alternation task.
(G) Compared with controls, 3-month-old Upf2 fb-KO mice were impaired in spontaneous alternation (n = 7/group, unpaired two-tailed t test, t = 2.95), which was not significantly different from the chance level (50%; n = 7/group, p = 0.11).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SEM.
Another trait commonly observed in children with ASD is behavioral inflexibility (American Psychiatric Association DSM Task Force, 2013). To test behavioral inflexibility in mice, we measured the willingness of mice to explore a new environment in a T-maze (Figure 3F). While exploring the maze, mice normally tend to alternate between the two arms. However, Upf2 fb-KO mice tended to repeatedly explore the same arm of the maze, an indication of behavioral inflexibility (Figure 3G). Hence, conditional deletion of Upf2 leads to deficits in social behavior and behavioral inflexibility.
Inhibition of Upf2-Dependent NMD Triggers Immune Activation
To identify the gene expression program controlled by NMD in an unbiased manner, we compared steady-state mRNA levels between Upf2 fb-KO and control mice. Deletion of Upf2 in the forebrain led to increased expression of approximately 300 genes (fold change ≥ 1 and false discovery rate [FDR] < 0.05; Figure S5A; Table S4]. Strikingly, Gene Ontology (GO) analysis identified a significant enrichment in immune-related genes, including the innate and adaptive immune system, antigen presentation and processing, and the complement cascade (Figure S5B).
Given the marked increase in expression of immune-related genes in the brains of Upf2 fb-KO mice, we further investigated the role of the immune response in these mice. Flow cytometry analyses revealed a significant increase in the total number of immune cells in the brain of Upf2 fb-KO mice compared with controls (Figure 4A). Specifically, Upf2 fb-KO mice exhibited a progressive increase in the number of CD3+ (T cells), CD11b+ (dendritic cells, macrophages, and microglia), B220+ (B cells), and Ly6G+ cells (neutrophils; Figures 4B and 4C; Figure S6A). In contrast, the numbers of immune cells in the spleen were similar in control and Upf2 fb-KO mice (Figures S6B–S6D), indicating that the immune response is specifically increased in the brain, where NMD is dysfunctional. In addition, we detected a significant increase in the pro-inflammatory cytokine RANTES (Figure 4D) and the T cell chemoattractants IP-10 (Figure 4E) and MIG (Figure S6E) only in the forebrain of Upf2 fb-KO mice. Moreover, when we stimulated T cells with forebrain homogenate from control or Upf2 fb-KO mice, we found that only homogenate from Upf2 fb-KO mice led to a significant T cell-proliferative response (Figure 4F). These data indicate that loss of Upf2-dependent NMD in the brain leads to immune activation. We also observed activation of the brain immune response, as evidenced by marked microgliosis (Iba1; Figure 4G) and astrogliosis (GFAP; Figure 4H) in the hippocampus and neocortex of Upf2 fb-KO mice. Thus, the loss of Upf2 results in an exacerbated neuroinflammatory response.
Figure 4. Immune Activation in the Brain of Upf2 fb-KO Mice.

(A) Compared with controls, Upf2 fb-KO mice exhibited a significant and progressive increase in the total number of immune cells in the brain (n = 3–7/group, two-way repeated-measures ANOVA, F2,22 = 7.91, p < 0.01 [between-group comparison: control versus Upf2 fb-KO 1 month t = 0.25, p > 0.99; 3 month t = 2.57, p = 0.05; 5 month t = 5.22]).
(B and C) The numbers of CD3+, Cd11b+, B220+, and Ly6G+ cells were normal in the brains of 1-month-old Upf2 fb-KO mice (B; n = 3 control, n = 5 Upf2 fb-KO, unpaired two-tailed t test, CD3+ t = 0.65, p = 0.58; CD11b+ t = 0.05, p = 0.96; B220+ t = 0.65, p = 0.54; Ly6G+ t = 0.48, p = 0.65) but significantly increased in 3-month-old Upf2 fb-KO mice (C; n = 4–7/group, unpaired two-tailed t test, CD3+ t = 2.78; CD11b+ t = 3.00; B220+ t = 4.92; Ly6G+ t = 1.63, p = 0.17).
(D and E) The pro-inflammatory cytokine RANTES (D; n = 3/group, one-way ANOVA, F7,16 = 6.69, p < 0.001 [between-group comparison: control versus Upf2 fb-KO cortex t = 3.85; hippocampus t = 4.32; midbrain t = 0.34, p > 0.99; cerebellum t = 0.45, p = 0.99]) and the T cell chemoattractant IP-10 (E; n = 3/group, one-way ANOVA, F7,16 = 6.80, p < 0.001 [between-group comparison: control versus Upf2 fb-KO cortex t = 3.78; hippocampus t = 4.35; midbrain t = 0.53, p = 0.98; cerebellum t = 0.43, p = 0.99]) were significantly increased in the cortex and hippocampus of 3-month-old Upf2 fb-KO mice.
(F) Control T cells were stimulated with brain homogenate from control and Upf2 fb-KO mice. Only the Upf2 fb-KO homogenate significantly increased T cell proliferation (n = 6/group, one-way ANOVA, F2,15 = 5.79, p < 0.01 [between-group comparison: resting versus control t = 1.46, p = 0.33; resting versus Upf2 fb-KO t = 3.39]).
(G and H) Upf2 fb-KO brains also exhibit increased microglial cell activation (G; Iba1+) and reactive astrogliosis (H; GFAP+) in the cortex and hippocampus. Scale bars indicate a length of 100 μm.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SEM.
See also Figures S5 and S6.
Reduction of Brain Inflammation Reverses the Synaptic and Behavioral Deficits in Upf2 fb-KO Mice
We hypothesized that the exacerbated activation of the immune response in the brain could lead to the synaptic and behavioral deficits observed in Upf2 fb-KO mice. To test this hypothesis, we treated mice with cyclophosphamide (CyP), an FDA-approved immunosuppressant that is used to treat diseases characterized by an aberrant immune response, including autoimmunity (Perini et al., 2008). Mice were treated with a low concentration of CyP at 1 month of age, when Upf2 fb-KO mice show normal behavior (Figure S4A). As expected, chronic treatment with a low concentration of CyP reduced the inflammatory response in the hippocampus of Upf2 fb-KO mice, leading to a decrease in immune cells (Figure 5A), reactive astrogliosis (GFAP), and microgliosis (Iba1; Figure S7A) in the brain. More importantly, CyP treatment significantly ameliorated contextual fear LTM in Upf2 fb-KO mice (Figure 5B). Results from several control experiments underscore the specificity of CyP’s action. Specifically, CyP did not affect contextual fear LTM in control littermates (Figure 5C) or freezing responses of naive Upf2 fb-KO mice (Figure 5B). Consistent with the behavioral results, CyP treatment also reversed the deficits in LTP in Upf2 fb-KO mice (Figure 5D) but had no effect in control mice (Figure 5E). Collectively, these results show that treatment with a low concentration of CyP reverses, at least in part, the deficits in LTM and synaptic plasticity in Upf2 fb-KO mice.
Figure 5. Treatment with a Low Dose of CyP Prevents Abnormal Immune Activation and Rescues Behavioral Deficits in Upf2 fb-KO Mice.
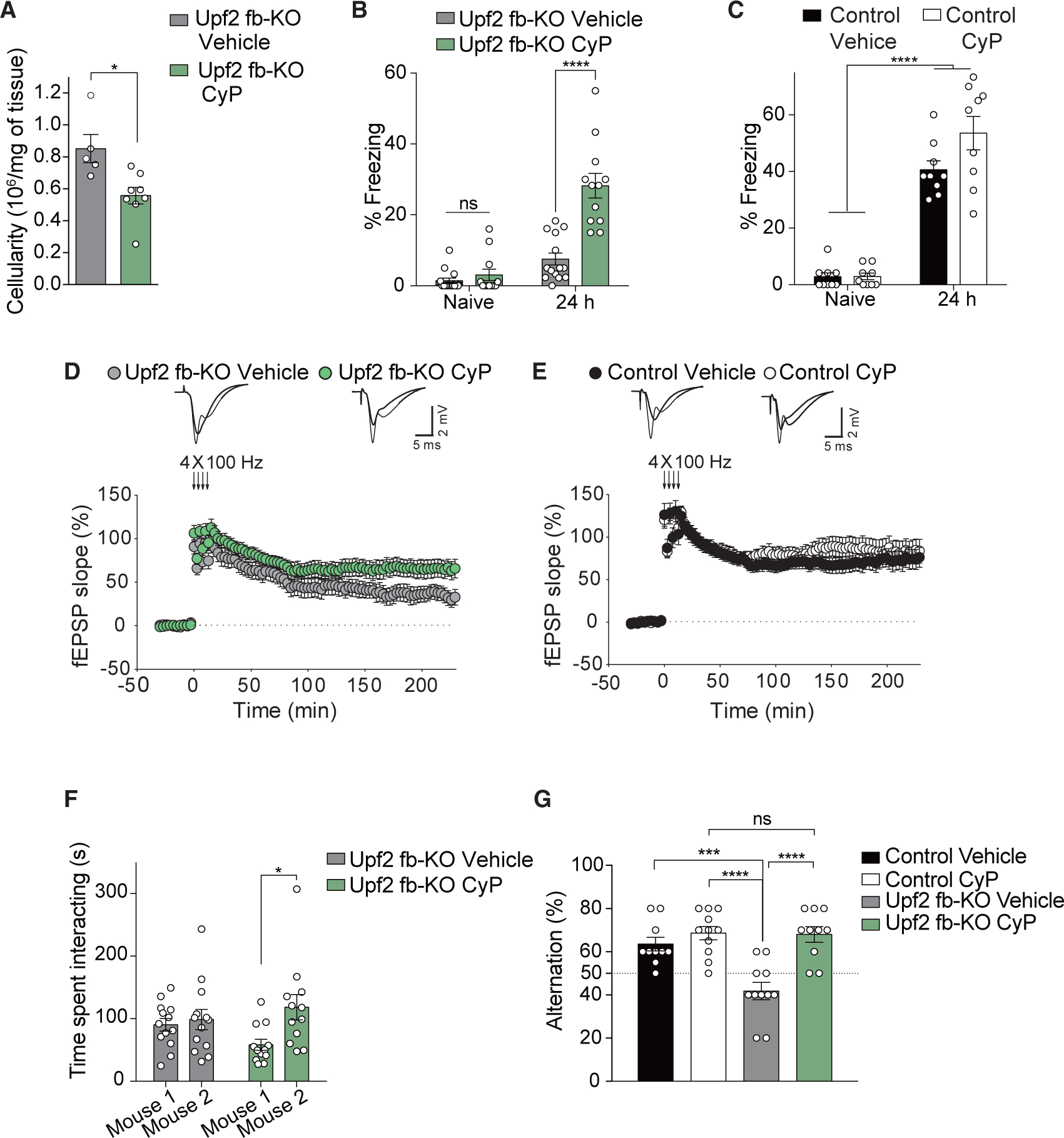
(A) Compared with vehicle-treated mice, chronic treatment of Upf2 fb-KO mice with CyP significantly decreased the number of immune cells in the brain (n = 5 vehicle, n = 8 CyP, unpaired two-tailed t test, t = 3.10).
