

Inorganic polyphosphate (polyP) is an evolutionarily ancient, widely conserved biopolymer required for stress resistance and pathogenesis in diverse bacteria, but we do not understand how its synthesis is regulated. In this work, I gained new insights into this process by characterizing the role of the transcriptional regulator DksA in polyP regulation in Escherichia coli and identifying previously unknown links between polyP synthesis and the stress-responsive alternative sigma factors FliA, RpoN, and RpoE.
KEYWORDS: polyphosphate, sigma factors, stress response, stringent response
ABSTRACT
Bacteria synthesize inorganic polyphosphate (polyP) in response to a variety of different stress conditions. polyP protects bacteria by acting as a protein-stabilizing chaperone, metal chelator, or regulator of protein function, among other mechanisms. However, little is known about how stress signals are transmitted in the cell to lead to increased polyP accumulation. Previous work in the model enterobacterium Escherichia coli has indicated that the RNA polymerase-binding regulatory protein DksA is required for polyP synthesis in response to nutrient limitation stress. In this work, I set out to characterize the role of DksA in polyP regulation in more detail. I found that overexpression of DksA increases cellular polyP content (explaining the long-mysterious phenotype of dksA overexpression rescuing growth of a dnaK mutant at high temperatures) and characterized the roles of known functional residues of DksA in this process, finding that binding to RNA polymerase is required but that none of the other functions of DksA appear to be necessary. Transcriptomics revealed genome-wide transcriptional changes upon nutrient limitation, many of which were affected by DksA, and follow-up experiments identified complex interactions between DksA and the stress-sensing alternative sigma factors FliA, RpoN, and RpoE that impact polyP production, indicating that regulation of polyP synthesis is deeply entwined in the multifactorial stress response network of E. coli.
IMPORTANCE Inorganic polyphosphate (polyP) is an evolutionarily ancient, widely conserved biopolymer required for stress resistance and pathogenesis in diverse bacteria, but we do not understand how its synthesis is regulated. In this work, I gained new insights into this process by characterizing the role of the transcriptional regulator DksA in polyP regulation in Escherichia coli and identifying previously unknown links between polyP synthesis and the stress-responsive alternative sigma factors FliA, RpoN, and RpoE.
INTRODUCTION
Bacteria, including the model enterobacterium Escherichia coli, have sophisticated regulatory systems that allow them to respond to changes in their environments. These systems are essential for survival of stressful or growth-inhibiting conditions, persistence in the environment, and pathogenesis (1–3). In E. coli, these include such well-studied systems as the general stress response driven by the alternative sigma factor RpoS (also called σS or σ38), which upregulates expression of genes involved in resisting starvation, reactive oxygen species, salt, and acid, among others (3, 4), and the stringent response, in which the small molecule alarmones guanosine-5′,3′-tetraphosphate and guanosine-5′,3′-pentaphosphate (collectively referred to as ppGpp) and the RNA polymerase-binding protein DksA act in coordination to slow growth and promote expression of genes involved in adaptation to various kinds of starvation stresses (5–7). Regulation of stress response systems is complex and interconnected, giving bacteria the capacity to survive in rapidly changing environments.
One long-known but poorly understood element of bacterial stress response is the production of inorganic polyphosphate (polyP), a linear biopolymer of phosphate up to 1,000 units in length that is synthesized by the widely conserved bacterial enzyme polyP kinase (PPK) (8, 9). In response to a variety of stressful changes in conditions, including nutrient limitation, heat, salt, and hypochlorous acid treatment, E. coli transiently accumulates large amounts of polyP (10–13). The physiological functions of polyP are not fully understood, but it is known to promote survival under different stress conditions by stabilizing unfolded proteins, chelating toxic metals, acting as a phosphate store, increasing translation fidelity, and as a second messenger that regulates the activity of a variety of proteins (12, 14–21). Importantly, in many bacterial pathogens, deleting the gene encoding PPK (ppk) results in the loss of the ability to cause disease (22–28), indicating that polyP metabolism may be a potential therapeutic target for use against bacterial infections (29–31).
We know surprisingly little about the mechanism by which polyP synthesis is regulated. Several different regulators have been implicated in modulating polyP production in E. coli under different stress conditions, including RpoS (10), the PhoB and PhoU regulators of phosphate transport (10, 13, 32, 33), and the ppGpp synthases RelA and SpoT (10, 13, 34), but no convincing mechanistic model has been developed that explains how any of these systems controls polyP accumulation or why different regulators appear to be required under different conditions. Transcription of the operon containing ppk and ppx, which encodes the exopolyphosphatase PPX, does not increase upon stress treatment in E. coli (11, 35), and while ppGpp potently inhibits degradation of polyP by PPX (34), neither PPX nor ppGpp is necessary for the induction of polyP by nutrient limitation stress (11, 12, 35). However, I have recently reported that the stringent response regulator DksA, which normally works in concert with ppGpp (5), is required for polyP synthesis under those conditions, and this phenotype can be reverted by deletion of the RNA polymerase-binding elongation factor GreA (11), indicating that there is an important role for transcription in polyP control.
In this work, I set out to understand the role of DksA in regulating polyP synthesis in more depth. I found that cellular polyP content can be modulated by changing the amount of DksA expression, and characterized the roles of known functional residues of DksA in this process. I also identified interactions between DksA and the alternative sigma factors FliA, RpoN, and RpoE that impact polyP synthesis, gaining new insights into how polyP regulation is linked to the broader stress response network of E. coli.
RESULTS
Overexpressing DksA enhances stress-induced polyP synthesis.
As I have previously reported (11), deletion of dksA prevents E. coli from accumulating polyP upon nutrient starvation stress (Fig. 1A). In that work, I found that deletion of greA in a dksA mutant reverts this phenotype and allows polyP to accumulate. However, the greA::cat+ allele used in that and many other studies (36–42) also deletes 109 bp upstream and 177 bp downstream of the greA coding sequence (36), potentially disrupting the neighboring yhbY gene (which encodes a ribosome assembly factor [43]), and the small RNA GraL (44, 45). To confirm that deletion of greA is the mutation responsible for allowing a dksA mutant to synthesize polyP, I therefore constructed a double mutant strain combining a ΔdksA1000::cat+ mutation (11) with the ΔgreA788::kan+ allele from the Keio collection (46), which does not interfere with neighboring gene sequences, and observed that this strain synthesized the same amount of polyP as the wild type after starvation stress (Fig. 1A). This confirmed that neither yhbY nor GraL is involved in regulation of polyP synthesis under these conditions. Interestingly, I also found that overexpressing dksA from a strong arabinose-inducible promoter (47) led to accumulation of twice as much polyP as was found in the wild-type strain (Fig. 1B), showing that cellular polyP levels can be tuned by modulating dksA expression, although under normal growth conditions the concentration of DksA in the cell is thought to be relatively stable (36). Overexpression of TraR, a protein from the F plasmid that mimics the transcriptional effects of DksA and ppGpp (48, 49), had a similar effect (Fig. 1C), complementing polyP synthesis in a dksA mutant and increasing polyP synthesis above wild-type levels in both wild-type and dksA greA mutant strains.
FIG 1.

Overexpressing DksA enhances stress-induced polyP synthesis, and multicopy suppression of a dnaK mutation by dksA is polyP dependent. (A) E. coli MG1655 wild-type and isogenic ΔdksA1000::cat+, ΔgreA788::kan+, and ΔdksA1000::cat+ ΔgreA788::kan+ strains were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (morpholinepropanesulfonic acid [MOPS] with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil) for 2 h (white circles) (n = 3, ± standard deviation [SD]). (B) MG1655 ΔaraA mutants containing either pBAD18 or pDKSA1 (dksA+) plasmids were grown at 37°C to an A600 of 0.2 to 0.4 in LB containing 2 g · liter−1 arabinose (black circles) and then shifted to minimal medium containing 2 g · liter−1 arabinose for 2 h (white circles) (n = 3, ±SD). (C) MG1655 or isogenic ΔdksA1000::cat+ or ΔdksA1000::cat+ ΔgreA788::kan+ mutants containing pRLG13077 (pTrc, VOC; vector-only control) or pRLG13078 (pTrc-TraR, traR+) were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium containing 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; black circles) and then shifted to minimal medium containing 1 mM IPTG for 2 h (white circles) (n = 3, ±SD). polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of the wild-type control for a given experiment (two-way repeated measures analysis of variance [ANOVA] with Holm-Sidak multiple-comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (D to F) E. coli MG1655 wild-type (dnaK+) and isogenic ΔdnaK52::cat+, Δppk-749, or Δppk-749 ΔdnaK52::cat+ strains containing plasmids pBAD18, pDKSA1 (dksA+), or pPPK30 (ppkG733A, encoding PPKE245K) were grown overnight at 30°C with shaking in LB containing 100 μg · ml−1 ampicillin, then diluted to an A600 of 0.01 in LB containing 100 μg · ml−1 ampicillin, 2 g · liter−1 arabinose, and, in the experiment shown in panel E, 1 mM MgCl2, and incubated with shaking in a Tecan Infinite M1000 plate reader at 40.5°C for 12 h (n = 3, ±SD).
Multicopy suppression of a dnaK mutation by dksA is polyP dependent.
DksA is now known to be a multifunctional protein involved in genome-wide transcriptional regulation, RNA chain elongation, transcription fidelity, preventing conflicts between RNA polymerase and the DNA replisome, DNA double-strand break repair, redox sensing, and preventing RNA polymerase arrest under nutritional stress (5, 50–55), but it was originally identified as a multicopy suppressor of the heat-sensitive growth phenotype caused by a null mutation in dnaK (hence the name “dksA,” for dnaK suppressor A) (56). DnaK (also known as Hsp70) is a protein-folding chaperone and central component of the E. coli response to proteotoxic stresses (57). Despite some speculation, the mechanism by which overexpression of DksA rescues growth of a dnaK mutant at high temperatures has never been established (5, 53, 58). polyP is a potent protein-stabilizing chemical chaperone (12, 59), and I wondered if the increase in polyP levels caused by DksA overexpression could account for its protective effect in a dnaK mutant. First, I tested the ability of a dksA overexpression plasmid to rescue the growth defect of a dnaK mutant at 40.5°C (Fig. 1D). As expected (56), it increased growth substantially, although not to wild-type levels. However, in a Δppk mutant background, in which no polyP synthesis is possible (60), overexpression of DksA provided no benefit (Fig. 1E). Expression of a hyperactive PPKE245K variant (35) also improved the growth of the dnaK mutant (Fig. 1F), indicating that increased polyP alone is able to exert a protective effect similar to that of DksA overexpression. Variability was quite high in these experiments, possibly due to the rapid accumulation of suppressors reported for dnaK mutants (61), but these results are consistent with the idea that overproduction of polyP is at least partially responsible for dksA’s multicopy suppression phenotype.
DksA requires interaction with RNA polymerase to regulate polyP synthesis.
A variety of dksA mutant alleles have been described that encode DksA variants with clearly defined effects on the functions of DksA. I used a panel of these to determine which functions of DksA are involved in regulating polyP synthesis by complementing a dksA null mutant with plasmids expressing different DksA variants (Fig. 2). I tested three variants with disruptions in the conserved aspartate residues necessary for the ability of DksA to regulate transcription by destabilizing promoter open complexes (62–64). A DksAD74E variant restored polyP synthesis to wild-type levels, but the more strongly defective DksAD74N variant (62), expression of which is also reported to be unable to rescue growth of a ΔdnaKJ mutant at 42°C (58), was able to only partially restore polyP synthesis. polyP synthesis in a strain expressing the DksAD71N D74N protein, reported to be completely defective in transcriptional control (63, 64), was highly variable but consistently lower than that that of the wild type. These results show that the active-site aspartates are involved in but are not essential for DksA’s role in activating polyP synthesis.
FIG 2.
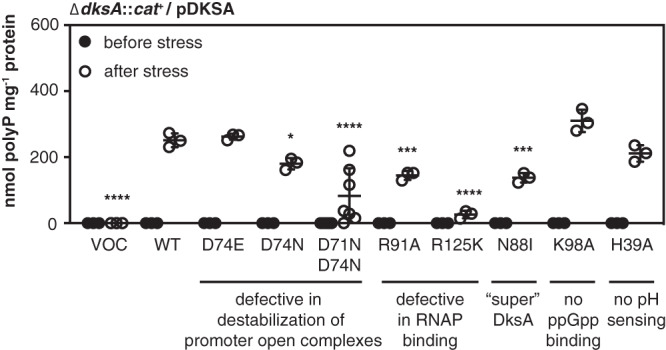
DksA requires interaction with RNA polymerase to regulate polyP synthesis. (A) MG1655 ΔdksA1000::cat+ mutants containing pBAD18 (VOC; vector-only control), pDKSA1 (dksA+, wild type [WT]), pDKSA2 (dksAC222A, D74E), pDKSA3 (dksAG220A C22T, D74N), pDKSA4 (dksAC271G G272C C273G, R91A), pDKSA5 (dksAC373A G374A C375A, R125K), pDKSA6 (dksAA263T C264T, N88I), pDKSA7 (dksAA292G A293C, K98A), pDKSA8 (dksAC115G A116C C117G, H39A), or pDKSA9 (dksAG211A C213T G220A C222T, D71N D74N) plasmids were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) containing 2 g · liter−1 arabinose and 100 μg · ml−1 ampicillin (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil) containing 2 g · liter−1 arabinose and 100 μg · ml−1 ampicillin for 2 h (white circles) (n = 3 to 7; ±SD). polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of the wild-type control for each experiment (mixed-effects model with Holm-Sidak multiple-comparison test; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001).
