

Abstract
Protein folding in the endoplasmic reticulum is an oxidative process that relies on protein disulfide isomerase (PDI) and endoplasmic reticulum oxidase 1 (ERO1). Over 30% of proteins require the chaperone PDI to promote disulfide bond formation. PDI oxidizes cysteines in nascent polypeptides to form disulfide bonds and can also reduce and isomerize disulfide bonds. ERO1 recycles reduced PDI family member PDIA1 using a FAD cofactor to transfer electrons to oxygen. ERO1 dysfunction critically affects several diseases states. Both ERO1 and PDIA1 are overexpressed in cancers and implicated in diabetes and neurodegenerative diseases. Cancer-associated ERO1 promotes cell migration and invasion. Furthermore, the ERO1-PDIA1 interaction is critical for epithelial-to-mesenchymal transition. Co-expression analysis of ERO1A gene expression in cancer patients demonstrated that ERO1A is significantly upregulated in lung adenocarcinoma (LUAD), glioblastoma and low-grade glioma (GBMLGG), pancreatic ductal adenocarcinoma (PAAD), and kidney renal papillary cell carcinoma (KIRP) cancers. ERO1A knockdown gene signature correlates with knockdown of cancer signaling proteins including IGF1R, supporting the search for novel, selective ERO1 inhibitors for the treatment of cancer. In this review, we explore the functions of ERO1 and PDI to support inhibition of this interaction in cancer and other diseases.
Keywords: Protein Folding, Targeted Therapy, Cancer, Endoplasmic Reticulum Oxidase, Protein Disulfide Isomerase, Gene Expression
1. Introduction
Reactive oxygen species (ROS) are partially reduced metabolites of oxygen with strong oxidizing capabilities that contribute to diseases related to cell metabolism, survival and death. There are two main classes of ROS: free radicals and non-radicals. Free radicals include hydroxyl radical (HO•), nitric oxide (•NO), peroxynitrite (ONOO−), superoxide anion (O2•−), nitrogen dioxide (•NO2), peroxyl radicals (ROO•), and lipid peroxyl (LOO•). Non-radicals include hydrogen peroxide (H2O2), singlet oxygen (1O2) and lipid peroxide (LOOH). The most well-understood ROS are hydrogen peroxide (H2O2), superoxide anion (O2•−), and hydroxyl radical (HO•) (Li, Jia, & Trush, 2016). At low doses, ROS maintain cellular homeostasis as secondary signaling molecules, and, at high concentrations, ROS can induce severe oxidative damage in proteins, lipids and DNA, earning their reputation as a double-edged sword (Cao & Kaufman, 2014; Puspita, Chung, & Shim, 2017; Sifuentes-Franco, Pacheco-Moisés, Rodríguez-Carrizalez, & Miranda-Díaz, 2017).
ROS are generated via both non-enzymatic and enzymatic reactions. ROS generated as byproducts of oxidative phosphorylation and ATP production during electron transfer reactions in the mitochondria are the major source (Fig. 1). ATP generation depends on the mitochondrial respiratory chain in the inner mitochondrial membrane, which consists of 5 major protein complexes (complex I, II, III, IV, and V) (Bolisetty & Jaimes, 2013). ATP is synthesized by F0F1-ATPase via the proton gradient across the membrane, and during this electron transfer, molecular oxygen accepts leaked electrons to form ROS (Dromparis & Michelakis, 2013). Complex I (NADH dehydrogenase subunits) and complex III (ubiquinol-cytochrome C reductase complex subunits) are the major sites of electron leak in the mitochondrial respiratory chain (Borek, Sarewicz, & Osyczka, 2008; Warnau, et al., 2018). Furthermore, complex II (succinate dehydrogenase) also plays a role in ROS generation, and mutation or dysfunction of complex II can enhance ROS production (Kausar, Wang, & Cui, 2018). In addition to the mitochondria, ROS are also produced by NADPH oxidases and xanthine oxidase in the cytoplasm, and generated in the endoplasmic reticulum (ER), peroxisome, and other organelles (Cao & Kaufman, 2014).
Fig. 1.
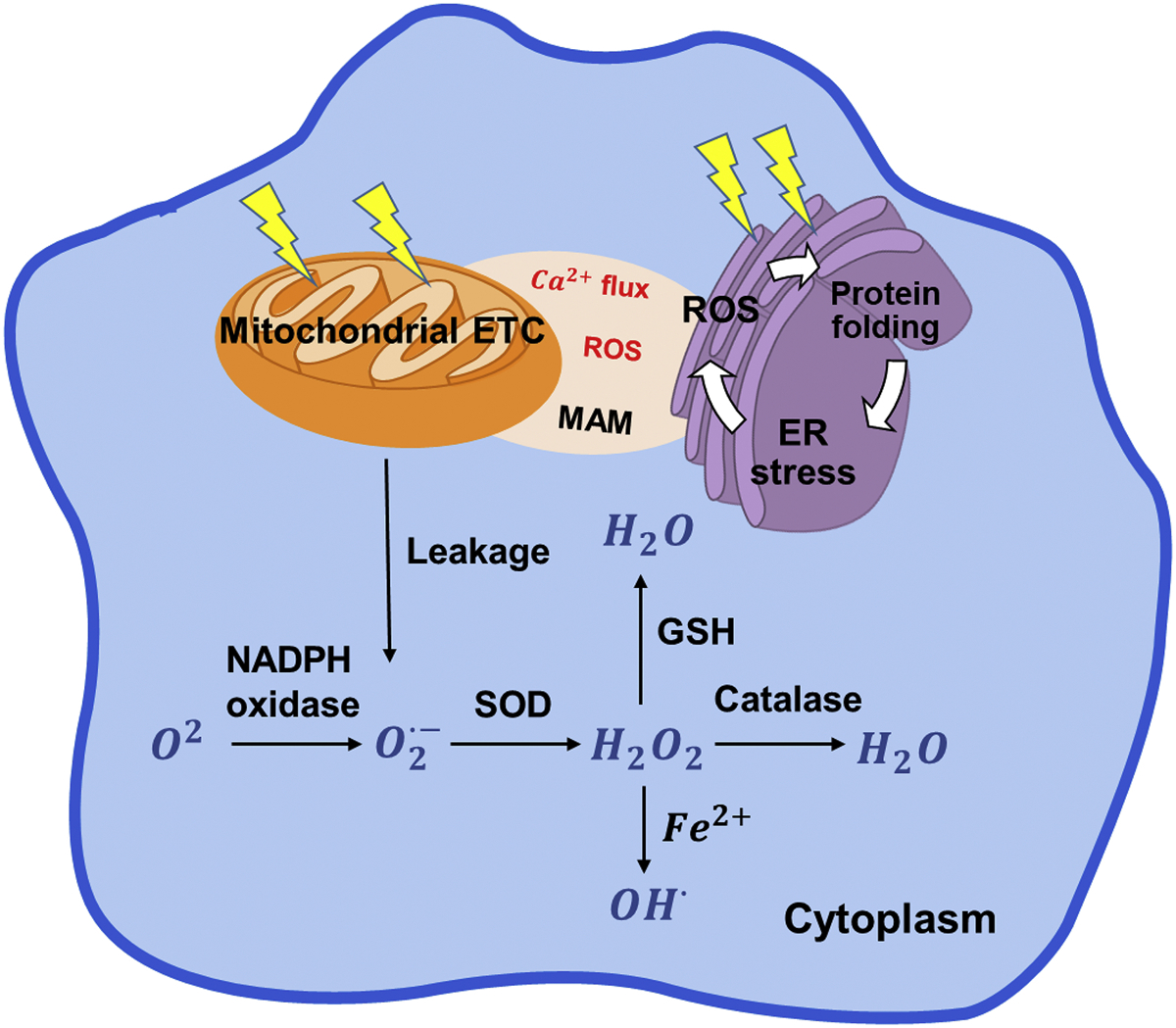
General ROS production in the cell cytoplasm, mitochondria and ER. The various types of ROS are generated based on cell requirements and extracellular stimuli. Mitochondria and ER are the two major organelles that control ROS signaling.
The purpose of this review is to address the dynamics of the ERO1-PDI interaction in intracellular ROS signaling and explore the rationale behind ERO1 inhibition in disease. ERO1 contributes up to 25% of the induced cellular ROS especially in heavily secretory cells (Chaudhari, Talwar, Parimisetty, Lefebvre d’Hellencourt, & Ravanan, 2014; Sevier & Kaiser, 2008; Tu & Weissman, 2004). ROS can act in several ways, including as signaling molecules to downstream pathways, and as cytotoxic elements. Furthermore, ERO1α (an ERO1 isoform) expression is upregulated in several cancers and plays an important role in the physiology of many diseases. These findings highlight a strong therapeutic potential for effectively targeting ERO1α and PDI as novel inhibitors become available.
2. Endoplasmic reticulum as an alternative source of ROS
Although mitochondria hold the reputation as the primary, ROS-inducing organelles of the cell, oxidative protein folding in the ER generates large quantities of ROS, and, in fact, the ER lumen contains more ROS than mitochondria (Bulleid & Ellgaard, 2011; Malinouski, Zhou, Belousov, Hatfield, & Gladyshev, 2011). In eukaryotes, the ER is responsible for secreted and membrane-bound protein folding and calcium storage. ROS are generated during oxidative protein folding, when molecular chaperones, such as protein disulfide isomerases (PDIs), form disulfide bonds in nascent polypeptides. Active site cysteine residues in PDI accept electrons from free thiols in nascent polypeptides to generate a disulfide bond. PDI then transfers the electrons to membrane-bound ERO1 to perform another cycle of catalysis (Cao & Kaufman, 2014). ERO1 further transfers the electrons to O2 and generates H2O2. H2O2 produced by ERO1 in the ER lumen is involved in signaling and oxidative protein folding via peroxiredoxin 4 (Tavender & Bulleid, 2010; Zito, Melo, et al., 2010). In response to ROS generation, antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, maintain redox homeostasis by converting ROS into water and oxygen. Other antioxidant molecules, including reduced glutathione (GSH), vitamin E, and NADH, can also inactivate ROS (Fig. 1) (Gomes, Silva, & de Oliveira, 2012; Tse, Yan, Chan, Tian, & Huang, 2016). In the ER, ROS can also be produced by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases downstream of PDIA1 activity (Laurindo, Araujo, & Abrahão, 2014).
The ER and mitochondria interact via a physical contact known as the mitochondria-associated ER membrane (MAM). The MAM was first identified in 1986 as a major site of phospholipid synthesis and transportation and is now known to be essential for calcium homeostasis, energy metabolism and other signaling pathways (Rieusset, 2018; Vance & Vance, 1986) (Csordas, Weaver, & Hajnoczky, 2018; Thoudam, Jeon, Ha, & Lee, 2016). The MAM allows exchange of Ca2+, ROS, lipids, and nutrients between the ER and the mitochondria, thus promoting cellular bioenergetics and metabolism (Fig. 1). High concentrations of ER Ca2+ are released at MAM sites and taken up into the mitochondria by the Ca2+ uniporter on the inner mitochondrial membrane (Csordás, Várnai, Golenár, Sheu, & Hajnóczky, 2012). ROS generated by either the ER or mitochondria in H2O2 nanodomains generated by cristae can localize at the MAM interface and perturb calcium signaling (Booth, Enyedi, Geiszt, Várnai, & Hajnóczky, 2016). Importantly, ERO1, a key regulator of oxidative stress in the ER, is enriched at the MAM interface and regulates calcium flux (Anelli, et al., 2012; Gilady, et al., 2010). ERO1 localization at the MAM interface demonstrates the role of redox signaling on calcium flux through the mitochondria. ERO1 does not cross the MAM but may exert its effect via diffusible H2O2. Dysfunction of the ER-mitochondrial crosstalk at the MAM has been implicated in neurological diseases such as Alzheimer’s and Parkinson’s, thus ERO1 function at this interface may emerge as a critical driver of MAM disfunction (Hetz & Mollereau, 2014).
The oxidizing environment of the ER is maintained by the GSH/GSSG ratio to facilitate protein folding (Fig. 2) (Cao & Kaufman, 2014; Chakravarthi, Jessop, & Bulleid, 2006). Correctly folded and processed proteins are transported out of the ER, while misfolded proteins can be either refolded or degraded via the ER-Associated Degradation (ERAD) pathway (Cao & Kaufman, 2014; Hwang & Qi, 2018). Therefore, the ER is equipped with a regulatory mechanism to accurately differentiate between unfolded/native, misfolded and correctly folded proteins. The rate of the reaction between PDI and GSSG or GSH is rapid, likely contributing to its role as an ER redox sensor (Lappi & Ruddock, 2011). In a disease state, misfolded and unfolded proteins can accumulate to trigger ER swelling and the ER stress response (Oakes & Papa, 2015). During ER stress, the GSH/GSSG ratio is disturbed, further increasing ROS production and distorting the ER redox environment (Tu & Weissman, 2004). In eukaryotes, the ER is more susceptible to oxidative stress due to limited antioxidant enzymes, which indicates its important role in the induction of redox imbalance and cellular stress (Santos, Tanaka, Wosniak, & Laurindo, 2009). ER stress stimulates the unfolded protein response (UPR), which can promote cell survival, or upon prolonged ER stress, apoptosis ensues (Zeeshan, Lee, Kim, & Chae, 2016). Furthermore, ER stress can induce calcium release from the ER into the cytosol. Mitochondria uptake the released Ca2+, causing physical and metabolic changes. For example, ROS induces the release of cytochrome c to the cytoplasm, inhibiting complex III activity and further inducing ROS in the form of a ubisemiquinone radical intermediate (Cao & Kaufman, 2014). Increased Ca2+ ions also enhance mitochondrial Krebs cycle dehydrogenases and nitric oxide synthase, both leading to the elevation of ROS. Most importantly, in a vicious cycle, the ER-induced mitochondrial ROS may accelerate release of Ca2+ from the ER to further increase mitochondrial oxidative stress (Cao & Kaufman, 2014). ERO1 silencing or pharmacological inhibition (EN460) blocks calcium re-uptake by mitochondria but not by the ER (Anelli, et al., 2012). Mitochondrial dysfunction alters ATP generation, which is required for protein folding and bond formation in the ER. Thus, mitochondrial dysfunction further aggravates ER stress (Cao & Kaufman, 2014).
Fig. 2.
