

SUMMARY
Ependymomas exist within distinct genetic subgroups, but the molecular diversity within individual ependymomas is unknown. We perform multiplatform molecular profiling of 6 spatially distinct samples from an ependymoma with C11orf95-RELA fusion. DNA methylation and RNA sequencing distinguish clusters of samples according to neuronal development gene expression programs that could also be delineated by differences in magnetic resonance blood perfusion. Exome sequencing and phylogenetic analysis reveal epigenomic intratumor heterogeneity and suggest that chromosomal structural alterations may precede accumulation of single-nucleotide variants during ependymoma tumorigenesis. In sum, these findings shed light on the oncogenesis and intratumor heterogeneity of ependymoma.
Graphical Abstract
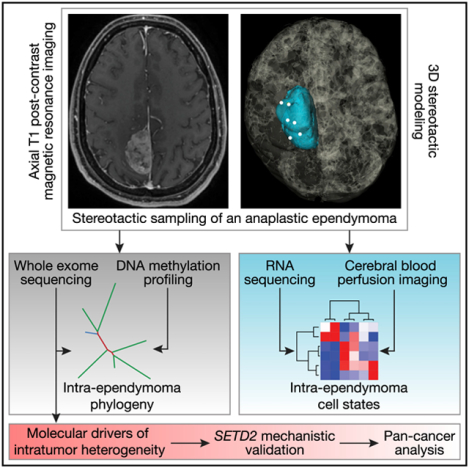
In Brief
Tumor heterogeneity poses a barrier to cancer treatment. Liu etal. investigate radiographically distinct regions of an ependymoma tumor using transcriptomic, genetic, and epigenomic profiling and discover axes of gene expression programs that recapitulate normal brain development in addition to phylogenies that shed light on the tumorigenesis of ependymoma.
INTRODUCTION
Ependymal tumors of the central nervous system have diverse demographic, anatomic, radiologic, clinical, and molecular characteristics (Pajtler et al., 2015). There are 9 molecular subgroups of ependymomas (Pajtler et al., 2015), and the majority of supratentorial ependymomas harbor a genetic fusion between the uncharacterized gene C11orf95 and the nuclear factor κB (NF-κB) transcriptional activator RELA (Pajtler et al., 2015; Parker et al., 2014). When expressed in neural stem cells, C11orf95-RELA is sufficient to induce ependymoma formation in vivo, suggesting that neural stem cells are a putative cell of origin for ependymal tumors (Ozawa et al., 2018; Taylor et al., 2005).
Molecular heterogeneity among ependymomas is well established (Capper et al., 2018; Johnson et al., 2010; Khatua et al., 2017; Modena etal., 2006; Pajtler et al., 2015; Parker etal., 2014; Witt et al., 2011). In contrast, intratumor molecular heterogeneity within individual ependymal tumors is incompletely understood. In other central nervous system malignancies, intratumorheterogeneity encompassing cell composition, immune infiltration, and somatic mutations contributes to tumor growth and resistance to therapy (Müller et al., 2017; Parker et al., 2016; Patel et al., 2014; Szerlip et al., 2012; Yung et al., 1982). Thus, we hypothesized that molecular heterogeneity may exist within ependymoma and that understanding ependymoma heterogeneity may shed light on tumor development, patterns of recurrence, or potential therapeutic targets. To test these hypotheses, we performed multiplatform molecular profiling of 6 spatially distinct samples from a supratentorial anaplastic ependymoma with C11orf95-RELA fusion. Our findings reveal significant intratumor heterogeneity and a molecular phylogeny containing regionally distinct stem-like, neuronal differentiation, and immune-enriched regions within a single tumor. We discover that stem-like regions of ependymal tumors can be delineated on preoperative magnetic resonance perfusion and find that chromosomal copy number changes are an early molecular event in ependymoma tumorigenesis. Finally, we identify a previously uncharacterized missense mutation in the histone methyltransferase SETD2 as an example of a potentially broader class of epigenomic modifiers of molecular heterogeneity within ependymoma.
RESULTS
Radiologic and Molecular Classification of Supratentorial Anaplastic Ependymoma with C11orf95-RELA Fusion
We collected 6 spatially distinct intraoperative samples from a 29-year-old male who initially presented to medical attention after experiencing a generalized seizure and was found to have a parasagittal mass on brain imaging (Figures 1A and 1B). Stereotactic analysis at the time of resection showed that samples were separated by a median of 1.7 cm (range 0.6–3.4 cm) and were closer to the periphery of the tumor than to the centroid (Figure S1). A separate sample that was designated for clinical pathology was diagnosed asanaplasticependymoma, World Health Organization (WHO) grade III, with a mitotic count of 15 mitoses per 10 high power fields, a Ki-67 labeling index of 20%, and strong L1CAM cytoplasmic and membrane staining, consistent with C11orf95-RELA fusion (Figure 1C). Targeted DNA sequencing using a Clinical-Lab-oratory-Improvement-Amendment-certified panel of 510 cancer-associated genes (Kline et al., 2017) showed multiple segmental copy number variations along chromosome 11q consistent with chromothripsis (Figure S2A). Break-apart DNA fluorescence in situ hybridization demonstrated rearrangement of RELA and C11orf95, but not YAP1 (Figure 1D), a gene fused to C11orf95 in a minority of supratentorial ependymomas (Parker et al., 2014).
Figure 1. Radiologic and Molecular Classification of a Supratentorial Anaplastic Ependymoma with C11orf95-RELA Fusion.
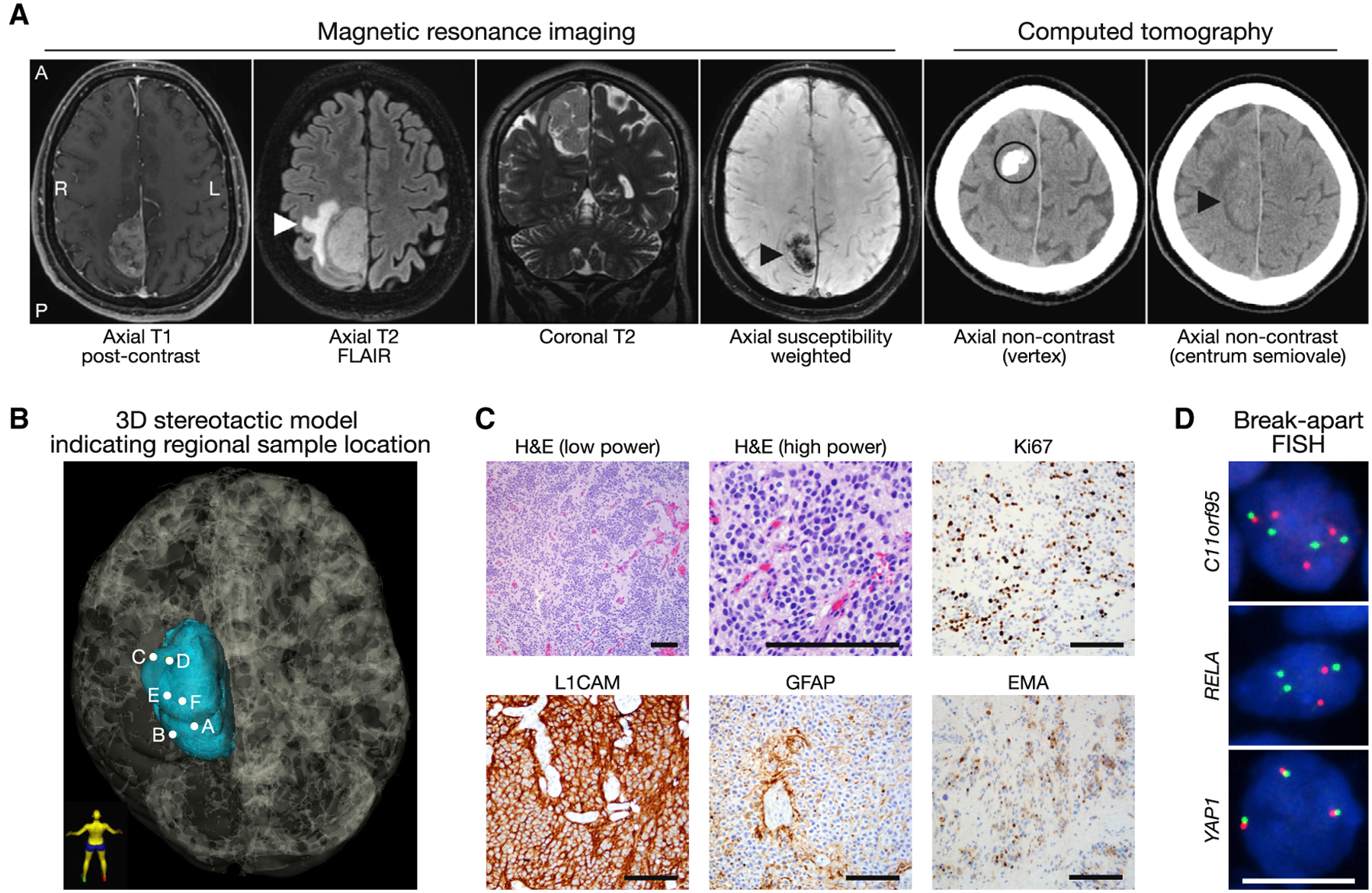
(A) Preoperative magnetic resonance imaging and computed tomography shows a right parasagittal heterogeneously enhancing mass with peritumoral edema (white arrowhead) and internal susceptibility (black arrowhead) that is distinct from a site of eccentric peripheral nodular calcification (black circle) and therefore suggestive of internal hemorrhage. Of note, a significant difference in head tilt and imaging gantry influences the appearance of tumor location as denoted by black arrowheads in images 4 and 6. A, anterior; FLAIR, fluid-attenuated inversion recovery; L, left; P, posterior; R, right.
