

Abstract
Purpose:
Blocking the function of myeloid-derived suppressor cells (MDSC) is an attractive approach for cancer immunotherapy. Having shown DC-HIL/GPNMB to be the T-cell-inhibitory receptor mediating the suppressor function of MDSCs, we evaluated the potential of anti-DC-HIL mAb as an MDSC-targeting cancer treatment.
Experimental Design:
Patients with metastatic cancer (n = 198) were analyzed by flow cytometry for DC-HIL or PDL1 expression on blood CD14+HLA-DRno/lo MDSCs. Their suppressor function was assessed by in vitro coculture with autologous T cells, and the ability of anti-DC-HIL or anti-PDL1 mAb to reverse such function was determined. Tumor expression of these receptors was examined histologically, and the antitumor activity of the mAb was evaluated by attenuated growth of colon cancers in mice.
Results:
Patients with metastatic cancer had high blood levels of DC-HIL+ MDSCs compared with healthy controls. Anti-DC-HIL mAb reversed the in vitro function in ~80% of cancer patients tested, particularly for colon cancer. Despite very low expression on blood MDSCs, anti-PDL1 mAb was as effective as anti-DC-HIL mAb in reversing MDSC function, a paradoxical phenomenon we found to be due to upregulated expression of PDL1 by T-cell-derived IFNγ in cocultures. DC-HIL is not expressed by colorectal cancer cells but by CD14+ cells infiltrating the tumor. Finally, anti-DC-HIL mAb attenuated growth of preestablished colon tumors by reducing MDSCs and increasing IFNγ-secreting T cells in the tumor microenvironment, with similar outcomes to anti-PDL1 mAb.
Conclusions:
Blocking DC-HIL function is a potentially useful treatment for at least colorectal cancer with high blood levels of DC-HIL+ MDSCs.
Introduction
Myeloid-derived suppressor cells (MDSC) are a relatively immature population of bone marrow (BM)-derived cells that can be sorted into monocytic (CD14+ CD15neg HIA-DRno/lo) and polymorphonuclear (CD14neg CD15+ HIA-DRno/lo) subsets (1, 2). In cancer-bearing hosts, MDSCs expand exponentially in blood and accumulate in many organs, where they can potently suppress T-cell function and promote cancer growth and dissemination (3). This exponential expansion of MDSCs in cancer patients was reported to associate with resistance to anti-CTLA4 and/or anti-PD1/PDL1 therapy (4, 5). A study of melanoma patients treated with anti-CTLA4 mAb correlated high blood MDSC levels at pretreatment with low survival rates and low blood CD8 T cells (6). Therefore, MDSCs are an attractive target for optimizing anticancer treatment. Indeed, cancer studies using animal models have documented benefits from depleting MDSCs or blocking their function (7, 8).
DC-HIL receptor is also known as GPNMB that associates with metastatic properties of tumor cells and angiogenesis (9-11). We discovered the DC-HIL receptor to be an immune checkpoint that inhibits T-cell activation via binding to syndecan-4 (SD4) expressed by activated T cells (12, 13). Other research groups also showed consistent results (14, 15). DC-HIL is constitutively expressed by antigen-presenting cells (APC) at very low levels in healthy controls, but this expression is remarkably upregulated by inflammatory signals in only some (but not all) APCs (16) and by tumor challenge particularly in MDSCs (17, 18). Some cancer cells also express DC-HIL/GPNMB at considerably variable levels (19, 20). Blocking the DC-HIL function using specific mAb, soluble recombinant proteins, or gene disruption worsened autoimmune response (21) while potentiating antitumor immunity in melanomabearing hosts (17, 18). Importantly, we showed DC-HIL on MDSCs to be a critical mediator of these cells' T-cell suppressor and cancer-promoting activities (17).
These data prompted us to assume that anti-DC-HIL mAb can be useful for MDSC-targeting approach. Here we evaluate the prevalence of expanded DC-HIL+ MDSC subpopulation among common solid cancers and the efficacy of anti-DC-HIL mAb to reverse the MDSC function in vitro, by comparing with anti-PDL1 mAb treatment, the established immunotherapy that has been recently approved by the FDA for metastatic lung cancer (22).
Materials and Methods
Study populations and specimens
Metastatic cancer patients (n = 198) with varying malignancies and healthy controls (n = 21; Supplementary Table S1) without immunologic conditions and/or immunotherapies were recruited through Tissue Resource, Harold C. Simmons Comprehensive Cancer Center at University of Texas Southwestern Medical Center. Blood and tissue specimens were collected through the Tissue Resource after informed consent was obtained (IRB-STU 032018-084). The study was conducted in accordance with the amended Declaration of Helsinki and the International Conference on Harmonization Guidelines.
Cell line
MC38 or CT26 is the colon adenocarcinoma cell line of C57BL/6 or BALB/c origin, respectively, which was obtained from Dr. Jeffrey Schlom, the National Cancer Institute (23) or from ATCC. These cells were maintained in DMEM containing 100 mL/L FCS with 100,000 U/L penicillin and 100 mg/L streptomycin, 1 mmol/L sodium pyruvate, 2 mmol/L l-glutamine, and 1 mmol/L nonessential amino acid solution.
mAbs
We established 3D5 mouse antihuman DC-HIL mAb (24) and UTX103 rabbit anti-mouse DC-HIL mAb (25). 3D5 IgG was produced by culturing the 3D5 mAb clone in serum-free media and purified by Protein A-agarose (Invitrogen). The chimeric IgG consisted of the V-regions of UTX103 rabbit IgG fused to the C-regions of mouse IgG1; it was produced by transient transfection of the heavy- and light-chain genes using ExpiCHO systems in serum-free media (Thermo-Fisher). mAb directed at human PD1 (MIH4), PDL1 (MIH1), or mouse PD1 (J43) were purchased from eBioscience; and anti-mouse PDL1 mAb (10F.9G2) from Bio X Cell.
Flow cytometry
Within 24 hours after collecting blood, peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Paque, treated with FcR blocking reagent (Militenyi Biotec), and incubated with 20 μg/mL 3D5 anti-DC-HIL or control anti-KLH mAb (both are mouse IgG1) and 1 μg/mL PE-anti-mouse IgG [F(ab′)2 fragment] (Jackson ImmunoResearch). After washing, cells were stained with APC-conjugated anti-HLA-DR, FITC-anti-CD14 Ab (each 5 μg/mL), PerCP/Cy5.5-conjugated anti-CD235ab (Biolegend) and analyzed for cell-bound fluorescence using FACS verse (BD Bioscience). Staining with anti-CD235ab was used to exclude nonnucleated red blood cells (RBC). CD14+ cells are sorted into HLA-DRneg, HLA-DRlo, and HLA-DRhi cells, with the first two fractions comprising monocytic MDSCs that were also positive for CD33 and CD11b (18).