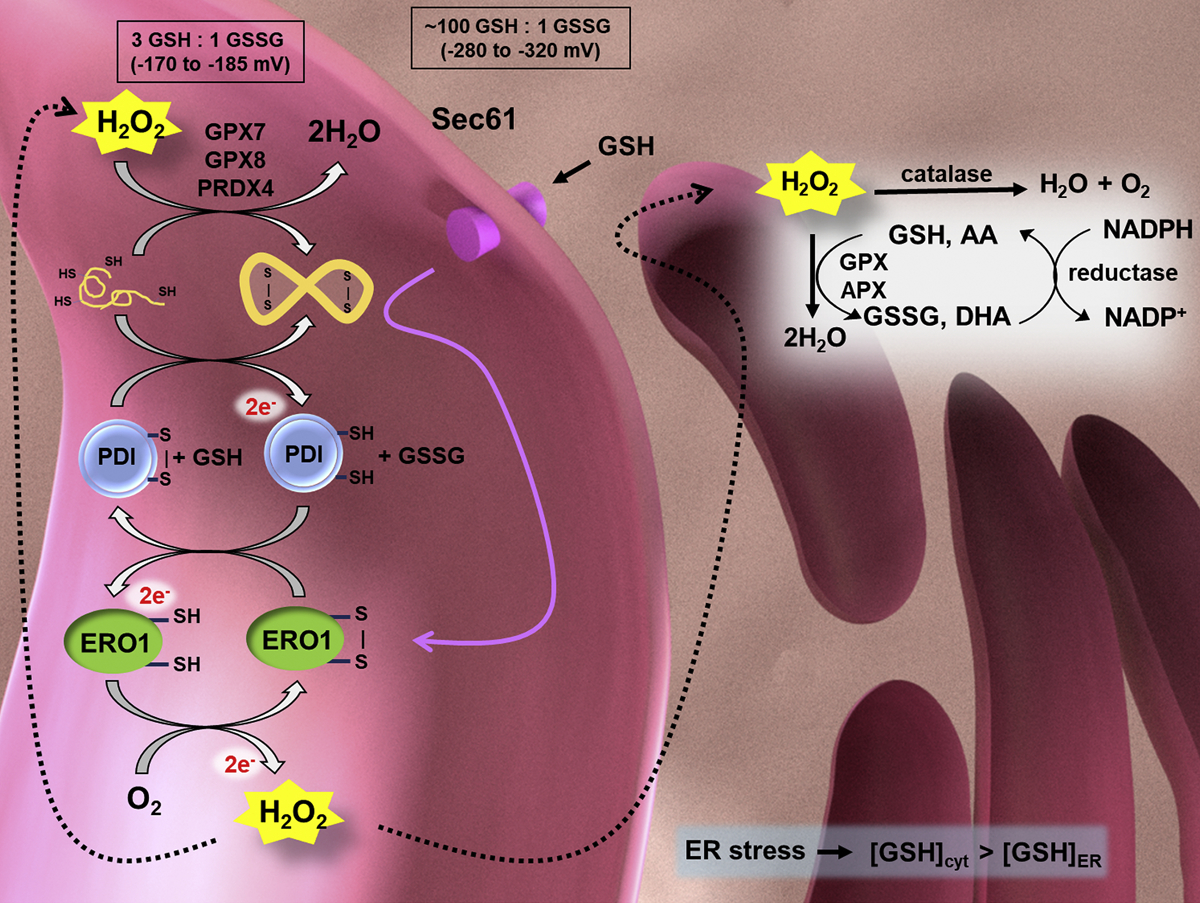
The ER (pink) is an oxidizing environment, maintained by redox sensor glutathione (GSH/GSSG), with a reduction potential much larger than that of the cytoplasm (ER ε: −170 to − 185 mV; cytoplasm ε: −280 to −320 mV). The oxidizing environment promotes nascent protein folding and disulfide bond formation. ERO1 is a key mediator of disulfide bond formation in the ER. Disturbed protein folding may cause ER stress and increased ER ROS production, further affecting the mitochondrial and cellular metabolism. Reduced polypeptides are oxidized by PDI, which transfers its electrons to ERO1. ERO1 is reoxidized by oxygen and produces H2O2. H2O2 is reduced through various mechanisms in the ER including catalase, glutathione peroxidases (GPX7 and GPX8), peroxiredoxin 4 (PRX4), and ascorbate peroxidase (APX). Background image created in Blender 2.79.
3. ERO1 function & link to PDIA1
Endoplasmic reticulum oxidoreductase 1 (ERO1) is an ER-resident thiol oxidoreductase responsible for catalyzing disulfide bond formation in nascent polypeptide substrates via electron transfer through protein disulfide isomerase (PDI) with oxygen acting as the final electron acceptor (Fig. 2) (Frand & Kaiser, 1999). The enzyme was first studied in Saccharomyces cerevisiae as an essential component of the oxidative folding machinery (Tu & Weissman, 2004). ERO1 is highly conserved and uses flavin adenine dinucleotide (FAD) as a coenzyme for electron transfer during oxidative folding. The ERO1/PDI oxidative folding pathway releases H2O2; thus, increased oxidative protein folding is a source of ER ROS.
Saccharomyces cerevisiae contains a single homolog, ERO1p, that is essential for growth. In mammals, there are two paralogs of ERO1, ERO1α and ERO1β, and these enzymes are two of many enzymes that perform similar functions, highlighting the importance of the oxidative folding pathway. ERO1α is ubiquitously expressed in most cells, whereas ERO1β is specifically expressed in cells of the pancreas and stomach (Dias-Gunasekara, et al., 2005). ERO1β is more active than ERO1α in vitro, and is highly expressed in the pancreas, emphasizing its importance in insulin biogenesis and glucose homeostasis (Zito, Chin, Blais, Harding, & Ron, 2010). ERO1α, encoded by the ERO1A gene, is induced by HIF1α (hypoxia-inducible factor 1α) and hypoxic conditions, whereas ERO1β is induced by the unfolded protein response (Cabibbo, et al., 2000; Gess, et al., 2003). Both isoforms are globular folds of alpha helices containing two essential CXXCXXC active sites and a regulatory loop region. Though the isoforms share 65.4% amino acid identity, ERO1β is missing the EF-hand calcium-binding motif contained in ERO1α. Furthermore, ERO1α contains two sites for N-glycosylation, Asp280 and Asp384. Crystal structures of human ERO1α were solved in 2010 (Fig. 3A) (Inaba, et al., 2010). The electron shuttle from reduced PDI to molecular oxygen facilitated by ERO1 is tightly regulated, and ERO1 activity is heavily dependent on the redox characteristics of PDI.
Fig. 3.
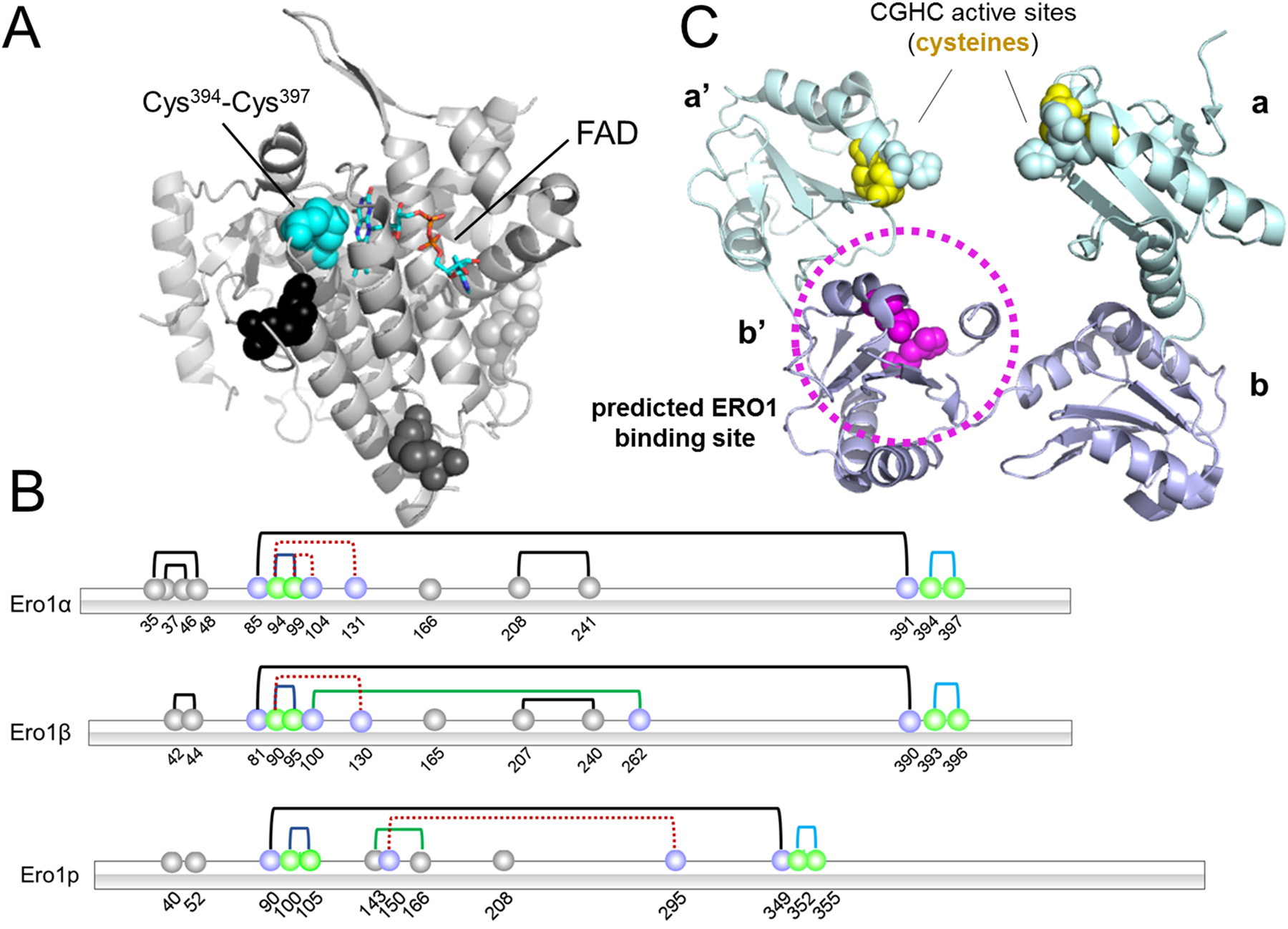
Structures of ERO1 and PDI. A) Hyperactive ERO1α (3AHQ) (Inaba, et al., 2010), with FAD moiety represented as a stick model. Blue spheres represent active site cysteines. Black, dark gray and light gray spheres indicate structural disulfides. B) A schematic of the disulfide bonds of ERO1α, ERO1β, and ERO1p (blue line - active site disulfides, pale orange line - flexible loop shuttle disulfides, black line - structural disulfides, dashed red line - regulatory cysteines (inactive ERO1), green line - auxiliary regulatory disulfides). ERO1p is a Saccharomyces cerevisiae homolog. C) Reduced PDI (4EKZ) is predicted to bind ERO1α via the substrate-binding pocket in the b’ domain (circled in magenta). Active site cysteines are depicted in yellow.
Tight regulation is crucial as unregulated ERO1 activity would lead to harmful concentrations of hydrogen peroxide, oxidative stress, and cell death. Active, partially oxidized ERO1α (OX1) can resist changes that could be induced by a reducing environment, whereas, inactive ERO1α (OX2) is readily reduced by dithiothreitol (Benham, van Lith, Sitia, & Braakman, 2013). Therefore, in the oxidizing environment of the ER, inactive, oxidized ERO1α is well-suited to donate a disulfide bond to PDI. ERO1 activity is regulated by disulfide bond combinations of four cysteines. Active ERO1α (OX1) contains a Cys94-Cys99 disulfide bond. ERO1α is inactivated when those cysteines form bonds with other cysteines in the protein, to form two disulfide bond pairs (OX2): Cys94-Cys131 and Cys99-Cys104 (Fig. 3B). ERO1β is similar, with a Cys90-Cys95 disulfide bond in the active form that is broken in the inactive form (OX: Cys90-Cys130). Upon reduction, ERO1α moves from the compact, inactive OX2 form to the more active OX1, and rapidly returns to the OX2 form when no longer needed (Benham, et al., 2013). A Cys81-Cys390 disulfide stabilizes ERO1β by linking the “loop cap” and “helical core.” A regulatory bond similar to the Cys94-Cys131 bond in ERO1α also exists as Cys90-Cys130 in inactive ERO1β (Wang, Zhu, & Wang, 2011). In general, ERO1β seems to be less tightly regulated than ERO1α and brings powerful oxidizing capacity when needed (Wang, et al., 2011).
An increase in protein folding for which the cell does not have biomolecular capacity could lead to toxic buildup of ROS molecules, ER oxidative stress, and apoptosis. The highly oxidizing environment of the ER is maintained by ERO1 and GSSG, though glutathione enters the ER in its reduced form and also provides potent reducing equivalents at high concentrations (Tu, Ho-Schleyer, Travers, & Weissman, 2000). In normal cells, H2O2 generation as a consequence of ERO1-mediated disulfide bond formation is tightly regulated, and H2O2 is quickly reduced by glutathione peroxidase 8 (GPX8) (Ramming, Hansen, Nagata, Ellgaard, & Appenzeller-Herzog, 2014). Overexpression of ERO1p increases ROS in yeast cells, and those ROS levels are exacerbated even further with GPX8 knockdown (Haynes, Titus, & Cooper, 2004; Ramming, et al., 2014). Thus, while ER ROS levels can exceed mitochondrial concentrations, increases in protein folding can be mitigated by overexpression of UPR-inducible GPX8 (Eletto, Chevet, Argon, & Appenzeller-Herzog, 2014). Furthermore, additional substrates besides O2 could act as electron acceptors because, while the enzyme has a high affinity for oxygen, it can also function under anaerobic conditions in yeast cells (Tu & Weissman, 2002). In cervical cancer, ERO1-dependent H2O2 promotes growth and migration via promotion of epithelial-to-mesenchymal transition (EMT) (Zhang, et al., 2019). Thus, ERO1 is a key modulator of redox homeostasis in the ER.
Although ERO1 is the major contributor to ROS generation in the ER, knockdown in normal cells is not detrimental. ERO1p knockdown in yeast (S. cerevisiae) impairs the redox environment of the ER and disrupts protein folding (Frand & Kaiser, 1998; Pollard, Travers, & Weissman, 1998). ERO1β knockdown in mice hinders proinsulin folding, while double knockdown of ERO1α and ERO1β in immunoglobulin-producing cells does not affect disulfide bond formation or secretion of immunoglobulin (Zito, Chin, et al., 2010). Thus, ERO1β plays a selective and important role in glucose homeostasis (Zito, Chin, et al., 2010). The modest effect of ERO1 knockout on phenotype indicates mammalian cells rely on redundant pathways for disulfide bond formation and ERO1-independent mechanisms. For example, ERO1-deficient cells still form disulfide bonds (Appenzeller-Herzog, et al., 2010). Knockout of ERO1α, ERO1β, and the antioxidant enzyme peroxiredoxin 4 (PRDX4) leads to a deficiency in procollagen maturation, depletion of ascorbic acid, and scurvy in mice (Zito, Hansen, Yeo, Fujii, & Ron, 2012). In cervical cancer cells, knockdown of ERO1 impaired tumorigenesis (Zhang, et al., 2019).
Whole body knockout of ERO1β specifically impairs pancreatic β cell function because ERO1β is expressed in the pancreas; in addition to the pancreas, ERO1β is selectively expressed in the testis, liver, appendix, thyroid, and pituitary gland (Pagani, et al., 2000; Zito, Chin, et al., 2010). This specific tissue expression may explain the fact that, in general, ERO1B was not expressed as highly as ERO1A in our TCGA pan-cancer RNA-Seq data survey. Figure 5 shows expression of ERO1A or ERO1B that has been z-score normalized per patient so that a z-score of 0 indicates average expression of all genes per patient. All TCGA cohorts had ERO1A median expression greater than zero, and nearly all cohorts had ERO1B median expression greater than zero. Median ERO1B expression was higher than median expression compared to ERO1A expression in PRAD, though the relevance for this has yet to be explored. ERO1B had the highest median expression in LIHC and PAAD; thus, ERO1β inhibition may be a potential treatment for pancreatic cancer. Furthermore, ERO1β knockout renders cells susceptible to ER stress; the capability of normal cells to withstand a level of ER stress may promote ERO1β as a selective inhibitor of tissue-specific cancers, such as pancreatic or prostate (Khoo, et al., 2011). However, because ERO1β plays such a significant role in insulin signaling, ERO1β inhibitors may cause side effects such as glucose intolerance. In addition, it has not yet been studied whether ERO1β would be expressed to compensate for the loss of ERO1α activity or if ERO1β is induced upon ER stress in tissues besides the ones in which basal expression is high. For example, in the progression of diabetes, as ER stress increases, ERO1B transcript expression decreases, unlike other UPR genes Bip and CHOP (Awazawa, et al., 2014). To consider the importance of ERO1β expression in cancer, we must establish 1) whether ERO1β expression is induced in cancerous tissues and 2) whether ERO1β expression induced by the UPR is critical for cancer progression. Based on the TCGA patient ERO1 expression data, median ERO1B expression is higher than average gene expression (zScore = 0) in most cancers assessed. Thus, it will be important to determine whether this higher than average expression is a critical driver of cancer progression.
Fig. 5.