(B) 3D stereotactic mapping of 6 samples (A–F) obtained for this study. Patient orientation is represented by the model in bottom left.
(C) Low- and high-power hematoxylin and eosin (H&E) images and immunohistochemical stains demonstrating strong L1CAM positivity, GFAP positivity in cytoplasmic processes of perivascular pseudorosettes, paranuclear dot-like and ring-like EMA positivity, and a Ki-67 labeling index of 20%, all features that support the diagnosis of anaplastic ependymoma, WHO grade III. Black scale bars, 100 μm.
(D) Break-apart fluorescence in situ hybridization (FISH) (red and green probes) shows rearrangement of C11orf95 and RELA, but not YAP1. Nuclei are shown in blue. White scale bar, 10 μm.
Cortical Development and Differentiation Gene Expression Programs Define Intratumor Heterogeneity in Ependymoma
We performed RNA sequencing (RNA-seq) of the 6 samples to delineate gene expression heterogeneity. Pooled analysis of all samples using two independent fusion gene detectors identified an in-frame fusion between exon 2 of C11orf95 and exon 2 of RELA (Figure S2B), corresponding to the pathogenic RELAFUS1 fusion found in the majority of supratentorial ependymomas (Parker et al., 2014). No other fusion genes were concordantly detected by both methods, and the 6 samples expressed similar levels of L1CAM, CCND1, C11orf95, and RELA (Figure S2C), each of which are specific markers of C11orf95-RELA ependymoma (Parker et al., 2014).
Principal-component analysis (PCA) was performed to separate the 6 samples based on gene expression variability (Figure 2A). The most variable genes in PCA space were used for hierarchical clustering, revealing 3 clusters of samples (Figure 2B). To identify drivers of intratumor heterogeneity, we ranked genes based on their loading scores for the first three principal components (Figure 2C). Gene Ontology analysis (Figure S2D) showed that genes in axis 1 (samples C and D) were enriched for the OLIG1 and OLIG2 co-expression networks (p = 0.01 and 0.04, respectively), which are implicated in stem-like identity in glioma (Aguirre-Cruz et al., 2004; Ligon et al., 2004, 2007) and early phases of glial cell development (Novitch et al., 2001; Zhou and Anderson, 2002). In contrast, genes in axis2 (samples A and E) were enriched for the TSHZ3 and LHX9 co-expression networks (p = 0.04 for each), which are involved in neuronal fate specification in normal development (Bertuzzi et al., 1999; Caubit et al., 2016; 2010; Liu etal., 2015; Peukert etal., 2011; Rétaux etal., 1999). Genes in axis3(samples B and F) were enriched for RELA target genes, including CXCL2 and HSPA1A (p < 0.0001 for each), which are suggestive of immune signaling (Hayden and Ghosh, 2008; Karin et al., 2002) and underlying activity of the C11orf95-RELA pathogenic fusion gene.
Figure 2. Cortical Development and Differentiation Gene Expression Programs Define Intratumor Heterogeneity in an Ependymoma.
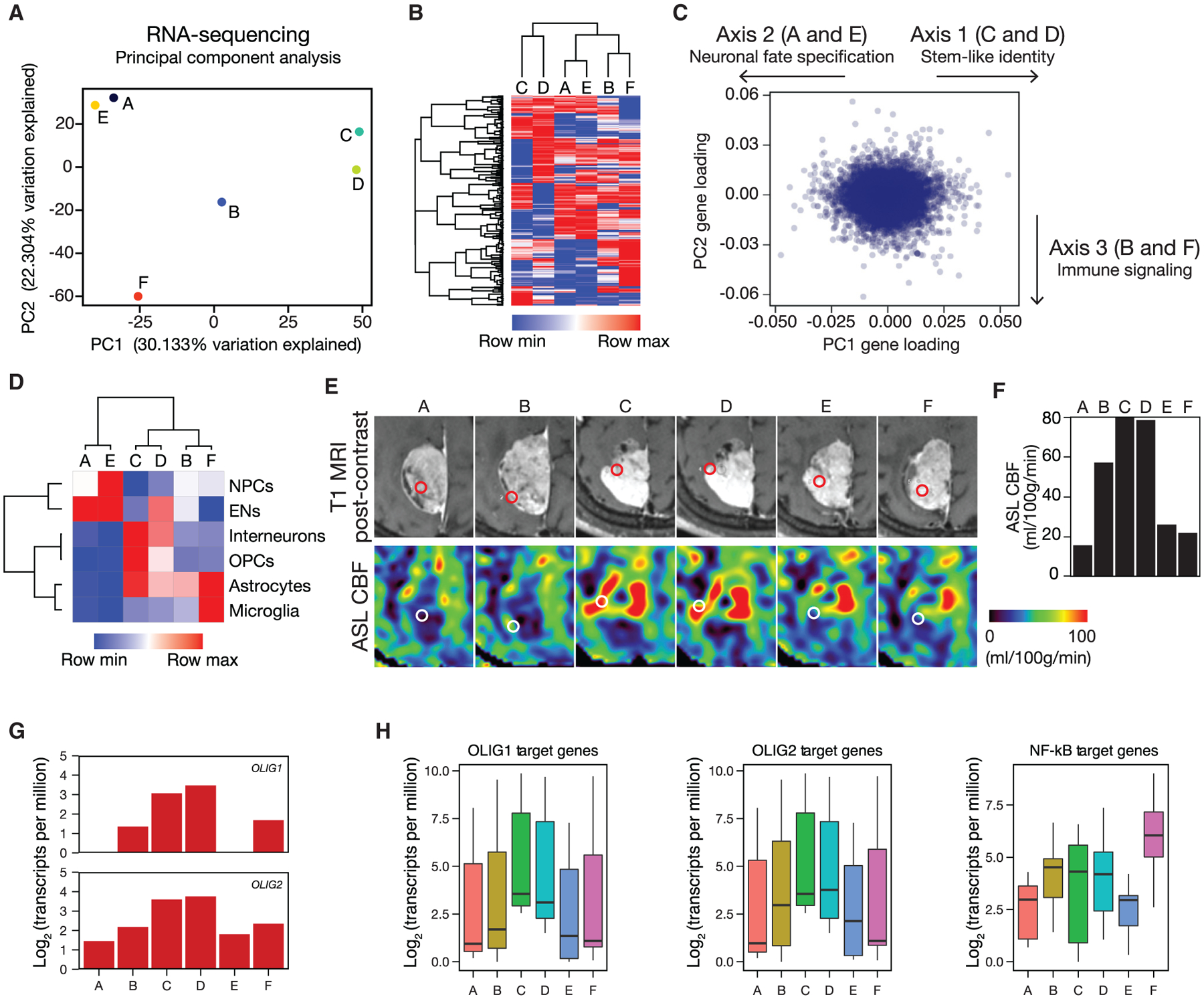
(A) Principal-component analysis (PCA) of RNA-seq data reveal that 52% percent of variation among regional samples is explained by the first two principal components. PC, principal component.
(B) Hierarchical clustering of samples using the top 250 most variable genes in RNA-seq identifies 3 subgroups.
(C) Scatterplot of gene loading scores in PCA with arrows indicating axes used for gene ranking.
(D) Gene expression heatmap of cell-type-specific genes in the developing human cerebral cortex (Zhong et al., 2018) recapitulates the same clusters as unbiased transcriptome-wide analysis. EN, excitatory neurons; NPCs, neural progenitor cells; OPCs, oligodendrocyte progenitor cells.
(E and F) Preoperative T1 post-contrast and arterial spin labeling perfusion (E) and quantitative arterial spin labeling perfusion (F) of stereotactically defined regions demonstrate enhanced cerebral blood flow in regions with stem-like identity (C and D). Red and white circles indicate sample locations. ASL, arterial spin labeling; CBF, cerebral blood flow; MRI, magnetic resonance imaging.
(G) OLIG1 and OLIG2, which are implicated in stem-like identity in glioma and early phases of glial cell development, are enriched in samples C and D (OLIG1 p = 0.007; OLIG2 p = 0.002; Student’s t test).
(H) Analysis of published OLIG1, OLIG2, and NF-κB target genes reveals enrichment of OLIG1 (p = 9.5 × 10−6) and OLIG2 (p = 0.00045) targets in stem-like samples C and D and enrichment of NF-κB targets in samples B and F(p = 0.00093), consistent with underlying activity of the C11oif95-RELA pathogenic fusion gene (Kolmogorov-Smirnov test).