T-cell suppression assays
CD14+HLA-DRneg MDSCs and T cells were freshly isolated from blood samples (~20 mL) of the same donor (26): PBMCs were depleted of HLA-DR+ cells using anti-HLA-DR-microbeads (Miltenyi Biotec). The pass-through (HLA-DRneg) fraction was sorted further into CD14+ and CD14neg subfractions using anti-CD14 beads; the former were considered MDSCs, whereas the latter were T cells (~90% CD3+). T cells (5 × 104 cells/well) were cocultured for 5 days with MDSCs at varying cell ratios and with anti-CD2/CD3/CD28 Ab-coated beads (Miltenyi Biotec; 1.5 beads per T cell) in microculture wells. IFNγ in the cocultures were assayed by ELISA (eBioscience). T-cell suppressor ability of MDSCs was expressed as percentage of suppression: 1 – b/a × 100%, where a is the IFNγ amounts in cultures of T cells only; and b is the IFNγ in cocultures at 1:1 ratio. To evaluate the effect of different mAb on MDSC function, mAb was added to 1:1 cocultures at a concentration of 50 μg/mL. MDSCs were pretreated with 50 μg/mL mAb for 30 minutes at 4°C, washed, and mixed with T cells. The ability of mAb to block MDSC function was expressed as percentage of restoration: c – b/a – b × 100%, where c is the IFNγ in 1:1 cocultures with mAb. Nonspecific restoration was factored by measuring the effect of anti-KLH or anti-CD14 mAb relative to cocultures without mAb (n = 45). Mouse MDSCs were similarly evaluated as before (17).
IHC staining
Serial sections of formalin-fixed tissues were deparaffinized, rehydrated, immersed in citrate buffer (pH 6.0), and microwaved for 15 minutes to retrieve antigens. Slides were incubated in 5% donkey, horse, or goat serum overnight at 4°C, stained for 1 hour with goat anti-GPNMB (1:1,000 dilution; R&D Systems), mouse anti-B7-H1/PDL1 (1:500 dilution; R&D Systems), mouse anti-Cytokeratin 20 (1:500 dilution; Invitrogen), rabbit anti-CD3 (1:500 dilution; Novus Bilogicals) or rabbit anti-CD14 (1:500 dilution; Novus Bilogicals). After washing, slides were incubated with biotin-secondary Ab and avidin-peroxidase solution (Vector), followed by color development (brown) by 3,3-diaminobenzidine and counterstaining with hematoxylin. These IHC-stained sections were examined for expression of receptors in a blinded-fashion. Frequency of receptor expression in total CD14+ cells was determined as follows: percentage of DC-HIL+ CD14+ cells was determined by the area of anti-DC-HIL-positive staining relative to that of anti-CD14-staining using image analysis. Data shown are the average of five separate microscopic views.
Animal studies
Female 6- to 8-week-old C57BL/6 and BALB/c mice (Harland Breeders) were housed in a pathogen-free facility and subjected to experimental procedures approved by the Institutional Animal Care Use Center at UT Southwestern. MC38 or CT26 cells (1 × 106) were injected subcutaneously into the right-shaved flank of mice. Tumor volume was measured (19). Six days later, mice were injected intraperitoneally with 200 μg mAb/mouse every 2 days until day 17 (total of six injections). On days 0, 9, 13, and 17, blood samples were collected from tail veins of mice (25 μL/mouse), stained with anti-CD11b and anti-Gr1 mAb (5 μg/mL), and determined by flow cytometry for percentage of MDSCs among PBMCs. In separate experiments, on day 13 (one day after the fourth injection), tumor and tumor-draining lymph nodes (DLN) were excised from treated mice and dissociated into single cells by Tumor Dissociation Kit (Miltenyi Biotec). Isolated cells were FcR-blocked and stained with mouse anti-mouse DC-HIL mAb or control IgG2b and 5 μg/mL PE-anti-mouse IgG [F(ab′)2 fragment]. After washing, cells were also stained with PE-cyanine7-anti-CD45 (BD Pharmingen), FITC-anti-CD11b, APC-anti-Gr1, PerCP-anti-Ly6c (eBioscience), and analyzed by flow cytometry. Regulatory T cells (Treg) were counted by flow cytometry for cells that were positive for CD4 (or CD8) and Foxp3. DLN cells (2 × 105/well) were cultures for 3 days in 96 microwells precoated with anti-CD3/CD28 (each 1 μg/mL). The culture supernatant and cells were harvested and assayed for secreted IFNγ amounts by ELISA and for IFNγ-secreting T cells by flow cytometry (27).
Statistical analysis
Statistical analyses were performed using Pearson correlation coefficients, TDIST, and Student t test for the in vitro assays.
Results
DC-HIL+ MDSCs are expanded in the blood of metastatic solid cancer patients
Having shown that metastatic melanoma patients display highly expanded DC-HIL+ MDSCs in the blood (17), we examined the prevalence of this blood index in other cancer types. Patients (n = 198) with metastatic forms of most common solid cancers were recruited; bladder (n = 4), breast (n = 11), colorectal (n = 64), kidney (n = 12), lung (n = 20), melanoma (n = 25), pancreatic (n = 37), or prostate cancer (n = 25; Supplementary Table S1). PBMCs from these patients were analyzed by flow cytometry for HLA-DR and CD14 expression using fluorescently labeled mAb. HLA-DRno/lo CD14+ monocytic MDSCs were sorted for DC-HIL expression (Fig. 1A) and characterized for percentage of MDSCs among PBMCs, percentage of DC-HIL-positivity among MDSCs, and percentage of DC-HIL+ MDSCs among PBMCs (Fig. 1B). There was significant expansion of HLA-DRno/lo CD14+ MDSCs in all of the cancers tested (P < 0.03), compared with healthy donors (a median of 0.6%, range of 0.1–2.7%). Among cancer types, lung and breast cancer patients showed lowest levels of blood MDSCs (median of 1.2% and 1.5%, respectively). For DC-HIL-positivity among MDSCs, the vast majority of patients in each cancer type exhibited very high DC-HIL-positivity (47–90% median), in contrast to 7.2% by healthy donors. Using percentage of DC-HIL+ MDSCs/PBMCs as the index, all cancer types tested showed significantly high-level DC-HIL expression, compared with healthy donors (P < 0.005; Supplementary Table S1). These results indicate that metastatic solid cancers are associated with expanded DC-HIL+ monocytic HLA-DRno/lo CD14+ MDSCs, with highest degree in colorectal, kidney, pancreatic and prostate cancers.
Figure 1.

Expansion of circulating DC-HIL+ MDSCs in patients with metastatic forms of most common solid cancers. A, PBMCs were isolated from blood samples of healthy donors (HD) or patients with metastatic cancer from bladder (BL), breast (BR), colon (CO), kidney (KI), lung (LU), melanoma (ME), pancreatic (PA), or prostate (PR) and examined by flow cytometry for expression of HAL-DR and CD14 (the left dot plots). CD14+ HLA-DRno/lo MDSCs (percentage in total PBMCs shown in a small window) were gated and determined for percentage of DC-HIL+ cells among total MDSCs. Representative data of each malignancy are shown. B, Flow cytometry data of each patient was calculated for percentage of MDSCs in total PBMCs, percentage of DC-HIL positivity in MDSCs, and percentage of DC-HIL+ MDSCs among PBMCs and plotted in a logarithmic or linear scale. Median (percentage) in each cancer type is shown in numerical number and by black lines. Number in the parenthesis indicates the sample size.
