

Abstract
The transcription factor NRF2 is one of the body’s major defense mechanisms, driving transcription of >300 antioxidant response element (ARE)-regulated genes that are involved in many critical cellular processes including redox regulation, proteostasis, xenobiotic detoxification, and primary metabolism. The transcription factor NRF2 and natural products have an intimately entwined history, as the discovery of NRF2 and much of its rich biology were revealed using natural products both intentionally and unintentionally. In addition, in the last decade a more sinister aspect of NRF2 biology has been revealed. NRF2 is normally present at very low cellular levels and only activated when needed, however, it has been recently revealed that chronic, high levels of NRF2 can lead to diseases such as diabetes and cancer, and may play a role in other diseases. Again, this “dark side” of NRF2 was revealed and studied largely using a natural product, the quassinoid, brusatol. In the present review, we provide an overview of NRF2 structure and function to orient the general reader, we will discuss the history of NRF2 and NRF2-activating compounds and the biology these have revealed, and we will delve into the dark side of NRF2 and contemporary issues related to the dark side biology and the role of natural products in dissecting this biology.
Graphical Abstract
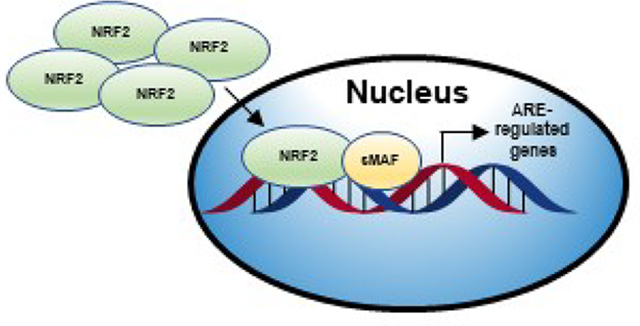
NRF2 is a protective transcription factor that has been intentionally activated by many natural products for chemoprevention, but it has also been shown aberrant NRF2 activation can lead to disease and natural products have also been used to inhibit the NRF2 pathway.
1. Introduction
Natural products have long served as chemical matter for the discovery and/or development of clinical therapies1–9. It has been posited that up to 40% of all new molecular entities submitted to the Food and Drug Administration (FDA) for approval are natural products, natural product-derived, or natural product inspired (containing a natural product pharmacophore) compounds. In the areas of cancer therapeutics and antibiotics, natural products make up 53% and 59%, respectively, of all FDA approved drugs4. But another important function of natural products has been in chemical biology, where natural products have served as potent and specific probes to dissect the physiologic and/or pathologic functions of biomolecules10, 11. Although small, synthetic compounds have certainly been important, the evolutionary processes and principles that have shaped natural products to have exquisite and privileged architectures and related biological functions has facilitated the significant contributions made by these important chemical entities5, 8, 12.
For many thousands of years, natural remedies for a variety of ailments have been employed13–17. Many of these traditional remedies remain without scientific explanation for their curative powers, but as natural products research has evolved so has the exploration of natural medicines and the organisms from which they are derived to both isolate active principles and to assign the mechanism(s) by which these active entities interact with their molecular target(s)18–23. Isolation of active ingredients is often carried out by extracting compounds from the sample and using some form of biological readout to guide purification of the desired natural product24–27. The assignment of the mode of action of a given natural product, that has been isolated in a phenotypical screen, is often a bottleneck in this process, but has seen application of many exciting and novel approaches to the solving of this task28. The assignment of a target(s) to natural products has often revealed new biology or even opened entirely new fields. This is certainly the case for the nuclear factor erythroid (NF-E2)-related factor 2 (NRF2) pathway29–37.
The NRF2 pathway, discussed in detail below, is the body’s primary defense against oxidative stress and many environmentally and/or intentionally introduced xenobiotic agents. In the 1960s and 1970s, it was discovered that phase 2 metabolic enzymes could be activated by phenolic antioxidants that had been used as preservatives in food and that this activation could also be carried out using natural products from plants. Animal models demonstrated that this activation had chemopreventive properties, meaning animals exposed to phase 2 activating compounds were less likely to develop tumors. This led to the search for compounds that could activate this response more effectively and hence potentially protect users of these compounds from cancer38–54. One of the early natural product discoveries was the isothiocyanate, sulforaphane (discussed in detail below), which was found to be present in cruciferous vegetables and especially in the seeds and sprouts of broccoli55, 56. The action of these anti-oxidant activating compounds ultimately led to the discovery of the anti-oxidant response element (ARE) that is found in the promoter region of the upregulated genes57, 58. This in turn led to the discovery of the transcription factor, NRF2, that binds the ARE and regulates these genes. Finally, the regulation of the NRF2 pathway was revealed and with this the mechanism of activation of NRF2 by chemopreventive agents was revealed. Thus, the observation of the action of the simple natural product sulforaphane led to the development of a large field of research that has expanded in many directions including the discovery of many other natural product activators and the discovery of some natural product pathway inhibitors. In addition, there have been and remain many clinical trials of NRF2 activating compounds initiated for a variety of indications and there is one NRF2 activator currently used in patients. The NRF2 field has now blossomed to >2000 publications per year with increasing growth every year (Fig. 1).
Figure 1. The NRF2 field is growing rapidly.

The graph shown lists the number of publications in pubmed by year.
In this review article, we will provide a detailed discussion of the NRF2 pathway. Many readers with primary interest in natural products might not have knowledge of the NRF2 field, so we feel this will help to orient readers. We will then discuss the structure and function of NRF2 and its key regulatory protein, Kelch-like ECH-associated protein 1 (KEAP1). Presently, there are very little structural data regarding NRF2, which we will highlight, but there have been several studies of KEAP1 domain structure at high resolution and a full-length low-resolution structure that have revealed critical mechanistic details and explained the mechanisms by which some activating compounds may operate. Because a number of recent reviews have been written with emphasis on NRF2 pathway modulators, including an outstanding review on natural products in this context, we will avoid a detailed discussion of these compounds and instead direct readers to these reviews35, 59–66. We will instead discuss NRF2-activating natural products or natural product-derived compounds based not as much on structural considerations but based on important principles of biology, biochemistry, or medicine they have revealed. In this context, we will focus primarily on compounds that regulate post-translational events, although we will discuss some post-translational modification modulators, transcriptional modulators, and translational modulators. We will not discuss epigenetic modulation as a mechanism to activate or deactivate NRF2 in any depth, but this is a rapidly growing field with important implications in the dark side of NRF267–69. In addition, recent work has indicated that dietary NRF2-modulating compounds can affect the gut microbiome, adding another layer of complexity to NRF2 research70. We will also discuss the dark side of NRF271. This is a more recent demonstration that the NRF2 pathway, like many stress-response pathways, can be hijacked by cells for nefarious purposes, allowing cells to survive the harsh conditions required for cancerous transformation, growth, and spread. Using the natural product, brusatol, it has been revealed that NRF2 inhibition might be a viable strategy to treat NRF2 addicted cancers either as a stand-alone therapy or an adjuvant to sensitize cancerous cells to first-line chemotherapies72. Moreover, brusatol has been used as a tool compound to reveal critical aspects of NRF2 biology as pertain to cancer and other diseases73, 74.
2. The canonical NRF2 pathway
NRF2 is a basic leucine zipper (bZip) transcription factor from the Cap ‘n’ Collar (CNC) family29, 75, 76. NRF2 is ubiquitously expressed in all cells, but under non-stressed, basal conditions, the level of NRF2 is kept low post translationally through the action of the ubiquitin proteasome system (UPS)77–81. In a non-stress situation, the Nrf2-ECH homology 2 (Neh2) domain of NRF2 (see Section 3) is bound by two molecules of the adapter protein KEAP1. The Kelch domain of KEAP1 has a recognition site for ETGE and ETGE-like motifs. The Neh2 domain of NRF2 has both an ETGE motif and a DLG motif. The ETGE motif binds one molecule of KEAP1 with high (8 nM) affinity and the DLG motif binds a second molecule of KEAP1 with lower affinity (500 nM)82–84. The two KEAP1 molecules form a homodimer via their BTB domains, which also form the binding site for Cullin3 (CUL3), bringing NRF2 into proximity to the CUL3-Really interesting new gene (RING)-box 1 (RBX1) E3 ubiquitin ligase complex. The stoichiometry of binding between KEAP1 and CUL3 is not known, with differing views, however, the structure of the CUL3-KEAP1 BTB complex is 1:185. The 2:1 KEAP1:NRF2 binding arrangement is critical for the correct function of this regulatory complex. Between the ETGE and DLG motifs of NRF2 are seven lysine residues, which are the sites of ubiquitylations77, so when both NRF2 motifs are bound to two KEAP1 molecules these lysines are correctly aligned and NRF2 is readily ubiquitylated. Polyubiquitylated NRF2 is then extracted from the KEAP1-CUL3-RBX1 complex through the action of the ATPase associated with various cellular activities (AAA+) chaperone, p97 and shuttled to the proteasome for degradation86. This post-translational regulatory system ensures low levels of NRF2 under non-stressed conditions (Fig. 2).
Figure 2. The canonical NRF2 pathway.

NRF2 is sequestered by the CUL3-RBX1 E3 ubiquitin ligase complex through the KEAP1 adapter protein, which binds the ETGE and DLG motifs of NRF2 in a 2:1 KEAP1:NRF2 ratio. A) Under basal, unstressed conditions, the CUL3 complex ubiquitylates NRF2 at one of the seven lysines residing between the ETGE and DLG motifs. B) Ubiquitylated NRF2 is then extracted from the CUL3 complex through the action of p97-UFD1-NPL4 mediated by UBXN7. C) Ubiquitylated NRF2 is transferred to the 26S proteasome, where it is destroyed. D) When cells are challenged with an oxidative or xenobiotic insult, one of the sensor cysteines of KEAP1 can become modified, which causes a structural rearrangement, releasing the DLG motif, and stopping subsequent ubiquitylation. Please see the text for alternate explanations and other models. E) The inhibited CUL3 complex blocks further NRF2 degradation, allowing NRF2 levels to rise in the cytosol. F) They then translocate to the nucleus, where they can bind to sMAF proteins and initiate ARE-regulated transcriptional programs.
When cells are exposed to oxidative or xenobiotic stress, the ubiquitylation of NRF2 is blocked, the levels of NRF2 rise, NRF2 translocates into the nucleus, and dimerizes with another bZip transcription factor, one of the small musculoaponeurotic fibrosarcoma (sMAF) proteins. This heterodimer binds AREs and ARE-regulated genes are activated to enhance cellular defense29, 33–35, 87. Through elegant mechanistic studies, Talalay and co-workers showed there are ten classes of molecules that activate the NRF2-ARE axis: (i) oxidizable diphenols, phenylenediamines, and quinones; (ii) Michael acceptors; (iii) isothiocyanates; (iv ) thio-carbamates; (v) trivalent arsenicals; (vi) dithiolethiones; (vii) hydroperoxides; (viii) vicinal dimercaptans; (ix) heavy metals; and (x) polyenes88–90. However, most of the natural products that are used to intentionally activate NRF2 and the canonical NRF2 activators are electrophilic in nature (i.e. sulforaphane, bardoxolone (semi-synthetic), curcumin, cinnamaldehyde, withaferin A). KEAP1 contains a series of cysteines (27 in humans), and most prominently for the present review, cysteine151 that act as sensors79, 81, 91–93. Cysteine151 has been shown to be surrounded by a collection of basic amino acids, bringing the pKa below 7 and increasing the nucleophilicity of this residue at physiologic pH94, 95. When an electrophilic compound is adducted to this sensor residue, an incompletely defined structural rearrangement takes place. There are two primary, not mutually exclusive, models that have been put forth to explain the effects of electrophiles: the hinge and latch model82, 83 and the CUL3 dissociation model96–99. In the hinge and latch model, electrophilic addition causes the lower affinity DLG-motif, the latch, to be released from KEAP1, while the ETGE motif, the hinge, remains attached. When the latch releases, the ubiquitylation of NRF2 is blocked, but NRF2 remains bound to the complex. In the CUL3 dissociation model, KEAP1 releases from CUL3, blocking ubiquitylation, but keeping KEAP1 bound to NRF2, blocking further NRF2 degradation. Both models then lead to inhibition of ubiquitylation of NRF2, an increase in the level of NRF2, nuclear translocation, dimerization with sMAF proteins, and subsequent transcriptional activation of ARE-containing NRF2 target genes (Fig. 2). Initially, it was thought NRF2 only regulated phase 2 metabolic enzymes, that is conjugating enzymes, however contemporary genetic experiments have revealed NRF2 controls many more (>300) genes including genes from phase 1 metabolism, phase 2 metabolism, transporters, protein quality control, redox regulation, transcriptional regulation, iron metabolism, and autophagy 33, 100. It is interesting to note, that despite the historical use of the term phase 2 response, the assay used to define the response, the Prochaska assay (see below), measures the activity of NQO1, an enzyme of phase 1 metabolism.
3. The structure and function of NRF2 and KEAP1
NRF2 is a complex multi-domain protein that is likely predominantly intrinsically disordered in cells. For this reason, there are very little structural data on NRF2. Biochemical and genetic experiments have assigned function to seven (Neh1–7) regions of NRF2, leading to functional domains, but none of these have been shown to have stand-alone tertiary structure and thus are perhaps not domains in the traditional sense of the word. Several structures of KEAP1 domains have been solved and these have provided a great deal of mechanistic insight, but a full-length structure or a co-structure with the full CUL3-RBX1 E3 ligase complex or with other proteins in the pathway have not been solved. Although there are certainly possibilities to activate or inhibit the NRF2 pathway independent of directly targeting KEAP1 (activation) or NRF2 (possible inhibition), the present discussion of structure will be confined to NRF2 and KEAP1.
3.1. NRF2
NRF2 is a 67 kDa bZip CNC transcription factor. The NRF2 domains, Neh1–7 (NRF2-ECH homology 1–7), and currently assigned functions are shown in Figure 3A. There is very little structural data on NRF2, and NRF2 is likely predominantly intrinsically disordered in the unbound state. Certainly, the nuclear magnetic resonance (NMR) structure of the Neh1 domain (https://www.rcsb.org), which is α-helical with large unstructured regions (Fig. 3B), compared to other structural data of dimeric, DNA-bound bZip transcription factors (Fig. 3C) argues for this region being intrinsically disordered101. The lack of structural data has surely stymied efforts to find natural products that modulate NRF2 function through direct targeting.
Figure 3. NRF2 domain architecture and structure.

A) NRF2 is comprised of seven domains termed Neh1–7 that have been defined according to biological function and homology to other protein domains. The numbering shown in the figure is for the human protein. Defined biological function of each of the domains is shown above the given domain and explained in greater detail in the text. B) The only structure of an unliganded domain of NRF2 is the NMR structure of the Neh1 domain shown (PDB ID 2LZ1). C) A crystal structure of the CNC bZip transcription factor MAFA as a potential model for the NRF2 Neh1 domain when bound to an obligate DNA-binding partner (PDB ID 4EOT). The high homology between bZip transcription factors argue this is a likely model for NRF2 in the active form, but to date, no structure has been solved.
Neh1, which lies near the C-terminus of NRF2 is the CNC-bZip domain. Although no structural data of the Neh1 domain bound to DNA exist, there is a monomeric NMR structure (Fig. 3B), showing an α-helical and disordered architecture, and there are a number of crystal structures for bZip transcription factors bound to DNA that are known and these are likely structurally similar to Neh1 when bound to sMAF proteins and ARE (Fig. 3C). In general, these DNA recognition elements contain a positively charged basic region that interacts with the negatively charged phosphate backbone of DNA and a series of leucines that form the hydrophobic dimerization domain, as implied by the name102–105. In the case of NRF2, it forms a heterodimeric structure with another bZIP transcription factor, one of three sMAF proteins, MAFF, MAFG, or MAFK76, 87, 106, 107. This heterodimeric structure then binds to an ARE with the consensus sequence 5’-TGA(G/C)NNNGC-3’, which is found in the promoter region of more than 300 identified genes100, 108. In addition to these functions, the nuclear localization sequence (NLS) and the nuclear export sequence are reported to be in the Neh1 domain109–111.
The Neh2 domain is at the N-terminus of NRF2 and is the KEAP1 interacting domain83, 112, 113. Two small peptides from this domain, one harboring the ETGE motif (Fig. 4A and B) and a second larger peptide harboring the DLG motif (Fig. 4C and D) bound to the KELCH domain of KEAP1 have been solved, but this is the extent of NRF2 x-ray crystallographic structural data (see below). The Neh2 domain contains the two essential KEAP1 recognition elements, the weak binding DLG motif and the tight binding ETGE motif that flank the seven critical lysines that are ubiquitylated by the CUL3 complex. Each of the two NRF2 recognition motifs binds to a single KEAP1 molecule and this weak-tight dual binding mode has been shown to be essential for the regulation of NRF2, with critical physiologic and pathologic implications (see Figure 3, above for regulatory considerations, and below for a more detailed discussion of physiologic and pathologic implications).
Figure 4. The KEAP1 Kelch domain bound to ETGE and DLG containing peptides.

A) and B) The ETGE motif binds to a series of positively charged amino acids in the KEAP1 Kelch domain. The ETGE forms a loop in the binding pose. Two views are shown: from the top A) and the side B). (PDB ID 5WFV). C) and D) The DLG containing peptide shows a pose like the ETGE, but shows fewer contacts, explaining the decreased affinity. The rest of the peptide forms an alpha-helical structure, but this is not known to be physiological or significant due to lack of larger structural data. Two views are shown: from the top C) and the side D). (PDB ID 3WN7).
The Neh3 domain is at the extreme C-terminus of NRF2 (Fig. 3A). Deletion of this domain does not affect dimerization, DNA-binding or localization of NRF2, but it does block NRF2’s transcriptional activity. Biochemical studies showed Neh3 interacts with chromo-ATPase/helicase DNA-binding protein (CHD6) and that this interaction is essential for expression of the NRF2 target gene NAD(P)H Quinone Dehydrogenase 1 (NQO1), arguing the Neh3 domain is a transactivation domain 114.
The Neh4 and Neh5 domains play a dual role in NRF2 function (Fig. 3A). The initially assigned function was transcriptional activation115–117. Deletion of either of these domains leads to a reduced expression of a number of NRF2 target genes, which was at least in part due to decreased binding to CREB (cAMP-response-element-binding protein)-binding protein (CBP) and Brahma-related gene 1 (BRG1)115, 118. In addition, it was demonstrated that NRF2 can be ubiquitylated by the endoplasmic reticulum associated E3 ubiquitin ligase, HMG-CoA Reductase Degradation (HRD1), followed by proteasome-mediated degradation and that this interaction is mediated through the Neh4/5 domains, although the precise mechanism and structural details of this interaction remain to be defined119.
NRF2 has also been shown to be regulated by E3 ubiquitin ligases other than the KEAP1-CUL3-RBX1 complex and HRD1. The Neh6 domain, like Neh2 or described Neh4/5 above, comprises another degron82, 120–123. The Neh6 domain can be phosphorylated by glycogen synthase kinase (GSK-3β), which recruits the E3 ubiquitin ligase, β-transducin repeat-containing protein (β-TrCP), leading to the degradation of NRF2 (Fig. 7). This will be discussed in greater detail in Section 7 (see below).
Figure 7. The conversion of glucoraphanin to sulforaphane by the plant enzyme Myrosinase.

Normally, in cruciferous vegetables, sulforaphane is in the glycosylated form. It is thought that when plants are attacked by herbivores, the level of Myrosinase increases, releasing sulforaphane and deterring the herbivore. This has important implications in the use of sulforaphane as a drug, since glucoraphanin is poorly bio-available. In the liver of mammals, glucoraphanin is reduced to glucoerucin. Both of these forms are substrates for Myrosinase. Once the carbohydrate is hydrolyzed, the resulting product undergoes a spontaneous Lossen rearrangement to the final, NRF2 activating isothiocyante.
The Neh7 domain was more recently described and early as well as some contemporary papers and figures do not show the Neh7 domain. However, biochemical and genetic data have shown that retinoic X receptor alpha (RXRα) is an NRF2 transcriptional repressor (see section 8.1 for further discussion). Biochemical experiments showed that the Neh7 domain of NRF2 directly interacts with RXRα, resulting in the negative regulation of NRF2 target genes (see Fig. 3A)124.
3.2. KEAP1
As discussed in Section 2, the KEAP1-CUL3-RBX1 E3 ligase complex is the best studied and the most critical NRF2 negative regulator. KEAP1 is a member of the BTB-Kelch family of proteins80, 125. To date, only a low-resolution cryo-electron microscopy (cryo-EM) structure of the complete KEAP1 protein has been reported126. However, several crystal structures of KEAP1 domains have been solved, including co-crystal structures with KEAP1-NRF2-ARE pathway activating molecules (Fig. 4, 5, and 6). In addition, co-crystal structures with NRF2 peptides comprising the ETGE and DLG motifs and with protein-protein interaction inhibitors (PPIs) have helped to discover and develop PPIs to activate the NRF2 pathway without the potential off-target actions of covalent activators (Fig. 4). Overall, KEAP1 has been shown to have three structural domains that will be discussed individually below (Fig. 5A). In addition, each of the domains has been assigned a function, but because no complete KEAP1 structure has been solved, some details of the mechanisms of modulation and domain communication remain incomplete.
Figure 5. KEAP1 domain architecture and structure.