(B and C) Unlike vehicle-treated mice, chronic treatment with CyP partially restored contextual fear LTM in Upf2 fb-KO mice (B; n = 14 vehicle, n = 12 CyP, two-way repeated-measures ANOVA, F1,48 = 22.04, p < 0.0001 [between-group comparison: naive versus 24 h vehicle t = 0.58, p = 0.81; naive versus 24 h CyP t = 7.22]) but had no effect in control mice (C; n = 9/group, two-way repeated-measures ANOVA, F1,16 = 2.81, p = 0.11 [between-group comparison: naive versus 24 h vehicle t = 9.12; naive versus 24 h CyP t = 12.22]).
(D and E) CyP treatment improved L-LTP in Upf2 fb-KO slices (D; n = 10/group, one-way ANOVA, F1,18 = 4.56) but had no effect on L-LTP in control slices (E; n = 9 vehicle, n = 10 CyP, one-way ANOVA, F1,17 = 0.02, p = 0.89). Superimposed single traces were recorded before and after 220 min tetani.
(F) In the three-chamber social behavior assay, CyP treatment rescued social novelty in Upf2 fb-KO mice (n = 13 vehicle, n = 12 CyP, two-way ANOVA, F1,23 = 5.73, p < 0.05 [between-group comparison: mouse 1 versus mouse 2 vehicle t = 0.43, p > 0.99; mouse 1 versus mouse 2 CyP t = 2.91]).
(G) In the T-maze spontaneous alternation task, CyP treatment reversed alternation performance (n = 10–11/group, two-way repeated-measures ANOVA, F1,38 = 10.16, p < 0.005 [between-group comparison: control vehicle versus Upf2 fb-KO vehicle t = 4.38, control CyP versus Upf2 fb-KO vehicle t = 5.55, Upf2 fb-KO vehicle versus Upf2 fb-KO CyP t = 5.29, control CyP versus Upf2 fb-KO CyP t = 0.13, p > 0.99]).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SEM.
See also Figure S7.
We next tested whether CyP treatment could also improve the social deficits and behavioral inflexibility in Upf2 fb-KO mice. CyP treatment was sufficient to reverse their deficits in preference for social novelty (Figure 5F) and did not affect sociability in Upf2 fb-KO mice (Figure S7B), which was normal in these mice. Moreover, CyP treatment fully reversed behavioral inflexibility because Upf2 fb-KO mice treated with CyP performed like control mice in the T-maze (Figure 5G). Hence, chronic treatment with a low concentration of CyP reverses social deficits and behavioral inflexibility in Upf2 fb-KO mice.
To corroborate that CyP’s beneficial effects in Upf2 fb-KO mice are due to dampening of the exacerbated immune response, Upf2 fb-KO mice were treated with minocycline (Mino), a tetracycline antibiotic known to reduce inflammation in the central nervous system (CNS) and used for the treatment of autoimmune disorders (Garrido-Mesa et al., 2013). Like with CyP treatment, mice were treated with Mino beginning at 1 month of age. We found that chronic treatment with Mino reduced the inflammatory response in the hippocampus of Upf2 fb-KO mice, as determined by a decrease in Iba1+ signal (Figure S7D). Similar to treatment with CyP, Mino treatment significantly improved contextual fear LTM in Upf2 fb-KO mice (Figure 6A) but had no effect on contextual fear LTM in control mice (Figure 6B). Mino treatment improved LTP (Figure 6C) and rescued preference for social novelty only in Upf2 fb-KO mice (Figures 6D and 6E). Again, sociability, which is normal in Upf2 fb-KO mice, was not affected by Mino treatment (FigureS7C). Last, behavioral inflexibility was completely reversed because Upf2 fb-KO mice treated with Mino performed like control mice in the T-maze (Figure 6F). Collectively, these results show that treatment with either CyP or Mino reverses, at least in part, the deficits in LTM and synaptic plasticity as well as behavioral inflexibility and social behavior deficits in Upf2 fb-KO mice.
Figure 6. Treatment with Mino Rescues Behavioral Deficits in Upf2 fb-KO Mice.
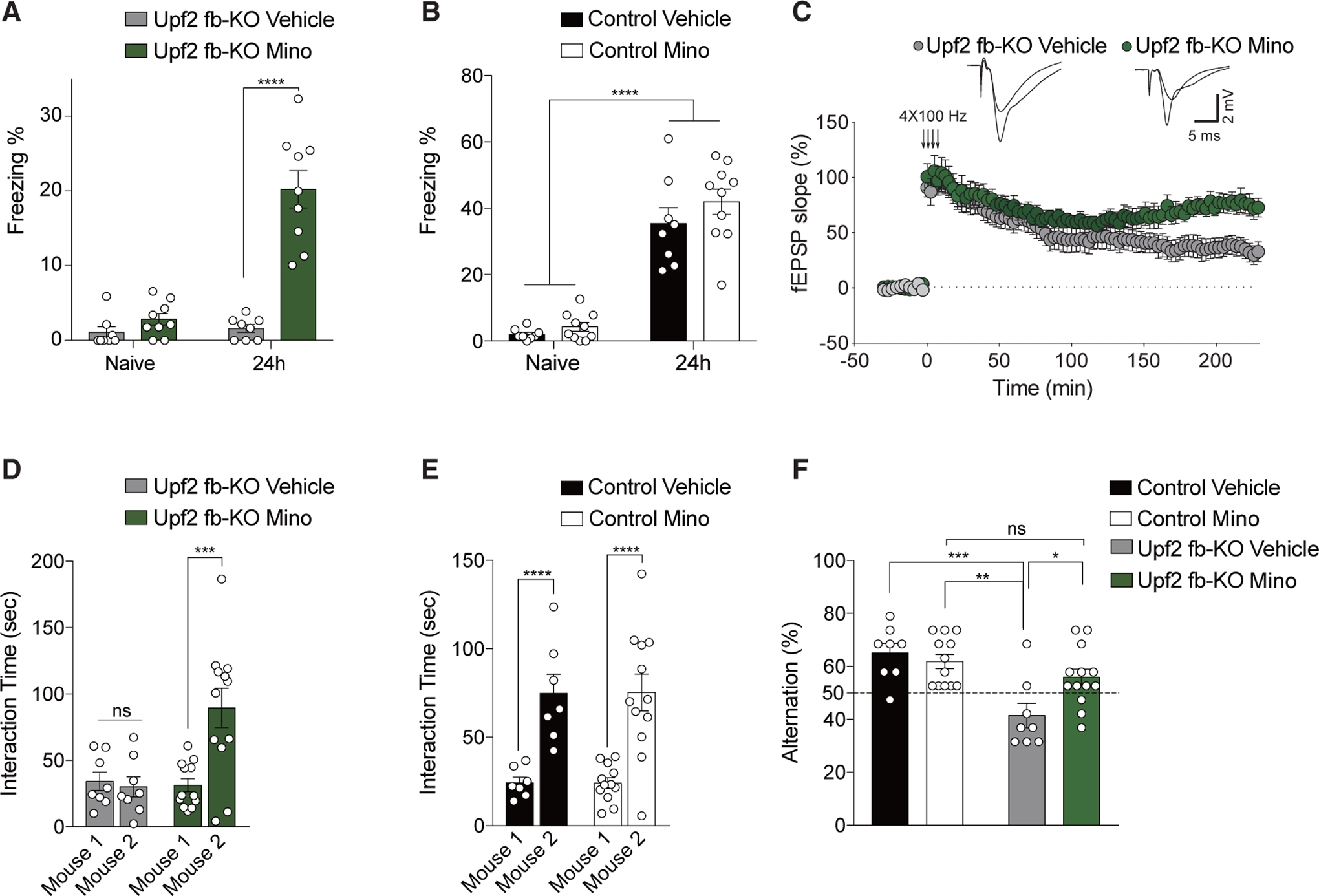
(A and B) Chronic treatment with Mino partially restored contextual fear LTM 24 h after training in Upf2 fb-KO mice (A; n = 8 vehicle, n = 9 Mino, two-way ANOVA, F1,15 = 37.69, p < 0.0001 [between-group comparison: vehicle versus Mino naive t = 0.86, p = 0.80; vehicle versus Mino 24 h t = 9.05]) but had no effect in control mice (B; n = 8 vehicle, n = 10 Mino, two-way ANOVA, F1,16 = 0.50, p = 0.45 [between-group comparison: naive versus 24 h vehicle t = 7.46; naive versus 24 h Mino t = 9.40]).
(C) Mino treatment improved L-LTP in Upf2 fb-KO slices (C; n = 10 vehicle, n = 7 Mino, one-way ANOVA, F1,13 = 26.26). Superimposed single traces were recorded before and after 220 min tetani.
(D and E) Mino treatment rescued social behavior in Upf2 fb-KO mice (D; n = 8 vehicle, n = 12 Mino, two-way ANOVA, F1,20 = 6.45, p < 0.05 [between-group comparison: mouse 1 versus mouse 2 vehicle t = 0.55, p > 0.99; Mino t = 4.37]) but had no effect in control mice (E; n = 7 vehicle, n = 12 Mino, two-way ANOVA, F1,20 = 0.01, p = 0.93 [between-group comparison: mouse 1 versus mouse 2 vehicle t = 5.70, Mino t = 6.39]).