Supratentorial ependymomas have been proposed to arise from radial glia neural stem cells (Taylor et al., 2005), and we discovered that developmental gene expression programs defined intratumor heterogeneity in ependymoma (Figures 2C and 2D). Thus, we hypothesized that clusters of samples would show molecular similarity to cell types in the developing brain (Nowakowski et al., 2017). To test that hypothesis, we quantified the mean expression signatures of marker genes for the major cell types in the developing cerebral cortex in each sample (Zhong etal., 2018). Consistent with PCA gene enrichment analysis, samples C and D were enriched for oligodendrocyte progenitor cell, astrocyte, and interneuron markers; samples A and E were enriched for neural progenitor cell and excitatory neuron markers; samples B and F were enriched for astrocyte markers; and sample F was further enriched for microglia markers (Figure 2D).
To determine whether preoperative imaging could distinguish between regions of molecular heterogeneity in ependymoma, we performed quantitative magnetic resonance imaging and compared differences between sample locations. Arterial spin labeling measurement of perfusion revealed that stem-like regions C and D exhibited elevated cerebral blood flow compared to other regions (Figures 2E and 2F). On diffusion tensor imaging, regions C and D had elevated fractional anisotropy, likely relating to differences in cellular architecture given the similar cellularity across the samples (Figures S2E, S2F, and S4D). Further, we found that sample F, which was closest to the tumor centroid (Figure S1A), had low cerebral blood flow (Figure 2E) and was enriched in HSPA1A by RNA-seq (Figure S2G), a transcriptional target of RELA that is implicated in prolonged hypoxia (Lewis et al., 1999; Rahat et al., 2011). Consistently, sample F was enriched in an immune gene expression program (Figures 2C and S2D) characteristic of microglia (Figure 2D), and hypoxia is an important regulator of immune activation (Lewis etal., 1999; Rahat et al., 2011). Cellular metabolism is also known to impact chromatin modification, and differential expression analysis revealed that the histone deacetylase gene HDAC9 was suppressed in samples C and D (Figure S3), the regions with the highest cerebral blood flow (Figures 2E and 2F).
To further investigate the putative stem-like markers identified in samples C and D, we investigated the expression of OLIG1 and OLIG2 and found that both were enriched in stem-like regions relative to other samples (Figure 2G). We also analyzed the expression levels of OLIG1 and OLIG2 target genes from the ARCHS4 database (Lachmann et al., 2018) that were highly variable in our RNA-seq data and found that these targets were also increased in samples C and D (Figure 2H). Conversely, we found elevated expression of highly variable RELA targets in samples B and F (Figure 2H), which is likely reflective of the underlying activity of the C11orf95-RELA pathogenic fusion gene (Kuleshov et al., 2016). These data are consistent with the hypothesis that regional variation in OLIG1 and OLIG2 expression and NF-kB pathway activation underlie intratumor heterogeneity in ependymoma. Moreover, our findings suggest that, in this ependymoma, intratumor heterogeneity is defined by neuronal development and immunologic gene expression programs that can be delineated by quantitative preoperative imaging characteristics.
Epigenomic Profiling of Intratumor Heterogeneity in Ependymoma Corroborates Neuronal Development Signatures
To validate our gene-expression-based clustering results, we applied PCA toward 850K DNA methylation profiles of the 6 samples. DNA-methylation-based clustering paralleled RNA-seq results and delineated regions C and D from the remaining regions in the first principal component (Figure S4A). Hierarchical clustering of the most variable methylation probes further revealed 3 clusters that were concordant to those obtained using RNA-seq (Figures 2B and 3A). Analysis of signature probes showed that sites of hypomethylation in samples C and D were associated with stem cell genes (false discovery rate [FDR] = 0.016; Figure S4B). Finally, we applied DNA methylation profiles to a methylation-based random forest classifier (Capper et al., 2018) to confirm that each sample unambiguously classified as RELA fusion ependymoma (Figure S4C).
Figure 3. Phylogenetic Analysis Identifies Early Chromosome Structural Alterations and Epigenomic Intratumor Heterogeneity in Ependymoma.
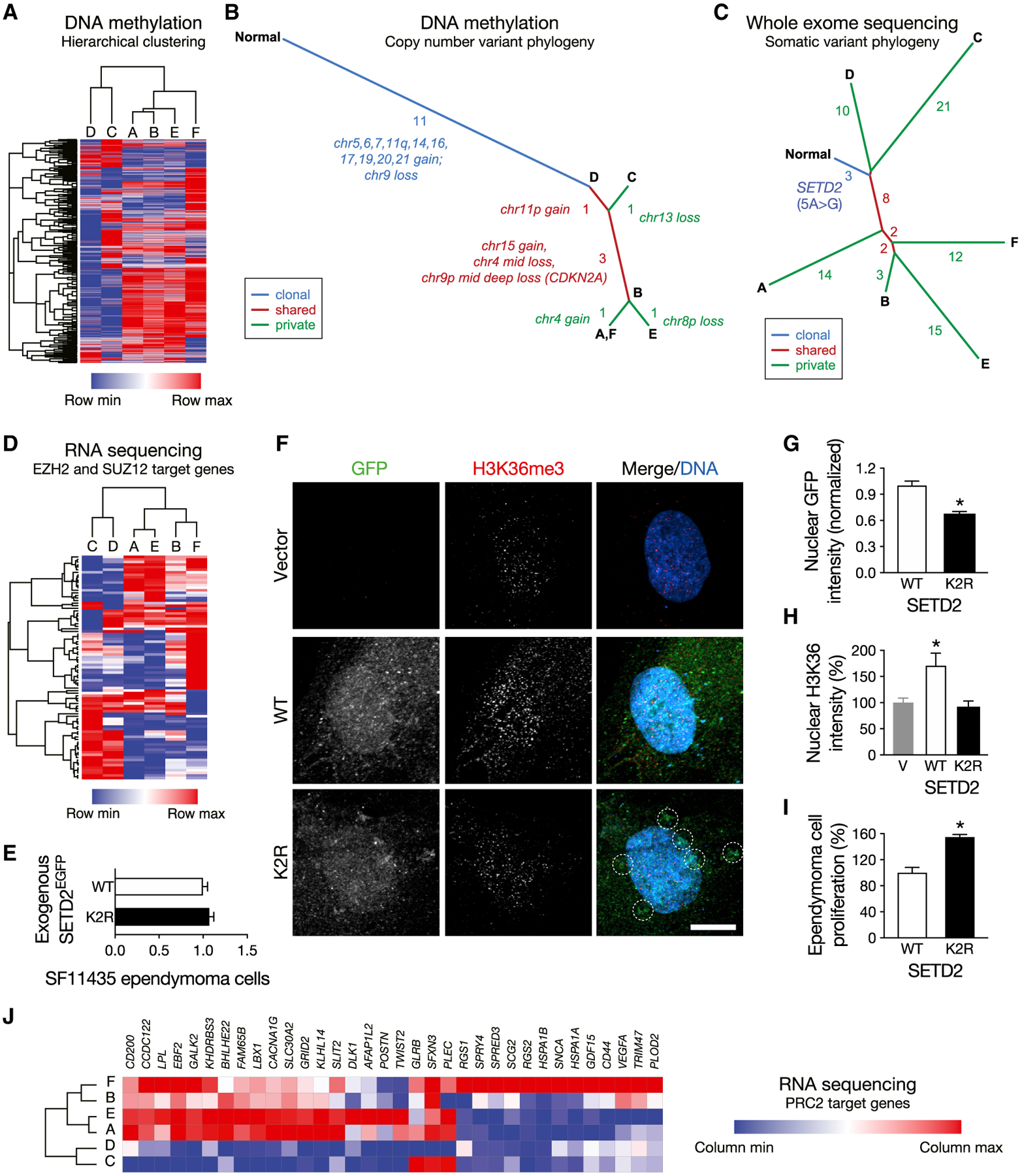
(A) Hierarchical clustering of regional samples using the top 2,000 most variable methylation probes closely recapitulates gene-expression-based clustering.
(B) Intratumor phylogeny based on clonal ordering of copy number variants derived from methylation analysis suggests that chromosomal structural alterations are an early event during ependymoma tumorigenesis. The number and identity of copy number variants defining each axis are indicated.
(C) Intratumor phylogeny based on clonal ordering of somatic variants suggests an epigenomic contribution to molecular heterogeneity. The number of somatic variants defining each axis is indicated.
(D) Hierarchical clustering restricted to highly variable EZH2 and SUZ12 target gene expression recapitulates the same clusters of samples as unbiased transcriptome-wide analysis.
(E) Confocal microscopy of total cellular fluorescence demonstrated equivalent expression of fluorescent fusions of SETD2K2R (K2R) and wild-type SETD2 (WT). (F-H) K2R fused to EGFP demonstrates diminished nuclear qualitative (F) and quantitative (G) localization and increased perinuclear aggregation (dotted circles) relative to WT fused to EGFP in SF11435 anaplastic ependymoma cells. Consistently, WT overexpression increases qualitative (F) and quantitative (H) nuclear H3K36me3 intensity relative to vector (V) and K2R overexpression. SETD2 constructs are shown in green, H3K36me3 chromatin marks are shown in red, and nuclei are marked with Hoechst 3342 in blue. Quantitative data are normalized to control. Scale bar, 10 μm. *p < 0.0001; Student’s t test.
(I) K2R overexpression in SF11435 cells increases cell proliferation relative to WT overexpression. Data are representative of 3 biologic replicates and are normalized to cell proliferation with WT overexpression. *p < 0.0001; Student’s t test.