A) KEAP1 is comprised of three structural domains the BTB domain, the IVR domain, and the Kelch domain. The numbering shown is for the human protein. The assigned functions of each of the domains is shown above each domain. Human KEAP1 has 27 cysteines that can work as sensors. The most important cysteine sensors are also shown. For a detailed discussion of domain and cysteine function, see the text. B) The BTB domain of KEAP1 forms a functional dimer to bind to a single NRF2 protein. This dual binding mode is essential for physiologic function. (PDB ID 4CXI). C) The BTB domain bound to the N-terminus of CUL3. (PDB ID 5NLB). D) The unliganded Kelch domain of KEAP1. (PDB ID 5WFV).
Figure 6. The KEAP1 BTB domain.

A) The apo BTB domain of KEAP1 showing Cys151 in green. B) The BTB domain of KEAP1 bound to the A ring of bardoxolone. This structure has been used to argue for dissociation of KEAP1 from the CUL3 complex upon activation by electrophiles. (PDB ID 4CXI). C) The KEAP1 BTB domain bound to CUL 3 with Cys150 highlighted in green. (PDB ID 4CXT). (PDB ID 5NLB).
KEAP1 is a cysteine-rich protein with 27 cysteines in the human KEAP1 protein80, 92, 125. A great deal of effort both in vitro and in vivo has been put forth to understand the significance, or lack thereof, of these cysteines in sensing various cellular insults. This has led to what is called the cysteine code – the rules that dictate how KEAP1 senses xenobiotic agents79, 93. In Figure 5A, the most important of these sensors are shown. As discussed, Cys151 is the major sensor for most of the molecules discussed in this review, it detects Michael acceptor containing molecules in addition to sensing nitric oxide and the thiocyanates. The mildly electrophilic sulfoxythiocarbamate alkyne derivative of sulforaphane revealed this compound modifies Cys273, Cys288, and Cys613. The cyclopentenone prostaglandins, alkenals, and nitro-oleic acid were shown to modify Cys288. Heavy metals were shown to be sensed by Cys226 and Cys613, which are close in space, allowing for chelation of the cations. Finally, Cys226, Cys622, and Cys624 have been shown to respond to H2O2127. Interestingly, the cysteine sensors are located primarily outside of the KELCH domain, despite this domain being the primary sight of NRF2 binding, as discussed below.
Near the N-terminus of KEAP1 is the Broad complex, Tramtrack, and Bric-à-Brac (BTB) domain (residues 61 to 179; Fig. 5A, B and C)). The structure of the BTB domain has been solved, including in complex with a natural product-derived compound (bardoxolone, see below, Fig. 6B). The dimeric BTB domain is shown in Figure 5B95 and the BTB domain bound to the N-terminus of CUL3 is shown in Figure 5C (http://www.rcsb.org/structure/5NLB). The BTB domain crystallized as a dimer mediated by helix 1 and a domain swapped β-sheet. The KEAP1 BTB domain is a protein-protein interaction domain that mediates formation of the KEAP1 homodimer and interaction with CUL3, however the structure of the CUL3-BTB domain is a 1:1 complex and this remains to be explained. In addition, the BTB domain contains Cys151 and a series of basic residues (His129, Lys131, Arg135, Lys150, and His154), which are near Cys151. The proximity of these basic amino acids decreases the pKa of Cys151, leading to a more active (more nucleophilic or more oxidation prone) sensor. Although Cys151 is not the only reactive cysteine in KEAP1, most of the natural products that have been shown to activate NRF2 signaling have been shown to form a covalent adduct with this cysteine and mutation to serine or another non-reactive amino acid eliminates activation (Fig. 5A)79.
Moving N-terminal to C-terminal, the intervening region (IVR) follows the BTB domain (residues 180 to 314; Fig. 5A). The IVR is also known as the BACK domain. There currently is not a structure of the KEAP1 IVR, but other homologous BACK domains have been solved92, 94. Based on homology, the IVR is predicted to be an α-helical domain as the other BACK domains that have been solved are all highly extended α-helical domains. In addition to connecting the BTB and Kelch domains, the IVR contains Cys273 and Cys288, which have been shown to be important for stress modulation, but seem to play only a minor role, if any, in responding to covalently binding natural products93. At the N-terminal side of the IVR is the 3-box motif, which connects the BTB and the IVR, likely mediating critical communication between the two. It has also been shown to facilitate the interaction between KEAP1 and CUL3128.
Finally, the most C-terminal domain (residues 315 to 598; Fig. 4, 5A and D) is a β-propeller structure called the Kelch domain129. As mentioned, a number of structures of this domain have been solved, including in complex with NRF2 peptides, which bind at the bottom of the “bowl-shaped” structure, engaging a series of positively charged residues (Fig. 4)83, 112, 113. These peptide-Kelch co-crystal structures have provided critical guidance to the development of non-covalent NRF2 activating compounds130–132. This will not be discussed extensively, as most of these PPIs are synthetic compounds, but the interested reader is directed towards a number of interesting publications on this matter, including an excellent recent analysis of the merits of each of the reported compounds and the traps that can befall the unwary when trying to discover compounds of this class, or really any class of inhibitor133, 134. However, we will discuss a recent comparison between covalent and non-covalent natural product activators, the geopyxins, that were reported by our group135.
4. KEAP1 targeting natural product NRF2 activators
The explosion of NRF2 research over the last decade or so has been quite remarkable (Fig. 1). The field had its beginning several decades prior to the discovery of the KEAP1-NRF2-ARE signaling axis, with the observation that small doses of polycyclic aromatic hydrocarbons protect against toxicity and carcinogenicity39, 40, 42. It was later found in animal tests that phenolic antioxidants, such as the commonly used food preservative, 2(3)-tert-butyl-4-hydroxyanisole (BHA), and compounds naturally in foods, when fed to rats in high doses, could protect the animals from carcinogens43–54, 136–140. These studies ultimately led to the field of chemoprevention, which was predicated on the intentional activation of phase 2 metabolic pathways, the so-called “phase 2 response”, to resist disease53, 54, 88, 90, 141, 142. The pioneering work of Paul Talalay and his research group set out to understand the mechanistic underpinnings of this activation of metabolic enzymes. Using an assay that measured the level of NQO1, called the Prochaska bioassay143, 144, Talalay and his team isolated 1-isothiocyanato-4R-(methyl-sulfinyl)butane – sulforaphane (SFN) from cruciferous vegetables, followed by many other natural products56, 145. It is pertinent to mention that the activity of NRF2/phase 2 activating compounds is measured by a parameter called CD, which is a measure of the amount of compound required to double the activity of NQO1 in Hepa 1c1c7 cells. Talalay observed that the compounds that led to activation of phase 2 metabolism were electrophiles or redox active compounds, pointing towards the engagement of a cysteine residue144.
Studies on chemoprevention also led to the search for the genetic element that regulated these phase 2 metabolic enzymes, ultimately revealing the ARE146. This responsive element was shown to be present in the promoter of the glutathione S transferase alpha 1 (Gst-Ya) gene in rats and to be responsive to phenolic antioxidants57, 58. This was followed by cloning of the ARE-responsive transcription factor, NRF2 76, 147 and to the generation of an Nrf2 knockout mouse75. Although the first report of an Nrf2−/− mouse was from the Kan group, shortly after this report, the Yamamoto group reported their own knockout mouse and distributed it to many research groups around the world, having a huge impact on the NRF2 field, this remains one of the critical reagents used in the study of NRF287. The next critical breakthrough in the NRF2 field was the discovery that KEAP1 binds to NRF2 and acts as a negative regulator125. This discovery led to a flurry of biochemical studies on KEAP1 demonstrating KEAP1-Cys151 is essential for sensing NRF2 activating compounds, supporting the observations of Talalay; KEAP1 is part of a CUL3 E3 ubiquitin ligase complex; and NRF2 is regulated post-translationally through ubiquitylation and degradation79,77, 78, 99.
4.1. Sulforaphane
Sulforaphane (SFN; 1-isothiocyanato-4-(methylsulfinyl)butane) is an isothiocyanate that is found in cruciferous vegetables such as broccoli, kale, and cabbage, with the highest concentration being found in broccoli sprouts. Normally, SFN is in the non-reactive glucoraphanin form until it is needed as a protective measure against herbivores, at which time it is hydrolyzed by Myrosinase (EC 3.2.1.147, thioglucoside glucohydrolase) to the aglycone148. The aglycone (thiohydroximate-O-sulfonate) then undergoes a spontaneous thio-Lossen-type rearrangement to generate the reactive isothiocyanate at neutral or basic (physiological) pH or the nitrile, sulforaphane nitrile (5-(methylsulfinyl)pentanenitrile), will form at acidic pH (Fig. 7). In mammalian liver, glucoraphanin is reduced to glucoerucin, which is also a substrate of Myrosinase and can be converted to erucin ((4-isothiocyanatobutyl)(methyl)sulfane) or erucin nitrile (5-(methylthio)pentanenitrile). The erucin compounds are readily converted to SFN or SFN nitrile via oxidation of the sulfur atom. Pharmacokinetic studies of SFN and glucoraphanin have shown that SFN has much better bioavailability, which has influenced therapeutic preparations and dosing strategies148–158. It is also interesting to note, that a series of epitionitriles have also been found to result from alkenyl glucosinolates (not shown in Fig. 7) and these also activate NRF2 in a KEAP1 dependent manner159.
Sulforaphane has become the gold standard by which all other covalent NRF2 activators are measured and has been used in thousands of studies as represented by the many publications reporting its use (2047 publications listed in PubMed). SFN is the most potent unmodified natural product NRF2 activator that has been described (CD = 0.23 μM) and it has been validated in many animal models using both wild-type animals and Nrf2−/− animals, demonstrating the protective action of SFN is through the NRF2 pathway 56, 149, 158, 160. In addition to rodent models, SFN has been in many clinical trials for various indications. The initial clinical studies with SFN were conducted in Qidong, China, which had a high incidence of liver cancer due to aflatoxin exposure160, 161. Presently, a search of clinical trials (clinicaltrials.gov) reveals trials for many indications including: schizophrenia, UV skin damage, autism, lung cancer, prostate cancer, COPD, asthma, bladder cancer, osteoarthritis, melanoma, breast cancer, type 2 diabetes mellitus, and pancreatic cancer, validating pre-clinical studies162–177. In addition, due to its protective role, SFN has been used as an adjuvant therapy to prevent side effects in a number of cancer drug trials178.
As discussed above, NRF2 and SFN are intimately connected, as SFN has been used to dissect many of the critical features of NRF2 biology and biochemistry62, 160. However, there have been conflicting studies about the mechanism by which SFN activates NRF2. There are studies that have shown SFN can react with Cys273 or 288 and other studies have shown SFN activity is dependent on Cys15179, 151, 179–186. It was subsequently shown that this discrepancy was due to the different conditions used in the conflicting studies. Based on the current understanding of the cysteine code, SFN activates NRF2 by binding to Cys151 of KEAP1 at physiologically relevant conditions. However, as shown, Cys151 is in the BTB domain (Fig. 5A), thus the mechanism by which SFN adduction to Cys151 leads to blocked ubiquitylation of NRF2 and signaling remains uncertain. A recent co-crystal structure of the BTB domain with bardoxolone (Fig. 6B) argued against release of the DLG motif from the Kelch domain, arguing instead for KEAP1 dissociation from CUL395. However, given the size difference between SFN and bardoxolone, it is perhaps possible both mechanisms are operational depending on the identity of the adduct. There are certainly data supporting both models95, 97, 187–193. Without a full structural characterization of KEAP1, KEAP1-CUL3, and KEAP1-NRF2, this remains an unresolved issue. Advances in cryo-electron microscopy may perhaps offer a solution to this.
4.2. Curcumin
Another important phytochemical is the polyphenol curcumin (Fig. 8A), isolated from Curcuma longa, which is what gives the spice turmeric its yellow color. Curcumin has long been used in Ayurveda to treat many diverse conditions and has even been the source of some scientific controversy leading to retraction of several papers reporting anti-cancer properties of curcumin. It has been shown that curcumin is a weak activator of NRF2 (maximum of 1.5-fold induction of an ARE-luciferase reporter in Beas-2B cells at 15 μM)194. In addition, curcumin has been reported to have poor pharmacological properties and has been shown to be active in many bio-assays, leading to it being referred to as a pan-assay interference (PAINS) compound195, 196. However, there has been interest in optimizing curcumin’s pharmacology and activity. This included work from our group that developed a synthetic derivative bis[2-hydroxybenzylidene]acetone (BHBA), which was shown to be more potent than curcumin at activating the KEAP1-NRF2-ARE axis in a variety of assays (Fig. 8B). It was shown to activate NRF2 in a KEAP1-Cys151 dependent manner, as expected for an electrophilic Michael acceptor containing compound. In addition, this derivative was shown to protect mice from lung cancer development when challenged with vinyl carbamate194. Curcumin (Fig. 8A) and its synthetic derivatives (BHBA and F10 in Fig. 8B and C) have been shown to be effective in treating or preventing other maladies, but so far, the poor pharmacology has prevented its use in the clinic178, 197–202. In addition, like other NRF2 activating compounds of this sort, curcumin activates many pathways with a complex mode of action203–205.
Figure 8. The NRF2 activators discussed in the text.

A) Curcumin from Curcuma longa. B) A curcumin derivative with more potent NRF2 activation and better pharmacological properties. C) A curcumin derivative with more potent NRF2 activation and better pharmacological properties. D) Cinnamaldehyde from Cinnamomum verum. E) Bixin from Bixa orellana. The red box is to differentiate natural product derived compounds from natural products.
4.3. Cinnamaldehyde
Cinnamaldehyde (Fig. 8D) is the aromatic aldehyde that gives cinnamon its pleasant taste and odor. This simple diterpene has been investigated for both anti-inflammatory and chemopreventive properties206–208. Our lab has carried out several investigations into the mode of action of cinnamaldehyde and its protective effects. We have shown that cinnamaldehyde can activate the NRF2 pathway and that this action can protect skin cells from photodamage209. We have also shown that this action can protect colon cells from genotoxic insults and azoxymethane/dextran sulfate-induced colon cancer formation210, 211. We demonstrated cinnamaldehyde can protect mice from diabetes in a streptozotocin-induced murine type 1 diabetes model177. These studies have been confirmed by others and expanded to include models of type 2 diabetes as well212–217. In addition, and in line with the effects of other NRF2-inducing compounds, cinnamaldehyde offers neuroprotection and protection from kidney damage caused by chronic kidney disease218, 219.
4.4. Bixin
Bixin (Fig. 8E) is an apocarotenoid derived from the seeds of the achiote tree (Bixa orellana) that is used as a spice in cooking and was demonstrated to induce the KEAP1-NRF2-ARE axis by adduction to KEAP1-Cys151 in vitro, although the chemical nature of the adduct is not yet known220. In these same studies, it was shown, in line with the data from cinnamaldehyde, that bixin protects SKH-1 mice from UV skin damage when applied directly to the skin. Moreover, using Nrf2+/+ and Nrf2−/− animals, this was shown to be an Nrf2 dependent process, as protection was only observed in the wild-type animals. Bixin was later shown to protect animals against ventilation induced lung injury (VILI) in a murine model. Animals treated by intraperitoneal injection of bixin showed normal lung morphology, decreased inflammation, and reduced oxidative DNA damage, all hallmarks of VILI221. Again, using Nrf2+/+ and Nrf2−/− mice, this response was shown to be Nrf2-dependent in vivo. Animal studies of cardiac injury caused by a high-fat diet demonstrated protection against cardiac dysfunction by inhibiting fibrosis, inflammation, and reactive oxygen induced damage. The reduction of inflammation was shown to be due to a decrease in the secretion of pro-inflammatory cytokines. These data were corroborated using an in vitro model of inflammation by treating cardiomyocytes with lipopolysaccharide to induce inflammation222. This of course argues for bixin as a safe means to reduce ROS and inflammation in a variety of disease states, in agreement with other NRF2 inducing compounds. Finally, a series of studies in vivo showed bixin can protect animals from lung damage caused by particles, offering a potential prophylaxis for people living in areas with heavy air pollution223, 224.
4.5. Withaferin A
Withaferin A is a steroidal lactone isolated from Withania somnifera (common name Ashwaganda or winter cherry), that has been used in Ayurveda for thousands of years and is reported to promote general well-being. This important compound was originally isolated in the 1960s and later shown to have a number of interesting activities including anti-cancer activity in a number of tumor lines, anti-inflammatory, immune-modulatory, anti-metastasis, and anti-angiogenesis225–229. The discovery of the anti-cancer activity led to a flurry of mode of action studies that indicated withaferin A can interact with a variety of pathways. As shown in Figure 9A, withaferin A contains both an A-ring and an E-ring Michael acceptor and a B-ring epoxide, all of which have the potential to interact with KEAP1 cysteines to activate NRF2 signaling. Several groups have reported activation of the NRF2 pathway by withaferin A and that it can be used to ameliorate a variety of effects in cellular and animal models of various disease states230–234. Interestingly, recent studies on the prevention of acetaminophen induced hepatotoxicity in a mouse model and follow up in vitro mechanistic studies indicated withaferin A can induce Nrf2 in a Keap1-dependent and in a PTEN/PI3K/Akt-dependent manner as discussed in section 7 below235. However, many other modes of action have been assigned to withaferin A. Our work and others have shown that withaferin A and some of its derivatives can inhibit the 20S core particle of the proteasome, which would also be expected to activate NRF2. In addition, we found that withaferin A can inhibit p97 activity, which also can inhibit NRF2 degradation86. Although withaferin A inhibited both p97 and the proteasome, a semi-synthetic-azido compound (Fig. 9B) was found to be selective for p97 and to still inhibit NRF2 degradation236–238. Thus, it seems the action of withaferin A and perhaps other withanolides is quite complex. Despite this complicated pharmacology, withaferin A remains of interest and has been in clinical trials for the treatment of schizophrenia and schizoaffective disorder.
Figure 9. Withaferin A and a semi-synthetic derivative.

A) Withaferin A (from Withania somnifera)has been assigned many modes of action, but it is a known NRF2 activator as verified by our lab, however the precise mechanism by which it activates NRF2 is more complex than simple Cys151 adduction (See text for further discussion). B) A semi-synthetic withaferin A derivative that does not inhibit the proteasome but inhibits p97 and activates NRF2. The red box is to differentiate natural product derived compounds from natural products.
5. NRF2 activators in neurological disorders
Given the critical role of oxidative stress in a variety of neurodegenerative states, it is perhaps not surprising NRF2 has been shown to be at the heart of many neurological diseases, as has been mentioned above and is reflected in neuro related clinical trials using NRF2 activating compounds. In fact, at present the only FDA approved NRF2 activating compound in the clinic is a multiple sclerosis drug, dimethyl fumarate (Fig. 11G; section 5.2). The topic of Nrf2 in neurodegeneration has been reviewed quite recently and the interested reader is directed to these reviews 36, 239, 240. However, as will be discussed in the section on the GSK-3β/NRF2/β-TrCP axis and in other publications on the topic, a number of natural products have been shown to be important in the dissection of the role of NRF2 in neurophysiology 241–253. As shown in Figure 10, there are many other natural products that have been used in studies of neuropathology and physiology. Carnosic acid (Fig. 10A), sulfuretin (Fig. 10B), and methysticin (Fig. 10C) were each shown to prevent cell death in cellular and in vivo Alzheimer’s disease (AD) models 254–256. Both resveratrol (Fig. 10D) and thymol (Fig. 10E) were also shown to protect neuronal function in an aging animal model and a high-fat diet animal model, respectively 257, 258. In addition, a number of recent papers have reported on the dual reactive oxygen species scavenging and NRF2 activating activity of a variety of natural compounds (Fig. 10F–O), and how these protect a neuronal cell line, however, the precise mechanistic details remain to be illuminated in many cases259–268. These exciting results and the FDA approval of DMF offer hope for NRF2 modulating compounds in treating patients with neurological disorders.
Figure 11. Natural product derived compounds.