(F) In the T-maze spontaneous alternation task, Mino treatment improved behavioral inflexibility in Upf2 fb-KO mice (n = 8 vehicle, n = 12–13 Mino, two-way repeated-measures ANOVA, F1,37 = 18.35, p < 0.001 [between-group comparison: Upf2 fb-KO vehicle versus Upf2 fb-KO Mino t = 2.97, control vehicle versus Upf2 fb-KO vehicle t = 4.38, control Mino versus Upf2 fb-KO vehicle t = 4.13, control Mino versus Upf2 fb-KO Mino t = 1.14, p > 0.99]).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SEM.
See also Figure S7.
Upf2 fb-KO Mice Display Vocal Communication Deficits that Are Reversed by Treatment with CyP or Mino
Currently, there are no effective pharmacological treatments for speech and language problems. Given that individuals with pathogenic variants in UPF2 show speech and language deficits, we examined whether Upf2 fb-KO mice have communication deficits by assessing ultrasonic vocalizations (USVs). Mice emit USVs in different contexts throughout development and in adulthood. Importantly, a reduced level of calling and/or an unusual calling pattern have been reported in several mouse models of ASD (Wöhr and Scattoni, 2013). To test whether vocal/communication behavior is impaired in Upf2 fb-KO mice, we recorded USVs from interacting, same-sex adult female mice (Figure 7A). Compared with control littermates, we found that Upf2 fb-KO mice emitted significantly fewer interaction-induced USVs (Figure 7B). USV calls can be classified into five main categories (Figure S7E; Ferhat et al., 2016). Among the USVs emitted from Upf2 fb-KO mice, the call patterns were strikingly different from those of controls and consisted of more complex calls that were louder and of longer duration (Figures 7C–7E). Consistent with the therapeutic benefit of CyP and Mino for the behavioral abnormalities in Upf2 fb-KO mice, we found that treatment with CyP or Mino also improved the vocal behavior deficits in Upf2 fb-KO mice, including call rate and call pattern (Figures 7C–7E). Thus, similar to the speech and language deficits observed in individuals carrying UPF2 variants, we found that impaired Upf2-dependent NMD in mice leads to vocal and communication problems. Importantly, the vocal communication problems in Upf2 fb-KO mice can be reversed through pharmacological dampening of the aberrant immune response.
Figure 7. Upf2 fb-KO Mice Display Abnormal USVs that Are Reversed by Treatment with CyP or Mino.

(A) A schematic of the same-sex social interaction assay used for USV recording. Two female mice from the same genotype group were placed in an open cage, and a microphone was placed above the middle of the cage to record calls from the mice.
(B) Quantification of the call rate in adult female-female interactions. Compared with controls, Upf2 fb-KO mice emitted significantly fewer calls (n = 12 pairs/24 mice control, n = 14 pairs/28 mice Upf2 fb-KO, one-way repeated-measures ANOVA, F3,37 = 5.19, p <0.01 [between-group comparison: control versus Upf2 fb-KO t = 3.15]). Chronic treatment with Mino rescued the call rate in Upf2 fb-KO mice to a similar level as in control mice (n = 12 pairs/24 mice control, n = 8 pairs/16 mice Mino, one-way repeated-measures ANOVA, F3,37 = 5.19, p <0.01 [between-group comparison: Upf2 fb-KO versus Upf2 fb-KO Mino t = 3.46]).
(C) Representative sonograms of USVs produced by adult female mice in all groups.
(D and E) Call types were classified into five major types: short, simple, complex, frequency jumps, and unstructured calls. For each group, the distribution (D) and quantification (E) of call types were plotted. Upf2 fb-KO mice emitted significantly more complex calls of longer duration and higher intensity compared with control pairs; fewer short and simple calls were recorded in Upf2 fb-KO same-sex social interaction compared with control pairs (n = 12 pairs/24 mice control, n = 14 pairs/28 mice Upf2 fb-KO, one-way ANOVA, F9,120 = 25.58, p < 0.0001 [between-group comparison: short calls t = 3.42; simple calls t = 4.39; complex calls t = 4.22; frequency jumps t = 1.62, p > 0.9999; unstructured calls t = 1.98, p > 0.99]). This abnormal call pattern was restored to one similar to controls after chronic treatment with CyP (n = 12 pairs/24 mice control, n = 7 pairs/14 mice CyP, one-way ANOVA, F9,95 = 22.99, p < 0.0001 [between group comparison: short calls t = 3.03, p = 0.14; simple calls t = 5.37; complex calls t = 3.95; frequency jumps t = 2.77, p = 0.31; unstructured calls t = 1.68, p = 0.99]) or Mino (n = 12 pairs/24 mice control, n = 8 pairs/16 mice Mino, one-way ANOVA, F9,100 = 23.88, p < 0.0001 [between-group comparison: short calls t = 1.42, p > 0.99; simple calls t = 4.75;complex calls t = 3.71; frequency jumps t = 1.24, p > 0.99; unstructured calls t = 1.22, p > 0.99]).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SEM.
See also Figure S7.
DISCUSSION
Protein Synthesis versus NMD in Cognitive Processes
It is widely believed that long-lasting forms of synaptic plasticity and memory require new protein synthesis (Costa-Mattioli et al., 2009; Kandel, 2001). Genetic inhibition of key transcription factors, including CREB, C/EBP, Egr, AP-1, and NPAS4, demonstrated that transcription (coupled with translation) is crucially required for memory formation (Alberini and Kandel, 2014; Silva et al., 1998). Moreover, we and others have deciphered some of the key translational control mechanisms involved in memory processes (Costa-Mattioli et al., 2009; Klann and Dever, 2004). Importantly, impaired protein synthesis control has been associated with neurodevelopmental dysfunction, as underscored by the identification of variants in genes encoding proteins that directly regulate translation rates in these disorders (Buffington et al., 2014; Sossin and Costa-Mattioli, 2019).
NMD is a protein synthesis-dependent process (Karousis and Muhlemann, 2019; Kervestin and Jacobson, 2012; Popp and Maquat, 2013). In mammalian cells, detection of PTCs by NMD occurs during the pioneer round of translation (Baker and Parker, 2004; Ishigaki et al., 2001). Consequently, inhibition of translation at the level of initiation or elongation, using either genetic or pharmacological agents, invariably blocks NMD because it prevents detection of PTC-containing mRNAs by the scanning ribosome (Baker and Parker, 2004; Karousis and Muhlemann, 2019; Kervestin and Jacobson, 2012; Popp and Maquat, 2013). Given that all manipulations that inhibit translation also impair NMD, it is not immediately clear whether the impairment in LTM consolidation (or long-lasting LTP) caused by inhibiting protein synthesis rates is due to (1) the inability to synthesize new proteins or (2) inhibition of NMD. Although our experiments are not able to differentiate between these two possibilities, our results showing that inhibition of NMD in both mice and flies blocks LTM storage may prompt the field to further study the role of NMD in synaptic plasticity and memory processes. In addition, it would be interesting to explore the function of NMD in neurological disorders in which translation is inhibited.
Impaired NMD Leads to Intellectual Disability, ASD, and Speech and Language Deficits
How impaired Upf2-mediated NMD leads to NDD is not yet understood (Jaffrey and Wilkinson, 2018). Here we generated a Upf2-deficient mouse model that recapitulates some of the salient aspects of the human phenotype associated with dysfunctional NMD. We found that Upf2 fb-KO mice exhibit severe impairments in learning and memory and corresponding deficits in synaptic plasticity (Figures 2A–2H). Similarly, genetic inhibition of Upf2 blocks LTM in flies (Figure 2J), supporting the notion that the role of Upf2 in memory formation is conserved through evolution. Moreover, mice lacking Upf3b exhibit impaired contextual fear memory (Huang et al., 2018). Thus, inhibition of NMD, either by deleting Upf2 or Upf3b, impairs LTM storage.
In addition, variants in UPF2 and other NMD core factors are strongly associated with NDD, including ASD (Nguyen et al., 2013). We found that Upf2 fb-KO mice exhibit endophenotypes associated with ASD, including social deficits and repetitive or restricted behaviors (Figures 3A–3G). Although the clinical presentations of ASD associated with dysfunctional NMD are highly heterogeneous, one of the major hallmarks is impairment of language development and verbal communication (Nguyen et al., 2013). Indeed, variants in additional genes shown to be causal for NDDs, including the FOXP transcription factors and CNTNAP2, have also been implicated in speech and language development (Fisher and Scharff, 2009; Hamdan et al., 2015; Morgan and Webster, 2018; Rodenas-Cuadrado et al., 2016). Our data describing UPF2 frameshift variants that impair NMD in cases with varied speech and language deficits (Figure 1) support the idea that mutation of UPF2 and, consequently, impaired NMD may be post-transcriptional causes of communication deficits. Similar to humans with variants in UPF2 displaying speech sounds disorders, mice lacking Upf2 exhibit abnormal USVs (Figure 7). It remains to be determined whether patients with variants in other NMD components, such as UPF3B, exhibit similar speech and language difficulties as those observed in individuals carrying UPF2 variants.
Impaired NMD Leads to Increased Inflammation
Our data demonstrate that the behavioral and neurophysiological phenotypes observed in Upf2 fb-KO mice can be attributed to increased brain inflammation; treatment with two FDA-approved anti-inflammatory drugs, CyP and Mino, ameliorated the behavioral and synaptic abnormalities in these mice. Thus, our results suggest that treatment with specific immune system suppressors could relieve selective behavioral symptoms in human NDDs in which NMD is dysfunctional. Both impaired mRNA metabolism (McMahon et al., 2016) and immune dysregulation (Boulanger and Shatz, 2004; Mostafa et al., 2013) have been implicated in NDDs, but little is known about the relationship between these two processes. Our finding that impaired NMD triggers an immune response in the brain of Upf2-deficient mice links these two axes of dysfunction. Consistent with our results, inhibition of NMD in tumor cells leads to immune-mediated inhibition of tumor growth in mice through the expression of new antigenic determinants and their immune-mediated rejection (Pastor et al., 2010). Moreover, in Arabidopsis, NMD deficiency results in aberrant growth caused by an autoimmune-like response (Riehs-Kearnan et al., 2012).