Mutation of the active site aspartates does not affect the binding affinity of DksA to RNA polymerase (62). Expression of a DksAR91A variant, which has an approximately 2-fold reduction in binding affinity but retains some DksA functions (54, 65, 66), led to only about half the amount of polyP production seen with wild-type DksA, while expression of DksAR125K, which has a severe 20-fold reduction in binding affinity (66), reduced polyP accumulation 10-fold. Curiously, expression of DksAN88I, a so-called “super DksA,” which binds to RNA polymerase about 4.5 times more tightly than wild-type DksA and has a correspondingly greater effect on expression of both positively and negatively regulated promoters (63), reduced polyP synthesis to a similar extent as the DksAR91A variant. These results indicate that DksA must bind to RNA polymerase to activate polyP synthesis but that either increases or decreases in its binding affinity can reduce the extent of that synthesis. This generally supports the hypothesis that DksA may influence polyP synthesis by controlling the access of other regulators (e.g., GreA) to the secondary channel of RNA polymerase (11, 39).
As expected, since a relA spoT mutant incapable of making ppGpp is able to synthesize wild-type levels of polyP under these conditions (11), expression of DksAK98A, which cannot bind ppGpp (65), allowed wild-type levels of polyP synthesis. DksA activity is sensitive to cytoplasmic pH (67), but disruption of the pH-sensing histidine residue (DksAH39A) also had no inhibitory effect on polyP synthesis. DksA contains a redox-sensing zinc finger domain (53, 68), but I was unable to test whether this plays a role in polyP synthesis in this set of experiments, since mutations disrupting this domain destabilize the protein and are equivalent to ΔdksA mutations (69).
Levels of ppk and ppx mRNA decrease after nutrient limitation stress.
I previously used reporter fusions to show that expression from the promoter of the ppk-ppx operon (Pppk) is decreased in the absence of either DksA or ppGpp but does not change significantly after nutrient limitation stress in wild-type, dksA, or dksA greA mutant strains (11). However, since both DksA and GreA can act as transcription elongation factors (51, 52, 70, 71), I hypothesized that the effect of dksA and greA mutations on polyP accumulation might result from changes in ppk or ppx transcript accumulation independent of transcription initiation from Pppk. I therefore used quantitative reverse transcription-PCR (qRT-PCR) to measure the amount of mRNA from three different points in the ppk-ppx operon (Fig. 3A) before and after nutrient limitation stress in wild-type, dksA, and dksA greA strains.
FIG 3.

Levels of ppk and ppx mRNA decrease after nutrient limitation stress. (A) Position of quantitative PCR (qPCR) amplicons within the ppk and ppx genes. (B, C) E. coli MG1655 wild-type and isogenic ΔdksA1000::cat+ and ΔdksA1000::cat+ ΔgreA788::kan+ strains were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) and then shifted to minimal medium (MOPS with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil) for 2 h. qRT-PCR was used to measure fold changes in transcript abundance at the indicated time points (n = 3, ±SD). In panel B, changes in expression for each amplicon are normalized to expression of that amplicon in the wild-type strain before stress treatment (t = 0 h). In panel C, changes in expression for each amplicon are normalized to expression of the ppkupstream amplicon in the same strain at each time point.
As I reported previously using promoter fusions (11), expression of ppk in the absence of stress was about 2-fold lower in the dksA and dksA greA mutants than that in the wild-type (Fig. 3B). Surprisingly, however, I found that in all three strains, levels of transcripts from both the 5′ and 3′ ends of the ppk gene, referred to here as ppkupstream (nucleotides 209 to 409 of ppk) and ppkdownstream (nucleotides 1530 to 1725 of ppk), rapidly decreased 4- to 16-fold after stress, and levels of ppx transcripts (nucleotides 510 to 695 of ppx) decreased more than 32-fold. Normalizing the levels of ppkdownstream and ppx transcripts to the amount of ppkupstream transcripts at each time point (Fig. 3C) showed that in the absence of stress, both ends of the ppk transcript are present at the same level, with 2- to 4-fold-fewer ppx transcripts present. After stress, however, levels of the ppkdownstream transcript drop 2- to 4-fold below the levels of the ppkupstream transcript, while the amounts of ppx transcript decrease more dramatically (8- to 32-fold). These results indicate that nutrient limitation stress has a significant effect on the accumulation of ppk and ppx transcripts and on their relative levels in the cell, and demonstrate previously unsuspected regulation of transcript elongation or mRNA stability in the ppk-ppx operon. However, since there were no dramatic differences in these patterns between wild-type, dksA, and dksA greA strains and certainly no effects that correlate with the respective polyP accumulation in those strains, these results do not account for how DksA and GreA regulate polyP accumulation during nutrient limitation.
In parallel with the above-described experiments, I also measured the levels of 16S rRNA (rrsD) at each time point after nutrient limitation (see Fig. S1 in the supplemental material). Amino acid starvation is a well-known inducer of the stringent response (72), in which DksA and ppGpp repress rRNA transcription (5), but the specific nutrient limitation stress used here (shift of exponentially growing cultures from LB to morpholinepropanesulfonic acid [MOPS] minimal medium containing 4 g · liter−1 glucose and 0.1 mM K2HPO4), originally described by Arthur Kornberg’s lab as a stress condition that robustly stimulates polyP synthesis (10), actually led to a 1.5- to 3-fold increase in rrsD expression. This is consistent with the lack of a role for ppGpp in polyP regulation under these conditions (11), but it does reopen the question of what roles ppGpp and DksA might play in polyP accumulation under conditions that do activate the classic stringent response. I note, however, that overproduction of ppGpp under nonstress conditions does not induce polyP synthesis (11).
Identifying genes regulated by DksA and GreA under nutrient limitation conditions.
I next used mRNA sequencing of wild-type, dksA, and dksA greA strains immediately before and 5 min after nutrient starvation stress to screen for genes whose expression patterns correlated, either positively or negatively, with the accumulation of polyP by these strains (see Data Set S1 and Fig. S2 in the supplemental material). Both DksA and GreA are global regulators (39), and nutrient limitation induced sweeping changes in gene expression in all three strains, so I found there to be a surprisingly large number of such genes. I used qRT-PCR to validate several of the most striking candidates (Fig. 4A to C). This reproduced the results of the RNA sequencing screen in all cases and confirmed that expression of genes involved in the regulation and synthesis of flagella (73, 74) (see Fig. S3A in the supplemental material) was inversely correlated with capacity for polyP synthesis, both before and after stress (Fig. 4A). Conversely, the expression of most genes involved in glycerol dissimilation (73, 75) (Fig. S3B and C) was positively correlated with capacity for polyP synthesis, at least before stress (Fig. 4B and C). Finally, the expression of the gadE operon, which encodes a central regulator of the E. coli acid stress response (73, 76) and the MdtEF multidrug exporter (77) (Fig. S3D) was inversely correlated with capacity for polyP synthesis both before and after stress. However, no mutations blocking the expression of operons encoding genes for flagella (Fig. 4D), glycerol dissimilation (Fig. 4E and F), or acid stress response (Fig. 4F) had any effect on polyP synthesis.
FIG 4.

Mutations disrupting dksA- and greA-regulated operons do not affect polyP accumulation. E. coli MG1655 wild-type and isogenic ΔdksA1000::cat+ and ΔdksA1000::cat+ ΔgreA788::kan+ strains (A to C), ΔflhD745::kan+, ΔfliA::kan+, ΔflgB742::kan+, ΔfliF1000::cat+, and ΔfliD770::kan+ strains (D), ΔglpA721::kan+, ΔglpD759::kan+, ΔglpF786::kan+, ΔglpT720::kan+, and ΔdhaL789::kan+ strains (E), or ΔglpE758::kan+, ΔglpG757::kan+, ΔgadE767::kan+, and ΔmdtE768::kan+ strains (F) were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4) for 2 h (white circles) (n = 3, ±SD). (A to C) qRT-PCR was used to measure fold changes in transcript abundance of the indicated genes before and 15 min after nutrient limitation, relative to expression in the wild-type strain in the absence of stress (n = 3, ±SD). (D to F) polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate gene expression significantly different from those of the wild-type control at each time point (two-way repeated-measures ANOVA with Holm-Sidak multiple-comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). No significant differences were found between polyP levels of any strain and that of the wild-type control.
Does overexpression of flagellar regulators inhibit polyP synthesis?
Although knocking out flagellar genes had no effect on polyP synthesis (Fig. 4D), this did not necessarily prove that the inhibition of polyP synthesis in a dksA mutant was not due to overexpression of one of the flagellar regulators, namely, FlhDC, FliA, or FliZ (74, 78–80). Expression of FlhDC responds to a wide variety of environmental stresses (80, 81) and activates expression of flagellar genes (including fliA and fliZ) (Fig. S3A), as well as a number of genes with other functions (74). Similarly, FliA (also known as σ28, σF, or RpoF) is a sigma factor that activates expression of many genes, not all of which are involved in motility (74). FliZ is a transcription factor, cotranscribed with FliA, that competes for promoter binding with RpoS, the master regulator of the general stress response in E. coli (4, 82), and so could directly or indirectly affect the expression of hundreds of genes (78, 79). A dksA mutant was hypermotile, as expected (83), and a dksA greA double mutant was almost completely nonmotile (Fig. 5A). Stringent mutations of RNA polymerase (84, 85) that lead, by an unknown mechanism presumably related to that of a dksA mutant, to reduced polyP accumulation (11) also had increased motility (see Fig. S4 in the supplemental material), despite growing substantially more slowly than the wild-type (86). However, overexpression of FlhDC, FliA, or FliZ did not repress polyP synthesis (Fig. 5B), nor was the inhibitory effect of a dksA mutation suppressed by flhDC or fliAZ mutations (Fig. 5C). Repression of polyP synthesis in a dksA mutant is therefore not due, directly or indirectly, to overproduction of any of the flagellar regulators. Surprisingly, though, mutation of either flhD or fliA did prevent overexpression of DksA from enhancing polyP synthesis (Fig. 5D), suggesting that the flagellar regulators might have an indirect effect on polyP regulation by DksA. This is not unprecedented, since work from several laboratories has shown indirect regulation by both ppGpp and DksA of promoters driven by alternative sigma factors, which is thought to depend on changes in the availability of RNA polymerase core enzyme due to DksA/ppGpp-dependent repression of rRNA transcription (87–92). FliA is abundant and has a high affinity for RNA polymerase (93–95), so deletion of fliA could plausibly have a substantial effect on the equilibrium among other sigma factors and core RNA polymerase (96, 97).
FIG 5.

Suppression of polyP synthesis in a dksA mutant is not due to overexpression of flagellar regulators. (A) E. coli MG1655 wild-type and isogenic ΔdksA1000::cat+ and ΔdksA1000::cat+ ΔgreA788::kan+ strains were inoculated into LB containing 0.25% agar and incubated at room temperature (representative of 3 independent experiments). (B to D) E. coli MG1655 wild-type or isogenic ΔdksA1000::cat+, ΔdksA1000::cat+ ΔflhD745::kan+, ΔdksA1000::cat+ ΔfliA::kan+, ΔflhD745::kan+, or ΔfliA::kan+ strains containing, where indicated, pBAD18 (vector-only control [VOC]) or the indicated pBAD18-derived plasmids were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) containing, when plasmids were present, 100 μg · ml−1 ampicillin (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil) containing 100 μg · ml−1 ampicillin for 2 h (white circles) (n = 3 or 4, ±SD). Experiments in panels B and D included 2 g · liter−1 arabinose in both rich and minimal media. polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of the wild-type control for each experiment (two-way repeated-measures ANOVA or mixed-effects model with Holm-Sidak multiple-comparison test; *, P < 0.05; ****, P < 0.0001). No significant differences (ns) were found among the three indicated strains in panel C or between the indicated VOC and pDKSA1-containing strains in panel D.
The role of alternative sigma factors in regulation of polyP accumulation.
