

Abstract
The availability of new psychoactive substances on the recreational drug market continues to create challenges for scientists in the forensic, clinical and toxicology fields. Phenmetrazine (3-methyl-2-phenylmorpholine) and an array of its analogs form a class of psychostimulants that are well documented in the patent and scientific literature. The present study reports on two phenmetrazine analogs that have been encountered on the NPS drug market following the introduction of 3-fluorophenmetrazine (3-FPM), namely 4-methylphenmetrazine (4-MPM) and 3-methylphenmetrazine (3-MPM). This study describes the syntheses, analytical characterization and pharmacological evaluation of the positional isomers of MPM. Analytical characterizations employed various chromatographic, spectroscopic and mass spectrometric platforms. Pharmacological studies were conducted in order to assess whether MPM isomers might display stimulant-like effects similar to the parent compound phenmetrazine. The isomers were tested for their ability to inhibit uptake or stimulate release of tritiated substrates at dopamine, norepinephrine and serotonin transporters using in vitro transporter assays in rat brain synaptosomes. The analytical characterization of three vendor samples revealed the presence of 4-MPM in two of the samples and 3-MPM in the third sample, which agreed with the product label. The pharmacological findings suggest that 2-MPM and 3-MPM will exhibit stimulant properties similar to the parent compound phenmetrazine, whereas 4-MPM may display entactogen properties more similar to MDMA. The combination of test purchases, analytical characterization, targeted organic synthesis and pharmacological evaluation of NPS and their isomers is an effective approach for the provision of data on these substances as they emerge in the marketplace.
Keywords: New psychoactive substances, psychostimulants, phenmetrazine, fluorophenmetrazine, monoamine transporters
1. Introduction
Phenmetrazine (3-methyl-2-phenylmorpholine; Preludin®) (Figure 1A) is a synthetic amphetamine derivative that consists of a phenylisopropylamine skeleton with the terminal amine incorporated into a morpholine ring.1 Phenmetrazine hydrochloride, the active ingredient of Preludin, was introduced as an anorectic medication in the 1950s along with its N-methylated analog phendimetrazine.2 Rothman et al. showed that phenmetrazine is a potent substrate-type releaser at dopamine transporters (DAT) and norepinephrine transporters (NET), with less potent effects at serotonin transporters (SERT).3 Phendimetrazine (Bontril®) (Figure 1A) is classified as a pro-drug that exerts its pharmacological effects via N-demethylation to form phenmetrazine.1,4,5 Phenmetrazine is listed as a Schedule II drug under the United Nations Convention on Psychotropic Substances 1971,2,6 whereas phendimetrazine is listed in Schedule IV of the same Convention.6 Phenmetrazine was removed from clinical use due to concerns about abuse and dependence. Nevertheless, following its removal from the clinical market, phenmetrazine remained a popular stimulant (‘prellies’) on the recreational drug market.
Figure 1.

A. Chemical structures of phenmetrazine, its N-methyl derivative phendimetrazine and the methylphenmetrazine (MPM) positional isomers. B. The synthesis pathway employed for the preparation of 2-, 3- and 4-MPM isomers. C. Structural representation of all four MPM enantiomers.
Phenmetrazine and an array of its analogs form a class of compounds that are well documented in the patent and scientific literature.1 Emerging evidence suggests that phendimetrazine may be an efficacious candidate treatment for cocaine dependence.7–9 However, recent reports indicate that scientific information about phenmetrazine and its analogs is being utilized by manufacturers of new psychoactive substances (NPS), which are offered for uncontrolled sale.10,11 The manipulation of the phenmetrazine structure by substitution on the phenyl or morpholine rings creates a variety of suitable candidates for the NPS market. 2-(3-Fluorophenyl)-3-methylmorpholine (3-fluorophenmetrazine; 3-FPM; PAL-593), 2-(4-fluorophenyl)-3-methylmorpholine (4-fluorophenmetrazine; 4-FPM; PAL-748), 2-(3-methylphenyl)-3-methylmorpholone (3-methylphenmetrazine; 3-MPM; PAL-773) and 2-(4-methylphenyl)-3-methylmorpholine (4-methylphenmetrazine; 4-MPM; PAL 747) are phenmetrazine analogs that first appeared in a patent application by Blough et al. in 2011.1 All of the aforementioned compounds were synthesized and pharmacological evaluations revealed that the compounds were substrate-type monoamine releasers with high selectivity towards DAT and NET. Furthermore, addition of a methyl group to the phenyl ring generally increased potency at SERT.1
In 2014, 3-FPM was first notified by the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) Early Warning System (EWS).12 A recent study on fluorophenmetrazine provided the analytical profile of all three positional isomers (2-, 3- and 4-FPM) and identified 3-FPM in a number of samples obtained from Internet vendors.11 Pharmacological studies on the three fluorophenmetrazine isomers revealed that 3-FPM and its positional isomers are substrate-type releasing agents at monoamine transporters with marked potency at DAT and NET, which suggests that these compounds might display abuse and dependence liability.13 A published case report found an association between the intravenous use of 3-FPM and acute kidney injury and critical limb ischaemia.14 3-FPM has also been detected in biological samples obtained from a multiple-drug fatality.15 In addition, a 2016 study identified the main in vivo phase I and II metabolites of 3-FPM in human and rat urine, and reported on the microbial biotransformation products of 3-FPM.10 A recent study by Bäckberg et al. described the non-fatal intoxications of several patients involving 3-FPM.16 3-FPM is still available for purchase on vendor websites.
The present study reports on two phenmetrazine analogs that have been encountered on the NPS drug market following the introduction of 3-FPM, namely 4-methylphenmetrazine (4-MPM) and 3-methylphenmetrazine (3-MPM) (Figure 1A). 4-MPM was notified by the EMCDDA EWS in October 2015.17 Subsequently, 3-MPM was notified by the EMCDDA EWS in July 2016.18 However, information about the 2-methylphenmetrazine (2-MPM) positional isomer (Figure 1A) could not be identified, which made it necessary to investigate the organic synthesis of the ortho-, meta- and para-substituted isomers to facilitate their analytical differentiation. Various chromatographic, spectroscopic and mass spectrometric platforms were employed followed by structural investigations using X-ray crystal structure analysis. In order to assess whether these methylphenmetrazine isomers might display stimulant-like effects similar to the parent compound, the isomers were tested for their ability to inhibit uptake and induce release of tritiated substrates at DAT, NET and SERT using in vitro transporter assays in rat brain synaptosomes. In these experiments, the parent compound phenmetrazine was included as a standard reference compound with known pharmacology.