Because of its potent immunosuppressive activity, CyP is the foundation of many conditioning regimens used to treat autoimmune disorders, including multiple sclerosis (Dezern et al., 2013; Mitwalli et al., 2011). However, treatment of autoimmune disorders with high doses of CyP can lead to adverse effects. Given this, we used a low dose of CyP that reduced the exacerbated immune response in the brain of Upf2 fb-KO mice (Figures 4A and S7A), reversed their synaptic (Figure 5D) and behavioral (Figures 5B, 5F, and 5G) deficits, but had no effect in control mice (Figures 5C, 5E, and 5G). In support of these findings, we also found that treatment with Mino, a broad-spectrum tetracycline antibiotic with potent anti-inflammatory effects (Garrido-Mesa et al., 2013), significantly improved LTM, LTP, social behavior deficits, and behavioral inflexibility in Upf2 fb-KO mice (Figures 6A, 6C, 6D, and 6F). In addition, both CyP and Mino reversed the abnormal USVs in Upf2 fb-KO mice (Figure 7). It will be interesting to examine whether these treatment regimens are also effective in other mouse models with vocalization and communication deficits.
In summary, our findings show that impaired Upf2-dependent NMD leads to learning and memory impairments, social deficits, behavioral inflexibility, and deficits in vocal communication. Unexpectedly, we found that the aberrant immune response in the brain caused by impaired NMD leads to behavioral neurophysiological deficits in Upf2 fb-KO mice. Last, our results hold promise that CyP or Mino, two FDA-approved anti-inflammatory drugs for which long-term safety and tolerability have been extensively documented, could be promptly repurposed for the treatment of neurodevelopmental disorders in which NMD is dysfunctional.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mauro Costa-Mattioli (costamat@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human participants
We studied three cases (Figure 1A) with pathogenic UPF2 variants. Age and sex for each case is listed in Figure 1A. The Human Research Ethics Committee of the Royal Children’s Hospital HREC37353 approved this study and informed consent was obtained. Participants completed face-to-face clinical measures outlined below. All were assessed by a clinical geneticist and speech and language pathologist. A standard case history and medical interview was conducted with the parents to confirm medical and developmental history (Mei et al., 2018; Morgan and Webster, 2018). Case 3 was non-verbal and was assessed using the Vineland Adaptive Behavior Scales interview format (Sparrow et al., 2005). For cases 1 and 2, evaluation of speech occurred across: articulation disorder (deficit at the phonetic, motoric level of speech, e.g., phonetic distortions such as a lisp), phonological disorder (deficit at the phonemic level, e.g., use of sound error patterns such as substituting /f/ for /th/ or weak syllable deletion, ‘mato’ for tomato), dysarthria (impaired neuromuscular execution of speech associated with disruption of tone and/or incoordination of movements, e.g., speech may sound slurred), and childhood apraxia of speech (impaired planning and programming of speech impacting on the ability to automatically and correctly sequence sounds and syllables) (Morgan et al., 2017; Morgan and Webster, 2018). Articulation and phonological deficits were identified with the Diagnostic Evaluation of Articulation and Phonology (Dodd et al., 2002). Features of childhood apraxia of speech, including syllable segregation, epenthesis, metathesis, omission errors, vowel errors and prosodic disturbance (Mei et al., 2018; Morgan et al., 2018) were assessed using transcriptions of the DEAP, the polysyllable single word test (Gozzard et al., 2008) and a 5-minute conversational speech sample. Dysarthria was evaluated based on the presence of oral tone or co-ordination disturbance impacting on oral motor function and speech as determined via the oral motor function using the Oral and Speech Motor Control Protocol (OSMCP) (Robbins and Klee, 1987) in addition to dysarthric features using the Mayo Clinic Dysarthria rating scale (Duffy, 2013). Deficits in receptive and expressive language for cases 1 and 2 were recorded based on the Clinical Evaluation of Language Fundamentals (IV) Core Language subtests (Semel et al., 2006). Literacy outcomes for case 2 were measured using the Word Reading and Spelling subtests of the Wide Range Achievement Test-4 (WRAT-4) (Wilkinson and Robertson, 2006). Case 3 could not read and literacy performance for case 1 was based on parent report of child’s reading ability relative to peers in the classroom. IQ was reported based on recent psychological reports and from testing on the Weschler Abbreviated Scales of Intelligence (Wechsler, 2011). While all three cases presented with a speech impairment, the specific diagnoses and severity of the communication phenotypes, as well as cognitive involvement and dysmorphology varied, as detailed below.
Case 1 was ascertained on the basis of his primary speech and language disorder for a gene discovery project, the ‘genetics of speech and language disorders’. Case 1 had a history of severe speech disorder characterized as childhood apraxia of speech with phonological errors throughout development as confirmed through past clinical reports from age 5 onward. At the age of testing for the current study (11 years), the speech disorder had resolved to a mild phonological disorder. Birth, development and medical history was otherwise uneventful with the exception of asthma, eczema and allergic rhinitis. He was literate, but was performing below average (mildly impaired) in reading and spelling, relative to peers. IQ and receptive language were better preserved than expressive language.
Case 2 was identified through the Deciphering Developmental Disorders (DDD) study (Firth et al., 2011). She was born at 36 weeks and had a history of cleft lip and palate and VACTERL syndrome. She had tracheesophageal fistula (repaired), atrioventricular septal defect (repaired in 2009 with residual mitral valve regurgitation), right hemifacial microsomia with a missing right zygomatic arch and a marginal mandibular nerve palsy, hemivertebrae T3 to T4, fused ribs, failure to thrive, and a gastrostomy and fundoplication inserted at 17 months of age (gastrostomy feeds still given at night at this age; oral food during the day). She had a moderate speech sound disorder, including articulation deficits associated with velopharyngeal incompetence due to cleft palate and a nasal fistula, but persistent delayed phonological errors (e.g., ‘f’ for ‘th’) unrelated to the cleft, and language impairment, were also present. IQ and receptive language were better preserved than expressive language. Case 2 was literate, but performing below average (mildly impaired) relative to peers.
Case 3, identified through the Victorian Clinical Genetics Service, had Autism spectrum disorder (ASD) diagnosis first confirmed at 3 years 1 month of age with the Autism Diagnostic Observation Schedule (ADOS) and the Childhood Autism Rating Scale (CARS) with reference to Diagnostic Statistical Manual of Mental Disorders – 4th Edition, Text Revision (DSM-IV-TR) criteria. The diagnostic assessment was conducted by an experienced multidisciplinary team consisting of a speech pathologist, psychologist and pediatrician. A follow up ADOS and Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV) were completed as part of a research assessment when the participant was 8 years 6 months of age, confirming his ASD diagnosis and co-occurring intellectual disability. In addition, Case 3 had macrocephaly and could produce only a handful of sounds as is at the pre-intentional stage of communication. Hence, Case 3 was unable to read or spell in line with his significant intellectual disability.
Lymphoblastoid Cell Lines (LCLs)
The Epstein-Barr virus–immortalized B cell lines (B-LCLs) used in this study were established from peripheral blood lymphocytes as described previously (Neitzel, 1986). Established lymphoblastoid cell lines from healthy controls and individuals carrying UPF2 variants were cultured in RPMI 1640 (GIBCO/BRL) supplemented with 10% FBS, 2 mM L-glutamine and 1% penicillin/streptomycin and grown at 37°C with 5% CO2.
Mice
All mice were housed and maintained in the animal facilities at Baylor College of Medicine. The mice were kept on a 12-h light/dark cycle, and the behavioral tests were always conducted during the light phase of the cycle. The mice had access to food and water ad libitum except during tests. Male and female mice were used in all studies, with the exception of ultrasonic vocalization (USV) measurements, and no differences between sexes were observed. Mice were randomly assigned to treatment and experimental conditions. All experiments used littermates as controls and were carried out and analyzed with the experimenter blinded to genotype and treatment. Animal care and experimental procedures were approved by the institutional animal care and use committee of Baylor College of Medicine, according to US National Institutes of Health Guidelines.
Upf2loxP/loxP mice (Weischenfeldt et al., 2008) were backcrossed for at least eight generations with C57BL/6J mice and subsequently crossed with CamK2α-Cre mice (Upf2+/+; Camk2α-Cre) (Dragatsis and Zeitlin, 2000). Upf2loxP/+; Camk2α-Cre mice were crossed to both Upf2loxP/loxP and Upf2loxP/+ mice. This breeding scheme generated the following experimental mice: Upf2 fb-KO (Upf2loxP/loxP; Camk2α-Cre mice) and three sets of control littermates (Upf2+/+ mice, Upf2+/+; Camk2α-Cre mice, and Upf2loxP/loxP mice) that were pooled and defined as the “control” group. Mice were weaned at the third postnatal week and genotyped by PCR. The Upf2 mutant and wild-type alleles were detected by PCR using the following primers: Upf2-forA (5′CCTCATTAATGTTGAGGAGTA-3′), Upf2-for1 (5′-TCCAGTTACAGTGATGAATTG-3′), and Upf2-revC (5′-ACTGAGTCTGGTTTCTAATAG-3′), which amplify a WT band of 391 bp and a Upf2 flox band of 482 bp. Cre expression was detected by PCR using the following primers: Cre-F3 (5′-TCCAGCAACATTGGGCC-3′) and Cre-R3 (5′-TCAGCTACACCAGAGACG-3′) which amplify a 432 bp fragment. All experiments were performed on three-month-old male and female mice unless otherwise stated.
Drosophila melanogaster
Flies were reared on a standard medium at 25 °C, 60% relative humidity, and a 12-h light/dark cycle. The following strains were obtained from Bloomington Stock Center (Indiana University); UAS-Upf2RNAi (#31095) and elav-GAL4 (#8765). The Upf2RNAi mutants were out-crossed onto a wild-type Canton-S background before behavioral experimentation. For behavioral studies, 50 flies, 3–5 days old and of mixed sex were aliquoted into food vials.
METHOD DETAILS
PCR and Sanger Sequencing
The UPF2 gene variant was amplified using gene-specific primers (see Table S5) designed to the reference human gene transcript (NM_080599; NCBI Gene; https://www.ncbi.nlm.nih.gov/). Amplification reactions were cycled using a standard protocol on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA). Bidirectional sequencing of all exons and flanking regions was completed with a BigDye™ v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer’s instructions. Sequencing products were resolved using an 3730xl DNA Analyzer (Applied Biosystems). All sequencing chromatograms were compared to published cDNA sequence; nucleotide changes were detected using Codon Code Aligner (CodonCode Corporation, Dedham, MA).
