

Abstract
The red palm weevil, Rhynchophorus ferrugineus, infests palm plantations, leading to large financial losses and soil erosion. Pest-host interactions are poorly understood in R. ferrugineus, but the analysis of genetic diversity and pest origins will help advance efforts to eradicate this pest. We sequenced the genome of R. ferrugineus using a combination of paired-end Illumina sequencing (150 bp), Oxford Nanopore long reads, 10X Genomics and synteny analysis to produce an assembly with a scaffold N50 of ~60 Mb. Structural variations showed duplication of detoxifying and insecticide resistance genes (e.g., glutathione S-transferase, P450, Rdl). Furthermore, the evolution of gene families identified those under positive selection including one glycosyl hydrolase (GH16) gene family, which appears to result from horizontal gene transfer. This genome will be a valuable resource to understand insect evolution and behavior and to allow the genetic modification of key genes that will help control this pest.
Subject terms: Ecology, Genetics, Evolutionary ecology, Computational biology and bioinformatics
Khaled Michel Hazzouri et al. study the red palm weevil, Rhynchophorus ferrugineus, which causes agricultural damage. They provide a genome assembly and report duplications of detoxifying and insecticide resistance genes as well as genes under positive selection like the glycosyl hydrolyase gene family. Their work provides genetic insights that can help control this pest.
Introduction
The order Coleoptera is the largest among insects and has over 400,000 species, which account for more than 20% of metazoans1 and include agriculture and forest pest species. Specialized interactions with host plants allowed their evolution as destructive herbivores and crop pests2,3. Rhynchophorus ferrugineus (R. ferrugineus) (Olivier 1790) is a Coleopteran pest in the Curculionoidea family whose larvae destroy palm trees worldwide. A native to Southeast Asia and Melanesia, its range has recently expanded due to accidental introductions into the Middle East, Mediterranean Basin, Caribbean, and USA4. It attacks more than 26 palm species belonging to 16 genera and has been classified as a serious pest on the A2 list according to the EPPO20085 (European and Mediterranean Plant Protection Organization). For instance, in 2009, the annual loss in the Arabian Gulf region, which accounts for 30% of the world date palm (Phoenix dactylifera) production, has been estimated at US$ 25.92 millions6. Symptoms of infestation are visible only after the tree has been severely damaged, thus destroying the tree beyond remediation before the pest is detected. This stealth lifestyle of the R. ferrugineus larva is enabled by its early migration to the heart of the date palm vascular system7.
In general, dry woody plants have limited sugar, nutrient, and mineral content because of the lignified nature of plant cell walls. However, palm trees are wet woody plants that have a very sugary sap. Pest species of woody plants have to be detoxified from secondary metabolites such as allelochemicals, which requires metabolic adaptation8,9 that also enables the pest to develop rapid metabolic resistance to other toxins, including insecticides. Indeed, multiple phytophagous beetles have increased activity of insecticide detoxifying enzymes, such as cytochrome P450s (CYPs), glutathione S-transferases (GSTs), and UDP-glycosyltransferases (UGTs)10–13, as well as xenobiotic transporters14,15. They also have plant cell wall degrading enzymes (PCWDEs) for cellulose, hemicellulose, or pectin16–18. In fact, some beetles appear to have acquired PCWDEs via horizontal gene transfer (HGT) from fungi or bacteria followed by gene duplication and expansion into multi-gene families19. In contrast, other wood-feeding insects such as termites, ants, and cockroaches host microbial symbionts that provide these metabolic activities20–23.
Despite recent investigations of gene expression in R. ferrugineus24–26, no genome was available. To address this gap, we performed whole genome sequencing and de novo assembly of its genome, transcriptome sequencing, genome annotation, and performed comparative genomic analyses with the mountain pine beetle (Dendroctonus ponderosae), the coffee berry borer (Hypothenemus hampei), the red flour beetle (Tribolium castaneum), and Drosophila melanogaster. We report the evolution of gene families in R. ferrugineus and demonstrate the duplication and amplification of detoxifying genes and insecticide resistance genes (e.g., Rdl). We document the origin of gene families by HGT events, such as that of the glycosyl hydrolase (GH16). We also estimate ancestral and recent effective population size of the species and investigate whether there was selective pressure on some gene families involved in the adaptation to life on date palm tissue.
This R. ferrugineus genome will be an essential resource to study the genetic diversity of the species and will allow genetic manipulation via CRISPR/Cas9. Self-propagating of deleterious genetic variants could spread through the R. ferrugineus population through gene drives, which could weaken or eliminate the ability of R. ferrugineus to infest and destroy date palm plantations, thus saving millions of dollars and years of labor as well as maintaining a stable food supply for vulnerable communities.
Results
Genome assembly, characterization, and annotation
The genome of R. ferrugineus (Fig. 1a) is the third Coleoptera genome sequenced in the Curculionoidea family and a distant relative of model insect species T. castaneum (a Tenebrionid that diverged 234 Mya) and D. melanogaster (a Diptera that diverged 294 Mya) (Fig. 1b). They are distributed in South East Asia and in the Middle East (Fig. 1c).
Fig. 1. Geographic and phylogenetic context.

a R. ferrugineus, male and female. The male weevil has a tuff of soft reddish brown hairs along the dorsal facet of the snout, which is absent in the female weevil. b Phylogenetic tree depicting the relationship between R. ferrugineus and other Coleoptera from the Curculionidae and Tenebrionidae families with Drosophila melanogaster as outgroup. c Geographic distribution of the native and invasive R. ferrugineus4 was plotted using R map package89,100.
The Supernova assembly was used to scaffold ABYSS assemblies from 150 bp Illumina paired-end data. This gave an improved assembly with N50 of 150.8 kb for female and 137.7 kb for male and an assembly size of ~780 Mb (male and female) (Table 1). Other methods were applied but did not yield significant improvements in assembly quality (Supplementary Fig. 1). The Nanopore long reads comprised 23 Gb of data with mean read length of ~2 kb (Supplementary Fig. 2) and an assembly of 474.4 Mb and a scaffold N50 of 79.8 kb. The hybrid Illumina paired-end (150 bp) and long-read Nanopore data generated an assembly of 789.9 Mb for female and 780.2 for male with an N50 ~2 Mb. Merging the two assemblies above did not improve the N50. A final chromosome-level assembly consisting of nine pseudochromosomes and an X chromosome was produced assuming syntenic relationships with the red flour beetle using Chromassemble (Fig. 2a). A summary of different assemblies generated in comparison to the red flour beetle is presented in Table 1. The final genome assembly of R. ferrugineus is deposited and available at NCBI and Dryad public databases (see “Data availability” section).
Table 1.
Assembly statistics of red palm weevil (R. ferrugineus) genome at different platforms in comparison to red flour beetle (T. castaneum).
| Species | Data | Sex | Size (Mbp)* | Assembly size (Mbp) | Scaffolds** | N50a/b (Mbp) | BUSCO*** C (%) |
|---|---|---|---|---|---|---|---|
| R. ferrugineus (M_v.1) | Illumina paired-end + 10x genomics | M | 696.3 ± 5.3 | 780.5 | 12,462 | 0.1377/0.024 | 84.6 |
| R. ferrugineus (F_v.1) | Illumina paired-end + 10x genomics | F | 726.2 ± 12.8 | 783.3 | 12,355 | 0.1508/0.029 | 84.6 |
| R. ferrugineus (M_v.2) | Oxford Nanopore | M | 696.3 ± 5.3 | 474.4 | 10,580 | 0.0798/0.017 | 83.2 |
| R. ferrugineus (M_v.3) | Hybrid assembly (Illumina + Oxford Nanopore) | M | 696.3 ± 5.3 | 780.2 | 4822 | 2.12 | 89.2 |
| R. ferrugineus (F_v.3) | Hybrid assembly (Illumina + Oxford Nanopore) | F | 726.2 ± 12.8 | 789.9 | 4788 | 2.02 | 89.2 |
| R. ferrugineus (M_pseudochr) | Synteny to red flour beetle | M | 696.3 ± 5.3 | 782.19 | 4812 | 64.11 | 92.6 |
| R. ferrugineus (F_pseudochr) | Synteny to red flour beetle | F | 726.2 ± 12.8 | 780.66 | 4515 | 60.8 | 91.9 |
| T. castaneum (red flour beetle) | Sanger + BACs + Genetic + maps + Illumina + BioNano | M + F | 204 | 165.9 | 6580 | 14.6/0.073 | 98.4 |
*Flow cytometry estimation.
**Scaffold numbers.
***Benchmarking Universal Single-Copy Orthologs.
aN50 scaffold.
bN50 contig.
Fig. 2. Comparative genomics and analysis of orthology.

a Synteny plot between the red palm weevil R. ferrugineus and the red flour beetle T. castaneum showing synteny going from low (yellow), medium (brown) to strong (green). b Vendiagram of shared and unique orthologues among R. ferrugineus, T. castaneum, D. ponderosae, H. hampei, and D. melanogaster, Orthologous groups (OGs) as well as the number of genes for each of the species is highlighted between brackets. c Enrichment of shared orthologues among the five species where the number of genes is shown as heatmap going from low (red) to high (purple) and significance with false discovery rate (FDR) depicted by the size of the bubble, small (low) to high (big). d The percentage of each transposable element is shown in each of the five species as a horizontal stacked barplot, with the genome size also being shown. The color depicts the different types of transposable elements such as DNA, cut and paste (orange), long terminal repeat (LTR) (green) as well as other satellite and simple repeats.