(J) PRC2 target genes are depleted in regions with higher allelic frequency of SETD2K2R (C and D), consistent with antagonism between H3K36me3 and PRC2 gene silencing.
Phylogenetic Analysis Identifies Early Chromosome Structural Alterations and Epigenomic Intratumor Heterogeneity in Ependymoma
To assess clonal relationships between the 6 samples, we performed phylogenetic ordering of copy number variation (CNV) profiles derived from methylation arrays. Eleven of 18 CNVs detected were common to all regions, although samples enriched in differentiation gene expression programs (A, B, E, and F) exhibited more CNVs than stem-like samples (C and D; Figure 3B; Table S1).
To determine whether differences in somatic variants may underlie intratumor heterogeneity in ependymoma, we performed exome sequencing of the 6 samples and matched normal DNA from peripheral blood. Consistent with diffusion tensor imaging (Figures S2E and S2F), cellularity and ploidy analyses from exome sequencing revealed little variability across ependymoma samples (cellularity range: 0.75–0.9; ploidy range: 3.4–3.5), suggesting equivalent proportions of tumor cells in each region (Figure S4D). Importantly, phylogenetic ordering of CNVs derived from exome sequencing was concordant with methylation-derived CNVs (Figure S4E; Table S1).
We identified 102 somatic variants (single-nucleotide variants and indels) from exome sequencing across all 6 samples, among which 3 were common to all samples and 75 were private to individual samples (Table S1). Forty-one variants were described in the COSMIC database (Forbes et al., 2017), and 15 were predicted to be pathogenic (Shihab et al., 2013). In contrast to CNVs, phylogenetic ordering of somatic variants revealed early divergence of the 6 samples (Figure 3C; Johnson et al., 2014). One of the earliest somatic variants that was detected in all samples was a 5A > G (p.K2R) missense substitution in the histone methyltransferase SETD2. Inactivating mutations in SETD2 are implicated in diverse pediatric brain tumors (Fontebasso et al., 2013), and we confirmed the SETD2K2R mutation in all tumor samples, but not in peripheral blood, by targeted DNA sequencing (Figure S4F). Remarkably, the highest mutant allele frequencies of SETD2K2R were detected in regions C (0.282) and D (0.352), the samples defined by stem cell transcriptional networks (Figure S2D).
To elucidate the molecular impact of SETD2K2R, we asked whether gene expression programs distinguishing clusters of samples were enriched for targets of epigenomic regulators. Genes in axis 2 (samples A and E) were enriched for targets of SUZ12 (p = 0.00001) and EZH2 (p = 0.001), both of which are critical components of the polycomb repressive complex 2 (PRC2) (Figure S4G). PRC2 is an epigenomic regulator that is antagonistic to SETD2 (Lachmann etal., 2010), and the allelic frequency of SETD2K2R was lower in samples defined by neuronal differentiation gene expression programs (A, B, E, and F; mean 0.244). Consistently, hierarchical clustering using only expression of highly variable PRC2 target genes recapitulated the same clusters as unbiased transcriptome-wide analysis (Figure 3D), suggesting an epigenomic contribution to intratumor molecular heterogeneity in ependymoma.
To investigate the functional impact of the K2R substitution, we overexpressed fluorescent fusions of wild-type SETD2 and SETD2 with the 5A > G substitution (SETD2K2R) in supratentorial anaplastic ependymoma cells with RELA fusion (Figure 3E). The K2R substitution is within the nuclear localization sequence of SET2, and consistently, SETD2K2R displayed diminished nuclear intensity and increased perinuclear aggregation compared to wild-type SETD2 (Figures 3F and 3G). Consistent with the chromatin-modifying function of SETD2, overexpression of wild-type SETD2 increased nuclear H3K36me3 intensity, but SETD2K2R did not (Figure 3H). Moreover, SETD2K2R overexpression increased cell proliferation and viability relative to wild-type SETD2 overexpression (Figure 3I).
SETD2 is the primary methyltransferase for histone H3 lysine 36 (H3K36), a chromatin mark associated with active transcriptional elongation (Kolasinska-Zwierz et al., 2009; Kouzarides, 2007). H3K36 methylation antagonizes histone H3 lysine (H3K27) methylation (Yuan et al., 2011), a chromatin mark that is induced by PRC2 and is associated with gene repression (Kouzarides, 2007). As nuclear localization of SETD2K2R is impaired (Figures 3F and 3G), we hypothesized that regions with higher allelic frequency of SETD2K2R would show greater gene silencing by PRC2. To test that hypothesis, we analyzed the expression of PRC2 target genes and found that many candidates were suppressed in regions with higher allelic frequency of SETD2K2R (C and D), consistent with antagonism between SETD2 transcriptional elongation and PRC2 gene silencing (Figure 3J). In sum, these data support epigenomic misregulation as a contributing factor to ependymoma cell proliferation and suggest that this paradigm may be particularly important in regions defined by stem cell transcriptional networks.
Mutation of the Epigenomic Regulator SETD2 Is Associated with Adverse Outcomes across All Cancers
Although SETD2 is not recurrently mutated in ependymoma (Mack et al., 2018), we hypothesized that mutation of epigenomic regulators may be a common feature of molecularly heterogeneous tumors and could therefore represent a biomarker of adverse outcomes in cancer. To test that hypothesis, we queried 42,199 samples across all human cancers and identified 1,548 tumors with SET2D2 mutations, representing a surprising 3.7% of all cases (Figure 4A; Gao et al., 2013). Consistent with the prognostic significance of SETD2 mutation in pediatric high-grade glioma (Fontebasso et al., 2013; Mackay et al., 2017), patients with SETD2mutant cancers exhibited significantly lower overall survival compared to patients with SETD2 wild-type cancers (Figure 4B). In contrast to ependymoma, genetic or transcriptomic alterations in SETD2 are identified in a remarkable 52% of renal cell carcinomas (Ricketts et al., 2018). Thus, we compared differentially expressed genes in 534 renal cell carcinomas with and without alterations in SETD2 and found that tumors with SETD2 alterations were enriched in heterogeneous gene expression programs that are normally reserved for CD4 T cells, fetal lung, brain cingulate gyrus, brain substantia nigra, and smooth muscle (Figure 4C). In sum, these data support a greater paradigm of epigenomic dysregulation as a regulator of intratumor heterogeneity and, in conjunction with our findings in ependymoma, suggest that regional differences in chromatin architecture may promote tumor growth and resistance to therapy.
Figure 4. Mutation of the Epigenomic Regulator SETD2 Is Associated with Adverse Outcomes across All Cancers.
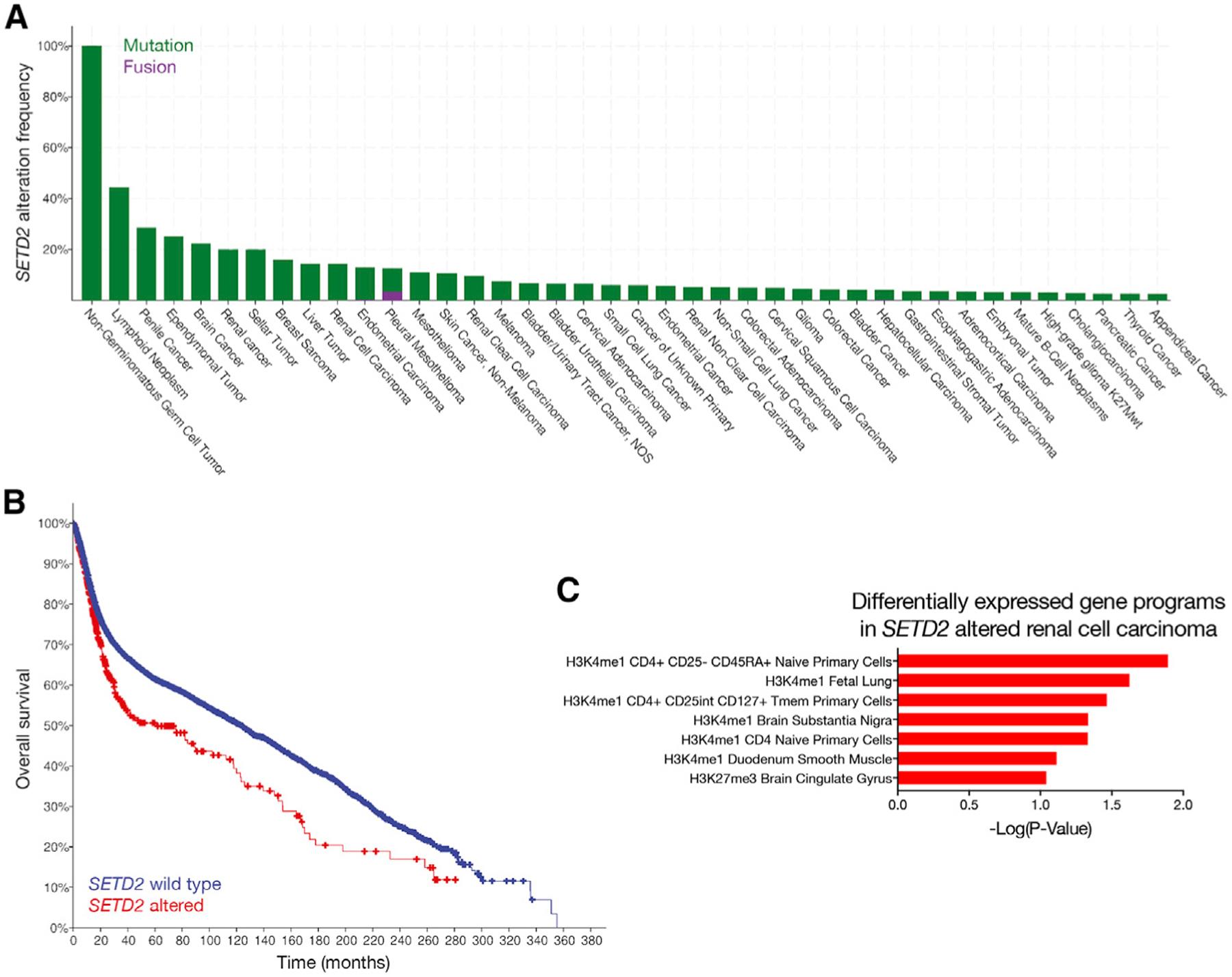
(A) Analysis of 42,199 samples from cBioPortal reveals 1,548 cancers with SETD2 alteration, representing 3.7% of all cases. The 40 most common cancers with SETD2 alterations are shown, with omission of 93 additional cancer types with known SETD2 alterations at lower frequencies.