Expansion of DC-HIL+ MDSCs is associated with colorectal cancer progression
Because colorectal cancer was the most common malignancy among our cases and because it was strongly associated with expansion of DC-HIL+ MDSCs, we analyzed the influence of patients' demographics, treatments, and cancer stages to DC-HIL expression (Supplementary Table S2). There was no gender-associated bias in the correlation. All therapies (radiation, chemotherapy, or gene-targeting therapy) showed no significant correlation with high DC-HIL expression (P > 0.2). Because expansion of MDSCs is shown to associate with tumor progression in mouse cancer models (28), we queried whether blood DC-HIL+ MDSC level and tumor burden were correlated. Patients with ≥4 ng/mL carcinoembryonic antigen (CEA; ref. 29) exhibited a significantly higher percentage of DC-HIL+ MDSCs/PBMCs than those with <4 ng/mL (P = 0.038; Supplementary Table S2). M1b patients showed a significantly higher indices compared with M1a cohorts (P = 0.0056), but indices for M0 versus M1a were not significantly different (P = 0.57). This apparent inconsistency was due to very high DC-HIL expression by the subgroup treated with radiation (n = 4) within the M0 cohort (Supplementary Table S3). Excluding that subgroup, the analysis rendered correlation between M0 and M1a to be significant (P = 0.019). Finally, DC-HIL was poorly expressed by stage I/II patients with colorectal cancer (0.16–0.48% DC-HIL+ MDSCs/ PBMCs; Supplementary Fig. S1). Taken together our findings indicate that DC-HIL expression correlates with colorectal cancer progression.
DC-HIL blockade restores the suppressed T-cell response caused by MDSCs
To understand the critical role of DC-HIL in suppressing T-cell response by MDSCs, we queried whether DC-HIL expression by MDSCs correlated with T-cell suppressor ability. Increasing doses of MDSCs purified from patients (CRC n = 17, pancreatic n = 15, and prostate cancer n = 10) were cocultured for 5 days with autologous T-cells at varying cell ratios, with costimulators (anti-CD2/CD3/CD28 Ab). T-cell activation was measured by IFN-γ secretion in the cultures, and suppressor ability assessed by percentage of suppression of IFN-γ production (Fig. 2A). For colorectal and pancreatic cancers, DC-HIL expression correlated positively with higher suppressor activity (R2 = 0.65/P = 0.0002 for CRC and R2 = 0.51/P = 0.004 for pancreatic cancer). However, this was not true for prostate cancers (R2 = 0.21 and P = 0.07). These data suggest heterogeneity among cancer types.
Figure 2.
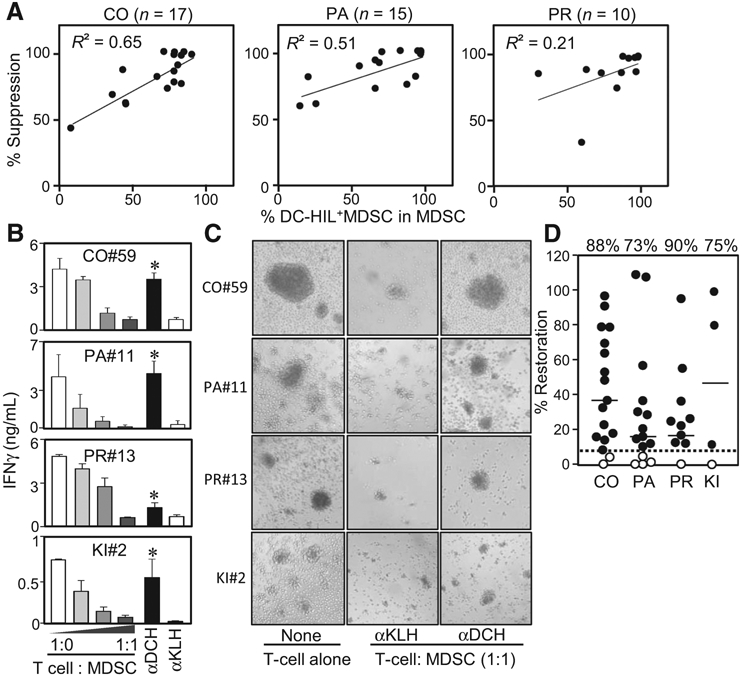
Anti-DC-HIL mAb restored the suppressed T-cell IFNγ response caused by MDSCs. A, Individual patients with colorectal (CO), pancreatic (PA), or prostate (PR) cancer was determined for percentage of DC-HIL+ cells among MDSCs and their ability to suppress T-cell IFNγ response, expressed as percentage of T-cell suppression at cocultures of a 1:1 cell ratio. Values (percentages) were plotted and analyzed for correlation coefficient R2. B, Representative data of T-cell restoration assays by 3D5 anti-DC-HIL mAb: MDSCs were isolated from the blood of indicated patients and cocultured with autologous T cells at different cell ratios with costimulations. 3D5 (αDCH) or control anti-KLH mAb (αKLH) was added to 1:1 cocultures. Five days after culturing, IFNγ amounts were determined and shown in median ± SD, n = 3. *, P < 0.05 compared with cocultures treated with anti-KLH mAb. αDCH, anti-DC-HIL mAb; αKLH, anti-KLH mAb; CO, colorectal, KI, kidney; PA, pancreatic; PR, prostate. C, Photos of day 3 cocultures are shown, with aggregates representing T-cell activation. αDCH, anti-DC-HIL mAb; αKLH, anti-KLH mAb; CO, colorectal, KI, kidney; PA, pancreatic; PR, prostate. D, Assays examining effects of 3D5 mAb on the T-cell suppressor function of MDSCs were performed with samples from patients with colorectal (CO; n = 17), pancreatic (PA; n = 15), prostate (PR; n = 10), and kidney (KI; n = 4) cancer. The ability to reverse the suppressor activity is expressed by percentage of restoration in IFNγ response; set IFNγ amounts in culture of T cells alone as 100%. Solid and dashed lines show median of percentage of restoration (shown on the top) in each cancer type and the cutoff value, respectively. Closed and open circles represent patients who showed higher and lower, respectively, than the cutoff value.
We examined the optimal concentration of mAb for blocking the MDSC function and found full receptor occupancy by 3D5 anti-DC-HIL or by anti-CD14 mAb at 50 μg/mL (Supplementary Fig. S2A and S2B). Using this dose, we assessed the ability of 3D5 mAb to reverse T-cell suppression by MDSCs from patients with CRC (n = 17), pancreatic (n = 15), prostate, (n = 10), or kidney cancer (n = 4; Fig. 2B). Addition of 3D5 mAb to the MDSC/T-cell cocultures (1:1 cell ratio) reversed MDSC suppressor activity of all 4 cancers, but to different degrees. Restoration of T-cell integrity was confirmed by microscopically observing their activation phenotype (cell aggregation) on day 3 cocultures (Fig. 2C). Since each patient displayed variation in IFN-γ response and suppressor activity, we evaluated effects of 3D5 mAb by percentage of restoration in IFN-γ response, in which IFN-γ amounts in T-cell cultures are set at 100%. To determine significance, we also set the cutoff value (6.4% for restoration) to represent the median of percentage of restoration shown by anti-KLH control IgG or anti-CD14 mAb; the latter binds to the surface of MDSCs, but with no effect on MDSC function (Supplementary Fig. S2C). Significant response to 3D5 mAb, which was higher than the cutoff, was noted for 15 of 17 CRC (88% response rate, and median of 35% restoration); 11 of 15 pancreatic cancer (73% and 16%); 9 of 10 prostate cancer (90% and 21%); and 3 of 4 kidney cancer (75% and 47%; Fig. 2D). These effects fluctuated within a considerably wide range, correlating poorly with percentage of DC-HIL-positivity among MDSCs. Overall, our data indicate DC-HIL blockade by 3D5 mAb to be an effective way of restoring the suppressed T-cell response caused by MDSCs, at least for colorectal, pancreatic, and prostate cancers.