2. Experimental
2.1. Reagents and standards
All reagents and dry solvents used in the syntheses were obtained from Sigma Aldrich Ltd (Arklow, Co. Wicklow, Ireland). 2- and 3-Methylpropiophenone were obtained from Fluorochem (Derbyshire, United Kingdom). 4-Methylpropiophenone was obtained from Sigma Aldrich Ltd (Arklow, Co. Wicklow, Ireland). Preparative silica gel thin layer chromatography plates (UV254, GF 20 x 20 cm, 2000 microns) were obtained from Analtech (Newark, NJ, USA). LC-MS grade solvents were obtained from Fisher Scientific (Dublin, Ireland). Three samples, two advertised as 4-methylphenmetrazine and one as 3-methylphenmetrazine, were obtained from two different Internet vendors.
2.2. Synthesis
2.2.1. 3-Methyl-2-(4-methylphenyl)morpholine (4-methylphenmetrazine, 4-MPM, PAL-747)
A solution of bromine (27 mmol, 4.37 g, 1.4 mL) in dichloromethane (26 mL) was added slowly to a solution of 4-methylpropiophenone (27 mmol, 4.0 g) in dichloromethane (26 mL). The mixture was stirred for 1 h, dried (anhydrous magnesium sulphate (MgSO4)) and the solvent removed to afford 2-bromo-1-(4-methylphenyl)propan-1-one as a colorless oil (20 mmol, 4.5 g). A portion of the oil, 0.295 g (1.3 mmol), ethanolamine (1.38 mmol, 0.084 g) and N,N-diisopropylethylamine (5.12 mmol, 0.672 g, 905 μL) were added to 8 x 2 mL Supelco micro reaction vessels and heated at 120-125 °C for 30 min and then allowed to cool to room temperature. The mixture from each reaction vessel was pooled and partitioned between aqueous hydrochloric acid (2 M) (25 mL) and diethyl ether (25 mL). The aqueous layer was washed repeatedly with diethyl ether and each aqueous fraction was pooled and made basic with aqueous sodium hydroxide (10 M). The basic aqueous fraction was then extracted with dichloromethane, the organic extract collected, dried (MgSO4), filtered and the volatiles removed under vacuum to afford the intermediates 2-((2-hydroxyethyl)amino)-1-(4-methylphenyl-)propan-1-one and 3-methyl-2-(4-methylphenyl)morpholin-2-ol as a ruby oil (5.8 mmol, 1.2 g). This oil was dissolved in methanol (12.5 mL) and sodium borohydride (13 mmol, 0.5 g) was slowly added. The mixture was stirred at room temp for 2 h and partitioned between water and dichloromethane. The organic fraction was collected, dried (MgSO4), filtered and the volatiles removed under vacuum to afford 2-((2-hydroxyethyl)amino)-1-(4-methylphenyl)propan-1-ol as a ruby oil (6.4 mmol, 1.3 g). This oil was dissolved in dichloromethane and 10 mL concentrated sulfuric acid was added and the mixture stirred overnight. The reaction was quenched with 250 mL water and made alkaline (10 M NaOH) and extracted into dichloromethane. The organic fraction was collected, dried (MgSO4), filtered and the volatiles removed under vacuum to give crude 4-methylphenmetrazine as a ruby oil (0.72 mmol, 0.137 g, 3%). Purification was conducted using preparative thin layer chromatography (TLC) using methanol/dichloromethane (9:1) as the mobile phase. The purified band was isolated from the TLC plate, dissolved in ethanol and centrifuged. Following centrifugation, the supernatant was collected, filtered, and the volatiles removed yielding the purified 4-methylphenmetrazine (0.2 mmol, 0.035 g, 1%).
2.2.2. Preparation of 4-methylphenmetrazine fumarate salt
To the purified sample (0.2 mmol, 0.035 g), dissolved in 0.5 mL methanol, was added fumaric acid (0.2 mmol, 0.021 g) dissolved in 1 mL methanol. The solvent was removed and addition of tert.-butyl methyl ether (TBME) afforded a white solid powder (0.1 mmol, 0.032 g). m.p. 158–160 °C. 1H NMR (DMSO) δ ppm 7.26–7.20 (m; 2 H; H-2’, 6’), 7.20–7.14 (m; 2 H; H-3’, 5’), 6.55 (s; 2 H; CH; fumarate CH), 4.12 (d; J = 9.4 Hz; 1 H; H-2), 3.94 (dt; J = 11.8, 2.5 Hz; 1 H; one H from CH2; H-6), 3.77–3.66 (m; 1 H; CH; one H from CH2; H-6), 3.10–2.97 (m; 3 H; H-3, 5), 2.30 (s; 3H; CH3 on phenyl ring) and 0.82 (d; J = 6.5 Hz;3H; CH3 on morpholine). 13C NMR (DMSO) δ ppm 167.09 (C=O; fumarate), 137.42 (C; C4’), 135.84 (C; C-1), 134.60 (CH; fumarate), 128.77 (CH2; C-3’, C-5’), 127.40 (CH2; C-2’, C-6’), 82.72 (CH; C-2), 65.14 (CH2; C-6), 54.66 (CH; C-3), 44.00 (CH2; C-5), 20.73 (CH3) and 15.89 (CH3). APCI-HRMS Found m/z 192.138822 (theory [M+H]+: C12H18NO+ m/z 192.138291, Δ = 2.8 ppm).
2.2.3. 3-Methyl-2-(3-methylphenyl)morpholine (3-methylphenmetrazine, 3-MPM, PAL-773)
The reaction was carried out as described above using 3-methylpropiophenone instead, yielding crude 3-methylphenmetrazine free base as a ruby oil (1.6 mmol, 0.305 g, 6%). Purification was conducted using preparative thin layer chromatography (TLC) using methanol/dichloromethane (9:1) as the mobile phase. The purified band was isolated from the TLC plate, dissolved in ethanol and centrifuged. Following centrifugation, the supernatant was collected, filtered, and the volatiles removed yielding the purified 3-methylphenmetrazine (0.8 mmol, 0.146 g, 3%). The freebase was converted to the fumarate salt yielding a white solid (0.5 mmol, 0.146g). m.p. 188–190 °C. 1H NMR (DMSO) δ ppm 7.27–7.06 (m; 4 H; H-2’, 4’, 5’, 6’), 6.53 (s; 2 H;fumarate CH), 4.04 (d; J = 9.2 Hz; 1 H; H-2), 3.91 (ddd; J = 11.5, 3.4, 1.4 Hz;2 H; H-6), 3.66 (td; J = 11.4, 3.4 Hz; 1 H; H-3), 3.06–2.86 (m; 2 H; H-5), 2.31 (s; 3 H; CH3 on phenyl ring) and 0.79 (d; J = 6.5 Hz; 3 H; CH3 on morpholine). 13C NMR (DMSO) δ ppm 167.92 (C=O; fumarate), 139.70 (C; C-1), 137.73 (C; C-3’),135.32 (CH; fumarate), 129.64 (CH; C-4’), 128.48 (CH; C-6’), 128.27 (CH; C-2’), 125.16 (CH; C-5’), 84.14 (CH; C-2), 66.26 (CH2; C-6), 55.46 (CH; C-3), 45.05 (CH2; C-5), 21.45 (CH3) and 16.98 (CH3). APCI-HRMS Found m/z 192.138879 (theory [M+H]+: C12H18NO+ m/z 192.138291, Δ = −3.1 ppm).
