

Abstract
Brain injury activates complex inflammatory signals in dying neurons, surviving neurons, and glia. Here, we establish that inflammation regulates the regeneration of photoreceptors in the zebrafish retina and determine the cellular expression and function of the inflammatory protease, matrix metalloproteinase 9 (Mmp‐9), during this regenerative neurogenesis. Following photoreceptor ablation, anti‐inflammatory treatment suppresses the number of injury‐induced progenitors and regenerated photoreceptors. Upon photoreceptor injury, mmp‐9 is induced in Müller glia and Müller glia‐derived photoreceptor progenitors. Deleting mmp‐9 results in over production of injury‐induced progenitors and regenerated photoreceptors, but over time the absence of Mmp‐9 compromises the survival of the regenerated cones. At all time‐points studied, the levels of tnf‐α are significantly elevated in mutant retinas. Anti‐inflammatory treatment in mutants rescues the defects in cone survival. These data provide a link between injury‐induced inflammation in the vertebrate CNS, Mmp‐9 function during neuronal regeneration and the requirement of Mmp‐9 for the survival of regenerated cones.
Keywords: cytokines, immunosuppression, microglia, Müller glia, proliferation, stem cell
Acute inflammation is necessary for photoreceptor regeneration in zebrafish.
MMP‐9 negatively regulates Müller glia‐derived progenitors during photoreceptor regeneration.
Mmp‐9 promotes the survival of regenerated cone photoreceptors.
1. INTRODUCTION
Inflammation modulates immune and nonimmune functions during tissue development, repair, and disease (Deverman & Patterson, 2009; Ekdahl, Kokaia, & Lindvall, 2009). Acute inflammation involves the secretion of proinflammatory cytokines and chemokines that recruit immune cells to the damaged tissue (Liddiard, Rosas, Davies, Jones, & Taylor, 2011; Nathan, 2002). In the central nervous system of adult mammals, acute inflammation can activate signaling cascades in stem and progenitor cells that stimulate proliferation and promote neurogenesis (Borsini, Zunszain, Thuret, & Pariante, 2015; Ekdahl et al., 2009; Kizil, Kyritsis, & Brand, 2015; Kyritsis et al., 2012; Kyritsis, Kizil, & Brand, 2014). Chronic inflammation, however, is detrimental, and results in secondary damage to neurons and, in severe cases, neurodegenerative disease (Amor, Puentes, Baker, & van der Valk, 2010). Proper regulation of inflammatory cascades in the central nervous system is critical both to maintain tissue homeostasis and repair brain injuries.
Unlike mammals, which possess a limited capacity for tissue regeneration, zebrafish have the ability to regenerate tissues and organs, such as fins, heart, brain, spinal cord, and retina (Gemberling, Bailey, Hyde, & Poss, 2013; Kizil, Kaslin, Kroehne, & Brand, 2012; Lenkowski & Raymond, 2014). Studies using zebrafish have identified cellular and molecular mechanisms which may unlock the regenerative potential of mammalian tissues (Martin & Poché, 2019; Ueki et al., 2015). Such mechanisms include inflammation. In zebrafish acute inflammation is both necessary and sufficient to induce neuronal regeneration (Caldwell et al., 2019; Kyritsis et al., 2012; White et al., 2017).
In the injured zebrafish retina, Müller glia serve as intrinsic stem cells that endow this nervous tissue the ability to regenerate (Bernardos, Barthel, Meyers, & Raymond, 2007; Fausett & Goldman, 2006). Neuronal injury and death stimulate Müller glia to initiate a transient gliotic response, followed by partial dedifferentiation and entry into the cell cycle (Raymond, Barthel, Bernardos, & Perkowski, 2006; Thomas, Ranski, Morgan, & Thummel, 2016). Müller glia then undergo a single asymmetric division to produce multipotent progenitors, which proliferate rapidly, migrate to areas of cell death and differentiate to replace the ablated neurons (Fimbel, Montgomery, Burket, & Hyde, 2007; Goldman, 2014; Gorsuch & Hyde, 2014; Lenkowski & Raymond, 2014; Nagashima, Barthel, & Raymond, 2013; Sherpa et al., 2008). In the zebrafish retina, paracrine and autocrine signaling play essential roles in facilitating this regenerative neurogenesis (Nelson et al., 2013; Zhao et al., 2014). Dying photoreceptors secrete Tnf‐α, to which Müller glia respond by reprogramming into a stem cell‐state, synthesizing Tnf‐α and entering into the cell cycle (Nelson et al., 2013). Also, leptin and Il‐6 family cytokines expressed by Müller glia and Müller glia‐derived progenitors are required for injury‐induced proliferation (Zhao et al., 2014).