Blocking PDL1 function can also reverse MDSC function via upregulated expression of its receptor induced by IFNγ secreted from T cells
We next compared the ability of anti-DC-HIL versus anti-PDL1 mAb to reverse MDSC function. We first examined the relative expression of DC-HIL versus PDL1 by blood MDSCs from colorectal (n = 21), pancreatic (n = 13), or prostate cancer patients (n = 8). All patients expressed markedly high levels of DC-HIL on MDSCs (median of 57% positivity), compared with PDL1 (2%; Fig. 3A). Among these patients, blood samples of some patients (colorectal, n = 2; pancreatic, n = 3; and prostate, n = 3) were used to compare the reversal ability of these mAb (Fig. 3B). We performed MDSC titration assays and T-cell restoration assays by anti-DC-HIL, anti-PDL1, combined mAb, or control anti-KLH mAb. Titration assays revealed all MDSCs to exhibit moderate to high suppressor activities. Five of eight patients (62.5%) exhibited high restoration by anti-DC-HIL and by anti-PDL1 mAb; two patients showed response to ether of the two mAb; and one patient (PA#31) showed no response to both mAb. Only PR#23 patient showed a better response for anti-DC-HIL mAb, and three patients showed a better response for anti-PDL1 mAb. Overall, anti-DC-HIL and anti-PDL1 mAb produced respective means of 19% and 37% restoration, with no statistically significant differences (P = 0.1; Fig. 3C). Combination blockades produced an even better percentage of restoration (48%). We then questioned why anti-PDL1 mAb produced equal-to-better responses despite very low expression of PDL1 by MDSCs. Because of PDL1 expression is known to be upregulated by IFNγ (30), we posited MDSCs to upregulate PDL1 expression during coculturing with costimulated T cells. Before culturing, MDSCs were 86% positive for DC-HIL and 2% for PDL1 (CO#79). After 3 days of culture, DC-HIL expression remained unchanged, whereas PDL1 dramatically rose to 56% (Fig. 3D). Because MDSC phenotypes became dull during cocultures, we used CD45+CD3neg phenotype to identify MDSCs. Similarly, PD1 expression on T cells was prominently increased from 5% to 92%, and SD4 from 2% to 30% (Fig. 3E). We then estimated the impact of upregulated PDL1 expression on MDSC function (Fig. 3F). Purified MDSCs were sorted into two batches; one regularly mixed with T cells and costimulators (anti-CD2/CD3/CD28 Ab-coated beads) in the continuous presence of mAb to cover the induced molecules; and the other pretreated with mAb, and unbound mAb washed out (to cover only preexisting molecules), followed by coculture with T cells/costimulators. Blood samples of seven patients were subjected to this analysis (Fig. 3F; Supplementary Table S4). All cases showed high DC-HIL (43–94%) and low PDL1 expression (0–17%). Consistently with Fig. 3B, the continuous presence of mAb in the cocultures produced 17% to 59% restoration by anti-DC-HIL mAb and higher percentage by anti-PDL1 mAb. For anti-DC-HIL mAb, percentage of restoration by mAb pretreatment was similar to the continuous treatment. However, we found obvious differences between these treatments with anti-PDL1 mAb. The continuous presence restored 24% to 76% of total suppressed IFNγ response, whereas pretreatment exhibited only 3% to 19%. These findings suggest restoration of T-cell response by anti-PDL1 mAb in this in vitro assay was not due to blocking preexisting molecules, but most likely a result of upregulated expression during coculture.
Figure 3.
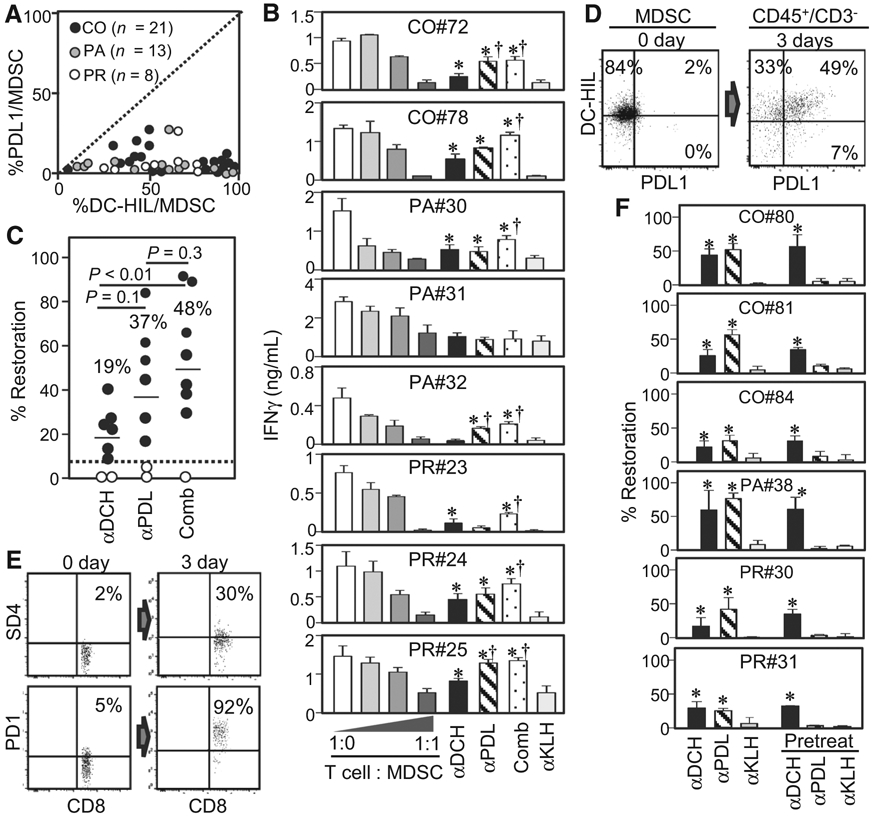
Comparison of anti-DC-HIL and anti-PDL1 mAb in expression and the ability to reverse MDSC function. A, MDSCs from indicated cancer patients were assayed for percentage of DC-HIL positivity versus percentage of PDL1 positivity, and their differential expression levels are plotted in a graph, with the dashed line displaying the same percentage between the two receptors. B, Similarly MDSC-T-cell suppression assays were performed with or without anti-DC-HIL (αDCH), anti-PDL1 (αPDL), combined (Comb), or anti-KLH mAb (αKLH), and mAb effects are shown by IFNγ amounts in the cocultures. C, Data are summarized in a scattered graph, with median percentage, the cutoff value (dashed line), and statistical significance P value between 2 groups. On days 0 and 3 post-coculturing MDSCs (D) and T cells (E), cells were harvested and examined for MDSC (or CD8 T cells) expression of DC-HIL (or SD4) and PDL1 (or PD1). MDSCs were gated for CD14+ HLA-DRno/lo on day 0 and for CD45+CD3neg on day 3. Dot plots are shown with percentage of positive cells within the population. αDCH, anti-DC-HIL mAb; αKLH, anti-KLH mAb; αPDL, anti-PDL1 mAb; CO, colorectal; Comb, combined; PA, pancreatic; PR, prostate. F, MDSCs isolated from varying cancer patients were treated with the continuous presence of mAb or pretreated with mAb before coculturing. *, P < 0.05 and †, P < 0.05 compared with αKLH and αDCH, respectively. αDCH, anti-DC-HIL mAb; αKLH, anti-KLH mAb; αPDL, anti-PDL1 mAb; CO, colorectal; Comb, combined; PA, pancreatic; PR, prostate.
