

Abstract
The inhibition of DNA binding of basic leucine zipper (B-ZIP) transcription factors is a clinically relevant molecular target. Our laboratory has previously reported two methods of inhibiting B-ZIP DNA binding in solution: 1) an arylstibonic acid compound that binds to the basic region, stabilizes the B-ZIP dimer, and prevents B-ZIP DNA binding and 2) dominant negative proteins, termed A-ZIPs, that heterodimerize with B-ZIP domains in a leucine zipper-dependent manner. To determine if these two agents also inhibit DNA binding in live cells, GFP-tagged B-ZIP domains and mCherry-tagged A-ZIP domains were transfected into NIH3T3 cells to assess protein localization and Fluorescence Recovery After nuclear Photobleaching (FRAP). FRAP, showed that all six GFP-B-ZIP domains examined recovered faster in the nucleus in the presence of drug that we interpret represents an inhibition of DNA binding. Faster recovery in the presence of the A-ZIP was leucine zipper dependent. The arylstibonic also induced a cytoplasmic localization of all B-ZIP domains while the A-ZIPs induced a leucine zipper-dependent cytoplasmic localization. Thus, the change in cellular localization of B-ZIP domains could be used as a high-throughput assay for inhibitors of B-ZIP DNA binding. Additionally, the arylstibonic acid compound was cytostatic in clear cell sarcoma cells, which express a chimera between the B-ZIP domain of ATF-1 and N-terminal activation domain of EWS but not in K562 cells that express a non-B-ZIP containing chimeric protein BCR-ABL. These studies suggest that arylstibonic acid compounds or other small molecules capable of inhibiting B-ZIP DNA binding could be valuable anticancer agents.
Keywords: B-ZIP domain VBP, FRAP arylstibonic acid antimony, DNA binding
Introduction
There are approximately 55 genes in vertebrate genomes that contain the dimeric B-ZIP domain (Vinson et al., 2002). These proteins function as homodimers or heterodimers to bind to DNA in a sequence-specific manner and regulate gene transcription (Hurst, 1995). The basic region mediates sequence specific DNA binding and the leucine zipper mediates parallel coiled-coil dimer formation. Despite the high homology between amino acid sequences of B-ZIP domains, there is wide diversity in recognition of specific DNA sequences (Fujii et al., 2000). The B-ZIP family of transcription factors regulates a variety of genes involved in cell growth and differentiation, some of which are activated in human cancers. For example, cJun, cMyc, and ATF1 are oncogenic transcription factors (Nesbit et al., 1999; Vogt, 2001; Zucman et al., 1993). In addition, upstream oncogenic mutations in serine-threonine kinase cascades can phosphorylate downstream nuclear B-ZIP transcription factors resulting in their chronic activation which makes them potential drug targets even though they themselves are not mutated (Darnell, 2002).
Inhibition of B-ZIP DNA binding has been studied using dominant negatives, termed A-ZIPs, which contain an amphipathic α-helical sequence that replaces the B-ZIP basic region (Vinson et al., 2002). The acidic extension forms a coiled-coil structure with the basic region in the B-ZIP|A-ZIP heterodimer and stabilizes the complex (Vinson et al., 2002). Dominant negatives to B-ZIP domains have had physiological effects in transgenic mouse models. For example, A-C/EBP expression in the epidermis of adult mice prevents skin papilloma formation. If A-C/EBP is expressed after papilloma formation, the papillomas regress (Oh et al., 2007). In contrast, A-CREB expression prevents papilloma formation but does not cause papillomas to regress (Rozenberg et al., 2009). A-Fos expression in mammalian cells reduces Ha-ras-mediated cellular transformation (Olive et al., 1997) whereas A-Fos expression in mouse epidermis converts papillomas to benign sebaceous adenomas and prevents conversion into carcinomas (Gerdes et al., 2006). These results suggest that inhibiting B-ZIP DNA binding is a clinically relevant molecular target.
Using a high-throughput fluorescent anisotropy screen, we previously identified NSC13778, an antimony-containing com- pound that binds to the basic region of the B-ZIP domain and prevents DNA binding (Rishi et al., 2005). We have examined 14 derivatives of the lead compound NSC13778 and identified NSC13746, a water-soluble compound that is more potent and promiscuous than NSC13778 (Rishi et al., in preparation1). However, the activity in vivo of these compounds has not been assessed. To address this, we investigated the effects of the antimony compound NSC13746 on B-ZIP domain mobility and localization in vivo using fluorescence imaging technologies. For this study we have constructed fluorescent fusion proteins of B-ZIP and A-ZIP domains (ATF2, C/EBPα, C/EBPβ, CREB, cFos, cJun, VBP). Fluorescence Recovery After Photobleaching (FRAP) is a microscopy technique that measures the movement of fluorescently labeled proteins into a region of the cell where the fluorescently labeled molecules have been irreversibly photobleached (Axelrod et al. 1976). The recovery of nuclear transcription factors into photobleached regions is thought to reflect the affinity of the transcription factors for specific and non-specific DNA binding sites (Mueller et al. 2008; Phair et al. 2004). B-ZIP recoveries in NIH-3T3 cells were faster in a dose-dependent manner in the presence of either NSC13746 or A-ZIPs, suggesting that they inhibit B-ZIP|DNA binding in live cells.
Incubation of NSC13746 with GFP-B-ZIP transfected cells caused a cytoplasmic localization of all B-ZIP domains, while cells co-transfected with B-ZIP and A-ZIP domains had a leucine zipper-dependent cytoplasmic localization. NSC13746 was non-toxic in the cells we examined except for a clear cell sarcoma that expresses the chimeric protein EWS-ATF1 where ATF1 is a B-ZIP domain (Zucman et al. 1993).
Materials and methods
Compounds –
The arylstibonic acids NSC13746, NSC13778 and NSC13748 that contain the antimony element were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute, Bethesda, MD (Fig. 2A).
Fig. 2. NSC13746 or A-ZIP causes a dose-dependent increase in the recovery (mobility) of GFP-VBP by FRAP.
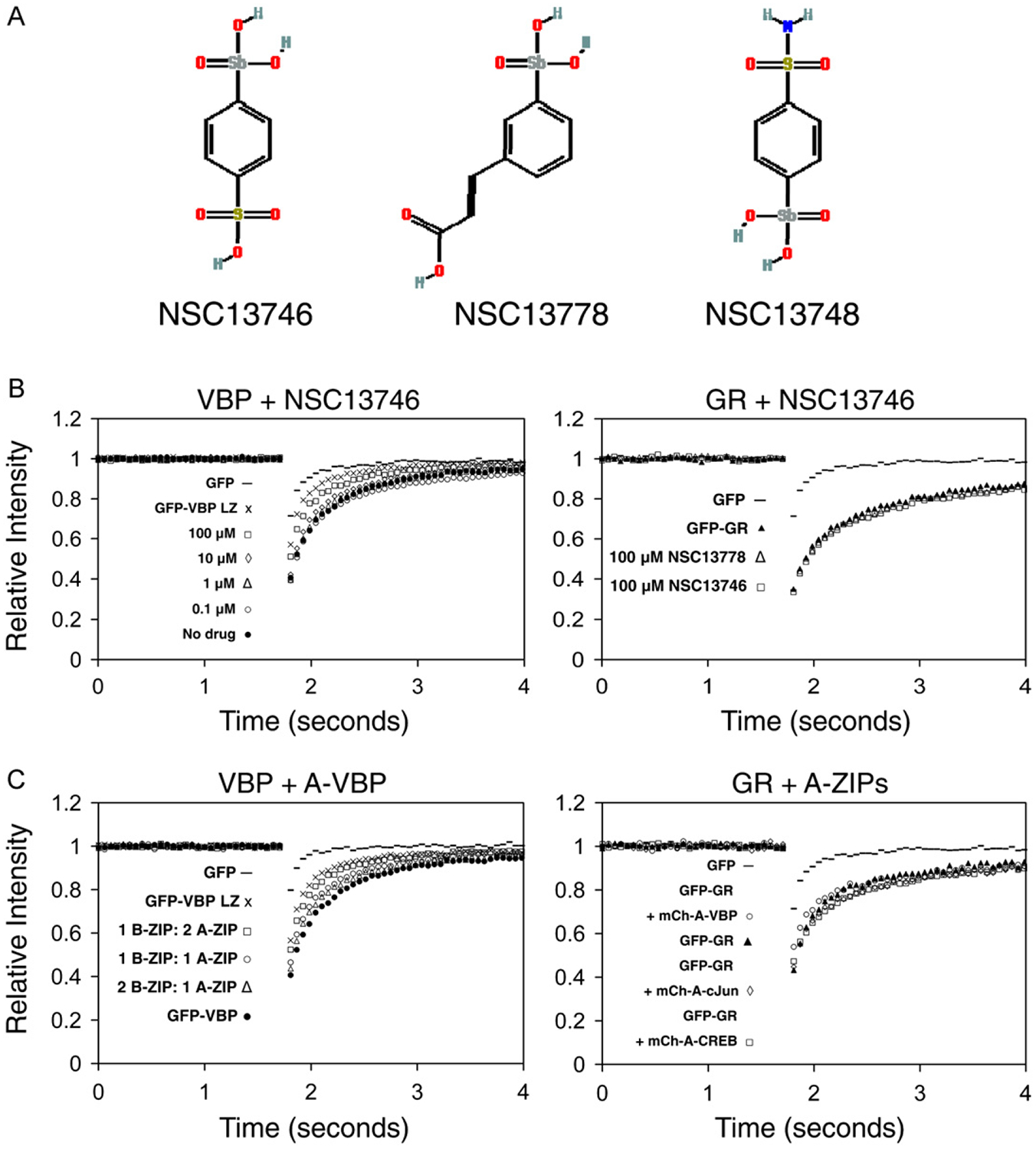
A) Structure of arylstibonic acid compounds. B) NIH3T3 cells were plated at a density of 6×104 on LabTekII chamber glass coverslips and were transiently transfected with GFP-VBP or GFP-Glucorticoid Receptor (GFP-GR). Cells incubated overnight with increasing concentrations of NSC13746 (0.1 μM to 100 μM), and were analyzed by FRAP on a Zeiss LSM 510 confocal microscope. For each FRAP experiment, at least 15 cells were imaged and the corrected normalized individual curves were averaged. All FRAP images presented in the paper are representative of at least 3 independent experiments in which at least 15 different cells were imaged in each experimental condition. Increasing concentrations of NSC13746 cause increased recovery of GFP-VBP while GFP-GR is not affected by the highest concentration (100 μM) of two different active drugs, NSC13746 and NSC13778. C) Cells were co-transfected with different ratios of GFP-VBP and mCherry-A-VBP and analyzed by FRAP. Increasing the amount of transfected A-ZIP (1 B-ZIP:1 A-ZIP or 1 B-ZIP:2 A-ZIP) causes faster recovery of GFP-VBP compared to the recovery of GFP-VBP alone. Increasing the amount of transfected B-ZIP (2 B-ZIP: 1 A-ZIP) does not change the recovery of GFP-VBP. GFP-GR is unaffected by co-transfection with different mCh-A-ZIPs.