2.2.4. 3-Methyl-2-(2-methylphenyl)morpholine (2-methylphenmetrazine, 2-MPM)
A solution of bromine (27 mmol, 4.37 g, 1.4 mL) in dichloromethane (26 mL) was added slowly to a solution of 2-methylpropiophenone (27 mmol, 4.0 g) in dichloromethane (26 mL). The mixture was stirred for 1 h, dried (MgSO4) and the solvent removed to afford 2-bromo-1-(2-methylphenyl)propan-1-one as a colorless oil (20 mmol, 4.5 g). A mixture of the 2-bromo-1-(2-methylphenyl)propan-1-one (4.4 mmol, 1.0 g) ethanolamine (16 mmol, 1.0 g) and N-methyl-2-pyrrolidone (0.052 mol, 5.14 g, 5 mL) was stirred at room temp. After 3 h, the reaction was quenched with water and made basic (10 M NaOH). The mixture was then extracted with ethyl acetate. The organic fraction was collected, dried (MgSO4), filtered and the volatiles removed under vacuum to afford the intermediates 2-((2-hydroxyethyl)amino)-1-(2-methylphenyl)propan-1-one and 3-methyl-2-(2-methylphenyl)morpholin-2-ol as a colorless oil (8 mmol, 1.70 g). The oil was dissolved in methanol (20 mL) and sodium borohydride (16 mmol, 0.6 g) was added. The mixture was stirred for 2 h at room temp. The mixture was partitioned between water and dichloromethane. The organic fraction was collected, dried (MgSO4), filtered and the volatiles removed were removed under vacuum to afford 2-((2-hydroxyethyl)amino)-1-(2-methylphenyl)propan-1-ol as a colorless oil (5.8 mmol, 1.201 g). A portion of this intermediate (1.4 mmol, 0.3 g) was dissolved in 4 mL dichloromethane and stirring commenced. To this, 8.5 mL of conc. sulfuric acid was slowly added followed by stirring for 4–5 h. The addition of 30 mL water followed and the mixture was made alkaline (10M NaOH) and extracted into dichloromethane. The organic fraction was collected, dried, filtered and the volatiles were removed under vacuum to give crude 2-methylphenmetrazine as a ruby oil (0.074 g, 0.4 mmol, 2%).
2.2.5. Preparation of 2-methylphenmetrazine fumarate salt
The sample was converted to the fumarate salt by dissolving 30 mg 2-MPM in 0.5 mL methanol followed by addition of fumaric acid (0.18 mmol, 0.020 g) dissolved in methanol (1 mL). The solvent was removed and the addition of TBME afforded a white solid powder (0.07 mmol, 0.020 g). m.p. 182–184 °C. 1H NMR (DMSO) δ ppm 7.37–3.32 (m; 1 H; H-5’), 7.24–7.11 (m; 3 H; H-3’, 4’, 6’), 6.52 (s; 2 H; fumarate CH), 4.44 (d; J = 9.4 Hz; 1 H; H-2), 3.91 (dd; J = 11.4, 3.1 Hz; 1 H; one H from CH2; H-6), 3.78–3.67 (m; 1 H; one H form CH2; H-6), 3.10–2.98 (m; 3 H; H-3 and H-5), 2.34 (s; 3 H; CH3 on phenyl ring) and 0.94–0.80 (m; 3H; CH3 on morpholine). 13C NMR (DMSO) δ ppm 167.58 (C=O; fumarate), 137.45 (C; C-2’), 135.55 (C; C-1), 134.89 (CH; fumarate), 130.04 (CH; C-3’, C-4’), 127.63 (CH; C-6’), 125.90 (CH; C-5’), 79.97 (CH; C-2), 65.79 (CH2; C-6), 55.12 (CH; C-3), 44.50 (CH2; C-5), 19.26 (CH3) and 15.84 (CH3). APCI-HRMS Found m/z 192.138787 (theory [M+H]+: C12H18NO+ m/z 192.138291, Δ = 2.6 ppm).
2.3. Instrumentation
2.3.1. Gas chromatography mass spectrometry
Samples were prepared to give a 1 mg/mL solution in methanol and analyzed on an Agilent 6890 N GC coupled to a 5975 Mass Selective Detector (Agilent, Little Island, Cork, Ireland). A HP-ULTRA 1 column (12 m x 0.2 m x 0.33 μm) was used with helium carrier gas at a constant flow of 0.8 mL/min and a spilt ratio of 1:1. The injector was set at 250 °C and the transfer line at 280 °C. The initial oven temperature was 50 °C, held for 2 min, then ramped at 10 °C/min to 100 °C with no hold time. The oven temperature was further ramped at 5 °C to 200 °C and then finally ramped at 20 °C to 295 °C with a hold time of 1 min. The mass spectra were collected after a 4.0 min solvent delay time. The ionization energy was set at 70 eV and the mass range was m/z 40–550. The total run time was 32.75 min.
2.3.2. Liquid chromatography mass spectrometry
LC-MS analyses were performed on an Agilent 1100 HPLC system equipped with a G13795 degasser, G1312A BinPump, a G1313A ALS and G1316A column oven (COLCOM) (Agilent, Little Island, Cork, Ireland). Separation was achieved using a Kinetex phenyl-hexyl column (2.6 μm, 100 x 2.10 mm) from Phenomenex (Macclesfield, Cheshire, United Kingdom). The analytes were eluted under isocratic conditions using a mobile phase of 95% water and 5% acetonitrile (both containing 0.1% formic acid). The Agilent LC-MSD settings were as follows: positive electrospray mode, capillary voltage 3500 V, drying gas (N2) 12 L/min at 350 °C, nebuliser gas (N2) pressure 50 psi, SIM m/z 192, fragmentor voltage 50 V and 150 V. Samples for LC-MS analysis were dissolved in acetonitrile/water (1:1, containing 0.1% formic acid) at a concentration of 10 μg/mL. The injection volume was 0.5 μL, flow rate was 0.2 mL/min and the column temperature was 30 °C. Total run time was 25 min.
2.3.3. High-resolution mass spectrometry
Atmospheric pressure chemical ionization (APCI) experiments were carried out on a Bruker micrOTOF-Q III mass spectrometer interfaced to a Dionex UltiMate 3000 LC. The Agilent tuning mix APCI-TOF was used for mass calibration. Masses were recorded over a range of m/z 100–1600. Operating conditions were as follows: capillary voltage 4000 V, corona 4000 nA, nebulizer gas 2.0 bar, drying gas flow rate 3.0 L/min, drying gas temperature 100–200 °C, vaporizer temperature 100–400 °C. MicroTof control and HyStar software were used for data analysis.