Based on their structure and specific substrates, matrix metalloproteinases (Mmps) are classified into five groups: gelatinases, collagenases, MTMMPs, stromelysins, and matrilysins (Vandooren, Van den Steen, & Opdenakker, 2013). Matrix metalloproteinase 9 (Mmp‐9) is a secreted gelatinase, first purified from neutrophils and monocytes stimulated by IL‐8 and IL‐1β (Masure, Proost, Van Damme, & Opdenakker, 1991; Opdenakker, Masure, Proost, Billiau, & van Damme, 1991). During tissue development and homeostasis, Mmp‐9 plays a prominent role by acting on extracellular molecules, including adhesion molecules, growth factors and cytokines (Bonnans, Chou, & Werb, 2014; Le, Xue, Castelnoble, & Jackson, 2007; Masure et al., 1991; Parks, Wilson, & López‐Boado, 2004; Vandooren, Van Damme, & Opdenakker, 2014; Vandooren, Van den Steen, et al., 2013), which can regulate proliferation, cellular migration and cellular differentiation. It is well established that inflammatory cytokines can induce the expression of Mmp‐9 (Nagase, Visse, & Murphy, 2006; Shubayev et al., 2006; Vandooren, Van den Steen, et al., 2013; Vecil et al., 2000). In turn, Mmp‐9 can cleave cytokine precursors and mature cytokines into activate and inactivate forms, respectively, revealing the complex role of Mmp‐9 in modulating inflammation (Parks et al., 2004). The complexities of Mmp‐9 function suggest its role during regeneration is likely tissue, substrate and injury‐context dependent (Ando, Shibata, Hans, Brand, & Kawakami, 2017; Hindi, Shin, Ogura, Li, & Kumar, 2013; Vandooren et al., 2014; Xue et al., 2017). The cellular expression and function of Mmp‐9 during photoreceptor regeneration in the zebrafish are unknown.
Here, we use zebrafish to determine the general role of inflammation and the cellular expression and function of the matrix metalloproteinase, Mmp‐9, during photoreceptor regeneration. We establish that in the adult retina inflammation governs the proliferation of Müller glia‐derived progenitors. We show that mmp‐9 is expressed in Müller glia, as they prepare to enter the cell cycle, and Müller glia‐derived progenitors. Using loss‐of‐function mutants, we determine that Mmp‐9 negatively regulates the proliferation of Müller glia‐derived progenitors. In mutants both unlesioned and lesioned retinas show elevated expression of the inflammatory cytokine, tnf‐α. Finally, we demonstrate that following photoreceptor regeneration, Mmp‐9 is required for the survival of regenerated cone photoreceptors.
2. MATERIALS AND METHODS
2.1. Animals
Wild‐type, AB strain zebrafish (Danio rerio; ZIRC, University of Oregon, Eugene, OR) were propagated, maintained, and housed in recirculating habitats at 28.5°C and on a 14/10‐h light/dark cycle. Embryos were collected after natural spawns, incubated at 28.5°C and staged by hours post fertilization (hpf). Adults were of either sex and used between 6 and 15 months of age. Ages of animals were matched within experiments. The transgenic reporter lines, Tg[gfap:EGFP] mi2002 and Tg[mpeg1:mCherry] gl23 were used to identify Müller glia and microglia, respectively (Bernardos & Raymond, 2006; Ellett, Pase, Hayman, Andrianopoulos, & Lieschke, 2011). All experimental protocols were approved by the University of Michigan's Institutional Animal Care and Use Committee (IACUC).