In addition to FliA and RpoS, E. coli has three other alternative sigma factors that also respond to environmental stresses and drive the expression of large numbers of genes, namely, RpoH (σ32 or σH), which controls chaperones and proteases in response to protein unfolding stresses (82, 98), RpoN (σ54 or σN), which responds to a number of signals, including nitrogen starvation (99), and RpoE (σ24 or σE), which responds to perturbations in the cell envelope (100). The only other sigma factor in E. coli, FecI (σ19), responds to ferric citrate and controls expression of only a single promoter (that of fecA) (101). Regulation of gene expression in response to changes in environmental conditions involves overlapping interactions among these sigma factors and other stress response pathways, including the stringent response (4, 78, 82, 83, 87–89, 91, 102). The observation that overexpression of the RpoS antagonist FliZ slightly increased polyP accumulation (Fig. 5B) and that a dksA fliA::kan+ mutant, which lacks both fliA and fliZ, appeared to contain even less polyP than a dksA mutant (Fig. 5C) (although that difference was not statistically significant in this set of experiments) supported the idea that alternative sigma factors might be playing a role in modulating polyP synthesis. While I have not found RpoS to be required for polyP accumulation in response to the nutrient limitation stress used here (11), others have reported roles for either RpoS or RpoN under some other stress conditions (10). Regardless, I wanted to know whether any of E. coli’s other major stress response pathways were interacting with DksA in the control of polyP synthesis. I therefore performed a series of epistasis experiments to determine the role of each of the above alternative sigma factors in polyP accumulation by E. coli in the presence and absence of DksA (Fig. 6).
FIG 6.

Roles of stress-sensing alternative sigma factors in polyP synthesis. E. coli MG1655 wild-type and isogenic ΔrpoS746, ΔdksA1000::cat+, and ΔrpoS746 ΔdksA1000::cat+ strains (A), ΔaraA zhf-50::Tn10(tet+), ΔaraA zhf-50::Tn10(tet+) sidB3, ΔaraA zhf-50::Tn10(tet+) ΔdksA1000::cat+, and ΔaraA zhf-50::Tn10(tet+) sidB3 ΔdksA1000::cat+ strains (B), ΔrpoN730, ΔrpoN730 ΔdksA1000::cat+ strains (C), or ΔrpoE1000::kan+ strains (D) were grown at 37°C or 30°C, as indicated, to an A600 of 0.2 to 0.4 in rich medium (LB) containing 100 μg · ml−1 ampicillin (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil) containing 100 μg · ml−1 ampicillin for 2 h (white circles) (n = 3 or 4, ±SD). Experiments in panel D included 10 μg · ml−1 erythromycin in both rich and minimal media. polyP concentrations are in terms of individual phosphate monomers. Unless specifically indicated, asterisks indicate polyP levels significantly different from those of the wild-type control for each experiment (two-way repeated-measures ANOVA with Holm-Sidak multiple-comparison test; ns, P > 0.05; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001).
As I previously reported (11), an rpoS mutant had no defect in polyP production under these conditions, and polyP levels in an rpoS dksA mutant were indistinguishable from those in a dksA mutant (Fig. 6A). E. coli mutants lacking rpoH are not viable above 20°C, although they rapidly accumulate suppressors that allow them to grow at 30°C or higher (103, 104). To assess the possible role of RpoH in polyP synthesis, I therefore used the defective rpoH allele sidB3, which contains much less RpoH than the wild type and which cannot induce the expression of heat shock proteins in response to elevated temperature but which grows stably at 30°C (61). polyP production in all strains (including MG1655 wild type; data not shown) was roughly 10-fold lower at 30°C than that at 37°C, indicating that temperature strongly influenced polyP production. The amounts of polyP produced under these conditions were close to the limit of detection of our polyP assay (105), making comparisons between strains difficult due to the relatively high contribution of background noise to variability, but the pattern of polyP production in sidB3 and sidB3 dksA double mutants did not suggest that sidB3 has a major effect on the dksA phenotype (Fig. 6B). Mutants lacking rpoN had a significant defect in polyP synthesis (Fig. 6C), as did an rpoN dksA double mutant. RpoE is essential in E. coli MG1655 under normal growth conditions but can be stably knocked out in the presence of subinhibitory concentrations of erythromycin (106, 107). Under these conditions, both the dksA and the rpoE mutant had significant polyP synthesis defects (Fig. 6D). Despite multiple attempts, I was unable to construct an rpoE dksA double mutant, indicating that these genes are synthetically lethal. Deletion of rsd, encoding a negative regulator of RpoD that facilitates sigma factor competition during the stationary phase (87, 108) had no effect on polyP synthesis (see Fig. S5 in the supplemental material).
Overexpressing DksA was able to restore polyP synthesis to the rpoN mutant but not to the rpoE mutant (Fig. 7A). Conversely, overexpression of RpoE increased polyP production in the wild type and the rpoN mutant but not in the dksA or, surprisingly, in the rpoE mutant (Fig. 7B). The meaning of this last result is unclear, but the ΔrpoE::kan+ mutant did not tolerate overexpression of rpoE very well and had a very long lag phase under even modest induction conditions (data not shown), possibly because of the lack of the RseA anti-sigma factor in this strain (109). However, since rpoE mutants are inviable at 42°C (110), I was unable to resolve the ΔrpoE::kan+ insertion (111) to test this idea. Nevertheless, the results of this experiment indicate that both rpoE and dksA are positive regulators of polyP synthesis upon nutrient limitation and depend on one another to have this effect but that overexpression of either gene rescues polyP synthesis in an rpoN mutant.
FIG 7.

Complementation of polyP-deficient mutants with ectopically expressed DksA and RpoE. E. coli MG1655 wild-type and isogenic ΔdksA1000::cat+, ΔrpoN730, or ΔrpoE1000::kan+ strains containing pBAD18 or the indicated pBAD18-derived plasmids were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) containing 100 μg · ml−1 ampicillin (black circles) and 0.2% (A) or 0.0125% (B) arabinose and then shifted to minimal medium (MOPS with no amino acids, 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil) containing 100 μg · ml−1 ampicillin and 0.2% (A) or 0.0125% (B) arabinose for 2 h (white circles) (n = 3 to 5, ±SD). Growth medium for rpoE mutants included 10 μg · ml−1 erythromycin in both rich and minimal media. polyP concentrations are in terms of individual phosphate monomers. Unless specifically indicated, asterisks indicate polyP levels significantly different from those of the wild-type control for each experiment (two-way repeated-measures ANOVA or mixed-effects model with Holm-Sidak multiple-comparison test; ns, P > 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
DISCUSSION
The purpose of this work was to characterize the role of DksA in regulating polyP synthesis in E. coli, with the ultimate goal of understanding how exposure to stressful environmental conditions leads to polyP accumulation. While I have made some progress toward this goal, the experiments presented here have also revealed several layers of unexpected complexity and emphasize the fact that polyP regulation is deeply entwined in the complicated multifactorial general stress response of E. coli.
While changes in DksA expression did not affect polyP levels under nonstress conditions, the amount of polyP produced after nutrient limitation correlated well with the amount of DksA being expressed (Fig. 1A and B). This also held true for expression of TraR, which has many of the same effects on RNA polymerase as DksA and ppGpp (48) (Fig. 1C). Experiments with dnaK knockouts indicated that the original multicopy suppression phenotype that gave DksA its name (56) is, in fact, probably due to the polyP overproduction that results from DksA overexpression (Fig. 1D to F). polyP overproduction could potentially also play a role in explaining other phenotypes associated with overexpression of dksA, including multicopy suppression of grpE, prc, yhhP, and mukB mutations (56, 112–114), although since DksA has many documented functions (5, 50–55), these would need to be tested on an individual basis. The ability of DksA to induce polyP accumulation was influenced its ability to bind RNA polymerase but was largely independent of other functional residues (Fig. 2), which may help to explain the suppression of the dksA mutant phenotype by a greA deletion (Fig. 1A). Both DksA and GreA bind in the secondary channel of RNA polymerase, and changes in the equilibrium between these two proteins at that site are thought to have regulatory consequences at certain promoters, although the details have not yet been fully worked out (36, 38, 39, 41, 42).
The nutrient shift stress used in this and earlier works (11, 13, 31, 35) was originally described by Arthur Kornberg’s lab (10) and is a robust and reproducible way to induce substantial polyP synthesis in E. coli. Despite its technical simplicity, however, the results of the experiments in this paper show that “nutrient limitation” leads to dramatic, genome-wide changes in gene expression, many of which, like polyP production, are strongly affected by dksA and greA mutations (see Data Set S1 and Fig. S2 in the supplemental material). These include up- or downregulation of operons regulated by many of the known stress-responsive regulators in E. coli, including multiple alternative sigma factors and transcription factors, and teasing apart which of these regulons directly influence polyP production is a significant challenge (Fig. 4 and 5). The assumption that this nutrient shift induces a stringent response (13) also appears to be wrong, since rRNA expression does not decrease after the shift (5) (Fig. S1). Typically, the ppGpp-dependent stringent response to amino acid starvation is induced in laboratory conditions by addition of serine hydroxamate or by specific isoleucine limitation (5, 115, 116), which is different from the simultaneous complete amino acid deprivation and phosphate limitation used here (10). As mentioned above, this may explain why ppGpp is not required for polyP synthesis under these conditions (11), but it reopens the question of what, if any, role ppGpp plays in polyP regulation under other stress conditions. polyP accumulation can reportedly be induced in E. coli by various kinds of starvation, serine hydroxamate, phosphate starvation or surplus, entry into the stationary phase, hypochlorous acid, salt, or heat (10, 12, 21, 59, 117). A signal common to all of these conditions is not obvious, but my results with alternative sigma factor mutants indicate that multiple stress-sensing mechanisms feed into control of polyP, either directly or indirectly.
E. coli encodes the following seven sigma factors: the housekeeping sigma RpoD and the alternative sigma factors RpoS, RpoN, RpoE, RpoH, FliA, and FecI (94, 118). Each alternative sigma factor drives expression of genes in response to particular environmental stressors (e.g., RpoS is active during the stationary phase, and RpoH is active during heat shock) (4, 82, 98), but there are sophisticated interactions among their regulons that are not fully understood. Expression of rpoN is activated by RpoE (119), expression of rpoE can be activated by RpoN or RpoS (120), both RpoN and RpoE activate transcription of rpoH (121, 122), and the FliA-dependent regulator FliZ inhibits expression of RpoS-dependent promoters (78, 79). DksA and ppGpp can also influence many of these interactions. Expression of fliA is inhibited by DksA (83), and DksA and ppGpp are required for transcription of at least some RpoN- and RpoE-dependent promoters (88, 102). Some of these effects are indirect and appear to depend on competition of sigma factors for core RNA polymerase (96, 97). For example, deletion of rpoN enhances expression from RpoS-dependent promoters, and the enhanced motility of an rpoS mutant is both FliA and RpoN dependent (89). Expression from weak (but not strong) RpoN-dependent promoters requires DksA and ppGpp in vivo but not in vitro (88), but ppGpp facilitates RpoS and RpoH competition with RpoD for RNA polymerase both in vitro and in vivo (87). No mechanistic model is currently available to explain all of these interactions, which appear to be both promoter and growth condition specific (102).
In light of this complexity, my results showing the impacts of DksA, RpoN, and RpoE on polyP production (Fig. 1, 6, and 7) must be interpreted cautiously. The effect of a fliA knockout on the DksA overexpression phenotype (Fig. 5D) hints at a role for sigma factor competition in that mutant background, but whether this is physiologically important in a wild-type strain is unclear. Complementation analysis (Fig. 7) shows that increasing expression of either RpoE or DksA can compensate for the loss of RpoN but that both RpoE and DksA are necessary for polyP accumulation. RpoN-dependent promoters are not thought to directly require DksA (88), but some RpoE-dependent promoters do (102). On the other hand, RpoN-dependent promoters do require enhancer proteins, of which E. coli has 11 (99). A mutant lacking the GlnG (NtrC) enhancer protein, reported to be required for polyP production upon nitrogen exhaustion (10), has no defect in polyP accumulation after nutrient limitation (11), but the others have not been tested yet. Future experiments will be needed to explore how the well-characterized inputs to the RpoN and RpoE regulons (99, 100, 123) contribute to polyP accumulation. Perhaps the most parsimonious model to explain the current data is that there is a signaling cascade leading to polyP synthesis in which an RpoN-dependent gene acts upstream of an RpoE/DksA-dependent gene, but without knowing which regulated genes directly affect polyP synthesis, it is difficult to make definitive conclusions about the nature of the regulatory network involved. Identifying this gene(s) is a major focus of my lab’s future directions.