2.3.4. Nuclear magnetic resonance spectroscopy
All analytes were prepared in deuterated dimethyl sulfoxide (DMSO-d6) at a concentration of 20 mg/mL. 1H (600 MHz) and 13C (150 MHz) spectra were recorded on a Bruker AV600 NMR spectrometer using a 5 mm TCI cryoprobe. 1H NMR spectra were referenced to an external TMS reference at δ = 0 ppm.
2.3.5. Gas chromatography solid-state infrared analysis
Samples were analyzed using a GC-solid phase-IR-system that consisted of an Agilent GC 7890B (Waldbronn, Germany) with probe sampler Agilent G4567A and a DiscovIR-GC™ (Spectra Analysis, Marlborough, MA, USA). The column eluent was cryogenically accumulated on a spirally rotating ZnSe disk cooled by liquid nitrogen. IR spectra were recorded through the IR-transparent ZnSe disk using a nitrogen-cooled MCT detector. GC parameters: injection in splitless mode with an injection port temperature set at 240 °C and a DB-1 fused silica capillary column (30 m × 0.32 mm i.d., 0.25 μm film thickness). The carrier gas was helium with a flow rate of 2.5 mL/min and the oven temperature program was as follows: 80 °C for 2 min, ramped to 290 °C at 20 °C/min, and held at for 20 min. The transfer line was heated at 280°C. Infrared conditions: oven temperature, restrictor temperature, disc temperature, and Dewar cap temperatures were 280 °C, 280 °C, −40 °C, and 35 °C, respectively. The vacuum was 0.2 mTorr, disc speed 3 mm/s, spiral separation was 1 mm, wavelength resolution 4 cm−1 and IR range 650–4000 cm−1. Acquisition time was 0.6 s/file with 64 scans/spectrum. Data were processed using GRAMS/AI Ver. 9.1 (Grams Spectroscopy Software Suite, Thermo Fischer Scientific, Dreieich, Germany) followed by implementation of the OMNIC Software, Ver. 7.4.127 (Thermo Electron Corporation, Dreieich, Germany).
2.3.6. Infrared spectroscopy
IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR with Universal ATR sampling accessory (Perkin Elmer, Waltham, MA, USA). The wavelength resolution was set to 2 cm−1. IR spectra were collected in a range of 650–4000 cm−1 with 16 scans per spectrum. The IR data were processed using Spectrum Perkin Elmer Version 6.3.4 Software (Perkin Elmer, Waltham, MA, USA).
2.3.7. X-ray crystallography
Data for the 4-MPM vendor sample were collected on a Bruker APEX DUO using Cu Kα radiation (λ = 1.54184 Å). The sample was mounted on a Mitegen cryoloop and data collected at 100(2) K (Oxford Cobra cryosystem). Bruker APEX19 software was used to collect and reduce data, determine the space group. The data was solved with XT structure solution program20 and refined with the XL refinement package21 using Olex2.22 Absorption corrections were applied using SADABS 2014.23 All non-hydrogen atoms were refined anisotropically. NH2 hydrogen atoms were located and freely refined. All other hydrogen atoms were assigned to calculated positions using a riding model with appropriately fixed isotropic thermal parameters. Crystal data and structure refinement parameters were as follows:
Representative 4-MPM vendor sample: C12H18NOCl, M (g/mol) = 227.72, SG = P212121 (no. 19), a (Å) = 6.7094(3), b (Å) = 7.5835(4), c (Å) = 24.6346(11), V (Å3) = 1253.43(10), Z = 4, T (K) = 100, μ(CuKα) (mm−1) = 2.492, Reflections measured = 21970, Reflections unique = 2336, Rint = 0.0341, Rsigma = 0.0189, S = 1.041, R1* (I > 2σ(I)) = 0.0239, wR2† (all data) = 0.0613, Flack parameter = 0.388(15), * = R1 = Σ||Fo|-|Fc||/Σ|Fo|, † = wR2 = [Σ[w(Fo2−Fc2)/Σ[w(Fo2)2]0.5
2.4. Monoamine transporter assays
Male Sprague-Dawley rats (250–300 g, Charles River Laboratories, Wilmington, MA, USA) were housed 2 per cage and maintained on a 12 h light-dark cycle. Food and water were provided ad libitum. Animal use procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and the Animal Care and Use Committee of the Intramural Research Program of the National Institute on Drug Abuse (Baltimore, MD, USA). Rats were euthanized by CO2 narcosis and brains were processed to yield synaptosomes as previously described.24–26 Briefly,rat caudate (for DAT assays) or whole brain minus cerebellum and caudate(for NET and SERT assays) was homogenized in ice-cold 10% sucrose. After 12 strokes with a Potter-Elvehjem homogenizer, the homogenates were centrifuged at 1,000 g for 10 min at 4°C and the supernatants (i.e., synaptosomal preparations) were retained on ice.
For uptake assays, synaptosomes were incubated with different concentrations of the test drugs in the presence of 5 nM [3H]dopamine, 10 nM [3H]norepinephrine or 5 nM [3H]5-HT for DAT, NET or SERT assays, respectively. To optimize uptake for a single transporter, unlabeled blockers were included that prevented uptake of [3H]transmitters by competing transporters (Supporting Information). The uptake assays were terminated by vacuum filtration and retained radioactivity was quantified by scintillation counting. For release assays, 9 nM [3H]-1-methyl-4-phenylpyridinium ([3H]MPP+) was used as the radiolabelled substrate for DAT and NET, whereas 5 nM [3H]5-HT was used as the radiolabelled substrate for SERT. All buffers used in the release assay methods contained 1 μM reserpine to block vesicular uptake of substrates. The selectivity of release assays was optimized for a single transporter by including unlabeled blockers to prevent the uptake of [3H]MPP+ or [3H]5-HT by competing transporters (Supporting information), and the non-selective monoamine oxidase inhibitor pargyline (50 μM) was added to prevent enzymatic degradation of monoamines.
Synaptosomes were preloaded with radiolabelled substrates in Krebs-phosphate buffer for 1 h (steady state). Release assays were initiated by adding 850 μL of preloaded synaptosomes to 150 μL of test drug. The release assays were terminated by vacuum filtration and retained radioactivity was quantified by scintillation counting.
Effects of test drugs on uptake or release were expressed as % of control uptake or % of maximum release, respectively. Maximum release (Emax) was defined as the release produced by tyramine at doses that evoke the efflux of all ‘releasable’ tritium by synaptosomes (10 μM tyramine for DAT and NET assay conditions, and 100 μM tyramine for SERT assay conditions). Effects of test drugs on uptake inhibition and release were analyzed by nonlinear regression using GraphPad Prism 7 (GraphPad Scientific, San Diego, CA). Dose-response values for uptake inhibition and release were fit to the equation Y(x) = Ymin + (Ymax − Ymin) / (1 + 10exp[(logP50 − logx)] x n), where x is the concentration of the compound tested, Y(x) is the response measured, Ymax is the maximal response, P50 is either IC50 (the concentration that yields half-maximal uptake inhibition) or EC50 (the concentration that yields half-maximal release), and n is the Hill Slope parameter.