A) Oleanolic acid is isolated in large quantity from olive (Olea europaea) waste and has been shown to have modest anti-inflammatory action but does not show NRF2 activation activity. B) Addition of a Michael acceptor to the A ring produced a μM NRF2 activating compound. C) Addition of a second Michael acceptor to the C ring led to an approximately order of magnitude increase in NRF2 activation activity, but it is not understood why. D) Electronic modulation of the A ring Michael acceptor gave another order of magnitude increase and the compound bardoxolone (CDDO), one of the most potent NRF2 activators known. Me Bardoxolone (more commonly CDDO-Me) only showed a modest increase in potency, but became orally bio-available, whereas bardoxolone must be injected. E) The imidazole variant has been in many studies but does not seem to be more efficacious. However, as discussed in the text, there are subtle differences between the activities of the varios CDDO derivatives for yet undescribed reasons. F) Omaveloxolone is a recent iteration from Reata Pharmaceuticals that is in clinical trials for several indications (see text). G) Dimethyl fumarate is a synthetic derivative of a primary metabolite but is included since it is the only compound to be used in humans that uses NRF2 activation as its primary proposed mode of action.
Figure 10. NRF2 activating compounds that have been used in neuroprotective studies.

In addition to these, other compounds discussed in other sections shown in other figures have been used in neuroprotective studies. A) Carnosic acid from Rosmarinus officinalis. B) Sulfuretin from Rhus verniciflua. C) Methysticin from Piper methysticum. D) Resveratrol. E) Thymol from Thymus vulgaris. F) 6-Dehydrogingerdione from Zingiber officinale. G) Xanthohumol from Humulus lupulus. H) Hydroxytyrosol from Olea europaea. I) 6-Shogaol from Zingiber officinale. J) Cardamonin from Alpinia katsumadae. K) Honokiol from Magnolia virginiana. L) Costunolide from Saussurea costus. M) Mangiferin from Mangifera indica. N) Chlorogenic acid from coffee. O) Lipoamide. Please see the text for details.
6. KEAP1 targeting natural product-derived NRF2 activators
In addition to the many natural products that have been shown to activate the KEAP1-NRF2-ARE axis, there are compounds that are of natural origin but have been optimized through medicinal chemistry efforts. We will focus our attention on two members of this class based on their important contributions to the development of NRF2 activating compounds as clinical candidates. The synthetic oleanane triterpenoids have been and are currently in numerous clinical trials, and the simple modified metabolite, dimethyl fumarate, is currently the only canonical NRF2 activator in the clinic. However, it is being constantly revealed that there are other clinical compounds that can activate the NRF2 pathway. In addition, it should be emphasized that the KEAP1-NRF2-ARE axis is not the sole target of these compounds, but that some of the important physiologic effects have been assigned to NRF2 using genetic and biochemical experiments.
6.1. Oleanane triterpenoids
Oleanolic acid (Fig. 11A) is found in many different plants and foods but is at very high levels in olive trees and most often isolated from olive pulp or leaves. It is perhaps this component of olive oil that is responsible for the salubrious benefits of the Mediterranean diet. In its parent form, oleanolic acid has modest anti-inflammatory activity. In an effort to optimize this anti-inflammatory potential, a series of synthetic oleanane triterpenoids were synthesized from oleanolic acid and evaluated for their ability to decrease NO synthesis269–273. In the parent form, oleanolic acid is not an activator of the NRF2 pathway, but studies on the synthetic oleanolic acid derivatives, inspired by the Michael acceptors that are required for potent activity, revealed a series of cyano enones that are the most potent NRF2 activating compounds reported274. The introduction of a Michael acceptor in the A ring gave CD = 3.9 μM (Fig. 11B; see above for the definition of CD), indicating the importance of this modification. Surprisingly, the introduction of another Michael acceptor into the C ring further increased the potency (Fig. 11C; CD = 0.28 μM). Addition of an electron withdrawing group to the A ring Michael acceptor further enhanced activity. A cyano group at this position gives the well-known bardoxolone (CDDO (2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid)), which has a further order of magnitude improvement in the activity (Fig. 11D; CD = 0.0023 μM). Making the methyl ester of the acid (CDDO-Me) gives a more modest improvement of activity (Fig. 11D; CD = 0.0010 μM), but this substitution makes the compound orally bio-available. Other bardoxolone derivatives (i.e. Fig. 11E and F) have also been made, and those that have been most extensively studied or are in clinical trials are shown. Importantly, it was found that the NRF2 activating activity correlated well with the anti-inflammatory activity274. The initial synthetic efforts allowed for the development of a biotinylated probe that was used to pull down potential targets of bardoxolone that could be analyzed by LC-MS, which revealed >500 targets in addition to KEAP1275, indicating the promiscuous nature of these compounds.
Despite the many targets identified, or perhaps because of, bardoxolone and its derivatives have shown a great deal of pre-clinical success and are being evaluated in several clinical trials. Initially, these compounds were synthesized to treat malignant cells and have been evaluated both in vitro and in vivo to treat cancers of blood, breast, ovaries, prostate, lungs, pancreas, colon, skin, and brain. These compounds have been evaluated as both standalone therapies and as adjuvants, mainly in combination with immunotherapies276–356. It must be pointed out that many of these studies attribute the activity of bardoxolone and its derivatives to an activity independent of NRF2 and at present this cannot be fully addressed. In addition, it is interesting to note that not all of the derivatives behave equivalently332. In addition to cancer treatment, CDDO and its derivatives have been used in chemoprevention, a more traditional role for an NRF2-activating compound281, 284, 287, 314, 353, 357–364.
In addition to the complex role played by bardoxolone in cancer, it has also been studied in several other disease contexts. Most of these are related to inflammation, perhaps more directly implicating the actions of the KEAP1-NRF2-ARE signaling axis. These include neurodegenerative proteinopathies such as Parkinson’s disease, Huntington’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis365–373, diseases of the eyes369, 374–379, the lungs376, 380–394, the heart392, 395–398, metabolic disorders399–401, liver diseases402–407, kidney disease408–413, and autoimmune disorders332, 414–417. It is interesting to note, that the bardoxolone derivative that is used in these studies is significant, as they have differential effects, again arguing for a much more complicated mode of action than simply activation of the KEAP1-NRF2-ARE axis. Despite the complexities and pleotropic effects of bardoxolone and its derivatives, these remain an interesting class of compounds that are in clinical trials for a number of indications including: chronic kidney diseases, pulmonary hypertension, type 2 diabetes mellitus, liver disease, and diabetic nephropathy in the case of CDDO-Me. In addition, omaveloxolone (Fig. 11F) is currently under clinical investigation for a variety of indications, including Friedreich’s ataxia, mitochondrial myopathies, immuno-oncology, and prevention of corneal endothelial cell loss following cataract surgery. As a final point, in 2013, CDDO-Me failed in phase 3 clinical trials due to cardio toxicity, there are a number of potential explanations for this418, but it remains these are promising compounds and continue to be investigated. In addition, this setback does not dampen enthusiasm for the NRF2 pathway as a promising therapeutic target.
6.2. Dimethyl fumarate (Fumaderm; Tecfidera)
Fumarate is a primary metabolite of the citric acid cycle and an oncometabolite (a metabolite that activates a cancer promoting pathway) that is upregulated in a rare kidney cancer that has a fumarate dehydratase deficiency419. Interestingly, a semisynthetic variant of fumarate, dimethylfumarate (DMF; Fig. 11G), is the only NRF2-modulating compound to enter the clinic. Originally, DMF had been approved in Europe to treat psoriasis and then in 2013, the US Food and Drug Administration (FDA) approved the use of DMF to treat relapsing multiple sclerosis (MS). Subsequent and/or concurrently with this event DMF, and the monomethyl variant (MMF), have been entered into several other clinical trials for pulmonary hypertension, brain cancer, lymphoma, psoriasis, rheumatoid arthritis, lupus erythematosus, and cutaneous T-cell lymphoma. The approval of DMF for MS has also spurred a flurry of other pre-clinical and mode of action studies on DMF. The preclinical studies have been on several indications with a seeming emphasis on neurodegenerative disorders, as discussed above. From the mode of action studies, it is clear that DMF and other fumarate derivatives, activate the NRF2 pathway and this can explain many of the benefits of DMF treatment, however, like many NRF2-activating compounds DMF does not have a straightforward mode of action and other, non-NRF2-mediated actions have been described. In all likelihood it is a combination of the NRF2-dependent and independent actions that ultimately lead to disease treatment, however further work is needed to fully understand this drug420–422.
7. A comparison between covalent and non-covalent activators – the geopyxins
As discussed extensively above in the section on electrophilic NRF2 activators, all the compounds in pre-clinical development, clinical assessment, or patient use have many potential targets and complex modes of action due to the nature of the reactive electrophilic moieties present. In addition, because these compounds likely modify many cysteines in the proteome, potentially leading to toxicity, there is growing interest in the discovery and development of non-covalent NRF2 activating compounds. This endeavor has produced many interesting studies and a flurry of structural studies including co-crystal structures of the ETGE and DLG motifs bound to the Kelch domain of KEAP1 (Fig. 4) that have guided the development of non-covalent protein-protein interaction inhibitors that block the interaction between, most likely, the DLG motif and the Kelch domain of KEAP1, mimicking the release of the latch in the hinge and latch model and leading to NRF2 activation. Nearly all these compounds have been synthetic compounds131, 423–427. However, our lab recently published a study of the ent-kaurane diterpenoid, geopyxin A, and its derivatives (Fig. 12). We found among the geopyxins many with Michael acceptors required KEAP1-Cys151 for their activity (Fig. 12A as one example), but we also found geopyxin F (Fig. 12B), which is devoid of a Michael acceptor, requires KEAP1, but does not require Cys151. This compound was not as potent as the covalent compounds but increased the expression of NRF2 with kinetics distinct from the other, electrophilic NRF2 activators in the series. We also showed that geopyxin F increased the half-life of NRF2 to a much greater extent than the other geopyxins. We showed that this non-covalent variant was KEAP1 dependent, but, unlike the other geopyxin, was not dependent on Cys151. Finally, and importantly, we found that geopyxin F conferred greater cellular protection on cells challenged with toxicants than either SFN or the other geopyxin and that this activity depended on the NRF2 pathway135. Presently, we are working to understand the mechanism by which geopyxin F activates NRF2. It seems, the most likely mechanism is by interacting with the Kelch domain of KEAP1, but we cannot rule out other mechanisms. In addition, given the modest activity of this compound, we are working to make more potent derivatives using a semi-synthetic strategy.
Figure 12. The geopyxins offer insight into the advantages of non-covalent NRF2 activation.

A) A series of ent-kaurane diterpenoids were shown to activate NRF2. Geopyxin C from Geopyxis aff. majalis, a fungus occurring in the lichen Pseudevernia intensa, was shown to potently activate NRF2 in a KEAP1-Cys151 manner. B) Geopyxin F from Geopyxis sp. AZ0066 inhabiting the lichen Pseudevernia intensa was shown to be a modest activator of the NRF2 pathway. However, geopyxin F was shown to activate NRF2 in a KEAP1-dependent, but Cys151-independent manner. Moreover, geopyxin F showed greater protection of cells against toxicants and that this protection was NRF2-dependent.
8. The GSK-3β/NRF2/β-TrCP signaling axis
NRF2 has recently been shown to be regulated by E3 ubiquitin ligases other than the KEAP1-CUL3-RBX1 complex and the Neh6 domain, like Neh4/5 described above, mediates one of these alternate (non-KEAP1-CUL3) degradative processes (Fig. 3A and 13; for a recent review see 428). The Neh6 domain contains a series of phosphodegron sequences that enhance binding of β-transducin repeat-containing protein (β-TrCP), a substrate adaptor for the S-Phase Kinase Associated Protein 1 (Skp1)-Cullin 1 (Cul1)-(Rbx1/Roc1) ubiquitin ligase complex 82, 120, 122. The sequence at the N-terminus of the Neh6 domain was shown to be phosphorylated by glycogen synthase kinase-3 (GSK-3), leading to enhanced NRF2 degradation 429–431. This was shown to have important potential application in NRF2-driven cancers 431. In human lung A549 cells which contain a KEAP1 mutation leading to high NRF2 levels, GSK-3 activation by inhibition of the AKT/PKB (protein kinase-B) pathway led to a decrease of NRF2 levels and sensitized cells to first line chemotherapy120.
Figure 13. The GSK-3β/NRF2/β-TrCP regulatory axis.

GSK-3β can phosphorylate the Neh6 domain of NRF2 making it an enhanced substrate for the CUL1/β-TrCP/RBX1 complex. GSK-3β is inhibited by Ser9 phosphorylation mediated by PKC or AKT/PKB, which are both activated by PDK1. AKT/PKB can also be activated by AMPK or inhibited by PHLPP2. PI3K converts PIP2 to PIP3, which activates PDK1. The action of PI3K can be reversed by PTEN. The letters A-D in the figure refer to sites of modulation by the compounds in Figure 14.
In addition to the traditional electrophilic NRF2 activating compounds, such as sulforaphane, several other natural products have played a critical role in dissecting the details of the GSK-3β/NRF2/β-TrCP axis. An abbreviated overview of this axis is shown in Figure 13. It should be pointed out, this figure is not meant to indicate each of these comes from the same signaling event, but for the sake of clarity, details have been removed. In Figure 13, the labels A-D are associated with a given group of molecules, indicate a branch of the axis that has been modulated by natural products, and correspond with the labeling from Figure 14. This is not meant to be a comprehensive list but illustrates some of the elements of this axis that have been revealed. An important early discovery, prior to dissection of this axis, was the discovery that the natural product wortmannin (Fig. 14A) from the fungi Penicillium funiculosum, a phosphoinositide 3-kinase (PI3K) inhibitor could reverse the effects of the synthetic NRF2 activating compound, tert-butylhydroquinone, in IMR-32 neuroblastoma cells 241. More recent studies have looked at the relationship between PI3K and NRF2 using natural products in liver (shikonin) and neuro protection (desoxo-narchinol A and narchinol B), but the mechanistic details were a bit vague and these were not PI3K inhibitors, but somehow activated the axis 252, 432. It was also shown that a polysaccharide from Abelmoschus esculentus mitigated type 2 diabetes symptoms in a mouse model through a similar activation of the PI3K axis without mechanistic details 433. Kaempferol was shown to have cardioprotective properties and this was thought to be due to NRF2 modulation through the PI3K/AKT/GSK-3β axis 434. A study using the protein kinase C (PKC) inhibitor, chelerythrine (Fig. 14B), showed inhibition of PKC could regulate NRF2 by deactivating GSK-3β (Fig. 13B), which was used to show the M1 muscarinic receptor activates NRF2 through the PKC pathway 245. The connection between PKC and NRF2 was further validated by showing sauchinone (Fig. 14B) can activate PKCδ, leading to GSK-3β inhibition and NRF2 activation to protect the liver against acetaminophen toxicity 435. NRF2 can also be positively regulated through activation of AMPK, which activates AKT and inhibits GSK-3β (Fig. 13C). The compounds shown in Figure 14C have been shown to activate NRF2 through activation of AMPK. Three of these compounds, nectandrin B, esculentoside A, and pterostilbene were shown to confer hepatoprotection through NRF2 upregulation 436–438. Two other AMPK activating compounds, butin and emodin, were shown to protect animals in ischemia reperfusion models 249, 439. In Figure 14D, a series of compounds are shown that activate AKT/PKB, but do not necessarily contain mechanistic details or the details will be discussed separately here 246, 251, 440–443. Interestingly, the bioflavonoid, morin, was shown to inhibit the phosphatase PHLPP2, which is a negative regulator of AKT/PKB, leading to NRF2 activation (Fig. 14D) and conferring protection in an acetaminophen challenge model 444–446. Finally, the peptide melittin from the honeybee (Apis mellifera) was shown to protect against myocarditis by increasing the expression of HDAC2 and activation of the GSK-3β/NRF2 axis 447. The relationship between epigenetic modifiers and NRF2 is an expanding area of research, but is beyond the present discussion 33, 448, 449. Finally, it is worth mentioning that many of the compounds described in this section likely have complex modes of action and many likely also activate NRF2 in a KEAP1-dependent manner. More rigorous investigations using KEAP1 knockout cells will solidify the conclusion that NRF2 activation by these compounds is through modulation of the GSK-3β/NRF2/β-TrCP signaling axis.
Figure 14. NRF2 modulating compounds that modulate the GSK-3β/NRF2/β-TrCP regulatory axis.

A) PI3K inhibitors that inhibit NRF2 by blocking PI3K. Wortmannin from Penicillium funiculosum. Despxo-narchinol A and narchinol B from Nardostachys jatamansi. Shikonin from Lithospermum erythrorhizon. Kaempferol from Brassica oleracea var. viridis. B) PKC modulators. Chelerythrine (Chelidonium majus) inhibits PKC and sauchinone (Saururus chinensis) activates PKC leading to inhibition of NRF2 and activation of NRF2, respectively. C) Compounds that activate AKT/PKB by increasing the activity of AMPK. Nectandrin B from Myristica fragrans. Emodin from Rheum hybridum. Esculentoside A from Phytolacca esculenta. Amentoflavone from Ginkgo biloba. Butin from Vernonia anthelmintica. Pterostilbene from blueberries. Apelin 13 from humans. D) Miscellaneous compounds that activate AKT/PKB by unknown mechanisms or through routes described in the text. 2-(penta-1,3-diynyl)-5-(3,4-dihydroxybut-1-ynyl)thiophene (PDDYT) from Echinops grijsii. Rosmarinic acid from Rosmarinus officinalis. Igalan from Inula helenium L. Oxymatrine from Sophora flavescens. Morin from Maclura pomifera. Totarol from Podocarpus totara. Melittin from honeybee (Apis mellifera) venom.
9. The dark side of NRF2 argues for the development of NRF2 inhibitors
After decades of chemopreventive research, data started to emerge indicating that persistent unregulated expression of NRF2 might have deleterious effects, the dark side of NRF2, which led to the discovery of the NRF2 pathway inhibitor, brusatol (see below)71, 72. In 2008, our group demonstrated that NRF2 promotes cancer, a concept that has been further supported by recent work from our lab and other’s demonstrating that once tumors have initiated, high levels of NRF2 promote tumor progression, metastasis, and chemoresistance. In addition, patients with high NRF2 levels in their tumor tissues have a higher risk of recurrence, increased incidence of chemoresistance, and overall poor prognosis450–454. Dysregulation of NRF2, resulting in high NRF2 expression, is common in many human cancers. Somatic NRF2/KEAP1 mutations that disrupt the NRF2-KEAP1 interaction and constitutively activate NRF2 are frequent in certain cancer types, particularly in lung cancer, where these mutations are present in up to one third of patients455–458. In fact, a recent genome-wide somatic point mutation saturation analysis of 21 tumor types, found that while only a few well-known cancer genes are significantly mutated across different tumor types, KEAP1 is significantly mutated in multiple cancer types, including lung, head and neck, and bladder (as a reference, classical cancer genes such as TP53, KRAS, BRAF and NRAS are also significantly mutated across four or more tumor types)459. Furthermore, in lung adenocarcinoma, KEAP1 is as frequently mutated (>30%) as the tumor suppressor gene TP53. In addition to the KEAP1 or NRF2 mutations that disrupt the NRF2-KEAP1 interaction, KEAP1 or CUL3 mutations that compromise KEAP1-CUL3 E3 ligase activity and result in high NRF2 expression have also been described460–463. High NRF2 expression can also be achieved by epigenetic silencing of KEAP1 through hypermethylation of its promoter464 or accumulation of oncometabolites that covalently modify KEAP1 and prevent NRF2 degradation419. NRF2 is also upregulated at the transcriptional level by aberrant signaling of KRAS, BRAF, and MYC oncogenes74, 465. An increasing number of studies have shown that high expression of NRF2 (through constitutive activation of NRF2) promotes cancer progression and resistance to treatment451, 466–468. More recently, we reported our novel findings suggesting that activation of NRF2 accelerates metastasis of existing tumors in mice469. Even though the mechanism by which NRF2 upregulates metastasis-associated proteins has not been established, mechanisms by which constitutive activation of NRF2 contributes to tumor progression and resistance have been demonstrated, including increased detoxification of chemotherapeutic agents, maintenance of reducing conditions, metabolic reprogramming, increased proliferation, maintenance of cancer stem cells, suppression of apoptosis, induction of autophagy, upregulation of the proteasome, and modification of protein synthesis470–478. Furthermore, knockdown or deletion of NRF2 decreases NRF2-addicted cancer cell proliferation and viability in vitro and impedes tumor growth in vivo471, 476, 479. This observation of the promotion of tumor development, growth, and metastasis by unregulated NRF2 led to the proposal of NRF2 as an oncogene and argues for the discovery and development of NRF2 inhibitors67, 72, 480, 481. A recent effort to define druggable targets in the NRF2 pathway has revealed potential new mechanisms of inhibiting NRF2 to combat NRF2 addicted cancers and especially chemo resistant cancers482.
9.1. All-trans retinoic acid
The first NRF2 pathway inhibitor to be discovered was all-trans retinoic acid (ATRA; Fig. 15A) as well as other retinoic acid receptor α (RARα) agonists483. Interestingly, there are conflicting reports of the effects of ATRA on the NRF2 pathway with claims of both activation and inhibition483–489. This conflict has been shown for a number of NRF2 pathway inhibitors, perhaps arguing that careful control of doses and models is critical to accurately assess these reagents490. This discrepancy not-withstanding, ATRA is an important probe molecule that revealed the role of retinoic X receptor alpha (RXRα) in the NRF2 pathway. In particular, it showed RXRα can bind to the Neh7 domain of NRF2 in the nucleus and negatively regulate transcription of ARE-regulated genes124. ATRA has also been shown to synergize with other cancer therapies, confirming the potential of an NRF2 inhibitor as either an adjuvant therapy or as a standalone cancer therapy485, 489.
Figure 15. The dark side of NRF2 has led to a search for NRF2 inhibitors.

