

Abstract
The higher order structure (HOS) of protein therapeutics is essential for drug safety and efficacy and can be evaluated by two-dimensional (2D) nuclear magnetic resonance (NMR) spectroscopy at atomic resolution. 1Hn −15N amide correlated and 1H-13C methyl correlated NMR spectroscopies at natural isotopic abundance have been demonstrated as feasible on protein therapeutics as large as monoclonal antibodies and show great promise for use in establishing drug substance structural consistency across manufacturing changes and in comparing a biosimilar to an originator reference product. Spectral fingerprints from 1Hn - 1Hα correlations acquired using 2D homonuclear proton-proton J-correlated NMR experiments provide an alternative and complementary approach for high-resolution assessment of the HOS of lower molecular weight (< 30 kDa) protein therapeutics. Here, we evaluate different pulse sequences (COSY, TOCSY and TACSY) used to generate proton-proton J-correlated NMR spectral fingerprints and appraise the performance of each method for application to protein therapeutic HOS assessment and comparability.
Keywords: 2D NMR, fingerprinting, biologic, higher order structure, COSY, TOCSY, COIN-TACSY
Introduction
For a protein pharmaceutical or biologic to be safe and effective, the protein must fold into its proper secondary, tertiary and quaternary structures. Taken together, these different levels of protein folding are termed the higher-order-structure (HOS) of a biologic. Robust analytical methods for precise and accurate measurement of the HOS of a biologic are therefore essential to ensure product quality in drug development, to establish comparability between drug products when manufacturing changes occur, and to develop biosimilar drugs. Two-dimensional (2D) heteronuclear magnetic resonance spectroscopy (NMR) has emerged as a promising tool for the assessment of the HOS of protein biotherapeutics at atomic resolution [1–5]. With modern NMR spectrometer hardware, pulse sequences that allow rapid data acquisition [6] and sparse data acquisition methods [7–9], 1Hn-15N amide correlated and 1H-13C methyl correlated NMR spectra of biotherapeutics can now be routinely acquired at the low natural abundance of 13C (1.11 %) and 15N (0.37 %) in experimental time-frames that are competitive with other analytical techniques [10–18]. The feasibility of the 2D NMR spectral fingerprinting, originally demonstrated by Aubin and co-workers through collection of a 2D 1H,15N spectral map of unlabeled granulocyte-macrophage colony-stimulating factor (GM-CSF) [10], now extends to protein therapeutics as large as monoclonal antibodies (~ 150 kDa) [12, 14, 17, 18].
Similarly, 2D proton-proton J-correlated techniques that generate 1Hn - 1Hα spectral correlation maps could be used to fingerprint the HOS of biologics of small to moderate molecular weight (< 25 kDa) and would have an advantage of correlating only highly sensitive 1H nuclei. Like the heteronuclear 2D spectra, the 1Hn and 1Hα cross peak chemical shifts in the homonuclear 2D spectra serve as reporters of the local structure of the correlated 1Hn and 1Hα atoms in the three dimensional space of the protein fold. Taken together, the pattern of cross peaks provide a unique fingerprint for the conformation of the protein biotherapeutic. Standard 2D proton-proton J-correlated NMR experiments, such as double quantum filtered correlation spectroscopy (DQF-COSY), in-phase correlation spectroscopy (IP-COSY, CLIP-COSY), and total correlation spectroscopy (TOCSY), can all generate 2D homonuclear correlation maps, but each has potential limitations that may preclude routine use for biopharmaceutical HOS fingerprinting. The DQF-COSY yields anti-phase cross peak correlations that can lead to severe signal cancellation as the proton line widths and cross peak overlap increase with larger protein biologics. In contrast, in-phase COSY methods, like CLIP- COSY and IP-COSY, add a symmetrical refocusing period to generate in-phase cross peaks and can have overall greater sensitivity and resolution relative to the DQF-COSY [19–21]. The TOCSY also yields in-phase correlations, is generally more sensitive than the IP-COSY, but use of isotropic mixing results in the observation of additional aliphatic signals from each amino acid spin system, generating a more complex and potentially overlapped spectral map. Similarly, 2D NOESY spectral regions, which have been reported for use in protein biologic fingerprinting [22], can suffer from even greater complexity and spectral overlap which can limit application. Glaser et al. [23] introduced a selective TOCSY pulse scheme, the combination of isotropic mixing and selective nutation tailored correlation spectroscopy (COIN-TACSY) that could address this issue by providing for selective, optimized transfer of magnetization during the TOCSY period. Using the TACSY pulse scheme, maximal and limited transfer of in-phase magnetization from 1Hn to 1Hα is achieved by decoupling the upfield aliphatic proton resonances using selective shape pulses during the isotropic mixing period. Here, we compare the performance of each of these homonuclear J-correlated experiments and discuss the practical utility of each method for application towards HOS characterization of protein pharmaceuticals.
Results and Discussion
2D J-correlated proton-proton correlation methods
Utilization of one-dimensional (1D) 1H spectra is routine for analysis of small molecule therapeutics and is becoming increasingly used for protein biologics [24–27]. However, since every 1H atom resonates in 1D 1H spectra, severe spectral overlap limits the resolution of the 1D method for comparability of protein biologics and spectral differences can be obscured by the mass of signal. Through spectral filtering by relaxation, diffusion, or through spectral processing techniques, 1D methods have been introduced that to some extent ameliorate the problem of signal overlap [24, 27]. As with the 2D heteronuclear NMR spectral fingerprint, resolving the 1D 1H signals in a second dimension through homonuclear J-correlation, provides an approach to resolve overlapped 1D 1H signals into a 2D spectral map.