GFP and mCherry Plasmids –
GFP fusions were constructed by engineering a new multiple cloning site into the pEGFP-C3 vector (Clontech, Mountain View, CA) (SFig. 1). The new multiple cloning site was inserted at the XhoI and BamHI sites in the original multiple cloning site of the vector. XhoI and BamHI (italics) were destroyed in the ligation. The new multiple cloning site included a phi 10 sequence (boldface) and BamHI, XhoI and HindIII restriction enzyme sites (underlined), respectively:

The mCherry plasmids were constructed by amplifying the mCherry sequence by PCR from a mCherry-pRSETB vector (Shaner et al. 2004). First, the C-terminal stop codon in mCherry was removed (TAA→TAC) and additional nucleotides were inserted to separate the EcoRI and HindIII restriction sites (SFig. 2). The following primers were used:
Forward primer at bp610–632 in mCherry-pRSETB (upstream of NcoI site):
5-GGCCCCGTAATGCAGAAGAAGA-3′;
Reverse primer at bp924–891 in mCherry-pRSETB (HindIII and EcoRI are underlined and the TAC is italicized):
5-GACGGGTCAAGCTTCCAGGGAGAATTCGTACTTGTACAGCTCG-TCC-3′. The PCR product was digested with NcoI and HindIII and ligated back into the mCherry-pRSETB vector at those restriction sites. Second, the modified mCherry sequence (stop codon removed) was isolated from the pRSETB background and substituted for GFP in the pEGFP-C3 vector containing new polylinker (SFig. 3). The mCherry sequence was isolated as two PCR fragments by using the following two pairs of primers:
Forward primer containing bp592–612 of pEGFP-C3 (bold-face; NheI site underlined) and bp199–207 of mCherry-pRSETB containing the initiating ATG (underlined) 5-CTAGCGCTACCGGTCGCCACCATGGTGAGCAAGGGCGAG-3′; Reverse primer at bp587–567 of mCherry-pRSETB: 5-GGCA-GCTTCACCTTGTAGATGA-3′;
Forward primer at bp522–539 of mCherry-pRSETB: 5-GCGT-GGTGACCGTGACCC-3′; Reverse primer at bp924–891 of mCherry-pRSETB (HindIII site underlined): 5′-GACGGGT-CAAGCTTCCAGGGAGAATTCGTACTTGTACAGCTCGTCC-3′.
The fragments were digested with NheI-PstI (primer set #1) or Pst-I-HindIII (primer set #2) and were cloned as a triple ligation into the pEGFP-C3 vector at the NheI and HindIII restriction enzymes sites. Third, a new polylinker was inserted into mCherry (stop codon removed, mCherry in the pEGFP-C3 vector backbone) at the EcoRI and HindIII restriction sites at the C-terminus of the mCherry sequence (SFig. 4). The new multiple cloning site included EcoRI (italics), a phi 10 sequence (boldface) and BamHI, XhoI and HindIII restriction enzyme sites (underlined), respectively:

The EcoRI (italics) site was destroyed during the ligation. The B-ZIP and A-ZIP domains of 10 different B-ZIP proteins (ATF2, C/EBPα, C/EBPβ, CREB, cFos, FosB, cJun, JunB, JunD, VBP) were sub-cloned as BamHI -HindIII fragments from prokaryotic expression vectors (Vinson et al., 2002) (Fig. 1). Leucine zipper domains were cloned as XhoI-HindIII fragments. All plasmids were sequenced, and protein expression levels were determined by Western blotting using a phi-10 antibody (Novagen, San Diego, CA) (data not shown). Proline mutants were constructed in the leucine zipper domain of VBP by site-directed mutagenesis with the following primers:
Fig. 1. GFP and mCherry B-ZIP, A-ZIP and Leucine Zipper fluorescent fusion protein construction.
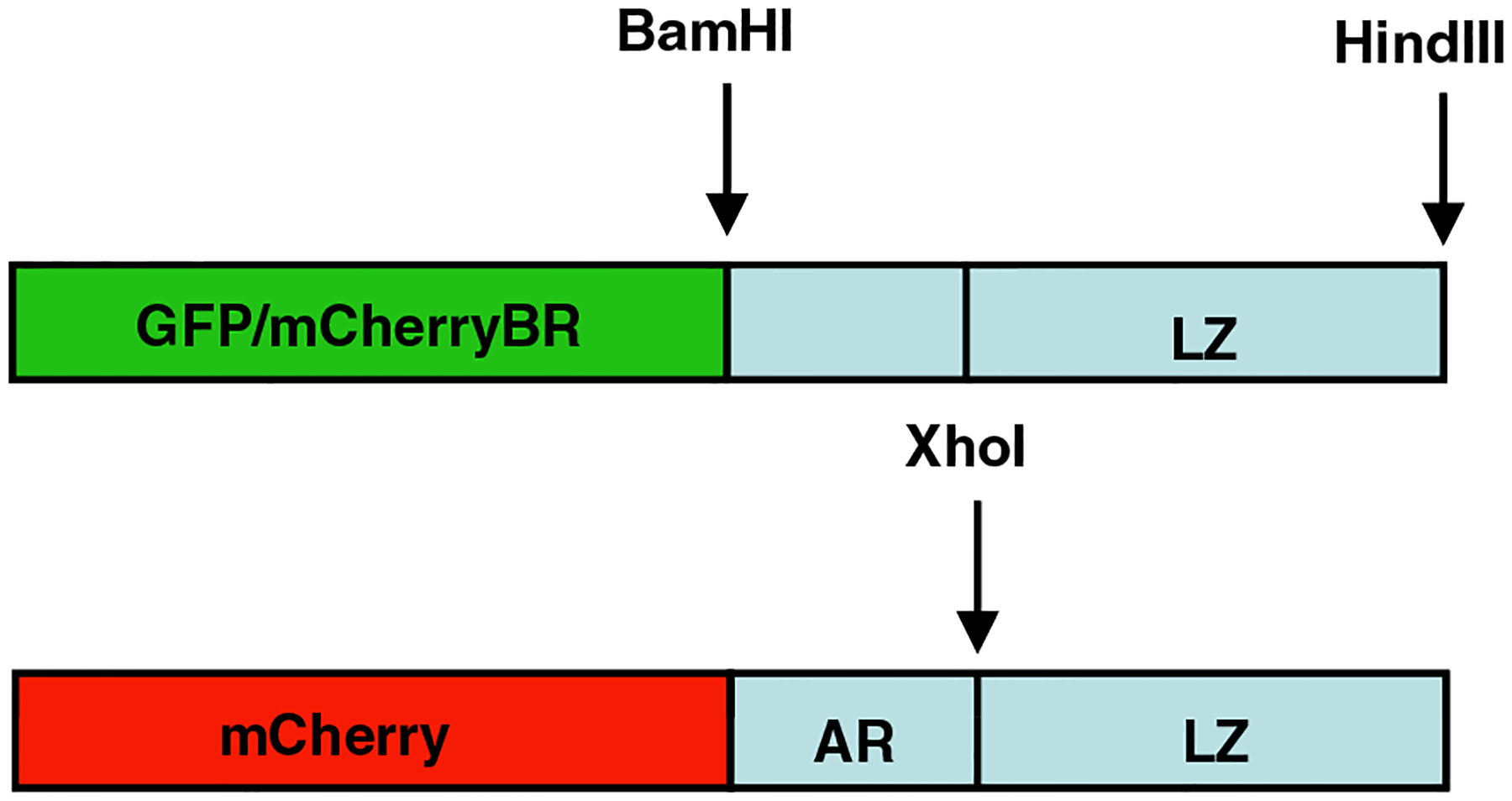
The B-ZIP, A-ZIP, or leucine zipper domain of different B-ZIP proteins (ATF2, C/EBPα, C/EBPβ, CREB, cFos, cJun, VBP) were sub-cloned into a GFP and an mCherry background.
Forward 5′-CAGGCGCTCGAGATTCGGGCAGCCTTCCCTGAGAAA-GAGAATACG-3′; Reverse 5′-GCAGCCTTCCCTGAGAAAGAGAATAC G-3′. The fragment was isolated by PCR, digested with Xho and HindIII and cloned into the modified GFP or mCherry vectors. Full length open reading frames for GFP or mCherry can be cloned as NheI/HindIII fragments. All constructs were sequenced using the C-terminal GFP sequencing primer from Clontech.
Cell Culture and Transfections –
NIH3T3 (ATCC # CRL-1658) cells were maintained in Dulbecco’s Modified Eagle Medium with high glucose (Invitrogen, Carlsbad, CA), 10% bovine calf serum (Hyclone, Logan, UT), 1% 100 mM Sodium Pyruvate and 1% Antibiotic/Antimycotic (Invitrogen, Carlsbad, CA) in a 5% CO2 incubator at 37 °C. Cells were plated at a density of 6 × 104 on LabTekII chamber glass coverslips (Nunc/Fischer Scientific, Pittsburgh, PA) and were transiently transfected with 0.5 μg DNA and 1.5 μl lipofectamine in serum-reduced OPTI-MEM media (Gibco-Invitrogen, Carlsbad, CA). EWS-ATF1 and K562 (chronic myelongenous leukemia) leukemia cell lines were maintained in RPMI media (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Lifeblood Medical, Adelphia, NJ) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA) in a 5% CO2 incubator at 37 °C.