It is clear from this and previous work that transcription of ppk itself does not increase upon polyP-stimulating stresses in E. coli (11, 35). In fact, while transcription initiation from the Pppk promoter stays fairly constant before and after stress (11, 35), I have now found that the levels of ppk and ppx mRNA transcripts drop precipitously after nutrient limitation (Fig. 3). The mechanism underlying this drop is currently unknown. It is reduced slightly by deletion of dksA, but the dksA and dksA greA mutants exhibit similar amounts of expression, so the regulation of ppk-ppx transcript elongation or mRNA stability cannot account for the differences in polyP accumulation in those mutants (Fig. 1A). Deletion of ppx does not eliminate stress regulation of polyP production (11, 12, 35). There must, therefore, be posttranscriptional regulation of PPK activity to account for increased polyP production. This, presumably, is what is being controlled by RpoN, RpoE, and DksA. The existence of ppk* mutations which change amino acids distant from the active site of PPK and result in massive overaccumulation of polyP in vivo (like PPKE245K) (Fig. 1F) (35) suggests that the PPK enzyme itself may be controlled by posttranslational or allosteric mechanisms, and work is ongoing in my lab to determine how PPK activity is modulated.
The idea that there must be an unknown factor or factors regulating PPK activity is not new. Kornberg hypothesized the existence of such a factor, which he called “X,” in 1998, to explain the various requirements for different regulators he observed under different stress conditions (10). My experiments have now added several new pieces of information to this old puzzle. The involvement of multiple stress-sensing systems helps to explain how so many different stress conditions can lead to polyP production and why different regulators may be required to respond to different stressors, but the focus now must be on identifying the mysterious factor “X” and understanding how it ties polyP regulation into the rest of the E. coli stress response machinery.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and molecular methods.
All strains and plasmids used in this study are listed in Table 1. I carried out DNA manipulations by standard methods (124, 125) in the E. coli cloning strain DH5α (Invitrogen). I grew E. coli at the indicated temperatures in lysogeny broth (LB) (126) containing 5 g · liter−1 NaCl and, where indicated, 1 mM MgCl2 (35). I added the following antibiotics when appropriate: ampicillin (100 μg · ml−1), chloramphenicol (17 or 34 μg · ml−1), erythromycin (10 μg · ml−1), kanamycin (25 or 50 μg · ml−1), rifampin (50 μg · ml−1), spectinomycin (50 μg · ml−1), or tetracycline (15 μg · ml−1).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Marker(s)b | Relevant genotypec | Source or referencea |
|---|---|---|---|
| E. coli strains | |||
| DH5α | F− λ− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 thi-1 gyrA96 relA1 | Invitrogen | |
| MC4100 ΔdnaK52 | Cmr Smr | F− λ− e14− [araD139]B/r Δ(argF-lac)169 flhD5301 Δ(fruK-yeiR)725(fruA25) relA1 rpsL150(Smr) rbsR22 Δ(fimB-fimE)632(::IS1) deoC1 ΔdnaK52::cat+ | 61, 130 |
| MT555 | Cmr | ΔmalE ΔhsdR ΔaraA::cat+ | J. Bardwell |
| MG1655 | F− λ− rph-1 ilvG− rfb-50 | 129 | |
| CAG18450 | Tcr | MG1655 zhf-50::Tn10(tet+) | 131, 132 |
| MJG0224 | MG1655 Δppk-749 | 12 | |
| MJG0344 | MG1655 ΔrpoS746 | 11 | |
| MJG0621 | MG1655 ΔaraA | 35 | |
| MJG0639 | Cmr | MG1655 ΔaraA::cat+ Δppk-749 | |
| MJG0640 | MG1655 ΔaraA Δppk-749 | ||
| MJG1205 | Rifr | MG1655 rpoB3449 (encoding RpoBΔAla532) | 11 |
| MJG1376 | Knr | MG1655 Δrsd-766::kan+ | |
| MJG1377 | Rifr | MG1655 rpoB3443 (encoding RpoBL533P) | |
| MJG1419 | Cmr | MG1655 ΔdksA1000::cat+ | 11 |
| MJG1448 | Tcr | MG1655 ΔaraA zhf-50::Tn10(tet+) sidB3 (encoding RpoH with 38 additional C-terminal amino acids) | |
| MJG1449 | Cmr | MG1655 ΔaraA ΔdnaK52::cat+ | |
| MJG1461 | Cmr | MG1655 ΔaraA Δppk-749 ΔdnaK52::cat+ | |
| MJG1559 | Knr | MG1655 ΔgreA788::kan+ | |
| MJG1561 | Cmr Knr | MG1655 ΔdksA1000::cat+ ΔgreA788::kan+ | |
| MJG1651 | Knr | MG1655 ΔglpA721::kan+ | |
| MJG1652 | Knr | MG1655 ΔglpD759::kan+ | |
| MJG1653 | Knr | MG1655 ΔglpF786::kan+ | |
| MJG1654 | Knr | MG1655 ΔglpT720::kan+ | |
| MJG1656 | Knr | MG1655 ΔdhaL789::kan+ | |
| MJG1666 | Cmr | MG1655 ΔfliF1000::cat+ | |
| MJG1698 | Knr | MG1655 ΔmdtE768::kan+ | |
| MJG1699 | Knr | MG1655 ΔglpG757::kan+ | |
| MJG1700 | Knr | MG1655 ΔglpE758::kan+ | |
| MJG1718 | Knr | MG1655 ΔgadE767::kan+ | |
| MJG1719 | Knr | MG1655 ΔflgB742::kan+ | |
| MJG1720 | Knr | MG1655 ΔflhD745::kan+ | |
| MJG1724 | Knr | MG1655 ΔfliA::kan+ | |
| MJG1730 | Knr | MG1655 ΔfliD770::kan+ | |
| MJG1750 | Cmr Knr | MG1655 ΔflhD745::kan+ ΔdksA1000::cat+ | |
| MJG1751 | Cmr Knr | MG1655 ΔfliA::kan+ ΔdksA1000::cat+ | |
| MJG1763 | Knr | MG1655 ΔrpoN730::kan+ | |
| MJG1765 | Tcr | MG1655 ΔaraA zhf-50::Tn10(tet+) | |
| MJG1766 | MG1655 ΔrpoN730 | ||
| MJG1767 | Emd Knr | MG1655 ΔrpoE1000::kan+ | |
| MJG1768 | Cmr | MG1655 ΔrpoS746 ΔdksA1000::cat+ | |
| MJG1769 | Cmr Tcr | MG1655 ΔaraA zhf-50::Tn10(tet+) ΔdksA1000::cat+ | |
| MJG1770 | Cmr Tcr | MG1655 ΔaraA zhf-50::Tn10(tet+) sidB3 ΔdksA1000::cat+ | |
| MJG1771 | Cmr | MG1655 ΔrpoN730 ΔdksA1000::cat+ | |
| Plasmids | |||
| pCas9-CR4 | Cmr | cas9+ cat+ | 137 |
| pKDsgRNA-ack | Spr | λ Red+ aadA+ CRISPR gRNA: ack | 137 |
| pKDsgRNA-sidB3 | Spr | λ Red+ aadA+ CRISPR gRNA: sidB3 | |
| pET-11a | Apr | bla+ | Novagen |
| pBAD18 | Apr | bla+ | 47 |
| pRLG13077 | Apr | bla+ | 48 |
| pRLG13078 | Apr | traR+ bla+ | 48 |
| pPPK7 | Apr | ppk+ bla+ | 12 |
| pPPK30 | Apr | ppkG733A (encoding PPKE245K) bla+ | |
| pDKSA1 | Apr | dksA+ bla+ | 11 |
| pDKSA2 | Apr | dksAC222A (encoding DksAD74E) bla+ | |
| pDKSA3 | Apr | dksAG220A C222T (encoding DksAD74N) bla+ | |
| pDKSA4 | Apr | dksAC271G G272C C273G (encoding DksAR91A) bla+ | |
| pDKSA5 | Apr | dksAC373A G374A C375A (encoding DksAR125K) bla+ | |
| pDKSA6 | Apr | dksAA263T C264T (encoding DksAN88I) bla+ | |
| pDKSA7 | Apr | dksAA292G A293C (encoding DksAK98A) bla+ | |
| pDKSA8 | Apr | dksAC115G A116C C117G (encoding DksAH39A) bla+ | |
| pDKSA9 | Apr | dksAG211A C213T G220A C222T (encoding DksAD71N D74N) bla+ | |
| pFLHDC1 | Apr | flhDC+ bla+ | |
| pFLIA1 | Apr | fliA+ bla+ | |
| pFLIZ1 | Apr | fliZ+ bla+ | |
| pRPOE1 | Apr | rpoE+ bla+ |
Unless otherwise indicated, all strains and plasmids were generated in the course of this work.
Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Emd, erythromycin dependence; Knr, kanamycin resistance; Rifr, rifampin resistance; Smr, streptomycin resistance; Spr, spectinomycin resistance; Tcr, tetracycline resistance.
gRNA, guide RNA.
Databases and primer design.
I obtained gene and protein sequences and metabolic pathway information from the Integrated Microbial Genomes database (127) and from EcoCyc (73) and obtained information about E. coli regulatory networks from RegulonDB (128). I designed PCR and sequencing primers with Web Primer (www.candidagenome.org/cgi-bin/compute/web-primer) and mutagenic primers with PrimerX (www.bioinformatics.org/primerx/index.htm). I designed all primers used for qPCR with Primer Quest (www.idtdna.com; parameter set “qPCR 2 primers intercalating dyes” for qRT-PCR primer design) and tested each primer pair to confirm specificity and amplification efficiencies of close to 1. These primers are listed in Table 2.
TABLE 2.
Primers used for quantitative RT-PCR
| Gene | Primer sequence |
|
|---|---|---|
| Forward | Reverse | |
| yqfB | GACGAGTCTGAATCGCACTT | TGTGTCTGACCGGGATAGAT |
| ppkupstream | ACTCTCATTCCCGCCATTTAC | CGTAATGTGCTGACGCAGATA |
| ppkdownstream | CCGCCGCCTATTGTATGAA | GCACGAATGTTGTCGCTAATG |
| ppx | CAGCTTTGCCCAGCTTTATTT | CGCCCATTTCCATTAACACTTC |
| flhD | CTACTTGCACAGCGTTTGATTG | GGAATCTTGCGTCAACTGAGTA |
| fliA | CCCTACAAGGAACGGCATTTA | TCGGCAATATCGATCCCTAAAC |
| flgB | GACCTCAACGCAACACATTC | ACGTTCATCATGCCTTTGATTT |
| fliF | GCCATTGTGCATCTGGTTTC | GCGACAGGATCGCTTCAATA |
| fliD | CGCAAACGGCGATTAAAGAC | GCTGGAAGAACTGACGGTATTA |
| glpA | CATGCTGGATGCCAAAGAAC | TGCGCAGATCGGCATATT |
| glpD | GGATTGTGGAAGCGGAAGAT | GTAGGCTTGCTTCTGGGTATG |
| glpF | GTTGCGTTGCAGCACTAAA | CGGCAACTTGTGAAACGATAAA |
| glpT | GCGCTTGCTATGCCTTATCT | CATGGCACAAAGCCCATAAAC |
| dhaL | GCTGACCACGGGCTAAATA | TCTTCCAGTGTCAGGCTTTG |
| glpE | GATATTCGCGATCCACAGAGTT | CCGTCAATGCTATAGACCACAT |
| gadE | AATCAATTCCCTGTCAGAGATCA | TTGATACTTTCTTTGCGGCTAAC |
Strain construction.
Unless otherwise indicated, all E. coli strains were derivatives of wild-type strain MG1655 (F− rph-1 ilvG− rfb-50) (129). I confirmed all chromosomal mutations by PCR. Strain MC4100 ΔdnaK52 {F− λ− e14− [araD139]B/r Δ(argF-lac)169 flhD5301 Δ(fruK-yeiR)725(fruA25) relA1 rpsL150(Smr) rbsR22 Δ(fimB-fimE)632(::IS1) deoC1 ΔdnaK52::cat+} (61, 130) was a gift from Ursula Jakob (University of Michigan), and strain CAG18450 [zhf-50::Tn10(tet+)] (131, 132) was a gift from Chuck Turnbough (University of Alabama at Birmingham).
I used P1vir transduction (11, 133) to move the ΔgreA788::kan+ allele from the Keio collection (46) into MG1655 and MJG1419 (ΔdksA1000::cat+) (11), generating strains MJG1559 (ΔgreA788::kan+) and MJG1561 (ΔdksA1000::cat+ ΔgreA788::kan+).
I generated strain MJG0639 (ΔaraA::cat+ Δppk-749) by P1vir transduction of the ΔaraA::cat+ allele from strain MT555 (ΔmalE ΔhsdR ΔaraA::cat+) (a gift from James Bardwell, University of Michigan) into MJG0224 (Δppk-749) (12). I then resolved the chloramphenicol resistance cassette in this strain (111), yielding strain MJG0640 (ΔaraA Δppk-749). I used P1vir transduction to move the ΔdnaK52::cat+ allele from strain MC4100 ΔdnaK52::cat+ into strains MJG0621 (ΔaraA) (35) and MJG0640 (ΔaraA Δppk-749), yielding strains MJG1449 (ΔaraA ΔdnaK52::cat+) and MJG1461 (ΔaraA Δppk-749 ΔdnaK52::cat+). Unless otherwise indicated, I grew all ΔdnaK52::cat+ strains at 30°C.