The utility of 1Hn - 1Hα COSY correlations as a fingerprint of a protein fold was recognized with the first applications of 2D NMR spectra to protein assignment and structure determination [28]. Acquisition of a 2D DQF-COSY on a hen egg white lysozyme (HEWL) test sample (Fig.1A) shows that the 1Hn - 1Hα fingerprint region can be defined as approximately 6.5 ppm – 10.0 ppm in the first dimension (1Hn region) and 3.0 ppm – 5.5 ppm in the second dimension (1Hα region). Supplemental material, Fig.S1A includes an expansion of the DQF- and IP-COSY spectra in the indirect dimension so that the data can be directly comparable to spectra generated by the TOCSY and COIN-TACSY experiments. While the performance of the DQF-COSY is reasonable for proteins that are less than approximately 25 kDa, the anti-phase cross peaks may lead to signal cancellation for higher molecular weight proteins and compromise the integrity of the spectral fingerprint. As has been previously noted, an in-phase COSY (e.g., IP- and CLIP-COSY) avoids the problem of signal cancellation by yielding in-phase correlations with higher sensitivity and greater resolution relative to the DQF-COSY experiment (Fig.1 B, Supplemental material, Fig.S1B). Using the test HEWL sample, the IP-COSY spectrum was acquired with four times fewer scans per increment to achieve comparable sensitivity to the DQF-COSY experiment. The primary disadvantage of the IP-COSY is the total constant time and refocusing period of greater than 50 ms, limiting its application to relatively small proteins (< 25 kDa) with sufficiently long transverse relaxation times. For larger protein biologics, such mAbs (~150 kDa) and even fragments of mAbs (i.e., Fab or Fc ~ 50 kDa), with shorter transverse relaxation times, magnetization coherence is attenuated during the refocusing periods, and broadened cross peaks are observed, reducing the overall utility of this method for larger proteins (Supplemental Material, Fig. S2A).
Fig. 1.
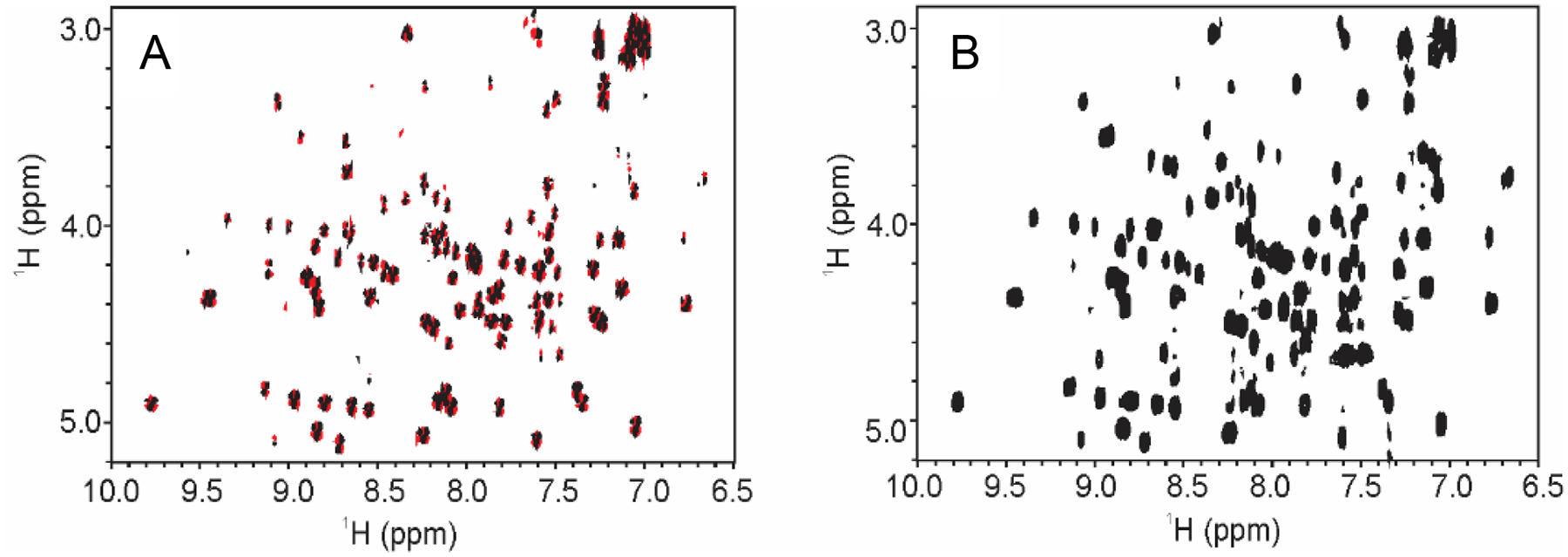
2D COSY-type spectral fingerprint of 1Hn - 1Hα region of HEWL collected at 600 MHz and 25 °C. (A) 2D DQF-COSY; (B) 2D IP-COSY. Positive contours shown in black and negative contours in red. The fingerprint region is defined as 6.5 ppm – 10.0 ppm (1Hn) and 2.5 ppm – 5.25 ppm (1Hα). See Material and Methods Section for acquisition and processing details.
As with the in-phase COSY, the TOCSY also yields in-phase cross peaks and eliminates the problem of signal cancellation observed in the DQF-COSY (Fig.2A, B; Supplemental Material, Fig.S3). The TOCSY is a standard tool for resonance assignment of complete spin systems as magnetization transfers efficiently from Spin A (SA) to SB to SC, and so on to yield a ‘total correlation’. In the TOCSY, the extent of magnetization transfer is governed by the magnitude of the J-couplings and the total mixing time, and increasing the mixing time will typically transfer magnetization farther along the spin system so long as there are appreciable spin-spin couplings. For each amino acid, the transfers give rise to cross peaks between 1Hn and other aliphatic proton resonances on the side chains within the same spin system. Due to these features, the TOCSY also provides a well-defined, spectral fingerprint of HOS, albeit a more complicated fingerprint since the 1Hn proton will have cross peaks to many other protons in each amino acid spin system. As a single cross peak type correlation (i.e., 1Hn - 1Hα) is typically sufficient for protein structural fingerprinting, the full 2D TOCSY generates a more complicated and potentially overlapping spectrum than is needed. For HOS comparability exercises, however, spectral analysis could be limited to the fingerprint region of interest. In addition, as with the IP-COSY, the TOCSY becomes less sensitive when applied to larger proteins with shorter transverse relaxation times, such as mAbs and fragments of mAbs.