Whole Exome Sequencing
Exome sequencing of Case 1 was performed using 3 μg of venous blood-derived genomic DNA from the affected proband and unaffected parents. Genomic DNA was sonicated to approximately 200 base pair (bp) fragments and adaptor-ligated to make a library for paired-end sequencing. Following amplification and barcoding, the libraries were hybridized to biotinylated complementary RNA oligonucleotide baits from the SureSelect XT Human All Exon + UTR v5 75Mb Kit (Agilent Technologies, Santa Clara, CA) and purified using streptavidin-bound magnetic beads as described previously (Zheng et al., 2011). Amplification was performed prior to paired-end sequencing on the Illumina HiSeq 2000 system (San Diego, CA) at 50-fold on-target depth. Exome sequencing reads were aligned with Novoalign version 3.02.00 (http://www.novocraft.com/) to the human genome assembly with ambiguous SNPs (hg19 dbSNP132-masked, UCSC Genome Browser). PCR duplicates were removed using MarkDuplicates from Picard (http://picard.sourceforge.net). Variant detection was performed with GATK HaplotypeCaller and variant annotation using ANNOVAR (Wang et al., 2010). Following exclusion of variation in known speech and language disorder genes, the variants were filtered based on a de novo model with variants considered if present only in the proband and with minor allele frequency greater than or equal to0.02. The highest quality variants were then selected by applying the following strict filters: read depth greater than or equal to 50, no filtering alerts/warnings as annotated by ANNOVAR, variant quality score greater than or equal to 50, variant mapping quality score greater than or equal to 50, and no indication that the variant is contained within a segmental duplicated region, as annotated by ANNOVAR.
Western Blotting
Protein samples (20 mg) were boiled in reducing SDS loading buffer and run on NuPAGE 4%–12% Bis-Tris gradient gels (Invitrogen). Gels were transferred to nitrocellulose membranes (Bio-Rad) and blocked with 5% skim milk, 5% BSA or 2% goat or horse serum in TBST. Primary antibodies were incubated overnight at 4°C and secondary HRP-conjugated (Abcam) antibodies for 1–2h at room temperature. Immunoreactivity was detected on ChemiDoc XRS+ (Bio-Rad) and analyzed using Image Lab Software (Bio-Rad). Primary antibodies used were goat anti-UPF2 (Santa Cruz), rabbit anti-RENT1/UPF1 (Bethyl), rabbit anti-UPF3B (Sigma) and mouse anti-β-ACTIN.
Chromosomal Microarray
SNP microarray processing was carried out according to the manufacturer’s recommendations using the HumanCytoSNP-12 300k array (Illumina, San Diego, CA). Analysis of the microarray data was performed using KaryoStudio (Illumina) software.
Analysis of NMD and DEGs in LCLs by RNaseq
All LCLs were cultured in parallel and mRNA isolated at the same time. PolyA+ mRNA was isolated from 7 controls, 3 individuals with UPF3B loss-of-function mutations (described in Tarpey et al., 2007; Nguyen et al., 2012), 3 individuals with UPF2 copy number variation [CNV; UPF2 gene deletions; Case 3 and two individuals from Nguyen et al., 2013 and an individual with a UPF2 point mutation (Case 1)]. Quality of the fastqc reads was checked using FastQC software package. RNA was subjected to paired-end RNaseq using Illumina NextSeq platform. The low-quality segments of the reads were trimmed using trim galore and an average of 58 million reads per sample (range 48–63 million per sample) and differentially expressed genes (DEGs) were identified between controls and each group (UPF3B, UPF2 CNV and UPF2 point mutation; see Figure S3). Normalization and gene differential expression analysis was performed using the DESeq2 pipeline. Prior to the differential expression analysis, the effect of cofounding variables was removed using the RUVSeq package. Specifically, we used the RUVg approach to calculate factors of unwanted variation using the least DEGs from a first pass GLM regression of the upper-quartile normalized counts based on the covariate of interest i.e the groups. Four factors of unwanted variation were removed (k = 4). Differential expression analysis was performed between controls and each patient group using DESeq2 and p values were adjusted to control for the false discovery rate (FDR) using the Benjamini-Hochberg method. A gene was considered to be differentially expressed with an adjusted p value < 0.05.
Quantitative reverse transcription PCR
Total RNA from LCLs and the hippocampus, cortex, and cerebellum of Upf2 fb-KO mice and their control littermates was extracted using TRIzol (Invitrogen) and the RNeasy Plus Universal Mini Kit (QIAGEN). For fly experiments, 200–500 heads were collected from WT Canton-S, Upf2RNAi/+, elav-GAL4 > Upf2RNAi, and Upf1 flies and total RNA was extracted using the above method. RNA (2.5 μg) was used to synthesize cDNA (Superscript VILO cDNA synthesis kit, Invitrogen) and RT-qPCR was performed using PowerUp SYBR Green Master Mix according to the manufacturer’s protocol (Invitrogen). Expression levels for each gene (primer set sequences listed in Table S5) were normalized to β-actin RNA and data are represented as fold-change relative to control levels. Significant differences were determined using unpaired t tests.
Behavioral Tests
Contextual fear conditioning
Fear conditioning was performed as previously described with some modifications (Huang et al., 2013; Zhu et al., 2011). Briefly, mice were first handled for 5 min for two consecutive days. On the training day, after 2 min in the conditioning chamber, mice received two pairings of a tone (2,800 Hz, 85 dB, 30 s) with a co-terminating foot shock (0.7 mA, 2 s), after which they remained in the chamber for an additional minute and were then returned to their home cages. 24 h after training, mice were tested for freezing (immobility except for respiration) in response to the training context (training chamber). Freezing behavior was automatically scored by FreezeView software (Actimetrics). The percentage of time spent freezing was taken as an index of learning and memory.
Morris Water Maze
Tests were performed in a circular pool (140 cm diameter) of opaque water as previously described (Huang et al., 2013; Zhu et al., 2011). In the hidden-platform version of the Morris water maze, mice were trained with four trials/day spaced 30 min apart for seven consecutive days as previously described. In each training trial, mice were released from a different starting point and could search for the hidden platform (10 cm in diameter) for 60 s. If a mouse did not find the platform within 60 s, it was guided to the platform and remained on the platform for 15 s before being removed from the pool. On day 8, memory was assessed in a probe trial during which the platform was removed, and the mice were allowed to search for 60 s. Data were acquired and analyzed using an automated video tracking system (HVS Image).
Three-chamber social test
Crawley’s three-chamber test for sociability and preference for social novelty was performed as described (Buffington et al., 2016; Sgritta et al., 2019). Age-, sex-, and weight- matched control mice were used as stranger mice and habituated to the test chamber for three sessions (20 min each), one or two days before the behavioral assay. On the test day, each test mouse was first given a 10 min habituation period during which it was allowed to freely explore a 60 × 40 × 23 cm Plexiglas arena divided into three equally sized, interconnected chambers (Left, Center, Right). Sociability was measured during a second ten-minute period in which the mouse was given a choice to interact with an empty, inanimate wire cup (“Empty”) or a wire cup containing a stranger mouse (“Mouse 1”). Time spent interacting with either the empty cup or the stranger mouse was recorded using the automated AnyMaze software. Empty cup placement in the Left or Right chamber during the sociability test was alternated between test mice to eliminate any confounds due to chamber bias. Preference for social novelty was assessed by introducing a second stranger mouse (“Mouse 2”) into the previously empty wire cup. Time spent interacting with either Mouse 1 or Mouse 2 was scored in real time by an experimenter blind to the genotype.
Reciprocal Social Interaction
Mice were placed in a 25 3 25 3 25 cm Plexiglas arena, to which they had not been previously habituated, with a stranger age- and sex-matched conspecific. We recorded the time a pair of mice engaged in social interaction (close following, touching, nose-to-nose sniffing, nose-to-anus sniffing, and/or crawling over/under each other). The human observer was blind to the genotype. Social behavior was analyzed with AnyMaze automated software.
T-maze spontaneous alternation task
The spontaneous alternation task was conducted as previously described (Deacon and Rawlins, 2006). The apparatus was a black wooden T-maze with walls 25 cm high and arms 30 cm long and 9 cm wide. A removable central partition was used during the sample phase but not the test phase of each trial. Guillotine doors were positioned at the entrance to each goal arm. At the beginning of the sample phase, both doors were raised, and the mouse was placed at the end of the start arm facing away from the goal arms. Each mouse was allowed to make a free choice between the two goal arms; after its tail had cleared the door of the chosen arm, the door was closed, and the mouse was allowed to explore the arm for 30 s. The mouse was then returned to the end of the start arm, with the central partition removed and both guillotine doors raised, signaling the beginning of the test phase. Again, the mouse was allowed to make a free choice between the two goal arms. This sequence (trial) was repeated 10 times per day for two days with each mouse. The percentage of alternation was averaged over the two days. Trials that were not completed within 90 s were terminated and disregarded during analysis.
USV
USVs were recorded between adult female mice as previously described with modifications (Ferhat et al., 2016). Briefly, mice were handled for three days prior to recording and were housed individually. During this time, they were also habituated to the test cage using the following protocol: mice were taken out from home cage and placed in a new cage with bedding; the microphone (Avisoft UltraSoundGate condenser microphone capsule CM16, Avisoft Bioacoustics, Berlin, Germany) was placed above the cage and mice were allowed to explore the test cage setup for 20 minutes per day before returning to their home cage. On the day of recording, a mouse was introduced into the test cage and allowed to explore for five minutes. Following this, a second age-, sex-, and genotype-matched mouse was introduced into the cage and USV recording began immediately (16-bit format, 300 kHz sampling frequency). USVs were recorded using Avisoft Bioacoustics UltraSoundGate 116Hb with Avisoft recorder. After four minutes of recording, both mice were returned to their home cage. USV recording was then analyzed by Avisoft SASLab Pro for spectrogram generation. USV call types were identified and quantified manually.