A) The first NRF2 pathway inhibitor to be revealed was all-trans retinoic acid (ATRA). However, this was not without controversy as some groups reported ATRA to be an NRF2 activator. In any case, ATRA revealed RXR-𝛼 as a negative regulator of NRF2 transcription and defined the Neh7 domain as the site of RXR-𝛼 binding. B) Brusatol is a quassinoid that inhibits the synthesis of NRF2 and is the most potent NRF2 pathway inhibitor known. Despite potential off-target effects, brusatol (Brucia javanica) has been used extensively to probe the NRF2 pathway and reveal the intricacies of the dark-side of NRF2. C) Brucein C (Brucia javanica) was found to be inactive in NRF2 pathway assays. D) Bruceantin (Brucea antidysenterica) has been shown to be more potent than brusatol at inhibiting NRF2 function. These three molecules, and others of the class, show interesting SAR related to the lipid ester. E) and F) Febrifugine (Dichroa febrifuga) and halofuginone, a semi-synthetic derivative of febrifuginone, were shown to block prolyl-tRNA synthetase, thus blocking NRF2 synthesis and confirmed some of the studies conducted by brusatol, cementing the importance of the discovery and development of an NRF2 inhibitor. G) Wogonin (Scutellaria baicalensis) has been shown to decrease NRF2 mRNA levels and to reverse chemoresistance. However, conflicting studies have shown this to be an NRF2 activating compound.
9.2. Brusatol
The first targeted attempt to discover an NRF2 inhibiting compound was carried out in our lab. Using an ARE-luciferase reporter cell line, we set out to discover compounds that decreased the level of the luciferase reporter from a series of natural product extracts. A plant extract derived from Brucea javanica showed the desired decrease in luciferase activity and the active principal was isolated using activity-guided fractionation, revealing the quassinoid, brusatol as a potent NRF2 pathway inhibitor, showing greater than 50% inhibition of luciferase activity at nM concentrations (Fig. 15B). Interestingly, the closely related compound, brucein C (Fig. 15C), did not show inhibition of NRF2 function. Bruceantin (Fig. 15D), however, was shown to be more potent than brusatol (data not shown). In each of the tested derivatives of brusatol, the only point of difference was the ester substitution on the C ring. Brusatol has proved to be an important probe molecule, allowing a detailed validation of the dark side of NRF2 hypothesis. Indeed, we went on to show that brusatol can sensitize NRF2-addicted491, 492 A549 lung cancer cells to the first line chemotherapeutic agents cisplatin and doxorubicin both in vitro and in vivo72, 73. This critical observation argues for the continued development of NRF2 inhibitors. In addition, we have employed brusatol to dissect the contribution of NRF2 to cancer development, growth, and metastasis. Using either brusatol or SFN, we showed in vivo that treatment with SFN prevents tumor formation by upregulating the NRF2 pathway, but that after tumors have been initiated, SFN increased the rate of tumor growth and facilitated metastasis. In contrast, treatment with brusatol increased the formation of tumors, but brusatol treatment after tumors have formed led to a slowing of tumor growth and blocked metastasis73. These critical studies argue for the importance of timing in the development of cancer treatment versus cancer prevention493.
Subsequent mode of action studies by our group confirmed what others have shown that brusatol, at least at higher concentrations, is a general translation inhibitor494–496. The precise biochemical mechanism for this is not known, but we were able to show that brusatol decreases the levels of short-lived proteins, such as NRF2, selectively over long-lived proteins, such as p97. This lack of a direct NRF2 targeting effect, however, does not lessen the importance of brusatol in the NRF2 field, as it has been used as an important probe in many studies of the dark side of NRF2, demonstrating NRF2-dependent effects. Therefore, brusatol remains the most potent NRF2 pathway inhibitor known497–503. Indeed, much like the many natural product NRF2 activators, such as SFN and bardoxolone, the multiple effects of brusatol do not limit its usefulness and the importance of this probe in this new area of NRF2 research.
9.3. Halofuginone
Halofuginone (Fig. 15F) is a semi-synthetic derivative of the quinazolinone alkaloid, febrifugine from the Chinese herb Dichroa febrifuga (Fig. 15E). Febrifuginone was discovered by the Yamamoto lab in a high-throughput screen using an ARE-luciferase reporter cell line to look for compounds that decreased the luciferase signal. Further screening of compounds with scaffolds like febrifuginone revealed the semi-synthetic derivative halofuginone as a more potent and less toxic NRF2 pathway inhibitor. Halofuginone was shown to decrease the level of the NRF2 protein at sub-μM levels. It was also shown to have selective cytotoxicity for NRF2 addicted cancer cell lines and to increase the efficacy of cisplatin in vivo, in an NRF2 dependent manner. Mode of action studies revealed halofuginone is an inhibitor of proline tRNA-synthetase, leading to a general inhibition of translation, similar to brusatol, and having greater effect on short-lived proteins504.
9.4. Wogonin
Wogonin (Fig. 15G) is a flavonoid isolated from the root of the Chinese skullcap (Scutellaria baicalensis) that has been shown to have anti-neoplastic activity against a variety of cancer cell lines. Initial reports on the relationship between wogonin and NRF2 were conducted in doxorubicin resistant MCF7 breast cancer cells. It was shown in these studies that increased NRF2 levels, and its downstream genes, correlated with resistance and that this could be mitigated by genetic knockdown of NRF2. Importantly, wogonin also decreased the level of NRF2 and imparted sensitivity to the cells 505. In agreement with these studies, wogonin was shown to block multidrug resistance in a leukemia cell line and cisplatin resistance in head and neck cancers through NRF2 inhibition506, 507. It was later shown that wogonin decreased NRF2 mRNA levels through the NFκB pathway in drug resistant myelogenous leukemia cells, sensitizing the cells508. However, it is important to indicate that there are contradictory studies, indicating wogonin is an NRF2 activating compound and this conundrum remains without explanation509–512.
10. Conclusions and future directions
The importance of natural products as drug discovery leads or as chemical biological tools has been revealed by many success stories in the clinic and a long list of publications. The importance of natural products is beautifully highlighted in the case of the NRF2-ARE cellular protective axis. The discovery of activation of phase 2 metabolic enzymes by natural products, the development of an assay to track down active principles, and the discovery of SFN as an NRF2 activating chemopreventive compound are all successes of natural product chemistry. This simple isothiocyanate has been used in thousands of studies that have revealed many of the secrets of the NRF2 pathway, a success of chemical biology. These biochemical, physiological, and pathological discoveries have paved the way for a series of drug discovery campaigns that have led to clinical trials and an important drug, DMF, that is now used to treat relapsing MS. This is the first intentional NRF2 activating compound to reach the clinic, but there are many other clinical trials and compounds holding promise for future therapies. These compounds seemingly defy our current view of drugs, as we pursue more and more selective compounds, the canonical NRF2 activating compounds display complex modes of action and seem to engage many targets. Certainly, the effects of the most advanced compounds SFN, bardoxolone and its derivatives, and DMF are due to both NRF2 dependent and independent actions, likely determined by dose, by disease being treated, and by formulation.
In addition to the discovery of the importance of NRF2 in cellular protection, more contemporary studies have revealed a dark side of NRF2. In this context, the protective powers of NRF2 can be hijacked by malignant cells to allow them to survive the harsh conditions required for cancerous transformation, growth, and metastasis. Indeed, it has been proposed that NRF2 might even be an oncogene and its suppressor, KEAP1, a tumor suppressor gene. In this area, like the protective side of NRF2, natural products, in particular the quasinoid brusatol, have verified this hypothesis and have shown the importance of the search for NRF2 inhibitors as either standalone or adjuvant therapies to treat NRF2 addicted cancers or perhaps cancers that have chemoresistance due to increased expression of NRF2.
Despite the many thousands of papers that have been published on NRF2, there are several critical questions that remain open and are intimately connected. First, there are virtually no structural data on NRF2, limited structural data on KEAP1, and no structural data on the CUL3-RBX1-KEAP1-NRF2 complex. This, of course is a daunting task, but with the incredible advances being seen in the cryo-EM field, it is likely within reach. Better structural data will likely reveal many of the critical mechanistic details about regulation of the pathway that remain controversial. Second, the structural data coupled with advanced drug discovery efforts are needed to produce better and more selective probe molecules and possibly clinical leads. There is no denying the importance of the existing NRF2 modulating compounds, but the pleotropic effects of these compounds have added some confusion to the field. This is seen in the importance of use and timing of use of given drugs and possible indications. At which point should an NRF2 activator be used versus an NRF2 inhibitor? What effects seen with current compounds are truly NRF2 related and which are due to off-target or different target effects? Better probe molecules will also help to define another important area of active research: the crosstalk between NRF2 and other signaling pathways. Many important connections have been made in recent years, but some of these conclusions have been challenging to validate due to non-specificity of the NRF2 probes used.
11. Acknowledgements
This work was supported by R01 ES023758 to E.C. and D.D.Z.
12 References
- 1.Newman DJ and Cragg GM, Future medicinal chemistry, 2009, 1, 1415–1427. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ and Cragg GM, Journal of natural products, 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newman DJ and Cragg GM, Marine drugs, 2014, 12, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman DJ and Cragg GM, Journal of natural products, 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 5.Cragg GM and Newman DJ, Biochimica et biophysica acta, 2013, 1830, 3670–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Z, Acta pharmaceutica Sinica. B, 2017, 7, 119–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown DG, Lister T and May-Dracka TL, Bioorganic & medicinal chemistry letters, 2014, 24, 413–418. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Li W, Yao H and Xu J, Fitoterapia, 2015, 103, 231–241. [DOI] [PubMed] [Google Scholar]
- 9.Cragg GM and Pezzuto JM, Medical principles and practice : international journal of the Kuwait University, Health Science Centre, 2016, 25 Suppl 2, 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlson EE, ACS chemical biology, 2010, 5, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong J, Current opinion in chemical biology, 2011, 15, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganesan A, Current opinion in chemical biology, 2008, 12, 306–317. [DOI] [PubMed] [Google Scholar]
- 13.Hesketh T and Zhu WX, BMJ (Clinical research ed.), 1997, 315, 115–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dias DA, Urban S and Roessner U, Metabolites, 2012, 2, 303–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen B, Cell, 2015, 163, 1297–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernardini S, Tiezzi A, Laghezza Masci V and Ovidi E, Natural product research, 2018, 32, 1926–1950. [DOI] [PubMed] [Google Scholar]
- 17.Wujastyk D and Smith FM, Journal, 2008. [Google Scholar]
- 18.Khan RA, Saudi pharmaceutical journal : SPJ : the official publication of the Saudi Pharmaceutical Society, 2018, 26, 739–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Y, Cobb RE and Zhao H, Current opinion in biotechnology, 2014, 30, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey AL, Edrada-Ebel R and Quinn RJ, Nature reviews. Drug discovery, 2015, 14, 111–129. [DOI] [PubMed] [Google Scholar]
- 21.Katz L and Baltz RH, Journal of industrial microbiology & biotechnology, 2016, 43, 155–176. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Zhu D and Shen Y, Drug discoveries & therapeutics, 2018, 12, 318–328. [DOI] [PubMed] [Google Scholar]
- 23.Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL and Metcalf WW, Nature chemical biology, 2014, 10, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bucar F, Wube A and Schmid M, Natural product reports, 2013, 30, 525–545. [DOI] [PubMed] [Google Scholar]
- 25.Sterner O, Methods in molecular biology (Clifton, N.J.), 2012, 864, 393–413. [DOI] [PubMed] [Google Scholar]
- 26.Sticher O, Natural product reports, 2008, 25, 517–554. [DOI] [PubMed] [Google Scholar]
- 27.Houssen WE and Jaspars M, Methods in molecular biology (Clifton, N.J.), 2012, 864, 367–392. [DOI] [PubMed] [Google Scholar]
- 28.La Clair JJ, Natural product reports, 2010, 27, 969–995. [DOI] [PubMed] [Google Scholar]
- 29.Itoh K, Tong KI and Yamamoto M, Free radical biology & medicine, 2004, 36, 1208–1213. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi M and Yamamoto M, Antioxidants & redox signaling, 2005, 7, 385–394. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki T and Yamamoto M, Free radical biology & medicine, 2015, 88, 93–100. [DOI] [PubMed] [Google Scholar]
- 32.Taguchi K, Motohashi H and Yamamoto M, Genes to cells : devoted to molecular & cellular mechanisms, 2011, 16, 123–140. [DOI] [PubMed] [Google Scholar]
- 33.Hayes JD and Dinkova-Kostova AT, Trends in biochemical sciences, 2014, 39, 199–218. [DOI] [PubMed] [Google Scholar]
- 34.Mathers J, Fraser JA, McMahon M, Saunders RD, Hayes JD and McLellan LI, Biochemical Society symposium, 2004, 157–176. [DOI] [PubMed] [Google Scholar]
- 35.Harder B, Jiang T, Wu T, Tao S, Rojo de la Vega M, Tian W, Chapman E and Zhang DD, Biochemical Society transactions, 2015, 43, 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidlin CJ, Dodson MB, Madhavan L and Zhang DD, Free radical biology & medicine, 2019, 134, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang DD, Drug metabolism reviews, 2006, 38, 769–789. [DOI] [PubMed] [Google Scholar]
- 38.Huggins C, Deuel TF and Fukunishi R, Biochemische Zeitschrift, 1963, 338, 106–113. [PubMed] [Google Scholar]
- 39.Huggins C, Ford E, Fukunishi R and Jensen EV, The Journal of experimental medicine, 1964, 119, 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huggins C and Fukunishi R, The Journal of experimental medicine, 1964, 119, 923–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huggins C and Fukunishi R, Arzneimittel-Forschung, 1964, 14, 834–836. [PubMed] [Google Scholar]
- 42.Huggins C, Grand L and Fukunishi R, Proceedings of the National Academy of Sciences of the United States of America, 1964, 51, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wattenberg LW, Cancer research, 1966, 26, 1520–1526. [PubMed] [Google Scholar]
- 44.Wattenberg LW, Gastroenterology, 1966, 51, 932–935. [PubMed] [Google Scholar]
- 45.Wattenberg LW, Journal of the National Cancer Institute, 1972, 48, 1425–1430. [PubMed] [Google Scholar]
- 46.Wattenberg LW, Journal of the National Cancer Institute, 1973, 50, 1541–1544. [DOI] [PubMed] [Google Scholar]
- 47.Wattenberg LW, Journal of the National Cancer Institute, 1974, 52, 1583–1587. [DOI] [PubMed] [Google Scholar]
- 48.Wattenberg LW, The American journal of digestive diseases, 1974, 19, 947–953. [DOI] [PubMed] [Google Scholar]
- 49.Wattenberg LW, Journal of the National Cancer Institute, 1975, 54, 1005–1006. [DOI] [PubMed] [Google Scholar]
- 50.Wattenberg LW, Cancer research, 1975, 35, 3326–3331. [PubMed] [Google Scholar]
- 51.Wattenberg LW, Advances in cancer research, 1978, 26, 197–226. [DOI] [PubMed] [Google Scholar]
- 52.Wattenberg LW and Leong JL, Cancer research, 1965, 25, 365–370. [PubMed] [Google Scholar]
- 53.Benson AM, Batzinger RP, Ou SY, Bueding E, Cha YN and Talalay P, Cancer research, 1978, 38, 4486–4495. [PubMed] [Google Scholar]
- 54.Benson AM, Cha YN, Bueding E, Heine HS and Talalay P, Cancer research, 1979, 39, 2971–2977. [PubMed] [Google Scholar]
- 55.Zhang Y, Kensler TW, Cho CG, Posner GH and Talalay P, Proceedings of the National Academy of Sciences of the United States of America, 1994, 91, 3147–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Talalay P, Cho CG and Posner GH, Proceedings of the National Academy of Sciences of the United States of America, 1992, 89, 2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rushmore TH, King RG, Paulson KE and Pickett CB, Proceedings of the National Academy of Sciences of the United States of America, 1990, 87, 3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rushmore TH and Pickett CB, The Journal of biological chemistry, 1990, 265, 14648–14653. [PubMed] [Google Scholar]
- 59.Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E and Zhang DD, Annual review of pharmacology and toxicology, 2019, 59, 555–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de la Vega MR, Dodson M, Chapman E and Zhang DD, Curr Opin Toxicol, 2016, 1, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar H, Kim IS, More SV, Kim BW and Choi DK, Natural product reports, 2014, 31, 109–139. [DOI] [PubMed] [Google Scholar]
- 62.Sova M and Saso L, Drug design, development and therapy, 2018, 12, 3181–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keum YS and Choi BY, Molecules (Basel, Switzerland), 2014, 19, 10074–10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robledinos-Anton N, Fernandez-Gines R, Manda G and Cuadrado A, Oxidative medicine and cellular longevity, 2019, 2019, 9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kensler TW, Wakabayashi N and Biswal S, Annual review of pharmacology and toxicology, 2007, 47, 89–116. [DOI] [PubMed] [Google Scholar]