(B) Patients from cBioPortal with SETD2 mutant cancers have lower overall survival compared to patients with SETD2 wild-type cancers (p = 0.000017; log rank test).
(C) Ontology analysis of differentially expressed genes in 276 renal cell carcinomas from cBioPortal with genetic or transcriptional alterations SETD2, compared to 258 renal cell carcinomas with wild-type SETD2, reveals heterogeneous gene expression programs that are normally reserved for CD4 T cells, fetal lung, brain cingulate gyrus, brain substantia nigra, and smooth muscle. In contrast, renal cell carcinomas with wild-type SETD2 are enriched in gene expression programs associated with penis foreskin primary cells and regulatory T (T reg) primary cells.
DISCUSSION
We find that transcriptional signatures of molecular compartments in this ependymoma recapitulate critical aspects of human cerebral cortex development. Remarkably, gene expression programs associated with oligodendrocyte progenitor cells, neural progenitor cells, and mature microglia are spatially distinct within this tumor. These data are consistent with the observation that supratentorial ependymomas are associated with gene expression profiles of the embryonic brain (Johnson et al., 2010) and are also enriched in neural differentiation genes, such as LHX2 and FOXG1 (Andreiuolo et al., 2010). Moreover, our results in ependymoma are reminiscent of embryonal brain tumors and other pediatric cancers, which are also associated with misactivation of signaling pathways that are critical for central nervous system development.
The SETD2K2R point substitution we report has been described in acute lymphoblastic leukemia, where its functional consequences are unknown (Mar et al., 2014). We hypothesize that regional differences in gene dosage regulate nuclear localization of SETD2, resulting in heterogeneous chromatin states within ependymoma that promote distinct developmental or differentiation gene expression programs to maintain or suppress a stem-like state, respectively. Indeed, H3K27 methylation at CpG islands can lead to a hypermethylated state that is associated with malignant transformation (Ohm et al., 2007; Schlesinger et al., 2007; Widschwendter et al., 2007). Consistently, we find that PRC2 activity is increased in ependymoma regions with higher allelic frequency of SETD2K2R. Notably, SETD2 is not recurrently mutated in ependymoma, but in the context of lower overall survival for SETD2 mutant cancers than SETD2 wild-type malignancies, we speculate that our discovery of SETD2 mutation as an epigenomic feature of molecular heterogeneity in an ependymoma may represent a foundational discovery supporting a greater paradigm in ependymoma and other tumors. Modeling these findings, however, is challenging. Reductionist cell culture systems are poorly suited to comprehensive investigations of intratumor heterogeneity, and primary tissues are not amenable to mechanistic studies that might identify molecular drivers of heterogeneity. Further, we cannot exclude the possibility that intratumor heterogeneity in RNA transcription or DNA methylation were a consequence of driver events not captured by our current study, such as mutation of non-coding regions within the genome (Weinhold et al., 2014). We acknowledge that profiling multiple regions from a single tumor will not fully reflect tumor heterogeneity in all ependymomas. Future studies will be aimed at extending our findings across additional ependymomas and correlating them with clinical outcomes. Nonetheless, epigenomic variability resulting in intratumor heterogeneity has been postulated for other malignancies (Ohlsson et al., 2003) and represents a compelling process that may have important implications for targeted therapies for ependymoma patients. Indeed, epigenomic regulators are frequently mutated in ependymoma (Huether et al., 2014), and epigenomic alterations define lethal pediatric ependymomas without identifiable recurrent mutations (Mack et al., 2014). Further, intratumor heterogeneity is a significant source of resistance to cancer treatment (Levine et al., 2019; Watkins and Schwarz, 2018), and histone modifiers function as drivers of cellular heterogeneity and resistance to molecular therapy (Hinohara et al., 2018). Thus, our discovery of a heterogeneously expressed epigenomic regulator of ependymoma cell proliferation suggests that such genes could play a crucial role in modulating tumor sensitivity to both traditional ependymoma therapies and molecular agents.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
SETD2 mutant constructs are available upon request. Cell lines (SF11435) are available upon request. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David R. Raleigh (david.raleigh@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
SF11435 cells (male) were maintained at 37°C in a 5% CO2 atmosphere with 21% oxygen, and grown in a 1:1 ratio of DMEM/F12 (Life Technologies, Carlsbad, CA) and Neurobasal medium (Life Technologies) supplemented with 5% FBS (Life Technologies), B-27 supplement without vitamin A (Life Technologies), N-2 supplement (Life Technologies), 1X GlutaMAX (Life Technologies), 1mM NEAA (Life Technologies), 100U/mL Anti-Anti (Life Technologies), 20ng/mL EGF (R&D systems, Minneapolis, MN), 20ng/mL FGF2 (Pepro-tech, Rocky Hill). Cell lines were validated using short-tandem repeat profiling at the UCSF Clinical Cancer Genomics Laboratory.
Tumor sample acquisition
A 29 year old male patient was taken to the operating room for a fronto-parietal parasagittal craniotomy for tumor resection. The preoperative stereotactic magnetic resonance images (MRI) were registered to physical space using the stereotactic neuronavigation system (BrainLab®, Munich, Germany). The accuracy of the registration was confirmed using anatomic landmarks. Surgical resection of the tumor was carried out in the usual manner. Throughout the resection, fresh tissue samples were obtained. The location of each sample (including a screenshot and DICOM coordinates) was collected using the stereotactic probe immediately prior to collecting the sample. Once collected from the single tumor, each of the six regional samples were immediately passed off the surgical field and placed into liquid nitrogen for subsequent analysis.
METHOD DETAILS
Quantitative imaging analysis
Preoperative MR imaging was performed on a 3.0 tesla scanner (Discovery; GE Healthcare, Waukesha, WI) using a neuronavigation protocol that included 3D volumetric structural sequences without and with gadolinium contrast as well as axial diffusion tensor imaging (DTI) and axial arterial spin labeling (ASL) perfusion imaging encompassing the brain.
The preoperative stereotactic MRI was imported into 3D Slicer v4.8.0. The brain and tumor were each segmented using the editor module, and the tumor volume was quantified using the label statistics module. DICOM coordinates collected from the stereotactic neuronavigation system were entered into the markups module and used to generate a 3D model of the tumor with sample sites. Regions of interest (ROI) were created around each point and used for downstream image analysis.
For radiographic characterization of the tumor and each spatially-defined sample, the 3D T1 post-gadolinium, DTI, and ASL images were transferred to a commercially available software system (AW Server; GE Healthcare) for diffusion and perfusion processing and analysis. Mean diffusivity (MD) and fractional anisotropy (FA) maps were generated on a voxel-by-voxel basis from the DTI imaging dataset. For ASL perfusion calculations, cerebral blood flow (CBF) maps were also created on a voxel-by-voxel basis. The 3D T1 post-gadolinoum images were aligned to the same axial location as the MD, FA, and CBF maps. To account for tissue shifts and sampling margin of error, 100 mm3 spherical ROIs were manually delineated about the six annotated sampling sites. Once validated, ROIs were transferred to MD, FA, and CBF maps to allow for collection of quantitative diffusion and perfusion metrics.
Sample distance metric analysis
DICOM files corresponding to the tumor and sample ROIs were imported using the oro.dicom package in the R statistical environment (Whitcher et al., 2011). Pairwise distances of samples from one another, as well the minimum pairwise distance from each sample to the tumor’s periphery and centroid, were calculated from DICOM coordinates. Heatmaps of sample distances were generated using the gplots R package.