Fluorescence Recovery After Photobleaching (FRAP) –
Cells were plated and observed in LabTekII chambers (Nunc/Fischer Scientific, Pittsburgh, PA). FRAP experiments were performed 24 h post-transfection on a Zeiss LSM 510 confocal microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY) with a 100X/1.3 N.A. oil-immersion objective. Cells were maintained on the microscope stage at 37 °C with an Air Stream incubator (ASI 400, Nevtek, Burnsville, VA). GFP was excited with the 488 nm laser line of a 40 mW Argon laser, and GFP emission was monitored with a LP505 filter above 505 nm. A nuclear region of interest was bleached using the 488-nm laser line at 89% laser power. A single iteration was used for the bleach pulse, which lasted for 44 ms. Cells were monitored every 59 ms for 23.511 s. To minimize the effect of photobleaching due to imaging, images were collected at 0.356% laser intensity. For quantification, the average fluorescent intensity was measured using software from Carl Zeiss Micro-Imaging Inc. Background fluorescence was measured in a random field outside of cells. For each time point, the fluorescent intensity of the bleached region of interest was corrected for the bleaching due to imaging and normalized to prebleach as Irel=(I(t) BG)/((Io−BG)*(T(t)−BG)), where Io is the background-corrected average intensity of the region of interest during prebleach, and T(t) is the background-corrected average fluorescence intensity of an unbleached region in the same cell. For each FRAP experiment, at least 15 cells were imaged and the corrected normalized individual curves were averaged. All FRAP images presented in the paper are representative of at least 3 independent experiments in which at least 15 different cells were imaged in each experimental condition.
Fluorescent Resonance Energy Transfer (FRET) –
FRET was used to directly measure the specificity of interaction between B-ZIP and A-ZIP proteins labeled with GFP and mCherry. Cells were prepared for live cell imaging as in FRAP, and a Zeiss 510 confocal microscope with the same objective, incubation and GFP excitation conditions defined for FRAP was also used for FRET. mCherry excitation was achieved with the 543 laser line of a 1 mW HeNe laser, and mCherry emission was monitored with an LP560 filter above 560 nm. Cells were imaged in multi-track mode at 0.75% argon laser power and 25% HeNe laser power. Sensitized emission was used to detect FRET. This was implemented using the Fc FRET macro within the Zeiss LSM software (Carl Zeiss MicroImaging, Inc, Thornwood, NY), which measures the fluorescence intensity from the appropriate images in the donor, acceptor and FRET channels of cells transfected with only the GFP fusion, only the mCherry fusion or both fusions, subtracts background from all of these images and then calculates the FRET efficiency based on the Youvan et al. (Paletzki et al., 2008) algorithm. For each experiment, at least 10 cells were imaged and 5 different regions of interest within each cell were analyzed. FRET efficiencies were compared in Microsoft Excel with a Student’s T-test (two sample assuming equal variance).
Chromatin Immunoprecipitation –
NIH3T3 cells were grown in 6-well tissue culture dishes. After cells reached ~90% confluency they were treated for 24 h with 100 μm of drug and lipofectamine or mock treated with lipofectamine only. Chromatin immunoprecipitation was performed as in (36), A single 330 bp fragment −364 to −34 bp relative to the transcription start site of the mouse c-fos promoter encompassing the SIE, SRE, C/EBP site, and CRE was amplified using primers previously described (37):
5′-GGCTGCAGCCGGCGAGCTG-3’ and 5′-AGAAGCGCTGTGAATG GATG-3’.
PCR was performed for 34 cycles (94 °C 15 s, 62 °C 30 s, 72 °C 30 s) using 10% of CREB ChIP DNA, 0.05% of input DNA and Sigma Red Extract-N-AMP PCR mix.
Cytotoxicity Assay –
EWS-ATF1 and K562 leukemia cells were plated at 10,000 cells per well in 96 well plates. PBS was plated around the perimeter of the 96-well plate to act as a humidifying chamber. NSC13746 (active) or NSC13748 (inactive) compound was added to the wells in 1:2 serial dilutions starting at 100 μM. Both drugs are water-soluble and were diluted in RPMI complete media. Cells incubated with the drugs in a 5% CO2 incubator at 37 °C for 96 h, after which 20 ml of CellTiter 96 Aqueous One Solution containing MTS was added to 100 μl of the cell/drug reaction (Promega, Madison, WI). Absorbance was recorded at 490 nm on a VersaMax microplate reader and analyzed with SoftMax Pro software (Molecular Devices, Sunnyvale, CA). All reactions were performed in triplicates, and data is representative of at least two independent experiments.
Soft Agar Assay –
Using 24 well tissue culture plates, warm 0.75% agarose (Lonza, Rockland, ME) in RPMI medium (Lonza) supplemented with 10% fetal bovine serum (HyClone) and 1% L-glutamine (HyClone) (Complete medium) was dispensed in 0.2 ml with a repeat pipetor. The agarose was allowed to solidify at room temperature. The second agarose layer composed of 0.4% hand-warmed agar and cells (EWS-ATF1 – 20,000 cells/well; K562 – 5,000 cells/well) in complete medium was dispensed in 0.2 ml. Plates were subsequently refrigerated for 5 min and placed in a 37° CO2 incubator overnight. After 24 h incubation, experimental compound (NSC13746 or NSC13748) was added at 100 μM, 50 μM, 25 μM, 12.5 μM, 6.25 μM or 0 μM in 0.2 ml of complete medium. Plates were incubated for the 13 days, and colonies between 60 and 300 μm were counted using an Oxfor Optronix Gelcount colony counter. Plates were run in duplicate, and data is representative of at least two independent experiments.
Results
The mobility of GFP-B-ZIP chimeras in cells after photobleaching increases in the presence of either an antimony compound or dominant negative A-ZIP domains.
Chimeric proteins containing Green Fluorescent Protein (GFP) and one of eight B-ZIP domains (ATF2, C/EBPα, C/EBP®, CREB, cJun, VBP, L-ZIP, cFos) were transiently transfected into NIH3T3 cells and nuclear mobility was monitored by FRAP. A chimera between VBP, a B-ZIP domain and GFP (GFP-VBP) had a recovery time after photobleaching of approximately 2 s (Fig. 2B), a timescale comparable to that reported for other B-ZIP domains (Mayr et al., 2005). To determine if small molecular weight antimony-containing arylstibonic acids that bind to the B-ZIP domain and inhibit DNA binding in solution (Rishi et al., 2005) were active in vivo, we determined if they changed GFP-VBP nuclear recovery after FRAP. We examined NSC13746 (Fig. 2A), a water-soluble arylstibonic acid identified in a screen of 14 antinomy derivates of NSC13778 that is more active and promiscuous than NSC13778 at inhibiting the DNA binding of different B-ZIP domains (Rishi et al., in preparation). NSC13746 caused a dose dependent increase in GFP-VBP nuclear recovery after FRAP (Fig. 2B). The increase in mobility as measured by the increase of fluorescence in the bleached area of the nucleus we interpret to represent inhibition of DNA binding that allows the protein to diffuse more quickly in the nucleosplasm. The nuclear recovery measured by FRAP of a structurally unrelated nuclear transcription factor, GFP labeled glucocorticoid receptor (GFP-GR), was unaffected after incubation with the highest concentration (100 μM) of two active compounds (NSC13746, NSC13778), demonstrating the specificity of NSC13746 in increasing the recovery of the GFP-VBP chimeric protein.
To investigate whether the increase in GFP-VBP recovery in the presence of NSC13746 represents inhibition of DNA binding, cells were co-transfected with GFP-VBP and mCherry-A-VBP, a dominant negative protein that preferentially binds to VBP and blocks DNA binding (Moll et al., 2000). Indeed, inhibition of GFP-VBP DNA binding by co-transfection with mCherry-A-VBP resulted in a similar increase in GFP-VBP recovery after FRAP as observed with NSC13746 (Fig. 2C). This effect was also dose dependent, as increasing the ratio of transfected A-ZIP to B-ZIP increased the B-ZIP recovery. Increasing the ratio of transfected B-ZIP to A-ZIP had no effect on the recovery of GFP-VBP, presumably because the excess B-ZIP can homodimerize, bind to DNA, and retain its normal recovery after FRAP. As a control to examine the specificity of mCherry-A-VBP at increasing the recovery of GFP-VBP, mCherry-A-VBP was co-transfected with the unrelated nuclear transcription factor, GFP-GR. mCherry-A-VBP did not affect the recovery of GFP-GR, indicating that specific binding of the A-ZIP to B-ZIP domains is responsible for the increased recovery times. These experiments suggest that the dose- dependent increase in GFP-VBP recovery by NSC13746 is due to the inhibition of VBP DNA binding in living cells.
FRAP of mutant GFP-VBP with DNA binding and leucine zipper dimerization changes.
To gain insight into the structural reasons for the increased recovery of VBP in the presence of NSC13746 after FRAP, we measured the recovery of GFP-VBP domain mutants by FRAP (Fig. 3). The VBP B-ZIP domain contains a basic region that mediates DNA binding and a leucine zipper region that mediates dimerization. A deletion mutant of the VBP basic region that has been shown to inhibit DNA binding (16) (GFP-VBP-LZ) had an even faster recovery. The slower recovery of GFP-VBP in the presence of either NSC13746 or A-VBP compared to GFP-VBP-LZ may be due to residual GFP-VBP DNA binding. Alternatively, a size difference between the longer B-ZIP|drug or B-ZIP|A-ZIP dimers compared to the shorter LZ dimers may result in their slower recovery. A GFP-VBP mutant containing two proline residues in the leucine zipper should structurally prevent dimerization and allow measurement of the recovery of a monomer. The recovery for this protein, GFP-VBP-Proline, was faster than the recovery of the GFP-VBP-LZ dimer and only slightly slower than monomeric GFP (Fig. 3). This suggests that the monomer can no longer bind DNA well, thus yielding a much faster FRAP recovery that is close to purely diffusing GFP. The slightly slower recovery of GFP-VBP-Proline relative to GFP alone could be a consequence of the increased size of the protein or some residual DNA binding of the monomer which has been observed in in vitro experiments with B-ZIP proteins (Kohler et al., 1999). Taken together, these data suggest that NSC13746 increases the recovery of GFP-VBP by inhibiting DNA binding.