Fig. 2.
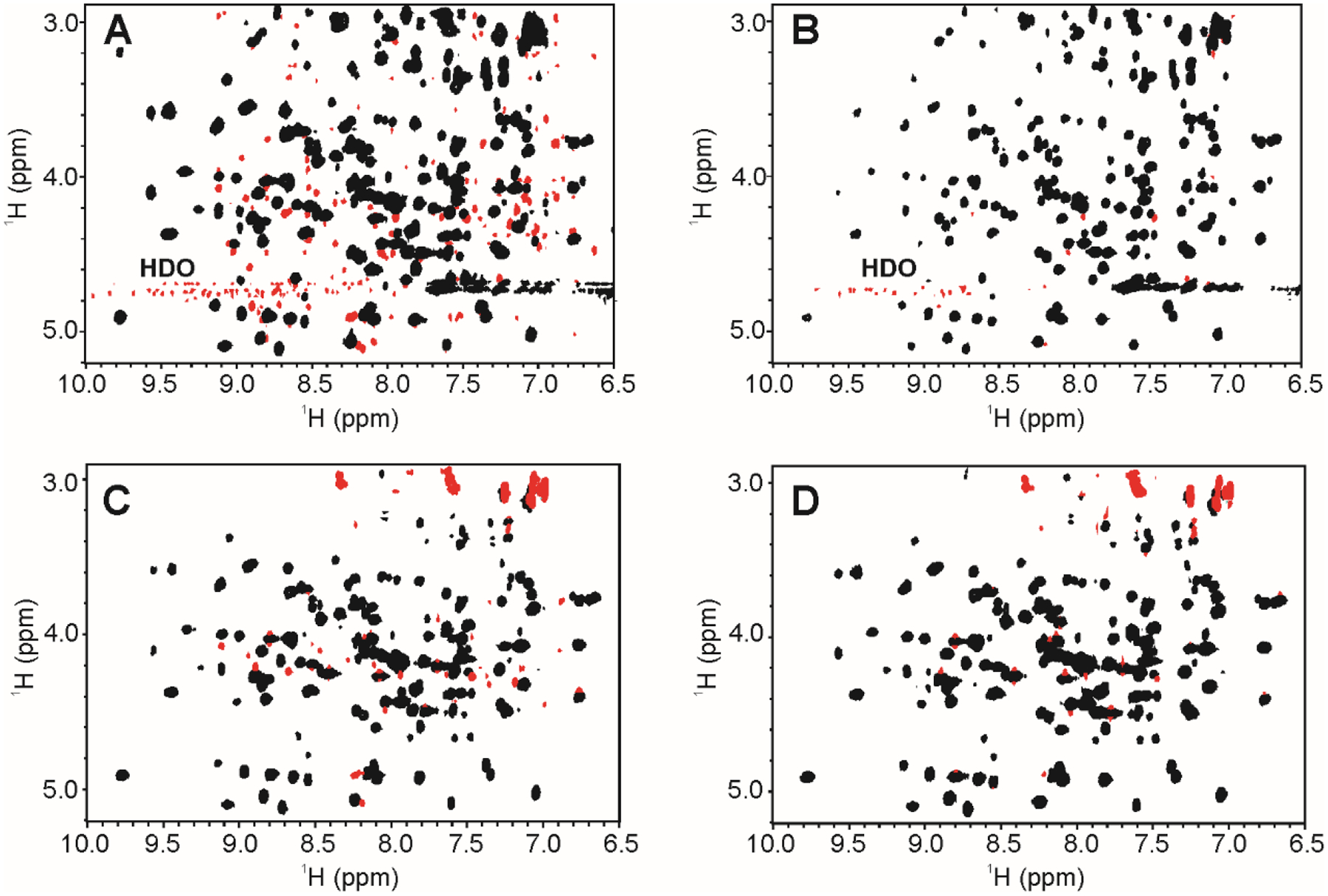
2D TOCSY-type spectral fingerprints of 1Hn - 1Hα region of HEWL collected at 600 MHz and 25 °C. (A) 2D 1H TOCSY using DIPSI-2; (B) 2D 1H clean TOCSY using DIPSI-2rc; (C) 2D 1H COIN-TACSY using DIPSI-2; (D) 2D 1H clean COIN-TACSY using DIPSI-2rc. Positive contours are in black and negative contours in red. ROE artifacts in panels A and C are removed by the addition of a relaxation compensation delay in the DIPSI-2rc pulse train in panels B and D. Residual negative peaks in the fingerprint region of panels B and D arose from signal truncation and apodization artifacts, and magnetization leakage due to incomplete decoupling by the TACSY pulse train (signals around 3 ppm). Additional negative artifacts in panels C and D are from magnetization leakage in the COIN-TACSY. The fingerprint region was defined as, 6.5 ppm – 10.0 ppm (Hn) and 2.5 ppm – 5.25 ppm (Hα). Expanded spectral plots with the upfield aliphatic region are shown in Supplemental Material, Fig.S2.