- 66.Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW and Dinkova-Kostova AT, Nature reviews. Drug discovery, 2019, 18, 295–317. [DOI] [PubMed] [Google Scholar]
- 67.Rojo de la Vega M, Chapman E and Zhang DD, Cancer cell, 2018, 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang KA and Hyun JW, Toxicological research, 2017, 33, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khor TO, Fuentes F, Shu L, Paredes-Gonzalez X, Yang AY, Liu Y, Smiraglia DJ, Yegnasubramanian S, Nelson WG and Kong AN, Cancer Prev Res (Phila), 2014, 7, 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bousquet MS, Ratnayake R, Pope JL, Chen QY, Zhu F, Chen S, Carney TJ, Gharaibeh RZ, Jobin C, Paul VJ and Luesch H, Free radical biology & medicine, 2020, 146, 306–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X-J, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK and Zhang DD, Carcinogenesis, 2008, 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA and Zhang DD, Proceedings of the National Academy of Sciences of the United States of America, 2011, 108, 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tao S, de la Vega MR, Chapman E, Ooi A and Zhang DD, Molecular carcinogenesis, 2017, DOI: 10.1002/mc.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK and Zhang DD, Cancer research, 2014, 74, 7430–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan K, Lu R, Chang JC and Kan YW, Proceedings of the National Academy of Sciences of the United States of America, 1996, 93, 13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Itoh K, Igarashi K, Hayashi N, Nishizawa M and Yamamoto M, Molecular and cellular biology, 1995, 15, 4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang DD, Lo SC, Cross JV, Templeton DJ and Hannink M, Molecular and cellular biology, 2004, 24, 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K and Yamamoto M, Molecular and cellular biology, 2004, 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang DD and Hannink M, Molecular and cellular biology, 2003, 23, 8137–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Itoh K, Mimura J and Yamamoto M, Antioxidants & redox signaling, 2010, 13, 1665–1678. [DOI] [PubMed] [Google Scholar]
- 81.Kansanen E, Kuosmanen SM, Leinonen H and Levonen A-L, Redox biology, 2013, 1, 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McMahon M, Thomas N, Itoh K, Yamamoto M and Hayes JD, The Journal of biological chemistry, 2004, 279, 31556–31567. [DOI] [PubMed] [Google Scholar]
- 83.Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S and Yamamoto M, Molecular and cellular biology, 2007, 27, 7511–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T and Yamamoto M, Molecular and cellular biology, 2006, 26, 2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Canning P, Sorrell FJ and Bullock AN, Free radical biology & medicine, 2015, 88, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tao S, Liu P, Luo G, Rojo de la Vega M, Chen H, Wu T, Tillotson J, Chapman E and Zhang DD, Molecular and cellular biology, 2017, DOI: 10.1128/mcb.00660-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M and Nabeshima Y, Biochemical and biophysical research communications, 1997, 236, 313–322. [DOI] [PubMed] [Google Scholar]
- 88.Prestera T, Zhang Y, Spencer SR, Wilczak CA and Talalay P, Advances in enzyme regulation, 1993, 33, 281–296. [DOI] [PubMed] [Google Scholar]
- 89.Dinkova-Kostova AT, Holtzclaw WD and Kensler TW, Chemical research in toxicology, 2005, 18, 1779–1791. [DOI] [PubMed] [Google Scholar]
- 90.Prestera T, Holtzclaw WD, Zhang Y and Talalay P, Proceedings of the National Academy of Sciences of the United States of America, 1993, 90, 2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suzuki T, Muramatsu A, Saito R, Iso T, Shibata T, Kuwata K, Kawaguchi SI, Iwawaki T, Adachi S, Suda H, Morita M, Uchida K, Baird L and Yamamoto M, Cell reports, 2019, 28, 746–758.e744. [DOI] [PubMed] [Google Scholar]
- 92.Dinkova-Kostova AT, Kostov RV and Canning P, Archives of biochemistry and biophysics, 2017, 617, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McMahon M, Lamont DJ, Beattie KA and Hayes JD, Proceedings of the National Academy of Sciences of the United States of America, 2010, 107, 18838–18843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A, Keates T, Thangaratnarajah C, Hojzan V, Ayinampudi V, Marsden BD, Gileadi O, Knapp S, von Delft F and Bullock AN, The Journal of biological chemistry, 2013, 288, 7803–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK, Qi H, Sweitzer T, Ward P and Davies TG, PloS one, 2014, 9, e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang M, An C, Gao Y, Leak RK, Chen J and Zhang F, Progress in neurobiology, 2013, 100, 30–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC and Freeman ML, Chemical research in toxicology, 2008, 21, 705–710. [DOI] [PubMed] [Google Scholar]
- 98.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD and Freeman ML, The Journal of biological chemistry, 2007, 282, 2529–2537. [DOI] [PubMed] [Google Scholar]
- 99.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K and Yamamoto M, Molecular and cellular biology, 2006, 26, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW and Biswal S, Nucleic acids research, 2010, 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu X, Guanga GP, Wan C and Rose RB, Biochemistry, 2012, 51, 9706–9717. [DOI] [PubMed] [Google Scholar]
- 102.Amoutzias GD, Robertson DL, Van de Peer Y and Oliver SG, Trends in biochemical sciences, 2008, 33, 220–229. [DOI] [PubMed] [Google Scholar]
- 103.Amoutzias GD, Veron AS, Weiner J 3rd, Robinson-Rechavi M, Bornberg-Bauer E, Oliver SG and Robertson DL, Molecular biology and evolution, 2007, 24, 827–835. [DOI] [PubMed] [Google Scholar]
- 104.Hurst HC, Protein profile, 1995, 2, 101–168. [PubMed] [Google Scholar]
- 105.Ellenberger T, Current opinion in structural biology, 1994, 4, 12–21. [Google Scholar]
- 106.Igarashi K, Itoh K, Motohashi H, Hayashi N, Matuzaki Y, Nakauchi H, Nishizawa M and Yamamoto M, The Journal of biological chemistry, 1995, 270, 7615–7624. [DOI] [PubMed] [Google Scholar]
- 107.Igarashi K, Kataoka K, Itoh K, Hayashi N, Nishizawa M and Yamamoto M, Nature, 1994, 367, 568–572. [DOI] [PubMed] [Google Scholar]
- 108.Venugopal R and Jaiswal AK, Proceedings of the National Academy of Sciences of the United States of America, 1996, 93, 14960–14965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F and Arinze IJ, The Journal of biological chemistry, 2008, 283, 8984–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zheng-Yu J, Hong-Xi C, Mei-Yang X, Ting-Ting Y, Jian-Min J, Jing-Jie H, Xiao-Ke G, Xiao-Jin Z, Qi-Dong Y and Hao-Peng S, Domain structure of Nrf2 and Keap1, 2013. [Google Scholar]
- 111.Li W, Yu SW and Kong AN, The Journal of biological chemistry, 2006, 281, 27251–27263. [DOI] [PubMed] [Google Scholar]
- 112.Lo SC, Li X, Henzl MT, Beamer LJ and Hannink M, The EMBO journal, 2006, 25, 3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fukutomi T, Takagi K, Mizushima T, Ohuchi N and Yamamoto M, Molecular and cellular biology, 2014, 34, 832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nioi P, Nguyen T, Sherratt PJ and Pickett CB, Molecular and cellular biology, 2005, 25, 10895–10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A and Yamamoto M, Genes to cells : devoted to molecular & cellular mechanisms, 2001, 6, 857–868. [DOI] [PubMed] [Google Scholar]
- 116.Ki SH, Cho IJ, Choi DW and Kim SG, Molecular and cellular biology, 2005, 25, 4150–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang J, Hosoya T, Maruyama A, Nishikawa K, Maher JM, Ohta T, Motohashi H, Fukamizu A, Shibahara S, Itoh K and Yamamoto M, The Biochemical journal, 2007, 404, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang J, Hosoya T, Maruyama A, Nishikawa K, Maher JM, Ohta T, Motohashi H, Fukamizu A, Shibahara S, Itoh K and Yamamoto M, The Biochemical journal, 2007, 404, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, Chapman E, Fang D and Zhang DD, Genes & development, 2014, 28, 708–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A and Hayes JD, Oncogene, 2013, 32, 3765–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cuadrado A, Free radical biology & medicine, 2015, 88, 147–157. [DOI] [PubMed] [Google Scholar]
- 122.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD and Cuadrado A, Molecular and cellular biology, 2011, 31, 1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rojo AI, Medina-Campos ON, Rada P, Zuniga-Toala A, Lopez-Gazcon A, Espada S, Pedraza-Chaverri J and Cuadrado A, Free radical biology & medicine, 2012, 52, 473–487. [DOI] [PubMed] [Google Scholar]
- 124.Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Li Y, Luo L, Hayes JD, Wang XJ and Tang X, Cancer research, 2013, 73, 3097–3108. [DOI] [PubMed] [Google Scholar]
- 125.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD and Yamamoto M, Genes & development, 1999, 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C and Yamamoto M, Proceedings of the National Academy of Sciences of the United States of America, 2010, 107, 2842–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Suzuki T, Muramatsu A, Saito R, Iso T, Shibata T, Kuwata K, Kawaguchi S.-i., Iwawaki T, Adachi S, Suda H, Morita M, Uchida K, Baird L and Yamamoto M, Cell reports, 2019, 28, 746–758.e744. [DOI] [PubMed] [Google Scholar]
- 128.Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, Hoggard T, Harper JW, White KP and Schulman BA, Molecular cell, 2009, 36, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li X, Zhang D, Hannink M and Beamer LJ, The Journal of biological chemistry, 2004, 279, 54750–54758. [DOI] [PubMed] [Google Scholar]
- 130.Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, Willems HM, Woolford AJ, Cottom JE, Kou JP, Yonchuk JG, Feldser HG, Sanchez Y, Foley JP, Bolognese BJ, Logan G, Podolin PL, Yan H, Callahan JF, Heightman TD and Kerns JK, Journal of medicinal chemistry, 2016, 59, 3991–4006. [DOI] [PubMed] [Google Scholar]
- 131.Marcotte D, Zeng W, Hus JC, McKenzie A, Hession C, Jin P, Bergeron C, Lugovskoy A, Enyedy I, Cuervo H, Wang D, Atmanene C, Roecklin D, Vecchi M, Vivat V, Kraemer J, Winkler D, Hong V, Chao J, Lukashev M and Silvian L, Bioorganic & medicinal chemistry, 2013, 21, 4011–4019. [DOI] [PubMed] [Google Scholar]
- 132.Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T, Ohishi M, Endo K, Uemura T, Nishito Y, Okuda S, Obata M, Kouno T, Imamura R, Tada Y, Obata R, Yasuda D, Takahashi K, Fujimura T, Pi J, Lee MS, Ueno T, Ohe T, Mashino T, Wakai T, Kojima H, Okabe T, Nagano T, Motohashi H, Waguri S, Soga T, Yamamoto M, Tanaka K and Komatsu M, Nature communications, 2016, 7, 12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pallesen JS, Tran KT and Bach A, Journal of medicinal chemistry, 2018, 61, 8088–8103. [DOI] [PubMed] [Google Scholar]
- 134.Tran KT, Pallesen JS, Solbak SMO, Narayanan D, Baig A, Zang J, Aguayo-Orozco A, Carmona RMC, Garcia AD and Bach A, Journal of medicinal chemistry, 2019, 62, 8028–8052. [DOI] [PubMed] [Google Scholar]
- 135.Liu P, Tian W, Tao S, Tillotson J, Wijeratne EMK, Gunatilaka AAL, Zhang DD and Chapman E, Cell chemical biology, 2019, DOI: 10.1016/j.chembiol.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Loub WD, Wattenberg LW and Davis DW, Journal of the National Cancer Institute, 1975, 54, 985–988. [PubMed] [Google Scholar]
- 137.Osterberg KA and Wattenberg LW, Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine (New York, N.Y.), 1965, 118, 477–479. [DOI] [PubMed] [Google Scholar]
- 138.Pearson WR, Windle JJ, Morrow JF, Benson AM and Talalay P, The Journal of biological chemistry, 1983, 258, 2052–2062. [PubMed] [Google Scholar]
- 139.Talalay P, Batzinger RP, Benson AM, Bueding E and Cha YN, Advances in enzyme regulation, 1978, 17, 23–36. [DOI] [PubMed] [Google Scholar]
- 140.Talalay P and Benson AM, Advances in enzyme regulation, 1982, 20, 287–300. [DOI] [PubMed] [Google Scholar]
- 141.Talalay P, BioFactors (Oxford, England), 2000, 12, 5–11. [DOI] [PubMed] [Google Scholar]
- 142.Kensler TW, Davidson NE, Groopman JD, Roebuck BD, Prochaska HJ and Talalay P, Basic life sciences, 1993, 61, 127–136. [DOI] [PubMed] [Google Scholar]
- 143.Prochaska HJ and Santamaria AB, Analytical biochemistry, 1988, 169, 328–336. [DOI] [PubMed] [Google Scholar]
- 144.Talalay P, De Long MJ and Prochaska HJ, Proceedings of the National Academy of Sciences of the United States of America, 1988, 85, 8261–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fahey JW, Dinkova-Kostova AT, Stephenson KK and Talalay P, Methods in enzymology, 2004, 382, 243–258. [DOI] [PubMed] [Google Scholar]
- 146.Mignotte V, Eleouet JF, Raich N and Romeo PH, Proceedings of the National Academy of Sciences of the United States of America, 1989, 86, 6548–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Moi P, Chan K, Asunis I, Cao A and Kan YW, Proceedings of the National Academy of Sciences of the United States of America, 1994, 91, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fahey JW, Holtzclaw WD, Wehage SL, Wade KL, Stephenson KK and Talalay P, PloS one, 2015, 10, e0140963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cornblatt BS, Ye L, Dinkova-Kostova AT, Erb M, Fahey JW, Singh NK, Chen MS, Stierer T, Garrett-Mayer E, Argani P, Davidson NE, Talalay P, Kensler TW and Visvanathan K, Carcinogenesis, 2007, 28, 1485–1490. [DOI] [PubMed] [Google Scholar]
- 150.Fahey JW, Wade KL, Stephenson KK, Panjwani AA, Liu H, Cornblatt G, Cornblatt BS, Ownby SL, Fuchs E, Holtzclaw WD and Cheskin LJ, Nutrients, 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hu C, Eggler AL, Mesecar AD and van Breemen RB, Chemical research in toxicology, 2011, 24, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Egner PA, Chen JG, Wang JB, Wu Y, Sun Y, Lu JH, Zhu J, Zhang YH, Chen YS, Friesen MD, Jacobson LP, Munoz A, Ng D, Qian GS, Zhu YR, Chen TY, Botting NP, Zhang Q, Fahey JW, Talalay P, Groopman JD and Kensler TW, Cancer Prev Res (Phila), 2011, 4, 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Egner PA, Kensler TW, Chen JG, Gange SJ, Groopman JD and Friesen MD, Chemical research in toxicology, 2008, 21, 1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, Ye L, Coady JL, Wang JB, Wu Y, Sun Y, Zhang QN, Zhang BC, Zhu YR, Qian GS, Carmella SG, Hecht SS, Benning L, Gange SJ, Groopman JD and Talalay P, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 2005, 14, 2605–2613. [DOI] [PubMed] [Google Scholar]
- 155.Fahey JW, Wade KL, Wehage SL, Holtzclaw WD, Liu H, Talalay P, Fuchs E and Stephenson KK, Molecular nutrition & food research, 2017, 61. [DOI] [PubMed] [Google Scholar]
- 156.Fahey JW, Wehage SL, Holtzclaw WD, Kensler TW, Egner PA, Shapiro TA and Talalay P, Cancer Prev Res (Phila), 2012, 5, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yagishita Y, Fahey JW, Dinkova-Kostova AT and Kensler TW, Molecules (Basel, Switzerland), 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shapiro TA, Fahey JW, Wade KL, Stephenson KK and Talalay P, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 2001, 10, 501–508. [PubMed] [Google Scholar]
- 159.Kelleher MO, McMahon M, Eggleston IM, Dixon MJ, Taguchi K, Yamamoto M and Hayes JD, Carcinogenesis, 2009, 30, 1754–1762. [DOI] [PubMed] [Google Scholar]
- 160.Dinkova-Kostova AT, Fahey JW, Kostov RV and Kensler TW, Trends in food science & technology, 2017, 69, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chen J, Zhu J, Wang G, Groopman JD and Kensler TW, Cancer biology & medicine, 2019, 16, 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bent S, Lawton B, Warren T, Widjaja F, Dang K, Fahey JW, Cornblatt B, Kinchen JM, Delucchi K and Hendren RL, Molecular autism, 2018, 9, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hashimoto K, Frontiers in pharmacology, 2018, 9, 1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hernandez-Rabaza V, Cabrera-Pastor A, Taoro-Gonzalez L, Malaguarnera M, Agusti A, Llansola M and Felipo V, Journal of neuroinflammation, 2016, 13, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hou TT, Yang HY, Wang W, Wu QQ, Tian YR and Jia JP, Journal of Alzheimer’s disease : JAD, 2018, 62, 1803–1813. [DOI] [PubMed] [Google Scholar]
- 166.Marchezan J, Winkler Dos Santos EGA, Deckmann I and Riesgo RDS, Neuroimmunomodulation, 2018, 25, 300–319. [DOI] [PubMed] [Google Scholar]
- 167.Pearson BL, Simon JM, McCoy ES, Salazar G, Fragola G and Zylka MJ, Nature communications, 2016, 7, 11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Singh K and Zimmerman AW, CNS & neurological disorders drug targets, 2016, 15, 597–601. [DOI] [PubMed] [Google Scholar]
- 169.Wang G, Fang H, Zhen Y, Xu G, Tian J, Zhang Y, Zhang D, Zhang G, Xu J, Zhang Z, Qiu M, Ma Y, Zhang H and Zhang X, Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology, 2016, 39, 901–907. [DOI] [PubMed] [Google Scholar]
- 170.Biswas S, Hwang JW, Kirkham PA and Rahman I, Current medicinal chemistry, 2013, 20, 1496–1530. [DOI] [PubMed] [Google Scholar]
- 171.Jiao Z, Chang J, Li J, Nie D, Cui H and Guo D, Molecular medicine reports, 2017, 16, 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jiang T, Huang Z, Lin Y, Zhang Z, Fang D and Zhang DD, Diabetes, 2010, 59, 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Long M, Rojo de la Vega M, Wen Q, Bharara M, Jiang T, Zhang R, Zhou S, Wong PK, Wondrak GT, Zheng H and Zhang DD, Diabetes, 2016, 65, 780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rojo de la Vega M, Dodson M, Chapman E and Zhang DD, Curr Opin Toxicol, 2016, 1, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schmidlin CJ, Dodson MB and Zhang DD, Archives of pharmacal research, 2019, DOI: 10.1007/s12272-019-01177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Whitman SA, Long M, Wondrak GT, Zheng H and Zhang DD, Experimental cell research, 2013, 319, 2673–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zheng H, Whitman SA, Wu W, Wondrak GT, Wong PK, Fang D and Zhang DD, Diabetes, 2011, 60, 3055–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Negrette-Guzman M, European journal of pharmacology, 2019, 859, 172513. [DOI] [PubMed] [Google Scholar]
- 179.Hong F, Freeman ML and Liebler DC, Chemical research in toxicology, 2005, 18, 1917–1926. [DOI] [PubMed] [Google Scholar]
- 180.Hong F, Sekhar KR, Freeman ML and Liebler DC, The Journal of biological chemistry, 2005, 280, 31768–31775. [DOI] [PubMed] [Google Scholar]