Fig. 3. GFP-VBP dimerization and DNA binding mutants by FRAP.
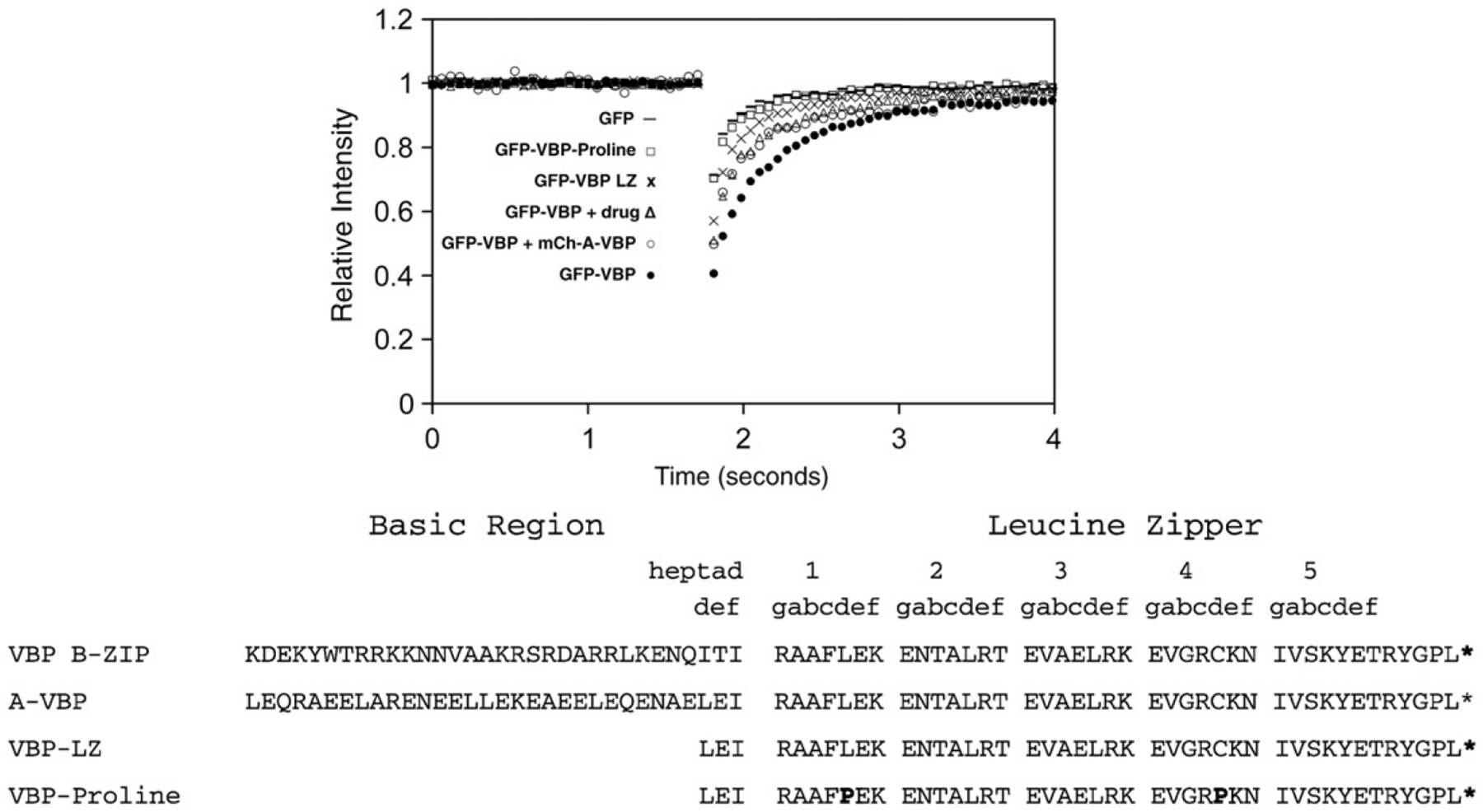
NIH3T3 cells were transiently transfected with plasmids containing mutations in the GFP-VBP B-ZIP domain and analyzed by FRAP as previously described. GFP-VBP (●) containing the basic region and leucine zipper region has the slowest recovery. Inhibition of VBP DNA binding by incubation with 100 μM NSC13746 (Δ) or co-transfection of GFP-VBP with mCherry-A-VBP (○) cause an increase in GFP-VBP recovery. Deletion of the basic region of VBP (GFP-VBP-LZ (x)) causes faster GFP-VBP recovery. The recovery of monomeric GFP-VBP-Proline (☐) is faster than all GFP-VBP dimers, but is slower than monomeric GFP.
NSC13746 inhibits CREB DNA binding by ChIP.
After demonstrating reasonable structural evidence using A-ZIP and leucine zipper mutants that FRAP could measure the ability of NSC13746 to inhibit B-ZIP DNA binding, we used Chromatin Immunoprecipitation (ChIP) as a more direct means to support this claim. Cells were treated for 24 h with 100 μM drug and lipofectamine and then analyzed by ChIP. Cells treated with NSC13746 showed a 30% reduction in CREB DNA binding compared to mock treated cells, while cells treated with NSC13748 were unaffected (Fig. 4). These results, along with analyses by FRAP, suggest that NSC13746 binds to the B-ZIP to inhibit DNA binding. Additional ChIP experiments will be performed with VBP and other B-ZIP domains.
Fig. 4. CREB binding to cFos promoter was inhibited by NSC13746 and not affected by NSC13782.
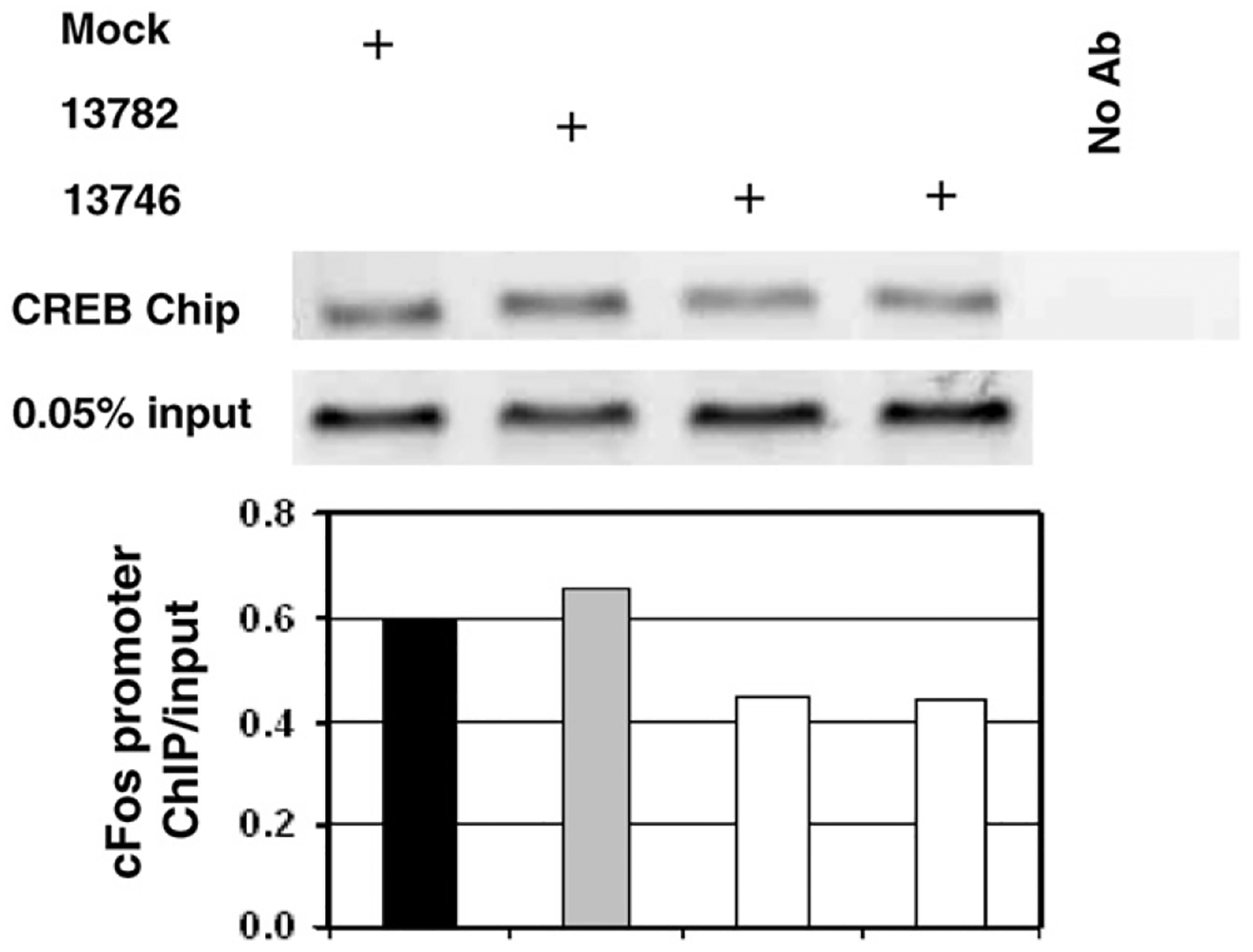
Cells were treated for 24 h with 100 μM of drug and lipofectamine or mock treated with lipofectamine only. Chromatin immunoprecipitation was used to detect CREB binding to cFos promoter in cells treated with NSC13746, NSC13782 or mock treated. Normalization to input level produced 30% lower level of band intensities in NSC13746 treated cells in comparison with mock treated cells indicating reduction of CREB binding. Treatment of cells with NSC13782 does not affect CREB binding.
NSC13746 increases the recovery of different B-ZIP domains. After observing a dose-dependent increase in recovery of GFP-VBP by NSC13746 (Fig. 2B), we investigated whether NSC13746 increased the recovery of additional B-ZIP domains. A FRAP analysis showed that the interaction of NSC13746 with GFP-CREB or GFP-C/EBPβ also caused an increase in the recovery of these B-ZIP domains (Fig. 5). These results indicate that NSC13746 is promiscuous and can inhibit the DNA binding of different B-ZIP domains in vivo.