3. Results and discussion
Three samples, two advertised as 4-MPM and one as 3-MPM, were obtained from two different Internet retailers and subjected to extensive characterization using a variety of analytical platforms. One of the vendor samples of 4-MPM consisted of crystals whereas the other was a powder. The 3-MPM vendor sample was in powder form. The identity of the vendor samples was found to be consistent with the information given on the product labels. In addition, the identity of each vendor sample was verified by targeted organic synthesis of each of the methylphenmetrazine positional isomers including the 2-MPM isomer. The presence of positional isomers remains a challenge for forensic scientists tasked with the unique identification of a specific NPS. In an attempt to overcome this challenge, it remains important to provide analytical data on all possible positional isomers so that the correct isomer can be identified and reported. Analytical details of both 3-MPM and 4-MPM have been reported by the European project Response.27,28 However, extensive analytical data on 4-MPM and its positional isomers appears to be lacking in the scientific literature.
The synthesis schemes employed for the preparation of 3-MPM and 4-MPM were adapted from McLaughlin et al. (Figure 1B). The synthesis involved the bromination of the methylphenmetrazine starting material, e.g. 4-methylpropiophenone (a), yielding 2-bromo-1-(4-methylphenyl)propan-1-one (b). This species was reacted with ethanolamine and N,N-diisopropylethylamine to give the intermediates 2-((2-hydroxyethyl)amino)-1-(4-methylphenyl-)propan-1-one and 3-methyl-2-(4-methylphenyl)morpholin-2-ol (c). Reduction to the alcohol (d) was achieved by reaction with sodium borohydride followed by reaction with concentrated sulfuric acid to aid cyclization and formation of the morpholine ring (e). This synthesis protocol was also followed for 2-MPM, however, sufficient yields for extensive characterization could not be achieved. Therefore, an alternative synthesis protocol utilizing N-methyl-2-pyrrolidone, instead of N,N-diisopropylethylamine, was developed which afforded the required 2-MPM product.29 Similar to synthesis of the fluorophenmetrazine positional isomers previously reported, it was found most practical to generate the fumarate salts to induce crystallization.11 Each methylphenmetrazine isomer contains two chiral centers, which yield the potential for four stereoisomers and two racemic mixtures (i.e., cis- and trans-racemates) (Figure 1C). As expected, the preparation of the methylphenmetrazine isomers were consistent with the prior synthesis of phenmetrazine where the formation of the more stable trans-isomer was reported.11,30,31 X-ray crystallography analysis of the 4-MPM vendor sample confirmed that the sample was consistent with the trans-form and that it was the hydrochloride salt. In addition, the chromatographic and NMR spectroscopy investigations confirmed that the vendor products were of high purity.
3.1. Analytical features
3.1.1. Gas chromatography mass spectrometry
Initial analysis of the underivatized isomers by GC-MS failed to result in separation between the 2- and 3-MPM isomers, however, separation from the 4-MPM isomer was achieved (Figure 2A). The electron ionization (EI) mass spectral data recorded for each isomer were identical (Figure 2C). A proposed fragmentation pathway for the underivatized methylphenmetrazine isomers under EI-MS conditions is shown in Figure 3A and, as expected, follows the same mass spectral pattern previously reported for fluorophenmetrazine positional isomers.11 In the EI-MS of each underivatized isomer, the molecular ion was detected at m/z 191. The loss of a diethylamine entity from the molecular ion leads to the formation of an oxonium ion at m/z 119 (C8H7O+). A further loss of CO from the oxonium species leads to the formation of the tropylium ion at m/z 91. Furthermore, the fragment observed at m/z 71 may be represented by the loss of 3-methylbenzaldehyde from the parent structure leading to the formation of a radical cation ion represented by C4H9+. A loss of a methyl group from this entity is thought to lead to the formation of a 2,3-dihydroazet-1-ium (C3H6N+) species at m/z 56. The fragment observed at m/z 42 may reflect the loss of an oxetane species from the parent molecule that caused the formation of 2H-azirin-1-ium species (C2H4N+) at m/z 42.
Figure 2.

Gas chromatographic (GC) separation achieved for the underivatized MPM isomers (A) and the TFAA-MPM derivatives (B). The electron ionization (EI) mass spectra recorded for underivatized 4-MPM and the TFAA-MPM derivatives are shown in Figure 2C and 2D, respectively.
Figure 3.

A. A proposed fragmentation pathway for underivatized MPM under EI-MS conditions. B. A proposed fragmentation pattern for the MPM-TFAA isomers under EI-MS conditions.
In an attempt to improve chromatographic separation, derivatization of the samples with trifluoroacetic anhydride (TFAA) was employed and dramatically improved the separation between all three isomers, with retention times of 15.85, 16.05 and 16.34 min recorded for 3-, 2- and 4-MPM isomers, respectively (Figure 2B). The EI-MS data for the methylphenmetrazine-TFAA isomers were identical (Figure 2D) and some of the fragments were consistent with those reported in previous studies on amphetamine/phenmetrazine-based compounds that have been derivatized with TFAA.11,32 The proposed fragmentation pattern for the methylphenmetrazine-TFAA isomers is outlined in Figure 3B. In the EI mass spectra of each methylphenmetrazine-TFAA isomer, the molecular ion was detected at m/z 287. The fragment observed at m/z 218 might have been the result of radical loss of CF3 via cleavage of the nitrogen in the morpholine ring. A further loss of carbon monoxide would be consistent with m/z 190. Two dominant fragments were observed at m/z 167 and m/z 70. The m/z 167 indicated a potential loss of the oxonium species, which is suggested to give rise to a TFAA-azetidine species (C6H8F3NO+). The base peak at m/z 70 may be accounted for by the loss of the ring substituted methylbenzyl alcohol, which is proposed to form an azete species (2-methyl-2,3-dihydroazete, C4H8N+). In addition, the loss of a TFAA-diethylamine entity from the molecular ion leads to the formation of an oxonium ion at m/z 119 (C8H7O+). The fragment at m/z 54 may reflect the formation of an aziridine species (C3H6N+). The GC-EI-MS data recorded for the vendor samples of 3- and 4-MPM (underivatized/derivatized) were consistent with the data of the respective synthesized reference standard (Supporting Information).