I moved the ΔglpA721::kan+, ΔglpD759::kan+, ΔglpF786::kan+, ΔglpT720::kan+, ΔmdtE768::kan+, ΔglpG757::kan+, ΔglpE758::kan+, ΔgadE767::kan+, ΔflgB742::kan+, ΔflhD745::kan+, ΔfliA::kan+ (no allele number provided in the Coli Genetic Stock Center database; http://cgsc.biology.yale.edu), and ΔfliD770::kan+ alleles from the Keio collection by P1vir transduction into MG1655, generating strains MJG1651 (ΔglpA721::kan+), MJG1652 (ΔglpD759::kan+), MJG1653 (ΔglpF786::kan+), MJG1654 (ΔglpT720::kan+), MJG1698 (ΔmdtE768::kan+), MJG1699 (ΔglpG757::kan+), MJG1700 (ΔglpE758::kan+), MJG1718 (ΔgadE767::kan+), MJG1719 (ΔflgB742::kan+), MJG1720 (ΔflhD745::kan+), MJG1724 (ΔfliA::kan+), and MJG1730 (ΔfliD770::kan+). I used P1vir transduction to move the ΔdksA1000::cat+ allele from MJG1419 into MJG1720 (ΔflhD745::kan+) and MJG1724 (ΔfliA::kan+), yielding strains MJG1750 (ΔflhD745::kan+ ΔdksA1000::cat+) and MJG1751 (ΔfliA::kan+ ΔdksA1000::cat+).
I replaced the fliF gene of strain MG1655 with a pKD3-derived chloramphenicol resistance cassette by recombineering (111), using primers 5′-GTT CCA CTT TGC CAA TAA CGC CGT CCA TAA TCA GCC ACG AGG TGC GCG ATG GTG TAG GCT GGA GCT GCT TC-3′ and 5′-TCG CCA ATG GTC ATC AGC AGG ATG ACG CTT TTA TCG GTG CCT GTC AGG TTA CAT ATG AAT ATC CTC CTT AG-3′ and yielding strain MJG1666 (ΔfliF1000::cat+). I amplified the ΔdhaL789::kan+ allele from the Keio collection with primers 5′-TTC TCA ATC ACC TTA CTG-3′ and 5′-CAA CTA TCA CTC ATT AAC-3′ and used the resulting product as a recombineering substrate in MG1655, yielding strain MJG1656 (ΔdhaL789::kan+).
I used oligonucleotide-directed recombineering (134) to construct a chromosomal rpoB3443 mutation (84–86, 135, 136) using the mutagenic primer 5′-AAG CCT GCA CGT TCA CGG GTC AGA CCG CCG GGA CCG GGG GCA GAG ATA CGA CGT TTG TGC GTA ATC TCA GAC A-3′, which contained four 5′ phosphorothioate linkages. This primer generates an rpoBC1593T, A1596C, T1598C, C1602T, A1605C allele, encoding RpoBL533P, with silent mutations in four codons adjacent to codon 533 to avoid mismatch repair. I transformed MG1655 with pKD46 (111), induced expression of λ Red recombinase, electroporated with 250 pmol of mutagenic primer, and selected recombinant colonies at 37°C on LB plates containing rifampin. I confirmed the sequence of rpoB alleles by PCR amplification and Sanger sequencing of a fragment of rpoB with primers 5′-GAT GTT ATG AAA AAG CTC-3′ and 5′-CTG GGT GGA TAC GTC CAT-3′. After curing pKD46 by growth at 37°C, this yielded strain MJG1377 (rpoB3443).
I used P1vir transduction to move the Δrsd-766::kan+ and ΔrpoN730::kan+ alleles from the Keio collection into MG1655, generating strains MJG1376 (Δrsd-766::kan+) and MJG1763 (ΔrpoN730::kan+), then resolved the kanamycin resistance cassette in MJG1763 (111), yielding strain MJG1766 (ΔrpoN730). I used P1vir transduction to move the ΔdksA1000::cat+ allele from MJG1419 into MJG0344 (ΔrpoS746) (11) and MJG1766 (ΔrpoN730), yielding strains MJG1768 (ΔrpoS746 ΔdksA1000::cat+) and MJG1771 (ΔrpoN730 ΔdksA1000::cat+).
I used Cas9-assisted recombineering (137) to construct a chromosomal sidB3 (61) mutation of strain CAG18450 [zhf-50::Tn10(tet+)] (131, 132) using the mutagenic primer 5′-ACT CTC ATC CAG GGT TCT CTG CTT AAT AGC GGG AAT TCG GCC TCG ATG GCA GCA CGC AAT TTT TTC ATC GCG T-3′, which contained four 5′ phosphorothioate linkages. This primer mutates the TAA stop codon of rpoH to a GAA glutamate codon (rpoHT853G) and makes silent mutations in 4 flanking codons to avoid mismatch repair. I confirmed the sequence of rpoH alleles by PCR amplification and Sanger sequencing of a fragment of rpoH and its 3′ downstream sequence with primers 5′-TGT CTT CCG ACG ACG ATT-3′ and 5′-GTA ACG CTT TAC CCT TTA-3′. Due to the temperature sensitivity of a sidB3 mutant (61), it was not possible to cure the pKDsgRNA-sidB3 protospacer targeting plasmid (see below) by growth at 42°C (137), so I used P1vir transduction to move sidB3 and the 60% linked zhf-50 tetracycline resistance marker (transposon Tn10 in the intergenic region between ugpB and ilvF) (131, 132) from the resulting strain into MJG0621 (ΔaraA) to generate strain MJG1448 [ΔaraA zhf-50::Tn10(tet+) sidB3]. I used P1vir transduction to move zhf-50 from CAG18450 to MJG0621 (ΔaraA) to generate strain MJG1765 [ΔaraA zhf-50::Tn10(tet+)]. I also used P1vir transduction to move the dksA1000::cat+ allele from MJG1419 into MJG1765 [ΔaraA zhf-50::Tn10(tet+)] and MJG1448 [ΔaraA zhf-50::Tn10(tet+) sidB3], yielding strains MJG1769 [ΔaraA zhf-50::Tn10(tet+) ΔdksA1000::cat+] and MJG1770 [ΔaraA zhf-50::Tn10(tet+) sidB3 ΔdksA1000::cat+]. I maintained all sidB3 strains at 30°C.
I replaced the rpoE gene of strain MG1655 with a pKD4-derived kanamycin resistance cassette by recombineering (111), using primers 5′-GCG TTT CGA TAG CGC GTG GAA ATT TGG TTT GGG GAG ACT TTA CCT CGG ATG GTG TAG GCT GGA GCT GCT TC-3′ and 5′-GTT GTT CTT TCT GCA TGC CTA ATA CCC TTA TCC AGT ATC CCG CTA TCG TCA CAT ATG AAT ATC CTC CTT AG-3′. This resulted in strain MJG1767 (ΔrpoE1000::kan+). I made multiple attempts to use P1vir transduction to move the ΔdksA1000::cat+ allele from MJG1419 into MJG1767 (ΔrpoE1000::kan+), without success. I constructed, maintained, and tested all rpoE mutant strains in medium containing 10 μg · ml−1 erythromycin (106).
Plasmid construction.
Plasmids pRLG13077 and pRLG13078 (48) were a gift from Richard Gourse and Saumya Gopalkrishnan (University of Wisconsin—Madison). pCas9-CR4 and pKDsgRNA-ack were gifts from Kristala Prather (Addgene plasmid no. 62655; http://n2t.net/addgene:62655; RRID Addgene_62655 and Addgene plasmid no. 62654; http://n2t.net/addgene:62654; RRID Addgene_62654) (137).
I replaced the single guide RNA (sgRNA) targeting sequence of plasmid pKDsgRNA-ack by round-the-horn cloning (137) with primer 5′-TAG CGG AAA TTA CGC TTC AAG TTT TAG AGC TAG AAA TAG CAA G-3′, yielding plasmid pKDsgRNA-sidB3.
I used the QuikChange site-directed mutagenesis method (Agilent Technologies), modified to use a single primer and 35 cycles of amplification, to mutate the pBAD18-derived (47) ppk+ plasmid pPPK7 (138) with primer 5′-GAA GCC AGC CTG ATG AAG TTG ATG TCT TCC-3′ to generate plasmid pPPK30, containing a ppkG733A allele encoding PPKE245K. I used the same procedure to mutate the pBAD18-derived dksA+ plasmid pDKSA1 (11) as follows: I used primer 5′-CAA CTT CCC GGA CCC GGT AGA ACG TGC AGC CCA GGA AGA AG-3′ to generate plasmid pDKSA2 (dksAC222A, encoding DksAD74E), I used primer 5′-CAA CTT CCC GGA CCC GGT AAA TCG TGC AGC CCA GGA AGA AG-3′ to generate plasmid pDKSA3 (dksAG220A C222T, encoding DksAD74N), I used primer 5′-CTC GAA CTG CGT AAC CGC GAT GCG GAG CGT AAG CTG ATC AAA AAG-3′ to generate plasmid pDKSA4 (dksAC271G G272C C273G, encoding DksAR91A), I used primer 5′-GTT GAA ATT GGT ATT CGC AAA CTG GAA GCG CGC CCG AC-3′ to generate plasmid pDKSA5 (dksAC373A G374A C375A, encoding DksAR125K), I used primer 5′-CAG CCT CGA ACT GCG TAT TCG CGA TCG CGA GCG TAA G-3′ to generate plasmid pDKSA6 (dksAA263T C264T, encoding DksAN88I), I used primer 5′-CGT AAG CTG ATC AAA GCG ATC GAG AAG ACG C-3′ to generate plasmid pDKSA7 (dksAA292G A293C, encoding DksAK98A), and I used primer 5′-GAA TGA AGC CCA GCT GGC GGC GTT CCG TCG TAT TCT GGA AG 3′ to generate plasmid pDKSA8 (dksAC115G A116C C117G, encoding DksAH39A). Finally, I mutated plasmid pDKSA3 with primer 5′ GCA GCC AAC TTC CCG AAT CCG GTA AAT CGT GCA G-3′ to generate plasmid pDKSA9 (dksAG211A C213T G220A C222T, encoding DksAD71N D74N).
I amplified the flhDC coding sequence (932 bp) from E. coli MG1655 genomic DNA with primers 5′-TTC GAA TTC AAG GAG ATA TAC ATA TGC ATA CCT CCG AGT TGC TGA AA-3′ and 5′-CTT AAG CTT TTA AAC AGC CTG TAC TCT CTG TTC A-3′, incorporating an AAG GAG ATA TAC AT ribosome binding site (RBS) sequence derived from pET-11a (Novagen), and cloned it into the EcoRI and HindIII sites of plasmid pBAD18 (47) to generate plasmid pFLHDC1. Similarly, I amplified the fliA and fliZ coding sequences (720 and 552 bp, respectively) with primers 5′-ACC GGT ACC AAG GAG ATA TAC ATA TGA ATT CAC TCT ATA CCG CTG AAG G-3′ and 5′-CTT AAG CTT TTA TAA CTT ACC CAG TTT AGT GCG T-3′ or 5′-TTC GAA TTC AAG GAG ATA TAC ATA TGA TGG TGC AGC ACC TGA AAA-3′ and 5′-CTT AAG CTT TTA ATA TAT ATC AGA AGA AGG CAG GCT GG-3′, generating products with the above RBS and, in the case of fliA, changing the native GTG start codon to an ATG. I cloned these products into the KpnI and HindIII or EcoRI and HindIII sites of pBAD18 to generate plasmids pFLIA1 and pFLIZ1. I amplified rpoE (576 bp) from E. coli MG1655 genomic DNA with primer pair 5′-TTT GAA TTC AAG GAG ATA TAC ATA TGA GCG AGC AGT TAA CGG A-3′ and 5′-AGA TCT AGA TCA ACG CCT GAT AAG CGG TT-3′, incorporating the pET-11a RBS, and cloned the resulting product into the EcoRI and XbaI sites of plasmid pBAD18 (47) to generate plasmid pRPOE1.
In vivo polyphosphate assay.
To induce polyP synthesis by nutrient limitation (10, 11, 35), I grew E. coli strains in 10 ml LB at 30°C or 37°C with shaking (200 rpm) to an A600 of 0.2 to 0.4. I harvested samples (1 ml; 50 to 100 μg of total protein) for polyP measurements as described below, then harvested 5 ml of each culture by centrifugation (5 min at 4,696 × g at room temperature), resuspended them in 5 ml phosphate-buffered saline (PBS) to rinse, and recentrifuged and resuspended them in 5 ml MOPS minimal medium (Teknova) (139) containing 4 g · liter−1 glucose, 0.1 mM K2HPO4, and 0.1 mM uracil. I incubated these cultures for 2 h at 30°C or 37°C with shaking and then collected additional samples for polyP measurements. Where indicated, I added arabinose (0.125 or 2 g · liter−1), isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM), or erythromycin (10 μg · liter−1) to both the LB and the MOPS media.