Tissue processing and immunohistochemistry
Immunohistochemical (IHC), and hematoxylin and eosin (H&E) stains were performed on formalin-fixed, paraffin embedded tissue sections at the University of California San Francisco Immunohistochemistry Laboratory and the University of California San Francisco Neuropathology Brain Tumor Center Biomarkers Laboratory. Primary antibodies used were as follows: MIB-1/Ki-67 (MIB-1, Dako, Glostrup, Denmark, 1:50 dilution), L1CAM (clone UJ127.11, Sigma, St Louis, MO, 1:3000 dilution), GFAP (Polyclonal, Dako, 1:3000 dilution) and EMA (clone GP1.4, Leica, Newcastle Upon Tyne, United Kingdom, prediluted). All staining was performed in Ventana or Leica Bond automated staining processors.
Fluorescence In Situ Hybridization (FISH)
Dual-color FISH was performed on 4 mm paraffin embedded tissue sections. Probes were derived from BAC clones (BACPAC Resources, Oakland, CA) and labeled with either AlexaFluor-488 or AlexaFluor-555 fluorochromes. BAC clones were used to develop break apart probes for the following genes: C11orf95 (CH17–67K13 & CH17–388O01), RELA (RP11–642F7 & CH17–211O12), and YAP1 (RP11–11N20 & RP11–1082I13). Probes were co-denatured with the target cells on a slide moat at 90°C for 12 minutes. The slides were incubated overnight at 37°C on a slide moat and then washed in 4M Urea/2xSSC at 25°C for 1 minute. Nuclei were counterstained with DAPI (200ng/ml, Vector Labs, Burlingame, CA) for viewing on an Olympus BX51 fluorescence microscope equipped with a 100-W mercury lamp; FITC, Rhodamine, and DAPI filters; 100X PlanApo (1.40) oil objective; and a Jai CV digital camera. Images were captured and processed using the Cytovision software (Leica Biosystems, Richmond, IL).
RNA-seq
RNA was isolated from tumor samples using the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN, Valencia, CA). Library preparation was performed using the TruSeq RNA Library Prep Kit v2 (RS-122- 2001, Illumina, San Diego, CA) and 50 bp single end reads were sequenced on an Illumina HiSeq 2500 to a mean of 7 million reads per sample at the Center for Advanced Technology at the University of California San Francisco. Quality control of FASTQ files was performed with FASTQ. Transcript abundances were quantified using Kallisto version 0.43.1 with the flags ‘-b 100-single-t 10 -l 200 -s50’ using GENCODE release 26, based on GRCh38 (Bray et al., 2016; Harrow etal., 2012; Kim etal., 2013). Principal component analysis was performed in the statistical environment R version 3.4.3 using the base command ‘prcomp’ with the parameters ‘center = TRUE, scale. = FALSE’ on log2 transformed transcripts per million (TPM) values with a pseudocount of 1. Genes were ranked by the absolute value of the maximum gene loading scores in PCA space among the first three principal components, which was determined based on diminishing variance explained in principal components four and greater. Hierarchical clustering was performed using complete linkage hierarchical clustering using the log2 transformed TPM values with pseudocount of 1. The top 250 most variable genes in principal components 1 to 3 were used for clustering, and cluster assignments were robust to both linkage method (average or complete linkage) and also number of genes used. Identical clusters were obtained using up to 10,000 variable genes for clustering analysis.
Analysis of developmental gene expression signatures was performed by obtaining cell type specific markers from Zhong et al. (2018) and overlapping this gene list with the top 2,500 most variable genes from PCA in our analysis. The mean TPM expression of the remaining cell type specific genes for each of the cell types described in Zhong et al. (2018) was calculated for each of the ependymoma regions. The log2 transformed TPM values were then clustered by complete linkage hierarchical clustering.
Gene ontology analysis was performed using Enrichr, and p values displayed were multiple hypothesis corrected adjusted p values (Chen et al., 2013). PRC2 target genes were obtained from the ChIP enrichment analysis (ChEA) database release 2016 (Lachmann et al., 2010) by filtering the gene lists for those containing EZH2, SUZ12, or EED in their titles.
Fusion gene analysis using RNA-seq reads was performed using two independent fusion gene detectors. JAFFA version 1.09 was executed using the flags ‘readLayout = single’ against the merged fastq files for all ependymoma regions (Davidson et al., 2015). The fusion detection function of Tophat version 2.1.0 was executed using the flags ‘–bowtie1–no-coverage-search–fusion-search’ against the merged fastq files for all ependymoma regions, using the GRCh38 reference annotation.
DNA Methylation Arrays
Genomic DNA was isolated from tumor samples using the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN, Valencia, CA). DNA (1000 ng) is bisulfite converted using the Zymo EZ DNA methylation kit (Zymo Research, Irvine, CA) according to the manufacturer’s recommendations. The amount of bisulfite-converted DNA as well as the completeness of bisuflite conversion for each sample are assessed using a panel of MethyLight-based real-time PCR quality control assays. Bisulfite-converted DNA is then used as a substrate for the Illumina EPIC BeadArrays. Each sample is whole genome amplified (WGA) and then enzymatically fragmented. Samples are then hybridized overnight to an 8-sample BeadArray, in which the WGA-DNA molecules anneal to locus-specific DNA oligomers linked to individual bead types. Adenine and thymine nucleotides are labeled with cy5 (red), while cytosine nucleotides are labeled with cy3 (green). BeadArrays are scanned and the raw signal intensities are extracted from the *.IDAT files using the ‘noob’ function in the minfi R package (Aryee et al., 2014; Fortin et al., 2017). Only probes with detection p < 0.05 in all samples were included for further analysis. Data were normalized using functional normalization (Fortin et al., 2014). Probes were filtered based on the following criteria: (i) removal of probes targeting the X and Y chromosomes (n = 11,551), (ii) removal of probes containing a common single nucleotide polymorphism (SNP) within the targeted CpG site or on an adjacent basepair (n = 24,536), and (iii) removal of probes not mapping uniquely to the hg19 human reference genome (n = 9,993). A total of 815,630 probes were retained for further analysis. Principal component analysis was performed in the statistical environment R version 3.4.3 using the base command ‘prcomp’ with the parameters ‘center = TRUE, scale. = FALSE’ against the normalized β enrichment values (β = methylated/[methylated+unmethylated]). Complete linkage hierarchical clustering was performed using the top 2,000 most variable probes, based on their absolute value of gene loading scores in the first three principal components, across all ependymoma samples. For probe-level differential methylation analysis, the limma Bioconductor package was used to fit a linear model accounting for the paired nature of the data with a FDR < 0.001 considered significant (Ritchie et al., 2015). β values were used for visualization of methylation levels and M values were used for statistical analysis (M = log2[methylated/unmethylated]) (Du et al., 2010). Sample type classification was performed using the random forest classifier of DNA methylation profiles on the Molecular Neuropathology web portal (Capper et al., 2018). CNV profiles from DNA methylation data were generated as described previously (Capper et al., 2018) using the conumee R package v.1.3.0. Segments with combined probe intensities greater than 0.05 or less than −0.05 log2 ratio compared to 2 sets of 50 control tissues with balanced chromosome profiles (Capper et al., 2018) were identified as either gain or loss, respectively.
Whole exome sequencing
Library preparation, exome capture and sequencing were performed at the Institute for Human Genetics at the University of California San Francisco. Sequencing libraries were prepared using the Kapa Hyper Prep Kit and exome capture was performed with the Nimblegen SeqCap EZ Human Exome Kit v. 3.0. Paired end sequencing with read length 100 base pairs was performed on the Illumina HiSeq4000. All subsequent data analysis was performed with the bcbio pipeline with default parameters [https://github.com/bcbio/bcbio-nextgen]. Reads were aligned with the Burrows-Wheeler aligner (Li and Durbin, 2009) to the reference human genome (build hg19). Only uniquely aligned reads were included for further processing with the Picard suite (http://broadinstitute.github.io/picard/) and the Genome Analysis Toolkit (McKenna et al., 2010) for de-duplication, local realignment and base quality score recalibration. Alignment quality metrics were calculated with the Picard suite. Somatic variants (point mutations, small indels) were identified from matched tumor-normal samples using Varscan2 (Koboldt et al., 2012), Freebayes (Garrison and Marth, 2012) and Vardict (Lai et al., 2016) at a significance threshold p < 0.05, and results from each algorithm were merged into a single Ensemble callset including only those significant hits identified in at least 2 callers. Hits were further filtered based on quality metrics using default parameters including (i) mean base quality > 25, (ii) > 5 reads in tumor and normal sample, (iii) > 10% variant reads in tumor, (iv) > 90% reference reads in normal, and (v) strand bias. Variants were annotated using Snpeff v4.3 (Cingolani et al., 2012)and were further filtered to only include those marked as high, moderate or low priority and occurring in protein coding or splice site locations. Annotations were obtained by querying the COSMIC database (Forbes et al., 2017). Ploidy, cellularity analysis, and chromosome segmentation was performed using Sequenza version 2.1.0 (Favero et al., 2015) using default parameters against reads above quality score of 20. Segmentation output from Sequenza were then used for generation of CNV profiles from whole exome sequencing data using default parameters (Favero et al., 2015).
Sanger sequencing
Samples were genotyped for the SETD2 mutation to validate whole exome sequencing reads. The first exon of SETD2 was PCR amplified using the GC-RICH PCR System (Roche Applied Science, Pleasanton, CA). PCR was performed with the SETD2-forward (5′-TGTAAAACGACGGCCAGTCCTGTTACTCCTCGCGCCG-3′) and SETD2-reverse primers (5′-CAGGAAACAGCTATGACCGGT CAAGCCAACAGCTGCAA-3′). The amplification profile was 40 cycles of denaturing at 95°C, annealing at 60°C, and extension at 72°C. PCR products were purified using the QIAGEN Gel Extraction kit, and submitted for Sanger sequencing with the M13-forward sequencing primer (5′-TGTAAAACGACGGCCAGT-3′).