Fig. 5. NSC13746 increases the recovery of different B-ZIP domains.
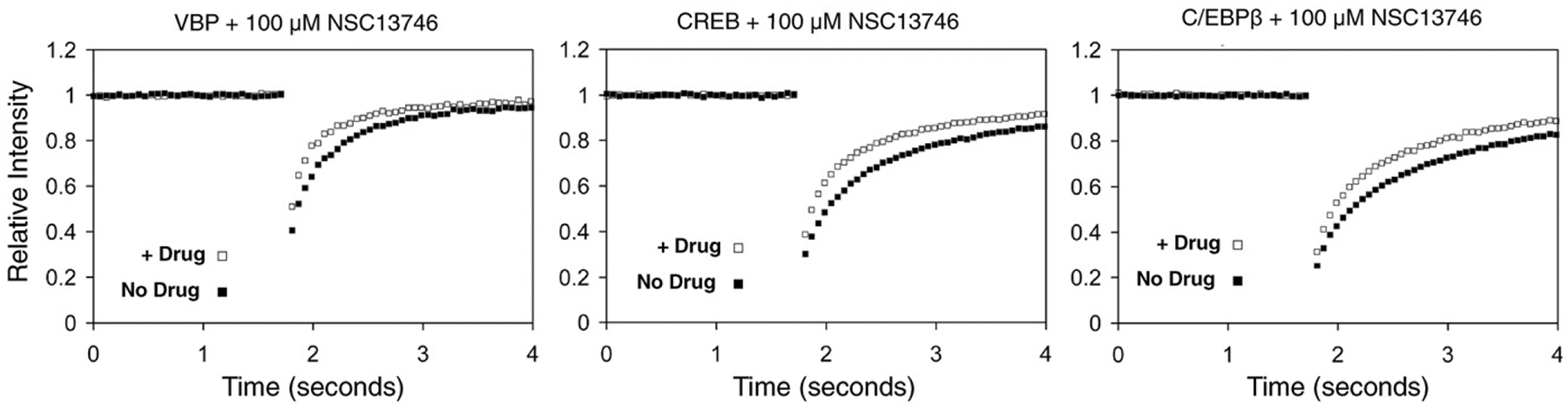
NIH3T3 cells were transiently transfected with different B-ZIP domains (GFP-VBP, GFP-CREB, GFP-C/EBPβ) and incubated overnight with 100 μM NSC13746. FRAP indicates that the antimony derivative NSC13746 causes an increase in the recovery of all B-ZIP domains.
FRAP and FRET of GFP-VBP with different dominant negative A-ZIP domains.
A-ZIP dominant negatives interact in a leucine zipper-specific manner with B-ZIP partners in vitro and block B-ZIP DNA binding (Acharya et al., 2006; Vinson et al., 2002). To examine whether B-ZIP and A-ZIP interact specifically in cells, NIH3T3 cells were co-transfected with GFP-VBP and 5 different mCherry-A-ZIP proteins (mCh-A-C/EBPα, mCh-A-C/EBPβ, mCh-A-CREB, mCh-A-cJun, mCh-A-VBP) and their interactions were analyzed by FRAP (Fig. 6A). Two A-ZIPs that do not interact with the VBP B-ZIP domain in vitro, mCherry-A-C/EBPα and mCherry-A-C/EBPβ, did not cause an increase in GFP-VBP recovery, suggesting that these dominant negatives preserve in vivo the specificity detected in vitro. However, mCherry-A-CREB and mCherry-A-cJun did increase the recovery of GFP-VBP even though A-CREB and A-cJun have not been previously shown to interact with the VBP B-ZIP domain in vitro.
Fig. 6. B-ZIP|A-ZIP interactions in vivo by FRAP and FRET.
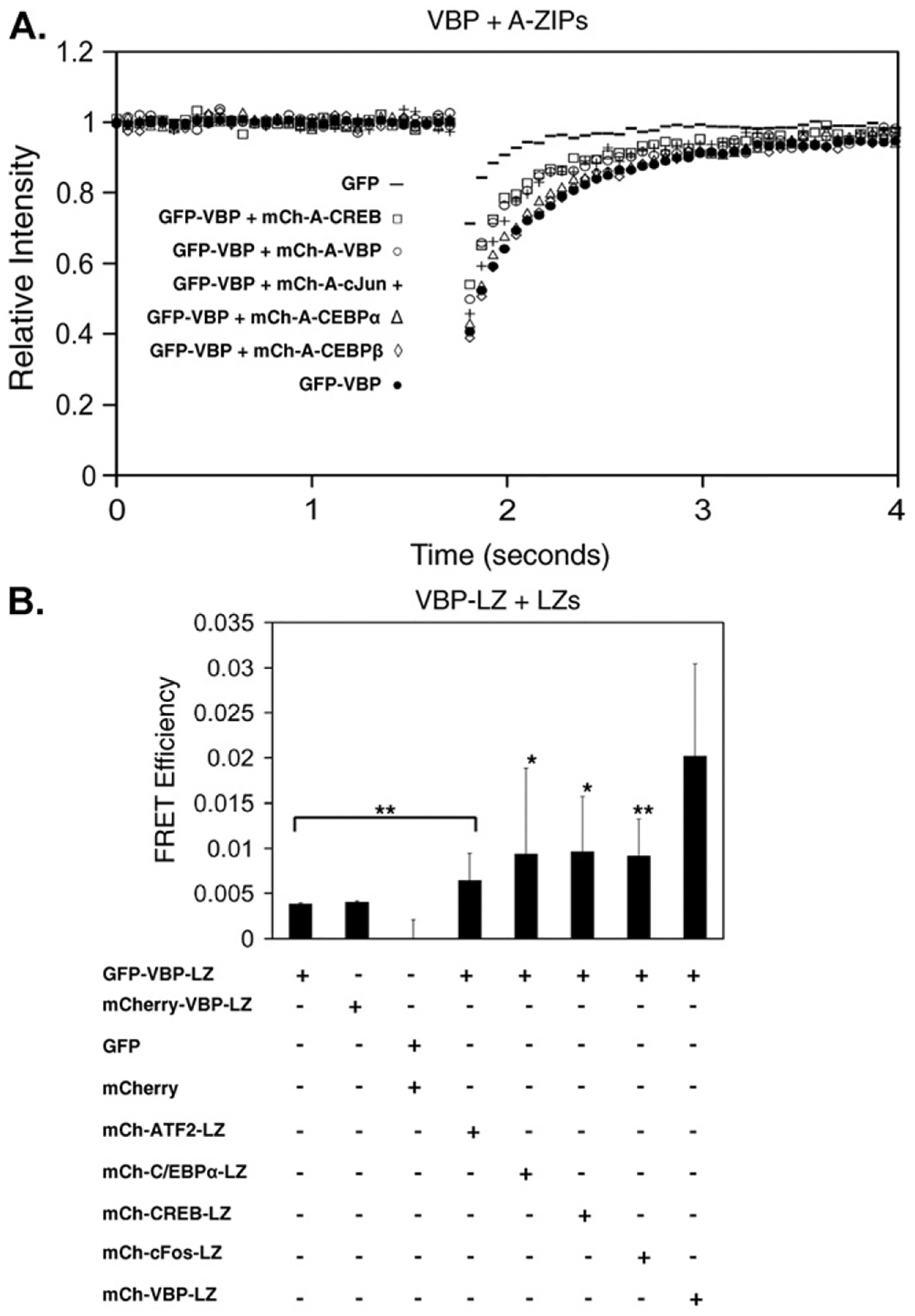
A) NIH3T3 cells were transiently co-transfected with GFP-VBP and 5 different mCherry-A-ZIPs and analyzed by FRAP. mCh-A-C/EBPα and mCh-A-C/EBPβ do not affect the recovery of GFP-VBP. mCh-A-VBP, mCh-A-CREB and mCh-A-cJun increase the recovery of GFP-VBP. B) NIH3T3 cells were transiently co-transfected with GFP-VBP-LZ and different mCh-LZ domains and analyzed by FRET. Cells were prepared for live cell imaging as in FRAP, and a Zeiss 510 confocal microscope with the same objective, incubation and GFP excitation conditions defined for FRAP was also used for FRET. mCherry excitation was achieved with the 543 laser line of a 1 mW HeNe laser, and mCherry emission was monitored with an LP560 filter above 560 nm. Cells were imaged in multi-track mode at 0.75% argon laser power and 25% HeNe laser power. Sensitized emission was used to detect FRET. The FRET efficiency between GFP-VBP-LZ with mCh-VBP-LZ homodimers was significantly greater than all other leucine zipper heterodimers (*p<0.05, **p<0.01). FRAP and FRET results are representative of at least 3 independent experiments in which at least 10–15 different cells were imaged in each experimental condition.
We examined the dimerization specificity of leucine zippers without the basic region to evaluate if dimerization specificity could be observed in vivo. A technique that directly measures protein-protein interactions is Fluorescence Resonance Energy Transfer, or FRET (Day and Schaufele, 2005). GFP and mCherry share significant spectral overlap, making them a good donor (GFP) and acceptor (mCherry) pair for FRET studies (Anderson et al., 2006; Tramier et al., 2006). Co-transfecting GFP-VBP-LZ with different mCherry-B-ZIP-LZ domains showed significantly more energy transfer between GFP-VBP-LZ and mCherry-VBP-LZ than between GFP-VBP-LZ and other mCherry-B-ZIP-LZ chimeras (Fig. 6B). However, the energy transfer between heterologous leucine zippers was significantly greater (0o0.05) than FRET between GFP and mCherry empty vectors, suggesting a non-specific transient leucine zipper interaction that is also observed by FRAP.
NSC13746 or A-ZIPs promote cytoplasmic localization of B-ZIP domains.