- 181.Eggler AL, Luo Y, van Breemen RB and Mesecar AD, Chemical research in toxicology, 2007, 20, 1878–1884. [DOI] [PubMed] [Google Scholar]
- 182.Luo Y, Eggler AL, Liu D, Liu G, Mesecar AD and van Breemen RB, Journal of the American Society for Mass Spectrometry, 2007, 18, 2226–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD and Darley-Usmar VM, The Biochemical journal, 2004, 378, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dinkova-Kostova AT, Holtzclaw WD and Wakabayashi N, Biochemistry, 2005, 44, 6889–6899. [DOI] [PubMed] [Google Scholar]
- 185.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H and Yamamoto M, Molecular and cellular biology, 2008, 28, 2758–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW and Talalay P, Proceedings of the National Academy of Sciences of the United States of America, 2004, 101, 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Baird L, Lleres D, Swift S and Dinkova-Kostova AT, Proceedings of the National Academy of Sciences of the United States of America, 2013, 110, 15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li Y, Paonessa JD and Zhang Y, PloS one, 2012, 7, e35122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Eggler AL, Gay KA and Mesecar AD, Molecular nutrition & food research, 2008, 52 Suppl 1, S84–94. [DOI] [PubMed] [Google Scholar]
- 190.Eggler AL, Small E, Hannink M and Mesecar AD, The Biochemical journal, 2009, 422, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Holland R, Hawkins AE, Eggler AL, Mesecar AD, Fabris D and Fishbein JC, Chemical research in toxicology, 2008, 21, 2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sekhar KR, Rachakonda G and Freeman ML, Toxicology and applied pharmacology, 2010, 244, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Baird L and Dinkova-Kostova AT, Biochemical and biophysical research communications, 2013, 433, 58–65. [DOI] [PubMed] [Google Scholar]
- 194.Shen T, Jiang T, Long M, Chen J, Ren DM, Wong PK, Chapman E, Zhou B and Zhang DD, Antioxidants & redox signaling, 2015, 23, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Baker M, Nature, 2017, 541, 144–145. [DOI] [PubMed] [Google Scholar]
- 196.Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF and Walters MA, Journal of medicinal chemistry, 2017, 60, 1620–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Li W, Pung D, Su ZY, Guo Y, Zhang C, Yang AY, Zheng X, Du ZY, Zhang K and Kong AN, Chemical research in toxicology, 2016, 29, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li W, Su ZY, Guo Y, Zhang C, Wu R, Gao L, Zheng X, Du ZY, Zhang K and Kong AN, Chemical research in toxicology, 2018, 31, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cui W, Min X, Xu X, Du B and Luo P, Journal of diabetes research, 2017, 2017, 3797802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Farzaei MH, Zobeiri M, Parvizi F, El-Senduny FF, Marmouzi I, Coy-Barrera E, Naseri R, Nabavi SM, Rahimi R and Abdollahi M, Nutrients, 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Paunkov A, Chartoumpekis DV, Ziros PG and Sykiotis GP, Antioxidants (Basel, Switzerland), 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pittala V, Vanella L, Salerno L, Romeo G, Marrazzo A, Di Giacomo C and Sorrenti V, Current medicinal chemistry, 2018, 25, 1577–1595. [DOI] [PubMed] [Google Scholar]
- 203.Shehzad A, Khan S and Sup Lee Y, Future oncology (London, England), 2012, 8, 179–190. [DOI] [PubMed] [Google Scholar]
- 204.Shehzad A and Lee YS, BioFactors (Oxford, England), 2013, 39, 27–36. [DOI] [PubMed] [Google Scholar]
- 205.Shehzad A, Wahid F and Lee YS, Archiv der Pharmazie, 2010, 343, 489–499. [DOI] [PubMed] [Google Scholar]
- 206.Chew EH, Nagle AA, Zhang Y, Scarmagnani S, Palaniappan P, Bradshaw TD, Holmgren A and Westwell AD, Free radical biology & medicine, 2010, 48, 98–111. [DOI] [PubMed] [Google Scholar]
- 207.Mateen S, Rehman MT, Shahzad S, Naeem SS, Faizy AF, Khan AQ, Khan MS, Husain FM and Moin S, European journal of pharmacology, 2019, 852, 14–24. [DOI] [PubMed] [Google Scholar]
- 208.Ose R, Tu J, Schink A, Maxeiner J, Schuster P, Lucas K, Saloga J and Bellinghausen I, Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2019, DOI: 10.1111/cea.13507. [DOI] [PubMed] [Google Scholar]
- 209.Wondrak GT, Cabello CM, Villeneuve NF, Zhang S, Ley S, Li Y, Sun Z and Zhang DD, Free radical biology & medicine, 2008, 45, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Long M, Tao S, Rojo de la Vega M, Jiang T, Wen Q, Park SL, Zhang DD and Wondrak GT, Cancer Prev Res (Phila), 2015, 8, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wondrak GT, Villeneuve NF, Lamore SD, Bause AS, Jiang T and Zhang DD, Molecules (Basel, Switzerland), 2010, 15, 3338–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chao LK, Chang WT, Shih YW and Huang JS, Toxicology and applied pharmacology, 2010, 244, 174–180. [DOI] [PubMed] [Google Scholar]
- 213.de Haan JB, Diabetes, 2011, 60, 2683–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sampath C, Sprouse JC, Freeman ML and Gangula PR, Free radical biology & medicine, 2019, 135, 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sartorius T, Peter A, Schulz N, Drescher A, Bergheim I, Machann J, Schick F, Siegel-Axel D, Schurmann A, Weigert C, Haring HU and Hennige AM, PloS one, 2014, 9, e92358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Subash Babu P, Prabuseenivasan S and Ignacimuthu S, Phytomedicine, 2007, 14, 15–22. [DOI] [PubMed] [Google Scholar]
- 217.Zhu R, Liu H, Liu C, Wang L, Ma R, Chen B, Li L, Niu J, Fu M, Zhang D and Gao S, Pharmacological research, 2017, 122, 78–89. [DOI] [PubMed] [Google Scholar]
- 218.Abou El-Ezz D, Maher A, Sallam N, El-Brairy A and Kenawy S, Neurochemical research, 2018, 43, 2333–2342. [DOI] [PubMed] [Google Scholar]
- 219.Choi BH, Kang KS and Kwak MK, Molecules (Basel, Switzerland), 2014, 19, 12727–12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tao S, Park SL, Rojo de la Vega M, Zhang DD and Wondrak GT, Free radical biology & medicine, 2015, 89, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tao S, Rojo de la Vega M, Quijada H, Wondrak GT, Wang T, Garcia JG and Zhang DD, Scientific reports, 2016, 6, 18760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Xu Z and Kong XQ, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2017, 89, 991–1004. [DOI] [PubMed] [Google Scholar]
- 223.Xue L, Zhang H, Zhang J, Li B, Zhang Z and Tao S, Toxicology research, 2018, 7, 258–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang H, Xue L, Li B, Tian H, Zhang Z and Tao S, Free radical biology & medicine, 2018, 126, 166–176. [DOI] [PubMed] [Google Scholar]
- 225.Berghe W, Sabbe L, Kaileh M, Haegeman G and Heyninck K, Biochemical pharmacology, 2012, 84, 1282–1291. [DOI] [PubMed] [Google Scholar]
- 226.Lee I-C and Choi BY, International journal of molecular sciences, 2016, 17, 290–290.26959007 [Google Scholar]
- 227.Chirumamilla CS, Perez-Novo C, Van Ostade X and Vanden Berghe W, The Proceedings of the Nutrition Society, 2017, 76, 96–105. [DOI] [PubMed] [Google Scholar]
- 228.Deocaris CC, Widodo N, Wadhwa R and Kaul SC, Journal of translational medicine, 2008, 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mirjalili MH, Moyano E, Bonfill M, Cusido RM and Palazon J, Molecules (Basel, Switzerland), 2009, 14, 2373–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Heyninck K, Sabbe L, Chirumamilla CS, Szarc Vel Szic K, Vander Veken P, Lemmens KJA, Lahtela-Kakkonen M, Naulaerts S, Op de Beeck K, Laukens K, Van Camp G, Weseler AR, Bast A, Haenen G, Haegeman G and Vanden Berghe W, Biochemical pharmacology, 2016, 109, 48–61. [DOI] [PubMed] [Google Scholar]
- 231.Jadeja RN, Urrunaga NH, Dash S, Khurana S and Saxena NK, Biochemical pharmacology, 2015, 97, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tekula S, Khurana A, Anchi P and Godugu C, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2018, 106, 1428–1440. [DOI] [PubMed] [Google Scholar]
- 233.Vaishnavi K, Saxena N, Shah N, Singh R, Manjunath K, Uthayakumar M, Kanaujia SP, Kaul SC, Sekar K and Wadhwa R, PloS one, 2012, 7, e44419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.White PT, Subramanian C, Motiwala HF and Cohen MS, Advances in experimental medicine and biology, 2016, 928, 329–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Palliyaguru DL, Chartoumpekis DV, Wakabayashi N, Skoko JJ, Yagishita Y, Singh SV and Kensler TW, Free radical biology & medicine, 2016, 101, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tao S, Tillotson J, Wijeratne EM, Xu YM, Kang M, Wu T, Lau EC, Mesa C, Mason DJ, Brown RV, La Clair JJ, Gunatilaka AA, Zhang DD and Chapman E, ACS chemical biology, 2015, 10, 1916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Vanden Berghe W, Sabbe L, Kaileh M, Haegeman G and Heyninck K, Biochemical pharmacology, 2012, 84, 1282–1291. [DOI] [PubMed] [Google Scholar]
- 238.Yang H, Wang Y, Cheryan VT, Wu W, Cui CQ, Polin LA, Pass HI, Dou QP, Rishi AK and Wali A, PloS one, 2012, 7, e41214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Dinkova-Kostova AT, Kostov RV and Kazantsev AG, The FEBS journal, 2018, 285, 3576–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sandberg M, Patil J, D’Angelo B, Weber SG and Mallard C, Neuropharmacology, 2014, 79, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lee JM, Hanson JM, Chu WA and Johnson JA, The Journal of biological chemistry, 2001, 276, 20011–20016. [DOI] [PubMed] [Google Scholar]
- 242.Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K and Nakashima K, FEBS letters, 2003, 546, 181–184. [DOI] [PubMed] [Google Scholar]
- 243.Rojo AI, Sagarra MR and Cuadrado A, Journal of neurochemistry, 2008, 105, 192–202. [DOI] [PubMed] [Google Scholar]
- 244.Rojo AI, Rada P, Egea J, Rosa AO, López MG and Cuadrado A, Mol Cell Neurosci, 2008, 39, 125–132. [DOI] [PubMed] [Google Scholar]
- 245.Espada S, Rojo AI, Salinas M and Cuadrado A, Journal of neurochemistry, 2009, 110, 1107–1119. [DOI] [PubMed] [Google Scholar]
- 246.Gao Y, Xu X, Chang S, Wang Y, Xu Y, Ran S, Huang Z, Li P, Li J, Zhang L, Saavedra JM, Liao H and Pang T, Toxicology and applied pharmacology, 2015, 289, 142–154. [DOI] [PubMed] [Google Scholar]
- 247.Gameiro I, Michalska P, Tenti G, Cores A, Buendia I, Rojo AI, Georgakopoulos ND, Hernandez-Guijo JM, Teresa Ramos M, Wells G, Lopez MG, Cuadrado A, Menendez JC and Leon R, Scientific reports, 2017, 7, 45701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Chen C, Li B, Cheng G, Yang X, Zhao N and Shi R, Neurochemical research, 2018, 43, 857–868. [DOI] [PubMed] [Google Scholar]
- 249.Park SY, Choi YW and Park G, J Pharm Pharmacol, 2018, 70, 525–535. [DOI] [PubMed] [Google Scholar]
- 250.Rong H, Liang Y and Niu Y, Free radical biology & medicine, 2018, 120, 114–123. [DOI] [PubMed] [Google Scholar]
- 251.Ge XH, Shao L and Zhu GJ, Metab Brain Dis, 2018, 33, 1869–1875. [DOI] [PubMed] [Google Scholar]
- 252.Kim KW, Yoon CS, Kim YC and Oh H, Neurotoxicity research, 2019, 35, 230–243. [DOI] [PubMed] [Google Scholar]
- 253.Duan J, Cui J, Yang Z, Guo C, Cao J, Xi M, Weng Y, Yin Y, Wang Y, Wei G, Qiao B and Wen A, Journal of neuroinflammation, 2019, 16, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lipton SA, Rezaie T, Nutter A, Lopez KM, Parker J, Kosaka K, Satoh T, McKercher SR, Masliah E and Nakanishi N, Cell death & disease, 2016, 7, e2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kwon SH, Ma SX, Hwang JY, Lee SY and Jang CG, Neuroscience, 2015, 304, 14–28. [DOI] [PubMed] [Google Scholar]
- 256.Fragoulis A, Siegl S, Fendt M, Jansen S, Soppa U, Brandenburg LO, Pufe T, Weis J and Wruck CJ, Redox biology, 2017, 12, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Csiszár A, Csiszar A, Pinto JT, Gautam T, Kleusch C, Hoffmann B, Tucsek Z, Toth P, Sonntag WE and Ungvari Z, J Gerontol A Biol Sci Med Sci, 2015, 70, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.FangFang, Li H, Qin T, Li M and Ma S, Metab Brain Dis, 2017, 32, 385–393. [DOI] [PubMed] [Google Scholar]
- 259.Hou Y, Li X, Peng S, Yao J, Bai F and Fang J, Journal of agricultural and food chemistry, 2019, 67, 8227–8234. [DOI] [PubMed] [Google Scholar]
- 260.Hou Y, Peng S, Li X, Yao J, Xu J and Fang J, ACS chemical neuroscience, 2018, 9, 3108–3116. [DOI] [PubMed] [Google Scholar]
- 261.Peng S, Hou Y, Yao J and Fang J, Food Funct, 2017, 8, 997–1007. [DOI] [PubMed] [Google Scholar]
- 262.Peng S, Hou Y, Yao J and Fang J, Food Funct, 2019, 10, 4143–4152. [DOI] [PubMed] [Google Scholar]
- 263.Peng S, Hou Y, Yao J and Fang J, BioFactors (Oxford, England), 2019, 45, 381–392. [DOI] [PubMed] [Google Scholar]
- 264.Peng S, Yao J, Liu Y, Duan D, Zhang X and Fang J, Food Funct, 2015, 6, 2813–2823. [DOI] [PubMed] [Google Scholar]
- 265.Peng S, Zhang B, Yao J, Duan D and Fang J, Food Funct, 2015, 6, 2091–2100. [DOI] [PubMed] [Google Scholar]
- 266.Yao J, Ge C, Duan D, Zhang B, Cui X, Peng S, Liu Y and Fang J, Journal of agricultural and food chemistry, 2014, 62, 5507–5518. [DOI] [PubMed] [Google Scholar]
- 267.Yao J, Peng S, Xu J and Fang J, BioFactors (Oxford, England), 2019, 45, 616–626. [DOI] [PubMed] [Google Scholar]
- 268.Yao J, Zhang B, Ge C, Peng S and Fang J, Journal of agricultural and food chemistry, 2015, 63, 1521–1531. [DOI] [PubMed] [Google Scholar]
- 269.Honda T, Gribble GW, Suh N, Finlay HJ, Rounds BV, Bore L, Favaloro FG Jr., Wang Y and Sporn MB, Journal of medicinal chemistry, 2000, 43, 1866–1877. [DOI] [PubMed] [Google Scholar]
- 270.Honda T, Honda Y, Favaloro FG Jr., Gribble GW, Suh N, Place AE, Rendi MH and Sporn MB, Bioorganic & medicinal chemistry letters, 2002, 12, 1027–1030. [DOI] [PubMed] [Google Scholar]
- 271.Honda T, Janosik T, Honda Y, Han J, Liby KT, Williams CR, Couch RD, Anderson AC, Sporn MB and Gribble GW, Journal of medicinal chemistry, 2004, 47, 4923–4932. [DOI] [PubMed] [Google Scholar]
- 272.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y and Sporn MB, Bioorganic & medicinal chemistry letters, 1998, 8, 2711–2714. [DOI] [PubMed] [Google Scholar]
- 273.Suh N, Honda T, Finlay HJ, Barchowsky A, Williams C, Benoit NE, Xie QW, Nathan C, Gribble GW and Sporn MB, Cancer research, 1998, 58, 717–723. [PubMed] [Google Scholar]
- 274.Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, Williams C, Risingsong R, Honda T, Gribble GW, Sporn MB and Talalay P, Proceedings of the National Academy of Sciences of the United States of America, 2005, 102, 4584–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Yore MM, Kettenbach AN, Sporn MB, Gerber SA and Liby KT, PloS one, 2011, 6, e22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ahmad R, Liu S, Weisberg E, Nelson E, Galinsky I, Meyer C, Kufe D, Kharbanda S and Stone R, Molecular cancer research : MCR, 2010, 8, 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Alabran JL, Cheuk A, Liby K, Sporn M, Khan J, Letterio J and Leskov KS, Cancer biology & therapy, 2008, 7, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Brookes PS, Morse K, Ray D, Tompkins A, Young SM, Hilchey S, Salim S, Konopleva M, Andreeff M, Phipps R and Bernstein SH, Cancer research, 2007, 67, 1793–1802. [DOI] [PubMed] [Google Scholar]
- 279.Chauhan D, Li G, Podar K, Hideshima T, Shringarpure R, Catley L, Mitsiades C, Munshi N, Tai YT, Suh N, Gribble GW, Honda T, Schlossman R, Richardson P, Sporn MB and Anderson KC, Blood, 2004, 103, 3158–3166. [DOI] [PubMed] [Google Scholar]
- 280.Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I and Safe S, Molecular pharmacology, 2005, 68, 119–128. [DOI] [PubMed] [Google Scholar]
- 281.Choi SH, Kim BG, Robinson J, Fink S, Yan M, Sporn MB, Markowitz SD and Letterio JJ, The Journal of clinical investigation, 2014, 124, 2472–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Deeb D, Brigolin C, Gao X, Liu Y, Pindolia KR and Gautam SC, Journal of carcinogenesis & mutagenesis, 2014, 5, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Deeb D, Gao X, Dulchavsky SA and Gautam SC, Anticancer research, 2007, 27, 3035–3044. [PubMed] [Google Scholar]
- 284.Deeb D, Gao X, Dulchavsky SA and Gautam SC, Journal of experimental therapeutics & oncology, 2008, 7, 31–39. [PubMed] [Google Scholar]
- 285.Deeb D, Gao X, Jiang H, Dulchavsky SA and Gautam SC, The Prostate, 2009, 69, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Deeb D, Gao X, Jiang H, Janic B, Arbab AS, Rojanasakul Y, Dulchavsky SA and Gautam SC, Biochemical pharmacology, 2010, 79, 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Deeb D, Gao X, Liu Y, Jiang D, Divine GW, Arbab AS, Dulchavsky SA and Gautam SC, Carcinogenesis, 2011, 32, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Deeb D, Gao X, Liu Y, Varma NR, Arbab AS and Gautam SC, Molecules (Basel, Switzerland), 2013, 18, 3250–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Deeb D, Gao X, Liu YB and Gautam SC, Journal of experimental therapeutics & oncology, 2012, 10, 51–64. [PMC free article] [PubMed] [Google Scholar]
- 290.Duan Z, Ames RY, Ryan M, Hornicek FJ, Mankin H and Seiden MV, Cancer chemotherapy and pharmacology, 2009, 63, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Elsawa SF, Novak AJ, Grote D, Konopleva M, Andreeff M, Witzig TE and Ansell SM, Leukemia research, 2008, 32, 1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Gao L, Wang Y, Xu Z, Li X, Wu J, Liu S, Chu P, Sun Z, Sun B, Lin Y, Peng J, Han G, Wang S and Tang Z, Apoptosis : an international journal on programmed cell death, 2015, 20, 1636–1650. [DOI] [PubMed] [Google Scholar]
- 293.Gao X, Deeb D, Hao J, Liu Y, Arbab AS, Dulchavsky SA and Gautam SC, Anticancer research, 2010, 30, 785–792. [PMC free article] [PubMed] [Google Scholar]
- 294.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky SA and Gautam SC, Journal of neuro-oncology, 2007, 84, 147–157. [DOI] [PubMed] [Google Scholar]
- 295.Gao X, Deeb D, Liu P, Liu Y, Arbab-Ali S, Dulchavsky SA and Gautam SC, Journal of experimental therapeutics & oncology, 2011, 9, 119–127. [PMC free article] [PubMed] [Google Scholar]
- 296.Gao X, Deeb D, Liu Y, Arbab AS, Divine GW, Dulchavsky SA and Gautam SC, Cancers (Basel), 2011, 3, 3353–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Gao X, Deeb D, Liu Y, Liu P, Zhang Y, Shaw J and Gautam SC, International journal of oncology, 2015, 47, 2100–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Gao X, Liu Y, Deeb D, Arbab AS, Guo AM, Dulchavsky SA and Gautam SC, Anticancer research, 2011, 31, 3673–3681. [PMC free article] [PubMed] [Google Scholar]