To generate a tailored TOCSY correlation of 1Hn - 1Hα with optimized sensitivity, the COIN-TACSY pulse scheme was developed to effectively reduce the number of coupled spins correlated in the isotropic mixing period [23]. The COIN-TACSY experiment works by decoupling aliphatic spins up field of the 1Hn - 1Hα region during the isotropic mixing period (Fig.2C, D; Supplemental Material, Fig.S3). The isotropic mixing in the COIN-TACSY was first implemented with a DIPSI-2 pulse train in accordance with literature precedent for application to small peptides [23]. The resulting spectrum of HEWL (Fig.2C) contained the expected TACSY transfer from 1Hn to 1Hα but also was significantly contaminated with cross relaxation (ROE) artifacts in the fingerprint region. The ROE artifacts, however, could be mitigated by modifying the original TACSY to include a relaxation compensation delay with the DIPSI-2rc pulse train [29]. The remaining negative peaks in the resulting COIN-TACSY fingerprint region of Fig.2D are artifacts from apodization. Again, as with the IP-COSY and TOCSY, the TACSY experiment becomes less sensitive when applied to larger proteins with shorter transverse relaxation times, and as such may limit its utility for fingerprinting of larger biopharmaceuticals. Nonetheless, the sensitivity afforded by the COIN-TACSY can allow the acquisition of fingerprints of mAb fragments (~ 50 kDa), albeit with more broader and overlapped signals than observed for the lower molecular weight protein biologics (Supplemental Material, Fig. S2B).
An advantageous feature of the COIN-TACSY is that it allows for the tailoring of the fingerprint based on the spectral map of a protein. Both the effective bandwidth and spectral offset of the decoupling shape pulse can be customized, although imperfections in decoupling of magnetization coherence transfer can lead to some up field 1HN-1Haliphatic cross peaks with reduced intensity (Supplemental Materials, Fig.S3C, D). The magnetization leakage, however, may again not be a significant hindrance for spectral comparability exercises as spectral analysis can be limited to the fingerprint region of interest. The pulse program for the COIN-TACSY with DIPSI- 2rc and details on the implementing this experiment are given in Supplemental Materials, Fig.S4.
To compare the relative S/N of each of the 2D J-correlated homonuclear experiments, two 1D slices were taken at 5.05 ppm and 4.07 ppm from the each of the corresponding 2D spectra (Fig.3, Supplemental Materials, Table S1). In general, the clean COIN-TACSY using DIPSI-2rc (red trace) was the most sensitive experiment, while the IP-COSY (black trace) was the least sensitive experiment. The other experiments achieved moderate levels of sensitivity. For the DQF-COSY, a direct comparison was not made due to the anti-phase nature of the cross peaks and the fact that this experiment needed to be run with four times the scans to achieve comparable sensitivity (data not shown). A listing of the parameters and experimental times are tabulated in Supplemental Materials, Table S2. While the overall measurement time was 3 hours for most experiments, experimental times can be further reduced if a non-uniform sampling (NUS) acquisition scheme is used or if reduced resolution can be tolerated. The rather modest effect of reduced resolution is highlighted by the IP-COSY spectra, for which only 208 total points could be collected due to the constant time delays. For this experiment, the total measurement time was only 35 minutes and the resolution of the spectrum is still sufficient for the purposes of fingerprinting the HOS.
Fig. 3.
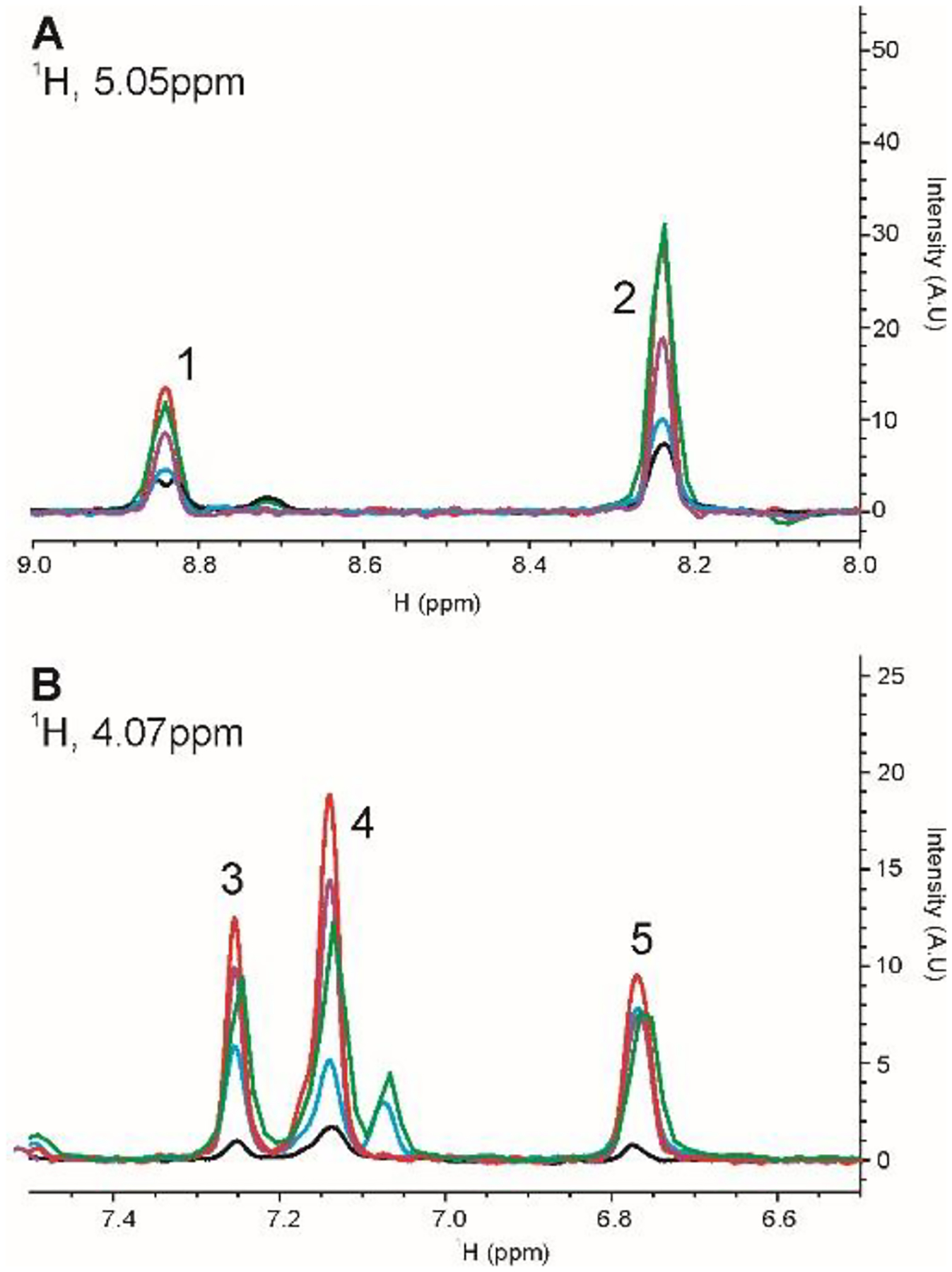
1D slices from 2D 1H homonuclear experiments on HEWL for the qualitative comparison of sensitivity of 2D 1H J-correlated methods. (A) Slice taken at 1H 5.05 ppm; (B) Slice taken at 1H 4.07 ppm. IP-COSY is black; TOCSY using DIPSI-2rc is blue; TOCSY using DIPSI-2 is green; COIN-TACSY using DIPSI-2rc is red; COIN-TACSY using DIPSI-2 is purple. Peak numbers are used in the tabulation of relative peak intensities in Supplemental Materials, Table S1.