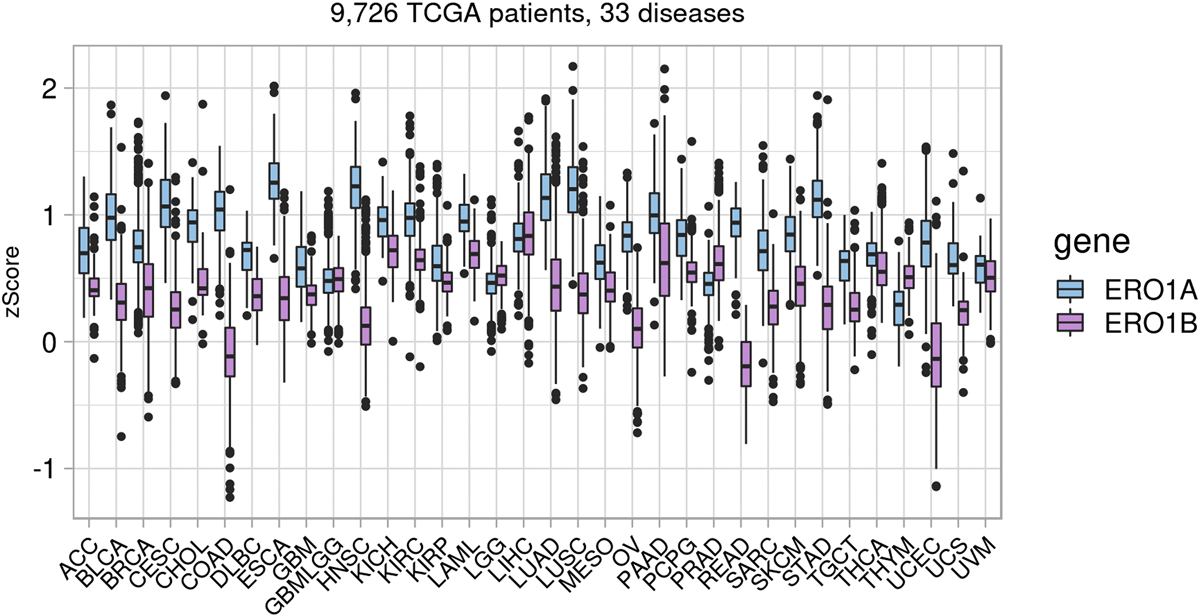
Pan-disease mRNA expression of ERO1A and ERO1B across 33 TCGA diseases. Z-scores were calculated per patient per disease. The majority of patients across all diseases show higher than average expression (z-score = 0) of ERO1A. Adrenocortical carcinoma (ACC), bladder urothelial carcinoma (BLCA), breast invasive carcinoma cohort (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney chromophobe (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), acute myeloid leukemia (LAML), brain low-grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), mesothelioma (MESO), ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), sarcoma (SARC), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), testicular germ cell tumors (TGCT), thyroid carcinoma (THCA), thymoma (THYM), uterine corpus endometrial carcinoma (UCEC), uterine carcinosarcoma (UCS), uveal melanoma (UVM).
ERO1α mediates rapid disulfide bond generation in oxidative protein folding via PDIA1 (Appenzeller-Herzog, et al., 2010). PDI is an abundant multifunctional oxidoreductase catalyzing disulfide bond formation, isomerization, and reduction (Shergalis & Neamati, 2017). In vascular smooth muscle cells where ROS are key signaling molecules, PDIA1 regulates NADPH oxidase activity (Laurindo, Fernandes, Amanso, Lopes, & Santos, 2008). PDIA1 (also known as PDI, but referred to in this review as PDIA1 for clarity), the canonical member of the PDI family of 22 members, is the major substrate of ERO1α. Additionally, ERO1β preferentially associates with PDIA1 and PDIp, among the other PDI family members (Wang, et al., 2011). PDIA1 contains catalytic a and a’ thioredoxin-like domains, a conserved CXXC active site, and b and b’ domains that are homologous to the a and a’ domains and also contain the thioredoxin-like fold, without the CXXC active site (Hatahet & Ruddock, 2009). ERO1α preferentially oxidizes the a’ domain of human PDIA1 over the a domain (Wang, et al., 2008). The cysteines in the CGHC active site of PDIA1 have high biochemical reduction potentials (−180 mV) and are poised to accept electrons from nascent polypeptides (Lundström & Holmgren, 1993). ERO1α interacts with PDIA1 via hydrophobic and electrostatic interactions in the b’ domain of PDIA1 (Fig. 3C). Specifically, Phe240, Phe249, and Phe304 in the b’ domain likely form non-covalent interactions with the β-hairpin region of ERO1α (Masui, Vavassori, Fagioli, Sitia, & Inaba, 2011). Furthermore, Val101 and Trp272 of ERO1α are necessary for PDI-ERO1 complex formation (Zhang, et al., 2019). These interactions position Cys397 and Cys400 in the a’ domain active site of PDIA1 near the disulfide-bonded cysteines in the shuttle loop of ERO1α and allow ERO1α to perform the disulfide exchange.
ERO1α activity is regulated by the redox state of PDI in the ER. An abundance of reduced PDIA1, or a PDIA1 molecule that has transferred its disulfide bond to an unfolded protein, can activate ERO1α by reducing the regulatory disulfide bonds (Appenzeller-Herzog, Riemer, Christensen, Sørensen, & Ellgaard, 2008). Reduced PDIA1 has a much higher affinity for ERO1α than oxidized PDIA1, and oxidation generates one molecule of H2O2 (Masui, et al., 2011). The generated H2O2 is typically scavenged by ER peroxidases PRDX4, GPX7, and GPX8, before it diffuses out of the cellular compartment (Ramming & Appenzeller-Herzog, 2013; Wang, Zhang, Niu, Sitia, & Wang, 2014). GPX7 and PRDX4 oxidize PDIA1 and catalyze disulfide bond formation independent of ERO1 (Zito, Melo, et al., 2010). Overexpression of ERO1 shifts PDIA1 towards an oxidized state, which promotes oxidation of PDIA1 substrates (Bhandary, Marahatta, Kim, & Chae, 2012). Inhibition of ERO1 may prevent or slow reoxidation of PDIA1 and impair protein folding in tumor cells.
Post-translational modifications on folding chaperones can have downstream signaling and protein folding consequences. For example, nitrosative stress generated by sustained levels of nitric oxide, often during chronic inflammation, can lead to S-glutathionylation of proteins (Hofseth, Hussain, Wogan, & Harris, 2003; Townsend, et al., 2009). Furthermore, S-nitrosylation of PDIA1 can lead to sulfinic acid (−SO2H) modification. S-glutathionylation is a post-translational modification on cysteine residues and an indicator of redox stress. Furthermore, nitrosative stress has been shown to be induced by electrophilic chemotherapy drugs, primarily affecting ER-located proteins (Townsend, 2007). Because ER protein folding relies on robust redox signaling, post-translational modifications arising from ER stress may have unexpected consequences. However, the ER is built to handle flux in redox signaling as part of its mechanisms for UPR activation. For example, S-glutathionylation of PDIA1 induced by nitrosative stress from O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl]1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO) activates the PERK and IRE1 branches of the UPR (Townsend, et al., 2009). PERK activation promotes protein degradation pathways, and IRE1 activation halts protein translation. Similarly, studies have shown that S-glutathionylation of PDIA1 by glutathione S-transferase P (GSTP) is cytoprotective when ER stress is induced (Ye, et al., 2017). S-glutathionylation, along with cysteine nitrosylation and sulfenilation, of PDIA1 was observed upon exposure to chemicals in cigarette smoke and also induced the UPR (Kenche, et al., 2016). Thus, S-glutathionylation of PDIA1 halts protein folding until cell homeostasis is restored.
While PDIA1 is the canonical ERO1 partner protein, other PDI family members have been shown to interact with ERO1. For example, ERp44 is a PDI family member that may have the ability to regulate ERO1α-PDIA1 complex formation and enable ERO1 retention in the ER (Anelli, et al., 2002; Masui, et al., 2011). ERp44 is induced during ER stress and associates with ERO1α at the same rate as PDIA1, however, its dissociation rate is faster (Masui, et al., 2011). ERp44 also interacts with inositol trisphosphate receptor and inhibits its function (Li, et al., 2009). Inositol trisphosphate receptor maintains balanced calcium flux between the ER and mitochondria and activity can be regulated by oncogenes to promote tumor growth. Hyperoxidation via increases in ROS levels disrupt the interaction between ERp44 and inositol trisphosphate receptor to lead to calcium release and apoptosis (Li, et al., 2009). ERO1 can also form mixed disulfides with PDI family members ERp57 and ERp72 (Appenzeller-Herzog, et al., 2010). ERp57 associates with ERO1α in vivo, suggesting ERO1α has the ability to oxidize ERp57 (Jessop, et al., 2007). ERp72, a PDI family member with a CGHC active site similar to ERp44 and PDIA1, also binds to ERO1α, likely in an active site-independent manner (Araki, et al., 2013). Therefore, while PDIA1 is the canonical ERO1 partner protein, other PDI family members may be involved in the link between ERO1 function and disease.
Because of the critical role of PDIA1 in protein folding, it has been linked to various diseases including cancer (Xu, Sankar, & Neamati, 2014), thrombosis (Bekendam & Flaumenhaft, 2016), Huntington’s disease (Zhou, et al., 2018), Parkinson’s disease (Cheng, Wang, & Wang, 2010), and diabetes (Zhang, Lai, Teodoro, & Volchuk, 2008). In several cancers, PDIA1 is overexpressed to meet the increased proliferation demands of the tumor. PDIA1 regulates integrin-mediated platelet function, thus, inhibition of PDIA1 has shown promise as a potential thrombosis inhibitor (Bekendam & Flaumenhaft, 2016). In neurodegenerative diseases, PDIA1 is overexpressed and causes apoptosis, and modulation of PDIA1 with small molecule inhibitors is neuroprotective in Huntington’s disease models (Conn, et al., 2004; Duennwald & Lindquist, 2008; Lee, et al., 2010; Zhou, et al., 2018). In diabetes, PDIA1 knockdown promotes proinsulin export and overexpression of PDIA1 leads to proinsulin retention in the organelle (Rajpal, Schuiki, Liu, Volchuk, & Arvan, 2012; Zhang, et al., 2008). Thus, PDIA1 seems to prevent insulin export and may be a promising target for inhibition in diabetes (Sun, et al., 2015; Zhang, et al., 2008). Because of the ubiquitous nature of PDIA1 function, the enzyme is an attractive drug target and multiple inhibitors and modulators have been identified for various indications; however, PDIA1 inhibitors are still in preclinical and early stage clinical studies. Pharmacological inhibition of PDIA1 by bisphenol A blocks ERO1α binding in the b’ domain and subsequently inhibits PDIA1-ERO1α-mediated disulfide bond formation (Okumura, et al., 2014). Furthermore, blocking the PDIA1-ERO1α interaction suppresses cervical cancer growth, thus providing validation for a b’ domain PDIA1 inhibitor in the context of cancer (Zhang, et al., 2019).
As ER-resident enzymes, both ERO1 and PDI are critically involved in the UPR. Under normal conditions, the UPR responds to the protein synthesis load and mediates the protein folding machinery by upregulating chaperones, halting protein synthesis, or initiating cell death (Hetz, 2012). ER stress, or an imbalance between the newly synthesized proteins and machinery expressed and ready to fold and secrete them, triggers the UPR via three major arms: protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6). The UPR affects downstream signaling in a complicated manner dependent on physiological conditions (Hetz, Chevet, & Harding, 2013). For example, in cancer, the UPR can be activated by the hypoxic environment and can contribute to tumor growth (Hetz, et al., 2013; Moenner, Pluquet, Bouchecareilh, & Chevet, 2007; Rouschop, et al., 2010).
Both ERO1α and ERO1β respond to ER stress; however, ERO1α is induced by hypoxic conditions, while ERO1β is induced by the UPR (Pagani, et al., 2000). ERO1α expression is regulated by the ER stress transcription factor CHOP (C/EBP homologous protein) (Marciniak, et al., 2004). CHOP is activated downstream of phosphorylation of eukaryotic initiation factor 2α under the PERK arm of the UPR and activates the translation of several ER stress response proteins (Fig. 4). Increased protein synthesis leads to an increase in ROS and ERO1α knockdown actually rescues cell death when CHOP and ATF4 are activated (Han, et al., 2013). However, under prolonged ER stress, increased disulfide bond formation can produce excess levels of H2O2 that can leak out of the ER and cause apoptosis (Tu & Weissman, 2004). It has been suggested that ERO1-mediated generation of H2O2 during ER stress leads to apoptosis; however, the origin of ROS may actually be the mitochondria (Bulleid & Ellgaard, 2011). Furthermore, overexpressing ERO1 in yeast did not increase ROS levels (Sevier, et al., 2007). ER stress leads to calcium release from the ER into the cytoplasm, increased mitochondrial calcium uptake, increased mitochondrial metabolism, and higher levels of ROS (Chaudhari, et al., 2014).
Fig. 4.
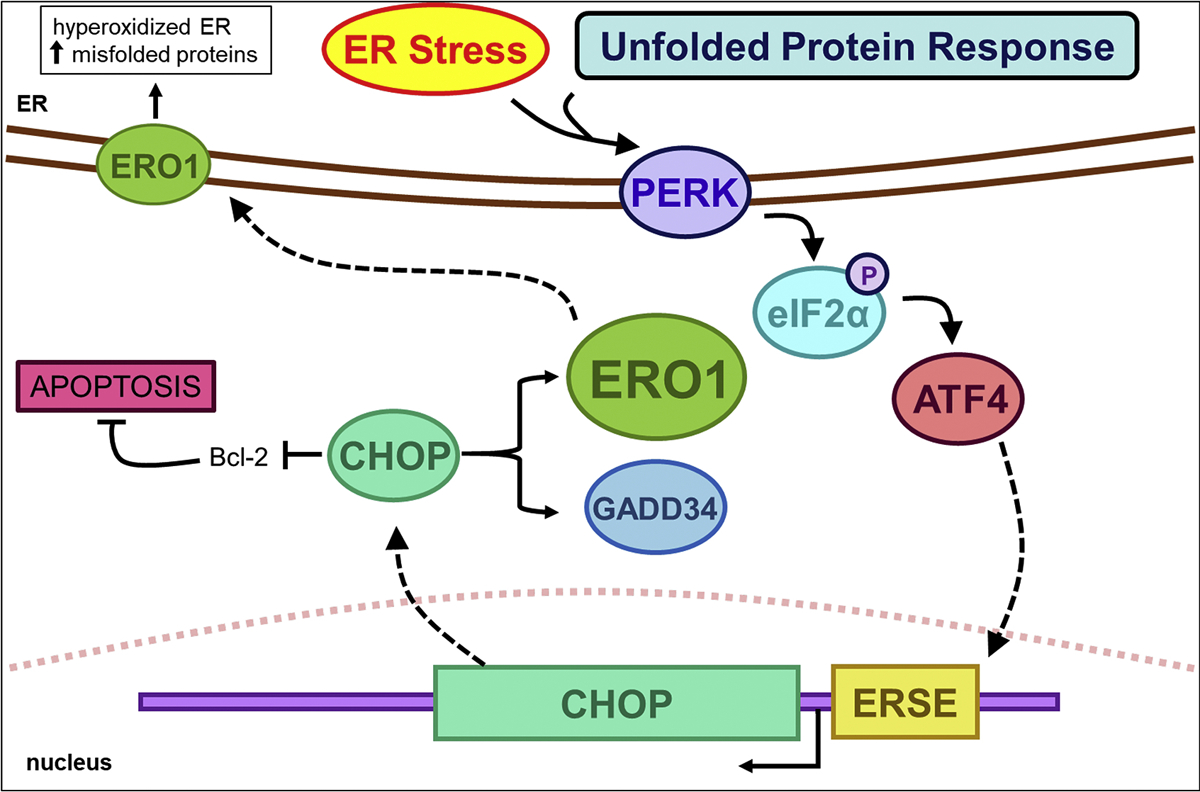
ER stress upregulates ERO1 via CHOP expression. ER stress and the unfolded protein response activate membrane-bound PERK. PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), which leads to induction of CHOP. CHOP expression promotes ER stress by inducing ERO1 and GADD34. ERO1α expression increases ROS levels in the ER, leading to hyperoxidation and an increase in misfolded proteins. Additionally, CHOP downregulates Bcl-2 to promote apoptosis.
A major role of the ER involves calcium storage, and research in recent years furthered our understanding of the role in calcium signaling. In addition to being highly oxidizing compared to other cellular compartments, the ER differs in that calcium concentrations are higher such that free calcium concentration is 100–800 μM (Carreras-Sureda, Pihán, & Hetz, 2018). ROS (O2•−, H2O2, and HO•) also communicate bidirectionally with calcium in a complex relationship. Calcium signaling is necessary for ROS production, and ROS can regulate calcium signaling (Gordeeva, Zvyagilskaya, & Labas, 2003; Görlach, Bertram, Hudecova, & Krizanova, 2015). In mitochondria, calcium promotes metabolism via both the tricarboxylic acid cycle (McCormack & Denton, 1993) and oxidative phosphorylation (Murphy, Kelleher, & Fiskum, 1990). In turn, ROS regulate calcium signaling by modulating plasma membrane and intracellular calcium channels and Ca2+ ATPases (Görlach, et al., 2015).