I measured intracellular polyP levels as previously described (105). Briefly, I harvested samples of bacterial cultures by centrifugation, resuspended them in 250 μl of 4 M guanidine isothiocyanate and 50 mM Tris-HCl (pH 7), lysed them by incubation for 10 min at 95°C, and immediately froze them at −80°C. After thawing at room temperature, I determined protein concentrations by Bradford assay (Bio-Rad) of 5-μl aliquots of each sample. I added 250 μl of 95% ethanol to the remaining sample and applied the resulting mixture to an EconoSpin silica spin column (Epoch Life Science), rinsed with 750 μl 5 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5 mM EDTA, 50% ethanol, and eluted with 150 μl 50 mM Tris-HCl (pH 8). I brought the eluate to 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 50 mM ammonium acetate with 1 μg of PPX1 exopolyphosphatase from Saccharomyces cerevisiae (140) in a final volume of 200 μl and incubated for 15 min at 37°C, then measured the resulting polyP-derived orthophosphate using a colorimetric assay (141) and normalized to total protein content. For all figures, I reported polyP concentrations in terms of individual phosphate monomers.
Quantitative RT-PCR.
I stressed E. coli strains by nutrient limitation as described above. At the indicated time points, I harvested 1 ml of cells by centrifugation and resuspended in RNAlater (Thermo Fisher) for storage at −20°C. I extracted RNA using the RiboPure RNA purification kit for bacteria (Ambion) following the manufacturer’s instructions, including the optional DNase treatment to remove contaminating genomic DNA. I used the SuperScript IV Vilo kit (Thermo Fisher) to reverse transcribe cDNA from mRNA, following the manufacturer’s instructions, including a control without reverse transcriptase for each reaction. I calculated changes in gene expression using the threshold cycle (2−ΔΔCT) method (142) with yqfB as an internal expression control. (Both nutrient limitation and mutation of dksA were expected to change the expression of rRNA genes [5], which are common housekeeping genes used to normalize qRT-PCR data in bacteria [143]. However, expression of cysG, hcaT, and idnT, other validated reference genes [144], was substantially impacted by dksA and/or greA mutations or by nutrient limitation stress [see Data Set S1 in the supplemental material]. To address this problem, I used yqfB instead, the expression of which did not change under the conditions tested here [Data Set S1].).
RNA sequencing.
I grew the indicated E. coli strains as described above for induction of polyP synthesis by nutrient limitation, and purified RNA from 2 ml of cells harvested immediately before and 5 min after stress using the RiboPure-Bacteria kit (Ambion) according to the manufacturer’s instructions. mRNA sequencing was performed by the UAB Heflin Center for Genomic Sciences, using an Illumina NextSeq 500 as described by the manufacturer (Illumina, Inc.). Briefly, the quality of the total RNA was assessed using an Agilent 2100 Bioanalyzer, and RNA with a RNA integrity number (RIN) of 7.0 or above was used for sequencing library preparation. The SureSelect strand-specific mRNA library kit was used according to the manufacturer’s instructions (Agilent). Library construction began with ribosome reduction with the RiboMinus protocol for Gram-negative and Gram-positive bacteria, as described by the manufacturer (Life Technologies). The resulting mRNA was randomly fragmented with cations and heat, followed by first-strand synthesis using random primers with inclusion of actinomycin D (2.4 ng · μl−1 final concentration). Second-strand cDNA production was done with standard techniques, the ends of the resulting cDNA were made blunt and A-tailed, and adaptors were ligated for amplification and indexing to allow for multiplexing during sequencing. The cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for Illumina library quantitation prior to cluster generation. Cluster generation was performed according to the manufacturer’s recommendations for onboard clustering (Illumina). Approximately 20 million single-end 75-bp reads were generated per sample, and I analyzed the resulting data using the Rockhopper 2 software package (145) and Morpheus (https://software.broadinstitute.org/morpheus).
Statistical analyses.
I used Prism version 8.3.1 for Macintosh (GraphPad Software) to perform statistical analyses, including two-way repeated-measures analysis of variance (ANOVA) with Holm-Sidak multiple-comparison tests. Repeated-measures ANOVA cannot handle missing values, so data sets with samples having different n numbers (e.g., Fig. 2) were analyzed with a mixed model that uses a compound symmetry covariance matrix and is fitted using restricted maximum likelihood (REML) (without Geisser-Greenhouse correction).
Data availability.
All strains and plasmids generated in the course of this work are available from the author upon request. RNA sequencing data have been deposited in NCBI’s Gene Expression Omnibus (146) and are accessible through GEO series accession number GSE144816.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by University of Alabama at Birmingham Department of Microbiology startup funds and NIH grant R35GM124590.
I have no conflicts of interest to declare.
I thank Avishek Mitra (UAB Department of Microbiology) and Saumya Gopalkrishnan, Rick Gourse, and Wilma Ross (University of Wisconsin—Madison) for helpful discussions of the data presented here.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Gottesman S. 2017. Stress reduction, bacterial style. J Bacteriol 199:e00433-17. doi: 10.1128/JB.00433-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fang FC, Frawley ER, Tapscott T, Vazquez-Torres A. 2016. Bacterial stress responses during host infection. Cell Host Microbe 20:133–143. doi: 10.1016/j.chom.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battesti A, Majdalani N, Gottesman S. 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65:189–213. doi: 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottesman S. 2019. Trouble is coming: signaling pathways that regulate general stress responses in bacteria. J Biol Chem 294:11685–11700. doi: 10.1074/jbc.REV119.005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gourse RL, Chen AY, Gopalkrishnan S, Sanchez-Vazquez P, Myers A, Ross W. 2018. Transcriptional responses to ppGpp and DksA. Annu Rev Microbiol 72:163–184. doi: 10.1146/annurev-micro-090817-062444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang B, Dai P, Ding D, Del Rosario A, Grant RA, Pentelute BL, Laub MT. 2019. Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nat Chem Biol 15:141–150. doi: 10.1038/s41589-018-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potrykus K, Murphy H, Philippe N, Cashel M. 2011. ppGpp is the major source of growth rate control in E. coli. Environ Microbiol 13:563–575. doi: 10.1111/j.1462-2920.2010.02357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jimenez J, Bru S, Ribeiro MP, Clotet J. 2017. Polyphosphate: popping up from oblivion. Curr Genet 63:15–18. doi: 10.1007/s00294-016-0611-5. [DOI] [PubMed] [Google Scholar]
- 9.Rao NN, Gómez-García MR, Kornberg A. 2009. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem 78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- 10.Ault-Riche D, Fraley CD, Tzeng CM, Kornberg A. 1998. Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol 180:1841–1847. doi: 10.1128/JB.180.7.1841-1847.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray MJ. 2019. Inorganic polyphosphate accumulation in Escherichia coli is regulated by DksA but not by (p)ppGpp. J Bacteriol 201:e00664-18. doi: 10.1128/JB.00664-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JC, Jakob U. 2014. Polyphosphate is a primordial chaperone. Mol Cell 53:689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao NN, Liu S, Kornberg A. 1998. Inorganic polyphosphate in Escherichia coli: the phosphate regulon and the stringent response. J Bacteriol 180:2186–2193. doi: 10.1128/JB.180.8.2186-2193.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray MJ, Jakob U. 2015. Oxidative stress protection by polyphosphate—new roles for an old player. Curr Opin Microbiol 24:1–6. doi: 10.1016/j.mib.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusano S, Ishihama A. 1997. Functional interaction of Escherichia coli RNA polymerase with inorganic polyphosphate. Genes Cells 2:433–441. doi: 10.1046/j.1365-2443.1997.13203301320330.x. [DOI] [PubMed] [Google Scholar]
- 16.McInerney P, Mizutani T, Shiba T. 2006. Inorganic polyphosphate interacts with ribosomes and promotes translation fidelity in vitro and in vivo. Mol Microbiol 60:438–447. doi: 10.1111/j.1365-2958.2006.05103.x. [DOI] [PubMed] [Google Scholar]
- 17.Shiba T, Tsutsumi K, Yano H, Ihara Y, Kameda A, Tanaka K, Takahashi H, Munekata M, Rao NN, Kornberg A. 1997. Inorganic polyphosphate and the induction of rpoS expression. Proc Natl Acad Sci U S A 94:11210–11215. doi: 10.1073/pnas.94.21.11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stumpf JD, Foster PL. 2005. Polyphosphate kinase regulates error-prone replication by DNA polymerase IV in Escherichia coli. Mol Microbiol 57:751–761. doi: 10.1111/j.1365-2958.2005.04724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grillo-Puertas M, Schurig-Briccio LA, Rodríguez-Montelongo L, Rintoul MR, Rapisarda VA. 2014. Copper tolerance mediated by polyphosphate degradation and low-affinity inorganic phosphate transport system in Escherichia coli. BMC Microbiol 14:72. doi: 10.1186/1471-2180-14-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varas M, Valdivieso C, Mauriaca C, Ortíz-Severín J, Paradela A, Poblete-Castro I, Cabrera R, Chávez FP. 2017. Multi-level evaluation of Escherichia coli polyphosphate related mutants using global transcriptomic, proteomic and phenomic analyses. Biochim Biophys Acta Gen Subj 1861:871–883. doi: 10.1016/j.bbagen.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Gross MH, Konieczny I. 2020. Polyphosphate induces the proteolysis of ADP-bound fraction of initiator to inhibit DNA replication initiation upon stress in Escherichia coli. Nucleic Acids Res doi: 10.1093/nar/gkaa217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cha SB, Rayamajhi N, Lee WJ, Shin MK, Jung MH, Shin SW, Kim JW, Yoo HS. 2012. Generation and envelope protein analysis of internalization defective Brucella abortus mutants in professional phagocytes, RAW 264.7. FEMS Immunol Med Microbiol 64:244–254. doi: 10.1111/j.1574-695X.2011.00896.x. [DOI] [PubMed] [Google Scholar]
- 23.Candon HL, Allan BJ, Fraley CD, Gaynor EC. 2007. Polyphosphate kinase 1 is a pathogenesis determinant in Campylobacter jejuni. J Bacteriol 189:8099–8108. doi: 10.1128/JB.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards MI, Michell SL, Oyston PC. 2008. An intracellularly inducible gene involved in virulence and polyphosphate production in Francisella. J Med Microbiol 57:1183–1192. doi: 10.1099/jmm.0.2008/001826-0. [DOI] [PubMed] [Google Scholar]
- 25.Ayraud S, Janvier B, Labigne A, Ecobichon C, Burucoa C, Fauchere JL. 2005. Polyphosphate kinase: a new colonization factor of Helicobacter pylori. FEMS Microbiol Lett 243:45–50. doi: 10.1016/j.femsle.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 26.Singh R, Singh M, Arora G, Kumar S, Tiwari P, Kidwai S. 2013. Polyphosphate deficiency in Mycobacterium tuberculosis is associated with enhanced drug susceptibility and impaired growth in guinea pigs. J Bacteriol 195:2839–2851. doi: 10.1128/JB.00038-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rashid MH, Rumbaugh K, Passador L, Davies DG, Hamood AN, Iglewski BH, Kornberg A. 2000. Polyphosphate kinase is essential for biofilm development, quorum sensing, and virulence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97:9636–9641. doi: 10.1073/pnas.170283397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim KS, Rao NN, Fraley CD, Kornberg A. 2002. Inorganic polyphosphate is essential for long-term survival and virulence factors in Shigella and Salmonella spp. Proc Natl Acad Sci U S A 99:7675–7680. doi: 10.1073/pnas.112210499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavez FP, Lagos CF, Reyes-Parada M, Guiliani N, Jerez CA. 2011. Polyphosphate synthesis as a target for novel antibiotics. Cei 7:163–168. doi: 10.2174/157340811798807605. [DOI] [Google Scholar]