Applications to Protein Biologics
The choice of a 2D NMR experiment will depend on the nature of the need for spectral fingerprinting a small peptide or protein biologic. If artifacts and/or extraneous cross peaks from spin diffusion are not tolerable, then the IP-COSY may be the experiment of choice. While the IP- COSY may be the less sensitive than other homonuclear experiments, its in-phase magnetization offers a distinct advantage over the DQF-COSY and allows a clear definition of the 1Hn- 1Hα region. On the other hand, if extraneous peaks can be tolerated, then the clean COIN-TACSY offers by far the most sensitive homonuclear correlated experiment. This sensitivity may be especially beneficial in cases when sample amounts are limited. With these considerations in mind, we collected IP-COSY and COIN-TACSY spectra for two biotherapeutically relevant molecules, Exendin-4, a 4.1 kDa peptide that has antidiabetic properties and Filgrastim, an 18.8 kDa protein that is used during cancer therapy to treat neutropenia. The Exendin-4 peptide shows limited structure in standard aqueous formulations, affording greater exchange between the amide protons and water. Although fewer cross peaks than might be expected are therefore observed in the 2D NMR spectra (Fig.4A, B), these spectral fingerprints can still be considered representative of its residual HOS fingerprint. In this case, addition of 30 % trifluoroethanol (TFE) to the Exendin-4 sample as a stabilizing agent of α-helical structure, results in a spectrum with a greater number of cross peaks as would be expected from a more structured peptide (Fig.4C).
Fig.4.
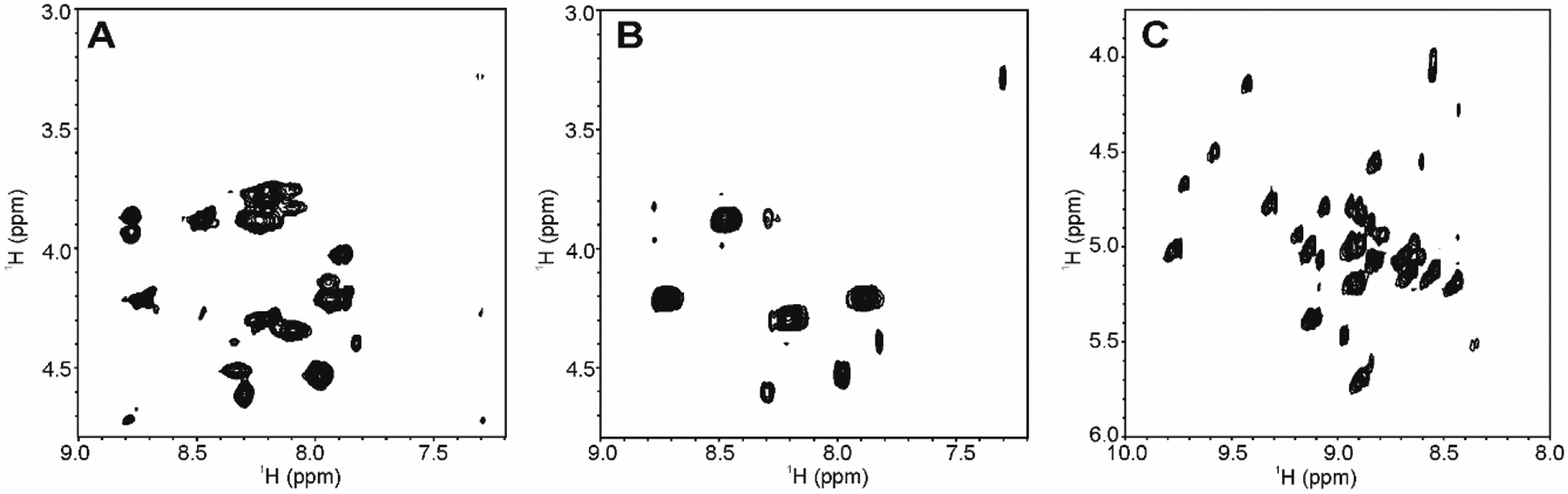
2D 1H spectral fingerprints of 1Hn - 1Hα region of Exendin-4 in 20 mM NaOAc-d3 pH 4.5, 97 % H2O, 3% D2O collected at 600 MHz and 25 °C. (A) 2D 1H COIN-TACSY using DIPSI-2rc; (B) 2D 1H IP-COSY; (C) 2D 1H IP-COSY in 70% water, 30% 2,2,2-trifluoroethanol-d2 (TFE). Only positive contours are shown since all cross peaks are in-phase. In Panels A and B, normal formulation conditions affords an unstructured state for Exendin-4. In panel C, 30% TFE causes the peptide to form an α-helix, affording the visualization of a greater number of cross peaks.