Electrophysiology
Electrophysiological recordings were performed as previously described (Huang et al., 2013; Stoica et al., 2011; Zhu et al., 2011). Briefly, horizontal hippocampal slices (350 μm) were cut with a Leica (VT 1000S) vibratome from the brains of control or Upf2 fb-KO mice in 4°C artificial cerebrospinal fluid (ACSF) and kept in ASCF at 22–24°C for at least 1 h before recording. Slices were maintained in an interface-type chamber perfused (2–3 mL min−1) with oxygenated ACSF (95% O2 and 5% CO2) containing 124 mM NaCl,2.0 mM KCl, 1.3 mM MgSO4, 2.5 mM CaCl2, 1.2 mM KH2PO4, 25 mM NaHCO3 and 10 mM glucose. Bipolar stimulating electrodes were placed in the CA1 stratum radiatum to excite Shaffer collateral and commissural fibers. Field excitatory postsynaptic potentials were recorded at 28–29°C with ACSF-filled micropipettes. The recording electrodes were placed in the stratum radiatum and the intensity of the 0.1-ms pulses was adjusted to evoke 30%–35% of maximal response. Tetanic LTP was induced by brief high-frequency trains (100 Hz, 1 s), applied in groups of four separated by 5-min intervals. A stable baseline of responses at 0.033 Hz was established for at least 30 min. To reduce day-to-day variations, on a given day, we recorded from control and Upf2 fb-KO slices or from slices treated with vehicle, CyP or Mino. Furthermore, in a given experiment, we recorded from control and Upf2 fb-KO slices in parallel from only one slice per genotype (in the same chamber to ensure uniformity in experimental conditions across groups). Thus, n values refer to both the number of slices and the number of mice.
In vivo compound administration
For CyP administration, cyclophosphamide monohydrate (Sigma) was freshly dissolved in saline and injected into one-month-old control and Upf2 fb-KO mice intraperitoneally (i.p.) at a dose of 100 mg per kg twice per week for three and a half weeks (total of seven injections). For Mino administration, minocycline hydrochloride (Sigma) was freshly dissolved in saline and injected into one-month-old control and Upf2 fb-KO mice i.p. at a dose of 50 mg per kg every other day for four weeks (total of 14 injections). For both drug treatments, mice were allowed to recover for two weeks before behavioral testing.
Drosophila aversive olfactory conditioning
The negatively reinforced olfactory learning procedure was carried out according to established protocols, with some modifications (Huang et al., 2013; Tully et al., 1994). Briefly, the training odorants were 0.2% 3-octanol (vol/vol, Sigma-Aldrich) and 0.12% 4-methylcyclohexanol (vol/vol, Sigma-Aldrich) diluted in mineral oil. For each training trial, flies were exposed to the first odorant (CS+) and twelve 1.25 s pulses of electric shock at 90 V for 1 min. This was followed by a 1-min presentation of the second odorant, which was not paired with shocks (CS−). Spaced training consisted of five training trials with an inter-trial interval of 15 min. Memory was then tested in a T-maze 24 h after the final training trial and the performance index calculated as the number of flies avoiding the shock-paired odor (CS−) minus the number of flies choosing the shock-paired odor (CS+) divided by the total number of flies (CS− + CS+): [(CS− ) – (CS + )]/[(CS − ) + (CS + )], as previously described 3-octanol 4-methylcyclohexanol alternated as CS+, and CS+, and the results were averaged. N = 1 is defined as the performance index average of two vials consisting of 50–60 flies per vial.
Immunohistochemistry (IHC)
For histology and IHC, mice were anesthetized and perfused intracardially with ice cold 0.1M phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. After further 4% PFA fixation overnight (at 4°C), brains were submitted to the Neurological Research Institute (NRI) neuropathology core facility for paraffin embedding and sectioning. For the immunostaining procedure, 6 mm thick sagittal sections were subjected to antigen retrieval in 10 mM citric acid. Subsequently, sections were blocked in 10% normal goat serum (30 min) and stained overnight (4°C) with primary antibodies [GFAP (1:500, Dako, Z0334), Iba1 (1:200, Wako, Cat. #019–19741). After primary and biotin conjugated secondary antibody application, sections were developed using the ABC kit with 3,3 diaminobenzidine tetrahydrochloride. Sections were counterstained with hematoxylin and mounted with Vectashield medium. Digital photos were taken using AxioVision4 imaging program (Carl Zeiss MicroImaging).
Immunofluorescence (IF)
Mice were deeply anesthetized with isoflurane and perfused transcardially with 10ml PBS followed by 30ml ice-cold 4% PFA. Brains were rapidly dissected and placed in 4% PFA at 4°C overnight, then transferred to 20% sucrose in 0.1M phosphate buffer (PB) at 4°C overnight for cryoprotection. 30 μm-thick sections were cut on a sliding blade microtome and then blocked to prevent nonspecific binding for 30 min in 0.1M PB containing 10% normal goat serum with 0.3% TX-100 (PBTgs). Floating sections were incubated in primary antibody diluted in PBTgs rocking at 4°C overnight. The next day, sections were washed 3× 5 min in PBTgs then incubated in secondary antibodies for 1h rocking in the dark at RT. Excess secondary antibody was washed off with 1× 5 min wash with each of PBTgs, 0.1M PB, and 0.05M PB prior to mounting. Sections were mounted to 2% gelatin-coated coverslips and then to slides with VECTASHIELD Antifade mounting media (Product H-1000). Imaging was performed on an AxioImager.Z2m microscope (Carl Zeiss MicroImaging) fitted with an AxioCam digital camera (Carl Zeiss MicroImaging). Images were acquired in AxioVision (Carl Zeiss MicroImaging). All images within a given dataset were collected at equivalent exposure times to allow for comparison of signal intensity. In some images, contrast and brightness were linearly adjusted using Photoshop (Adobe). Image processing was applied uniformly across all images within a dataset.
Primary antibodies used for immunofluorescence included mouse anti-GFAP (Sigma clone G-A-5, Product G3893, 1:2000 dilution) and rabbit anti-Iba-1 (WAKO, Product 019–19741, 1:400 dilution). Secondary antibodies used to visualize the primaries were goat anti-mouse AlexaFluor 594 (ThermoScientific, Product A11032, 1:1000 dilution) and goat anti-rabbit AlexaFluor 488 (ThermoScientific, Product A11034, 1:1000 dilution). Hoechst stain (1:10:000 in the penultimate wash buffer) or DAPI in the mounting media (Vectashield) was used to mark nuclei.
Immune cell isolation and flow cytometry
Mice were transcardially perfused with 1X PBS. Brains were collected, weighed, subjected to collagenase (2mg/ml, Roche) digestion for 1 hour and CNS infiltrating cells were isolated by gradient centrifugation using 37/70% Percoll gradient (GE Healthcare). A single cell suspension from spleens was prepared by mechanical dissociation using a cell strainer (BD Falcon) followed by RBC lysis using ACK lysis buffer (Quality Biological, Inc). Cells were counted and after Fc blocking stained for flow cytometric analysis. The following antibodies were used: CD3-PE.Cy7 (BD Biosciences, #561100), B220 (BD Biosciences, #553087), Cd11b-APC.Cy7 (BD Biosciences, #561039) and Lys6G-PE (BD Biosciences, #551461). Flow cytometry was performed using LSR Fortessa (Becton Dickinson). Data were collected with FACSDiva and analyzed with FlowJo software. Statistical significance between groups was determined by one-way ANOVA followed by Sidak’s multiple comparison’s test.
Cytokine and chemokine measurements
Cytokine and chemokine concentrations (pg/ml) were determined using the Milliplex Mouse 32-Plex Cytokine detection kit (Milli-pore), as per manufacturer’s instructions. The 32-panel consisted of the following analytes: Eotaxin, G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-1, IL-13, IL-15, IL-17, IP-10, KC, LIF, LIX, MCP-1, M-CSF, MIG, MIP-1α, MIP-1β, MIP-2, RANTES, TNFα and VEGF). The brain regions of interest were dissected and homogenized in a buffer containing 20mM Tris HCl (pH 7.5), 150mM NaCl, 0.05% Tween-20, 1mM PMSF, and protease inhibitor cocktail. The concentration of analytes was measured simultaneously in each sample using a multiplexed immunoassay, with each sample being run in duplicate. Statistical significance between groups was determined by one-way ANOVA followed by Sidak’s multiple comparison’s test.
T cell proliferation assay
Splenocytes were isolated from Upf2 fb-KO mice and littermate controls using Histopaque-1077 density gradients (Sigma-Aldrich, MO, USA) and immediately plated (105 cells/well) in a flat-bottom 96-well plates (Corning). Cells were stimulated with control or Upf2 fb-KO forebrain homogenate. T lymphocyte proliferation was measured by thymidine incorporation. Briefly, cells were incubated for a total of 72 h (37°C, 5% CO2) in the conditions described above. [3H] thymidine (1 μCi/well) was added to each well 16–18h before harvesting of DNA on glass fiber filters and measurement of incorporated [3H] thymidine with a β-scintillation counter (Beckman Coulter, Brea, CA). Statistical significance for genotype and stimulation effects was determined by two-way repeated-measures ANOVA followed by Sidak’s multiple comparison’s test.
Microarray analysis
Total RNA was extracted from the forebrain of control and Upf2 fb-KO mice (three months of age) using TRIzol (Invitrogen) in biological triplicates. These samples were submitted to the Bioinformatics and Expression Analysis core facility (BEA) at Karolinska Instituet and samples were labeled and hybridized to the Affymetrix Mouse Exon 1.0 ST microarray according to the instructions of the manufacturer. The resulting data were summarized and normalized using robust multiarray average (rma) using the rma function in affy 1.48.0 and R version 3.2.3 (www.r-project). Updated probe set definitions (Entrez gene v 20) (Dai et al., 2005) were used, as these improve precision and accuracy for DNA micrarrays (Sandberg and Larsson, 2007). The reproducibility using hierarchical clustering of correlations obtained for all genes in all sample to sample comparisons was assessed (Spearman correlations were used). Samples clustered according to biological class, which is consistent with good reproducibility. The random variance model (RVM) modified t test (Wright and Simon, 2003) was used to identify differential expression and the resulting p values were adjusted for multiple testing using Benjamini-Hochberg False Discovery Rate (FDR) (Benjamini and Hochberg, 1995). Genes with an FDR < 5% and a log2 fold-change ≥ 0.75 were considered differentially expressed. PANTHER version 12.0 was utilized to conduct Gene Over-representation Testing on upregulated genes using Gene Ontology Biological Process Complete annotation. Bonferroni correction for multiple testing was applied.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification is described in Method Details and in figure legends. Number of samples is described in the figure legends. Unless otherwise stated, statistical analysis was performed using GraphPad Prism 8 and the used statistical tests are defined in the figure legend. For behavioral experiments, statistics were based on the two-sided unpaired Student’s t test or one- or two-way ANOVA with Bonferroni post hoc analysis to correct for multiple comparisons, unless otherwise indicated. In all instances, statistical significance was defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data throughout the paper are displayed as mean ± s.e.m. No outlier analysis was performed, or data points excluded. Sample size was chosen according to that used for similar experiments in previously published literature. Experimenters were blinded whenever possible to experimental condition during data acquisition or quantification.