Phylogenetic analysis
Binary mutation calls were used to build a distance matrix across all samples using the Manhattan distance metric, including a normal tissue sample for which all mutations were absent. A phylogenetic tree was constructed using an ordinary least-squares minimum evolution approach (Desperand Gascuel, 2002) from the APE R package (Paradis et al., 2004). Each branch of the resulting tree was annotated with the total number of mutations attributed to that branch.
SETD2 cloning
Full-length SETD2 was initially cloned into a CMV-Eos2.1 vector backbone to generate SETD2-Eos2.1. A custom G-block was used to generate the coding sequence of the N-terminal 503 amino acids of SETD2 (Integrated DNA Technologies, Coralville, IA), which was fused to the coding sequence C-terminal 2062 amino acids from SETD2-GFP (Addgene, Cambridge, MA) by restriction ligation of NheI and AgeI sites. An internal missense mutation (L1962P) was corrected using theQuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) per the manufacturer’s protocol with the following primers: 5′-GACGCTGAAATAGAGCCCA AAGAGAGCAACGGC-3′ and 5′-GCCGTTGCTCTCTTTGGGCTCTATTTCAGCGTC-3′. The K2R substitution was also introduced using the QuikChange Lightning Site-Directed Mutagenesis Kit per the manufacturer’s protocol with the following primers: 5′-TCCGC TAGCGCCACCATGAGGCAGCTGCAGCCGCAGCCGCCTCC-3′ and 5′-GGAGGCGGCTGCGGCTGCAGCTGCCTCATGGTGGCGC TAGCGGA-3′. For experimentation, the complete SETD2 coding sequences either with or without the K2R substitution were moved to pLMJ1, a viral vector suitable for mammalian transduction which affixed a C-terminal EGFP tag onto each SETD2 construct.
Microscopy and proliferation assays
For microscopy, SF11435 cells were grown on glass coverslips and transduced with either wild-type SETD2-EGFP or SETD2K2R-EGFP using polybrene at 70% confluency. Forty-eight hours post transfection, cells were fixed in 4% PFA for 8 minutes, blocked in 2.5% FBS, 200mM glycine, and 0.1% Triton X-100 in PBS for 30 minutes, incubated with anti-GFPandanti-H3K36me3 (Abcam, Cambridge, UK) primary antibodies overnight at 4°C, washed, and incubated with Alexa Fluor secondary antibodies (Thermo Fischer Scientific, Waltham, MA) for 1 hour at room temperature. Hoechst 3342 (Life Technologies) was added to the secondary antibody inclubation to mark DNA. Following a final wash, cells were mounted in ProLong Diamond Antifade Mountant (Thermo Fischer Scientific), and immunofluorescence images were collected using a Zeiss 780 confocal microscope system (Carl Zeiss AG, Oberkochen, Germany).
For cell proliferation assays, SF11435 cells were grown in a 96-well plate and transduced with either wild-type SETD2-EGFP or SETD2K2R-EGFP using polybrene at 50% confluency. Forty-eight hours post-transfection, cell proliferation was assayed using the CellTiter 96 Non-Radioactive Cell Proliferation Assay and a GloMax Discovery plate reader per the manufacturer’s protocols (Promega, Madison, WI).
QUANTIFICATION AND STATISTICAL ANALYSIS
For Figures 2A–2C, 3A, S3, and S4A analysis is described above in section “DNA Methylation Arrays.”
For Figures 2D, 3D, and 3J, analysis is described above in section “RNA-seq.”
For Figure 2H, boxplots demonstrate median with first and third quartiles, whiskers represent 1.5*IQR. Tests of significance using the non-parametric Kolmogorov-Smirnov test, described in figure legends, to account for non-normally distributed data.
For Figure 3B, analysis is described above in section “Phylogenetic Analysis.”
For Figures 3C and S4E, analysis is described above in section “Whole exome sequencing.”
For Figure 3F, Signal intensities were quantified from 2D maximum intensity projections using ImageJ by outlining nuclei and measuring the integrated density of SETD2 fluorescent fusion proteins relative to total cellular integrated density.
For Figures 3E and 3G–3I, data are representative of 3 biologic replicates and are normalized to cells with WT overexpression, except that nuclear H3K36me3 nuclear intensity was normalized to cells transfected with empty vector. Tests of significance performed with Student’s t test, described in figure legends.
For Figure 4, SETD2 mutational status was queried on the cBioportal in 42,199 pan-cancer samples (curated set of all non-redundant samples) (Gao et al., 2013). Samples were then stratified by those with WT SETD2 or those with any alteration in SETD2 (including in-frame, missense, truncating, fusion, amplification, or deep deletion mutation). Overall survival was performed using Kaplan-Meier analysis, and significance calculated using the log rank test. Described in main text and figure legends.
For Figure S1, analysis is described in above section “Sample distance metric analysis.”
For Figures S2D and S4G Gene ontology analysis was performed using Enrichr, and p values displayed were multiple hypothesis corrected adjusted p values (Chen et al., 2013).
For Figure S4B, Gene ontology analysis of differentially methylated regions was performed using the Genomic Regions Enrichment of Annotations Tool (GREAT) using default settings.
DATA AND CODE AVAILABILITY
Methylation array data are deposited at GEO accession GEO: GSE142320. RNA-seq and whole exome-seq data are deposited at SRA accession PRJNA597052.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MIB-1/Ki-67 | Dako | GA626 |
| L1CAM | Sigma | UJ127.11 |
| GFAP | Dako | GA524 |
| EMA | Leica | GP1.4 |
| GFP | Abcam | ab6556 |
| H3K36me3 | Abcam | ab9050 |
| Biological Samples | ||
| Anaplastic ependymoma samples | UCSF Brain Tumor Center | N/A |
| Critical Commercial Assays | ||
| TruSeq RNA Library Prep Kit v2 | Illumina | RS-122- 2001 |
| Nimblegen SeqCap EZ Human Exome Kit v3.0 | Roche | 06465684001 |
| CellTiter 96 Non-Radioactive Cell Proliferation Assay | Promega | G4000 |
| Deposited Data | ||
| DNA Methylation Array | This paper | GEO: GSE142320 |
| RNA-seq data | This paper | SRA: PRJNA597052 |
| WES data | This paper | SRA: PRJNA597052 |
| Experimental Models: Cell Lines | ||
| Human: SF11435 ependymoma cell culture | UCSF Clinical Cancer Genomics Laboratory | SF11435 |
| Oligonucleotides | ||
| G-blocks | IDT | N/A |
| Genotyping: SETD2 forward 5′-TGTAAAACGACGG CCAGTCCTGTTACTCCTCGCGCCG-3′ | This paper | N/A |
| Genotyping: SETD2 reverse 5′-CAGGAAACAGCTA TGACCGGTCAAGCCAACAGCTGCAA-3′). | This paper | N/A |
| SETD2 L1962P correction forward: 5′-GACGCTGA AATAGAGCCCAAAGAGAGCAACGGC-3′ | This paper | N/A |
| SETD2 L1962P correction reverse: 5′-GCCGTTGC TCTCTTTGGGCTCTATTTCAGCGTC-3′ | This paper | N/A |
| SETD2 K2R mutagenesis forward: 5′-TCCGCTAGCG CCACCATGAGGCAGCTGCAGCCGCAGCCGCCTCC-3′ | This paper | N/A |
| SETD2 K2R mutagenesis reverse: 5′-GGAGGCGGCT GCGGCTGCAGCTGCCTCATGGTGGCGCTAGCGGA-3′ | This paper | N/A |
| Recombinant DNA | ||
| SETD2-EGFP | This paper | N/A |
| SETD2K2R-EGFP | This paper | N/A |
| C11orf95 | BACPAC Resources | CH17–67K13; CH17–388O01 |
| RELA | BACPAC Resources | RP11–642F7; CH17–211O12 |
| YAP1 | BACPAC Resources | RP11–11N20; RP11–1082I13 |
| Software and Algorithms | ||
| 3D Slicer v4.8.0 | https://www.slicer.org | https://www.slicer.org |
| R v3.4.3 | https://www.r-project.org | https://www.r-project.org |
| oro.dicom v0.5.3 | Whitcher et al., 2011 | https://cran.r-project.org/web/packages/oro.dicom |
| Kallisto v0.43.1 | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| JAFFA v1.09 | Davidson et al., 2015 | https://github.com/Oshlack/JAFFA/ |
| Tophat v2.1.0 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/ |
| Minfi v1.30.0 | Aryee et al., 2014; Fortin et al., 2017 | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| Limma v3.40.0 | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Conumee v1.3.0 | https://bioconductor.org/packages/release/bioc/html/conumee.html | https://bioconductor.org/packages/release/bioc/html/conumee.html |
| Bcbio | https://github.com/bcbio/bcbio-nextgen | https://github.com/bcbio/bcbio-nextgen |
| Picard suite | http://broadinstitute.github.io/picard | http://broadinstitute.github.io/picard |
| Genome Analysis Toolkit | McKenna et al., 2010 | https://software.broadinstitute.org/gatk/ |
| Varscan2 v2.4.0 | Koboldt et al., 2012 | http://dkoboldt.github.io/varscan/ |
| Freebayes | Garrison and Marth, 2012 | https://github.com/ekg/freebayes |
| Vardict | Lai et al., 2016 | https://github.com/AstraZeneca-NGS/VarDict |
| Snpeff v4.3 | Cingolani et al., 2012 | http://snpeff.sourceforge.net |
| Sequenza v2.1.0 | Favero et al., 2015 | https://cran.r-project.org/web/packages/sequenza/index.html |
| Ape v5.0 | https://cran.r-project.org/web/packages/ape | https://cran.r-project.org/web/packages/ape |
Highlights.