3.1.2. Liquid chromatography mass spectrometry
Satisfactory separation of all three methylphenmetrazine isomers was achieved with retention times of 13.06 min, 16.70 min and 17.33 min for 2-MPM, 3-MPM and 4-MPM, respectively (Figure 4A). The 2-MPM isomer was completely separated from the other two positional isomers, however, only partial separation was achieved for the 3- and 4-MPM isomers. Electrospray ionization (ESI) single quadrupole mass spectra were obtained from in-source collision induced dissociation (CID) at increased fragmentor voltage (150 V) (Figure 4B) and the suggested dissociation pathways are shown in Figure 4C. The fragmentation pattern of the MPM isomers were consistent with the fragmentation pattern also recorded for the FPM isomers.11 The protonated molecule [M+H]+ was present in all spectra at m/z 192. The formation of m/z 174 might have represented a loss of methanol from the protonated molecule, presumably consistent with C12H16N+. The m/z 148 ion might have reflected a loss of ethylene oxide from the protonated molecule to form an aziridine species. The product ion at m/z 131 may be represented by the formation a stabilized allylic cation and might have formed following the loss of ethenamine (C2H5N) from m/z 174 and/or the loss of NH3 from the aziridine species at m/z 148. The fragments at m/z 105 and 91 may be represented by the formation of a methylium and tropylium species, respectively. The LC-ESI-MS data recorded for the vendor samples of 3- and 4-MPM were consistent with the data recorded for the respective synthesized reference standard (Supporting Information).
Figure 4.

A. HPLC separation achieved for all three methylphenmetrazine isomers. B. The product ion spectra obtained from in-source collision-induced dissociation (CID) at increased fragmentor voltage (150 V). C. A suggested dissociation pathway for the 4-MPM isomer.
3.1.3. X-ray crystal analysis
4-MPM (vendor sample) crystallizes in the orthorhombic space group P212121. Crystallographic and refinement data are listed in the experimental section and a full numbering scheme figure is shown in the Supporting Information section. The two chiral centres C5 and C6 are shown as R (Figure 5), although the Flack parameter indicates that the model is not enantiopure. The solid state structure of 4-MPM is very similar to that of the parent phenmetrazine hydrochloride (PMHCl).33 An overlay of the morpholine ring shows only minor differences in hydrogen atom placement with a difference in the twist of the phenyl to morpholine rings (plane twist in: 1, 72°; PMHCl ca. 77° and ca. 94° in phendimetrazine bitartrate34 (Supporting Information). In solution, the structure has also been elucidated by NMR, which indicates that H5 and H6 are also trans to each other (J = 9.9 Hz, full assignment given in Supporting Information) also giving the threo conformation seen in the solid state. In 4-MPM, as in PMHCl, there are significant intermolecular N…Cl hydrogen bonds with d(N…Cl) = 3.1243(16) and 3.0768(16)i Å with angles of 159(2) and 173(2) ° respectively (symmetry codes used to generate equivalent atoms: (i) x+1/2,−y+3/2,−z+2). The crystal packing in 4-MPM is quite different to that in PMHCl. The 4-MPM molecules interdigitate and pack head to tail in layers ca. 15 Å wide parallel to the c-axis, with the Cl atoms in a staggered arrangement (Supporting Information). There are further weaker C…Cl (3.6–3.74 Å) and C…O (C10…O4 3.512(2) Å) intermolecular interactions which are also seen in an analysis of the Hirshfeld surface (Supporting Information).
Figure 5.
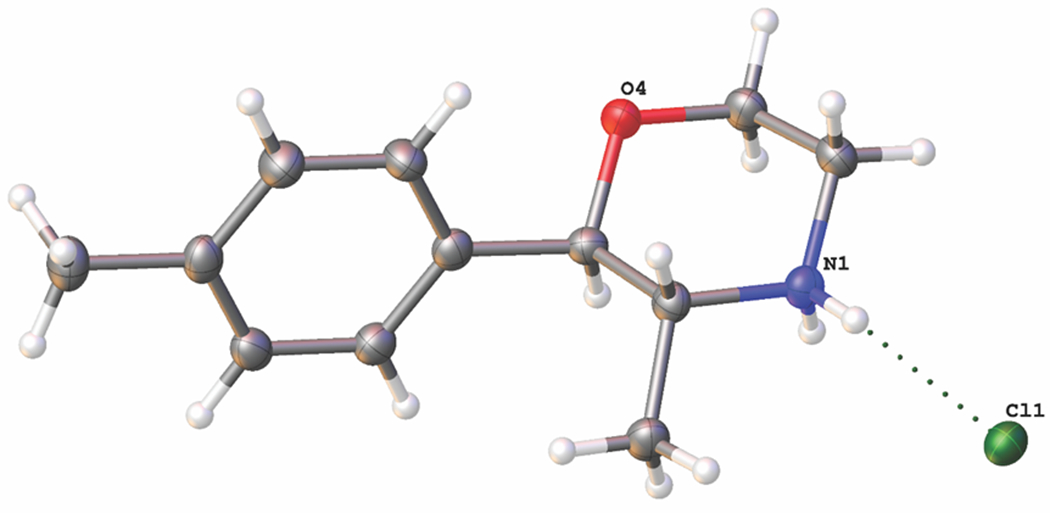
Molecular structure achieved for 4-MPM vendor sample using x-ray crystal structure analysis.
3.2. Pharmacology
As a first step in characterizing the monoamine transporter activity of the compounds, we tested the effects of phenmetrazine, 2-MPM, 3-MPM and 4-MPM in uptake inhibition assays. The dose-response curves are depicted in Figure 6 and corresponding IC50 values are given in Table 1. Phenmetrazine, 2-MPM and 4-MPM were efficacious uptake blockers at DAT, NET and SERT with IC50 values in the low μM range, whereas 3-MPM was substantially weaker at DAT and SERT (Table 1). 4-MPM was about 3.5-fold more potent than 2-MPM at DAT, with IC50 values of 1.93 and 6.74 μM, respectively. In addition, 4-MPM was nearly 10-fold more potent than 2-MPM and phenmetrazine at SERT. All compounds were fully efficacious uptake blockers at NET with potencies ranging from 1.2 to 5.2 μM for phenmetrazine and 3-MPM. The results from the uptake inhibition experiments demonstrated that phenmetrazine analogs interact with monoamine transporters, but uptake assays cannot differentiate between ligands acting as non-transportable uptake blockers versus those acting as transportable substrates. Thus, we next tested the compounds in transporter release assays which can detect the activity of substrate-type releasing agents.
Figure 6.

Dose-response effects of phenmetrazine (PM), 2-MPM, 3-MPM and 4-MPM on inhibition of [3H]neurotransmitter uptake at DAT, NET and SERT in rat brain synaptosomes. For uptake assays, 5 nM [3H]dopamine, 10 nM [3H]norepinephrine or 5 nM [3H]5-HT were incubated with different concentrations of test drugs under assay conditions optimized for DAT, NET or SERT, respectively. Data are expressed as % of control uptake (mean ± SD) for n = 3 experiments performed in triplicate.
Table 1.