2.2. Light lesions
To kill photoreceptors, fish were dark adapted, then exposed to high intensity light (ca. 100,000 lx) for 30 min, followed by exposure to constant light (ca. 30.000 lx) for 72 hr (Taylor et al., 2015). After 72 hr fish were returned to the recirculating habitats and normal light/dark cycle.
2.3. Quantitative real‐time PCR
Total RNA was extracted from dissected whole retinas using TRIzol (Invitrogen, Carlsbad, CA). Reverse transcription to cDNA was performed using 1 μg RNA (Qiagen QuantiTect Reverse Transcription kit; Venlo, Netherlands), and each qPCR reaction was performed with 6 ng cDNA and Bio‐Rad IQ SYBR Green Supermix (Bio‐Rad CFX384 Touch Real Time PCR Detection System; Hercules, CA). Three independent biological replicates were collected at each time point, and each replicate contained six homogenized retinas from three fish (18 retinas/time point). Each biological replicate was run in triplicate. For each time point, average mRNA levels were represented as fold change, calculated using the DDCT method and normalized to the housekeeping gene, gpia. Primers used for qPCR analyses are listed in Table 1.
Table 1.
Primer sequences
| Name | Sequence |
|---|---|
| gpia | F) 5′‐TCCAAGGAAACAAGCCAAGC‐3′ |
| R) 5′‐TTCCACATCACACCCTGCAC‐3′ | |
| mmp‐9 | F) 5′‐TGATGTGCTTGGACCACGTAA‐3′ |
| R) 5′‐ACAGGAGCACCTTGCCTTTTC‐3′ | |
| tnf‐α | F) 5′‐GCGCTTTTCTGAATCCTACG‐3′ |
| R) 5′‐TGCCCAGTCTGTCTCCTTCT‐3′ | |
| tnf‐β | F) 5′‐CCTCAGACCACGGAAAAGT‐3′ |
| R) 5′‐GCCCTGTTGGAATGCCTGAT‐3′ | |
| il‐6 | F) 5′‐TCTTTCCCTCTTTTCCTCCTG‐3′ |
| R) 5′‐TCAACTTCTCCAGCGTGATG‐3′ | |
| il‐8 | F) 5′‐GTCGCTGCATTGAAACAGAA‐3′ |
| R) 5′‐AGGGGTCCAGACAGATCTCC‐3′ | |
| nfκb1 | F) 5′‐ACCAGACTGTGAGCGTGAAG‐3′ |
| R) 5′‐CGCAAGTCCTACCCACAAGT‐3′ | |
| nfκb2 | F) 5′‐CATATGTCCCACACAATCAAGAC‐3′ |
| R) 5′‐AGCCACCATAATGATCTGGAA‐3′ | |
| stat3 | F) 5′‐GAGGAGGCGTTTGGCAAA‐3′ |
| R) 5′‐TGTGTCAGGGAACTCAGTGTCTG‐3′ | |
| gfap | F) 5′‐AGGCTCATGTGAAGAGGAGCATAG‐3′ |
| R) 5′‐CCTCATTATGGCAGATCCTTCCTC‐3′ |
2.4. Dexamethasone treatment
A previously described protocol was used that efficiently suppresses the immune system in zebrafish (Kyritsis et al., 2012; see also Bollaerts et al., 2019). Briefly, fish were housed in system water with 15 mg/L dexamethasone (Sigma‐Aldrich, Corp, D1756) diluted in 0.1% MetOH. Dexamethasone‐containing and control solutions were changed daily, and fish were fed brine shrimp every other day.