DATA AND CODE AVAILABILITY
Data Resources
The whole-exome human sequencing data cannot be made accessible due to IRB restrictions. The accession number for the control and Upf2 fb-KO mouse microarray data reported in this paper is NCBI GEO: GSE81591.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-Cy7 Hamster Anti-Mouse CD3e | BD Biosciences | Cat. #: 561100 RRID: AB_10562036 |
| FITC Rat Anti-Mouse CD45R/B220 | BD Biosciences | Cat. #: 553087 RRID: AB_394617 |
| APC-Cy7 Rat Anti-CD11b | BD Biosciences | Cat. #: 561039 RRID: AB_2033993 |
| PE Rat Anti-Mouse Ly-6G | BD Biosciences | Cat. #: 551461 RRID: AB_394208 |
| Mouse Monoclonal Anti-Glial Fibrillary Acidic Protein (GFAP) | Sigma-Aldrich | Cat. #: G3893 RRID: AB_477010 |
| Rabbit Polyclonal Anti-Glial Fibrillary Acidic Protein (GFAP) | Dako | Cat. #: Z0334 RRID: AB_10013382 |
| Rabbit Polyclonal Anti-lba1 | Wako | Cat. #: 019–19741 RRID: AB_839504 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat. #: A-11032 RRID: AB_2534091 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat. #: A-11034 RRID: AB_2576217 |
| Goat Polyclonal Anti-UPF2 | Santa Cruz | Cat. #: sc-20227 RRID: AB_2272706 |
| Rabbit Polyclonal Anti-RENT1 | Bethyl Laboratories | Cat. #: A301–902A RRID: AB_1524122 |
| Rabbit Polyclonal Anti-UPF3B | Sigma-Aldrich | Cat.#: SAB2102656 RRID: AB_10605424 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3-Octanol | Sigma-Aldrich | Cat. #: 218405 |
| 4-Methylcyclohexanol, mixture of cis and trans | Sigma-Aldrich | Cat. #: 153095 |
| Paraformaldehyde | Sigma-Aldrich | Cat. #: P6148 |
| Triton-100X | Bio-Rad | Cat. #: 1610407 |
| Cyclophosphamide monohydrate | Sigma-Aldrich | Cat. #: 239785 |
| Minocycline hydrochloride | Sigma-Aldrich | Cat. #: M9511 |
| Critical Commercial Assays | ||
| BigDye v3.1 Terminator Cycle Sequencing Kit | Applied Biosystems | Cat. #: 4337455 |
| SureSelect XT Human All Exon + UTR v5 75Mb Kit | Agilent Technologies | |
| RNAeasy Plus Universal Mini Kit | QIAGEN | Cat. #: 73404 |
| Maxwell RSC Simply RNA Cell Kit | Promega | Cat. #: AS1390 |
| Superscript IV VILO Master Mix | Thermo Fisher Scientific | Cat. #: 11756050 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat. #: A25777 |
| MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel - Premixed 32 Plex - Immunology Multiplex Assay | Millipore Sigma | Cat.#: MCYTMAG-70K-PX32 |
| Deposited Data | ||
| Affymetrix Mouse Exon 1.0 ST microarray | This paper | GEO:GSE81591 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster. RNAi of Upf2: y1 v1; P {TRiP.JF01560}attP2 | Bloomington Drosophila Stock Center | BDSC: 31095 FlyBase: FBst0031095 |
| D. melanogaster: elav-GAL4: P{GAL4-elav.L} | Bloomington Drosophila Stock Center | BDSC: 8765 FLyBase: FBtp0000743 |
| Upf2 conditional knockout mice | Weischenfeldt et al., 2008 | Upf2floxed/floxed |
| CamKII-Cre mice | The Jackson Laboratory | JAX: 027400 |
| Oligonucleotides | ||
| See Table S5 for the list of oligos | N/A | |
| Software and Algorithms | ||
| Prism 8 v8.1.2 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FreezeView Software | Actimetrics | https://www.coulbourn.com/category_s/277.htm |
| Image Lab Software v6.0.1 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?lD=KRE6P5E8Z |
| PANTHER v12.0 | Tang and Thomas, 2016 | http://www.pantherdb.org |
| Avisoft SASLab Pro V5.2.13 | Avisoft Bioacoustics | http://www.avisoft.com/soundanalysis.htm |
| CodonCode Aligner | CodonCode Corporation | https://www.codoncode.com/aligner/ |
| Novoalign v3.02.00 | Novooraft Technologies | http://www.novocraft.com/products/novoalign/ |
| MarkDuplioates (Picard) | Broad Institute | https://broadinstitute.github.io/picard/command-line-overview.html |
| FastQC | Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR aligner v2.5.3a | Cold Spring Harbor Laboratories | https://github.com/alexdobin/STAR |
| Trim Galore | Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| RUVSeq v1.16.1 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| StepOne™ Software | Applied Biosystem (Life Technology-ThermoFisher Scientific) | https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/StepOne-and-StepOnePlus-Real-Time-PCR-System.html |
| DESeq2 v1.22.2 | European Molecular Biology Laboratory | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| KaryoStudio Software | Illumina | https://support.illumina.com/downloads/karyostudio_software_v14.html |
| RSubread Package v1.5.2 | The Walter and Eliza Hall Institute of Medical Research | http://subread.sourceforge.net |
Highlights.
Humans carrying novel variants in UPF2 exhibit speech and language deficits
Upf2-deficient mice and flies have impaired NMD and exhibit behavioral deficits
Inhibition of Upf2-dependent NMD triggers immune activation in mice
Reduction of brain inflammation reverses synaptic and behavioral deficits
ACKNOWLEDGMENTS
We thank the individuals carrying UPF variants and their families for participating in our research program. We thank members of the Costa-Mattioli laboratory for comments on the manuscript. This work was supported by the NIH (NIMH 096816 and NINDS 076708 to M.C.M.); Sammons Enterprises (to M.C.M.); the Intellectual Disability Research Center (P30HD024064), IDDRC Neuropathology Core (NICHD, U54HD083092), BCM Cytometry and Cell Sorting Core (RR024574, AI036211, and CA125123), and T32 award HL007676 from the NIH (to R.H.); a centre grant from the Novo Nordisk Foundation (to B.P.); and the Australian State of Victoria Government Operational Infrastructure Support Program and the Australian National Health and Medical Research Council Independent Research Institute Infrastructure Support Scheme (IRIISS). J.G. was supported by Australian NHMRC grants 1155224 and 1091593. L.J. was supported by Australian Research Council fellowship DE160100620. A.M., M.H., D.A., I.E.S., and M.B. are funded by NHMRC Centre of Research Excellence (1116976) and project (1127144) grants. A.M. is funded by an NHMRC practitioner fellowship (1105008). M.H. is funded by a career development fellowship (ID 1063799). I.E.S. is funded by a practitioner fellowship (1006110). M.B. is funded by a senior research fellowship (ID 1102971).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2019.08.027.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alberini CM, and Kandel ER (2014). The regulation of transcription in memory consolidation. Cold Spring Harb. Perspect. Biol 7, a021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association, DSM-5 Task Force (2013). In Diagnostic and statistical manual of mental disorders: DSM-5, 5th ed (American Psychiatric Publishing, Inc.). [Google Scholar]
- Avery P, Vicente-Crespo M, Francis D, Nashchekina O, Alonso CR, and Palacios IM (2011). Drosophila Upf1 and Upf2 loss of function inhibits cell growth and causes animal death in a Upf3-independent manner. RNA 17, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KE, and Parker R (2004). Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr. Opin. Cell Biol 16, 293–299. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300. [Google Scholar]
- Boulanger LM, and Shatz CJ (2004). Immune signalling in neural development, synaptic plasticity and disease. Nat. Rev. Neurosci 5, 521–531. [DOI] [PubMed] [Google Scholar]
- Buffington SA, Huang W, and Costa-Mattioli M (2014). Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci 37, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, and Costa-Mattioli M (2016). Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 165, 1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin A, Hu H, Rynearson SG, Hollien J, Yandell M, and Metzstein MM (2014). In vivo determination of direct targets of the nonsense-mediated decay pathway in Drosophila. G3 (Bethesda) 4, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, and Sonenberg N (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. (2005). Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM, and Rawlins JN (2006). T-maze alternation in the rodent. Nat. Protoc 1, 7–12. [DOI] [PubMed] [Google Scholar]
- Dezern AE, Styler MJ, Drachman DB, Hummers LK, Jones RJ, and Brodsky RA (2013). Repeated treatment with high dose cyclophosphamide for severe autoimmune diseases. Am. J. Blood Res 3, 84–90. [PMC free article] [PubMed] [Google Scholar]
- Dodd B, Hua Z, Crosbie S, Holm A, and Ozanne A (2002). Diagnostic Evaluation of Articulation and Phonology (London: Psychological Corporation; ). [Google Scholar]
- Dragatsis I, and Zeitlin S (2000). CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis 26, 133–135. [DOI] [PubMed] [Google Scholar]
- Duffy JR (2013). Motor Speech Disorders: Substrates, Differential Diagnosis, And Management, Third Edition (Elsevier; ). [Google Scholar]
- Ehninger D, Li W, Fox K, Stryker MP, and Silva AJ (2008). Reversing neurodevelopmental disorders in adults. Neuron 60, 950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferhat AT, Torquet N, Le Sourd AM, de Chaumont F, Olivo-Marin JC, Faure P, Bourgeron T, and Ey E (2016). Recording Mouse Ultrasonic Vocalizations to Evaluate Social Communication. J. Vis. Exp Published online June 5, 2016 10.3791/53871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, and Wright CF; DDD Study (2011). The Deciphering Developmental Disorders (DDD) study. Dev. Med. Child Neurol 53, 702–703. [DOI] [PubMed] [Google Scholar]
- Fisher SE, and Scharff C (2009). FOXP2 as a molecular window into speech and language. Trends Genet 25, 166–177. [DOI] [PubMed] [Google Scholar]
- Garrido-Mesa N, Zarzuelo A, and Gálvez J (2013). Minocycline: far beyond an antibiotic. Br. J. Pharmacol 169, 337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzard H, Baker E, and McCabe P (2008). Requests for clarification and children’s speech responses: Changing ‘pasghetti’ to ‘spaghetti’. Child Lang. Teach. Ther 24, 249–263. [Google Scholar]
- Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, He F, and Jacobson A (2015). Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu. Rev. Genet 49, 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, and Jacobson A (2015). Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu. Rev. Genet 49, 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjević K,Roman G, and Costa-Mattioli M (2013). mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci 16, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Shum EY, Jones SH, Lou CH, Dumdie J, Kim H, Roberts AJ, Jolly LA, Espinoza JL, Skarbrevik DM, et al. (2018). A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry 23, 1773–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y, Li X, Serin G, and Maquat LE (2001). Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 106, 607–617. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, and Wilkinson MF (2018). Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat. Rev. Neurosci 19, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Karam R, Lou CH, Kroeger H, Huang L, Lin JH, and Wilkinson MF (2015). The unfolded protein response is shaped by the NMD pathway. EMBO Rep 16, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis ED, and Muhlemann O (2019). Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb. Perspect. Biol 11, a032862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S, and Jacobson A (2012). NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol 13, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E, and Dever TE (2004). Biochemical mechanisms for translational regulation in synaptic plasticity. Nat. Rev. Neurosci 5, 931–942. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Shoubridge C, Antar C, Nguyen LS, Van Esch H, Kleefstra T, Briault S, Fryns JP, Hamel B, Chelly J, et al. (2010). Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 15, 767–776. [DOI] [PubMed] [Google Scholar]
- Malenka RC (1994). Synaptic plasticity in the hippocampus: LTP and LTD. Cell 78, 535–538. [DOI] [PubMed] [Google Scholar]
- McMahon JJ, Miller EE, and Silver DL (2016). The exon junction complex in neural development and neurodevelopmental disease. Int. J. Dev. Neurosci 55, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, and Dietz HC (2001). Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet 10, 99–105. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Batshaw ML, and Hoffman EP (2012). Genomics, intellectual disability, and autism. N. Engl. J. Med 366, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei C, Fedorenko E, Amor DJ, Boys A, Hoeflin C, Carew P, Burgess T, Fisher SE, and Morgan AT (2018). Deep phenotyping of speech and language skills in individuals with 16p11.2 deletion. Eur. J. Hum. Genet 26, 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, and Dietz HC (2004). Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet 36, 1073–1078. [DOI] [PubMed] [Google Scholar]
- Metzstein MM, and Krasnow MA (2006). Functions of the nonsense-mediated mRNA decay pathway in Drosophila development. PLoS Genet 2, e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner B, and Klein D (2016). Loss of recent memory after bilateral hippocampal lesions: memory and memories-looking back and looking forward.J. Neurol. Neurosurg. Psychiatry 87, 230. [DOI] [PubMed] [Google Scholar]
- Mitwalli AH, Al Wakeel JS, Hurraib S, Aisha A, Al Suwaida A, Alam A, Hammad D, Sulimani F, Memon NA, Askar A, et al. (2011). Comparison of high and low dose of cyclophosphamide in lupus nephritis patients: a long-term randomized controlled trial. Saudi J. Kidney Dis. Transpl 22, 935–940. [PubMed] [Google Scholar]
- Morgan AT, and Webster R (2018). Aetiology of childhood apraxia of speech: A clinical practice update for paediatricians. J. Paediatr. Child Health 54, 1090–1095. [DOI] [PubMed] [Google Scholar]
- Morgan A, Ttofari Eecen K, Pezic A, Brommeyer K, Mei C, Eadie P, Reilly S, and Dodd B (2017). Who to Refer for Speech Therapy at 4 Years of Age Versus Who to “Watch and Wait”? J. Pediatr 185, 200–204.e1. [DOI] [PubMed] [Google Scholar]
- Morgan AT, Haaften LV, van Hulst K, Edley C, Mei C, Tan TY, Amor D, Fisher SE, and Koolen DA (2018). Early speech development in Koolen de Vries syndrome limited by oral praxis and hypotonia. Eur. J. Hum. Genet 26, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort M, Ivanov D, Cooper DN, and Chuzhanova NA (2008). A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat 29, 1037–1047. [DOI] [PubMed] [Google Scholar]
- Mostafa GA, Shehab AA, and Al-Ayadhi LY (2013). The link between some alleles on human leukocyte antigen system and autism in children. J. Neuroimmunol 255, 70–74. [DOI] [PubMed] [Google Scholar]
- Neitzel H (1986). A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum. Genet 73, 320–326. [DOI] [PubMed] [Google Scholar]
- Neves G, Cooke SF, and Bliss TV (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci 9, 65–75. [DOI] [PubMed] [Google Scholar]
- Nguyen LS, Jolly L, Shoubridge C, Chan WK, Huang L, Laumonnier F, Raynaud M, Hackett A, Field M, Rodriguez J, et al. (2012). Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol. Psychiatry 17, 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LS, Kim HG, Rosenfeld JA, Shen Y, Gusella JF, Lacassie Y, Layman LC, Shaffer LG, and Gécz J (2013). Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neurodevelopmental disorders. Hum. Mol. Genet 22, 1816–1825. [DOI] [PubMed] [Google Scholar]
- Nguyen LS, Wilkinson MF, and Gecz J (2014). Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci. Biobehav. Rev 46, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor F, Kolonias D, Giangrande PH, and Gilboa E (2010). Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 465, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perini P, Calabrese M, Rinaldi L, and Gallo P (2008). Cyclophosphamide-based combination therapies for autoimmunity. Neurol. Sci 29 (Suppl 2), S233–S234. [DOI] [PubMed] [Google Scholar]
- Popp MW, and Maquat LE (2013). Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet 47, 139–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Letunic I, Raes J, Bork P, and Izaurralde E (2005). Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA 11, 1530–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehs-Kearnan N, Gloggnitzer J, Dekrout B, Jonak C, and Riha K (2012). Aberrant growth and lethality of Arabidopsis deficient in nonsense-mediated RNA decay factors is caused by autoimmune-like response. Nucleic Acids Res 40, 5615–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, and Klee T (1987). Clinical assessment of oropharyngeal motor development in young children. J. Speech Hear. Disord 52, 271–277. [DOI] [PubMed] [Google Scholar]
- Rodenas-Cuadrado P, Pietrafusa N, Francavilla T, La Neve A, Striano P, and Vernes SC (2016). Characterisation of CASPR2 deficiency disorder–a syndrome involving autism, epilepsy and language impairment. BMC Med. Genet 17, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, and Sur M (2015). Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350, aab3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, and Larsson O (2007). Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics 8, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semel E, Wiig EH, and Secord WA (2006). Clinical Evaluation of Language Fundamentals, Fourth Edition (Pearson; ). [Google Scholar]
- Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA, and Costa-Mattioli M (2019). ). Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 101, 246–259.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, and Kida S (1998). CREB and memory. Annu. Rev. Neurosci 21, 127–148. [DOI] [PubMed] [Google Scholar]
- Sossin WS, and Costa-Mattioli M (2019). Translational Control in the Brain in Health and Disease. Cold Spring Harb. Perspect. Biol 11, a032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow SS, Cicchetti D, and Balla DA (2005). Vineland Adaptive Behavior Scales, 2nd Edition manual (NCS Pearson, Inc.). [Google Scholar]
- Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, and Costa-Mattioli M (2011). Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. USA 108, 3791–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, and Thomas PD (2016). PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinforma. Oxf. Engl 32, 2230–2232. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, et al. (2007). Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet 39, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoren LA, Nørgaard GA, Weischenfeldt J, Waage J, Jakobsen JS, Damgaard I, Bergström FC, Blom AM, Borup R, Bisgaard HC, and Porse BT (2010). UPF2 is a critical regulator of liver development, function and regeneration. PLoS ONE 5, e11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomblin B (2011). Co-morbidity of autism and SLI: kinds, kin and complexity. Int. J. Lang. Commun. Disord 46, 127–137. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, and Tonegawa S (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Tully T, Preat T, Boynton SC, and Del Vecchio M (1994). Genetic dissection of consolidated memory in Drosophila. Cell 79, 35–47. [DOI] [PubMed] [Google Scholar]
- Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, and Arraiano CM (2007). Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 35, 7651–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D (2011). Wechsler Abbreviated Scale of Intelligence, Second Edition (Pearson; ). [Google Scholar]
- Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Mönch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, and Porse BT (2008). NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson GS, and Robertson GJ (2006). Wide Range Achievement Test 4 (Lutz: Psychological Assessment Resources; ). [Google Scholar]
- Wöhr M, and Scattoni ML (2013). Behavioural methods used in rodent models of autism spectrum disorders: current standards and new developments. Behav. Brain Res 251, 5–17. [DOI] [PubMed] [Google Scholar]
- Wright GW, and Simon RM (2003). A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics 19, 2448–2455. [DOI] [PubMed] [Google Scholar]
- Xu X, Zhang L, Tong P, Xun G, Su W, Xiong Z, Zhu T, Zheng Y, Luo S, Pan Y, et al. (2013). Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin. Genet 83, 560–564. [DOI] [PubMed] [Google Scholar]
- Zheng J, Miller KK, Yang T, Hildebrand MS, Shearer AE, DeLuca AP, Scheetz TE, Drummond J, Scherer SE, Legan PK, et al. (2011). Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss (DFNA4). Proc. Natl. Acad. Sci. USA 108, 4218–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjević K, et al. (2011). Suppression of PKR promotes network excitability and enhanced cognition by interferon-g-mediated disinhibition. Cell 147, 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Resources
The whole-exome human sequencing data cannot be made accessible due to IRB restrictions. The accession number for the control and Upf2 fb-KO mouse microarray data reported in this paper is NCBI GEO: GSE81591.