The R. ferrugineus karyotype comprises ten autosomes and a pair of sex chromosomes X and yp27 with males having X and yp and females XX. We identified 54 scaffolds that match the X chromosomes of the red flour beetle (Supplementary Fig. 3). The diversity of these scaffolds was low compared to the nine autosomes (Supplementary data file 1). Nucleotide diversity (pi) was 0.009 ± 0.006, on the X, whereas the autosomes had a diversity of 0.012 ± 0.006. However, the parachute yp scaffolds were hard to detect because of the degeneracy of this Y chromosome. We managed to identify scaffolds in the assembly that are likely part of the yp sex chromosome, however, more data will be needed such as a genetic map, in order to generate linkage groups and be able to orient anchored scaffolds on all of these pseudochromosomes and generate full oriented chromosomes.
The genome size of R. ferrugineus based on flow cytometry of two batches of individuals (five males or five females) was estimated to be between 696.3 and 726.2 Mbp (Supplementary Fig. 4). Kmer-based estimates of genome size were 626 for males and 603 Mb for females (Supplementary Fig. 5). These estimate are much larger than the genome size of D. ponderosae (257.091 Mb) and of H. hampei (151.27 Mb), which may be explained by greater transposable element content (see below). The R. ferrugineus genome has a G + C content of ~32%, which is similar to the other insect species28. We also assembled the mitochondrial genome that was 16,074 bp, with 38 genes typical of insect mitochondrial genomes, including 13 protein coding genes, two ribosomal RNAs (rRNA), 23 transfer RNAs (tRNA) and an A + T rich region (Supplementary Fig. 6, Supplementary data file 3).
Annotation of the R. ferrugineus genome
We used the GeneMark prediction method to annotate the genome, resulting in 45,876 and 45,615 gene models in the two sequenced groups of individuals. Augustus de novo gene prediction gave rise to 27,119/27,116 gene models. EVM gene prediction used 149,007 insect transcripts and 114,218 insect protein models, resulting in 64,091/63,695 gene models. By combining all predictions, we annotated 25,567 good quality gene models. Non-coding tRNA gene prediction resulted in 1024 tRNA genes. Eighty percent of predicted proteins were found against the NCBI-Insecta database while ~78% of proteins shared homology in the UniProt database. 8,726 (34%) proteins were annotated against the KEGG pathway database (Supplementary data files 4–8).
The quality of the initial assembly was assessed by comparing the genome against the BUSCO arthropoda database. Approximately 84.6% (1404/1658) of complete BUSCO gene models (567 Complete and single-copy and 837 Complete and duplicated) were annotated in the assembled male weevil genome. Approximately 12% of BUSCO gene models were found in a fragmented form and ~15% were not annotated in either genome. CEGMA analysis identified ~88.31% (219/248) of ultra-conserved Core Eukaryotic Genes (CEGs) in the genome. The pseudochromosome assembly through synteny with T. castaneum showed improved N50 as well as BUSCO scores (Table 1).
The genome of R. ferrugineus has 25,567 genes. The average exon length was 230 bp with an average of 4.4 exons per gene (maximum: 56) while the average intron length was 681 bp. In contrast, the genome of D. ponderosae has 14,166 genes with an average exon length of 151 bp, an average of 8.3 exons per gene (maximum 85) and an average intron length of 1488 bp. H. hampei has 10,213 genes, with an average intron length of 1023 bp and an average exon length of 101 bp (6.3 exons per gene, maximum 62). T. castaneum has 14,467 genes, with an average exon length of 162 bp and an average intron length of 105 bp (Supplementary Fig. 7).
Transposable element content
Inter-species variation of genome size is known to be the result of amplification, deletion, and rearrangements of repetitive DNA sequences29. As a result, the size of the genome is a function of repeat dynamics but also of the average size of introns and many other factors30. Comparative analysis of the landscape of transposable elements (TE) (Fig. 2b, Supplementary Fig. 8, Supplementary data file 2) of R. ferrugineus showed that its genome has more repeats (45.24%; 354,353,149 bp) compared to D. ponderosae (16.41%; 41,501,291 bp), and H. hampei (16.79%; 25,391,186 bp). These percentages of TE content vary in Diptera from 6% in the Antarctic midge (Belgica antarctica) to 58% in Anopheles gambiae, while the Hymenoptera honeybee (Apis mellifera) and turnip sawfly (Athalia rosae) have less than 6%. The Orthoptera migratory locust (Locusta migratoria) has 58% of its genome occupied by TEs. The most abundant transposable element in R. ferrugineus is transposase-mediated cleavage (Tc) mariner (class II “cut and paste” DNA transposon) (Fig. 2b).
Ontology analysis of the R. ferrugineus proteome
Orthologous analysis using the proteins predicted in R. ferrugineus against those of H hampei, D. ponderosae, T. castaneum, and D. melanogaster showed 4733 orthologous groups that are shared among all these species (Fig. 2c). Molecular function ontology of predicted proteins in the shared clustered group shows enrichments of genes encoding hydrolase, metal-ion binding, oxidoreductase, peptidase, and transferase, which is also found in other insect-plant systems31,32 (Fig. 2c, Supplementary data file 9). Genes annotated as metal-ion binding and oxidoreductase activity include several alcohol dehydrogenases as well as many cytochromes and CYPs, while genes with transferase activity (e.g., methyl group, glycosyl group, acyl group, phosphorus-containing, or other groups) included glutathione transferases and UGTs. Hydrolase activity genes including peptidases, serine proteases, serine/threonine phosphatases were also annotated. Structural constituents of chitin-based cuticle are also enriched and include insect cuticular proteins. Biological processes of this shared ontology were mostly enriched for signal transduction processes and lipid metabolism (Supplementary data file 9, Fig. 2d).
The Venn diagram in Fig. 2b shows that there are 1033 cluster groups; representing 2931 R. ferrugineus proteins that do not share direct orthology with other proteins (orphans). However 1554 (564 cluster groups) of these proteins still have recognizable functional domains. GO enrichment analysis of these clusters identified stress-activated protein kinase signaling cascades (JNK signaling pathway involved in insect immunity), replication fork processing and DNA recombination (including transposase activity) (Supplementary Table 1).
Transcription factors (TFs) play a vital role in controlling gene regulation and many diverse physiological processes in insects. Our comparative orthologous results show that R. ferrugineus has more bHLH, homeobox, Zf-C2H2 main families similar to other beetles in the study (Supplementary Fig. 9). Signaling and metabolic orthologous counts of protein involved in different pathway are summarized in Supplementary data file 10, which highlights some increase of protein in R. ferrugineus involved in the degradation of aromatic compounds, immunity, and metabolism of xenobiotics by cytochrome P450 pathways.
Evolution of genes families in R. ferrugineus
By clustering all the proteins into orthologous groups, we managed to identify R. ferrugineus-specific clusters and signatures of expanded and contracted gene families. For the expanded and contracted gene families, the CAFÉ results (Fig. 3a) are represented by the ultrametric tree where the average expansion/contraction is depicted by the radius of node circles. This tree highlights more expansion in R. ferrugineus compared to the other species in the Curculionidae family. There are 2942 expanded and 1797 contracted gene families in R. ferrugineus (https://github.com/LKremer/CAFE_fig) [–count_all_expansions] (Supplementary data files 11 and 12). There are 38 significant (P < 0.001) expanded and 4 contracted gene families (Table 2; Supplementary data file 13; zipped families). Below are examples of expanded/contracted families in R. ferrugineus in comparison with other members of the Curculionidae family (H. hampei and D. ponderosae).
Fig. 3. Evolutionary inferences of gene family sizes.

a We used CAFE (Computational Analysis of gene Family Evolution) to infer the size change of gene families. This summary tree shows the average expansion/contraction (radius of node circles), the number of expanded contracted families (±), and the estimated gene gain/loss rates (blue: low rate; red: high rate). b Phylogeny of Pheromone: Odorant biding protein (PBP:OBP) using H. hampei (yellow), T. castaneum (green), D. ponderosae (blue), and R. ferrugineus (red) showing the expansion and the diversity of this gene family. Colors correspond to colored dots in (a). c Vendiagram depicting the shared and unique families of PBP:OBP among the five species in the study.
Table 2.
Summary of gene families (expanded/contracted) in R. ferrugineus.
| Gene families | Pfam | Function |
|---|---|---|
| Variant SH3 domain | PF07653, PF14604 | Signal transduction related to cytoskeletal organization |
| PDZ domain | PF00595, PF17820 | Signaling complex including neuronal synapses |
| Guanylate kinase | PF00625 | Cell proliferation |
| GPCR proteolysis site | PF01825, PF16489 | Mediate cell adhesion |
| Secretin family | PF00002 | Immune system |
| Galactose binding lectin domain | PF02140 | Immune system |
| RNase H-like domain | PF17919 | Immune system |
| Integrase core domain | PF00665 | Required for integration of viral DNA into host |
| Helix-loop-helix DNA-binding domain | PF00010 | Developmental processes |
| Hairy orange | PF07527 | Cell differentiation, embryonic patterning and other biological processes |
| SAM domain | PF07647, PF00536 | Repressors of target gene expression and RTK signaling |
| HMG box | PF00505 | Immune system |
| BAH domain | PF01426 | Chromatin biology |
| Glutathione S-transferase | PF00043, PF13417 | Development of insecticide resistance |
| N-terminal of Par3 | PF12053 | Cell polarity |
| Ion transport protein | PF00520 | Signaling in all sensory modalities |
| Cyclic nucleotide-binding domain | PF00027 | Cellular processes |
| PBP/GOBP family | PF01395 | Odorant detection |
| RPEL repeat | PF02755 | Actin binding |
| Sorbin homologous domain | PF02208 | Signal transduction |
| Polo kinase | PF12474 | Required for cytokinesis |
| RhoGEF domain | PF00621 | Adaptation of cells to environmental signals |
| Cadherin domain | PF00028 | Play a role in morphogenesis |
| Odorant receptor | PF02949 | Odorant perception |
| Cytochrome P450 | PF00067 | Detoxification of natural and external chemical |
| Phorbol esters domain | PF00130 | Pheromone response and communication |
| BTB/POZ domain | PF16017 | Leg and antenna segmentation, sex differentiation, color sexual dimorphism |
| Autophagy-related protein | PF10377 | Metamorphosis |
| GNS1/SUR4 family | PF01151 | Glucose-signaling pathway |
| OAR domain | PF03826 | Correct morphogenesis of the limbs and cranium |
| Homeobox domain | PF00046 | Control of development and cell fate |
| Zinc carboxypeptidase | PF00246 | Protein digestion |
| Carboxypeptidase activation peptide | PF02244 | Protein digestion |
Bold color: expanded families.