2.5. Immunohistochemistry
Tissues were fixed overnight at 4°C in 0.1 M phosphate buffered 4% paraformaldehyde, cryoprotected with 20% sucrose, and embedded in optical cutting temperature (OCT) medium (Sakura Finetek USA, Torrance, CA). Immunohistochemistry (IHC) was performed as previously described (Taylor et al., 2015). Briefly, 10‐μm‐thick sections were collected such that the optic nerve head was included, and sections were mounted onto glass slides. Sections were washed in phosphate buffer saline with 0.5% Triton‐x (PBST) and incubated with 20% heat‐inactivated normal sheep serum in PBST for 2 hr (NSS; Sigma‐Aldrich, Corp.). Primary antibodies were applied overnight at 4°C. Sections were then washed with PBST and incubated in secondary antibodies for 1 hr at room temperature. Prior to IHC for BrdU, sections were immersed in 100°C sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 30 min and cooled at room temperature for 20 min. Sections were then subjected to IHC as described above.
Immunohistochemistry performed on whole retinas was conducted as described previously (Nagashima, Hadidjojo, Barthel, Lubensky, & Raymond, 2017). Prior to IHC, retinas were dissected from dark‐adapted animals, flattened with radial relaxing cuts, fixed at 4°C overnight in 4% paraformaldehyde in 0.1 M phosphate buffer with 5% sucrose. Retinas were then placed in boiling sodium citrate buffer for 5 min and allowed to cool for 5 min. Retinas were incubated for 2 hr in 10% normal goat serum, 1% Tween, 1% Triton X‐100, 1% DMSO, in phosphate buffered saline with 0.1% sodium azide. Primary and secondary antibody were diluted in 2% normal goat serum, 1% Tween, 1% Triton X‐100, 1% DMSO, in phosphate buffered saline with 0.1% sodium azide and incubations were performed overnight at room temperature. After washing in phosphate buffered saline with 1% Tween, 1% Triton X‐100, 1% DMSO, retinas were mounted on glass slides, photoreceptor side down, in ProLong Gold anti‐fade reagent (Life Technologies Corp., Eugene, OR). Antibodies used in this study and their concentrations are listed in Table 2.
Table 2.
Antibody list
| Primary antibodies | Company | Dilution |
|---|---|---|
| Mouse monoclonal anti‐GFP | Milipore | 1:200 |
| MAB3580 | ||
| Mouse monoclonal anti‐Actin | Calbiochem | 1:1000 |
| Mouse monoclonal anti‐Zn5 | ZIRC; zfin.Org/ZDB‐ATB‐081002‐19 | 1:200 |
| Mouse monoclonal anti‐Zpr1 (anti‐Arrestin‐3) | ZIRC; zfin.Org/ZDB‐ATB‐081002‐43 | 1:200 |
| Mouse monoclonal anti‐Zpr‐3 | ZIRC; zfin.Org/ZDB‐ATB‐081002‐45 | 1:200 |
| Mouse anti‐HPC1 | ZIRC; zfin.org/ZDB‐ATB 130225–1 | 1:200 |
| Mouse anti‐Zrf1 | ZIRC; zfin.Org/ZDB‐ATB‐081002‐46 | 1:1000 |
| Mouse monoclonal ZO‐1A‐12 | Innovative research cat# 33–9,100, RRID:AB_87181 | 1:200 |
| Mouse anti‐BrdU | BD biosciences Ca# 347580 | 1:100 |
| Rat anti‐BrdU | Abcam cat# ab6326, RRID:AB_305426 | 1:200 |
| Polyclonal anti‐GNAT2 | MBL Ca# PM075 | 1:1000 |
| Mouse monoclonal anti‐1D1 (ZF Rhodoposin) | Hyatt et al., 1996 | 1:1000 |
| Gift from Dr. Jim Fadool (FSU) | ||
| Mouse monoclonal anti‐4C4 (zebrafish microglia) | Raymond et al., 2006 | 1:200 |
| Hitchcock lab | ||
| Polyclonal anti‐mCherry antibody | Abcam ab167453 | 1:200 |
| Secondary antibodies | ||
| Alexa Fluor goat anti‐mouse 488, 555, or 647 | Invitrogen | 1:500 |
| Alexa Fluor goat anti‐rabbit 488, 555, or 647 | Invitrogen | 1:500 |
| Alexa Fluor goat anti‐rat 488, 555, or 647 | Invitrogen | 1:500 |
2.6. TUNEL staining
TUNEL staining was performed using In Situ Cell Death Detection Kit, TMR red (Sigma‐Aldrich). Briefly, following immunocytochemistry, slides were treated with PBS containing 1% sodium citrate/ 1% TritonX‐100 at 4°C, were rinsed with PBS, and them were incubated with TUNEL reaction cocktail for 30 min at 37°C.