B-ZIP domains localize to the nucleus and form homodimers or heterodimers (Fig. 7) (Williams et al., 1997). The individual B-ZIP domain subnuclear localization shows various patterns: some are excluded from the nucleolus (ATF2, CREB, cFos), others preferentially go to nucleolus (cJun), whereas others localize to the heterochromatin (C/EBPα, C/EBPβ) and a final group shows no preference (VBP) (Bhoumik et al., 2004; Demarco et al., 2006; Hu et al., 2002; Liu et al., 2006; Schaufele et al., 2001; Tang and Lane, 1999). One exception to the nuclear localization of the B-ZIP domain is cFos, which does not contain a nuclear localization signal and localizes to the nucleus only as a heterodimer with cJun or ATF2 (Supplementary Fig. 5B, C) (Chida et al., 1999). To investigate the effect of inhibiting B-ZIP DNA binding on cellular localization of the different B-ZIP domains, NIH3T3 cells were imaged after incubation of GFP- B-ZIP-transfected cells with 100 μM NSC13746 or co-transfection with different A-ZIP domains linked to mCherry. Incubating any GFP-B-ZIP domain with 100 μM NSC13746 caused cytoplasmic localization (Fig. 7). In contrast, 100 μM NSC13746 did not cause GFP-GR to localize to the cytoplasm demonstrating the specificity of NSC13746 for a subset of nuclear transcription factors. To determine whether the cytoplasmic localization of the B-ZIP domain was due to inhibition of DNA binding, changes in cellular localization were examined in cells co-transfected with GFP-B-ZIPs and mCh-A-ZIPs. Dominant negative A-ZIP domains caused a cytoplasmic localization of specific B-ZIP partners with the exception of GFP-cFos, which is normally expressed throughout the cell (Fig. 7). Cross-reactivity between the C/EBP family (C/EBPα and C/EBPβ), and AP1 family (cFos, cJun, ATF2) confirm previous interactions demonstrated in vitro (Newman and Keating, 2003; Vinson et al. 2002). The cellular localization of GFP-GR was not affected by different mCherry-A-ZIPs. These results highlight the specificity of the A-ZIP dominant negatives to cause cytoplasmic localization of the B-ZIP domain. The difference in FRET results where some interactions are observed between A-ZIP and B-ZIP proteins that not observed in vitro and the cytoplasmic localizations that recapitulate what is observed in vitro may reflect that FRET is identifying transient interactions that are not robust enough to be productive as monitored by cytoplasmic localization.
Fig. 7. Inhibition of B-ZIP DNA binding by NSC13746 or A-ZIP causes B-ZIP cytoplasmic localization.
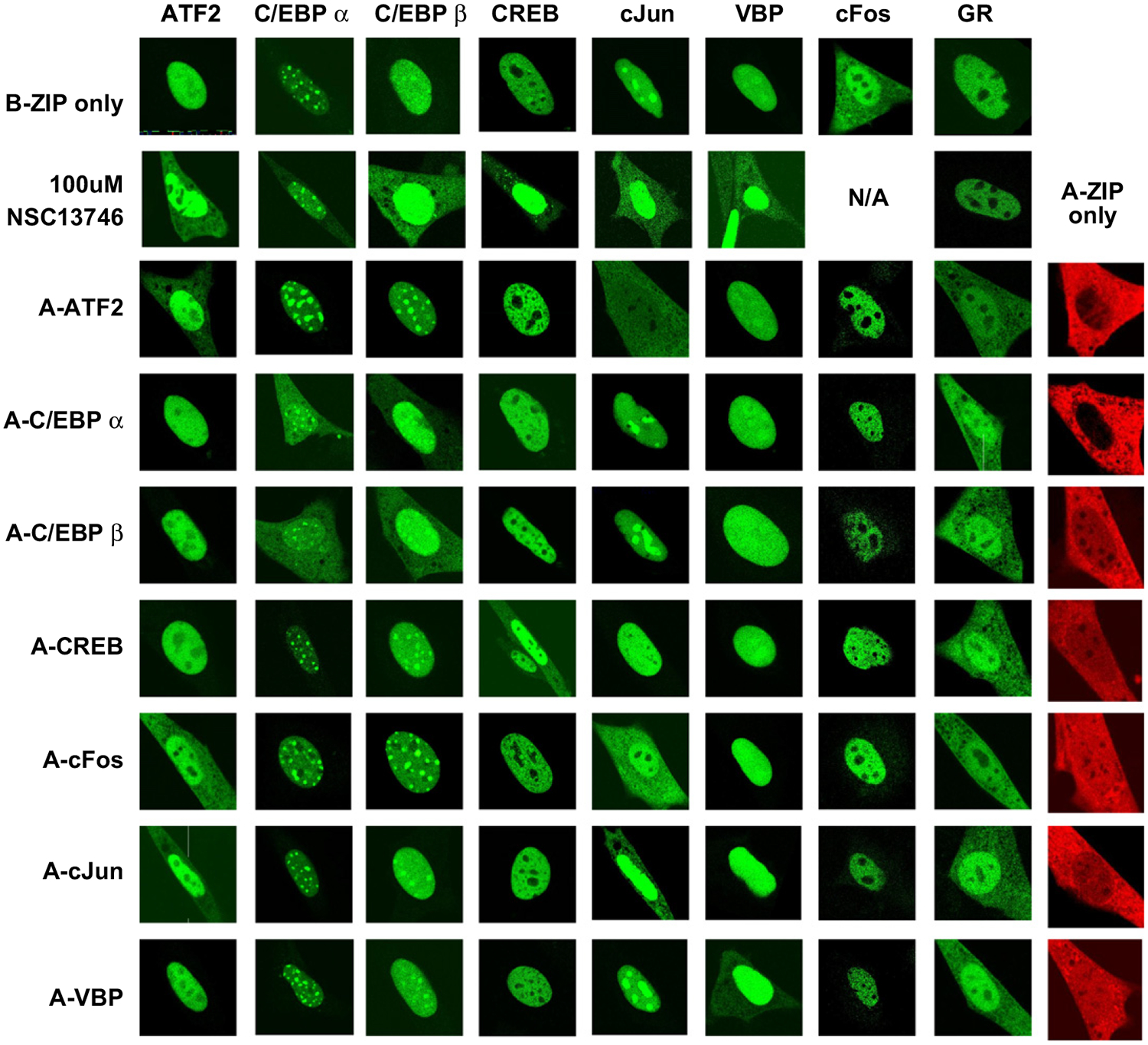
Cellular transfections were performed as described previously. Images were taken using a Zeiss 510 confocal microscope and are representative of at least 3 independent experiments in which at least 10–15 different cells were imaged in each experimental condition. GFP-B-ZIP domains localize to the nucleus, with the exception of cFos which does not homodimerize. mCherry-A-ZIPs (in red) are localized throughout the cell. Incubating GFP-B-ZIPs with100 μM of NSC13746 caused a cytoplasmic localization for all B-ZIPs. A matrix of GFP-B-ZIP and mCherry-A-ZIP co-transfections show an A-ZIP-dependent cytoplasmic localization of the B-ZIP partner.
The growth of a clear cell sarcoma cell line containing a EWS-ATF1chimeric protein is suppressed by NSC13746.
Because NSC13746 promiscuously disrupts the DNA binding of all B-ZIP domains examined in living cells (Figs. 5 and 7), one may expect it to have a widespread and potentially cytotoxic effect on cell viability. However, NSC13746 was not toxic to NIH3T3 cells at concentrations as high as 1 mM (data not shown) suggesting that B-ZIP transcription factors may not be completely inhibited, are not active in these cells, or their activity is not critical to cell survival.
To investigate the potential for NSC13746 to inhibit the growth of cells, we used a cell line whose viability is dependent on a B-ZIP domain. Several human cancers contain chimeric proteins containing a B-ZIP domain. One chimera is an E2A-HLF fusion where HLF is a member of the PAR family of B-ZIP proteins. In this case, a dominant negative suppressor to HLF causes rapid apoptosis (Inaba et al., 1996). An additional example is a clear cell sarcoma containing an oncogenic gene fusion of EWS-ATF1. EWS acts as an N-terminal trans-activating domain and ATF1 is a B-ZIP member of the CREB family and tethers the chimeric protein to DNA (Zucman et al., 1993). A cell line derived from a human clear cell sarcoma patient containing the EWS-ATF1 chimeric protein was used in a cell growth assay. A 96-h incubation with increasing concentrations of NSC13746 caused up to 25% decrease in cell growth, whereas the inactive antimony compound (NSC13748) did not affect cell growth (Fig. 8). The K562 leukemia cells containing a non-B-ZIP oncogenic gene fusion, BCR/abl, were not affected by active (NSC13746) or inactive drug (NSC13748), suggesting that the inhibition of cell growth in EWS-ATF1 cells is caused by specific targeting of the B-ZIP domain by NSC13746.
Fig. 8. NSC13746 inhibits the growth of EWS-ATF1 containing clear cell sarcoma cells.