Regular color: contracted families.
In insects, perception of the environmental cues is mainly guided by chemical signals. The red palm weevil is an invasive species and similar to other insects, it relies mostly on its olfactory system for food foraging and for mating. It uses resistance mechanisms for detoxification of plant secondary metabolites and xenobiotic33. We highlight some gene families that are expanded in this context.
Odorant receptor (ORs)
Olfactory gene families are involved in pheromone and odorant detection. They allow R. ferrugineus to locate infected palm trees that emit volatiles in the air, as well as the male aggregate pheromone released to coordinate explosive attacks33. The R. ferrugineus genome contains 46 OBP:PBP odorant/pheromone binding proteins (PF01395) and 80 ORs (PF02949). By comparison, D. ponderosae has 40 OBP:PBP and 57 ORs and H. hampei has 36 OBP:PBP and 16 ORs. In the same context, the generalist honeybees (Apis mellifera and A. cerana) have 21 OBP:PBP and 175 ORs for broad olfactory perception of pheromones blends and floral odorants. Similarly, T. castaneum, which is a pest for a broad range of dried stored products, has 56 OBP:PBP and 265 ORs34.
A phylogenetic tree generated from the alignment of the different PBP:PBP shows their divergence, diversity, and expansion (Fig. 3b). There are two subclasses that are shared among all the species (Fig. 3c). There are ten unique OBP:PBP subclasses in R. ferrugineus (Fig. 3c; see also Supplementary data file 14). Two of the unique subclasses PBP:OBP in R. ferrugineus are important for host plant discrimination and to sense nutrient sources (with subclass 11)35, while subclass 4 appears to be involved in male-specific pheromone production36. The similar numbers of OBPs in R. ferrugineus and in D. ponderosae, which are involved in the transport of odorants to ORs, is in contrast with the significant higher number of ORs in R. ferrugineus.
Basic Helix-loop-helix (bHLH) DNA-binding proteins
bHLH transcription factors play important roles in different developmental processes37,38. We identified an expansion of the Myc-type bHLH genes with a Pfam domain (PF00010) that are involved in cell proliferation/differentiation, sterol metabolism, adipocyte formation, and expression of glucose-responsive genes39,40. In this class of bHLHs, R. ferrugineus has seven members that contain a Sterol-sensing domain of the SREBP family, compared to D. ponderosae and H. hampei that only have four. In Drosophila, and mice, glucose can activate genes via the transcription factor ChREBP, an ortholog of the seven SREBP bHLHs found in the weevil (PF00010), to induce the utilization of glucose and de novo lipogenesis41.
Glutathione S-transferase
GST is a large gene family that is involved in the detoxification of plant secondary metabolites. In R. ferrugineus, we identified 47 cytosolic GST genes, which is more than the 40 GST in H. hampei and 43 in D. ponderosae, the 38 GST found in D. melanogaster or 36 GST in T. castaneum. The higher number of GST in R. ferrugineus might reflect their need to detoxify diverse toxins, because their host range includes 26 species of palms. There are 6 subclasses of GSTs in R. ferrugineus with 4 members of the Delta family, 25 Epsilon, 8 Omega, 3 Theta, 4 Sigma and 1 Zeta. There is a correlation between the amplification of GST genes and resistance to insecticides42,43.
We analyzed duplications/deletions, inversions and tandem duplications in R. ferrugineus (Supplementary data files 15 and 16). Our results show more duplication, in particular more tandems duplications (2243) relative to the other beetles in our study (Fig. 4a) (Fisher’s exact test P < 0.001). The intersected results of the structural variants (SV) with the R. ferrugineus genome annotation revealed several genes families important in detoxification of secondary metabolites and insecticides that were tandemly duplicated such as GST and P450 (Fig. 4b, Supplementary Fig. 10). Thirty-eight of the 45 R. ferrugineus GST mapped to 4 of the 10 pseudochromosomes (Fig. 4b). Tandem duplication is a general feature for GSTs, including in the beetles. In R. ferrugineus, the GSTs are divided into clusters, according to their subclasses. Four Delta are in a cluster on pseudochromosome 2 and 25 Epsilon are in two clusters on pseudochromosome 1 and 4 (Fig. 4b). In D. melanogaster, the ten Epsilon subclasses as well as the Delta genes are all in tandem44. In T. castaneum, the two members of the Delta subclass are in tandem and genes in the Epsilon class are in tandem in two clusters on chromosome 2 and 345.
Fig. 4. Structural variation.

a Distribution of the different classes of structural variants (SVs) (duplication, deletion, inversion, and tandem duplication) in the five species studied depicted in the horizontal boxplot with error bars represents standard deviation (s.d). The x axis represents the length of the structural variants with less than 100 kb. The counts of the SVs are represented in a column on the right side of the plot. b Circos plot depicting nucleotide diversity distribution across the different pseudochromosomes from the outer track of the plot. From the inside duplications, as the second track represented as the normalized read depth, in red bubble. The size of the bubble represents the size of the duplication in kilobases (kb). The inner part of the Circos is the annotation of the tandem duplication of glutathione S-transferase (GST) highlighting the cluster of Epsilon and delta subclasses. c Alignment of the portion of Dieldrin gene Rdl surrounding the equivalent residue 301 in insect species, showing copy number variation and the relative amino acid variation at the 301 site. Bombyx mori contains three Rdl orthologs, with a different residue at 301 (1 = Ala, 2 = Ser, 3 = Gln). Both aphid species contain two Rdl orthologs: Acyrthosiphon pisum and Myzus persicae (1 = Ala, 2 = Ser). Rhynchophorus ferrugineus (1 = Ala, 2 = Ser, 3 = Thr). D. mel, D. melanogaster; A. mel, Apis mellifera; A. aeg Aedes aegypti, T. cas Tribolium castaneum, B.mor Bombyx mori, A. pis Acyrthosiphon pisum, M. per Myzus persicae, Rhynchophorus ferrugineus R.fer_(1,2,3). d Depth of the coverage plot for two individuals showing the duplication (in red) of the Rdl gene.
Cytochrome P450
The Cytochrome gene family encompasses oxidases and cytochrome P450 (CYP450) monooxygenases. It has a wide diversity of functions for steroid hormone synthesis, which is important for the development and reproduction of insects, to the metabolism of chemicals that play a role in host plant adaptation and survival in toxic environments. A number of related P450 proteins control these processes with the number and rate of expansion of Cytochromes dependent on the species physiology and the environmental in which a species lives. In R. ferrugineus, the cytochrome P450 family is composed of 120 genes compared to 88 in D. melanogaster (http://flybase.org/). CYP6 and CYP12 (mitochondrial) are the most expanded gene families and R. ferrugineus has 104 CYP6 genes (and 16 CYP12), almost 5 times the 23 genes found in D. melanogaster, with a more specific expansion of subfamilies CYP6A (14 genes), CYP6G (9 genes) and CYP6D (5 genes), which have been associated with insecticide resistance46,47. The number of these genes is even higher in H. hampei (116 CYP6 and 12 CYP12) and in D. ponderosae (117 CYP6 and 8 CYP12) showing that a dramatic expansion occurred in the Curculionidae in which it could mediate resistance to insecticide33. All pseudochromosomes, except #10, had genes for P450s. Only 8 P450 genes were found individually located, while the other 112 are tandem duplicates (Supplementary Fig. 10).
Other gene families, including several immunity gene families (e.g., secretin family) (Supplementary data file 11), were also expanded, suggesting that the R. ferrugineus immune system responds differently to the challenge of the life inside a tree.
Glycosyl hydrolase and carboxypeptidase genes
The genome-wide comparison of digestion-related genes such as proteases suggests that they have undergone a major expansion in Diptera, Lepidoptera, and Coleoptera, but not in Hymenoptera or Hemiptera48. In contrast to T. castaneum that has 62 carboxypeptidase genes, R. ferrugineus has only 13. There are 20 in D. ponderosae and 30 in H. hampei. However, the genome of R. ferrugineus encodes 70 glycosyl hydrolase genes, which might be important for the hydrolysis of the rich sugar content in the phloem sap that is rich in sugar but lacks starch. T. castaneum that feeds on starch-rich grains has a high number of α-amylase genes (12) compared to D. ponderosae that has 8 genes and H. hampei that has 6 genes. In contrast, we found 17 α-amylase genes and one chitin synthase CHS2 gene in R. ferrugineus.