- 299.Gao X, Liu Y, Deeb D, Liu P, Liu A, Arbab AS and Gautam SC, Anticancer research, 2013, 33, 215–221. [PMC free article] [PubMed] [Google Scholar]
- 300.Goldsmith KC and Hogarty MD, Cancer letters, 2005, 228, 133–141. [DOI] [PubMed] [Google Scholar]
- 301.Han SS, Peng L, Chung ST, DuBois W, Maeng SH, Shaffer AL, Sporn MB and Janz S, Molecular cancer, 2006, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hughes DT, Martel PM, Kinlaw WB and Eisenberg BL, Cancer investigation, 2008, 26, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hyer ML, Croxton R, Krajewska M, Krajewski S, Kress CL, Lu M, Suh N, Sporn MB, Cryns VL, Zapata JM and Reed JC, Cancer research, 2005, 65, 4799–4808. [DOI] [PubMed] [Google Scholar]
- 304.Hyer ML, Shi R, Krajewska M, Meyer C, Lebedeva IV, Fisher PB and Reed JC, Cancer research, 2008, 68, 2927–2933. [DOI] [PubMed] [Google Scholar]
- 305.Ikeda T, Kimura F, Nakata Y, Sato K, Ogura K, Motoyoshi K, Sporn M and Kufe D, Cell death and differentiation, 2005, 12, 523–531. [DOI] [PubMed] [Google Scholar]
- 306.Ikeda T, Nakata Y, Kimura F, Sato K, Anderson K, Motoyoshi K, Sporn M and Kufe D, Molecular cancer therapeutics, 2004, 3, 39–45. [PubMed] [Google Scholar]
- 307.Ikeda T, Sporn M, Honda T, Gribble GW and Kufe D, Cancer research, 2003, 63, 5551–5558. [PubMed] [Google Scholar]
- 308.Inoue S, Snowden RT, Dyer MJ and Cohen GM, Leukemia, 2004, 18, 948–952. [DOI] [PubMed] [Google Scholar]
- 309.Ito Y, Pandey P, Place A, Sporn MB, Gribble GW, Honda T, Kharbanda S and Kufe D, Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research, 2000, 11, 261–267. [PubMed] [Google Scholar]
- 310.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S and Kufe D, Molecular pharmacology, 2001, 59, 1094–1099. [DOI] [PubMed] [Google Scholar]
- 311.Jeong SA, Kim IY, Lee AR, Yoon MJ, Cho H, Lee JS and Choi KS, Oncotarget, 2015, 6, 21173–21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ji Y, Lee HJ, Goodman C, Uskokovic M, Liby K, Sporn M and Suh N, Molecular cancer therapeutics, 2006, 5, 1452–1458. [DOI] [PubMed] [Google Scholar]
- 313.Jutooru I, Chadalapaka G, Abdelrahim M, Basha MR, Samudio I, Konopleva M, Andreeff M and Safe S, Molecular pharmacology, 2010, 78, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kim EH, Deng C, Sporn MB, Royce DB, Risingsong R, Williams CR and Liby KT, Cancer Prev Res (Phila), 2012, 5, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kim EH, Deng CX, Sporn MB and Liby KT, Cancer Prev Res (Phila), 2011, 4, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kim KB, Lotan R, Yue P, Sporn MB, Suh N, Gribble GW, Honda T, Wu GS, Hong WK and Sun SY, Molecular cancer therapeutics, 2002, 1, 177–184. [PubMed] [Google Scholar]
- 317.Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M and Ruvolo PP, Leukemia, 2005, 19, 1350–1354. [DOI] [PubMed] [Google Scholar]
- 318.Konopleva M, Tsao T, Estrov Z, Lee RM, Wang RY, Jackson CE, McQueen T, Monaco G, Munsell M, Belmont J, Kantarjian H, Sporn MB and Andreeff M, Cancer research, 2004, 64, 7927–7935. [DOI] [PubMed] [Google Scholar]
- 319.Konopleva M, Tsao T, Ruvolo P, Stiouf I, Estrov Z, Leysath CE, Zhao S, Harris D, Chang S, Jackson CE, Munsell M, Suh N, Gribble G, Honda T, May WS, Sporn MB and Andreeff M, Blood, 2002, 99, 326–335. [DOI] [PubMed] [Google Scholar]
- 320.Konopleva M, Zhang W, Shi YX, McQueen T, Tsao T, Abdelrahim M, Munsell MF, Johansen M, Yu D, Madden T, Safe SH, Hung MC and Andreeff M, Molecular cancer therapeutics, 2006, 5, 317–328. [DOI] [PubMed] [Google Scholar]
- 321.Koschmieder S, D’Alo F, Radomska H, Schoneich C, Chang JS, Konopleva M, Kobayashi S, Levantini E, Suh N, Di Ruscio A, Voso MT, Watt JC, Santhanam R, Sargin B, Kantarjian H, Andreeff M, Sporn MB, Perrotti D, Berdel WE, Muller-Tidow C, Serve H and Tenen DG, Blood, 2007, 110, 3695–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kress CL, Konopleva M, Martinez-Garcia V, Krajewska M, Lefebvre S, Hyer ML, McQueen T, Andreeff M, Reed JC and Zapata JM, PloS one, 2007, 2, e559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Lapillonne H, Konopleva M, Tsao T, Gold D, McQueen T, Sutherland RL, Madden T and Andreeff M, Cancer research, 2003, 63, 5926–5939. [PubMed] [Google Scholar]
- 324.Leal AS, Sporn MB, Pioli PA and Liby KT, Carcinogenesis, 2016, 37, 1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Liao D, Liu Z, Wrasidlo WJ, Luo Y, Nguyen G, Chen T, Xiang R and Reisfeld RA, Cancer research, 2011, 71, 5688–5696. [DOI] [PubMed] [Google Scholar]
- 326.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A and Sporn MB, Cancer research, 2005, 65, 4789–4798. [DOI] [PubMed] [Google Scholar]
- 327.Liby K, Risingsong R, Royce DB, Williams CR, Ma T, Yore MM and Sporn MB, Cancer Prev Res (Phila), 2009, 2, 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Liby K, Risingsong R, Royce DB, Williams CR, Yore MM, Honda T, Gribble GW, Lamph WW, Vannini N, Sogno I, Albini A and Sporn MB, Clinical cancer research : an official journal of the American Association for Cancer Research, 2008, 14, 4556–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA and Sporn MB, Cancer research, 2007, 67, 2414–2419. [DOI] [PubMed] [Google Scholar]
- 330.Liby K, Voong N, Williams CR, Risingsong R, Royce DB, Honda T, Gribble GW, Sporn MB and Letterio JJ, Clinical cancer research : an official journal of the American Association for Cancer Research, 2006, 12, 4288–4293. [DOI] [PubMed] [Google Scholar]
- 331.Liby KT, Royce DB, Risingsong R, Williams CR, Maitra A, Hruban RH and Sporn MB, Cancer Prev Res (Phila), 2010, 3, 1427–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Liby KT and Sporn MB, Pharmacological reviews, 2012, 64, 972–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Ling X, Konopleva M, Zeng Z, Ruvolo V, Stephens LC, Schober W, McQueen T, Dietrich M, Madden TL and Andreeff M, Cancer research, 2007, 67, 4210–4218. [DOI] [PubMed] [Google Scholar]
- 334.Melichar B, Konopleva M, Hu W, Melicharova K, Andreeff M and Freedman RS, Gynecologic oncology, 2004, 93, 149–154. [DOI] [PubMed] [Google Scholar]
- 335.Pedersen IM, Kitada S, Schimmer A, Kim Y, Zapata JM, Charboneau L, Rassenti L, Andreeff M, Bennett F, Sporn MB, Liotta LD, Kipps TJ and Reed JC, Blood, 2002, 100, 2965–2972. [DOI] [PubMed] [Google Scholar]
- 336.Place AE, Suh N, Williams CR, Risingsong R, Honda T, Honda Y, Gribble GW, Leesnitzer LM, Stimmel JB, Willson TM, Rosen E and Sporn MB, Clinical cancer research : an official journal of the American Association for Cancer Research, 2003, 9, 2798–2806. [PubMed] [Google Scholar]
- 337.Ray DM, Morse KM, Hilchey SP, Garcia TM, Felgar RE, Maggirwar SB, Phipps RP and Bernstein SH, Experimental hematology, 2006, 34, 1202–1211. [DOI] [PubMed] [Google Scholar]
- 338.Riccioni R, Senese M, Diverio D, Riti V, Mariani G, Boe A, LoCoco F, Foa R, Peschle C, Sporn M and Testa U, Leukemia research, 2008, 32, 1244–1258. [DOI] [PubMed] [Google Scholar]
- 339.Ryu K, Susa M, Choy E, Yang C, Hornicek FJ, Mankin HJ and Duan Z, BMC cancer, 2010, 10, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G and Bishayee A, Cancer letters, 2014, 346, 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Shishodia S, Sethi G, Konopleva M, Andreeff M and Aggarwal BB, Clinical cancer research : an official journal of the American Association for Cancer Research, 2006, 12, 1828–1838. [DOI] [PubMed] [Google Scholar]
- 342.So JY, Lin JJ, Wahler J, Liby KT, Sporn MB and Suh N, PloS one, 2014, 9, e107616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.So JY, Wahler JE, Yoon T, Smolarek AK, Lin Y, Shih WJ, Maehr H, Uskokovic M, Liby KT, Sporn MB and Suh N, Cancer Prev Res (Phila), 2013, 6, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Suh N, Roberts AB, Birkey Reffey S, Miyazono K, Itoh S, ten Dijke P, Heiss EH, Place AE, Risingsong R, Williams CR, Honda T, Gribble GW and Sporn MB, Cancer research, 2003, 63, 1371–1376. [PubMed] [Google Scholar]
- 345.Tabe Y, Konopleva M, Kondo Y, Contractor R, Tsao T, Konoplev S, Shi Y, Ling X, Watt JC, Tsutsumi-Ishii Y, Ohsaka A, Nagaoka I, Issa JP, Kogan SC and Andreeff M, Cancer biology & therapy, 2007, 6, 1967–1977. [DOI] [PubMed] [Google Scholar]
- 346.To C, Ringelberg CS, Royce DB, Williams CR, Risingsong R, Sporn MB and Liby KT, Carcinogenesis, 2015, 36, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Townson JL, Macdonald IC, Liby KT, Mackenzie L, Dales DW, Hedley BD, Foster PJ, Sporn MB and Chambers AF, Clinical & experimental metastasis, 2011, 28, 309–317. [DOI] [PubMed] [Google Scholar]
- 348.Tsao T, Kornblau S, Safe S, Watt JC, Ruvolo V, Chen W, Qiu Y, Coombes KR, Ju Z, Abdelrahim M, Schober W, Ling X, Kardassis D, Meyer C, Schimmer A, Kantarjian H, Andreeff M and Konopleva M, Cancer research, 2010, 70, 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Vannini N, Lorusso G, Cammarota R, Barberis M, Noonan DM, Sporn MB and Albini A, Molecular cancer therapeutics, 2007, 6, 3139–3146. [DOI] [PubMed] [Google Scholar]
- 350.Wang YY, Yang YX, Zhao R, Pan ST, Zhe H, He ZX, Duan W, Zhang X, Yang T, Qiu JX and Zhou SF, Drug design, development and therapy, 2015, 9, 993–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Wang YY, Zhe H and Zhao R, Molecular cancer, 2014, 13, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Wang YY, Zhou S, Zhao R, Hai P and Zhe H, American journal of translational research, 2016, 8, 1695–1707. [PMC free article] [PubMed] [Google Scholar]
- 353.Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB and Kensler TW, Molecular cancer therapeutics, 2007, 6, 154–162. [DOI] [PubMed] [Google Scholar]
- 354.Zhao Y, Huo M, Xu Z, Wang Y and Huang L, Biomaterials, 2015, 68, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Zou W, Chen S, Liu X, Yue P, Sporn MB, Khuri FR and Sun SY, Cancer biology & therapy, 2007, 6, 1614–1620. [DOI] [PubMed] [Google Scholar]
- 356.Zou W, Liu X, Yue P, Zhou Z, Sporn MB, Lotan R, Khuri FR and Sun SY, Cancer research, 2004, 64, 7570–7578. [DOI] [PubMed] [Google Scholar]
- 357.Cao M, Onyango EO, Williams CR, Royce DB, Gribble GW, Sporn MB and Liby KT, Pharmacological research, 2015, 100, 135–147. [DOI] [PubMed] [Google Scholar]
- 358.Sogno I, Vannini N, Lorusso G, Cammarota R, Noonan DM, Generoso L, Sporn MB and Albini A, Recent results in cancer research. Fortschritte der Krebsforschung. Progres dans les recherches sur le cancer, 2009, 181, 209–212. [DOI] [PubMed] [Google Scholar]
- 359.Suh N, Wang Y, Honda T, Gribble GW, Dmitrovsky E, Hickey WF, Maue RA, Place AE, Porter DM, Spinella MJ, Williams CR, Wu G, Dannenberg AJ, Flanders KC, Letterio JJ, Mangelsdorf DJ, Nathan CF, Nguyen L, Porter WW, Ren RF, Roberts AB, Roche NS, Subbaramaiah K and Sporn MB, Cancer research, 1999, 59, 336–341. [PubMed] [Google Scholar]
- 360.Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, Silkworth JB, Taguchi K, Yamamoto M, Williams CR, Liby KT, Sporn MB, Sutter TR and Kensler TW, Carcinogenesis, 2009, 30, 1024–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Eaton DL and Schaupp CM, Cancer Prev Res (Phila), 2014, 7, 653–657. [DOI] [PubMed] [Google Scholar]
- 362.Livingstone MC, Johnson NM, Roebuck BD, Kensler TW and Groopman JD, Molecular carcinogenesis, 2017, 56, 2382–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Taguchi K, Takaku M, Egner PA, Morita M, Kaneko T, Mashimo T, Kensler TW and Yamamoto M, Toxicological sciences : an official journal of the Society of Toxicology, 2016, 152, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, Honda T, Gribble GW, Sporn MB and Kensler TW, Cancer research, 2006, 66, 2488–2494. [DOI] [PubMed] [Google Scholar]
- 365.Gupta K, Patani R, Baxter P, Serio A, Story D, Tsujita T, Hayes JD, Pedersen RA, Hardingham GE and Chandran S, Cell death and differentiation, 2012, 19, 779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Tran TA, McCoy MK, Sporn MB and Tansey MG, Journal of neuroinflammation, 2008, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Wang B, Liu Q, Shan H, Xia C and Liu Z, Biochemistry and cell biology = Biochimie et biologie cellulaire, 2015, 93, 351–358. [DOI] [PubMed] [Google Scholar]
- 368.Wei Y, Gong J, Yoshida T, Eberhart CG, Xu Z, Kombairaju P, Sporn MB, Handa JT and Duh EJ, Free radical biology & medicine, 2011, 51, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Xu Z, Cho H, Hartsock MJ, Mitchell KL, Gong J, Wu L, Wei Y, Wang S, Thimmulappa RK, Sporn MB, Biswal S, Welsbie DS and Duh EJ, Journal of neurochemistry, 2015, 133, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, Liby KT, Williams C, Royce D, Risingsong R, Musiek ES, Morrow JD, Sporn M and Beal MF, PloS one, 2009, 4, e5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Dumont M, Wille E, Calingasan NY, Tampellini D, Williams C, Gouras GK, Liby K, Sporn M, Nathan C, Flint Beal M and Lin MT, Journal of neurochemistry, 2009, 109, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M and Beal MF, Free radical biology & medicine, 2010, 49, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, Liby KT, Risingsong R, Sporn M, Beal MF and Kiaei M, Free radical biology & medicine, 2011, 51, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Wang L, Kondo N, Cano M, Ebrahimi K, Yoshida T, Barnett BP, Biswal S and Handa JT, Free radical biology & medicine, 2014, 70, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Pitha-Rowe I, Liby K, Royce D and Sporn M, Investigative ophthalmology & visual science, 2009, 50, 5339–5347. [DOI] [PubMed] [Google Scholar]
- 376.Abel EL, Bubel JD, Simper MS, Powell L, McClellan SA, Andreeff M, MacLeod MC and DiGiovanni J, Toxicology and applied pharmacology, 2011, 255, 176–183. [DOI] [PubMed] [Google Scholar]
- 377.Himori N, Yamamoto K, Maruyama K, Ryu M, Taguchi K, Yamamoto M and Nakazawa T, Journal of neurochemistry, 2013, 127, 669–680. [DOI] [PubMed] [Google Scholar]
- 378.Kuriyan AE, Lehmann GM, Kulkarni AA, Woeller CF, Feldon SE, Hindman HB, Sime PJ, Huxlin KR and Phipps RP, Experimental eye research, 2012, 94, 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Xue P, Hu X, Powers J, Nay N, Chang E, Kwon J, Wong SW, Han L, Wu TH, Lee DJ, Tseng H and Ko CC, Biochemical and biophysical research communications, 2019, 511, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Cantin AM, Proceedings of the American Thoracic Society, 2010, 7, 368–375. [DOI] [PubMed] [Google Scholar]
- 381.Chen T, Mou Y, Tan J, Wei L, Qiao Y, Wei T, Xiang P, Peng S, Zhang Y, Huang Z and Ji H, International immunopharmacology, 2015, 25, 55–64. [DOI] [PubMed] [Google Scholar]
- 382.Ferguson HE, Kulkarni A, Lehmann GM, Garcia-Bates TM, Thatcher TH, Huxlin KR, Phipps RP and Sime PJ, American journal of respiratory cell and molecular biology, 2009, 41, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Hogan CM, Thatcher TH, Sapinoro RE, Gurell MN, Ferguson HE, Pollock SJ, Jones C, Phipps RP and Sime PJ, PPAR research, 2011, 2011, 318134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Kulkarni AA, Thatcher TH, Hsiao HM, Olsen KC, Kottmann RM, Morrissette J, Wright TW, Phipps RP and Sime PJ, PloS one, 2013, 8, e63798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP and Sime PJ, PloS one, 2011, 6, e15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Mathis BJ and Cui T, Advances in experimental medicine and biology, 2016, 929, 291–314. [DOI] [PubMed] [Google Scholar]
- 387.Nagashima R, Kosai H, Masuo M, Izumiyama K, Noshikawaji T, Morimoto M, Kumaki S, Miyazaki Y, Motohashi H, Yamamoto M and Tanaka N, Journal of immunology (Baltimore, Md. : 1950), 2019, 202, 1331–1339. [DOI] [PubMed] [Google Scholar]
- 388.Nichols DP, Ziady AG, Shank SL, Eastman JF and Davis PB, American journal of physiology. Lung cellular and molecular physiology, 2009, 297, L828–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Olsen KC, Epa AP, Kulkarni AA, Kottmann RM, McCarthy CE, Johnson GV, Thatcher TH, Phipps RP and Sime PJ, American journal of respiratory cell and molecular biology, 2014, 50, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Reddy NM, Suryanaraya V, Yates MS, Kleeberger SR, Hassoun PM, Yamamoto M, Liby KT, Sporn MB, Kensler TW and Reddy SP, American journal of respiratory and critical care medicine, 2009, 180, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML and Blackwell TS, PloS one, 2010, 5, e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D, Yates MS, Kombairaju P, Yamamoto M, Liby KT, Sporn MB, Gabrielson KL, Champion HC, Tuder RM, Kensler TW and Biswal S, Proceedings of the National Academy of Sciences of the United States of America, 2009, 106, 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW and Biswal S, Biochemical and biophysical research communications, 2006, 351, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Wang YY, Zhang CY, Ma YQ, He ZX, Zhe H and Zhou SF, Drug design, development and therapy, 2015, 9, 3163–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Bubb KJ, Kok C, Tang O, Rasko NB, Birgisdottir AB, Hansen T, Ritchie R, Bhindi R, Reisman SA, Meyer C, Ward K, Karimi Galougahi K and Figtree GA, Free radical biology & medicine, 2017, 108, 585–594. [DOI] [PubMed] [Google Scholar]
- 396.Ichikawa T, Li J, Meyer CJ, Janicki JS, Hannink M and Cui T, PloS one, 2009, 4, e8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Tian C, Gao L, Zhang A, Hackfort B and Zucker IH, The Journal of pharmacology and experimental therapeutics, 2019, DOI: 10.1124/jpet.119.261792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Xing Y, Niu T, Wang W, Li J, Li S, Janicki JS, Ruiz S, Meyer CJ, Wang XL, Tang D, Zhao Y and Cui T, PloS one, 2012, 7, e44899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Furusawa Y, Uruno A, Yagishita Y, Higashi C and Yamamoto M, Genes to cells : devoted to molecular & cellular mechanisms, 2014, 19, 864–878. [DOI] [PubMed] [Google Scholar]
- 400.Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW and Yamamoto M, Molecular and cellular biology, 2013, 33, 2996–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Yang N, Tang Q, Qin W, Li Z, Wang D, Zhang W, Cao X, Lu Y, Ge X, Sun H and Shen P, EBioMedicine, 2019, 45, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Getachew Y, Cusimano FA, Gopal P, Reisman SA and Shay JW, Toxicological sciences : an official journal of the Society of Toxicology, 2016, 149, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, Taguchi K, Yamamoto M and Kensler TW, Toxicological sciences : an official journal of the Society of Toxicology, 2008, 104, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Reisman SA, Ward KW, Klaassen CD and Meyer CJ, Xenobiotica; the fate of foreign compounds in biological systems, 2013, 43, 571–578. [DOI] [PubMed] [Google Scholar]