In the case of Filgrastim, greater spectral complexity and more overlap is observed due to a greater number of residues as compared to the Exendin-4 or HEWL samples (Fig.5). Nevertheless, the IP-COSY run with 208 total points in t1 due to the constant time evolution, provided a reasonably well-resolved fingerprint spectra (Fig.5A). Additional resolution could potentially be afforded through use of the CLIP-COSY which is not limited by a constant time t1 acquisition, although, these gains will be modest for a protein of the size of Filgrastim. As expected, acquisition of the Filgrastim fingerprint (Fig.5B) with the COIN-TACSY experiment run with 1,024 total points in t1 afforded greater sensitivity than the in-phase COSY and slightly better resolution, showing the advantages of this approach for application to spectral fingerprinting of biologics in the 10 kDa - 25 kDa size range.
Fig.5.
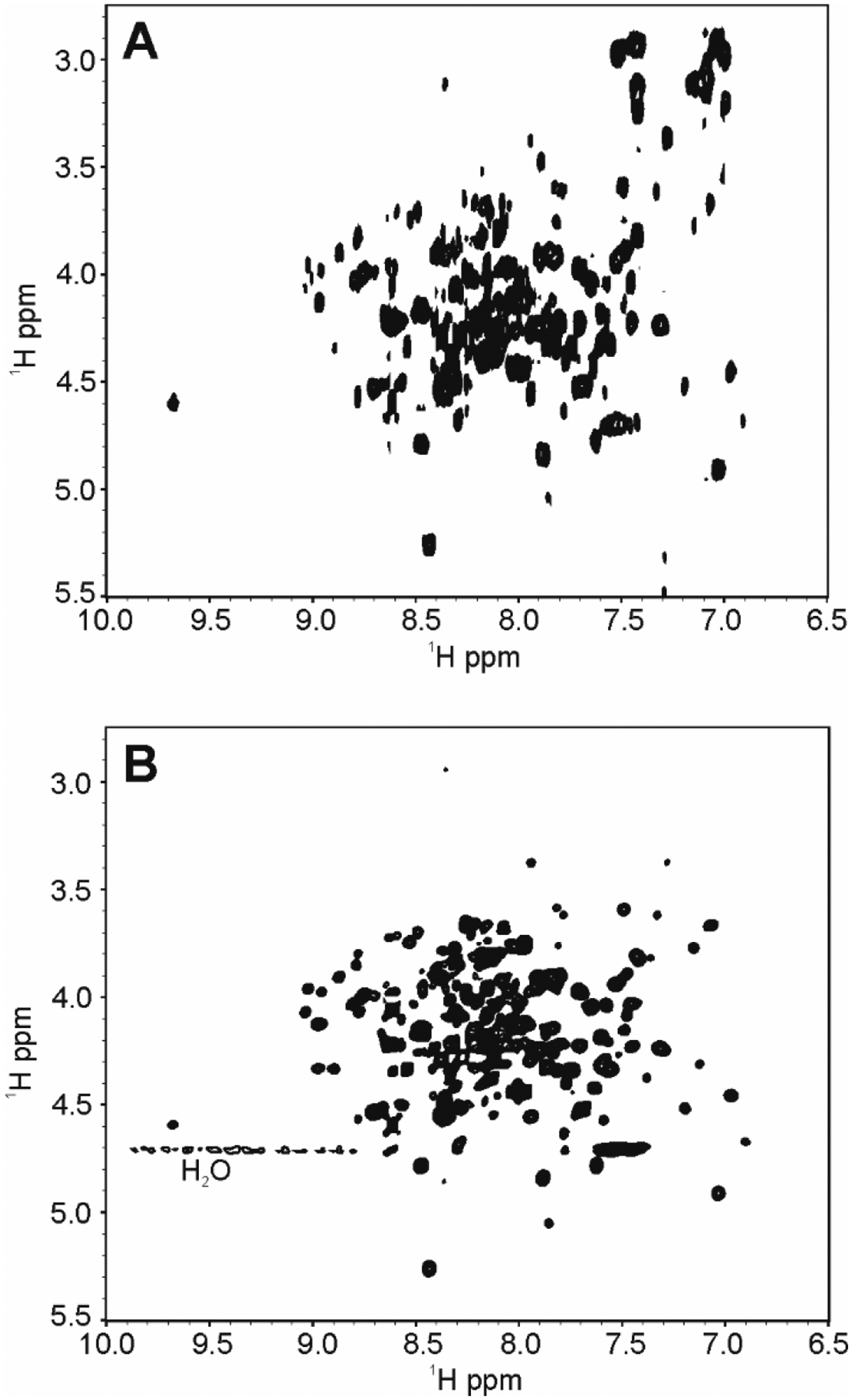
2D 1H spectral fingerprints of 1Hn - 1Hα region of Filgrastim in 20 mM NaCl, 10 mM NaOAc-d3 pH 4.0 collected at 600 MHz and 25 °C. (A) 2D IP-COSY and (B) 2D COIN-TACSY using DIPSI-2rc. Only positive cross peaks are shown, since all peaks are in-phase.