Calcium signaling and ROS interact in many cellular compartments, including the mitochondria, ER, nucleus, and Golgi apparatus, but the extent of signaling is tissue specific. In the ER, sarco/endoplasmic-reticulum Ca2+ ATPase proteins use ATP to import calcium into the ER lumen. Increased ROS can increase sarco/endoplasmic-reticulum Ca2+ ATPase activity, in turn increasing calcium levels in the ER. ER calcium is released passively through ryanodine receptors and inositol 1,4,5-triphosphate-receptors (IP3R). ERO1α regulates calcium release by regulating IP3R (Anelli, et al., 2012). ERO1α expression increases passive calcium flux, which can lead to apoptosis (Li, et al., 2009). Many ER chaperones use calcium as a cofactor to aid protein folding, thus calcium depletion can affect protein folding. Calcium-binding chaperones include PDIA1, calnexin, calreticulin, GRP78, and GRP94 (Coe & Michalak, 2009). PDIA1 binds calcium with high capacity (19 mol Ca2+/mol protein) and may regulate ER calcium homeostasis (Primm & Gilbert, 2000). Crosstalk between the ER and mitochondria via MAMs is regulated by the flow of calcium. Within the ER and second to the rough ER, MAMs have the highest concentration of ER chaperones and oxidoreductases that may aid in regulating calcium flux (Gilady, et al., 2010; Hayashi, Rizzuto, Hajnoczky, & Su, 2009). ERO1α localizes at MAMs under oxidizing conditions (Anelli, et al., 2012; Gilady, et al., 2010). MAM signaling is important in biological processes including lipid biosynthesis, cell death, and macroautophagy (Phillips & Voeltz, 2016).
4. Clinical implications & disease relevance
ERO1 knockout in mammals is not as lethal as it is in yeast, suggesting that mammalian systems have evolved redundant pathways to compensate for ERO1 loss. Evolution and redundancy of several oxidative folding enzymes explains the viability of ERO1 KO mice. Abnormalities associated with dysfunctional ERO1 include cardiac conduction irregularities in ERO1α mutant mice (Chin, et al., 2011) and problems with insulin folding and secretion (Zito, Chin, et al., 2010). In adult worms, attenuated expression of ero-1 promotes viability and prolongs lifespan (Curran & Ruvkun, 2007). However, ERO1-related ROS generation may promote cancer growth through EMT (Zhang, et al., 2019).
Although the knockout phenotype is mild, ERO1 dysfunction may stimulate ER stress and lead to diseases related to oxidative stress and ER malfunction. For example, ERO1 may impact the folding and secretion of insulin in pancreatic beta cells and malfunction could lead to diabetes (Nardai, et al., 2005; Wang, et al., 1999). ERO1α overexpression decreases mutant proinsulin-G(B32)V misfolding in a model of mutant Ins-gene-induced diabetes of youth. Similarly, ERO1α expression can reduce ER stress caused by proinsulin misfolding (Wright, et al., 2013). Furthermore, ERO1α is responsible for adiponectin secretion in adipocytes (Qiang, Wang, & Farmer, 2007). Adiponectin is a hormone that is protective against type 2 diabetes and cardiovascular disease (Matsuzawa, 2005). ERp44 forms a covalent bond with adiponectin to promote its retention in the secretory pathway, and ERO1α can release adiponectin, likely because it is a competitive binder of ERp44 (Cortini & Sitia, 2010; Wang, et al., 2007). Thus, in this context, knockout or inhibition of ERO1α could lead to potential side effects caused by improper folding and release of functional adiponectin. ERO1β deficiency in mice causes problems with insulin folding and production to contribute to a diabetic phenotype (Zito, Chin, et al., 2010). Furthermore ERO1β deficiency is linked to ER stress-induced cell death, and hampers insulin secretion by delaying proinsulin folding (Khoo, et al., 2011). ERO1β expression is lower in the islets of mouse models of diabetes, and expression decreases with age (Awazawa, et al., 2014). However, in the diabetic mouse model, increasing ERO1β expression led to ER stress and impaired insulin secretion; thus, the dysfunction caused by lower levels of ERO1 expression may be part of a complex pathway that ERO1β cannot rescue (Awazawa, et al., 2014). ERO1β deficiency may be linked with the pathogenesis of type 2 diabetes (Marselli, et al., 2010). ERO1α is primarily in the oxidized state in diabetic mice, possibly indicating ERO1 inactivation (Awazawa, et al., 2014; Nardai, et al., 2005). In other mouse models of diabetes, in addition to promoting cell survival and decreasing oxidative damage, CHOP deletion decreased ERO1α expression, reflecting the role of ERO1α as a target of CHOP (Song, Scheuner, Ron, Pennathur, & Kaufman, 2008). Thus, ERO1α activation may be a strategy to improve the oxidative folding capacity of the ER in diabetes.
There are some links between ERO1α and the immune system. For example, ERO1α overexpression promotes tumor growth and inhibits T cell response (Tanaka, et al., 2015). In breast cancer, ERO1α upregulates programmed cell death ligand 1, the immune response ligand that blocks antitumor response by programmed cell death 1 (Tanaka, et al., 2017). Additionally, ERO1α expression affects the function and oxidative folding of major histocompatibility complex class I H chain (Kukita, et al., 2015). Furthermore, in macrophages, ER stress-induced apoptosis may occur via ERO1α-induced inositol 1,4,5-triphosphate (IP3) receptor activation (Li, et al., 2009). Thus, ERO1 has a complex role in immuno-oncology, but may be a therapeutic target for inhibition in some cases.
5. Relevance of ERO1 in cancer
ERO1α RNA expression is upregulated significantly in several cancers and may indicate sensitivity to ERO1α inhibitors. Furthermore, ERO1α overexpression is associated with poor prognosis of cervical (Zhang, et al., 2019), gastric (Seol, et al., 2016), breast (Kutomi, et al., 2013), lung (Endoh, et al., 2004), hepatocellular (Yang, et al., 2018), and multiple myeloma (Hayes, et al., 2019) cancers, and knockdown of ERO1α in mice inhibited tumor growth (Kutomi, et al., 2013). To further uncover the role of ERO1α across multiple cancers, we surveyed ERO1A mRNA expression using The Cancer Genome Atlas (TCGA) patient data (Lee, Palm, Grimes, & Ji, 2015). Patient RNA-Seq data was downloaded from the GDAC Firehose (https://gdac.broadinstitute.org/), log2 transformed, and z-score normalized per patient. Across all 33 diseases (9726 unique patient samples), median ERO1A expression is higher than average gene expression (> z-score = 0), indicating that ERO1A is being consistently and actively expressed in cancer patient cohorts (Fig. 5). We observed that esophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), and lung squamous cell carcinoma (LUSC) patients tended to express ERO1A the most; thus, inhibitors may be effective in these diseases. Median patient ERO1A expression was higher than median patient ERO1B expression in 28 of 33 (85%) diseases reinforcing its greater relevance to cancer (Fig. 5).
To further identify ERO1α-related signaling programs and pathways, we analyzed ERO1A association with cancer patient reduced survival and disease progression. We identified four diseases in which ERO1A expression was significantly associated with reduced survival and disease progression (tumor stage, grade, or glioma type). As previously described by Shergalis et al., TCGA disease cohorts were stratified into patient groups with high and low ERO1A expression; differences in survival of patient groups were evaluated using a log-rank statistic and confirmed using the GDAC Firebrowse mRNA coxph survival analysis (Center, 2016; Shergalis, Bankhead, Luesakul, Muangsin, & Neamati, 2018). High ERO1A expression was associated with reduced survivability in glioma (GBMLGG), kidney renal papillary cell carcinoma (KIRP), lung adenocarcinoma (LUAD), and pancreatic ductal adenocarcinoma (PAAD) patients (Fig. 6). ERO1A expression in these disease cohorts was also associated with tumor progression endpoints (e.g. stage, grade). For example, LUAD patients with higher pathological stage (III and IV) had significantly higher ERO1A expression (Wilcoxon p = 0.0067) than patients with lower pathological stage (I and II). Glioblastoma patients with more aggressive disease than lower grade glioma patients expressed significantly higher ERO1A (Wilcoxon p = 5e-15). These observations are supported by previous studies (Endoh, et al., 2004; Kutomi, et al., 2013; Zhang, et al., 2019). We also note that no TCGA diseases had significant associations with ERO1B expression or reduced survival and disease progression using the criteria described above.
Fig. 6.
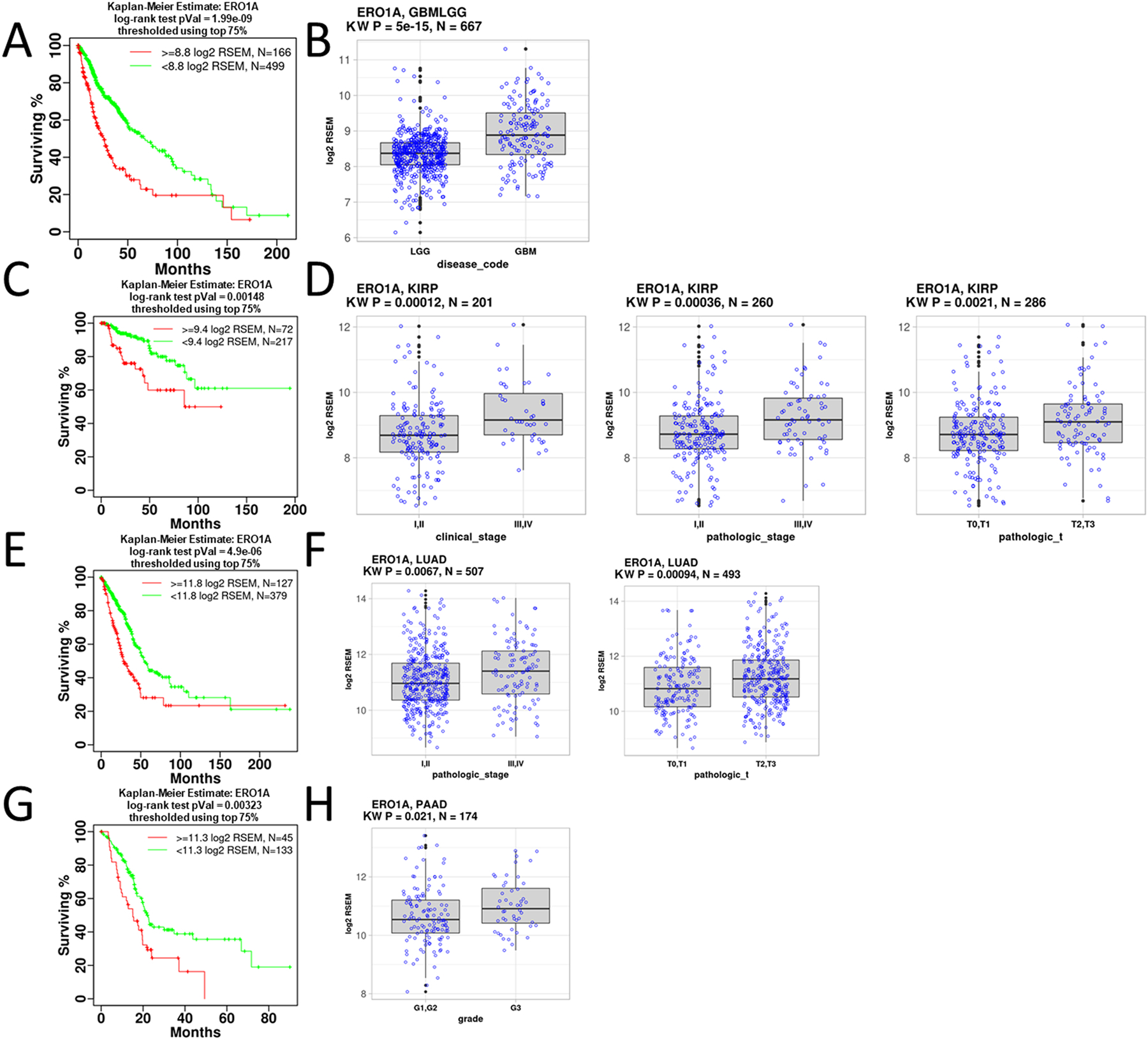
Analysis of ERO1α in cancer. A) Significant reduction in survival is observed for patients with high expression of ERO1α in glioma. B) ERO1A mRNA expression is significantly higher in more aggressive GBM gliomas than LGG. C) Significant reduction in survival is observed for patients with high ERO1A expression in KIRP. D) ERO1A mRNA expression is significantly higher in higher stages of KIRP. E) Significant reduction in survival is observed for patients with high ERO1A expression in LUAD. F) ERO1A mRNA expression is significantly higher in higher stages of LUAD. G) Significant reduction in survival is observed for patients with high ERO1A expression of in PAAD. H) ERO1A mRNA expression is significantly higher in increasing grades of PAAD. Kruskal-Wallis (KW) and survival analysis statistics were calculated using the R statistical programming language (Gravendeel, et al., 2009; Madhavan, et al., 2009).
We performed a co-expression analysis to identify transcriptional programs involving ERO1α in diseases for which ERO1A expression was associated with patient survival and tumor progression. Using Gene Set Enrichment Analysis (GSEA), we identified fourteen common gene sets across LUAD, GBMLGG, KIRP, and PAAD (Fig. 7) that were significantly enriched for genes correlating with ERO1A expression. Included in the common gene set sets was the Hallmark hypoxia gene set. HIF1α regulates ERO1α expression, and hypoxic tumors may be more sensitive to ERO1α inhibitors (May, et al., 2004). Additionally, inhibition of ERO1 activity may prevent tumor growth in hypoxic tumors as ERO1α was identified as an adaptive response element in VEGF-mediated angiogenesis (May, et al., 2004). Identifying the link between hypoxia and ERO1α expression provided validation that this analysis could robustly identify key connections with canonical ERO1α-related pathways. This analysis also implicated ERO1α expression with cell cycle regulation, E2F signaling, epithelial-to-mesenchymal transition, and interleukin 6 JAK (Janus kinase)-STAT3 (signal transduction and transcription 3) signaling and provides additional hypotheses regarding the link between ERO1α, patient survival, and disease progression. The link between EMT and ERO1 has been previously established in cervical cancer, and the ERO1-PDIA1 interaction was identified to contribute to epithelial-to-mesenchymal transition in cervical cancer (Zhang, et al., 2019). On the other hand, ERO1 overexpression in osteosarcoma enhanced the cytotoxicity of microtubule-targeting agent CYT997 (lexibulin) and inhibition decreased cell death (Wang, et al., 2019). The link between ERO1α and interleukin 6 JAK-STAT3 signaling was demonstrated in hepatocellular carcinoma (HCC). Knockdown of ERO1α blocked STAT3 phosphorylation in cancer cells (Yang, et al., 2018). STAT3 is a critical signaling transducer responsible for cancer cell growth and migration. The link between ERO1α, cell cycle, and E2F1 may indicate a connection between p53 function and ER stress, or more directly, ERO1 activity. Tumor suppressor p53 function is inhibited by ER stress (Mahdi, Rizvi, & Parveen, 2016). However, ERO1α function is not directly under the regulation of p53 transcription (May, et al., 2004). Additionally, our co-expression analysis strengthens the connection between ERO1α and known pathways, such as EMT and STAT3 signaling, and provides rationale for further studying the relationship between ERO1α and E2F signaling.
Fig. 7.