IC50 potency values for phenmetrazine and its methyl ring-substituted analogs to inhibit [3H]neurotransmitter uptake at DAT, NET and SERT in rat brain synaptosomes
| Drug | [3H]Dopamine uptake at DAT IC50 (nM) |
[3H]Norepinephrine uptake at NET IC50 (nM) |
[3H]5-HT uptake at SERT IC50 (nM) |
DAT/SERT ratio |
|---|---|---|---|---|
| Phenmetrazine | 933 ± 57 | 1163 ± 197 | 3575 ± 836 | 3.8 |
| 2-MPM | 6586 ± 1311 | 2595 ± 633 | 4011 ± 317 | 0.6 |
| 3-MPM | >10,000 | 5210 ± 1650 | >10,000 | n.d. |
| 4-MPM | 1926 ± 207 | 1933 ± 392 | 408 ± 17 | 0.2 |
Data are mean ± SD for N=3 experiments performed in triplicate.
DAT/SERT ratio = (DAT IC50−1)/(SERT IC50−1); higher value indicates greater DAT selectivity.
Figure 7 shows the effects of 2-MPM, 3-MPM and 4-MPM on release of [3H]MPP+ via DAT and NET, and [3H]serotonin via SERT. The corresponding EC50 values are presented in Table 2. Consistent with prior findings, we found that phenmetrazine is a potent releaser at DAT and NET, with negligible activity at SERT. The data obtained for the 2-, 3- and 4-MPM isomers exhibited sigmoidal dose-response curves indicative of fully efficacious release activity across all three transporters (Figure 7; Table 2). Importantly, the addition of a methyl moiety to the phenmetrazine molecular structure increased the relative potency at SERT. Moving the methyl group from the ortho- or meta- position to the para- position leads to a substantial increase in the selectively for SERT, so much so that 4-MPM is essentially a non-selective monoamine releaser, with similar potency at DAT (227 nM), NET (62 nM) and SERT (86 nM). In this regard, the release data are generally consistent with the uptake inhibition data, which show greater relative potency toward SERT for 4-MPM when compared to 2- and 3-MPM. The non-selective profile of monoamine release produced by 4-MPM is similar to the club drug 3,4-methylenedioxymethamphetamine (MDMA) and the synthetic cathinone 3,4-methylenedioxymethcathinone (methylone).24 The unique psychoactive properties of MDMA reside in its more potent substrate activity at SERT when compared to more classic stimulants like amphetamine and cathinone35, which display potent substrate activity at DAT and NET but not at SERT. Previous studies on the pharmacological activity of 2-, 3- and 4-fluorophenmetrazine (FPM) found that addition of the fluorine atom to the phenyl ring increased activity at SERT, but none of the FPM isomers were able to affect SERT releasing activity to the same extent as 4-MPM.13 Overall, these findings are in agreement with previous studies, which demonstrate increased steric bulk of the phenyl ring substituent increases activity at SERT relative to DAT for amphetamine type compounds.36,37
Figure 7.

Dose-response effects of phenmetrazine (PM), 2-MPM, 3-MPM and 4-MPM on release of [3H]substrates by DAT, NET and SERT in rat brain synaptosomes. For release assays, synaptosomes were preloaded with 9 nM [3H]MPP+ for DAT and NET or 5 nM [3H]5-HT for SERT, then incubated with different concentrations of test drugs to evoke release via reverse transport. Data are expressed as % of maximal release (mean ± SD) for n = 3 experiments performed in triplicate.
Table 2.
EC50 potency values for phenmetrazine and its methyl ring-substituted analogs to release [3H]MPP+ at DAT or NET, or [3H]5-HT at SERT in rat brain synaptosomes
| Drug | Release at DAT EC50 (nM) [% Emax] | Release at NET EC50 (nM) [% Emax] | Release at SERT EC50 (nM) [% Emax] | DAT/SERT ratio |
|---|---|---|---|---|
| Phenmetrazine | 70 ± 9 [101] | 29 ± 4 [95] | >10,000 | >142.0 |
| 2-MPM | 374 ± 38 [104] | 102 ± 12 [91] | 1758 ± 336 [101] | 4.7 |
| 3-MPM | 1321 ± 218 [110] | 509 ± 73 [90] | 5383 ± 947 [111] | 4.1 |
| 4-MPM | 227 ± 30 [99] | 62 ± 9 [90] | 86 ± 12 [88] | 0.4 |
Data are mean ± SD for N=3 experiments performed in triplicate.
% Emax is defined as % of maximal releasing response induced by 10 μM tyramine for DAT and NET or 100 μM tyramine for SERT.
DAT/SERT ratio = (DAT EC50−1)/(SERT EC50−1); higher value indicates greater DAT selectivity.
4. Conclusion
The availability of NPS on the recreational drug market continues to create challenges for scientists in the forensic, clinical and toxicology fields. This report provides comprehensive analytical and pharmacological data on ortho-, meta- and para-substituted methylphenmetrazine isomers. The combination of test purchases, analytical characterization, targeted organic synthesis and pharmacological evaluation of NPS and their isomers is an effective approach for the provision of data on these substances as they emerge in the marketplace. The analytical characterization of three vendor samples revealed the presence of 4-MPM in two of the samples and 3-MPM in the third sample, which agreed with the product labels. The pharmacological findings suggest that 2-MPM and 3-MPM will exhibit stimulant properties similar to the parent compound phenmetrazine, whereas 4-MPM may display entactogen properties more similar to MDMA.
Supplementary Material
References
- 1.Blough BE, Rothman R, Landavazo A, Page KM, Decker AM. Preparation of phenylmorpholines and analogs thereof. Patent No.WO 2011/146850 A1 Research Triangle Institute, USA, 2011.
- 2.Barceloux DG. Medical Toxicology of Drug Abuse – Synthesized Chemicals and Psychoactive Plants. John Wiley & Sons, Hoboken, NJ, USA, 2012. [Google Scholar]
- 3.Rothman RB, Katsnelson M, Vu N, et al. Interaction of the anorectic medication, phendimetrazine, and its metabolites with monoamine transporters in rat brain. Eur J Pharmacol 2002;447(1):51–57. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph GR, Miksic JR, Levitt MJ. GLC determination of phedimetrazine in human plasma, serum, or urine. J. Pharm. Sci 1983;72:519–521. [DOI] [PubMed] [Google Scholar]
- 5.Beckett AH, Salami MA. A note on the identification of N-hydroxyphenmetrazine as a metabolic product of phendimetrazine and phenmetrazine. J. Pharm. Pharmacol 1972; 24: 900–902. [DOI] [PubMed] [Google Scholar]
- 6.United Nations. Convention on Psychotropic Substances, 1971. https://www.unodc.org/pdf/convention_1971_en.pdf. Accessed February 28, 2017.