DC-HIL is expressed by tumor-infiltrating CD14+ cells, but not by colorectal cancer cells
Expression of immune checkpoints by the cancer cells has been using as a predictive marker for response to their respective blocking Ab (31). We thus examined DC-HIL (or PDL1) expression in colorectal cancer tissues (n = 5) of patients using IHC. Strong staining for DC-HIL was observed in cancer tissues of patient #19, #26, #35, and #40, and lower expression for patient #21, but no expression was noted in colorectal tissues of a healthy individual (Fig. 4A). We determined expression of these receptors in cancer cells (stained for cytokeratin 20) and in tumor-stromal CD14+ cells (as surrogate of MDSCs), and compared expression of these receptors (Supplementary Table S5). DC-HIL expression was noted into 21% to 95% of total CD14+ cells, but there was nearly no expression at all in cancer cells (<5%). Similarly, tumor expression of PDL1 was barely detectable, except for patient #19 showing 50% positivity. Unlike DC-HIL, PDL1 expression by CD14+ cells was significantly lower (at most <20%). Thus, expression of these receptors in CD14+ cells may be inversely proportional (Fig. 4B). Because these patients were already assayed for percentage of blood DC-HIL+ MDSCs/PBMCs, we queried whether emergence of DC-HIL+CD14+ cells in cancer tissue correlated with blood DC-HIL+ MDSC levels. We found a significant positive correlation (R2 = 0.88, P = 0.0007), but not with PDL1 expression (Fig. 4C and D). Our data may suggest that blood MDSCs infiltrate the tumor microenvironment without undergoing phenotypic changes in expression of DC-HIL versus PDL1, except CO#21 patient whose tissue-CD14+ cells showed the converse phenotype (DC-HILlo and PDL1hi).
Figure 4.

DC-HIL and PDL1 expression in colorectal tissues of patients with cancer. A, Serial sections of tumor biopsies from patients with colorectal cancer (CO; n = 5) or a healthy donor (HD) were IHC stained for expression of indicated markers (shown in brawn) or counter stained with H&E. Histologic examination was performed under light microscope (10× magnification, a scale bar of 200 μm). Closed triangles show the location of cancer cells. B–D, Percentage of positivity for DC-HIL versus PDL1 among tissue-resident CD14+ cells (t-CD14) was determined, and correlations between these two receptors and between t-CD14 and blood MDSCs (b-MDSCs) are analyzed in a graph, with correlation coefficient R2.
DC-HIL blockade produces treatment benefits for colorectal cancer in a preclinical animal model
We first examined the immunologic phenotype of mouse MC38 colon cancer and of tumor-stromal cells. MC38 cells constitutively express high-levels of PDL1, but almost no expression of DC-HIL (Fig. 5A). This was confirmed by immunoblotting (Supplementary Fig. S3A). By contrast, tumor-infiltrating MDSCs expressed DC-HIL (Fig. 5B): Gr1+Ly6C+ MDSCs within the tumor microenvironment were sorted into three subsets: M1 (Gr1intLy6Chi monocytic MDSC), M2 (Gr1loLy6Clo non-monocytic and non-granulocytic MDSC), and M3 (Gr1hiLy6Clo granulocytic MDSC; ref. 32). M1 and M2 subsets expressed both receptors at similarly high-levels, but M3 subset expressed highly PDL1 and lowly DC-HIL. In other organs, MDSCs in spleen and DLNs were absent of DC-HIL expression (Supplementary Fig. S4); those in blood and BM expressed DC-HIL more than PDL1 (Fig. 5C and D). Thus, both DC-HIL and PDL1 are expressed by all MDSCs in many organs of mice with MC38 tumor, but they differed in the tumor expression. We then examined effects of DC-HIL blockade on MC38 tumor growth by injecting anti-DC-HIL mAb into mice starting on day 6 after implanting tumor cells subcutaneously and every 2 days a total of six injections (Fig. 5E). Because low-dose implantation (1–2 × 105 cells/mouse; ref. 33) did not produce established tumors in our hands, we used high dose (1 × 106 cells/mouse). Anti-DC-HIL mAb attenuated MC38 tumor growth (60% inhibition on day 17 vs. control mice). This tumor growth was also attenuated by injection of anti-PDL1 mAb (26% reduction on day 17). Thus, anti-DC-HIL treatment produced better outcomes than anti-PDL1 under this protocol. We also examined effects of infused mAb on circulating MDSC expansion. Blood MDSCs expanded progressively up to 20% on day 17 in control mice, but anti-DC-HIL treatment prevented MDSC expansion, more efficaciously than anti-PDL1 treatment (Fig. 5F). Combining the two mAb produced no synergistic antitumor effects for MC38 tumor (Supplementary Fig. S5). Anti-DC-HIL treatment produced similar antitumor activity as anti-PDL1 for another colon cancer CT26 line also devoid of DC-HIL expression and from a different genetic background (Supplementary Fig. S4A). Unlike the case for MC38 tumor, combination treatment using the two mAb produced better outcomes than treatment with either mAb alone (Supplementary Fig. S3B). Finally, we found no detectable adverse effects of anti-DC-HIL mAb treatment on blood cells of treated mice (Supplementary Fig. S6).
Figure 5.
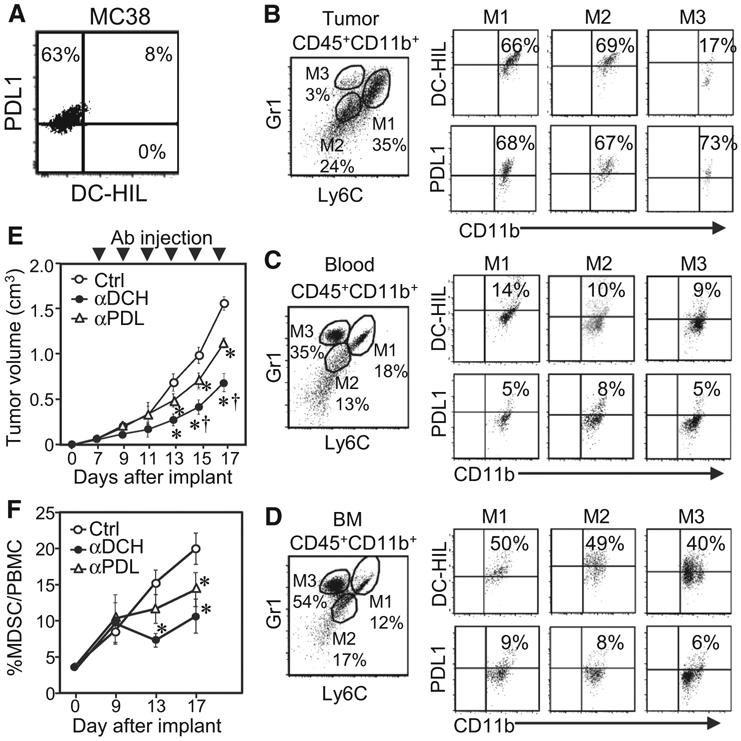
Infusion of anti-DC-HIL mAb retarded MC38 tumor growth and reduced frequency of DC-HIL+ MDSCs in tumor. A, Expression of DC-HIL and PDL1 on MC38 cells was assayed by flow cytometry. B–D, Cells isolated from tumors (B), blood (C), or BM (D) of MC38 tumor (~1.5 cm)-bearing mice were Ab stained and gated for CD45+CD11b+ cells (to exclude tumor cells), which were sorted into M1 (Ly6ChiGr1hi), M2 (Ly6CloGr1lo), and M3 (Ly6CloGr1hi) subsets and examined by flow cytometry for DC-HIL or PDL1 expression. E and F, On day 6 post-subcutaneous implantation of MC38 cells, mice (n = 5) were given intraperitoneal injection of UTX103 anti-DC-HIL mAb (αDCH), anti-PDL1 mAb (αPDL), or control IgG (Ctrl) every 2 days for a total of 6 injections. Tumor volume was measured every 2 days (E), and percentage of CD11b+Gr1+ MDSCs in PBMCs of blood on indicated days was determined (F). *, P < 0.01 and †, P < 0.01 compared with Ctrl and aPDL, respectively.