Gene families under positive selection
We conducted phylogenetic tests for selection in R. ferrugineus by aligning CDS sequences from each gene family to their homologs in D. ponderosae and H. hampei. We found evidence of positive selection in 115 gene families (Supplementary data file 17). These functions of these families range from calcium channels (e.g., TRPM) to xenobiotic metabolism (e.g., CYP450, UDP-glucuronosyltransferase), to odorant-binding proteins. One interesting gene family that is under positive selection is family 16 of glycoside hydrolase (GH16)49 that supports selection for this family (P = 0.003) (Table 3). Another gene family showing signature of positive selection is the TRPM transient receptor potential ion channels (Supplementary Table 2), and the GABA-gated chloride channel subunit encoded by the Rdl gene (Fig. 4c, Supplementary data file 17) that have been shown to be responsible for insecticide resistance in many insect species50,51. In R. ferrugineus, we identified a 10 kb duplicated region containing a WT copy of Rdl and a second copy with the A30S point mutations as well as another A301T (Fig. 4d). The frequency of the mutation using 50 individual transcriptomes is high (60%) (Supplementary Table 3).
Table 3.
Parameter estimates and likelihood scores for glycoside hydrolase (GH16) gene under models of variable ω ratios.
| Nested model pairs | dN/dSb | Parameter estimatesc | PSS (*P > 95%; **P > 99%)d | Likelihood |
|---|---|---|---|---|
| M0:one-ratio (1)a | 0.0871 | ω = 0.0871 | −12,014.227 | |
| M3: discrete (5) | 0.2194 |
p0 = 0.335, p1 = 0.353, (p2 = 0.298) ω0 = 0.011, ω1 = 0.082, ω2 = 0.251 |
3 I 0.999** 6 W 0.979* 9 I 0.999** |
−11,619.479 |
| M1: neutral (1) | 0.3076 |
p0 = 0.764, p1 = 0.235 ω0 = 0.094, ω1 = 1 |
−11,844.804 | |
| M2: selection (3) | 0.3076 |
p0 = 0.764, p1 = 0.085, (p2 = 0.149) ω0 = 0.094, (ω1 = 1), ω2 = 1 |
3 I 0.956* 6 W 0.730 9 I 0.945 |
−11,844.804 |
| M7: beta (2) | 0.1221 | p = 0.605, q = 3.970 | −11,628.839 | |
| M8: beta + ω > 1(4) | 0.2225 |
p0 = 0.989, (p1 = 0.010) p = 0.628, q = 4.589, ω = 10.628 |
3 I 0.999** 6 W 0.972* 9 I 0.998** |
−11,617.599 |
| M8a: beta + ω = 1(4) | 0.1281 |
p0 = 0.652, (p1 = 0.028) p = 0.652, q = 5.181, ω = 1 |
−11,621.768 |
| Gene | Modele | P value | ||
|---|---|---|---|---|
| GH16 | M3 vs M0 | 2.86508957212e−167 | ||
| GH16 | M2 vs M1 | 1 | ||
| GH16 | M8 vs M7 | 1.31253379663e−05 | ||
| GH16 | M8 vs M8a | 0.00387888988444 | ||
aThe number of free parameters in the ω distribution.
bAverage ratio dN/dS of all sites for the GH16 gene alignment.
cThe number in parentheses are not free parameters.
dNumber of positively selected sites.
eLikelihood ratio test statistics for models of variable selective pressure among codons.
Ancestral and present effective population size (Ne)
We applied PSMC analysis (https://github.com/lh3/psmc) to evaluate population dynamics of R. ferrugineus from 8 million years before present (Ma) to 10,000 years before present (ka). Assuming a generation time of ~4 months, we estimated a per nucleotide per generation mutation rate of 0.89e−10 (Supplementary data file 18 (zipped files for each pseudochromosome)). We estimated the peak of Ne at 5.5 10+06, which occurred approximately around 1.2 million years before present (Ma). This expansion was followed by a population decline from the middle to the end of Pleistocene (Fig. 5).
Fig. 5. Demographic history of R. ferrugineus.
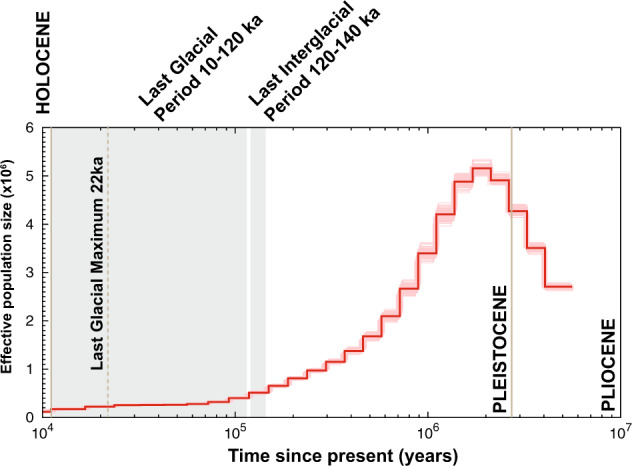
PSMC analysis was applied on the genomic sequences of R. ferrugineus converted to demographic units assuming a generation time of 4 month (g = 0.3 years) and a substitution rate of µ = 0.89 × 10−10. The x axis represents time before present in years in log scale and the y axis is the effective population size. The bold red curve shows the estimate of the original data and the shaded red curves are estimate for 100 bootstrapped sequences.
In order to test for the present effective population size, we used the gene for Rdl, which is known to have two single nucleotide mutations in the same codon that confer resistance to insecticides in other insects50 (A→S) (Fig. 4c). The frequency of the mutation (A→S) in the population was determined (Supplementary Table 3) to be ~0.6. Assuming both bp mutations are required for the resistance, and a base pair mutation rate similar to the red flour beetle of ≈2.70e−10, this gives a mutation rate of ≈5.39e−10. If we apply the haploid algorithm to the data52, we can estimate diversity θ = 0.005 (giving ns = 100 (2 chromosomes and 50 individuals), nm = 68) and a recent effective population size of 2.3 10+06.
Horizontal gene transfer (HGT)
Genes involved in plant cell wall degradation found in some insects were often horizontally transferred from bacteria53. CAZy (Carbohydrate active enzymes (http://www.cazy.org)) are the enzymes that collectively assemble and degrade oligo- and polysaccharides. We identified 224 GHs assigned to 19 families in R. ferrugineus (Supplementary Table 4).
Models of horizontal transfer suggest that successful acquisition of a horizontally transferred gene requires that the gene be active and maintained, and likely be under positive selection54. GH16 in R. ferrugineus is under positive selection and responsible for the hydrolysis of β-1,3 glycans, which are found in the phloem of plants as callose55 and are a main component of the date palm sap56.
GH16 could thus have originated from HGT from bacteria or fungi, although endogenous eukaryotic β-1,3-glucanases such as Gram-negative binding proteins (GNBPs/βGRP) already exist in insects57. We investigated whether GH16 was also horizontally transferred into the R. ferrugineus genome. Phylogenetic analysis shows that GH16 from R. ferrugineus clusters monophyletically with D. ponderosae but is distantly related to T. castaneum GH16 (Fig. 6a, Supplementary Fig. 11). No match was found in H. hampei. The clustering illustrates also the duplication of the GH16 in R. ferrugineus. The phylogeny also shows clear separation of the eukaryotic GNBPs57 (Fig. 6a) found in many order. In addition, this cluster was closely related to a gene both in a γ-proteobacterium as well as in the fungus Pisolitus microcarpus, which did not allow us to identify its origin from a fungus or a bacterium (Fig. 6a). Using RNAseq, we validated the expression of these GH16 genes (Fig. 6b). We also used a fragment from one of the GH16 genes and used PCR to validate its presence in the genome in different parts of the weevil to rule out contamination coming from the gut microbiota of R. ferrugineus (Fig. 6c, Supplementary Fig. 12).
Fig. 6. Horizontal gene transfers in R. ferrugineus (HGT).

a Phylogeny of the some of the different glycosyl hydrolase (GH16) (e.g., M019946-T1) that are horizontally transferred and clustered with other beetles, which are highlighted in green. The Yellow cluster highlight the Eukaryotic Gram-negative-biding protein, similar to GH16, but already exists in insects. The gray cluster highlights the close microorganisms (bacteria and fungi) to the horizontally transferred hydrolase. While the orange cluster shows the distantly cluster of microorganisms. b The schematics about HGT depicted by acquisition, intron gain, and duplication and diversification. We show a GH16 with seven introns gains as well as expression on the y axis shown as coverage of RNAseq reads, where the surrounding shows DNA and non-LTR transposable elements. c Gel electrophoresis of a 258 bp fragment of one GH16 to validate the presence in the genome and rule out gut microbial contamination.
GH16 could have been acquired from a bacterium or from a fungus since the closest fungal relative showed no introns. If the gene was acquired once in the last common ancestor, GH16 should share the same exon/intron structure in D. ponderosae, in R. ferrugineus, and in T. castaneum. However, GH16 has no introns in D. ponderosae and has three in T. castaneum. In R. ferrugineus, the number of introns varies from 2 to 7, suggesting independent and subsequent acquisition of introns after HGT. We evaluated the expression of the different GH16 in R. ferrugineus, which showed higher gene expression for the genes with more introns (Supplementary Table 5). This suggests that the introns were acquired after HGT followed by gene duplication.
Discussion
The genome of R. ferrugineus provides insights into the behavior of the species. It is the largest beetle genome of the Curculionidae family sequenced to date with an estimated genome of around 720 Mb. It shows a high synteny with T. castaneum although the two species diverged 236 Mya ago58 and has low diversity on the X chromosome, likely due to the suppression of recombination as shown in other insects59.