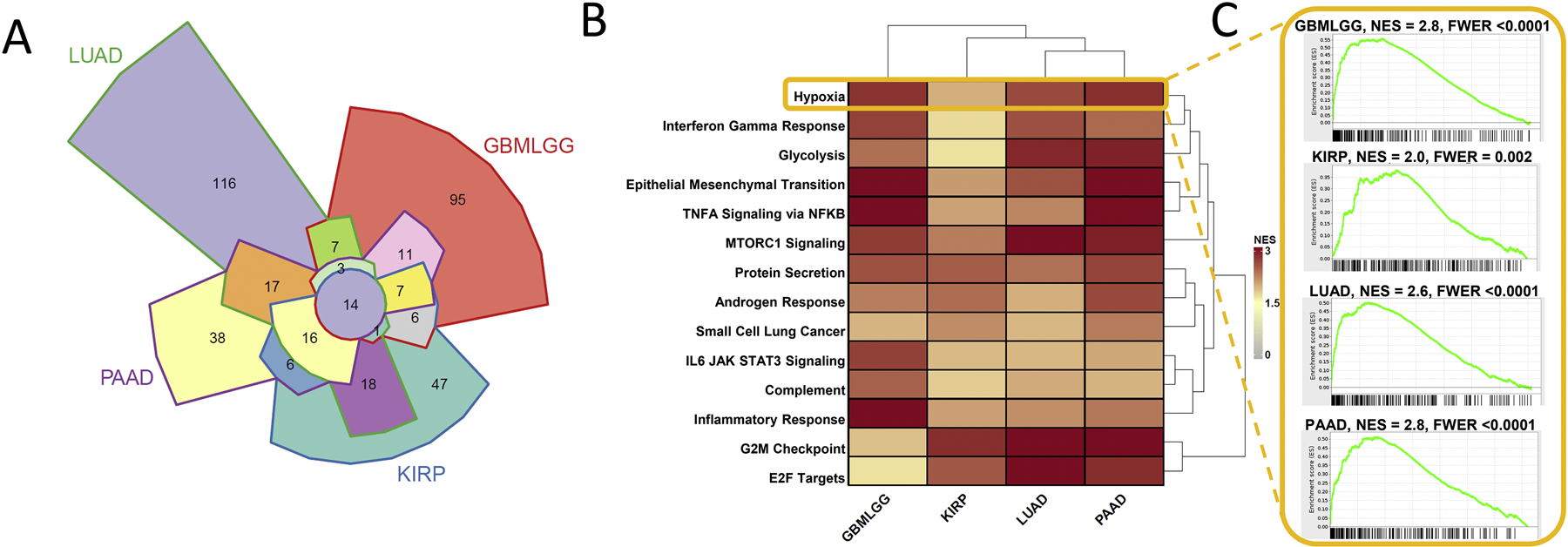
Common gene sets enriched for genes that are co-expressed with ERO1A. A) Gene set enrichment analysis (GSEA) was used to identify enriched pathways with genes that are co-expressed with ERO1A. A Chow-Ruskey diagram shows overlap of significant gene sets correlated with ERO1A expression within LUAD, GBMLGG, PAAD, and KIRP TCGA diseases. Fourteen gene sets commonly enriched among the four diseases are shown in the heatmap. B) GSEA was used to identify enriched pathways with genes that are correlated with ERO1α. Heatmap coloring indicates normalized enrichment score (NES). C) GSEA running sum statistic visualizations are shown for the Hallmark gene set, Hypoxia, that was significantly enriched in GBMLGG, KIRP, LUAD, and PAAD TCGA diseases. All common gene sets were Hallmark except for the KEGG “Small Cell Lung Cancer” gene set. GSEAv2.2.3 was used with v6 gene sets sourced from MSigDB. 10,000 gene set permutations were performed using weighted mode scoring and Pearson metric (Subramanian, et al., 2005). Only genes with evidence of expression in > 50 % of a disease patient population were considered.
To identify genes that were most highly correlated with ERO1A expression, we compared ERO1A to all other genes using Pearson correlation and ranked genes based on their correlation with ERO1A. Using the top 300 ERO1A co-expressed genes in LUAD, GBMLGG, PAAD, and KIRP TCGA diseases, we identified 10 common genes across the four diseases (Fig. 8A). P4HA1 (Prolyl 4-hydroxylase subunit alpha-1) was the top gene co-expressed with ERO1A in the four diseases studied (Fig. 8B). P4HA1 is part of a protein complex with PDIA1 that catalyzes the hydroxylation of proline to promote collagen maturation, important for cancer progression and metastasis. The P4H complex (collagen prolyl 4-hydroxylase) is responsible for stabilizing HIF1α, which enhances angiogenesis. Because tumor cells grow rapidly, they have increased metabolic demands and consume higher rates of oxygen. Higher oxygen consumption leads to hypoxic tumor conditions and activation of the HIF1α pathway to promote cell survival. Furthermore, inhibitors of P4H sensitize breast cancer cells to docetaxel and doxorubicin (Xiong, et al., 2018). Similarly, knockdown of P4HA1 prevented collagen synthesis and neovascularization in glioma (Zhou, et al., 2017), and its expression could serve as a prognostic biomarker for high grade glioma (Hu, et al., 2017). Additionally, HIF1α can activate P4HA1 and promote extracellular matrix remodeling (Gilkes, Bajpai, Chaturvedi, Wirtz, & Semenza, 2013). P4HA1 is activated by transcriptional repressors EZH2 and CtBP1 through miR-124 downregulation (Chakravarthi, et al., 2014). In ovarian cancer, an upstream repressor of P4HA1, miR-122, is typically downregulated, and overexpression can prevent epithelial to mesenchymal transition (Duan, et al., 2018). The link between ERO1α and P4HA1, rather than P4HB (PDIA1), emphasizes the important role of ERO1α in hypoxia signaling in cancer.
Fig. 8.

A) Chow-Ruskey diagram shows overlap of the top 100 genes correlated with ERO1A expression within GBMLGG, KIRP, PAAD, and LUAD TCGA diseases. B) P4HA1 was one of the top co-expressed genes in common across GBMLGG, KIRP, PAAD, and LUAD. Gene log2RSEM expression values are shown in scatter plots.
Using the top 100 ERO1A-correlated genes in GBMLGG, a ClueGO network was used to visualize gene ontology (GO) biological process gene set enrichment and highlight ERO1α-related GO categories (Bindea, et al., 2009). We identified ERO1A co-expressed genes involved in cell-matrix adhesion, VEGFR signaling pathway, and COPII vesicle coating (Fig. 9). COPII vesicle coating functions in concert with ER protein trafficking. Co-expression of ERO1α with proteins involved in COPII vesicle coating points to its role in lipid trafficking and biosynthesis, although this relationship has yet to be explored in detail. Co-expressed genes were also involved in cell-matrix adhesion and VEGR signaling. In HCC, ERO1α was demonstrated to trigger angiogenesis via the S1PR1/STAT3/VEGF-A signaling pathway (Yang, et al., 2018). Thus, our findings are consistent with previous research on ERO1α function.
Fig. 9.
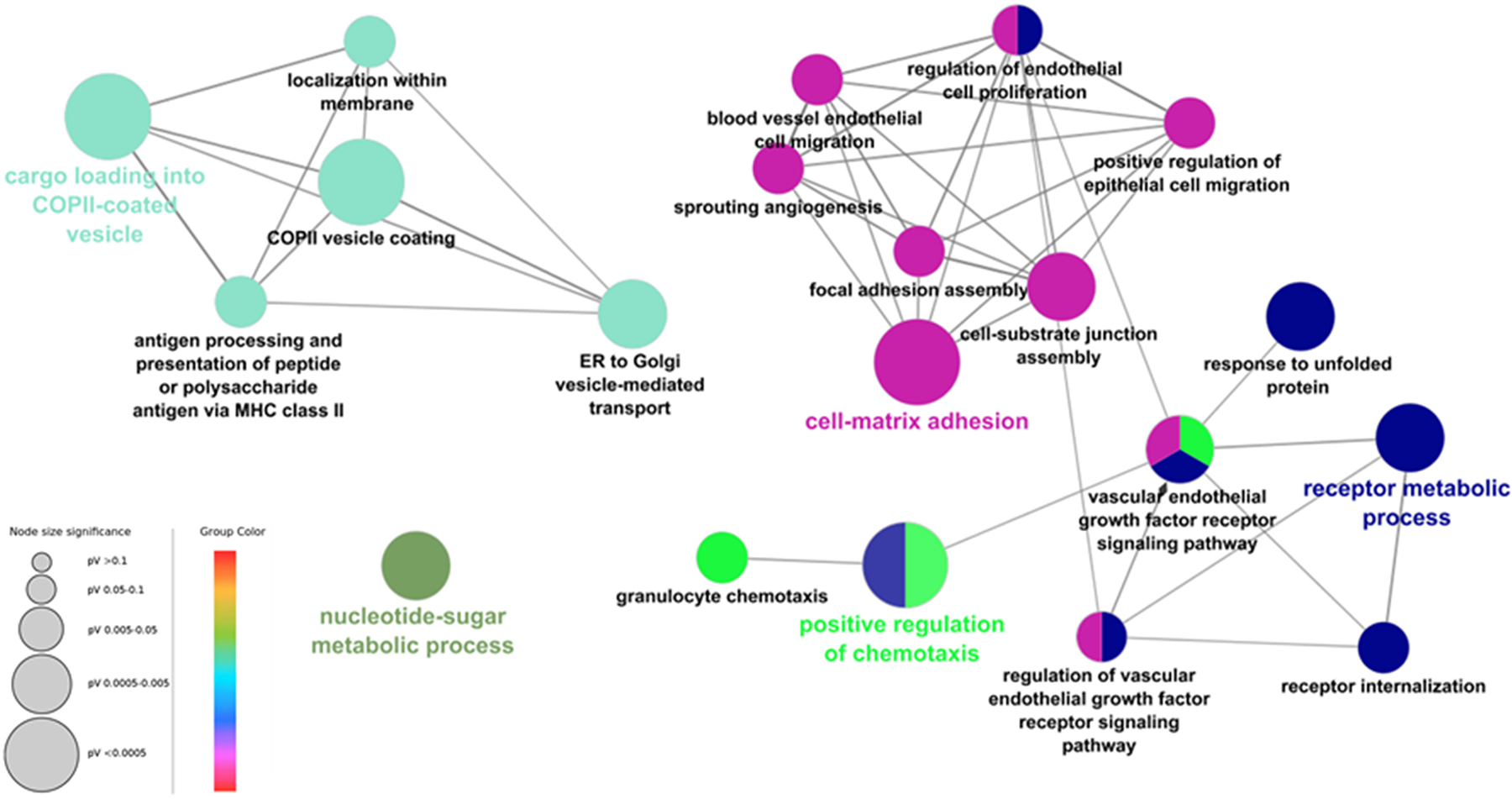
GO pathways associated with top 100 genes correlated with ERO1 expression in TCGA glioma generated by ClueGO. Terms with Bonferroni corrected p values < 0.05 are shown.
6. ERO1α knockdown associations with Connectivity Map analysis
We evaluated ERO1α knockdown data from Connectivity Map (CMap), a reference database containing gene expression profiles from cancer cells treated by drugs or gene modification (knockdown or overexpression) (Lamb, et al., 2006). CMap is widely used to find connections between drugs, genes, and diseases in order to understand the drug mechanisms and identify drug repurposing opportunities (Musa, et al., 2017; Qu & Rajpal, 2012). We first analyzed the CMap ERO1α knockdown data to find novel biological pathway connections. We focused on lung cancer cell lines because ERO1α expression is associated with reduced LUAD patient survival.
The Touchstone database in CMap contains ERO1α knockdown data in two non-small cell lung cancer cell lines, A549 and HCC515. We examined the correlated signatures across perturbagen classes, compounds, gene knockdown and gene overexpression to generate a hypothesis about ERO1α-related genes, pathways and pharmacological activities. All perturbagen types were ranked by their connectivity score in each cell line, and perturbagens with a score over + 90 or below − 90 were considered a strong connection. Among the top 50 correlated compounds, ERO1α knockdown showed a strong positive connectivity score with 8 diverse classes of compounds in both cell lines (Table S1, Fig. 10A). These classes of compounds may have novel connections to ERO1α. Among them are p38 MAPK inhibitor, JAK inhibitor, and EGFR inhibitor, which relate to kinase signaling and cell growth, suggesting these inhibitors induce a similar gene expression profile as ERO1α knockdown (Fig. 10B). This also suggests that ERO1α knockdown may affect kinase signaling, such as JAK, p38, and other protein kinases. Similar gene expression profiles from CMap can be used to generate hypotheses about their similar pathways and synergistic anti-cancer combinations (Hassane, et al., 2010; Qu & Rajpal, 2012). Thus, this CMap analysis demonstrated a link between ERO1α and kinase pathways. ERO1α inhibition or knockdown may be lethal to cancer cells when combined with EGFR inhibitors, JAK inhibitors, or other kinase inhibitors. We did not observe common negative compound perturbagen hits between the two cell lines (Table S2).
Fig. 10.
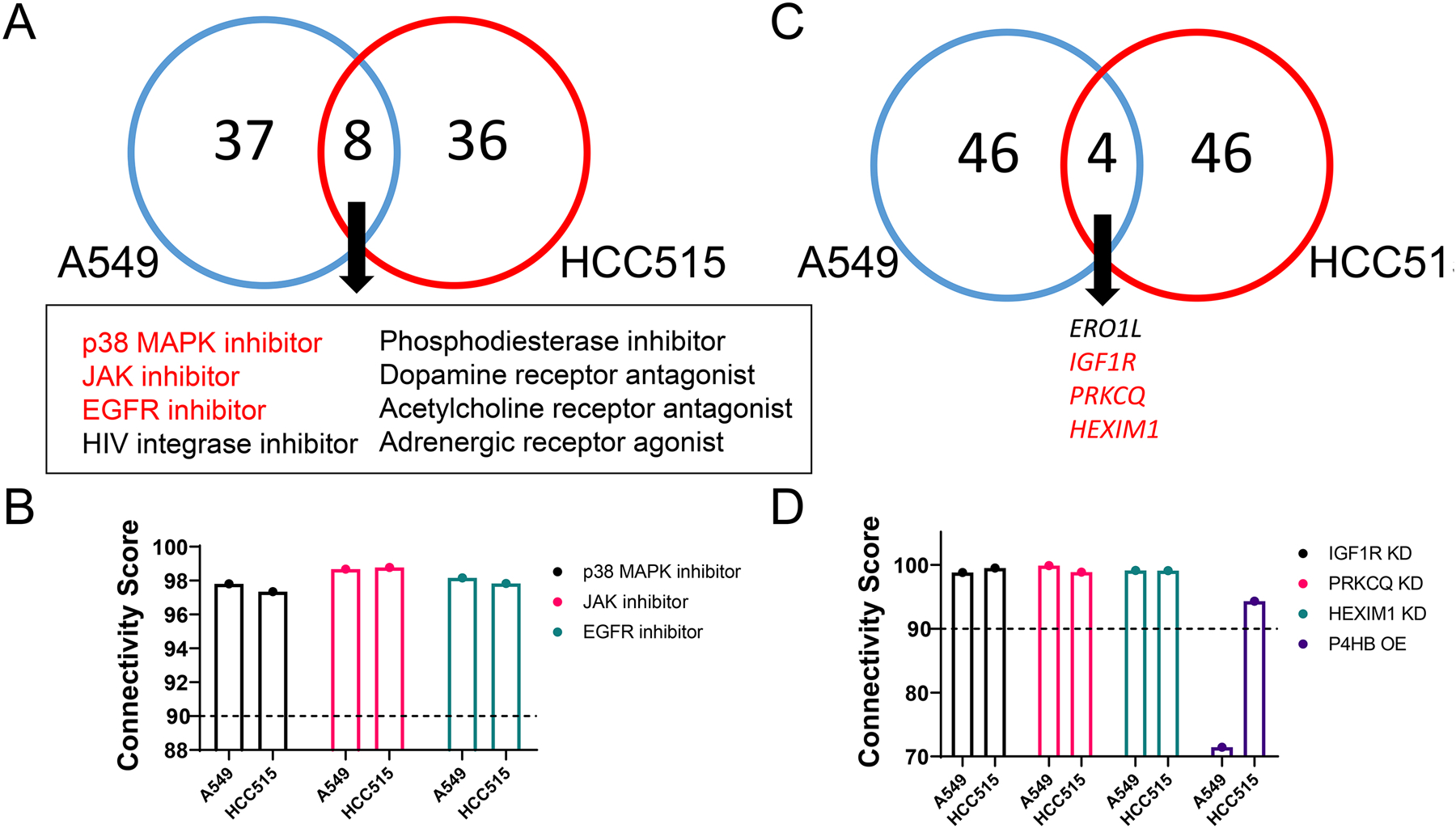
Highly connected CMap perturbagens reveal potential genes and signaling pathways with which ERO1α is involved in A549 and HCC515 cell lines. A) Eight classes of compounds are shared between A549 and HCC515 cell lines among the top 50 positively connected compound perturbagens. B) Select compound perturbagens with high connectivity score (≥ +90) in two lung cancer cell lines. C) Four gene KD perturbagens are shared between two cell lines among the top 50 positively connected hits. D) Select gene KD profiles with high connectivity score (≥ +90) in two lung cancer cell lines. P4HB overexpression is included since it is positively correlated with ERO1A knockdown in the HCC515 cell line.