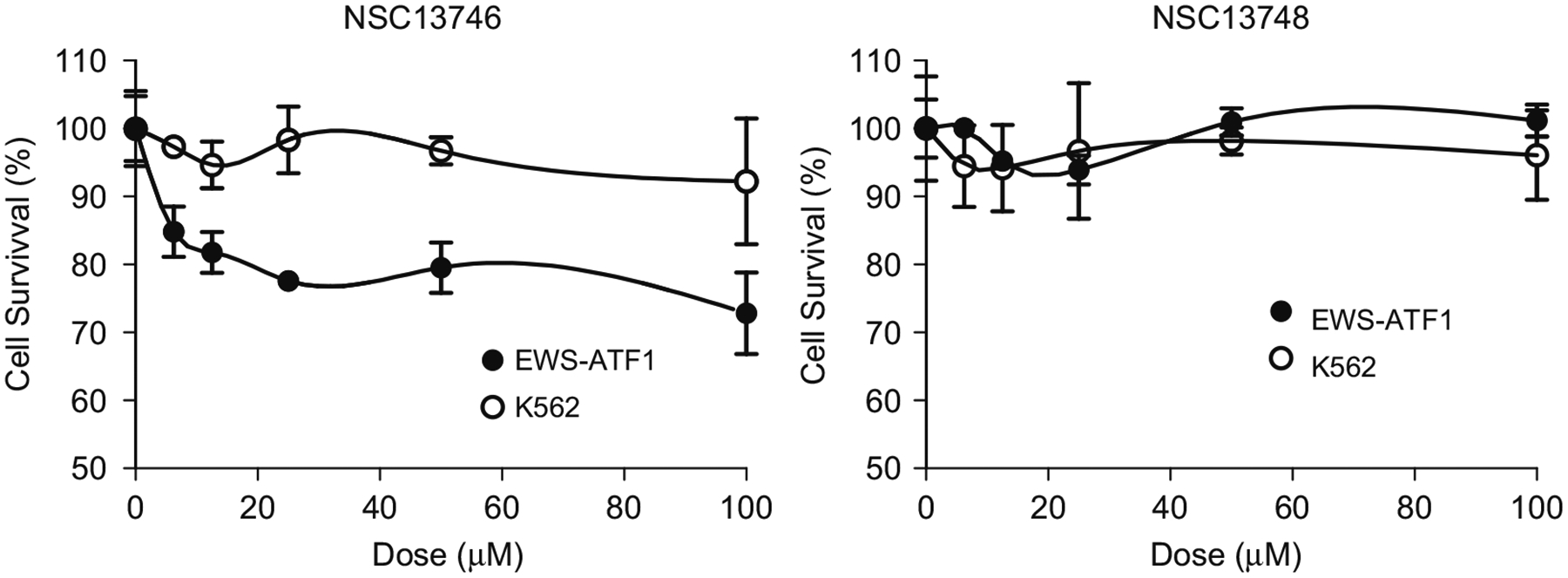
EWS-ATF1 and K562 leukemia cells were plated at 10,000 cells per well in 96 well plates. NSC13746 (active) or NSC13748 (inactive) compound was added to the wells in 1:2 serial dilutions starting at 100 μM. Cells incubated with the drugs in a 5% CO2 incubator at 37 °C for 96 h, after which CellTiter 96 Aqueous One Solution containing MTS was added and absorbance was recorded at 490 nm on a VersaMax microplate reader. NSC13746 caused ~25% growth inhibition in EWS-ATF1 cells while K562 leukemia cells (BCR-ABL) were unaffected. All reactions were performed in triplicates, and data is representative of at least two independent experiments.
Further supporting these results, increasing doses of NSC13746 inhibited colony formation in soft agar of EWS-ATF1 clear cell sarcoma cells by over 50% while NSC13748 again was not active (Fig. 9). Neither compound inhibited growth of the K562 leukemia cell line. These results demonstrate that the promiscuous inhibition of B-ZIP transcription factors could be therapeutically useful in cancer cells where oncogenesis is driven by a B-ZIP protein.
Fig. 9. NSC13746 inhibits colony formation of EWS-ATF1 cells.
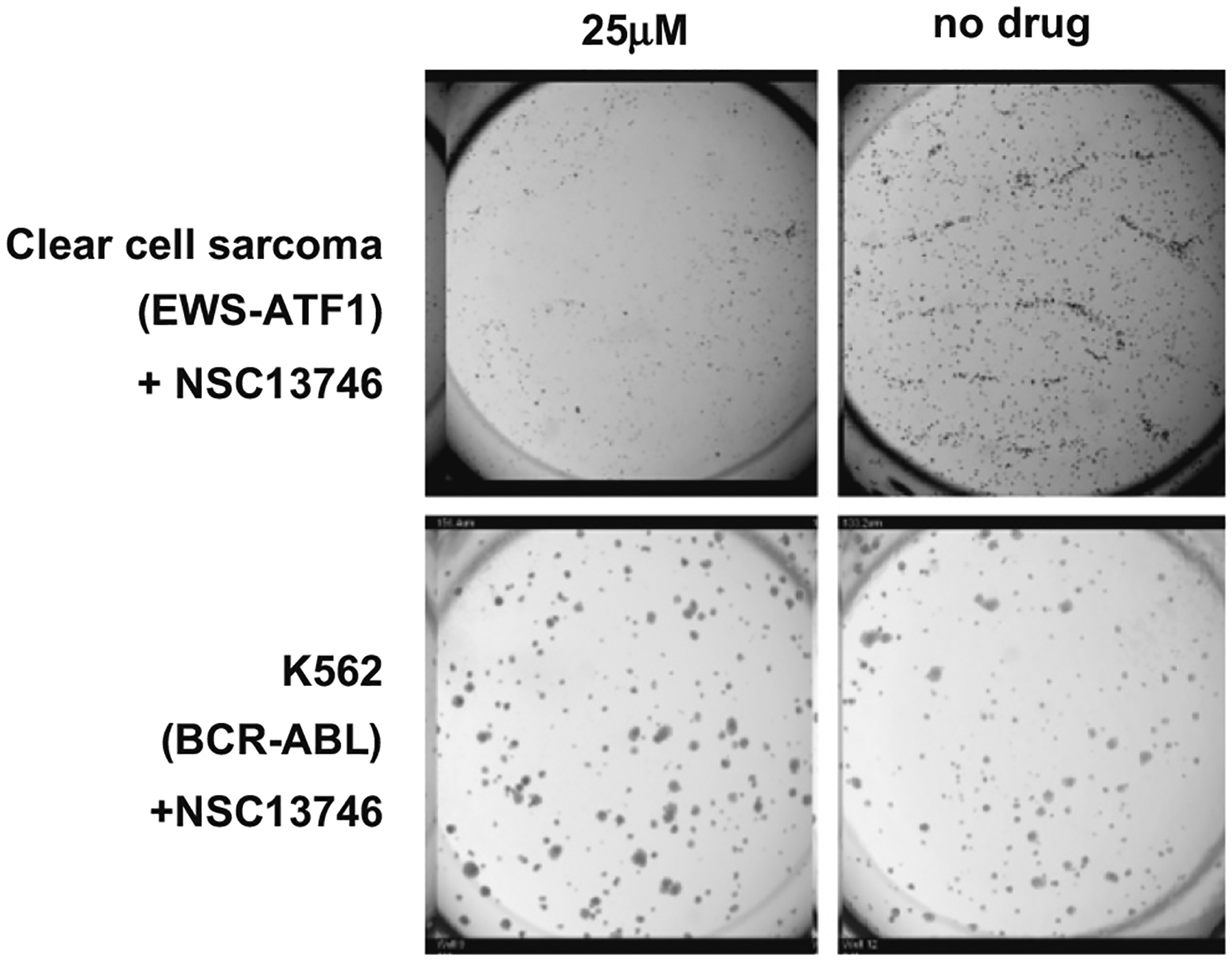
Cells were plated at 20,000 cells/well (EWS-ATF1) and 5,000 cells/well (K562) in 24-well tissue culture plates on top of an agarose layer. After 24 h incubation in a 5% CO2 incubator at 37 °C, 25 μM antimony compound was added and plates were incubated for another 13 days. Colonies between 60 and 300 μm were counted using an Oxfor Optronix Gelcount colony counter. NSC13746 inhibited approximately 50% of colony formation in EWS-ATF1 cells while K562 (BCR-ABL) colony formation was unaffected. Plates were run in duplicate, and data is representative of at least two independent experiments.
Discussion
Inhibition of B-ZIP DNA binding has been suggested to be a clinically relevant molecular target (Darnell, 2002). In a previous study, we identified the small molecule antimony compound (NSC13778) as a potent inhibitor of B-ZIP DNA binding (Rishi et al., 2005). In the present study, we evaluated the effects of NSC13746, a water soluble analogue of NSC13778, on B-ZIP mobility and localization in living cells and compared these findings to well-characterized dominant negatives that selectively inhibit B-ZIP DNA binding (Vinson et al., 2002). A FRAP analysis showed that NSC13746 caused a dose-dependent increase in the recovery of GFP-VBP, as well as GFP-CREB and GFP-C/EBPβ. This increase in recovery of different B-ZIP domains by NSC13746 is caused by inhibition of DNA binding, since dominant negative A-ZIPs which have been shown to specifically interact with B-ZIP domains to inhibit DNA binding (Acharya et al., 2006) caused similar increases in B-ZIP recovery by FRAP. In addition, NSC13746 inhibits B-ZIP DNA binding as directly measured by ChIP. Deleting the basic region from GFP-VBP caused even faster recovery of the B-ZIP protein, highlighting the importance of the basic region for the decreased recovery of the B-ZIP domain and providing further support that the increased recovery of the B-ZIP domains in the present of NSC13746 or A-ZIPs is a consequence of the inhibition of DNA binding. Together these results demonstrate that both the A-ZIPs and NSC13746 exerted in vivo effects on DNA binding that were comparable to their previously measured in vitro properties.
The in vivo microscopy assays also allowed us to probe some of the cellular consequences of inhibiting B-ZIP DNA binding with either NSC13746 or A-ZIPs that were not appreciated from previous in vitro analyses. We found that NSC13746 caused a partial cytoplasmic localization of all B-ZIP domains but not the GR domain. The A-ZIP dominant negative proteins in contrast caused a cytoplasmic localization of a B-ZIP partner that was leucine zipper dependent. The acidic extension was necessary to induce the cytoplasmic localization, as co-transfection of cJun B-ZIP with different leucine zippers had no effect on the localization of cJun (Supplementary Fig. 5A). The localization of some of the GFP-B-ZIP protein to the cytoplasm in the presence of the A-ZIP could be because the B-ZIP|A-ZIP heterodimer inactivates the nuclear localization sequence (NLS) in the basic region. This mechanism may prevent the B-ZIP|A-ZIP heterodimer from entering the nucleus. Cytoplasmic localization could also be directly related to inhibition of B-ZIP DNA binding by the acidic extension by inactivating the B-ZIP|A-ZIP heterodimer and targeting this complex for export into the cytoplasm.
The B-ZIP|A-ZIP interaction specificity measured by FRAP showed an interaction of VBP with A-CREB and A-cJun, two dominant negatives that did not cause cytoplasmic localization of VBP. In addition, FRET detected heterologous leucine zipper energy transfer that was greater than energy transfer between empty GFP and mCherry vectors. Because FRAP and FRET measure molecular interactions on a timescale of seconds, these techniques may highlight transient, non-specific interactions between the B-ZIP and A-ZIP domains. In contrast, pictures of the cells were taken 16 h after transfection, allowing the drug or A-ZIP to have long term affects on B-ZIP DNA binding and cellular localization. Clearly, it is only the specific interaction of a B-ZIP domain with its A-ZIP partner that ultimately leads to cytoplasmic localization.