The increased number of genes in R. ferrugineus genome compared to other beetles results from the expansion of gene families. In contrast, the average intron length in R. ferrugineus is lower when compared to other Curculionoidea, which is not consistent with the established correlation between genome size and intron length60 (Supplementary Fig. 6). This suggests relaxed selection and an increase in deleterious TEs that represent 45% of the weevil genome, leading to structural variations such as duplications, deletions, inversions, and translocations61–63.
Orthologous analysis shows enrichment of genes with transferase and hydrolase activity that are important for detoxification, xenobiotic metabolism, and digestion, while feeding on different host plants17,64,65. The genome shows expansion of gene families important for chemoreception, food intake and for dealing with a hostile environment (ORs and bHLHs transcription factor (TF)). This suggests that the large number of host plants for this generalist species leads to diversification of ORs. The function of the expanded family of bHLHs transcription factor is still unknown, but members of this family regulate glucose metabolism and the production of pheromones66,67 involved in the massive invasion of trees and promote mating. TFs are essential in orchestrating many physiological processes and their identification will help the growing entomologist, to invest more in this understudied filed by using emerging research methods to study their regulatory functions. Furthermore, it will prompt to investigate more their contributions in pest control and in general human health.
The vast array of GST and CYP450 genes in insects represents the largest repertoire of detoxification genes known. In particular, the Epsilon and Delta subclasses of GSTs are involved in insect response to environmental conditions14, as well as in xenobiotic and insecticide resistance14. For instance, A. gambiae and A. aegypti are able to metabolize DDT by GST epsilon2–268,69. We propose that the history of insecticide application for the control of R. ferrugineus70 has led to resistance mediated by amplification of GST33. It is known that the P450 families evolve through duplication and diversification71,72. Our structural variation results suggest that P450 in R. ferrugineus also arose through localized tandem duplication73,74. R. ferrugineus has undergone different insecticides treatments70, and this explains the rapid evolution and duplication of GST and P450 monooxygenases as well as the TRPM family that could promote insecticide resistance (Supplementary data file 18). The recent duplication of the gene that encodes the Rdl channel that is the target of cyclodiene and phenylpyrazole insecticides, likely results from their use against the native and invasive R. ferrugineus75. Insecticide resistance can be accomplished via gene duplication by increasing the Rdl gene product or to adaptive mutations in Rdl that prevent the action of the insecticides without affecting its essential role50. The resistance to cyclodiene dieldrin in Drosophila is due to a single amino acid replacement, A30S50, a mutation that was subsequently identified in Rdl orthologs in different resistant insect species (Fig. 4c). This classical example of parallel evolution of the Rdl gene shows that it is a hotspot of evolution (Fig. 4c). Our data show that positive selection is currently acting on a mutation in Rdl in R. ferrugineus that is found at high frequency through the population, but it is not clear whether this mutation(s) promote resistance. This mutation might explain the inefficacy of cyclodiene dieldrin insecticides to control R. ferrugineus.
In R. ferrugineus, the contraction of carboxypeptidase genes may reflect the feeding strategy of its larvae that spend most of their lives inside the trunk of the date palm, chewing and sucking the sugar-rich sap of the soft tissue. This lifestyle minimizes the requirements for digestion of proteins. The 17 α-amylase genes we found likely help the larva to ingest the starch-rich date palm stem, which is the point of entry for adults during invasion. R. ferrugineus has only one chitin synthase CHS2 gene that produces the peritrophic matrix, a chitin layer that lines the midgut and protects the epithelium from damage caused by rough food particles, digestive enzymes as well as ingestion of pathogens. In contrast to sucking species like R. ferrugineus48, these genes are expanded in species feeding on diverse grains (T. castaneum has 3 genes) or woody substrates (D. ponderosae has 5 and H. hampei has 2).
The PSMC analysis suggests that R. ferrugineus experienced an increase in Ne during the early Pleistocene, which could reflect an expansion and spread of the range of the ancestral population. The duration of the last glacial period and the transition to Holocene was, for most of the species, associated with dramatic population changes associated with reduced Ne. The mid to the end of Pleistocene experienced glacial-interglacial cycling and around 900 ka, there is an evidence that the two southern oceans experienced sluggish thermohaline overturn76, which might have led to the contraction of the range of the Pacific and India coconut trees (Cocos nucifera) in isolated refugia. This scenario is consistent with the distribution of the two native species R. ferrugineus and R. vulneratus4 as the insect-tree relationship must have resulted in the contraction of the weevil population.
Population genetic analysis could enable the establishment of the possible route(s) of invasion from South East Asia to the Mediterranean and to the Arabian Gulf. For example, the recent (1985)77 R. ferrugineus introduction to the United Arab Emirates likely originated in a date palm offshoot from an infected country78, which created a bottleneck effect for a period of time, followed by an increase in the effective population size that likely reduced drift and increased the effect of selection at fixing beneficial mutations79. The difference in the calculated effective population size and the actual number census population size (actual numbers of animals present) (~108) in R. ferrugineus is likely caused by overlapping generations80,81.
GH16 genes are under positive selection and provide a significant advantage to the weevil to efficiently process its food: Since the larvae are embedded inside the truck of the date palm tree, these genes are likely very important for larval digestion and survival. Our phylogenetic clustering with D. ponderosae and T. castaneum suggests that GH16 in R. ferrugineus originated from HGT that occurred in one of the common ancestors of beetles, although we cannot rule out its independent acquisition. After HGT, natural selection played a role in maintaining and fixing GH16 in the population of R. ferrugineus and later expanding it by gene duplication. Another example of GH specialization is in the western corn rootworm (Coleoptera Diabrotica virgifera virgifera) that has a close relationship to maize82. GH16 was shown to be horizontally transferred from bacteria in the Antarctic springtail, Cryptopygus antarcticus83. Other phytophagous beetles and some leaf beetles also acquired plant cell degrading enzymes via HGT, such as GH28, GH4519,84.
This high-quality whole genome assembly provides a foundation that will make it feasible to genetically modify the insect in order to potentially control this pest by editing genes important for the reproduction of R. ferrugineus through the eventual release in the population of mutants successfully tested in field trials.
Methods
R. ferrugineussamples
Male and female adult (Fig. 1a) were collected from an infested field in Al Ain, UAE region (Fig. 1c), flash frozen and maintained on dry ice for sample extraction, library preparation, and sequencing. High molecular weight DNA was extracted. Oxford Nanopore and 10X Genomics libraries were generated as well as two Hiseq 2500 (2× 150 bp) libraries following standard protocols before sequencing. These data generated 190 Gb of raw sequencing reads with ~80× coverage. RNA extraction was done on 50 individuals. Library construction was done using a TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA, USA) and sequenced in a 150 bp PE run on an Illumina HiSeq 2500 platform. The transcriptome data were used for genome annotation and expression analysis. In addition, available short-reads archive (SRA) RNAseq libraries were used (SRX096967, SRX096966, SRX096965, SRX096462, SRX096968, SRX096969, SRX096970, SRX096971, and SRX096972) for different developmental stages (egg, larvae, and pupae) in the annotation and expression analysis.
Genome size estimation
We used two methods to estimate the size of the R. ferrugineus genome. For flow cytometry, we dissected whole brains and placed them in 1 mL Galbraith buffer85 along with the head of one Drosophila virilis female. The mixture was grounded to release the nuclei and filtered through 40-µL-nylon mesh being mixed with a vortex, and stained staining with 25 μL propidium iodide for 3.0 h at 4 °C. The relative fluorescence of 2 C nuclei from the sample and standard were measured with a Beckman/Counter CytoFLEX flow cytometer. The amount of DNA in each sample was determined as the ratio of the average relative fluorescence of the diploid sample nuclei divided by the relative fluorescence of the diploid nuclei of the standard, divided by the amount of DNA in the D. virilis standard. Linearity of the CytoFLEX was verified by estimating comparable genome size using the 4 C peak of the sample and standard. Six biological replicates and three technical replicates were tested.
We also used a Kmer-based approach to estimate genome size and heterozygosity using Illumina Hiseq 2500 (2× 150 bp) paired-end sequencing data86.
Genome assembly and annotation
Genome assembly was done using a combination of 10X genomics and Illumina Hiseq 2500. The results were combined with Oxford nanopore. The final assembly was done using syntheny to the T. castaneum (see Supplementary methods in Supplementary information). Genome annotation was carried out using Funannotate (https://github.com/nextgenusfs/funannotate), a gene prediction pipeline (see Supplementary methods in Supplementary information). We compared R. ferrugineus genome features to other beetles’ annotation (D. ponderosae, H. hampei and T. castaneum) using gt stat command from genometools87 and extracted information from their annotation file format GFF3(s) associated genomic features (genes, exons, CDS, intron) length and counts. We annotated TFs using (http://bioinfo.life.hust.edu.cn/AnimalTFDB/) and signaling and metabolic proteins using KEGG pathways reconstruction (https://www.genome.jp/kegg/tool/map_pathway.html).
Genome characterization of repeats
We used RepeatModeler a de novo prediction analysis (http://www.repeatmasker.org) to construct a transposable element library specific for the species. Repeat-Masker (4.0.7)88 was used for TE identification and classification using the generated library by RepeatModeler. Repeats with percentage genome content for D. ponderosae, H. hampei, and male/ female R. ferrugineus was generated. Repeat Landscape of the different classes (LTR and non-LTR, DNA-TE) is plotted for male and female of R. ferrugineus using R89.