We also identified genes that, when knocked down, have a similar signature as ERO1α knockdown in two lung cancer cell lines. In particular, there were three highly significant genes (except ERO1L) among the top 50 knockdown perturbagens: IGF1R, PRKCQ, and HEXIM1 (Table S3, Fig. 10C, D). IGF1R (insulin-like growth factor 1 receptor) is a receptor that binds insulin-like growth factor, has tyrosine kinase activity, and is involved in the PI3K/AKT and Ras/MAPK pathways. IGF1R is overexpressed in tumors and mediates tumor proliferation, invasion and metastasis (Riedemann & Macaulay, 2006). As a membrane-bound protein, IGF1R requires processing through the endoplasmic reticulum in order to function properly. PRKCQ (protein kinase c theta isoform) is a member of the Protein Kinase C (PKC) family, another critical membrane-bound protein family of signaling kinases. The role of PKC in cancer is complex, but overexpression of PRKCQ can promote tumor growth in triple-negative breast cancer (Byerly, Halstead-Nussloch, Ito, Katsyv, & Irie, 2016). PKCs are connected downstream of IGF1R/PI3K signaling via activation by phosphoinositide-dependent kinase 1, further confirming the relevance of ERO1α to IGF1R signaling pathways. HEXIM1 (hexamethylene bisacetamide inducible 1) is a member of the 7SK small nuclear ribonucleoprotein complex that inhibits positive transcription elongation factor b and control mRNA synthesis (Michels, et al., 2004). HEXIM1 has also been demonstrated as a robust pharmacodynamic marker for bromodomain and extraterminal domain inhibitors (Lin, et al., 2017). Furthermore, HEXIM1 is downregulated during cancer progression, and expression sensitizes prostate cancer cells to anti-androgens. HEXIM1 induces expression of the histone demethylase KDM5B (lysine-specific demethylase 5B) to inhibit histone methylation (Yeh, et al., 2014). It stabilizes p53 tumor suppressor by associating with the C-terminal negative domain (Lew, et al., 2012). In addition to the three significantly connected perturbagens in CMap, P4HB overexpression is positively correlated with ERO1α knockdown in HCC515 cells (ranked 127 out of the 215 hits with a connectivity score > +90), suggesting there may be a compensatory expression mechanism (Table S4, Fig. 10D).
A STRING (Search Tool for the Retrieval of Interacting Genes) protein interaction network was constructed to highlight proteins associated with ERO1α (ERO1A) (Fig. 11) (Szklarczyk, et al., 2015). ERO1α protein-protein interactions primarily included protein folding and cell redox homeostasis pathways. Many of the interactions are with PDI family members (PDIA1, PDIA3, PDIA4, PDIA6, ERP29, and TXNDC5). Insulin and GRP78 are also closely associated with ERO1α.
Fig. 11.
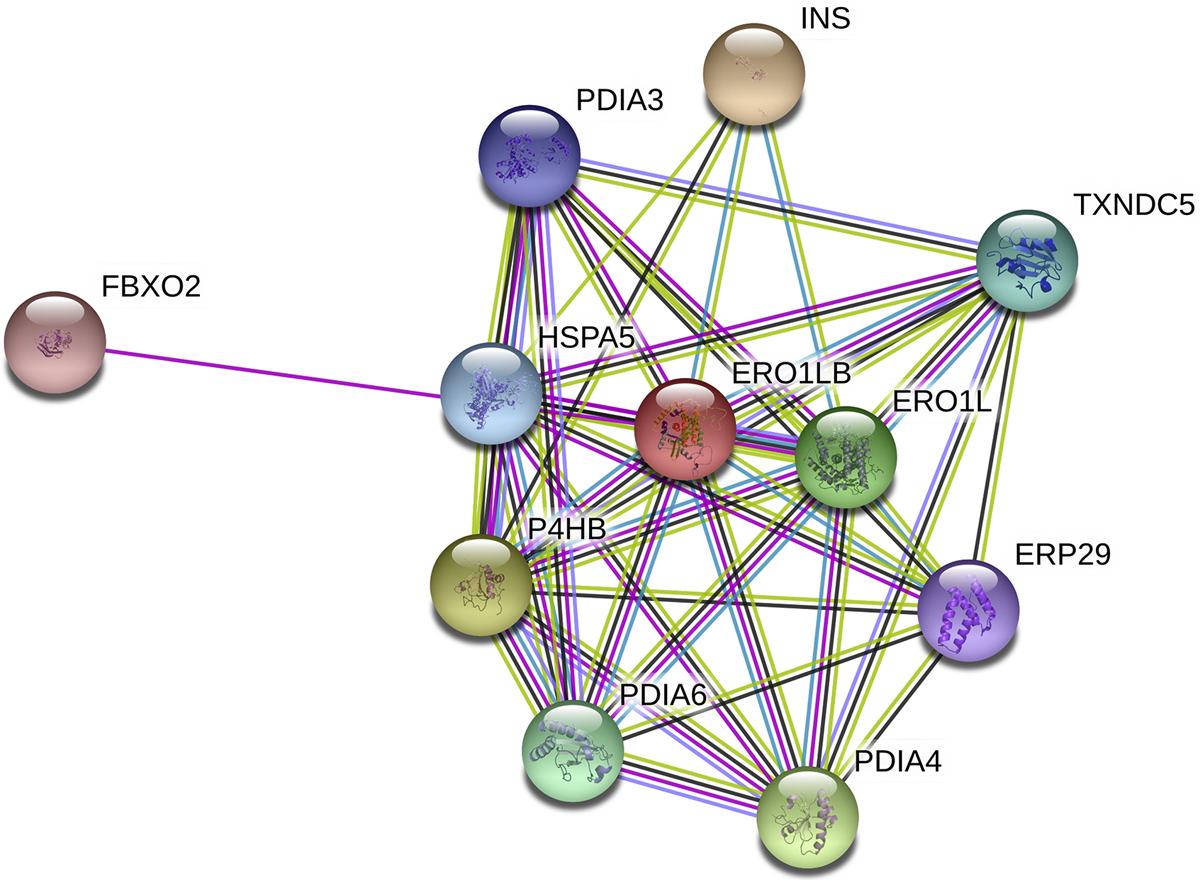
STRING analysis of protein interactions with ERO1L. ERO1LB, ERO1-like protein beta; ERP29, endoplasmic reticulum resident protein 29; FBXO2, F-box only protein 2; HSPA5, 78 kDa glucose-regulated protein; INS, insulin; P4HB, protein disulfide isomerase A1; PDIA3, protein disulfide isomerase A3; PDIA4, protein disulfide isomerase A4; PDIA6, protein disulfide isomerase A6; TXNDC5, thioredoxin domain-containing protein 5.
Three compounds have been reported to inhibit ERO1 (Fig. 12). Erodoxin was selective for yeast ERO1 compared to mouse ERO1α (IC50 > 400 μM) (Costanzo, et al., 2010). Another series of ERO1p inhibitors was identified through virtual screening techniques with biochemical IC50 values as low as 3 μM (Chu, Chen, Yang, & Tang, 2011). Probe molecules EN460 and QM295 were identified from a biochemical high throughput screen to inhibit ERO1α-mediated reduction of thioredoxin A with IC50 values around 1.9 μM (Blais, et al., 2010). EN460 interacts reversibly with the reduced, active form of ERO1α, displacing the FAD moiety. EN460 inhibition of ERO1α prevents H2O2 production when ERO1α function is stimulated. EN460 also prevents replication of the chikunguya virus (Langsjoen, et al., 2017). However, EN460 is not selective for ERO1 and also inhibits other FAD-containing enzymes including monoamine oxidase A, monoamine oxidase B, and LSD-1 (Hayes, et al., 2019). Recently, the interaction between ERO1α and PDIA1 was demonstrated to depend on Val101 of ERO1α; thus, inhibition of the ERO1α-PDIA1 interaction around Val101 may be a potential anti-cancer strategy (Zhang, et al., 2019). One hypothesis may be that ERO1α inhibition would block PDIA1 re-oxidation, and thus be another strategy for selective PDIA1 inhibition. Because this interaction involves the conserved a’ domain of PDIA1, it is possible that inhibiting the ERO1α-PDIA1 interaction may disrupt associations between ERO1α and other PDI family members, such as ERp46 and P5 (PDIA6). ERp46 is critical for prostate cancer progression (Duivenvoorden, Hopmans, Austin, & Pinthus, 2017), and P5 activates the Wnt/β-catenin pathway in HeLa cells (Gao, et al., 2016). Thus, inhibiting this interaction may have a broader application to cancers other than cervical cancer. However, the development of a pan inhibitor may lead to unexpected side effects because of the broad, complex roles these proteins play in different tissues. Thus, careful consideration of off-target effects should be monitored when developing ERO1α inhibitors. Furthermore, in vivo toxicity to tissues such as pancreas and liver should be observed closely to identify undesired consequences of off-target ERO1α complex inhibition.
Fig. 12.
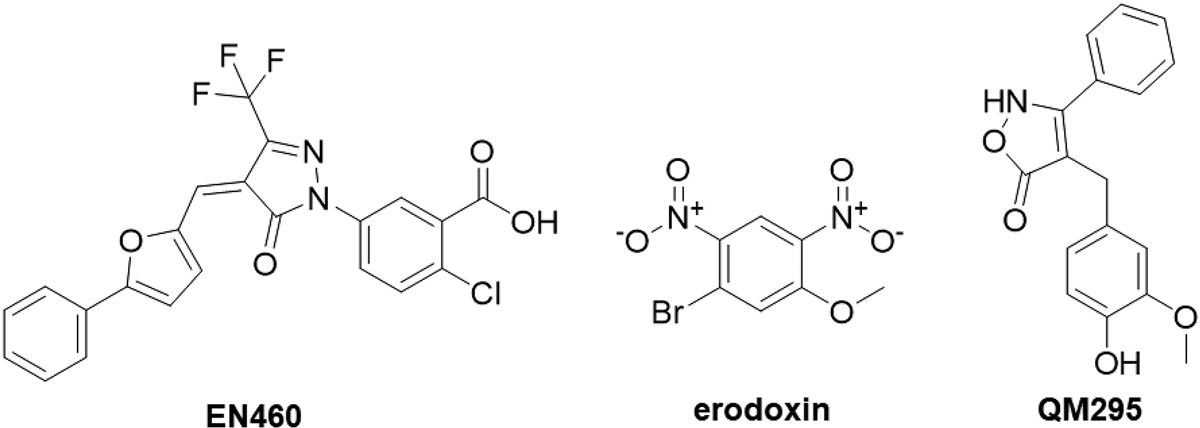
Reported ERO1 inhibitors
Inhibition of PDIA1 has been linked to many diseases and a number of inhibitors have been discovered. In more recent years, PDIA1 has been studied in the context of cardiovascular diseases, particularly in thrombus formation mechanisms (Cho, Furie, Coughlin, & Furie, 2008; Jasuja, et al., 2012), but is also linked to cancer (Xu, et al., 2014), neurodegenerative diseases (Uehara, et al., 2006), diabetes (Rajpal, et al., 2012), and viral entry (Stolf, et al., 2011). Polymorphisms in P4HB have been linked to Cole Carpenter syndrome (Porntaveetus, Theerapanon, Srichomthong, & Shotelersuk, 2018). PDI overexpression has been linked to several cancers, including glioblastoma, lymphoma, kidney, ovarian, prostate and lung cancers (Xu, et al., 2014). In highly secretory cancers such as multiple myeloma, PDI inhibitors have been particularly effective (Robinson, et al., 2018; Vatolin, et al., 2016). A broad class of electrophilic compounds have been identified that target the PDIA1 active site cysteines (Cole, et al., 2018; Kyani, et al., 2018; Xu, et al., 2012). PDIA1 inhibitors that do not bind in the active site are in high demand, likely because these types of inhibitors are more selective for PDIA1 inhibition. Furthermore, a well-designed b’ domain inhibitor has the potential to block the ERO1α-PDIA1 interaction (Zhang, et al., 2019). PDIA1 has multiple binding sites, including the substrate-binding pocket in the b’ domain and hormone reservoirs for estradiol and TH hormone (Primm & Gilbert, 2000). Bepristat 1a binds in the b’ substrate binding pocket and exhibits antithrombotic properties (Bekendam, et al., 2016). Furthermore, BAP2, a chalcone-containing PDIA1 and PDIA2 inhibitor, was reported to bind in a pocket in the b’xa’ domain (Yang, et al., 2019). Additionally, isoquercetin, in Phase II clinical trials to decrease thrombosis in at risk advanced cancer patients (Bekendam, et al., 2016; Zwicker, et al.), and analogues bind in the b’x domain (Lin, et al., 2015). CRISPR/Cas9-mediated PDIA1 knockout prevents ROS production induced by lipopolysaccharide (LPS) and inactivates the NF-κB pathway (Xiao, et al., 2018). PDIA1 has been linked to a major source of ROS, Nox (Laurindo, et al., 2008), and PDIA1 overexpression leads to an increase in ROS levels via increased Nox activity (Gimenez, et al., 2019; Santos, Stolf, et al., 2009). Furthermore, ROS production by PDIA1 in leukocytes was found to be redox-dependent; oxidized PDIA1 stimulated ROS production, whereas reduced PDIA1 did not (de A. Paes, et al., 2011). Studies have not yet demonstrated whether b’ domain inhibitors of PDI prevent ERO1α interaction.
7. Conclusions
Roughly 25 % of intracellular ROS originate from the ER. Thus, the ER is a critical, yet underrepresented ROS-producing organelle. Most of the generated ROS remain within the organelle, without leaking out, and are reduced by high concentrations of ER antioxidants (Tu & Weissman, 2004). Furthermore, during the unfolded protein response, the ER is resistant to redox imbalances, supporting that ROS do not leak from the ER (Avezov, et al., 2013). However, increased ROS levels are observed in cells overexpressing ERO1, and, in some disease states, ROS leakage may occur (Yang, et al., 2018). Thus, ROS remain critical signaling molecules in the ER. Increasing evidence (including results presented in this paper) demonstrates that ERO1α expression correlates with survival and is upregulated in several cancers, despite the existence of compensatory oxidative folding pathways in the cell. ERO1 function is essential in yeast systems; however, mammalian cells can survive ERO1 knockout, indicating redundant pathways for disulfide formation. This provides an opportunity to develop tumor-selective inhibitors, as normal cells may have the mechanisms to cope with ERO1 inhibition, while tumor cells overexpressing ERO1 may suffer. As interest in developing ERO1 inhibitors grows, more selective inhibitors are needed as probe molecules. The discovery that the PDI-ERO1α interaction is critical for cervical cancer progression validates the development of inhibitors with anticancer potential.
Overall, overexpression of ERO1α suggests cancer cells depend on its expression and dual inhibition of ERO1 and PDI may be a lethal combination in several diseases. However, the field has lacked selective ERO1 inhibitors to explore inhibition in in vivo disease models. Selective ERO1 inhibitors may be able to discriminate between cancerous cells and healthy cells because of the reliance of cancer cells on ERO1 expression. Furthermore, our bioinformatics analysis demonstrated that ERO1 inhibitors may be effective in lung cancer, uncovering a potential avenue of therapeutic value to be explored. Thus, this space represents novel opportunity with enormous potential for the development of first-in-class inhibitors of ERO1α.
Supplementary Material
Acknowledgements
This work was supported by a grant from the NIH (CA193690).