- 7.Bauer CT, Negus SS, Blough BE, Banks ML. Cocaine-like discriminative stimulus effects of phendimetrazine and phenmetrazine in rats. Behavioural pharmacology 2016;27(2–3 Spec Issue):192–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolin BL, Stoops WW, Sites JP, Rush CR. Abuse potential of oral phendimetrazine in cocaine-dependent individuals: implications for agonist-like replacement therapy. Journal of addiction medicine 2016;10(3):156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czoty PW, Blough BE, Fennell TR, Snyder RW, Nader MA. Attenuation of cocaine self-administration by chronic oral phendimetrazine in rhesus monkeys. Neuroscience 2016;324:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mardal M, Miserez B, Bade R, et al. 3-Fluorophenmetrazine, a fluorinated analogue of phenmetrazine: Studies on in vivo metabolism in rat and human, in vitro metabolism in human CYP isoenzymes and microbial biotransformation in Pseudomonas Putida and wastewater using GC and LC coupled to (HR)-MS techniques. J Pharm Biomed Anal 2016;128:485–495. [DOI] [PubMed] [Google Scholar]
- 11.McLaughlin G, Morris N, Kavanagh PV, et al. Test purchase, synthesis and characterization of 3-fluorophenmetrazine (3-FPM) and differentiation from its ortho- and para-substituted isomers. Drug Test Anal 2016; 9:369–377. [DOI] [PubMed] [Google Scholar]
- 12.European Monitoring Centre for Drug and Drug Addiction and Europol. EMCDDA–Europol 2014 Annual Report on the implementation of Council Decision 2005/387/JHA. In accordance with Article 10 of Council Decision 2005/387/JHA on the information exchange, risk assessment and control of new psychoactive substances. 2015. http://www.emcdda.europa.eu/system/files/publications/1018/TDAN15001ENN.pdf. Accessed November 21, 2017.
- 13.Mayer FP, Burchardt NV, Decker AM, et al. Fluorinated phenmetrazine “legal highs” act as substrates for high-affinity monoamine transporters of the SLC6 family. Neuropharmacology 2017. doi: 10.1016/j.neuropharm.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fawzy M, Wong-Morrow WS, Beaumont A, Farmer CKT. Acute kidney injury and critical limb ischaemia associated with the use of the so called “legal high” 3-fluorophenmetrazine. CEN case reports. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellefsen KN, Taylor EA, Simmons P, Willoughby V, Hall BJ. Multiple Drug-Toxicity Involving Novel Psychoactive Substances, 3-Fluorophenmetrazine and U-47700. Journal of analytical toxicology 2017:1–6. [DOI] [PubMed] [Google Scholar]
- 16.Backberg M, Westerbergh J, Beck O, Helander A. Adverse events related to the new psychoactive substance 3-fluorophenmetrazine - results from the Swedish STRIDA project. Clinical toxicology (Philadelphia, Pa) 2016;54(9):819–825. [DOI] [PubMed] [Google Scholar]
- 17.European Monitoring Centre for Drug and Drug Addiction and Europol. EMCDDA–Europol 2015 Annual Report on the implementation of Council Decision 2005/387/JHA. In accordance with Article 10 of Council Decision 2005/387/JHA on the information exchange, risk assessment and control of new psychoactive substances. 2016. http://www.emcdda.europa.eu/system/files/publications/2880/TDAS16001ENN.pdf. Accessed November 21, 2017.
- 18.European Monitoring Centre for Drug and Drug Addiction and Europol. EMCDDA–Europol 2016 Annual Report on the implementation of Council Decision 2005/387/JHA. In accordance with Article 10 of Council Decision 2005/387/JHA on the information exchange, risk assessment and control of new psychoactive substances. 2017. http://www.emcdda.europa.eu/system/files/publications/4724/TDAN17001ENN_PDFWEB.pdf. Accessed November 21, 2017. [Google Scholar]
- 19.Bruker APEX v2012.12-0, Bruker AXS Inc., Madison, Wisconsin, USA. [Google Scholar]
- 20.Sheldrick GM. Acta Cryst 2015;A71:3–8. [Google Scholar]
- 21.Sheldrick GM. Acta Cryst 2008;A64:112–122. [Google Scholar]
- 22.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann HJ. OLEX2: a complete structure solution, refinement and analysis program. Appl. Cryst 2009;42: 339–341. [Google Scholar]
- 23.SADABS. 2014. Bruker AXS Inc., Madison, Wisconsin, USA; Sheldrick, G. M. University of Göttingen, Germany. [Google Scholar]
- 24.Baumann MH, Ayestas MA Jr., Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 2012;37(5):1192–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothman RB, Vu N, Partilla JS, et al. In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J. Pharmacol. Exp. Ther 2003; 307:138–145. [DOI] [PubMed] [Google Scholar]
- 26.Solis E, Partilla JS, Sakloth F, et al. N-Alkylated Analogs of 4-Methylamphetamine (4-MA) Differentially Affect Monoamine Transporters and Abuse Liability. Neuropsychopharmacology 2017;42(10):1950–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.European Project Response. Analytical Report on 4-Methylphenmetrazine.https://www.policija.si/apps/nfl_response_web/seznam.php. Accessed November 22, 2017.
- 28.European Project Response. Analytical Report on 3-Methylphenmetrazine. https://www.policija.si/apps/nfl_response_web/seznam.php. November 22, 2017.
- 29.Cao G, Hu A, Xiao XR. Asymmetric synthesis, crystal structure, and antidepressant activity of 2-aryl-3-alkyl-5-methyl-2-morpholinol hydrochlorides. Can J Chem 2007;85:29–36. [Google Scholar]
- 30.Clarke FH. cis- and trans-3-methyl-2-phenylmorpholine. J. Org. Chem 1962;27(9):3251–3253. [Google Scholar]
- 31.Tscharner CJ, Warwick RI. Process for the conversion of cis-2-phenyl-3-methylmorpholine to trans-2-phenyl-3-methylmorpholine. Patent No. US 3282936A Geigy Chemical Corporation; New York, USA, 1966.
- 32.Kumazawa T, Hara K, Hasegawa C, et al. Fragmentation pathways of trifluoroacetyl derivatives of methamphetamine, amphetamine and methylenedioxyphenylalkylamine designer drugs by gas chromatography/mass spectrometry. Int. J. Spectrosc 2011; DOI: 10.1155/2011/318148. [DOI] [Google Scholar]
- 33.Carlstrom D, Hacksell I. The crystal structure of phenmetrazine hydrochloride. Acta Cryst 1974;30:2477–2480. [Google Scholar]
- 34.Glaser R, Adin I, Drouin M, Michel A. Solid-state structure of (+)-phenmetrazine bitartarate, an anorexic drug. Struct. Chem 1994; 5:197–203. [Google Scholar]
- 35.Bonano JS, Banks ML, Kolanos R, et al. Quantitative structure-activity relationship analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 2015;172(10):2433–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakloth F, Kolanos R, Mosier PD, et al. Steric parameters, molecular modeling and hydropathic interaction analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 2015;172(9):2210–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farré M, Abanades S, Roset PN, et al. Pharmacological interaction between 3,4-methylenedioxymethamphetamine (ecstasy) and paroxetine: pharmacological effects and pharmacokinetics. J Pharmacol Exp Ther 2007; 323(3):954–962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.