Phylogenetic analysis and gene family evolution
Predicted proteins encoded by the four insects genomes with GeneBank assembly accessions (GCA_000001215.4; GCA_000002335.3; GCA_000355655.1; GCA_001012855.1) and the one from R. ferrugineus were filtered to keep the longest isoforms using a script provided in CAFÉ. Orthologs in the R. ferrugineus genome in comparisons to three beetles’ genomes as well as D. melanogaster were generating using Orthofinder with default parameters. A phylogenetic tree using 439 single-copy ortholog groups was generated in orthofinder after supplying –m MSA (command line to generate alignment file). The number of conserved sites was calculated for the concatenated alignments.
Using the phylogenetic tree generated above, we have generated a calibrated species tree using the software r8s (http://loco.biosci.arizona.edu/r8s/) and the analysis was done using the penalized likelihood method and the TN algorithm. D. melanogaster and the mountain pine beetle (D. ponderosae) were chosen as calibration points using (http://www.timetree.org). We used CAFE90 program version 3.1 for gene family expansion/contraction across the phylogeny as well as estimated the gene gain/loss rates varying lambda (maximum likelihood value of the birth and death parameter) value across the branches where each branch has assigned unique lambda and the best value was obtained using iterative calculation. Significant size variance of expansion and contractions of gene families was identified using 1000 random samples and a p value of 0.01 and deviated branches were identified using Viterbi algorithm implemented in CAFE with a p value of 0.05. Phylogenetic tree was build using an online tool (http://www.phylogeny.fr/simple_phylogeny.cgi) using protein alignment of an expanded family using matches from the different species analyzed.
Structural variation
Normalized Read-depth variation analysis was performed using CNVnator91 (version 0.2.7). Aligned bams were used as input for CNVnator to extract read alignment information. A bin size of 1 kb was used in the intermediate processing of the bams as well as when calling variants. A table of duplication and deletion is generated. We discarded any duplication/deletion more than >100 kb as well, as hits that span gaps and beginning of a scaffold. Tandem duplication was screened using the software SoftV92. (See Supplementary methods in Supplementary information). We intersect duplication and tandem with the annotation for male and female and looked at genes that overlap the structural variant. Duplication/Tandem duplication was plotted using Circos93.
Horizontally transferred gene families under positive selection
We investigated the extent of HGT using similar approach as in Nowel et al.94 (see Supplementary methods in Supplementary information).
We looked if there were any selective pressures on different gene families by aligning sequences from R. ferrugineus, the mountain pine beetle and coffee borer. We ran the Fustr95 with command Codeml from PAML96. A list of gene families under positive selection is reported. We highlighted two examples; one is the glycoside hydrolase (GH16) gene and the other is the transient receptor potential ion channels (TrpmM) and estimated parameter and likelihood scores for under models of variable ω (dN/dS) ratios. dS represents synonymous rate while dN non-synonymous rate. In the absence of evolutionary pressure this ratio = 1, under purifying selection it is <1 and under positive selection it is >1.
Phylogenetic analysis of GH16 was done using a combination of BLAST97 and available CAZy databases (http://www.cazy.org/GH16_unclassified.html) in order to gain insight into its evolutionary history. We looked at the potential donor HGT microorganism, using the build up phylogenetic tree of the target with the most probable species that share maximum homology with the GH16 domain using blast to microbiome.
Ancestral and recent effective population size (Ne)
The mutation rate for R. ferrugineus was estimated comparatively using the red flour beetle genome assembly (T. castaneum), downloaded from NCBI public database (https://www.ncbi.nlm.nih.gov/genome/?term=txid7070[orgn]), Tcas5.234 (Tribolium Genome Sequencing Consortium). The genome was split into individual chromosomes and the R. ferrugineus genome was aligned to each one of the red flour beetle chromosomes using LastZ v1.04.0098, applied with the following parameters: –ydrop = 9400, –hspthresh = 4500, –gappedthresh = 3000 and –notransition. The number of nmatch (matches) and nmismatch (mismatches) was used in the output format options, respectively. Any matches or mismatches were not considered if they are classified as N/n or if there is an alignment gap. The mutation rate (per nucleotide per year, u) was calculated using the equation: (number of mismatches/total length)/2t, where t is the divergence time between the red flour beetle and R. ferrugineus, which is estimated around 236 Mya57.
We ran PSMC (https://github.com/lh3/psmc) analysis using consensus genome sequence (fastq) that was filtered for coverage and sequencing errors. For each R. ferrugineus sample, we used samtools to generate the consensus autosomal fastq using the “mpileup” command. SNP calling on single individual was done using samtools pipeline, which is independent of population frequencies and not assume Hardy–Weinberg equilibrium. We masked sites when read depth of a site is less than third the average depth genome. These criteria represent the default setting in PSMC when analyzing highly covered genome, which is our case.
We adjusted some parameters (−t, −p and −r) and set the upper limit of TMRCA to 5 with the −t option, −r option to 1 (θ/ρ). The analysis of effective population was inferred using 24 free atomic time intervals (4 + 24 × 2 + 4 + 6 + 10) and this was set with −p option. We performed 100 bootstrap replicates to check for variance in effective population size (Ne). This was done on a 5 Mb sequences obtained from the consensus genome using the splifa command in PSMC. We applied the mutation rate calculated above. Diversity estimate π (Pi) and θ (Watterson) was generated from the whole genome of the R. ferrugineus male and female using angsd99 version 0.917 and plotted using Circos93.
For recent effective population size (Ne), we followed the method in Khatri and Burt52. We calculated a recent Ne, where we used only the knowledge of the number of independent recurrent origins and the frequency of the beneficial allele in the population, without a prior knowledge of the strength of selection and age of mutation. In this analysis, we used the Rdl gene, which is known to have two point mutations in the same codon that confer resistance to insecticides, (A→S mutations) and the frequency is used from the data.
Statistics and reproducibility
All statistics was done using available packages and reproducibility can be accomplished using the same command lines mentioned in the methods, where we used for most of the analysis publicly available softwares and online tool for plotting with adjusted parameters when appropriate or kept with default parameters to suit the different types of analysis.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We would like to thank all the personnel of the Khalifa center for Genetic Engineering and Biotechnology (KCGEB) who made this phase one of the project come true. A special thanks to Mr. Sajid at KCGEB for taking pictures of R. ferrugineus. This project was funded by the Khalifa Center for Genetic Engineering and Biotechnology (KCGEB) and by the New York University Abu Dhabi Research Institute (G-1205C and G-1205i).
Author contributions
K.A., C.D. and K.M.H. designed the experiments. K.M.H., D.N. and N.S. performed the analysis. M.D., B.K. and A.M. collected the R. ferrugineus samples and extracted high molecular weight DNA. J.J.S. did the flow cytometry analysis. K.M.H., K.A. and C.D. wrote the manuscript.
Data availability
The Illumina reads Hiseq 2500 (2× 150 bp as well as the 10x Genomics data) were deposited at NCBI short archive under SRA accession PRJNA524026 (Biosample accession SAMN10995380). The RNA sequencing data as well as the Oxford Nanopore were deposited at NCBI short archive under BioProject (PRJNA600770, SRA ID SUB6799681). The final genome, is deposited at NCBI GeneBank (https://www.ncbi.nlm.nih.gov/genbank/wgs_update/) under accession (JABAOJ000000000), and supplementary data files, mitochondria genome as well as HGT validation are deposited at Dryad database (https://datadryad.org/stash/share/yyBU31Aj2_n2H9QYCPkG-hoZcYf63-2OToegnkmBcV8).
Competing interests
The authors declare they have no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Claude Desplan, Email: cd38@nyu.edu.
Khaled M. A. Amiri, Email: k.amiri@uaeu.ac.ae
Supplementary information
Supplementary information is available for this paper at 10.1038/s42003-020-1060-8.
References
- 1.Hutchinson GE. Homage to Santa Rosalia or why are there so many kinds of animals? Am. Naturalist. 1959;93:145–159. [Google Scholar]
- 2.Mckenna DD, et al. The beetle tree of life reveals that Coleoptera survived end-P ermian mass extinction to diversify during the Cretaceous terrestrial revolution. Syst. Entomol. 2015;40:835–880. [Google Scholar]
- 3.McKenna DD, Sequeira AS, Marvaldi AE, Farrell BD. Temporal lags and overlap in the diversification of weevils and flowering plants. Proc. Natl Acad. Sci. USA. 2009;106:7083–7088. doi: 10.1073/pnas.0810618106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rugman-Jones PF, Hoddle CD, Hoddle MS, Stouthamer R. The lesser of two weevils: molecular-genetics of pest palm weevil populations confirm Rhynchophorus vulneratus (Panzer 1798) as a valid species distinct from R. ferrugineus (Olivier 1790), and reveal the global extent of both. PLoS ONE. 2013;8:e78379. doi: 10.1371/journal.pone.0078379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.EPPO. List of biological control agents widely used in the EPPO region. EPPO Bull. 2002;32:447–461. [Google Scholar]
- 6.El-Sabea AM, Faleiro J, Abo-El-Saad MM. The threat of red palm weevil Rhynchophorus ferrugineus to date plantations of the Gulf region in the Middle-East: an economic perspective. Outlooks Pest Manag. 2009;20:131–134. [Google Scholar]
- 7.Giblin-Davis, R. M. Borers of palms. In Insects on Palms (eds. Howard, F. W., Moore, D, Giblin-Davis, R. M., & Abad, R. G.) 267–304. (CABI Publishing, Wallingford, GB, 2001).