Abbreviations:
- ATF6
activating transcription factor 6
- Cmap
Connectivity Map
- CHOP
C/EBP homologous protein
- EMT
epithelial-to-mesenchymal transition
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERO1
endoplasmic reticulum oxidase 1
- ERp
endoplasmic reticulum protein
- ESCA
esophageal carcinoma
- FAD
flavin adenine dinucleotide
- FBXO2
F-box only protein 2
- GBMLGG
glioma
- GO
gene ontology
- GPX
glutathione peroxidase 8
- GSEA
Gene Set Enrichment Analysis
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HCC
hepatocellular carcinoma
- HEXIM1
hexamethylene bisacetamide inducible 1
- HIF1α
hypoxia-inducible factor 1α
- HNSC
head and neck squamous cell carcinoma
- GRP78
78 kDa glucose-regulated protein
- IGF1R
insulin-like growth factor 1 receptor
- IL6
interleukin 6
- IP3R
inositol 1,4,5-triphosphate-receptors
- IRE1α
inositol-requiring protein 1α
- JAK
Janus kinase
- KIRP
kidney renal papillary cell carcinoma
- KW
Kruskal-Wallis
- LUAD
lung adenocarcinoma
- LUSC
lung squamous cell carcinoma
- MAM
mitochondrial-associated membrane
- P4HA1
prolyl 4-hydroxylase subunit alpha-1
- P4HB
protein disulfide isomerase A1
- PAAD
pancreatic ductal adenocarcinoma
- PDI
protein disulfide isomerase
- PERK
RNA-like ER kinase
- PRDX4
peroxiredoxin 4
- PRKCQ
Protein Kinase C theta isoform
- ROS
reactive oxygen species
- STAT3
signal transducer and activator of transcription 3
- STRING
Search Tool for the Retrieval of Interacting Genes
- TCGA
The Cancer Genome Atlas
- TXNDC5
thioredoxin domain-containing protein 5
- UPR
unfolded protein response
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, & Sitia R (2002). ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO Journal, 21, 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, … Sitia R (2012). Ero1α regulates Ca2 fluxes at the endoplasmic reticulum–mitochondria interface (MAM). Antioxidants & Redox Signaling, 16, 1077–1087. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Christensen B, Sørensen ES, & Ellgaard L (2008). A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO Journal, 27, 2977–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Zito E, Chin K-T, Ron D, Spiess M, & Ellgaard L (2010). Disulphide production by Ero1α–PDI relay is rapid and effectively regulated. EMBO Journal, 29, 3318–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Iemura S-I, Kamiya Y, Ron D, Kato K, Natsume T, & Nagata K (2013). Ero1- α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. Journal of Cell Biology, 202, 861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avezov E, Cross BCS, Kaminski Schierle GS, Winters M, Harding HP, Melo EP, … Ron D (2013). Lifetime imaging of a fluorescent protein sensor reveals surprising stability of ER thiol redox. Journal of Cell Biology, 201, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awazawa M, Futami T, Sakada M, Kaneko K, Ohsugi M, Nakaya K, … Ueki K (2014). Deregulation of pancreas-specific oxidoreductin ERO1 in the pathogenesis of diabetes mellitus. Molecular and Cellular Biology, 34, 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekendam RH, Bendapudi PK, Lin L, Nag PP, Pu J, Kennedy DR, … Flaumenhaft R (2016). A substrate-driven allosteric switch that enhances PDI catalytic activity. Nature Communications, 7, 12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekendam RH, & Flaumenhaft R (2016). Inhibition of protein disulfide isomerase in thrombosis. Basic & Clinical Pharmacology and Toxicology, 119 Suppl 3, 42–48. [DOI] [PubMed] [Google Scholar]
- Benham AM, van Lith M, Sitia R, & Braakman I (2013). Ero1-PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Philosophical Transactions of the Royal Society B: Biological Sciences, 368, 20110403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandary B, Marahatta A, Kim H-R, & Chae H-J (2012). An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. International Journal of Molecular Sciences, 14, 434–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, … Galon J (2009). ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics, 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais JD, Chin K-T, Zito E, Zhang Y, Heldman N, Harding HP, … Ron D (2010). A small molecule inhibitor of endoplasmic reticulum oxidation 1 (ERO1) with selectively reversible thiol reactivity. Journal of Biological Chemistry, 285, 20993–21003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolisetty S, & Jaimes EA (2013). Mitochondria and reactive oxygen species: Physiology and pathophysiology. International Journal of Molecular Sciences, 14, 6306–6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth DM, Enyedi B, Geiszt M, Várnai P, & Hajnóczky G (2016). Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Molecular Cell, 63, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek A, Sarewicz M, & Osyczka A (2008). Movement of the iron–sulfur head domain of cytochrome bc1 transiently opens the catalytic Qo site for reaction with oxygen†. Biochemistry, 47, 12365–12370. [DOI] [PubMed] [Google Scholar]
- Bulleid NJ, & Ellgaard L (2011). Multiple ways to make disulfides. Trends in Biochemical Sciences, 36, 485–492. [DOI] [PubMed] [Google Scholar]
- Byerly J, Halstead-Nussloch G, Ito K, Katsyv I, & Irie HY (2016). PRKCQ promotes oncogenic growth and anoikis resistance of a subset of triple-negative breast cancer cells. Breast Cancer Research, 18, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery MR, Bulleid NJ, & Sitia R (2000). ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. Journal of Biological Chemistry, 275, 4827–4833. [DOI] [PubMed] [Google Scholar]
- Cao SS, & Kaufman RJ (2014). Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & Redox Signaling, 21, 396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras-Sureda A, Pihán P, & Hetz C (2018). Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium, 70, 24–31. [DOI] [PubMed] [Google Scholar]
- Center, B. I. T. G. D. A. (2016). Correlation between mRNAseq expression and clinical features. Broad Institute of MIT and Harvard. [Google Scholar]
- Chakravarthi BV, Pathi SS, Goswami MT, Cieslik M, Zheng H, Nallasivam S, … Varambally S (2014). The miR-124-prolyl hydroxylase P4HA1-MMP1 axis plays a critical role in prostate cancer progression. Oncotarget, 5, 6654–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthi S, Jessop CE, & Bulleid NJ (2006). The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Reports, 7, 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, & Ravanan P (2014). A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Frontiers in Cellular Neuroscience, 8, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Wang L, & Wang C-C (2010). Domain a’ of protein disulfide isomerase plays key role in inhibiting alpha-synuclein fibril formation. Cell Stress Chaperones, 15, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K-T, Kang G, Qu J, Gardner LB, Coetzee WA, Zito E, … Ron D (2011). The sarcoplasmic reticulum luminal thiol oxidase ERO1 regulates cardiomyocyte excitation-coupled calcium release and response to hemodynamic load. FASEB Journal, 25, 2583–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Furie BC, Coughlin SR, & Furie B (2008). A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. Journal of Clinical Investigation, 118, 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Chen X, Yang Y, & Tang Y (2011). Identification of small molecular inhibitors for Ero1p by structure-based virtual screening. Bioorganic & Medicinal Chemistry Letters, 21, 1118–1121. [DOI] [PubMed] [Google Scholar]
- Coe H, & Michalak M (2009). Calcium binding chaperones of the endoplasmic reticulum. General Physiology and Biophysics, 28 Spec No Focus, F96–F103. [PubMed] [Google Scholar]
- Cole KS, Grandjean JMD, Chen K, Witt CH, O’Day J, Shoulders MD, … Weerapana E (2018). Characterization of an A-site selective protein disulfide isomerase A1 inhibitor. Biochemistry, 57, 2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn KJ, Gao W, McKee A, Lan MS, Ullman MD, Eisenhauer PB, … Wells JM (2004). Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Research, 1022, 164–172. [DOI] [PubMed] [Google Scholar]
- Cortini M, & Sitia R (2010). From antibodies to adiponectin: role of ERp44 in sizing and timing protein secretion. Diabetes Obes Metab, 12 Suppl 2, 39–47. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, … Boone C (2010). The genetic landscape of a cell. Science, 327, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Várnai P, Golenár T, Sheu S-S, & Hajnóczky G (2012). Calcium transport across the inner mitochondrial membrane: Molecular mechanisms and pharmacology. Molecular and Cellular Endocrinology, 353, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Weaver D, & Hajnoczky G (2018). Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol, 28, 523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran SP, & Ruvkun G (2007). Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genetics, 3, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de A Paes AM, Veríssimo-Filho S, Guimarães LL, Silva ACB, Takiuti JT, Santos CXC, … Lopes LR (2011). Protein disulfide isomerase redox-dependent association with p47phox: Evidence for an organizer role in leukocyte NADPH oxidase activation. Journal of Leukocyte Biology, 90, 799–810. [DOI] [PubMed] [Google Scholar]
- Dias-Gunasekara S, Gubbens J, van Lith M, Dunne C, Williams JAG, Kataky R, … Benham AM (2005). Tissue-specific expression and dimerization of the endoplasmic reticulum oxidoreductase Ero1beta. Journal of Biological Chemistry, 280, 33066–33075. [DOI] [PubMed] [Google Scholar]
- Dromparis P, & Michelakis ED (2013). Mitochondria in vascular health and disease. Annual Review of Physiology, 75, 95–126. [DOI] [PubMed] [Google Scholar]
- Duan Y, Dong Y, Dang R, Hu Z, Yang Y, Hu Y, & Cheng J (2018). MiR-122 inhibits epithelial mesenchymal transition by regulating P4HA1 in ovarian cancer cells. Cell Biology International, 42, 1564–1574. [DOI] [PubMed] [Google Scholar]
- Duennwald ML, & Lindquist S (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes & Development, 22, 3308–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duivenvoorden WCM, Hopmans SN, Austin RC, & Pinthus JH (2017). Endoplasmic reticulum protein ERp46 in prostate adenocarcinoma. Oncol Lett, 13, 3624–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletto D, Chevet E, Argon Y, & Appenzeller-Herzog C (2014). Redox controls UPR to control redox. Journal of Cell Science, 127, 3649–3658. [DOI] [PubMed] [Google Scholar]
- Endoh H, Tomida S, Yatabe Y, Konishi H, Osada H, Tajima K, … Mitsudomi T (2004). Prognostic model of pulmonary adenocarcinoma by expression profiling of eight genes as determined by quantitative real-time reverse transcriptase polymerase chain reaction. Journal of Clinical Oncology, 22, 811–819. [DOI] [PubMed] [Google Scholar]
- Frand AR, & Kaiser CA (1998). The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Molecular Cell, 1, 161–170. [DOI] [PubMed] [Google Scholar]
- Frand AR, & Kaiser CA (1999). Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Molecular Cell, 4, 469–477. [DOI] [PubMed] [Google Scholar]
- Gao H, Sun B, Fu H, Chi X, Wang F, Qi X, … Shao S (2016). PDIA6 promotes the proliferation of HeLa cells through activating the Wnt/beta-catenin signaling pathway. Oncotarget, 7, 53289–53298. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gess B, Hofbauer K-H, Wenger RH, Lohaus C, Meyer HE, & Kurtz A (2003). The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1- Lα. European Journal of Biochemistry, 270, 2228–2235. [DOI] [PubMed] [Google Scholar]
- Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, & Simmen T (2010). Ero1α requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones, 15, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, & Semenza GL (2013). Hypoxia- inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. Journal of Biological Chemistry, 288, 10819–10829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gimenez M, Veríssimo-Filho S, Wittig I, Schickling BM, Hahner F, Schürmann C, … Lopes LR (2019). Redox activation of Nox1 (NADPH oxidase 1) involves an intermolecular disulfide bond between protein disulfide isomerase and p47 phox in vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 39, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes EC, Silva AN, & de Oliveira MR (2012). Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid Med Cell Longev, 2012, 756132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordeeva AV, Zvyagilskaya RA, & Labas YA (2003). Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry, 68, 1077–1080. [DOI] [PubMed] [Google Scholar]
- Görlach A, Bertram K, Hudecova S, & Krizanova O (2015). Calcium and ROS: A mutual interplay. Redox Biology, 6, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, … French PJ (2009). Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Research, 69, 9065–9072. [DOI] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin Y-H, Gildersleeve R, Shan J, … Kaufman RJ (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nature Cell Biology, 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassane DC, Sen S, Minhajuddin M, Rossi RM, Corbett CA, Balys M, … Jordan CT (2010). Chemical genomic screening reveals synergism between parthenolide and inhibitors of the PI-3 kinase and mTOR pathways. Blood, 116, 5983–5990. [DOI] [PubMed] [Google Scholar]
- Hatahet F, & Ruddock LW (2009). Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxidants & Redox Signaling, 11, 2807–2850. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, & Su T-P (2009). MAM: More than just a housekeeper. Trends in Cell Biology, 19, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes KE, Batsomboon P, Chen W-C, Johnson BD, Becker A, Eschrich S, … Hazlehurst LA (2019). Inhibition of the FAD containing ER oxidoreductin 1 (Ero1) protein by EN-460 as a strategy for treatment of multiple myeloma. Bioorganic & Medicinal Chemistry, 27, 1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Titus EA, & Cooper AA (2004). Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Molecular Cell, 15, 767–776. [DOI] [PubMed] [Google Scholar]
- Hetz C (2012). The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nature Reviews Molecular Cell Biology, 13, 89–102. [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, & Harding HP (2013). Targeting the unfolded protein response in disease. Nature Reviews Drug Discovery, 12, 703–719. [DOI] [PubMed] [Google Scholar]
- Hetz C, & Mollereau B (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nature Reviews Neuroscience, 15, 233–249. [DOI] [PubMed] [Google Scholar]
- Hofseth LJ, Hussain SP, Wogan GN, & Harris CC (2003). Nitric oxide in cancer and chemoprevention. Free Radic Biol Med, 34, 955–968. [DOI] [PubMed] [Google Scholar]
- Hu WM, Zhang J, Sun SX, Xi SY, Chen ZJ, Jiang XB, … Sai K (2017). Identification of P4HA1 as a prognostic biomarker for high-grade gliomas. Pathology - Research and Practice, 213, 1365–1369. [DOI] [PubMed] [Google Scholar]
- Hwang J, & Qi L (2018). Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends in Biochemical Sciences, 43, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Masui S, Iida H, Vavassori S, Sitia R, & Suzuki M (2010). Crystal structures of human Ero1α reveal the mechanisms of regulated and targeted oxidation of PDI. EMBO Journal, 29, 3330–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, … Flaumenhaft R (2012). Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. Journal of Clinical Investigation, 122, 2104–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop CE, Chakravarthi S, Garbi N, Hämmerling GJ, Lovell S, & Bulleid NJ (2007). ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO Journal, 26, 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausar S, Wang F, & Cui H (2018). The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells, 7, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenche H, Ye ZW, Vedagiri K, Richards DM, Gao XH, Tew KD, … Blumental-Perry A (2016). Adverse outcomes associated with cigarette smoke radicals related to damage to protein-disulfide isomerase. J Biol Chem, 291, 4763–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo C, Yang J, Rajpal G, Wang Y, Liu J, Arvan P, & Stoffers DA (2011). Endoplasmic reticulum oxidoreductin-1-like β (ERO1lβ) regulates susceptibility to endoplasmic reticulum stress and is induced by insulin flux in β-cells. Endocrinology, 152, 2599–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukita K, Tamura Y, Tanaka T, Kajiwara T, Kutomi G, Saito K, … Sato N (2015). Cancer-associated oxidase ERO1-α regulates the expression of MHC class I molecule via oxidative folding. Journal of Immunology, 194, 4988–4996. [DOI] [PubMed] [Google Scholar]
- Kutomi G, Tamura Y, Tanaka T, Kajiwara T, Kukita K, Ohmura T, … Hirata K (2013). Human endoplasmic reticulum oxidoreductin 1-α is a novel predictor for poor prognosis of breast cancer. Cancer Science, 104, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyani A, Tamura S, Yang S, Shergalis A, Samanta S, Kuang Y, … Neamati N (2018). Discovery and mechanistic elucidation of a class of protein disulfide isomerase inhibitors for the treatment of glioblastoma. ChemMedChem, 13, 164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, … Golub TR (2006). The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Langsjoen RM, Auguste AJ, Rossi SL, Roundy CM, Penate HN, Kastis M, … Weaver SC (2017). Host oxidative folding pathways offer novel anti-chikungunya virus drug targets with broad spectrum potential. Antiviral Research, 143, 246–251. [DOI] [PubMed] [Google Scholar]
- Lappi A-K, & Ruddock LW (2011). Reexamination of the role of interplay between glutathione and protein disulfide isomerase. Journal of Molecular Biology, 409, 238–249. [DOI] [PubMed] [Google Scholar]
- Laurindo FRM, Araujo TLS, & Abrahão TB (2014). Nox NADPH oxidases and the endoplasmic reticulum. Antioxidants & Redox Signaling, 20, 2755–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurindo FRM, Fernandes DC, Amanso AM, Lopes LR, & Santos CXC (2008). Novel role of protein disulfide isomerase in the regulation of NADPH oxidase activity: Pathophysiological implications in vascular diseases. Antioxidants & Redox Signaling, 10, 1101–1113. [DOI] [PubMed] [Google Scholar]