Antitumor activity of anti-DC-HIL mAb is due mostly to blocking MDSC function
To address MDSC targeting of anti-DC-HIL mAb, we analyzed immunologic changes in the tumor microenvironment (TME) and DLN. With respect to TME, anti-DC-HIL mAb reduced total MDSCs, including M1 and M2 (but not M3) subsets (Fig. 6A). By contrast, anti-PDL1 mAb produced just a marginal decline in MDSCs. Anti-DC-HIL markedly increased total CD8 T cells, whereas anti-PDL1 had a small effect (Fig. 6B). To evaluate the balance of negative and positive signals in T-cell immunity, we examined ratios of CD4 or CD8 T cells versus MDSCs or Tregs (Fig. 6C-E; Supplementary Fig. S7). Anti-DC-HIL mAb treatment resulted in an increase in CD8 (or CD4) T cells relative to MDSCs within the TME, without changing Tregs. By contrast, anti-PDL1 mAb significantly reduced Tregs (P = 0.048), while also increasing the ratio of CD8 (or CD4) relative to Tregs. Anti-DC-HIL treatment also amplified the IFNγ response within DLN; increased IFNγ amounts and number of IFNγ-secreting CD8 T cells. By contrast, anti-PDL1 had weak effects (Fig. 6F and G). Finally, we tested influence of anti-DC-HIL to the suppressor activity of MDSCs isolated from treated mice (Fig. 6H). MDSCs from control IgG-treated mice were highly potent suppressors, inhibiting up to 70% of the IFNγ response at cell ratio of 1:0.25 (T cell:MDSC). Compared with control IgG, MDSCs from mice treated with anti-DC-HIL were less potent. MDSCs from anti-PDL1-treated mice were slightly weaker suppressors than IgG-treated MDSCs, but at a considerably higher activity than those from anti-DC-HIL-treated mice. Thus, anti-DC-HIL treatment decreased levels and suppressor ability of MDSCs, while enhancing IFN-γ response of CD8 T cells, but with no impact on Tregs. This beneficial effect was not due to blocking the function of tumor-DC-HIL. These preclinical studies support the potential usefulness of functionally blockading DC-HIL on MDSCs in the treatment of metastatic cancers, particularly of the colorectal type.
Figure 6.
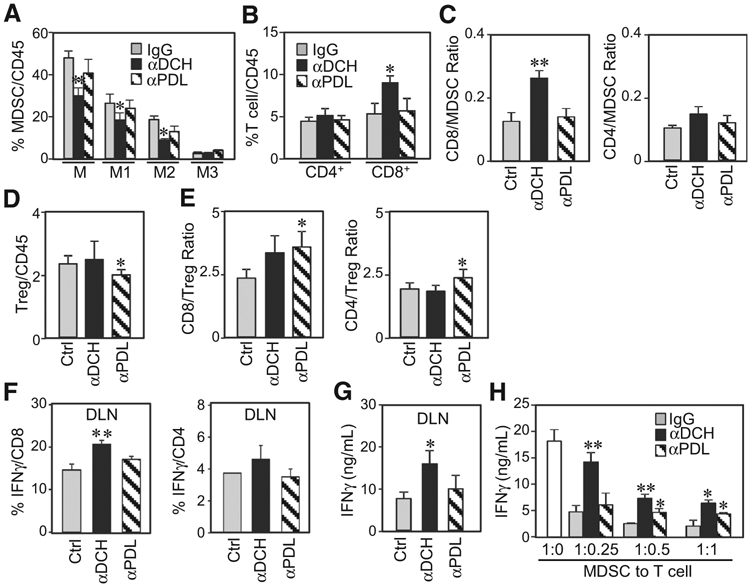
Anti-DC-HIL treatment decreased MDSCs, while increasing CD8 T cells in microenvironments of tumors and draining lymph nodes. CD45+ cells isolated from tumors (A–E) or draining lymph nodes (F and G) of mice treated with control IgG (Ctrl), anti-DC-HIL (αDCH), or anti-PDL1 (αPDL) mAb were determined by flow cytometry for percentage of MDSCs (A), CD4 and CD8 (B), their IFNγ-secreting T cells (F), or Tregs (D). Ratios of CD4 or CD8 T cells to MDSCs (C) or to Tregs (E) were calculated. IFNγ amounts in the DLN were measured (G). H, MDSCs purified from tumors of mice treated with mAb were assayed for their suppressor activity by coculturing with T cells from tumor-free mice in the presence of anti-CD3/CD28 Ab. IFNγ response was determined. *, P < 0.05 and **, P < 0.01 compared with Ctrl.
Discussion
We showed the prevalence of expanded DC-HIL+ MDSCs in the blood of cancer patients (n = 198) with metastatic forms of eight most common solid cancers. This blood index is the more reliable correlate of cancer progression than expansion of the entire monocytic MDSCs (percentage of MDSC/PBMC) used by other investigators (34, 35). Our expression studies using DC-HIL+ MDSC as an index showed significant difference between metastatic cancer patients and healthy controls 10- to 1,000-fold greater than values for percentage of MDSC/PBMC, and particularly in cases of breast, colorectal, kidney, melanoma, and prostate cancers. Given the high correlation between DC-HIL-positivity and the suppressor capacity for colorectal cancer and pancreatic cancer, DC-HIL expression distinguishes immunosuppressive cells from the functionally heterogeneous population of MDSCs including cells that are weakly immunosuppressive or not at all. In sum, the expanded DC-HIL+ MDSC subset may reflect the immunosuppressive milieu in cancer patients.