- 8.Zhu F, Moural TW, Nelson DR, Palli SR. A specialist herbivore pest adaptation to xenobiotics through up-regulation of multiple Cytochrome P450s. Sci. Rep. 2016;6:20421. doi: 10.1038/srep20421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guillet G, Lavigne M-È, Philogène BJ, Arnason JT. Behavioral adaptations of two phytophagous insects feeding on two species of phototoxic Asteraceae. J. Insect Behav. 1995;8:533–546. [Google Scholar]
- 10.Panini M, Manicardi GC, Moores G, Mazzoni E. An overview of the main pathways of metabolic resistance in insects. Invertebr. Survival J. 2016;13:326–335. [Google Scholar]
- 11.AlJabr AM, Hussain A, Rizwan-ul-haq M. Toxin-Pathogen synergy reshaping detoxification and antioxidant defense mechanism of Oligonychus afrasiaticus (McGregor) Molecules. 2018;23:1978. doi: 10.3390/molecules23081978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplanoglu E, Chapman P, Scott IM, Donly C. Overexpression of a cytochrome P450 and a UDP-glycosyltransferase is associated with imidacloprid resistance in the Colorado potato beetle, Leptinotarsa decemlineata. Sci. Rep. 2017;7:1–10. doi: 10.1038/s41598-017-01961-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Q, et al. Heterologous expression of a Glyoxalase I gene from sugarcane confers tolerance to several environmental stresses in bacteria. PeerJ. 2018;6:e5873. doi: 10.7717/peerj.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Schuler MA, Berenbaum MR. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev. Entomol. 2007;52:231–253. doi: 10.1146/annurev.ento.51.110104.151104. [DOI] [PubMed] [Google Scholar]
- 15.Dermauw W, et al. A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae. Proc. Natl Acad. Sci. USA. 2013;110:E113–E122. doi: 10.1073/pnas.1213214110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vatanparast M, Hosseininaveh V, Ghadamyari M, Sajjadian SM. Plant cell wall degrading enzymes, pectinase and cellulase, in the digestive system of the red palm weevil, Rhynchophorus ferrugineus (Coleoptera: Curculionidae) Plant Prot. Sci. 2014;50:190–198. [Google Scholar]
- 17.Pauchet Y, Wilkinson P, Chauhan R. Diversity of beetle genes encoding novel plant cell wall degrading enzymes. PLoS ONE. 2010;5:e15635. doi: 10.1371/journal.pone.0015635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scully ED, et al. Metagenomic profiling reveals lignocellulose degrading system in a microbial community associated with a wood-feeding beetle. PLoS ONE. 2013;8:e73827. doi: 10.1371/journal.pone.0073827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirsch R, et al. Horizontal gene transfer and functional diversification of plant cell wall degrading polygalacturonases: key events in the evolution of herbivory in beetles. Insect Biochem. Mol. Biol. 2014;52:33–50. doi: 10.1016/j.ibmb.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Ohtoko K, et al. Diverse genes of cellulase homologues of glycosyl hydrolase family 45 from the symbiotic protists in the hindgut of the termite Reticulitermes speratus. Extremophiles. 2000;4:343–349. doi: 10.1007/s007920070003. [DOI] [PubMed] [Google Scholar]
- 21.Todaka N, et al. Phylogenetic analysis of cellulolytic enzyme genes from representative lineages of termites and a related cockroach. PLoS ONE. 2010;5:e8636. doi: 10.1371/journal.pone.0008636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sethi A, et al. A GHF7 cellulase from the protist symbiont community of Reticulitermes flavipes enables more efficient lignocellulose processing by host enzymes. Arch. insect Biochem. Physiol. 2013;84:175–193. doi: 10.1002/arch.21135. [DOI] [PubMed] [Google Scholar]
- 23.Calderón-Cortés N, Quesada M, Watanabe H, Cano-Camacho H, Oyama K. Endogenous plant cell wall digestion: a key mechanism in insect evolution. Annu. Rev. Ecol. Evol. Syst. 2012;43:45–71. [Google Scholar]
- 24.Yin A, et al. Transcriptomic study of the red palm weevil Rhynchophorus ferrugineus embryogenesis. Insect Sci. 2015;22:65–82. doi: 10.1111/1744-7917.12092. [DOI] [PubMed] [Google Scholar]
- 25.Antony B, et al. Identification of the genes involved in odorant reception and detection in the palm weevil Rhynchophorus ferrugineus, an important quarantine pest, by antennal transcriptome analysis. BMC Genom. 2016;17:69. doi: 10.1186/s12864-016-2362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, et al. A large-scale gene discovery for the red palm weevil Rhynchophorus ferrugineus (Coleoptera: Curculionidae) Insect Sci. 2013;20:689–702. doi: 10.1111/j.1744-7917.2012.01561.x. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett AC, Rananavare H. Karyotype and sperm of the red palm weevil (Coleoptera: Curculionidae) Ann. Entomological Soc. Am. 1983;76:1011–1013. [Google Scholar]
- 28.Li F, et al. Insect genomes: progress and challenges. Insect Mol. Biol. 2019;28:739–758. doi: 10.1111/imb.12599. [DOI] [PubMed] [Google Scholar]
- 29.Biemont, C. Genome Size Evolution: Within-species Variation in Genome Size (Nature Publishing Group, 2008). [DOI] [PubMed]
- 30.Kelly LJ, et al. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. N. Phytologist. 2015;208:596–607. doi: 10.1111/nph.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ragland GJ, et al. Differences in performance and transcriptome-wide gene expression associated with R hagoletis (Diptera:Tephritidae) larvae feeding in alternate host fruit environments. Mol. Ecol. 2015;24:2759–2776. doi: 10.1111/mec.13191. [DOI] [PubMed] [Google Scholar]
- 32.Eyres I, et al. Differential gene expression according to race and host plant in the pea aphid. Mol. Ecol. 2016;25:4197–4215. doi: 10.1111/mec.13771. [DOI] [PubMed] [Google Scholar]
- 33.Antony B, Johny J, Aldosari SA. Silencing the odorant binding protein RferOBP1768 reduces the strong preference of palm weevil for the major aggregation pheromone compound ferrugineol. Front. Physiol. 2018;9:252. doi: 10.3389/fphys.2018.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium TGS. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452:949. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- 35.Mack PD, Kapelnikov A, Heifetz Y, Bender M. Mating-responsive genes in reproductive tissues of female Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 2006;103:10358–10363. doi: 10.1073/pnas.0604046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarov-Blat L, So WV, Liu L, Rosbash M. The Drosophila takeout gene is a novel molecular link between circadian rhythms and feeding behavior. Cell. 2000;101:647–656. doi: 10.1016/s0092-8674(00)80876-4. [DOI] [PubMed] [Google Scholar]
- 37.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5:226. doi: 10.1186/gb-2004-5-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu A, et al. A genome-wide identification and analysis of the basic helix-loop-helix transcription factors in the ponerine ant, Harpegnathos saltator. BMC Evolut. Biol. 2012;12:165. doi: 10.1186/1471-2148-12-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moriyama M, et al. Ecdysteroid promotes cell cycle progression in the Bombyx wing disc through activation of c-Myc. Insect Biochem. Mol. Biol. 2016;70:1–9. doi: 10.1016/j.ibmb.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 41.Sillam-Dussès D, et al. The role of the glucose-sensing transcription factor carbohydrate-responsive element-binding protein pathway in termite queen fertility. Open Biol. 2016;6:160080. doi: 10.1098/rsob.160080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Syvanen M, Zhou Z, Wharton J, Goldsbury C, Clark A. Heterogeneity of the glutathione transferase genes encoding enzymes responsible for insecticide degradation in the housefly. J. Mol. Evol. 1996;43:236–240. doi: 10.1007/BF02338831. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Z-H, Syvanen M. A complex glutathione transferase gene family in the housefly Musca domestica. Mol. Gen. Genet. 1997;256:187–194. doi: 10.1007/s004380050560. [DOI] [PubMed] [Google Scholar]
- 44.Walters KB, Grant P, Johnson DL. Evolution of the GST omega gene family in 12 Drosophila species. J. Heredity. 2009;100:742–753. doi: 10.1093/jhered/esp043. [DOI] [PubMed] [Google Scholar]
- 45.Shi H, et al. Glutathione S-transferase (GST) genes in the red flour beetle, Tribolium castaneum, and comparative analysis with five additional insects. Genomics. 2012;100:327–335. doi: 10.1016/j.ygeno.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 46.Bergé J, Feyereisen R, Amichot M. Cytochrome P450 monooxygenases and insecticide resistance in insects. Philos. Trans. R. Soc. Lond. Ser. B: Biol. Sci. 1998;353:1701–1705. doi: 10.1098/rstb.1998.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adolfi A, et al. Functional genetic validation of key genes conferring insecticide resistance in the major African malaria vector, Anopheles gambiae. Proc. Natl Acad. Sci. USA. 2019;116:25764–25772. doi: 10.1073/pnas.1914633116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue J, et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014;15:521. doi: 10.1186/s13059-014-0521-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genta FA, Bragatto I, Terra WR, Ferreira C. Purification, characterization and sequencing of the major β-1, 3-glucanase from the midgut of Tenebrio molitor larvae. Insect Biochem. Mol. Biol. 2009;39:861–874. doi: 10.1016/j.ibmb.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Remnant EJ, et al. Gene duplication in the major insecticide target site, Rdl, in Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 2013;110:14705–14710. doi: 10.1073/pnas.1311341110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dale R, et al. Identification of ion channel genes in the Acyrthosiphon pisum genome. Insect Mol. Biol. 2010;19:141–153. doi: 10.1111/j.1365-2583.2009.00975.x. [DOI] [PubMed] [Google Scholar]