- Lee H, Palm J, Grimes SM, & Ji HP (2015). The Cancer Genome Atlas Clinical Explorer: A web and mobile interface for identifying clinical-genomic driver associations. Genome Medicine, 7, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park U-J, & Gwag BJ (2010). Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Experimental & Molecular Medicine, 42, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew QJ, Chia YL, Chu KL, Lam YT, Gurumurthy M, Xu S, … Chao S-H (2012). Identification of HEXIM1 as a positive regulator of p53. Journal of Biological Chemistry, 287, 36443–36454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Mongillo M, Chin K-T, Harding H, Ron D, Marks AR, & Tabas I (2009). Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. Journal of Cell Biology, 186, 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Jia Z, & Trush MA (2016). Defining ROS in biology and medicine. Reactive Oxygen Species (Apex, N. C.), 1, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, … Furie B (2015). Quercetin-3-rutinoside inhibits protein disulfide isomerase by binding to its b′x domain. Journal of Biological Chemistry, 290, 23543–23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Huang X, Uziel T, Hessler P, Albert DH, Roberts-Rapp LA, … Shen Y (2017). HEXIM1 as a robust pharmacodynamic marker for monitoring target engagement of BET family bromodomain inhibitors in tumors and surrogate tissues. Molecular Cancer Therapeutics, 16, 388–396. [DOI] [PubMed] [Google Scholar]
- Lundström J, & Holmgren A (1993). Determination of the reduction-oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry, 32, 6649–6655. [DOI] [PubMed] [Google Scholar]
- Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, & Buetow K (2009). Rembrandt: Helping personalized medicine become a reality through integrative translational research. Molecular Cancer Research, 7, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi AA, Rizvi SH, & Parveen A (2016). Role of endoplasmic reticulum stress and unfolded protein responses in health and diseases. Indian Journal of Clinical Biochemistry, 31, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinouski M, Zhou Y, Belousov VV, Hatfield DL, & Gladyshev VN (2011). Hydrogen peroxide probes directed to different cellular compartments. PLoS One, 6, e14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, … Ron D (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & Development, 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, … Weir GC (2010). Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One, 5, e11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui S, Vavassori S, Fagioli C, Sitia R, & Inaba K (2011). Molecular bases of cyclic and specific disulfide interchange between human ERO1α protein and protein-disulfide isomerase (PDI). Journal of Biological Chemistry, 286, 16261–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa Y (2005). Adiponectin: Identification, physiology and clinical relevance in metabolic and vascular disease. Atheroscler Suppl, 6, 7–14. [DOI] [PubMed] [Google Scholar]
- May D, Itin A, Gal O, Kalinski H, Feinstein E, & Keshet E (2004). Ero1-Lα plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene, 24, 1011–1020. [DOI] [PubMed] [Google Scholar]
- McCormack JG, & Denton RM (1993). Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Developmental Neuroscience, 15, 165–173. [DOI] [PubMed] [Google Scholar]
- Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, … Bensaude O (2004). Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO Journal, 23, 2608–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moenner M, Pluquet O, Bouchecareilh M, & Chevet E (2007). Integrated endoplasmic reticulum stress responses in cancer. Cancer Research, 67, 10631–10634. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Kelleher JK, & Fiskum G (1990). Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. Journal of Biological Chemistry, 265, 10527–10534. [PubMed] [Google Scholar]
- Musa A, Ghoraie LS, Zhang S-D, Glazko G, Yli-Harja O, Dehmer M, … Emmert-Streib F (2017). A review of connectivity map and computational approaches in pharmacogenomics. Briefings in Bioinformatics, 18, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardai G, Stadler K, Papp E, Korcsmáros T, Jakus J, & Csermely P (2005). Diabetic changes in the redox status of the microsomal protein folding machinery. Biochemical & Biophysical Research Communications, 334, 787–795. [DOI] [PubMed] [Google Scholar]
- Oakes SA, & Papa FR (2015). The role of endoplasmic reticulum stress in human pathology. Annual Review of Pathology: Mechanisms of Disease, 10, 173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura M, Kadokura H, Hashimoto S, Yutani K, Kanemura S, Hikima T, … Inaba K (2014). Inhibition of the functional interplay between endoplasmic reticulum (ER) oxidoreduclin-1α (Ero1α) and protein-disulfide isomerase (PDI) by the endocrine disruptor bisphenol A. Journal of Biological Chemistry, 289, 27004–27018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M, Fabbri M, Benedetti C, Fassio A, Pilati S, Bulleid NJ, … Sitia R (2000). Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. Journal of Biological Chemistry, 275, 23685–23692. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, & Voeltz GK (2016). Structure and function of ER membrane contact sites with other organelles. Nature Reviews Molecular Cell Biology, 17, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard MG, Travers KJ, & Weissman JS (1998). Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Molecular Cell, 1, 171–182. [DOI] [PubMed] [Google Scholar]
- Porntaveetus T, Theerapanon T, Srichomthong C, & Shotelersuk V (2018). Cole-Carpenter syndrome in a patient from Thailand. American Journal of Medical Genetics Part A, 176, 1706–1710. [DOI] [PubMed] [Google Scholar]
- Primm TP, & Gilbert HF (2000). Hormone binding by protein disulfide isomerase, a high capacity hormone reservoir of the endoplasmic reticulum. Journal of Biological Chemistry, 276, 281–286. [DOI] [PubMed] [Google Scholar]
- Puspita L, Chung SY, & Shim J-W (2017). Oxidative stress and cellular pathologies in Parkinson’s disease. Molecular Brain, 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Wang H, & Farmer SR (2007). Adiponectin secretion is regulated by SIRT1 and the endoplasmic reticulum oxidoreductase Ero1-Lα. Molecular and Cellular Biology, 27, 4698–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu XA, & Rajpal DK (2012). Applications of Connectivity Map in drug discovery and development. Drug Discovery Today, 17, 1289–1298. [DOI] [PubMed] [Google Scholar]
- Rajpal G, Schuiki I, Liu M, Volchuk A, & Arvan P (2012). Action of protein disulfide isomerase on proinsulin exit from endoplasmic reticulum of pancreatic β-cells. Journal of Biological Chemistry, 287, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramming T, & Appenzeller-Herzog C (2013). Destroy and exploit: Catalyzed removal of hydroperoxides from the endoplasmic reticulum. International Journal of Cell Biology, 2013, 180906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramming T, Hansen HG, Nagata K, Ellgaard L, & Appenzeller-Herzog C (2014). GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radical Biology and Medicine, 70, 106–116. [DOI] [PubMed] [Google Scholar]
- Riedemann J, & Macaulay VM (2006). IGF1R signalling and its inhibition. Endocrine- Related Cancer, S33–S43. [DOI] [PubMed] [Google Scholar]
- Rieusset J (2018). The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death & Disease, 9, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RM, Reyes L, Duncan RM, Bian H, Reitz AB, Manevich Y, … Dolloff NG (2018). Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia, 33, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, … Wouters BG (2010). The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. Journal of Clinical Investigation, 120, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CXC, Stolf BS, Takemoto PVA, Amanso AM, Lopes LR, Souza EB, … Laurindo FRM (2009). Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. Journal of Leukocyte Biology, 86, 989–998. [DOI] [PubMed] [Google Scholar]
- Santos CXC, Tanaka LY, Wosniak J, & Laurindo FRM (2009). Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxidants & Redox Signaling, 11, 2409–2427. [DOI] [PubMed] [Google Scholar]
- Seol S-Y, Kim C, Lim JY, Yoon SO, Hong SW, Kim JW, … Cho JY (2016). Overexpression of endoplasmic reticulum oxidoreductin 1-α (ERO1L) is associated with poor prognosis of gastric cancer. Cancer Research and Treatment, 48, 1196–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, & Kaiser CA (2008). Ero1 and redox homeostasis in the endoplasmic reticulum. Biochimica et Biophysica Acta, 1783, 549–556. [DOI] [PubMed] [Google Scholar]
- Sevier CS, Qu H, Heldman N, Gross E, Fass D, & Kaiser CA (2007). Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell, 129, 333–344. [DOI] [PubMed] [Google Scholar]
- Shergalis A, Bankhead A 3rd, Luesakul U, Muangsin N, & Neamati N (2018). Current challenges and opportunities in treating glioblastoma. Pharmacological Reviews, 70, 412–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shergalis A, & Neamati N (2017). Protein Disulfide Isomerase In Choi S(Ed.), Encyclopedia of Signaling Molecules (pp. 4200–4211). New York, NY: Springer New York. [Google Scholar]
- Sifuentes-Franco S, Pacheco-Moisés FP, Rodríguez-Carrizalez AD, & Miranda-Díaz AG (2017). The role of oxidative stress, mitochondrial function, and autophagy in diabetic polyneuropathy. Journal of Diabetes Research, 2017, 1673081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Scheuner D, Ron D, Pennathur S, & Kaufman RJ (2008). Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. Journal of Clinical Investigation, 118, 3378–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolf BS, Smyrnias I, Lopes LR, Vendramin A, Goto H, Laurindo FRM, … Santos CXC (2011). Protein disulfide isomerase and host-pathogen interaction. Scientific World Journal, 11, 1749–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, … Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Cui J, He Q, Chen Z, Arvan P, & Liu M (2015). Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Molecular Aspects of Medicine, 42, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, … von Mering C (2015). STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research, 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kajiwara T, Torigoe T, Okamoto Y, Sato N, & Tamura Y (2015). Cancer-associated oxidoreductase ERO1-α drives the production of tumor-promoting myeloid-derived suppressor cells via oxidative protein folding. Journal of Immunology, 194, 2004–2010. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kutomi G, Kajiwara T, Kukita K, Kochin V, Kanaseki T, … Tamura Y (2017). Cancer-associated oxidoreductase ERO1-α promotes immune escape through up- regulation of PD-L1 in human breast cancer. Oncotarget, 8, 24706–24718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavender TJ, & Bulleid NJ (2010). Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. Journal of Cell Science, 123, 2672–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoudam T, Jeon JH, Ha CM, & Lee IK (2016). Role of Mitochondria-Associated Endoplasmic Reticulum Membrane in Inflammation-Mediated Metabolic Diseases. Mediators Inflamm, 2016, 1851420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM (2007). S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol Interv, 7, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Xiong Y, Bowers RR Jr., Hutchens S, & Tew KD (2009). Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res, 69, 7626–7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse G, Yan BP, Chan YW, Tian XY, & Huang Y (2016). Reactive Oxygen Species, Endoplasmic Reticulum Stress and Mitochondrial Dysfunction: The Link with Cardiac Arrhythmogenesis. Front Physiol, 7, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Ho-Schleyer SC, Travers KJ, & Weissman JS (2000). Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science, 290, 1571–1574. [DOI] [PubMed] [Google Scholar]
- Tu BP, & Weissman JS (2002). The FAD- and O(2)-dependent reaction cycle of Ero1- mediated oxidative protein folding in the endoplasmic reticulum. Molecular Cell, 10, 983–994. [DOI] [PubMed] [Google Scholar]
- Tu BP, & Weissman JS (2004). Oxidative protein folding in eukaryotes: Mechanisms and consequences. Journal of Cell Biology, 164, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi Z-Q, Gu Z, Ma Y, … Lipton SA (2006). S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature, 441, 513–517. [DOI] [PubMed] [Google Scholar]
- Vance JE, & Vance DE (1986). Specific pools of phospholipids are used for lipoprotein secretion by cultured rat hepatocytes. Journal of Biological Chemistry, 261, 4486–4491. [PubMed] [Google Scholar]
- Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D, … Reu FJ (2016). Novel protein disulfide isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Research, 76, 3340–3350. [DOI] [PubMed] [Google Scholar]
- Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, … Izumi T (1999). A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. Journal of Clinical Investigation, 103, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li S-J, Sidhu A, Zhu L, Liang Y, Freedman RB, & Wang C-C (2008). Reconstitution of human Ero1-Lα/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a’ domains of protein-disulfide isomerase. Journal of Biological Chemistry, 284, 199–206. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang L, Niu Y, Sitia R, & Wang CC (2014). Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1alpha to promote oxidative protein folding. Antioxid Redox Signal, 20, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhu L, & Wang C-C (2011). The endoplasmic reticulum sulfhydryl oxidase Ero1β drives efficient oxidative protein folding with loose regulation. Biochemical Journal, 434, 113–121. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yin F, Xu J, Zhang T, Wang G, Mao M, … Cai Z (2019). CYT997(Lexibulin) induces apoptosis and autophagy through the activation of mutually reinforced ER stress and ROS in osteosarcoma. Journal of Experimental Clinical Cancer Research, 38, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZV, Schraw TD, Kim JY, Khan T, Rajala MW, Follenzi A, & Scherer PE (2007). Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol Cell Biol, 27, 3716–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnau J, Sharma V, Gamiz-Hernandez AP, Di Luca A, Haapanen O, Vattulainen I, … Kaila VRI (2018). Redox-coupled quinone dynamics in the respiratory complex I. Proceedings of the National Academy of Sciences of the United States of America, 115, E8413–E8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J, Birk J, Haataja L, Liu M, Ramming T, Weiss MA, … Arvan P (2013). Endoplasmic reticulum oxidoreductin-1α (Ero1α) improves folding and secretion of mutant proinsulin and limits mutant proinsulin-induced endoplasmic reticulum stress. Journal of Biological Chemistry, 288, 31010–31018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Li C, Gu M, Wang H, Chen W, Luo G, … Sheng P (2018). Protein disulfide isomerase silence inhibits inflammatory functions of macrophages by suppressing reactive oxygen species and NF-κB pathway. Inflammation, 41, 614–625. [DOI] [PubMed] [Google Scholar]
- Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, … Xu R (2018). Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nature Communications, 9, 4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, … Neamati N (2012). Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proceedings of the National Academy of Sciences of the United States of America, 109, 16348–16353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Sankar S, & Neamati N (2014). Protein disulfide isomerase: A promising target for cancer therapy. Drug Discovery Today, 19, 222–240. [DOI] [PubMed] [Google Scholar]
- Yang S, Shergalis A, Lu D, Kyani A, Lu Z, Ljungman M, & Neamati N (2019). Design, synthesis, and biological evaluation of novel allosteric protein disulfide isomerase inhibitors. Journal of Medicinal Chemistry, 62, 3447–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Yang C, Yu F, Ding W, Hu Y, Cheng F, … Rao J (2018). Endoplasmic reticulum resident oxidase ERO1-Lalpha promotes hepatocellular carcinoma metastasis and angiogenesis through the S1PR1/STAT3/VEGF-A pathway. Cell Death & Disease, 9, 1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZW, Zhang J, Ancrum T, Manevich Y, Townsend DM, & Tew KD (2017). Glutathione S-transferase P-mediated protein S-glutathionylation of resident endoplasmic reticulum proteins influences sensitivity to drug-induced unfolded protein response. Antioxid Redox Signal, 26, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh IJ, Song K, Wittmann BM, Bai X, Danielpour D, & Montano MM (2014). HEXIM1 plays a critical role in the inhibition of the androgen receptor by anti-androgens. Biochemical Journal, 462, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeshan H, Lee G, Kim H-R, & Chae H-J (2016). Endoplasmic reticulum stress and associated ROS. International Journal of Molecular Sciences, 17, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lai E, Teodoro T, & Volchuk A (2008). GRP78, but not protein-disulfide isomerase, partially reverses hyperglycemia-induced inhibition of insulin synthesis and secretion in pancreatic β-cells. Journal of Biological Chemistry, 284, 5289–5298. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li T, Zhang L, Shangguan F, Shi G, Wu X, … Wang L (2019). Targeting the functional interplay between endoplasmic reticulum oxidoreductin-1α and protein disulfide isomerase suppresses the progression of cervical cancer. EBioMedicine, 41, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li G, Kaplan A, Gaschler MM, Zhang X, Hou Z, … Duan W (2018). Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Human Molecular Genetics, 27, 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Jin G, Mi R, Zhang J, Zhang J, Xu H, … Liu F (2017). Knockdown of P4HA1 inhibits neovascularization via targeting glioma stem cell-endothelial cell transdifferentiation and disrupting vascular basement membrane. Oncotarget, 8, 35877–35889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E, Chin K-T, Blais J, Harding HP, & Ron D (2010). ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. Journal of Cell Biology, 188, 821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E, Hansen HG, Yeo GSH, Fujii J, & Ron D (2012). Endoplasmic reticulum thiol oxidase deficiency leads to ascorbic acid depletion and noncanonical scurvy in mice. Molecular Cell, 48, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E, Melo EP, Yang Y, Wahlander Å, Neubert TA, & Ron D (2010). Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Molecular Cell, 40, 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker JI, Schlechter BL, Stopa JD, Liebman HA, Aggarwal A, Puligandla M, … Flaumenhaft R CATIQ Investigators. (2019). Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight, 4, 125851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.