- 30.Saha SB, Verma V. 2013. In silico analysis of Escherichia coli polyphosphate kinase (PPK) as a novel antimicrobial drug target and its high throughput virtual screening against PubChem library. Bioinformation 9:518–523. doi: 10.6026/97320630009518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dahl JU, Gray MJ, Bazopoulou D, Beaufay F, Lempart J, Koenigsknecht MJ, Wang Y, Baker JR, Hasler WL, Young VB, Sun D, Jakob U. 2017. The anti-inflammatory drug mesalamine targets bacterial polyphosphate accumulation. Nat Microbiol 2:16267. doi: 10.1038/nmicrobiol.2016.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morohoshi T, Maruo T, Shirai Y, Kato J, Ikeda T, Takiguchi N, Ohtake H, Kuroda A. 2002. Accumulation of inorganic polyphosphate in phoU mutants of Escherichia coli and Synechocystis sp. strain PCC6803. Appl Environ Microbiol 68:4107–4110. doi: 10.1128/aem.68.8.4107-4110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirota R, Motomura K, Nakai S, Handa T, Ikeda T, Kuroda A. 2013. Stable polyphosphate accumulation by a pseudo-revertant of an Escherichia coli phoU mutant. Biotechnol Lett 35:695–701. doi: 10.1007/s10529-012-1133-y. [DOI] [PubMed] [Google Scholar]
- 34.Kuroda A, Murphy H, Cashel M, Kornberg A. 1997. Guanosine tetra- and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J Biol Chem 272:21240–21243. doi: 10.1074/jbc.272.34.21240. [DOI] [PubMed] [Google Scholar]
- 35.Rudat AK, Pokhrel A, Green TJ, Gray MJ. 2018. Mutations in Escherichia coli polyphosphate kinase that lead to dramatically increased in vivo polyphosphate levels. J Bacteriol 200:e00697-17. doi: 10.1128/JB.00697-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rutherford ST, Lemke JJ, Vrentas CE, Gaal T, Ross W, Gourse RL. 2007. Effects of DksA, GreA, and GreB on transcription initiation: insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. J Mol Biol 366:1243–1257. doi: 10.1016/j.jmb.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marr MT, Roberts JW. 2000. Function of transcription cleavage factors GreA and GreB at a regulatory pause site. Mol Cell 6:1275–1285. doi: 10.1016/S1097-2765(00)00126-X. [DOI] [PubMed] [Google Scholar]
- 38.Potrykus K, Vinella D, Murphy H, Szalewska-Palasz A, D’Ari R, Cashel M. 2006. Antagonistic regulation of Escherichia coli ribosomal RNA rrnB P1 promoter activity by GreA and DksA. J Biol Chem 281:15238–15248. doi: 10.1074/jbc.M601531200. [DOI] [PubMed] [Google Scholar]
- 39.Vinella D, Potrykus K, Murphy H, Cashel M. 2012. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J Bacteriol 194:261–273. doi: 10.1128/JB.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Traverse CC, Ochman H. 2018. A genome-wide assay specifies only GreA as a transcription fidelity factor in Escherichia coli. G3 (Bethesda) 8:2257–2264. doi: 10.1534/g3.118.200209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aberg A, Fernández-Vázquez J, Cabrer-Panes JD, Sánchez A, Balsalobre C. 2009. Similar and divergent effects of ppGpp and DksA deficiencies on transcription in Escherichia coli. J Bacteriol 191:3226–3236. doi: 10.1128/JB.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aberg A, Shingler V, Balsalobre C. 2008. Regulation of the fimB promoter: a case of differential regulation by ppGpp and DksA in vivo. Mol Microbiol 67:1223–1241. doi: 10.1111/j.1365-2958.2008.06115.x. [DOI] [PubMed] [Google Scholar]
- 43.Gagarinova A, Stewart G, Samanfar B, Phanse S, White CA, Aoki H, Deineko V, Beloglazova N, Yakunin AF, Golshani A, Brown ED, Babu M, Emili A. 2016. Systematic genetic screens reveal the dynamic global functional organization of the bacterial translation machinery. Cell Rep 17:904–916. doi: 10.1016/j.celrep.2016.09.040. [DOI] [PubMed] [Google Scholar]
- 44.Dylewski M, Ćwiklińska M, Potrykus K. 2018. A search for the in trans role of GraL, an Escherichia coli small RNA. Acta Biochim Pol 65:141–149. doi: 10.18388/abp.2018_2562. [DOI] [PubMed] [Google Scholar]
- 45.Potrykus K, Murphy H, Chen X, Epstein JA, Cashel M. 2010. Imprecise transcription termination within Escherichia coli greA leader gives rise to an array of short transcripts, GraL. Nucleic Acids Res 38:1636–1651. doi: 10.1093/nar/gkp1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gopalkrishnan S, Ross W, Chen AY, Gourse RL. 2017. TraR directly regulates transcription initiation by mimicking the combined effects of the global regulators DksA and ppGpp. Proc Natl Acad Sci U S A 114:E5539–E5548. doi: 10.1073/pnas.1704105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blankschien MD, Potrykus K, Grace E, Choudhary A, Vinella D, Cashel M, Herman C. 2009. TraR, a homolog of a RNAP secondary channel interactor, modulates transcription. PLoS Genet 5:e1000345. doi: 10.1371/journal.pgen.1000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, Herman C, Wang JD. 2010. The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell 141:595–605. doi: 10.1016/j.cell.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Mooney RA, Grass JA, Sivaramakrishnan P, Herman C, Landick R, Wang JD. 2014. DksA guards elongating RNA polymerase against ribosome-stalling-induced arrest. Mol Cell 53:766–778. doi: 10.1016/j.molcel.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furman R, Sevostyanova A, Artsimovitch I. 2012. Transcription initiation factor DksA has diverse effects on RNA chain elongation. Nucleic Acids Res 40:3392–3402. doi: 10.1093/nar/gkr1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JS, Liu L, Fitzsimmons LF, Wang Y, Crawford MA, Mastrogiovanni M, Trujillo M, Till JKA, Radi R, Dai S, Vazquez-Torres A. 2018. DksA-DnaJ redox interactions provide a signal for the activation of bacterial RNA polymerase. Proc Natl Acad Sci U S A 115:E11780–E11789. doi: 10.1073/pnas.1813572115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myka KK, Kusters K, Washburn R, Gottesman ME. 2019. DksA-RNA polymerase interactions support new origin formation and DNA repair in Escherichia coli. Mol Microbiol 111:1382–1397. doi: 10.1111/mmi.14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satory D, Gordon AJ, Wang M, Halliday JA, Golding I, Herman C. 2015. DksA involvement in transcription fidelity buffers stochastic epigenetic change. Nucleic Acids Res 43:10190–10199. doi: 10.1093/nar/gkv839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang PJ, Craig EA. 1990. Identification and characterization of a new Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J Bacteriol 172:2055–2064. doi: 10.1128/jb.172.4.2055-2064.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Genest O, Wickner S, Doyle SM. 2019. Hsp90 and Hsp70 chaperones: collaborators in protein remodeling. J Biol Chem 294:2109–2120. doi: 10.1074/jbc.REV118.002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chandrangsu P, Wang L, Choi SH, Gourse RL. 2012. Suppression of a dnaKJ deletion by multicopy dksA results from non-feedback-regulated transcripts that originate upstream of the major dksA promoter. J Bacteriol 194:1437–1446. doi: 10.1128/JB.06726-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoo NG, Dogra S, Meinen BA, Tse E, Haefliger J, Southworth DR, Gray MJ, Dahl JU, Jakob U. 2018. Polyphosphate stabilizes protein unfolding intermediates as soluble amyloid-like oligomers. J Mol Biol 430:4195–4208. doi: 10.1016/j.jmb.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crooke E, Akiyama M, Rao NN, Kornberg A. 1994. Genetically altered levels of inorganic polyphosphate in Escherichia coli. J Biol Chem 269:6290–6295. [PubMed] [Google Scholar]
- 61.Bukau B, Walker GC. 1990. Mutations altering heat shock specific subunit of RNA polymerase suppress major cellular defects of E. coli mutants lacking the DnaK chaperone. EMBO J 9:4027–4036. doi: 10.1002/j.1460-2075.1990.tb07624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JH, Lennon CW, Ross W, Gourse RL. 2012. Role of the coiled-coil tip of Escherichia coli DksA in promoter control. J Mol Biol 416:503–517. doi: 10.1016/j.jmb.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blankschien MD, Lee JH, Grace ED, Lennon CW, Halliday JA, Ross W, Gourse RL, Herman C. 2009. Super DksAs: substitutions in DksA enhancing its effects on transcription initiation. EMBO J 28:1720–1731. doi: 10.1038/emboj.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondary channel–structural framework for ppGpp-DksA synergism during transcription. Cell 118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 65.Ross W, Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, Gourse RL. 2016. ppGpp binding to a site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol Cell 62:811–823. doi: 10.1016/j.molcel.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parshin A, Shiver AL, Lee J, Ozerova M, Schneidman-Duhovny D, Gross CA, Borukhov S. 2015. DksA regulates RNA polymerase in Escherichia coli through a network of interactions in the secondary channel that includes sequence insertion 1. Proc Natl Acad Sci U S A 112:E6862–71. doi: 10.1073/pnas.1521365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Furman R, Danhart EM, NandyMazumdar M, Yuan C, Foster MP, Artsimovitch I. 2015. pH dependence of the stress regulator DksA. PLoS One 10:e0120746. doi: 10.1371/journal.pone.0120746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henard CA, Tapscott T, Crawford MA, Husain M, Doulias PT, Porwollik S, Liu L, McClelland M, Ischiropoulos H, Vazquez-Torres A. 2014. The 4-cysteine zinc-finger motif of the RNA polymerase regulator DksA serves as a thiol switch for sensing oxidative and nitrosative stress. Mol Microbiol 91:790–804. doi: 10.1111/mmi.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crawford MA, Tapscott T, Fitzsimmons LF, Liu L, Reyes AM, Libby SJ, Trujillo M, Fang FC, Radi R, Vazquez-Torres A. 2016. Redox-active sensing by bacterial DksA transcription factors is determined by cysteine and zinc content. mBio 7:e02161-15. doi: 10.1128/mBio.02161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belogurov GA, Artsimovitch I. 2015. Regulation of transcript elongation. Annu Rev Microbiol 69:49–69. doi: 10.1146/annurev-micro-091014-104047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Borukhov S, Polyakov A, Nikiforov V, Goldfarb A. 1992. GreA protein: a transcription elongation factor from Escherichia coli. Proc Natl Acad Sci U S A 89:8899–8902. doi: 10.1073/pnas.89.19.8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 73.Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martínez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Peralta-Gil M, Subhraveti P, Velázquez-Ramírez DA, Weaver D, Collado-Vides J, Paulsen I, Karp PD. 2017. The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res 45:D543–D550. doi: 10.1093/nar/gkw1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fitzgerald DM, Bonocora RP, Wade JT. 2014. Comprehensive mapping of the Escherichia coli flagellar regulatory network. PLoS Genet 10:e1004649. doi: 10.1371/journal.pgen.1004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin EC. 1976. Glycerol dissimilation and its regulation in bacteria. Annu Rev Microbiol 30:535–578. doi: 10.1146/annurev.mi.30.100176.002535. [DOI] [PubMed] [Google Scholar]
- 76.Hommais F, Krin E, Coppee JY, Lacroix C, Yeramian E, Danchin A, Bertin P. 2004. GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli. Microbiology 150:61–72. doi: 10.1099/mic.0.26659-0. [DOI] [PubMed] [Google Scholar]
- 77.Deng Z, Shan Y, Pan Q, Gao X, Yan A. 2013. Anaerobic expression of the gadE-mdtEF multidrug efflux operon is primarily regulated by the two-component system ArcBA through antagonizing the H-NS mediated repression. Front Microbiol 4:194. doi: 10.3389/fmicb.2013.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pesavento C, Becker G, Sommerfeldt N, Possling A, Tschowri N, Mehlis A, Hengge R. 2008. Inverse regulatory coordination of motility and curli-mediated adhesion in Escherichia coli. Genes Dev 22:2434–2446. doi: 10.1101/gad.475808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pesavento C, Hengge R. 2012. The global repressor FliZ antagonizes gene expression by σS-containing RNA polymerase due to overlapping DNA binding specificity. Nucleic Acids Res 40:4783–4793. doi: 10.1093/nar/gks055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soutourina OA, Bertin PN. 2003. Regulation cascade of flagellar expression in Gram-negative bacteria. FEMS Microbiol Rev 27:505–523. doi: 10.1016/S0168-6445(03)00064-0. [DOI] [PubMed] [Google Scholar]
- 81.Patrick JE, Kearns DB. 2012. Swarming motility and the control of master regulators of flagellar biosynthesis. Mol Microbiol 83:14–23. doi: 10.1111/j.1365-2958.2011.07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishihama A. 2018. Building a complete image of genome regulation in the model organism Escherichia coli. J Gen Appl Microbiol 63:311–324. doi: 10.2323/jgam.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 83.Lemke JJ, Durfee T, Gourse RL. 2009. DksA and ppGpp directly regulate transcription of the Escherichia coli flagellar cascade. Mol Microbiol 74:1368–1379. doi: 10.1111/j.1365-2958.2009.06939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 85.Zhou YN, Jin DJ. 1998. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci U S A 95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin DJ, Gross CA. 1989. Characterization of the pleiotropic phenotypes of rifampin-resistant rpoB mutants of Escherichia coli. J Bacteriol 171:5229–5231. doi: 10.1128/jb.171.9.5229-5231.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jishage M, Kvint K, Shingler V, Nystrom T. 2002. Regulation of sigma factor competition by the alarmone ppGpp. Genes Dev 16:1260–1270. doi: 10.1101/gad.227902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bernardo LM, Johansson LU, Solera D, Skarfstad E, Shingler V. 2006. The guanosine tetraphosphate (ppGpp) alarmone, DksA and promoter affinity for RNA polymerase in regulation of sigma-dependent transcription. Mol Microbiol 60:749–764. doi: 10.1111/j.1365-2958.2006.05129.x. [DOI] [PubMed] [Google Scholar]