- 52.Khatri BS, Burt A. Robust estimation of recent effective population size from number of independent origins in soft sweeps. Mol. Biol. Evol. 2019;36:2040–2052. doi: 10.1093/molbev/msz081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pauchet Y, Heckel DG. The genome of the mustard leaf beetle encodes two active xylanases originally acquired from bacteria through horizontal gene transfer. Proc. R. Soc. B: Biol. Sci. 2013;280:20131021. doi: 10.1098/rspb.2013.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blaxter M. Symbiont genes in host genomes: fragments with a future? Cell Host Microbe. 2007;2:211–213. doi: 10.1016/j.chom.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 55.Bacic A, Harris PJ, Stone BA. Structure and function of plant cell walls. Biochem. Plants. 1988;14:297–371. [Google Scholar]
- 56.Fang C, et al. Hydrothermal pretreatment of date palm (Phoenix dactylifera L.) leaflets and rachis to enhance enzymatic digestibility and bioethanol potential. Biomed. Res. Int. 2015;2015:216454. doi: 10.1155/2015/216454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henrissat B, Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1993;293:781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wheat CW, Wahlberg N. Phylogenomic insights into the Cambrian explosion, the colonization of land and the evolution of flight in Arthropoda. Syst. Biol. 2013;62:93–109. doi: 10.1093/sysbio/sys074. [DOI] [PubMed] [Google Scholar]
- 59.Natri HM, Shikano T, Merilä J. Progressive recombination suppression and differentiation in recently evolved neo-sex chromosomes. Mol. Biol. Evolution. 2013;30:1131–1144. doi: 10.1093/molbev/mst035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Canapa A, Barucca M, Biscotti MA, Forconi M, Olmo E. Transposons, genome size, and evolutionary insights in animals. Cytogenetic Genome Res. 2015;147:217–239. doi: 10.1159/000444429. [DOI] [PubMed] [Google Scholar]
- 61.Burns KH, Boeke JD. Human transposon tectonics. Cell. 2012;149:740–752. doi: 10.1016/j.cell.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 63.Pritham EJ, Feschotte C, Wessler SR. Unexpected diversity and differential success of DNA transposons in four species of entamoeba protozoans. Mol. Biol. Evol. 2005;22:1751–1763. doi: 10.1093/molbev/msi169. [DOI] [PubMed] [Google Scholar]
- 64.Heidel-Fischer HM, Vogel H. Molecular mechanisms of insect adaptation to plant secondary compounds. Curr. Opin. insect Sci. 2015;8:8–14. doi: 10.1016/j.cois.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 65.Faddeeva-Vakhrusheva A, et al. Gene family evolution reflects adaptation to soil environmental stressors in the genome of the collembolan Orchesella cincta. Genome Biol. Evolu. 2016;8:2106–2117. doi: 10.1093/gbe/evw134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krupp JJ, et al. Social experience modifies pheromone expression and mating behavior in male Drosophila melanogaster. Curr. Biol. 2008;18:1373–1383. doi: 10.1016/j.cub.2008.07.089. [DOI] [PubMed] [Google Scholar]
- 67.Gunawardena NE, et al. Host attractants for red weevil, Rhynchophorus ferrugineus: Identification, electrophysiological activity, and laboratory bioassay. J. Chem. Ecol. 1998;24:425–437. [Google Scholar]
- 68.Ortelli F, Rossiter LC, Vontas J, Ranson H, Hemingway J. Heterologous expression of four glutathione transferase genes genetically linked to a major insecticide-resistance locus from the malaria vector Anopheles gambiae. Biochemical J. 2003;373:957–963. doi: 10.1042/BJ20030169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lumjuan N, et al. The role of the Aedes aegypti Epsilon glutathione transferases in conferring resistance to DDT and pyrethroid insecticides. Insect Biochem. Mol. Biol. 2011;41:203–209. doi: 10.1016/j.ibmb.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Wakil W, et al. Resistance to commonly used insecticides and phosphine fumigant in red palm weevil, Rhynchophorus ferrugineus (Olivier) in Pakistan. PLoS ONE. 2018;13:e0192628. doi: 10.1371/journal.pone.0192628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gotoh, O. Evolution and differentiation of P-450 genes in Cytochrome P-450 (eds Omura, T., Ishimura, Y., & Fujii-Kuriyama, Y.) 207–223 (Kodansha, Tokyo, 1993).
- 72.Feyereisen, R. Evolution of Insect P450 (Portland Press Ltd., 2006). [DOI] [PubMed]
- 73.Yu L, et al. Characterization and expression of the cytochrome P450 gene family in diamondback moth, Plutella xylostella (L.) Sci. Rep. 2015;5:8952. doi: 10.1038/srep08952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lao S-H, et al. Genomic and transcriptomic insights into the cytochrome P450 monooxygenase gene repertoire in the rice pest brown planthopper, Nilaparvata lugens. Genomics. 2015;106:301–309. doi: 10.1016/j.ygeno.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 75.Aldawood, A., Alsagan, F., Altuwariqi, H., Almuteri, A. & Rasool, K. Red palm weevil chemical treatments on date palms in Saudi Arabia: results of extensive experimentations. In Colloque méditerranéen sur les ravageurs des palmiers, Nice, France, 16–18 Janvier 2013 (Association Française de Protection des Plantes (AFPP), 2013).
- 76.Diester-Haass, L., Billups, K., Lear, C. Productivity changes across the mid-Pleistocene climate transition. Earth-Sci. Rev. 179. 10.1016/j.earscirev.2018.02.016 (2018).
- 77.Ferry M, Gomez S. The red palm weevil in the Mediterranean area. Palms. 2002;46:172–178. [Google Scholar]
- 78.El-Ezaby, F., Khalifa, O., & El-Assal, A. Integrated pest management for the control of red palm weevil Rhynchphorus ferrugineus Oliv in the United Arab Emirates, Eastern region, Al Ain. In Proceedings of 1st International Conference on Date Palms, Mar (1998).
- 79.Petit N, Barbadilla A. Selection efficiency and effective population size in Drosophila species. J. Evolut. Biol. 2009;22:515–526. doi: 10.1111/j.1420-9101.2008.01672.x. [DOI] [PubMed] [Google Scholar]
- 80.Felsenstein J. Inbreeding and variance effective numbers in populations with overlapping generations. Genetics. 1971;68:581. doi: 10.1093/genetics/68.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murphy S, Briscoe B. The red palm weevil as an alien invasive: biology and the prospects for biological control as a component of IPM. Biocontrol News Inf. 1999;20:35N–46N. [Google Scholar]
- 82.Eyun S-I, et al. Molecular evolution of glycoside hydrolase genes in the western corn rootworm (Diabrotica virgifera virgifera) PLoS ONE. 2014;9:e94052. doi: 10.1371/journal.pone.0094052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song JM, et al. Molecular and biochemical characterizations of a novel arthropod endo-β-1, 3-glucanase from the Antarctic springtail, Cryptopygus antarcticus, horizontally acquired from bacteria. Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol. 2010;155:403–412. doi: 10.1016/j.cbpb.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 84.McKenna DD, et al. Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle–plant interface. Genome Biol. 2016;17:227. doi: 10.1186/s13059-016-1088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220:1049–1051. doi: 10.1126/science.220.4601.1049. [DOI] [PubMed] [Google Scholar]
- 86.Vurture GW, et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33:2202–2204. doi: 10.1093/bioinformatics/btx153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gremme G, Steinbiss S, Kurtz S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput Biol. Bioinform. 2013;10:645–656. doi: 10.1109/TCBB.2013.68. [DOI] [PubMed] [Google Scholar]
- 88.Tempel, S. Using and understanding RepeatMasker. In Mobile Genetic Elements (Springer, 2012).
- 89.Team RC. R: A language and environment for statistical computing. Computing10.1890/0012-9658(2002)083[3097:CFHIWS]2.0.CO (2013).
- 90.De Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 91.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bartenhagen C, Dugas M. Robust and exact structural variation detection with paired-end and soft-clipped alignments: SoftSV compared with eight algorithms. Brief. Bioinform. 2016;17:51–62. doi: 10.1093/bib/bbv028. [DOI] [PubMed] [Google Scholar]
- 93.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nowell RW, et al. Comparative genomics of bdelloid rotifers: Insights from desiccating and nondesiccating species. PLoS Biol. 2018;16:e2004830. doi: 10.1371/journal.pbio.2004830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cole TJ, Brewer MS. FUSTr: a tool to find gene Families Under Selection in Transcriptomes. PeerJ. 2018;6:e4234. doi: 10.7717/peerj.4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 97.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 98.Harris, R. S. Improved pairwise Alignmnet of genomic DNA. Ph.D. Thesis, The Pennsylvania State University (2007).
- 99.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinform. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Giblin-Davis, R. M., Faleiro, J. R., Jacas, J. A., Peña, J. E., & Vidyasagar, P. Biology and management of the red palm weevil, Rhynchophorus ferrugineus. In Peña JE (ed) Potential Invasive Pests of Agricultural Crops. CABI 1–34 (2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The Illumina reads Hiseq 2500 (2× 150 bp as well as the 10x Genomics data) were deposited at NCBI short archive under SRA accession PRJNA524026 (Biosample accession SAMN10995380). The RNA sequencing data as well as the Oxford Nanopore were deposited at NCBI short archive under BioProject (PRJNA600770, SRA ID SUB6799681). The final genome, is deposited at NCBI GeneBank (https://www.ncbi.nlm.nih.gov/genbank/wgs_update/) under accession (JABAOJ000000000), and supplementary data files, mitochondria genome as well as HGT validation are deposited at Dryad database (https://datadryad.org/stash/share/yyBU31Aj2_n2H9QYCPkG-hoZcYf63-2OToegnkmBcV8).