Our ultimate goal was to evaluate the efficacy of DC-HIL blockade in reversing the T-cell suppressor function of MDSCs expanded in cancer patients. As cited for patients with CRC, DC-HIL is highly expressed (median of 35%, n = 5) by monocytic MDSCs and poorly expressed (median of 10%, n = 5) by granulocytic MDSCs. By contrast, PDL1 is expressed poorly by both of these MDSC subsets (<5% by either; Supplementary Fig. S8). We were able to recapitulate the suppressor function of monocytic MDSCs (but not granulocytic MDSCs) in ex vivo cultures, similar to what was shown in a previous report (36). Using similar ex vivo cultures, we were able to determine the suppressor function of mouse granulocytic MDSCs (37). Given these findings, we decided to focus on monocytic MDSCs. In this regard, we used the PBMC fraction of peripheral blood for FACS analysis, in which most granulocytic MDSCs were removed. It should be noted that granulocytic MDSCs have been reported to contribute importantly to cancer-induced immunosuppression, in particular for prostate cancer (38).
Given that GPNMB/DC-HIL is also expressed by lung and breast cancer cells (39-41), the specific mAb has been using as a vehicle to deliver the cytotoxin to these tumor cells, called Glembatumumab vedotin (CDX-011), which consists of a fully humanized CR011 mAb conjugated to a highly potent antimitotic agent, monomethyl auristatin E (MMAE; ref. 42). Clinical trials of this agent for treating metastatic breast cancer reported that the overall response rate was 13% and median of progression-free survival was 9.1 weeks (43-45); and clinical study for treating metastatic melanoma reported objective response rate of 25% to 35% (46). However, this antibody–drug conjugate therapy may not be able to deplete MDSCs because these cells appear insensitive to MMAE, which is toxic to mitotic cells (47). We showed that the drug-unconjugated forms of 3D5 anti-DC-HIL/GPNMB mAb neutralizes the detrimental effects of MDSCs in many malignancies. Moreover, our preclinical animal studies revealed that the therapeutic effects of anti-DC-HIL mAb on MC38 and CT26 colon cancers cannot be ascribed to tumor expression of DC-HIL. Our findings thus provide valuable information for employing anti-DC-HIL/GPNMB mAb as an immune checkpoint blocker, rather than tumor-targeting therapy.
Although the majority of cancer patients displayed a significant in vitro response to anti-DC-HIL treatment of MDSCs, percentage of restored IFNγ response showed a poor correlation with percentage of DC-HIL-positivity among MDSCs. These data imply diversity in the T-cell-inhibitory mechanisms of MDSCs (26). In this regard, MDSCs utilize the two major pathways to inhibit T-cell function; first, direct cell–cell contact that causes receptor–ligand interactions leading to activation of T-cell-inhibitory mechanisms; and second, preexisting high-production of soluble inhibitory factors (e.g., NO, ROS, and urea) by MDSCs (48). This pathway does not require the cell–cell contact. We thus speculate that the suppressor function of MDSCs producing high levels of soluble inhibitory factors may be less dependent on the DC-HIL pathway.
Using anti-PDL1 mAb, the current industry standard for immune checkpoint blockers, as a comparative control, we were able to gauge an equivalent potential for anti-DC-HIL mAb as cancer immunotherapy. These mAbs seem to exert their beneficial effects via different mechanisms; anti-DC-HIL mAb targets DC-HIL+ MDSCs, whereas anti-PDL1 mAb targets PDL1+ tumor cells and PDL1+ Tregs (49, 50). Unlike the case for DC-HIL as cited above, we were surprised poor PDL1 expression by MDSCs. And yet, anti-PDL1 mAb was able to reverse MDSC function and restore T-cell function to similar or higher than anti-DC-HIL mAb. This apparent discrepancy was accounted for by upregulated PDL1 expression caused by secretion of IFNγ from T cells in our cultures. These data suggest the possibility that T-cell-inhibitory mechanisms may switch from DC-HIL to the PDL1 pathway in cases associated with an abundance of IFNγ within the tumor microenvironment.
Although evaluation of the actual efficacy of anti-DC-HIL/GPNMB mAb for cancer treatment remains to be executed, our data from in vitro studies of MDSCs and animal studies provide strong support for its promise as a new immune checkpoint blocker that may find utility as monotherapy or in combination with anti-PDL1 therapy or other modalities. Finally, one advantage of anti-DC-HIL therapy over other treatments might be that blood DC-HIL+ MDSC level may serve as a useful and easily accessible index for identifying the most responsive patients.
Supplementary Material
Translational Relevance.
Myeloid-derived suppressor cells (MDSC) are highly potent suppressors of T-cell function, and their exponential expansion in patients with metastatic cancer correlates with suboptimal efficacy of various anticancer treatments, including immune checkpoint therapy. Therefore, depleting MDSCs or blocking their suppressor function is a logical modality to investigate for optimizing cancer immunotherapy. We report that the most common metastatic malignancies are associated with significantly elevated blood levels of DC-HIL+ MDSCs (P < 0.005); these MDSCs potently suppress the function of autologous T cells; anti-DC-HIL mAb restores T-cell integrity; and anti-DC-HIL treatment shifts the tumor microenvironment from the immunosuppressive to the inflammatory in preclinical studies. These in vitro data not only provide a rationale for developing DC-HIL blockade as an anticancer treatment but also suggest high levels of DC-HIL+ MDSCs to be a potential index for identifying the most responsive subjects to this anti-DC-HIL treatment.
Acknowledgments
The authors thank Irene Dougherty for technical support and acknowledge the assistance of The University of Texas Southwestern Medical Center Tissue Resource, a shared resource of the Harold C. Simmons Comprehensive Cancer Center, which is supported in part by the National Cancer Institute under award number 5P30 CA 142543. This study was supported by Bridging the Gap: Early Translational Research Awards of Cancer Prevention Research Institute of Texas (CPRIT DP150051).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
Y. Arriaga reports receiving speakers bureau honoraria from AstraZeneca. K. Courtney is a consultant/advisory board member for Janssen. No potential conflicts of interest were disclosed by the other authors.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci 2014;1319:47–65. [DOI] [PubMed] [Google Scholar]
- 3.Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res 2015;128:95–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017;543:728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 2017;52:71–81. [DOI] [PubMed] [Google Scholar]
- 6.Kitano S, Postow MA, Ziegler CG, Kuk D, Panageas KS, Cortez C, et al. Computational algorithm-driven evaluation of monocytic myeloid-derived suppressor cell frequency for prediction of clinical outcomes. Cancer Immunol Res 2014;2:812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tobin RP, Davis D, Jordan KR, McCarter MD. The clinical evidence for targeting human myeloid-derived suppressor cells in cancer patients. J Leukoc Biol 2017;102:381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wesolowski R, Duggan MC, Stiff A, Markowitz J, Trikha P, Levine KM, et al. Circulating myeloid-derived suppressor cells increase in patients undergoing neo-adjuvant chemotherapy for breast cancer. Cancer Immunol Immunother 2017;66:1437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiorentini C, Bodei S, Bedussi F, Fragni M, Bonini SA, Simeone C, et al. GPNMB/OA protein increases the invasiveness of human metastatic prostate cancer cell lines DU145 and PC3 through MMP-2 and MMP-9 activity. Exp Cell Res 2014;323:100–11. [DOI] [PubMed] [Google Scholar]