- 89.Dong T, Yu R, Schellhorn H. 2011. Antagonistic regulation of motility and transcriptome expression by RpoN and RpoS in Escherichia coli. Mol Microbiol 79:375–386. doi: 10.1111/j.1365-2958.2010.07449.x. [DOI] [PubMed] [Google Scholar]
- 90.Szalewska-Palasz A, Johansson LUM, Bernardo LMD, Skärfstad E, Stec E, Brännström K, Shingler V. 2007. Properties of RNA polymerase bypass mutants: implications for the role of ppGpp and its co-factor DksA in controlling transcription dependent on σ54. J Biol Chem 282:18046–18056. doi: 10.1074/jbc.M610181200. [DOI] [PubMed] [Google Scholar]
- 91.Costanzo A, Nicoloff H, Barchinger SE, Banta AB, Gourse RL, Ades SE. 2008. ppGpp and DksA likely regulate the activity of the extracytoplasmic stress factor σE in is by both direct and indirect mechanisms. Mol Microbiol 67:619–632. doi: 10.1111/j.1365-2958.2007.06072.x. [DOI] [PubMed] [Google Scholar]
- 92.Brown L, Gentry D, Elliott T, Cashel M. 2002. DksA affects ppGpp induction of RpoS at a translational level. J Bacteriol 184:4455–4465. doi: 10.1128/jb.184.16.4455-4465.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jishage M, Iwata A, Ueda S, Ishihama A. 1996. Regulation of RNA polymerase sigma subunit synthesis in Escherichia coli: intracellular levels of four species of sigma subunit under various growth conditions. J Bacteriol 178:5447–5451. doi: 10.1128/jb.178.18.5447-5451.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maeda H, Fujita N, Ishihama A. 2000. Competition among seven Escherichia coli sigma subunits: relative binding affinities to the core RNA polymerase. Nucleic Acids Res 28:3497–3503. doi: 10.1093/nar/28.18.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kundu TK, Kusano S, Ishihama A. 1997. Promoter selectivity of Escherichia coli RNA polymerase σF holoenzyme involved in transcription of flagellar and chemotaxis genes. J Bacteriol 179:4264–4269. doi: 10.1128/jb.179.13.4264-4269.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grigorova IL, Phleger NJ, Mutalik VK, Gross CA. 2006. Insights into transcriptional regulation and sigma competition from an equilibrium model of RNA polymerase binding to DNA. Proc Natl Acad Sci U S A 103:5332–5337. doi: 10.1073/pnas.0600828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mauri M, Klumpp S. 2014. A model for sigma factor competition in bacterial cells. PLoS Comput Biol 10:e1003845. doi: 10.1371/journal.pcbi.1003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arsene F, Tomoyasu T, Bukau B. 2000. The heat shock response of Escherichia coli. Int J Food Microbiol 55:3–9. doi: 10.1016/s0168-1605(00)00206-3. [DOI] [PubMed] [Google Scholar]
- 99.Riordan JT, Mitra A. 2017. Regulation of Escherichia coli pathogenesis by alternative sigma factor N. EcoSal Plus 7 doi: 10.1128/ecosalplus.ESP-0016-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barchinger SE, Ades SE. 2013. Regulated proteolysis: control of the Escherichia coli σE-dependent cell envelope stress response. Subcell Biochem 66:129–160. doi: 10.1007/978-94-007-5940-4_6. [DOI] [PubMed] [Google Scholar]
- 101.Maeda H, Jishage M, Nomura T, Fujita N, Ishihama A. 2000. Two extracytoplasmic function sigma subunits, σE and σFecI, of Escherichia coli: promoter selectivity and intracellular levels. J Bacteriol 182:1181–1184. doi: 10.1128/jb.182.4.1181-1184.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gopalkrishnan S, Nicoloff H, Ades SE. 2014. Co-ordinated regulation of the extracytoplasmic stress factor, sigmaE, with other Escherichia coli sigma factors by (p)ppGpp and DksA may be achieved by specific regulation of individual holoenzymes. Mol Microbiol 93:479–493. doi: 10.1111/mmi.12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou YN, Kusukawa N, Erickson JW, Gross CA, Yura T. 1988. Isolation and characterization of Escherichia coli mutants that lack the heat shock sigma factor sigma 32. J Bacteriol 170:3640–3649. doi: 10.1128/jb.170.8.3640-3649.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kusukawa N, Yura T. 1988. Heat shock protein GroE of Escherichia coli: key protective roles against thermal stress. Genes Dev 2:874–882. doi: 10.1101/gad.2.7.874. [DOI] [PubMed] [Google Scholar]
- 105.Pokhrel A, Lingo JC, Wolschendorf F, Gray MJ. 2019. Assaying for inorganic polyphosphate in bacteria. J Vis Exp (143):e58818. doi: 10.3791/58818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Daimon Y, Narita S, Akiyama Y. 2015. Activation of toxin-antitoxin system toxins suppresses lethality caused by the loss of σE in Escherichia coli. J Bacteriol 197:2316–2324. doi: 10.1128/JB.00079-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Konovalova A, Grabowicz M, Balibar CJ, Malinverni JC, Painter RE, Riley D, Mann PA, Wang H, Garlisi CG, Sherborne B, Rigel NW, Ricci DP, Black TA, Roemer T, Silhavy TJ, Walker SS. 2018. Inhibitor of intramembrane protease RseP blocks the σE response causing lethal accumulation of unfolded outer membrane proteins. Proc Natl Acad Sci U S A 115:E6614–E6621. doi: 10.1073/pnas.1806107115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jishage M, Ishihama A. 1998. A stationary phase protein in Escherichia coli with binding activity to the major sigma subunit of RNA polymerase. Proc Natl Acad Sci U S A 95:4953–4958. doi: 10.1073/pnas.95.9.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S. 1997. Modulation of the Escherichia coli σE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol Microbiol 24:355–371. doi: 10.1046/j.1365-2958.1997.3601713.x. [DOI] [PubMed] [Google Scholar]
- 110.Rouviere PE, De Las Penas A, Mecsas J, Lu CZ, Rudd KE, Gross CA. 1995. rpoE, the gene encoding the second heat-shock sigma factor, sigma E, in Escherichia coli. EMBO J 14:1032–1042. doi: 10.1002/j.1460-2075.1995.tb07084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bass S, Gu Q, Christen A. 1996. Multicopy suppressors of prc mutant Escherichia coli include two HtrA (DegP) protease homologs (HhoAB), DksA, and a truncated R1pA. J Bacteriol 178:1154–1161. doi: 10.1128/jb.178.4.1154-1161.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ishii Y, Yamada H, Yamashino T, Ohashi K, Katoh E, Shindo H, Yamazaki T, Mizuno T. 2000. Deletion of the yhhP gene results in filamentous cell morphology in Escherichia coli. Biosci Biotechnol Biochem 64:799–807. doi: 10.1271/bbb.64.799. [DOI] [PubMed] [Google Scholar]
- 114.Yamanaka K, Mitani T, Ogura T, Niki H, Hiraga S. 1994. Cloning, sequencing, and characterization of multicopy suppressors of a mukB mutation in Escherichia coli. Mol Microbiol 13:301–312. doi: 10.1111/j.1365-2958.1994.tb00424.x. [DOI] [PubMed] [Google Scholar]
- 115.Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. 2008. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T. 2008. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schurig-Briccio LA, Farias RN, Rintoul MR, Rapisarda VA. 2009. Phosphate-enhanced stationary-phase fitness of Escherichia coli is related to inorganic polyphosphate level. J Bacteriol 191:4478–4481. doi: 10.1128/JB.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gruber TM, Gross CA. 2003. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol 57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- 119.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the σE stress response in related genomes. PLoS Biol 4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Klein G, Stupak A, Biernacka D, Wojtkiewicz P, Lindner B, Raina S. 2016. Multiple transcriptional factors regulate transcription of the rpoE gene in Escherichia coli under different growth conditions and when the lipopolysaccharide biosynthesis is defective. J Biol Chem 291:22999–23019. doi: 10.1074/jbc.M116.748954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Janaszak A, Majczak W, Nadratowska B, Szalewska-Palasz A, Konopa G, Taylor A. 2007. A σ54-dependent promoter in the regulatory region of the Escherichia coli rpoH gene. Microbiology 153:111–123. doi: 10.1099/mic.0.2006/000463-0. [DOI] [PubMed] [Google Scholar]
- 122.Erickson JW, Gross CA. 1989. Identification of the sigma E subunit of Escherichia coli RNA polymerase: a second alternate sigma factor involved in high-temperature gene expression. Genes Dev 3:1462–1471. doi: 10.1101/gad.3.9.1462. [DOI] [PubMed] [Google Scholar]
- 123.Sineva E, Savkina M, Ades SE. 2017. Themes and variations in gene regulation by extracytoplasmic function (ECF) sigma factors. Curr Opin Microbiol 36:128–137. doi: 10.1016/j.mib.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 125.Guo B, Bi Y. 2002. Cloning PCR products: an overview. Methods Mol Biol 192:111–119. doi: 10.1385/1-59259-177-9:111. [DOI] [PubMed] [Google Scholar]
- 126.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. doi: 10.1128/JB.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 40:D115–22. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Santos-Zavaleta A, Salgado H, Gama-Castro S, Sánchez-Pérez M, Gómez-Romero L, Ledezma-Tejeida D, García-Sotelo JS, Alquicira-Hernández K, Muñiz-Rascado LJ, Peña-Loredo P, Ishida-Gutiérrez C, Velázquez-Ramírez DA, Del Moral-Chávez V, Bonavides-Martínez C, Méndez-Cruz C-F, Galagan J, Collado-Vides J. 2019. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res 47:D212–D220. doi: 10.1093/nar/gky1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 130.Paek KH, Walker GC. 1987. Escherichia coli dnaK null mutants are inviable at high temperature. J Bacteriol 169:283–290. doi: 10.1128/jb.169.1.283-290.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev 53:1–24. doi: 10.1128/MMBR.53.1.1-24.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nichols BP, Shafiq O, Meiners V. 1998. Sequence analysis of Tn10 insertion sites in a collection of Escherichia coli strains used for genetic mapping and strain construction. J Bacteriol 180:6408–6411. doi: 10.1128/JB.180.23.6408-6411.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Silhavy TJ, Berman ML, Enquist LW (ed). 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 134.Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. 2007. Recombineering: in vivo genetic engineering in E. coli, S. Enterica, and beyond. Methods Enzymol 421:171–199. doi: 10.1016/S0076-6879(06)21015-2. [DOI] [PubMed] [Google Scholar]
- 135.Jin DJ, Cashel M, Friedman DI, Nakamura Y, Walter WA, Gross CA. 1988. Effects of rifampicin resistant rpoB mutations on antitermination and interaction with nusA in Escherichia coli. J Mol Biol 204:247–261. doi: 10.1016/0022-2836(88)90573-6. [DOI] [PubMed] [Google Scholar]
- 136.Jin DJ, Walter WA, Gross CA. 1988. Characterization of the termination phenotypes of rifampicin-resistant mutants. J Mol Biol 202:245–253. doi: 10.1016/0022-2836(88)90455-x. [DOI] [PubMed] [Google Scholar]
- 137.Reisch CR, Prather K. 2017. Scarless Cas9 assisted recombineering (no-SCAR) in Escherichia coli, an easy-to-use system for genome editing. Curr Protoc Mol Biol 117:31.8.1–31.8.20. doi: 10.1002/cpmb.29. [DOI] [PubMed] [Google Scholar]
- 138.Gray MJ, Wholey W-Y, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JCA, Jakob U. 2014. Polyphosphate is a primordial chaperone. Mol Cell 53: 689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. doi: 10.1128/JB.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wurst H, Kornberg A. 1994. A soluble exopolyphosphatase of Saccharomyces cerevisiae: purification and characterization. J Biol Chem 269:10996–11001. [PubMed] [Google Scholar]
- 141.Christ JJ, Blank LM. 2018. Enzymatic quantification and length determination of polyphosphate down to a chain length of two. Anal Biochem 548:82–90. doi: 10.1016/j.ab.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 142.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 143.Rocha DJ, Santos CS, Pacheco LG. 2015. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek 108:685–693. doi: 10.1007/s10482-015-0524-1. [DOI] [PubMed] [Google Scholar]
- 144.Zhou K, Zhou L, Lim Q, Zou R, Stephanopoulos G, Too HP. 2011. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol Biol 12:18. doi: 10.1186/1471-2199-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tjaden B. 2015. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol 16:1. doi: 10.1186/s13059-014-0572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All strains and plasmids generated in the course of this work are available from the author upon request. RNA sequencing data have been deposited in NCBI’s Gene Expression Omnibus (146) and are accessible through GEO series accession number GSE144816.