Conclusion
In summary, 2D J-correlated proton-proton experiments have been surveyed for application in generating 1Hn - 1Hα spectral fingerprints of protein therapeutic structure. In comparing the different modes for acquisition of the 1Hn - 1Hα spectral map, the COIN-TACSY, modified with a clean DISPI sequence, was found to be the best candidate for structural mapping of protein therapeutics in the 10 kDa - 25 kDa size range due to its potential for selective correlation and superior sensitivity. The TACSY sequence, however, did suffer from some upfield spectral artifacts due to imperfections in the pulse sequence. If such artifacts are not tolerable and sample concentration is not limiting, then the in-phase COSY becomes a ‘cleaner’ alternative to the TACSY, albeit with considerably lower sensitivity. In general, relatively rapid acquisition times make these 2D NMR experiments another practical approach to spectral fingerprinting of protein therapeutics in the lower molecular weight range (MW < 25 kDa), that can complement the 2D heteronuclear correlated methods. In yielding correlating spectra to 1Hα protons and, if desired, other aliphatic side chain protons, these methods provide access to additional backbone and side chain nuclear spin probes of the protein HOS. Lastly, the 1Hn - 1Hα spectral maps generated using these methods can be similarly analyzed using well-established statistical tools, which have previously been applied to datasets of 2D correlated heteronuclear NMR spectra to assess structural comparability [2, 5, 22, 30, 31].
Materials and methods1
Sample Preparation
Hen egg white lysozyme (HEWL, Sigma-Aldrich, INC) sample was dissolved in 5 mM sodium deuteroacetate (Cambridge Isotope Labs, INC), pH 4.0 with 3% D2O. The final concentration of HEWL was 1 mM. Exendin-4 (Prospec Protein Specialists) was constituted in 20 mM sodium deuteroacetate, pH 4.5 with 3 % D2O or 15 mM sodium phosphate pH 5.9 in 30% trifluoroethanol, 67% water, and 3% D2O. The final concentration for both Exendin-4 samples was 1.5 mM. Filgrastim was prepared as previously reported [14] and dialyzed into a final buffer of 50 mM NaCl, 10 mM NaOAc-d3 pH 4.0 with 3% D2O.
NMR Spectroscopy
Data were acquired on a three-channel Bruker Biospin AVANCE III 600 MHz spectrometer equipped with an actively shielded z-axis gradient triple resonance TCI cryoprobe. The 2D DQF- COSY, 2D standard and clean TOCSY using both DIPSI-2 and DIPSI-2rc, and 2D COIN-TACSY were acquired with a sweep width of 9,600 Hz in both dimensions, 2690 × 1024 total points, and 8 scans per increment, except for the DQF-COSY, for which 32 scans per increment were collected. Selected experimental parameters and measurement times are tabulated in Supplemental Materials, Table S2. The 1H transmitter was placed at 4.70 ppm. Total experimental time was 12 h for the DQF-COSY and 3 h for all other TOCSY experiments. For the TOCSY experiments, the isotropic mixing period was achieved using the DIPSI-2 [32] or DIPSI-2rc [29] pulse trains, covering a bandwidth of 38,460 Hz. For the clean TOCSY, a relaxation compensating delay of 41.6 μs was used. The total mixing period was 80 ms. In a similar fashion, the 2D IP-COSY was acquired with the same parameters as above, except that only 208 total points were collected in t1 due to the constant time delay. The experimental time for the IP-COSY was 35 min. All spectra were recorded at 25 °C.
Unless otherwise indicated in the figure captions, the COIN-TACSY [23] was implemented using an e.SNOB pulse [33] with an offset of −2,100 Hz and an effective bandwidth of 2,100 Hz (Fig.S4). The original COIN-TACSY was implemented with the DIPSI-2 pulse train covering a bandwidth of 25,640 Hz and a total mixing period of 83 ms. For the clean COIN-TACSY, a DIPSI-2rc pulse train was used with an effective bandwidth of 38,460 Hz and a relaxation compensating delay of 41.6 μs. The total mixing period was set to 84.7 ms. All other acquisition parameters are the same as the standard TOCSY experiment.
All spectra collected using uniform acquisition and processed using NMRPipe [34]. Data were apodized with a shifted sine bell function and zero-filled prior to Fourier Transform. Spectra were visualized using NMRFAM-Sparky software. [35]
Supplementary Material
Acknowledgement
The authors acknowledge support, in part, from the W.M. Keck Laboratories for Biomolecular NMR and the NIST Biomanufacturing Initiative.
Footnotes
Note
Certain commercial equipment, instruments, and materials are identified in this paper in order to specify the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment identified is necessarily the best available for the purpose.
References
- 1.Ghasriani H, et al. , Precision and robustness of 2D-NMR for structure assessment of filgrastim biosimilars. Nat Biotechnol, 2016. 34(2): p. 139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Japelj B, et al. , Biosimilar structural comparability assessment by NMR: from small proteins to monoclonal antibodies. Sci Rep, 2016. 6: p. 32201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiss R, Fizil A, and Szantay C Jr., What NMR can do in the biopharmaceutical industry. J Pharm Biomed Anal, 2018. 147: p. 367–377. [DOI] [PubMed] [Google Scholar]
- 4.Pandya A, et al. , An Evaluation of the Potential of NMR Spectroscopy and Computational Modelling Methods to Inform Biopharmaceutical Formulations. Pharmaceutics, 2018. 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinson RG, et al. , Enabling adoption of 2D-NMR for the higher order structure assessment of monoclonal antibody therapeutics. MAbs, 2019. 11(1): p. 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schanda P, Kupce E, and Brutscher B, SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR, 2005. 33(4): p. 199–211. [DOI] [PubMed] [Google Scholar]