Epigenomic, transcriptomic, and exome profiles and phylogenies within an ependymoma
Spatially distinct clusters reflect neuronal development gene expression programs
Quantitative magnetic resonance imaging characteristics identify stem-like regions
Mutant histone methyltransferase SETD2 enhances proliferation in ependymoma cells
ACKNOWLEDGMENTS
We would like to thank and acknowledge David A. Solomon, MD, PhD for providing targeted DNA sequencing data and analysis using a Clinical-Laboratory-Improvement-Amendment-certified panel of 510 cancer-associated genes. D.R.R. is supported by NIH NCI award K08 CA212279-01. S.T.M. is supported by NIH NCI award F32 CA213944-01.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.018.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aguirre-Cruz L, Mokhtari K, Hoang-Xuan K, Marie Y, Criniere E, Tailli-bert S, Lopes M, Delattre J-Y, and Sanson M (2004). Analysis of the bHLH transcription factors Olig1 and Olig2 in brain tumors. J. Neurooncol 67, 265–271. [DOI] [PubMed] [Google Scholar]
- Andreiuolo F, Puget S, Peyre M, Dantas-Barbosa C, Boddaert N, Philippe C, Mauguen A, Grill J, and Varlet P (2010). Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Neuro-oncol. 12, 1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, and Irizarry RA (2014). Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertuzzi S, Porter FD, Pitts A, Kumar M, Agulnick A, Wassif C, and Westphal H (1999). Characterization of Lhx9, a novel LIM/homeobox gene expressed by the pioneer neurons in the mouse cerebral cortex. Mech. Dev 81, 193–198. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, et al. (2018). DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caubit X, Thoby-Brisson M, Voituron N, Filippi P, Bevengut M, Faralli H, Zanella S, Fortin G, Hilaire G, and Fasano L (2010).Teashirt 3 regulates development of neurons involved in both respiratory rhythm and airflow control. J. Neurosci 30, 9465–9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caubit X, Gubellini P, Andrieux J, Roubertoux PL, Metwaly M, Jacq B, Fatmi A, Had-Aissouni L, Kwan KY, Salin P, et al. (2016). TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat. Genet 48, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY,Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson NM, Majewski IJ, and Oshlack A (2015). JAFFA: high sensitivity transcriptome-focused fusion gene detection. Genome Med. 7, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desper R, and Gascuel O (2002). Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J. Comput. Biol 9, 687–705. [DOI] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, and Lin SM (2010). Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Schwartzentruber J, Khuong-Quang D-A, Liu X-Y, Sturm D, Korshunov A, Jones DTW, Witt H, Kool M, Albrecht S, et al. (2013). Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 125,659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (D1), D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, and Hansen KD (2014). Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J-P,Triche TJ Jr., and Hansen KD (2017). Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 33, 558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, and Marth G (2012). Haplotype-based variant detection from short-read sequencing. arXiv, arXiv:1207.3907v2. https://arxiv.org/abs/1207.3907. [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocin-ski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S (2008). Shared principles in NF-kappaB signaling. Cell 132, 344–362. [DOI] [PubMed] [Google Scholar]
- Hinohara K, Wu H-J, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, Madsen T, Fassl A, Egri SB, Papanastasiou M, et al. (2018). KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell 34, 939–953.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. (2014). The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun 5, 3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SES, White E, Eden C, et al. (2010). Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. (2014). Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, and Li Z-W (2002). NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301–310. [DOI] [PubMed] [Google Scholar]
- Khatua S, Ramaswamy V, and Bouffet E (2017). Current therapy and the evolving molecular landscape of paediatric ependymoma. Eur. J. Cancer 70, 34–41. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, Aboian M, Cha S, Raleigh DR, Braunstein S, et al. (2017). Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro-oncol. 19, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, and Ahringer J (2009). Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet 41, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, and Ma’ayan A (2010). ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, and Ma’ayan A (2018). Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9, 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, and Dry JR (2016). VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Jenkins NA, and Copeland NG (2019). The roles of initiating truncal mutations in human cancers: the order of mutations and tumor cell type matters. Cancer Cell 35, 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JS, Lee JA, Underwood JC, Harris AL, and Lewis CE (1999). Macrophage responses to hypoxia: relevance to disease mechanisms. J. Leukoc. Biol 66, 889–900. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, and Rowitch DH (2004). The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J. Neuropathol. Exp. Neurol 63, 499–509. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. (2007). Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Merkle FT, Gandhi AV, Gagnon JA, Woods IG, Chiu CN, Shi-mogori T, Schier AF, and Prober DA (2015). Evolutionarily conserved regulation of hypocretin neuron specification by Lhx9. Development 142, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, et al. (2014). Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, Erkek S, Federation A, Song A, Lee C, et al. (2018). Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. (2017). Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE, Silverman LB, et al. (2014). Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun 5, 3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modena P, Lualdi E, Facchinetti F, Veltman J, Reid JF, Minardi S, Janssen I, Giangaspero F, Forni M, Finocchiaro G, et al. (2006). Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J. Clin. Oncol 24, 5223–5233. [DOI] [PubMed] [Google Scholar]
- Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, Watchmaker PB, Yagnik G, Di Lullo E, Malatesta M, et al. (2017). Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 18, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Chen AI, and Jessell TM (2001). Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31, 773–789. [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson R, Kanduri C, Whitehead J, Pfeifer S, Lobanenkov V, and Feinberg AP (2003). Epigenetic variability and the evolution of human cancer. Adv. Cancer Res 88, 145–168. [DOI] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. (2007). A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet 39, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Arora S, Szulzewsky F, Juric-Sekhar G, Miyajima Y, Bolouri H, Yasui Y, Barber J, Kupp R, Dalton J, et al. (2018). Ade novo mouse model of C11orf95-RELA fusion-driven ependymoma identifies driver functions in addition to NF-κB. Cell Rep. 23, 3787–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, et al. (2015). Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27, 728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, and Strimmer K (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, Lee R, Tatevossian RG, Phoenix TN, Thiruvenkatam R, et al. (2014). C11orf95-RELA fusions drive oncogenic NF-kB signalling in ependymoma. Nature 506, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker NR, Hudson AL, Khong P, Parkinson JF, Dwight T, Ikin RJ, Zhu Y, Cheng ZJ, Vafaee F, Chen J, et al. (2016). Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci. Rep 6, 22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert D, Weber S, Lumsden A, and Scholpp S (2011). Lhx2 and Lhx9 determine neuronal differentiation and compartition in the caudal forebrain by regulating Wnt signaling. PLoS Biol. 9, e1001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahat MA, Bitterman H, and Lahat N (2011). Molecular mechanisms regulating macrophage response to hypoxia. Front. Immunol 2, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rétaux S, Rogard M, Bach I, Failli V, and Besson MJ (1999). Lhx9: a novel LIM-homeodomain gene expressed in the developing forebrain. J. Neurosci 19, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R, et al. ; Cancer Genome Atlas Research Network (2018). The Cancer Genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 23, 3698. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. (2007). Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 39, 232–236. [DOI] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Day INM, and Gaunt TR (2013). Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29, 1504–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, et al. (2012). Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 109, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, et al. (2005). Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8, 323–335. [DOI] [PubMed] [Google Scholar]
- Watkins TBK, and Schwarz RF (2018). Phylogenetic quantification ofintra-tumor heterogeneity. Cold Spring Harb. Perspect. Med 8, a028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold N, Jacobsen A, Schultz N, Sander C, and Lee W (2014). Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet 46, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcher B, Schmid VJ, and Thornton A (2011). Working with the DICOM and NIfTI data standards in R. J. Stat. Soft 44, 1–29. [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, and Laird PW (2007). Epigenetic stem cell signature in cancer. Nat. Genet 39, 157–158. [DOI] [PubMed] [Google Scholar]
- Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, Benner A, Hielscher T, Milde T, Remke M, et al. (2011). Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, and Zhu B (2011). H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem 286, 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung WK, Shapiro JR, and Shapiro WR (1982). Heterogeneous chemo-sensitivities of subpopulations of human glioma cells in culture. Cancer Res. 42, 992–998. [PubMed] [Google Scholar]
- Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, Zhang H, Li L, Sun L, Pan N, et al. (2018). A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528. [DOI] [PubMed] [Google Scholar]
- Zhou Q, and Anderson DJ (2002). The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109, 61–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Methylation array data are deposited at GEO accession GEO: GSE142320. RNA-seq and whole exome-seq data are deposited at SRA accession PRJNA597052.