- 10.Maric G, Annis MG, Dong Z, Rose AA, Ng S, Perkins D, et al. GPNMB cooperates with neuropilin-1 to promote mammary tumor growth and engages integrin alpha5beta1 for efficient breast cancer metastasis. Oncogene 2015;34:5494–504. [DOI] [PubMed] [Google Scholar]
- 11.Taya M, Hammes SR. Glycoprotein non-metastatic melanoma protein B (GPNMB) and cancer: a novel potential therapeutic target. Steroids 2018; 133:102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung JS, Dougherty I, Cruz PD Jr, Ariizumi K. Syndecan-4 mediates the coinhibitory function of DC-HIL on T cell activation. J Immunol 2007; 179:5778–84. [DOI] [PubMed] [Google Scholar]
- 13.Chung JS, Sato K, Dougherty II, Cruz PD Jr, Ariizumi K. DC-HIL is a negative regulator of T lymphocyte activation. Blood 2007;109: 4320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutknecht M, Geiger J, Joas S, Dorfel D, Salih HR, Muller MR, et al. The transcription factor MITF is a critical regulator of GPNMB expression in dendritic cells. Cell Commun Signal 2015;13:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarzbich MA, Gutknecht M, Salih J, Salih HR, Brossart P, Rittig SM, et al. The immune inhibitory receptor osteoactivin is upregulated in monocyte-derived dendritic cells by BCR-ABL tyrosine kinase inhibitors. Cancer Immunol Immunother 2012;61:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shikano S, Bonkobara M, Zukas PK, Ariizumi K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem 2001;276:8125–34. [DOI] [PubMed] [Google Scholar]
- 17.Chung JS, Tamura K, Cruz PD Jr, Ariizumi K. DC-HIL-expressing myelomonocytic cells are critical promoters of melanoma growth. J Invest Dermatol 2014;134:2784–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turrentine J, Chung JS, Nezafati K, Tamura K, Harker-Murray A, Huth J, et al. DC-HIL+ CD14+ HLA-DRno/low cells are a potential blood marker and therapeutic target for melanoma. J Invest Dermatol 2014; 134:2839–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomihari M, Chung JS, Akiyoshi H, Cruz PD Jr, Ariizumi K. DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res 2010;70: 5778–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rose AA, Pepin F, Russo C, Abou Khalil JE, Hallett M, Siegel PM. Osteoactivin promotes breast cancer metastasis to bone. Mol Cancer Res 2007;5: 1001–14. [DOI] [PubMed] [Google Scholar]
- 21.Chung JS, Tamura K, Akiyoshi H, Cruz PD Jr, Ariizumi K. The DC-HIL/syndecan-4 pathway regulates autoimmune responses through myeloid-derived suppressor cells. J Immunol 2014;192:2576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sgambato A, Casaluce F, Sacco PC, Palazzolo G, Maione P, Rossi A, et al. Anti PD-1 and PDL-1 immunotherapy in the treatment of advanced non-small cell lung cancer (NSCLC): a review on toxicity profile and its management. Curr Drug Saf 2016;11:62–8. [DOI] [PubMed] [Google Scholar]
- 23.Tirapu I, Arina A, Mazzolini G, Duarte M, Alfaro C, Feijoo E, et al. Improving efficacy of interleukin-12-transfected dendritic cells injected into murine colon cancer with anti-CD137 monoclonal antibodies and alloantigens. Int J Cancer 2004;110:51–60. [DOI] [PubMed] [Google Scholar]
- 24.Chung JS, Bonkobara M, Tomihari M, Cruz PD Jr, Ariizumi K. The DC-HIL/syndecan-4 pathway inhibits human allogeneic T-cell responses. Eur J Immunol 2009;39:965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung JS, Yudate T, Tomihari M, Akiyoshi H, Cruz PD Jr, Ariizumi K. Binding of DC-HIL to dermatophytic fungi induces tyrosine phosphorylation and potentiates antigen presenting cell function. J Immunol 2009; 183:5190–8. [DOI] [PubMed] [Google Scholar]
- 26.Cao LY, Chung JS, Teshima T, Feigenbaum L, Cruz PD Jr, Jacobe HT, et al. Myeloid-derived suppressor cells in psoriasis are an expanded population exhibiting diverse T-cell-suppressor mechanisms. J Invest Dermatol 2016; 136:1801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Letsch A, Scheibenbogen C. Quantification and characterization of specific T-cells by antigen-specific cytokine production using ELISPOT assay or intracellular cytokine staining. Methods 2003;31:143–9. [DOI] [PubMed] [Google Scholar]
- 28.Kano A Tumor cell secretion of soluble factor(s) for specific immunosuppression. Sci Rep 2015;5:8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem 2001;47:624–30. [PubMed] [Google Scholar]
- 30.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 2004;64:1140–5. [DOI] [PubMed] [Google Scholar]
- 31.Shukuya T, Carbone DP. Predictive markers for the efficacy of anti-PD-1/PD-L1 antibodies in lung cancer. J Thorac Oncol 2016;11:976–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato Y, Shimizu K, Shinga J, Hidaka M, Kawano F, Kakimi K, et al. Characterization of the myeloid-derived suppressor cell subset regulated by NK cells in malignant lymphoma. Oncoimmunology 2015;4: e995541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214: 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother 2012;61:1373–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One 2013;8:e57114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res 2016;22:3924–36. [DOI] [PubMed] [Google Scholar]
- 37.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008;181: 5791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hossain DM, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin Cancer Res 2015;21:3771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose AA, Grosset AA, Dong Z, Russo C, Macdonald PA, Bertos NR, et al. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin Cancer Res 2010;16:2147–56. [DOI] [PubMed] [Google Scholar]
- 40.Kuan CT, Wakiya K, Dowell JM, Herndon JE 2nd, Reardon DA, Graner MW, et al. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin Cancer Res 2006;12(7 Pt 1):1970–82. [DOI] [PubMed] [Google Scholar]
- 41.Tse KF, Jeffers M, Pollack VA, McCabe DA, Shadish ML, Khramtsov NV, et al. CR011, a fully human monoclonal antibody-auristatin E conjugate, for the treatment of melanoma. Clin Cancer Res 2006;12:1373–82. [DOI] [PubMed] [Google Scholar]
- 42.Keir CH, Vahdat LT. The use of an antibody drug conjugate, glembatumumab vedotin (CDX-011), for the treatment of breast cancer. Expert Opin Biol Ther 2012;12:259–63. [DOI] [PubMed] [Google Scholar]
- 43.Yardley DA, Weaver R, Melisko ME, Saleh MN, Arena FP, Forero A, et al. EMERGE: a randomized phase II study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotein NMB-expressing breast cancer. J Clin Oncol 2015;33:1609–19. [DOI] [PubMed] [Google Scholar]
- 44.Vaklavas C, Forero A. Management of metastatic breast cancer with second-generation antibody-drug conjugates: focus on glembatumumab vedotin (CDX-011, CR011-vcMMAE). BioDrugs 2014;28:253–63. [DOI] [PubMed] [Google Scholar]
- 45.Bendell J, Saleh M, Rose AA, Siegel PM, Hart L, Sirpal S, et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with locally advanced or metastatic breast cancer. J Clin Oncol 2014;32:3619–25. [DOI] [PubMed] [Google Scholar]
- 46.Ott PA, Hamid O, Pavlick AC, Kluger H, Kim KB, Boasberg PD, et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with advanced melanoma. J Clin Oncol 2014;32: 3659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frey AB. Myeloid suppressor cells regulate the adaptive immune response to cancer. J Clin Invest 2006;116:2587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Yu Y, Yang S, Zeng B, Zhang Z, Jiao G, et al. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol Immunother 2009;58:687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009;206:3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.