- 405.Sharma RS, Harrison DJ, Kisielewski D, Cassidy DM, McNeilly AD, Gallagher JR, Walsh SV, Honda T, McCrimmon RJ, Dinkova-Kostova AT, Ashford MLJ, Dillon JF and Hayes JD, Cellular and molecular gastroenterology and hepatology, 2018, 5, 367–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S, Liby KT, Sporn MB, Yamamoto M and Kensler TW, European journal of pharmacology, 2009, 620, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Xu D, Chen L, Chen X, Wen Y, Yu C, Yao J, Wu H, Wang X, Xia Q and Kong X, Cell death & disease, 2017, 8, e2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Aminzadeh MA, Reisman SA, Vaziri ND, Khazaeli M, Yuan J and Meyer CJ, Xenobiotica; the fate of foreign compounds in biological systems, 2014, 44, 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Aminzadeh MA, Reisman SA, Vaziri ND, Shelkovnikov S, Farzaneh SH, Khazaeli M and Meyer CJ, Redox biology, 2013, 1, 527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Huang Z, Mou Y, Xu X, Zhao D, Lai Y, Xu Y, Chen C, Li P, Peng S, Tian J and Zhang Y, Journal of medicinal chemistry, 2017, 60, 8847–8857. [DOI] [PubMed] [Google Scholar]
- 411.Nagasu H, Sogawa Y, Kidokoro K, Itano S, Yamamoto T, Satoh M, Sasaki T, Suzuki T, Yamamoto M, Wigley WC, Proksch JW, Meyer CJ and Kashihara N, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2019, DOI: 10.1096/fj.201900217R, fj201900217R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Nezu M, Souma T, Yu L, Suzuki T, Saigusa D, Ito S, Suzuki N and Yamamoto M, Kidney international, 2017, 91, 387–401. [DOI] [PubMed] [Google Scholar]
- 413.Shelton LM, Lister A, Walsh J, Jenkins RE, Wong MH, Rowe C, Ricci E, Ressel L, Fang Y, Demougin P, Vukojevic V, O’Neill PM, Goldring CE, Kitteringham NR, Park BK, Odermatt A and Copple IM, Kidney international, 2015, 88, 1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Fitzpatrick LR, Stonesifer E, Small JS and Liby KT, Inflammopharmacology, 2014, 22, 341–349. [DOI] [PubMed] [Google Scholar]
- 415.Pareek TK, Belkadi A, Kesavapany S, Zaremba A, Loh SL, Bai L, Cohen ML, Meyer C, Liby KT, Miller RH, Sporn MB and Letterio JJ, Scientific reports, 2011, 1, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Wei HJ, Pareek TK, Liu Q and Letterio JJ, Scientific reports, 2017, 7, 9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Wu T, Ye Y, Min SY, Zhu J, Khobahy E, Zhou J, Yan M, Hemachandran S, Pathak S, Zhou XJ, Andreeff M and Mohan C, Arthritis & rheumatology (Hoboken, N.J.), 2014, 66, 3129–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Zhang DD, Antioxidants & redox signaling, 2013, 19, 517–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ, Zhou M, Gardie B, Molinie V, Richard S, Tan PH, Teh BT and Furge KA, Cancer cell, 2011, 20, 511–523. [DOI] [PubMed] [Google Scholar]
- 420.Hosseini A, Masjedi A, Baradaran B, Hojjat-Farsangi M, Ghalamfarsa G, Anvari E and Jadidi-Niaragh F, Journal of cellular physiology, 2019, 234, 9943–9955. [DOI] [PubMed] [Google Scholar]
- 421.Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F and Alexandre J, Medicinal research reviews, 2019, 39, 1923–1952. [DOI] [PubMed] [Google Scholar]
- 422.Montes Diaz G, Hupperts R, Fraussen J and Somers V, Autoimmunity reviews, 2018, 17, 1240–1250. [DOI] [PubMed] [Google Scholar]
- 423.Tran KT, Pallesen JS, Solbak SMO, Narayanan D, Baig A, Zang J, Aguayo-Orozco A, Carmona R, Garcia A and Bach A, Journal of medicinal chemistry, 2019, DOI: 10.1021/acs.jmedchem.9b00723. [DOI] [PubMed] [Google Scholar]
- 424.Jain AD, Potteti H, Richardson BG, Kingsley L, Luciano JP, Ryuzoji AF, Lee H, Krunic A, Mesecar AD, Reddy SP and Moore TW, European journal of medicinal chemistry, 2015, 103, 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Richardson BG, Jain AD, Potteti HR, Lazzara PR, David BP, Tamatam CR, Choma E, Skowron K, Dye K, Siddiqui Z, Wang YT, Krunic A, Reddy SP and Moore TW, Journal of medicinal chemistry, 2018, 61, 8029–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Wang L, Lewis T, Zhang Y-L, Khodier C, Magesh S, Chen L, Inoyama D, Chen Y, Zhen J, Hu L, Beamer LJ, Faloon PW, Dandapani S, Perez JR, Munoz B, Palmer M and Schreiber S, Journal, 2010. [PubMed] [Google Scholar]
- 427.Bresciani A, Missineo A, Gallo M, Cerretani M, Fezzardi P, Tomei L, Cicero DO, Altamura S, Santoprete A, Ingenito R, Bianchi E, Pacifici R, Dominguez C, Munoz-Sanjuan I, Harper S, Toledo-Sherman L and Park LC, Archives of biochemistry and biophysics, 2017, 631, 31–41. [DOI] [PubMed] [Google Scholar]
- 428.Hayes JD, Chowdhry S, Dinkova-Kostova AT and Sutherland C, Biochemical Society transactions, 2015, 43, 611–620. [DOI] [PubMed] [Google Scholar]
- 429.Salazar M, Rojo AI, Velasco D, de Sagarra RM and Cuadrado A, The Journal of biological chemistry, 2006, 281, 14841–14851. [DOI] [PubMed] [Google Scholar]
- 430.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobon-Velasco JC, Devijver H, Garcia-Mayoral MF, Van Leuven F, Hayes JD, Bertho G and Cuadrado A, Molecular and cellular biology, 2012, 32, 3486–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Rojo AI, Rada P, Mendiola M, Ortega-Molina A, Wojdyla K, Rogowska-Wrzesinska A, Hardisson D, Serrano M and Cuadrado A, Antioxidants & redox signaling, 2014, 21, 2498–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Li H, Chen Y, Zhang J, Chen X, Li Z, Liu B and Zhang L, Molecules (Basel, Switzerland), 2018, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Liao Z, Zhang J, Liu B, Yan T, Xu F, Xiao F, Wu B, Bi K and Jia Y, Molecules (Basel, Switzerland), 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Zhang L, Guo Z, Wang Y, Geng J and Han S, Drug Dev Res, 2019, 80, 294–309. [DOI] [PubMed] [Google Scholar]
- 435.Kay HY, Kim YW, Ryu DH, Sung SH, Hwang SJ and Kim SG, Br J Pharmacol, 2011, 163, 1653–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Song JS, Kim EK, Choi YW, Oh WK and Kim YM, Toxicology and applied pharmacology, 2016, 307, 138–149. [DOI] [PubMed] [Google Scholar]
- 437.Wang L, Zhang S, Cheng H, Lv H, Cheng G and Ci X, Free radical biology & medicine, 2016, 101, 401–412. [DOI] [PubMed] [Google Scholar]
- 438.Fan X, Wang L, Huang J, Lv H, Deng X and Ci X, Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology, 2018, 49, 1943–1958. [DOI] [PubMed] [Google Scholar]
- 439.Duan J, Guan Y, Mu F, Guo C, Zhang E, Yin Y, Wei G, Zhu Y, Cui J, Cao J, Weng Y, Wang Y, Xi M and Wen A, Scientific reports, 2017, 7, 41491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Shi J, Zhang X and Jiang H, Molecules (Basel, Switzerland), 2010, 15, 5273–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Cui HY, Zhang XJ, Yang Y, Zhang C, Zhu CH, Miao JY and Chen R, Neural regeneration research, 2018, 13, 2119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Lee KM, Shin JM, Chun J, Song K, Nho CW and Kim YS, Journal of biochemical and molecular toxicology, 2019, 33, e22297. [DOI] [PubMed] [Google Scholar]
- 443.Feng X, Guan W, Zhao Y, Wang C, Song M, Yao Y, Yang T and Fan H, Journal of cellular physiology, 2019, 234, 18994–19009. [DOI] [PubMed] [Google Scholar]
- 444.Mathur A, Rizvi F and Kakkar P, Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association, 2016, 89, 19–31. [DOI] [PubMed] [Google Scholar]
- 445.Rizvi F, Mathur A and Kakkar P, Apoptosis : an international journal on programmed cell death, 2015, 20, 1296–1306. [DOI] [PubMed] [Google Scholar]
- 446.Rizvi F, Mathur A, Krishna S, Siddiqi MI and Kakkar P, Redox biology, 2015, 6, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Wang T, Zhang J, Xiao A, Liu W, Shang Y and An J, Biochemical and biophysical research communications, 2016, 480, 126–131. [DOI] [PubMed] [Google Scholar]
- 448.Silva-Palacios A, Ostolga-Chavarría M, Zazueta C and Königsberg M, Ageing research reviews, 2018, 47, 31–40. [DOI] [PubMed] [Google Scholar]
- 449.Wu TY, Khor TO, Lee JH, Cheung KL, Shu L, Chen C and Kong AN, Curr Drug Metab, 2013, 14, 688–694. [DOI] [PubMed] [Google Scholar]
- 450.Tao S, Rojo de la Vega M, Chapman E, Ooi A and Zhang DD, Mol Carcinog, 2017, DOI: 10.1002/mc.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Satoh H, Moriguchi T, Takai J, Ebina M and Yamamoto M, Cancer research, 2013, 73, 4158–4168. [DOI] [PubMed] [Google Scholar]
- 452.Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H, Igarashi K, Ebina M, Shibata T and Yamamoto M, Cancer research, 2016, 76, 3088–3096. [DOI] [PubMed] [Google Scholar]
- 453.Bauer AK, Cho HY, Miller-Degraff L, Walker C, Helms K, Fostel J, Yamamoto M and Kleeberger SR, PloS one, 2011, 6, e26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Wang Y, Wang Y, Zhang Z, Park JY, Guo D, Liao H, Yi X, Zheng Y, Zhang D, Chambers SK and Zheng W, Oncotarget, 2016, 7, 10363–10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.N. Cancer Genome Atlas Research, Nature, 2012, 489, 519–525.22960745 [Google Scholar]
- 456.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, Peters BA, Pujara K, Cordes S, Davis DP, Carlton VE, Yuan W, Li L, Wang W, Eigenbrot C, Kaminker JS, Eberhard DA, Waring P, Schuster SC, Modrusan Z, Zhang Z, Stokoe D, de Sauvage FJ, Faham M and Seshagiri S, Nature, 2010, 466, 869–873. [DOI] [PubMed] [Google Scholar]
- 457.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV and Biswal S, PLoS medicine, 2006, 3, e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Hayes JD and McMahon M, Trends in biochemical sciences, 2009, 34, 176–188. [DOI] [PubMed] [Google Scholar]
- 459.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES and Getz G, Nature, 2014, 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, Walter V, Zheng N, Hayes DN and Major MB, Cancer research, 2014, 74, 808–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R, Anema J, Craig D, Carpten J, Teh BT and Furge KA, Cancer research, 2013, 73, 2044–2051. [DOI] [PubMed] [Google Scholar]
- 462.Dorr C, Janik C, Weg M, Been RA, Bader J, Kang R, Ng B, Foran L, Landman SR, O’Sullivan MG, Steinbach M, Sarver AL, Silverstein KAT, Largaespada DA and Starr TK, Molecular Cancer Research, 2015, 13, 1238–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Martinez VD, Vucic EA, Thu KL, Pikor LA, Hubaux R and Lam WL, BioMed Research International, 2014, 2014, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Wang R, An J, Ji F, Jiao H, Sun H and Zhou D, Biochemical and biophysical research communications, 2008, 373, 151–154. [DOI] [PubMed] [Google Scholar]
- 465.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, Wistuba II, Minna JD, DeBerardinis RJ and Cantley LC, Nat Genet, 2015, 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Jaramillo MC and Zhang DD, Genes & development, 2013, 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Lau A, Villeneuve NF, Sun Z, Wong PK and Zhang DD, Pharmacological research, 2008, 58, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK and Zhang DD, Carcinogenesis, 2008, 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, Zheng Y, Liao X, Wang Y, Liao Q, Li W, Tang Z, Tong Q, Wang X, Fang F, Rojo de la Vega M, Ouyang Q, Zhang DD, Yu S and Zheng H, Science translational medicine, 2016, 8, 334ra351. [DOI] [PubMed] [Google Scholar]
- 470.Bao LJ, Jaramillo MC, Zhang ZB, Zheng YX, Yao M, Zhang DD and Yi XF, International journal of clinical and experimental pathology, 2014, 7, 1502–1513. [PMC free article] [PubMed] [Google Scholar]
- 471.Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Ohlund D, Wright K, Filippini D, Lee EJ, Da Silva B, Schoepfer C, Wilkinson JE, Buscaglia JM, DeNicola GM, Tiriac H, Hammell M, Crawford HC, Schmidt EE, Thompson CB, Pappin DJ, Sonenberg N and Tuveson DA, Cell, 2016, 166, 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon MK, Madriaga A, Kiss von Soly S, Kumar A, Parlati F, Yakes FM, Shawver L, Le Moigne R, Anderson DJ, Rolfe M and Wustrow D, Journal of medicinal chemistry, 2015, 58, 9480–9497. [DOI] [PubMed] [Google Scholar]
- 473.Duru N, Gernapudi R, Zhang Y, Yao Y, Lo P-K, Wolfson B and Zhou Q, Cancer Letters, 2015, 369, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T and Yamamoto M, Neoplasia, 2011, 13, 864–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Singh A, Bodas M, Wakabayashi N, Bunz F and Biswal S, Antioxidants & redox signaling, 2010, 13, 1627–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA and Tuveson DA, Nature, 2011, 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, Nasipuri P, Krausz KW, Wakabayashi N, Dewi R, Boros LG, Gonzalez FJ, Gabrielson E, Wong KK, Girnun G and Biswal S, The Journal of clinical investigation, 2013, 123, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M and Motohashi H, Cancer Cell, 2012, 22, 66–79. [DOI] [PubMed] [Google Scholar]
- 479.Murakami S and Motohashi H, Free radical biology & medicine, 2015, 88, 168–178. [DOI] [PubMed] [Google Scholar]
- 480.Jaramillo MC and Zhang DD, Genes & development, 2013, 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA and Tuveson DA, Nature, 2011, 475, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Matthews JH, Liang X, Paul VJ and Luesch H, ACS chemical biology, 2018, 13, 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Wang XJ, Hayes JD, Henderson CJ and Wolf CR, Proceedings of the National Academy of Sciences of the United States of America, 2007, 104, 19589–19594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Cheng X, Qian W, Chen F, Jin Y, Wang F, Lu X, Lee SR, Su D and Chen B, Molecular medicine reports, 2019, 20, 2294–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Furfaro AL, Piras S, Domenicotti C, Fenoglio D, De Luigi A, Salmona M, Moretta L, Marinari UM, Pronzato MA, Traverso N and Nitti M, PloS one, 2016, 11, e0152465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Kim D, Choi BH, Ryoo IG and Kwak MK, Cell death & disease, 2018, 9, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Molina-Jijon E, Rodriguez-Munoz R, Namorado Mdel C, Bautista-Garcia P, Medina-Campos ON, Pedraza-Chaverri J and Reyes JL, The Journal of nutritional biochemistry, 2015, 26, 441–454. [DOI] [PubMed] [Google Scholar]
- 488.Sapiro JM, Monks TJ and Lau SS, American journal of physiology. Renal physiology, 2017, 313, F1200–f1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Valenzuela M, Glorieux C, Stockis J, Sid B, Sandoval JM, Felipe KB, Kviecinski MR, Verrax J and Buc Calderon P, British journal of cancer, 2014, 111, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Zhu J, Wang H, Chen F, Fu J, Xu Y, Hou Y, Kou HH, Zhai C, Nelson MB, Zhang Q, Andersen ME and Pi J, Free radical biology & medicine, 2016, 99, 544–556. [DOI] [PubMed] [Google Scholar]
- 491.Kitamura H and Motohashi H, Cancer science, 2018, DOI: 10.1111/cas.13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Hammad A, Namani A, Elshaer M, Wang XJ and Tang X, Cancer letters, 2019, DOI: 10.1016/j.canlet.2019.09.016. [DOI] [PubMed] [Google Scholar]
- 493.Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E and Zhang DD, Annual review of pharmacology and toxicology, 2018, DOI: 10.1146/annurev-pharmtox-010818-021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Willingham W 3rd, Considine RT, Chaney SG, Lee KH and Hall IH, Biochemical pharmacology, 1984, 33, 330–333. [DOI] [PubMed] [Google Scholar]
- 495.Willingham W Jr., Stafford EA, Reynolds SH, Chaney SG, Lee KH, Okano M and Hall IH, Biochimica et biophysica acta, 1981, 654, 169–174. [DOI] [PubMed] [Google Scholar]
- 496.Harder B, Tian W, La Clair JJ, Tan AC, Ooi A, Chapman E and Zhang DD, Molecular carcinogenesis, 2016, DOI: 10.1002/mc.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Avila-Carrasco L, Majano P, Sanchez-Tomero JA, Selgas R, Lopez-Cabrera M, Aguilera A and Gonzalez Mateo G, Frontiers in pharmacology, 2019, 10, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Cai SJ, Liu Y, Han S and Yang C, Cell & bioscience, 2019, 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Lee JH, Rangappa S, Mohan CD, Sethi G, Lin ZX, Rangappa KS and Ahn KS, Biomolecules, 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Liu X, Xu H, Zhang Y, Wang P and Gao W, Journal of cellular biochemistry, 2019, 120, 10556–10563. [DOI] [PubMed] [Google Scholar]
- 501.Murakami Y, Sugiyama K, Ebinuma H, Nakamoto N, Ojiro K, Chu PS, Taniki N, Saito Y, Teratani T, Koda Y, Suzuki T, Saito K, Fukasawa M, Ikeda M, Kato N, Kanai T and Saito H, BMC cancer, 2018, 18, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Park SH, Kim JH, Ko E, Kim JY, Park MJ, Kim MJ, Seo H, Li S and Lee JY, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2018, DOI: 10.1096/fj.201800011R, fj201800011R. [DOI] [Google Scholar]
- 503.Turpaev K, Krizhanovskii C, Wang X, Sargsyan E, Bergsten P and Welsh N, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2019, 33, 3510–3522. [DOI] [PubMed] [Google Scholar]
- 504.Tsuchida K, Tsujita T, Hayashi M, Ojima A, Keleku-Lukwete N, Katsuoka F, Otsuki A, Kikuchi H, Oshima Y, Suzuki M and Yamamoto M, Free Radical Biology and Medicine, 2017, 103, 236–247. [DOI] [PubMed] [Google Scholar]
- 505.Zhong Y, Zhang F, Sun Z, Zhou W, Li ZY, You QD, Guo QL and Hu R, Molecular carcinogenesis, 2013, 52, 824–834. [DOI] [PubMed] [Google Scholar]
- 506.Xu X, Zhang Y, Li W, Miao H, Zhang H, Zhou Y, Li Z, You Q, Zhao L and Guo Q, Biochemical pharmacology, 2014, 92, 220–234. [DOI] [PubMed] [Google Scholar]
- 507.Kim EH, Jang H, Shin D, Baek SH and Roh JL, Apoptosis : an international journal on programmed cell death, 2016, 21, 1265–1278. [DOI] [PubMed] [Google Scholar]
- 508.Xu X, Zhang X, Zhang Y, Yang L, Liu Y, Huang S, Lu L, Kong L, Li Z, Guo Q and Zhao L, Scientific reports, 2017, 7, 39950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 509.Wang L, Li C, Sreeharsha N, Mishra A, Shrotriya V and Sharma A, J Photochem Photobiol B, 2020, 204, 111775. [DOI] [PubMed] [Google Scholar]
- 510.Bei W, Jing L and Chen N, Colloids Surf B Biointerfaces, 2020, 185, 110635. [DOI] [PubMed] [Google Scholar]
- 511.Wang YS, Cho JG, Hwang ES, Yang JE, Gao W, Fang MZ, Zheng SD and Yi TH, Appl Biochem Biotechnol, 2018, 184, 1073–1093. [DOI] [PubMed] [Google Scholar]
- 512.Khan NM, Haseeb A, Ansari MY, Devarapalli P, Haynie S and Haqqi TM, Free radical biology & medicine, 2017, 106, 288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]