NSC13746 is a promiscuous compound that inhibits the B-ZIP DNA binding of many B-ZIP proteins. A non-specific inhibitor has the risk of exerting off-target effects in a disease system, however, this promiscuity may also be an advantage therapeutically because such an inhibitor can target more than one transcription factor. Non-selective kinase inhibitors, for example, have been shown to be effective in certain cancers (Wilhelm et al., 2006).
The B-ZIP structural class of transcription factors regulates a variety of genes in different cells involved in cell growth, differentiation and oncogenesis. Provocatively, inhibiting the DNA binding of all B-ZIP domains by NSC13746 was not toxic to NIH3T3 cells. B-ZIP transcription factors may become critically active only during development or in pathological states like cancer. For example, expressing a dominant negative to C/EBP in the basal epidermis of adult mouse skin has minimal effects. However, following a two-stage chemical carcinogenesis protocol, A-CEBP expression prevents skin papilloma formation or causes existing papillomas to regress (Oh et al., 2007).
Oncogenic conversion resulting from translocation events often involves transcription factors (Zucman et al., 1993). Child- hood cancers can be formed by translocation events leading to gene fusions (Berman and Look, 2007). One of the best known examples is the Philadelphia chromosome translocation t(9:22) leading to the gene fusion BCR-ABL that causes chronic myelogenous leukemia (Heisterkamp et al., 1983). Clear cell sarcoma is another example and is caused by a t(12;22)(q13;q12) translocation resulting in the oncogenic gene fusion of EWS-ATF1. EWS acts as an N-terminal trans-activating domain and ATF1 is a B-ZIP domain variant of CREB (Zucman et al., 1993). In our studies, a cell-based growth assay using an EWS-ATF1 patient-derived clear cell sarcoma cell line showed a 25% decrease in cell growth and 50% inhibition of colony formation when incubated with 100 μM NSC13746. The clinical efficacy of NSC13746 will require studies to analyze the effect of the compound on cells that have been transformed with other oncogenes and to understand why the in vivo concentrations are much higher than those used in vitro. Thus, NSC13746 or its analogs may be useful as therapeutic agents in disease systems containing activated B-ZIP transcription factors that are essential for pathological cell proliferation.
These studies demonstrate that arylstibonic acid compounds can enter living cells and function to inhibit B-ZIP DNA binding, a clinically relevant molecular target in systems with activated B-ZIP domains. Additional compounds will be evaluated for increased potency and specificity, potentially using cytoplasmic localization as an assay to screen more inhibitors of transcription factor-DNA binding.
Supplementary Material
Acknowledgements
We thank Dr. Diana Stavreva for the GFP-GR plasmid and Alan Epstein, Department of Pathology, USC Keck School of Medicine for the EWS-ATF1 cell line. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. This research was conducted in partial fulfillment of the Johns Hopkins University/National Cancer Institute, Center for Cancer Research degree in Molecular Targets and Drug Discovery Technologies (Heyerdahl, SL).
Abbreviations:
- B-ZIP domain
basic leucine zipper domain
- CREB
CRE-binding protein
- VBP
vitellogenin gene-binding protein
- C/EBP
CAAT/enhancer binding protein
- AP1
activator protein 1
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescence resonance energy transfer
Footnotes
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ejcb.2009.11.029.
Rishi, in preparation – This manuscript will investigate inhibition of B-ZIP DNA binding in vitro and in vivo by 14 derivatives of the lead compound NSC13778.
References
- Acharya A, Rishi V, Moll J, Vinson C, 2006. Experimental identification of homodimerizing B-ZIP families in Homo sapiens. J. Struct. Biol 155, 130–139. [DOI] [PubMed] [Google Scholar]
- Anderson KI, Sanderson J, Gerwig S, Peychl J, 2006. A new configuration of the Zeiss LSM 510 for simultaneous optical separation of green and red fluorescent protein pairs. Cytometry A 69, 920–929. [DOI] [PubMed] [Google Scholar]
- Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW, 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 16, 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman JN, Look AT, 2007. Targeting transcription factors in acute leukemia in children. Curr. Drug. Targets 8, 727–737. [DOI] [PubMed] [Google Scholar]
- Bhoumik A, Jones N, Ronai Z, 2004. Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci USA 101, 4222–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chida K, Nagamori S, Kuroki T, 1999. Nuclear translocation of Fos is stimulated by interaction with Jun through the leucine zipper. Cell Mol. Life Sci 55, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE Jr., 2002. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2, 740–749. [DOI] [PubMed] [Google Scholar]
- Day RN, Schaufele F, 2005. Imaging molecular interactions in living cells. Mol. Endocrinol 19, 1675–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarco IA, Periasamy A, Booker CF, Day RN, 2006. Monitoring dynamic protein interactions with photoquenching FRET. Nat. Methods 3, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Shimizu T, Toda T, Yanagida M, Hakoshima T, 2000. Structural basis for the diversity of DNA recognition by bZIP transcription factors. Nat. Struct. Biol 7, 889–893. [DOI] [PubMed] [Google Scholar]
- Gerdes MJ, Myakishev M, Frost NA, Rishi V, Moitra J, Acharya A, Levy MR, Park SW, Glick A, Yuspa SH, Vinson C, 2006. Activator protein-1 activity regulates epithelial tumor cell identity. Cancer Res 66, 7578–7588. [DOI] [PubMed] [Google Scholar]
- Heisterkamp N, Stephenson JR, Groffen J, Hansen PF, de Klein A, Bartram CR, Grosveld G, 1983. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 306, 239–242. [DOI] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, Kerppola TK, 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798. [DOI] [PubMed] [Google Scholar]
- Hurst HC, 1995. Transcription factors 1: bZIP proteins. Protein. Profile 2, 101–168. [PubMed] [Google Scholar]
- Inaba T, Inukai T, Yoshihara T, Seyschab H, Ashmun RA, Canman CE, Laken SJ, Kastan MB, Look AT, 1996. Reversal of apoptosis by the leukaemia-associated E2A-HLF chimaeric transcription factor. Nature 382, 541–544. [DOI] [PubMed] [Google Scholar]
- Kohler JJ, Metallo SJ, Schneider TL, Schepartz A, 1999. DNA specificity enhanced by sequential binding of protein monomers. Proc. Natl. Acad Sci. USA 96, 11735–11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Deng X, Shyu YJ, Li JJ, Taparowsky EJ, Hu CD, 2006. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. Embo. J 25, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr BM, Guzman E, Montminy M, 2005. Glutamine rich and basic region/ leucine zipper (bZIP) domains stabilize cAMP-response element-binding protein (CREB) binding to chromatin. J. Biol. Chem 280, 15103–15110. [DOI] [PubMed] [Google Scholar]
- Moll JR, Olive M, Vinson C, 2000. Attractive interhelical electrostatic interactions in the proline- and acidic-rich region (PAR) leucine zipper subfamily preclude heterodimerization with other basic leucine zipper subfamilies. J. Biol. Chem 275, 34826–34832. [DOI] [PubMed] [Google Scholar]
- Mueller F, Wach P, McNally JG, 2008. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys. J 94, 3323–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV, 1999. MYC oncogenes and human neoplastic disease. Oncogene 18, 3004–3016. [DOI] [PubMed] [Google Scholar]
- Newman JR, Keating AE, 2003. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 300, 2097–2101. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Rishi V, Orosz A, Gerdes MJ, Vinson C, 2007. Inhibition of CCAAT/ enhancer binding protein family DNA binding in mouse epidermis prevents and regresses papillomas. Cancer Res 67, 1867–1876. [DOI] [PubMed] [Google Scholar]
- Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C, 1997. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J. Biol. Chem 272, 18586–18594. [DOI] [PubMed] [Google Scholar]
- Paletzki RF, Myakishev MV, Polesskaya O, Orosz A, Hyman SE, Vinson C, 2008. Inhibiting activator protein-1 activity alters cocaine-induced gene expression and potentiates sensitization. Neuroscience 152, 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, Brown DT, Hager G, Bustin M, Misteli T, 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell Biol 24, 6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishi V, Potter T, Laudeman J, Reinhart R, Silvers T, Selby M, Stevenson T, Krosky P, Stephen AG, Acharya A, Moll J, Oh WJ, Scudiero D, Shoemaker RH, Vinson C, 2005. A high-throughput fluorescence-anisotropy screen that identifies small molecule inhibitors of the DNA binding of B-ZIP transcription factors. Anal Biochem 340, 259–271. [DOI] [PubMed] [Google Scholar]
- Rozenberg J, Rishi V, Orosz A, Moitra J, Glick A, Vinson C, 2009. Inhibition of CREB function in mouse epidermis reduces papilloma formation. Mol. Cancer Res 7, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaufele F, Enwright JF 3rd, Wang X, Teoh C, Srihari R, Erickson R, MacDougald OA, Day RN, 2001. CCAAT/enhancer binding protein alpha assembles essential cooperating factors in common subnuclear domains. Mol. Endocrinol 15, 1665–1676. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY, 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Tang QQ, Lane MD, 1999. Activation and centromeric localization of CCAAT/ enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev 13, 2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramier M, Zahid M, Mevel JC, Masse MJ, Coppey-Moisan M, 2006. Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photobleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells. Microsc. Res. Tech 69, 933–939. [DOI] [PubMed] [Google Scholar]
- Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR, Bonovich M, 2002. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell Biol 22, 6321–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt PK, 2001. Jun, the oncoprotein. Oncogene 20, 2365–2377. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S, 2006. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov 5, 835–844. [DOI] [PubMed] [Google Scholar]
- Williams SC, Angerer ND, Johnson PF, 1997. C/EBP proteins contain nuclear localization signals imbedded in their basic regions. Gene Expr 6, 371–385. [PMC free article] [PubMed] [Google Scholar]
- Zucman J, Delattre O, Desmaze C, Epstein AL, Stenman G, Speleman F, Fletchers CD, Aurias A, Thomas G, 1993. EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 4, 341–345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.