- 7.Palmer MR, et al. , Performance tuning non-uniform sampling for sensitivity enhancement of signal- limited biological NMR. J Biomol NMR, 2014. 58(4): p. 303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaglio F, et al. , Non-Uniform Sampling for All: More NMR Spectral Quality, Less Measurement Time. Am Pharm Rev, 2017. 20(4). [PMC free article] [PubMed] [Google Scholar]
- 9.Robson S, et al. , Nonuniform Sampling for NMR Spectroscopy. Methods Enzymol, 2019. 614: p. 263–291. [DOI] [PubMed] [Google Scholar]
- 10.Aubin Y, Gingras G, and Sauve S, Assessment of the three-dimensional structure of recombinant protein therapeutics by NMR fingerprinting: demonstration on recombinant human granulocyte macrophage-colony stimulation factor. Anal Chem, 2008. 80(7): p. 2623–7. [DOI] [PubMed] [Google Scholar]
- 11.Jin X, et al. , Heteronuclear NMR as a 4-in-1 analytical platform for detecting modification-specific signatures of therapeutic insulin formulations. Anal Chem, 2014. 86(4): p. 2050–6. [DOI] [PubMed] [Google Scholar]
- 12.Arbogast LW, Brinson RG, and Marino JP, Mapping monoclonal antibody structure by 2D 13C NMR at natural abundance. Anal Chem, 2015. 87(7): p. 3556–61. [DOI] [PubMed] [Google Scholar]
- 13.Aubin Y, et al. , Monitoring Effects of Excipients, Formulation Parameters and Mutations on the High Order Structure of Filgrastim by NMR. Pharm Res, 2015. 32(10): p. 3365–75. [DOI] [PubMed] [Google Scholar]
- 14.Arbogast LW, et al. , 2D (1)H(N), (15)N Correlated NMR Methods at Natural Abundance for Obtaining Structural Maps and Statistical Comparability of Monoclonal Antibodies. Pharm Res, 2016. 33(2): p. 462–75. [DOI] [PubMed] [Google Scholar]
- 15.Arbogast LW, Brinson RG, and Marino JP, Application of Natural Isotopic Abundance (1)H-(1)(3)C- and (1)H-(1)(5)N-Correlated Two-Dimensional NMR for Evaluation of the Structure of Protein Therapeutics. Methods Enzymol, 2016. 566: p. 3–34. [DOI] [PubMed] [Google Scholar]
- 16.Hodgson DJ and Aubin Y, Assessment of the structure of pegylated-recombinant protein therapeutics by the NMR fingerprint assay. J Pharm Biomed Anal, 2017. 138: p. 351–356. [DOI] [PubMed] [Google Scholar]
- 17.Singh SM, et al. , Effect of Polysorbate 20 and Polysorbate 80 on the Higher-Order Structure of a Monoclonal Antibody and Its Fab and Fc Fragments Probed Using 2D Nuclear Magnetic Resonance Spectroscopy. J Pharm Sci, 2017. 106(12): p. 3486–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodgson DJ, Ghasriani H, and Aubin Y, Assessment of the higher order structure of Humira(R), Remicade(R), Avastin(R), Rituxan(R), Herceptin(R), and Enbrel(R) by 2D-NMR fingerprinting. J Pharm Biomed Anal, 2019. 163: p. 144–152. [DOI] [PubMed] [Google Scholar]
- 19.Xia Y, et al. , IP-COSY, a totally in-phase and sensitive COSY experiment. Magn Reson Chem, 2005. 43(5): p. 372–9. [DOI] [PubMed] [Google Scholar]
- 20.Koos MR, et al. , CLIP-COSY: A Clean In-Phase Experiment for the Rapid Acquisition of COSY-type Correlations. Angew Chem Int Ed Engl, 2016. 55(27): p. 7655–9. [DOI] [PubMed] [Google Scholar]
- 21.Kakita VMR and Bharatam J, Real-time homonuclear broadband decoupled pure shift COSY. Magn Reson Chem, 2018. 56(10): p. 963–968. [DOI] [PubMed] [Google Scholar]
- 22.Zuperl S, et al. , Chemometric approach in quantification of structural identity/similarity of proteins in biopharmaceuticals. J Chem Inf Model, 2007. 47(3): p. 737–43. [DOI] [PubMed] [Google Scholar]
- 23.Carlomagno T, Prasch T, and Glaser SJ, COIN TACSY, a Novel Approach to Tailored Correlation Spectroscopy. Journal of Magnetic Resonance, 2001. 149(1): p. 52–57. [Google Scholar]
- 24.Poppe L, et al. , Profiling formulated monoclonal antibodies by (1)H NMR spectroscopy. Anal Chem, 2013. 85(20): p. 9623–9. [DOI] [PubMed] [Google Scholar]
- 25.Visser J, et al. , Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs, 2013. 27(5): p. 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorgel F, et al. , Comparability of biosimilar filgrastim with originator filgrastim: protein characterization, pharmacodynamics, and pharmacokinetics. BioDrugs, 2015. 29(2): p. 123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franks J, et al. , Spin Diffusion Editing for Structural Fingerprints of Therapeutic Antibodies. Anal Chem, 2016. 88(2): p. 1320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wüthrich K, NMR of proteins and nucleic acids. 1986.
- 29.Cavanagh J and Rance M, Suppression of cross-relaxation effects in TOCSY spectra via a modified DIPSI-2 mixing sequence. Journal of Magnetic Resonance (1969), 1992. 96(3): p. 670–678. [Google Scholar]
- 30.Amezcua CA and Szabo CM, Assessment of higher order structure comparability in therapeutic proteins using nuclear magnetic resonance spectroscopy. J Pharm Sci, 2013. 102(6): p. 1724–1733. [DOI] [PubMed] [Google Scholar]
- 31.Arbogast LW, et al. , Multivariate Analysis of Two-Dimensional (1)H, (13)C Methyl NMR Spectra of Monoclonal Antibody Therapeutics To Facilitate Assessment of Higher Order Structure. Anal Chem, 2017. 89(21): p. 11839–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaka AJ, Lee CJ, and Pines A, Iterative schemes for bilinear operators; application to spin decoupling. Journal of Magnetic Resonance (1969), 1988. 77(2): p. 274–293. [Google Scholar]
- 33.Kupce E, Boyd J, and Campbell ID, Short selective pulses for biochemical applications. J Magn Reson B, 1995. 106(3): p. 300–3. [DOI] [PubMed] [Google Scholar]
- 34.Delaglio F, et al. , NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR, 1995. 6(3): p. 277–93. [DOI] [PubMed] [Google Scholar]
- 35.Lee W, Tonelli M, and Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics, 2014. 31(8): p. 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.