2.7. Labeling dividing cells
In adults, dividing cells were labeled with BrdU by housing animals in system water containing 5 mM BrdU for 24 hr (Gramage, D'Cruz, Taylor, Thummel, & Hitchcock, 2015).
2.8. In situ hybridization
In situ hybridizations were performed as previously described (Luo et al., 2012). Digoxigenin (DIG)‐labeled riboprobes for rhodopsin and pde6c were generated from full‐length cDNA clones (Luo et al., 2012; Ochocinska & Hitchcock, 2007), whereas riboprobe for mmp‐9 was generated by PCR amplification using primers containing the T3 or T7 promoter sequences (David & Wedlich, 2001). mmp‐9 (F): 5′ TAATACGACTCACTATAGGGGATTCTTCTACTTCTGCCGGG 3′ mmp‐9 (R): 5′ AATTAACCCTCACTAAAGGGCTTAATAAATTTGTAAACAAG 3′.
Briefly, 10‐μm‐thick sections were hybridized with riboprobes at 55°C, incubated with an alkaline‐phosphatase‐conjugated anti‐DIG antibody and visualized using Fast Red TR/Naphthol AS‐MX (SIGMAFAST) as the enzymatic substrate. When in situ hybridizations were combined with BrdU IHC, sections were removed from the fast red solutions, rinsed and post‐fixed in buffered 4% paraformaldehyde for 10 min then processed for BrdU IHC as described above.
2.9. Production of custom antibodies
Antibodies specific to zebrafish Mmp‐9 were generated by Pocono Rabbit Farm & Laboratory (PRF&L, Canadensis, PA) as previously described (Calinescu, Raymond, & Hitchcock, 2009). A 24 amino acid C‐terminal peptide was used as the immunogen, CDIDGIQYLYGPRTGPEPTAPQPR; NCBI: AY151254. Polyclonal antibodies were affinity purified and confirmed by ELISA (data not shown). Western blots performed with retinal proteins using pre‐ and post‐immune sera confirmed the post‐immune serum detected a 76 kDa band, the predicted size for Mmp‐9. Western blot analysis and known cleavage sites on pro‐Mmp9 suggest these antibodies recognize both the pro‐ and active forms of Mmp‐9 (Vandooren, Geurts, Martens, Van den Steen, & Opdenakker, 2013).
2.10. Western blot analysis
Protein samples were obtained from whole retinas homogenized in RIPA lysis buffer (ThermoFisher Scienific) containing protease and phosphatase inhibitor cocktail (5872S; Cell Signaling Technology, Danvers, MA). Proteins were separated in a 12% Mini‐PROTEAN TGX Precast gel (BioRad) and transferred to a polyvinylidene difluoride (PVDF) membrane (GenHunter Corp., Nashville, TN). To block non‐specific staining, membranes were incubated in 5% nonfat dry milk in Tris buffered saline containing 0.3% Tween‐20 (TBST) for 2 hr. Membranes were incubated with the antibodies‐containing solution overnight at 4°C. Blots were then washed in TBST and incubated with horseradish peroxidase‐conjugated secondary IgG (1:1000) for 1 hr at room temperature. Antibody‐bound protein was visualized using Pierce ECL Western blotting substrate (32,106; ThermoFisher Scienific, Waltham, MA). To visualize loading controls, blots were also stained with antibodies against Actin. Images were captured using the Azure C500 (Azure Biosystems, Dublin, CA). Densitometry of protein bands was performed using ImageJ software (https://imagej.nih.gov/ij/).
2.11. Zymography
Protein samples were prepared as described for the Western blot analyses. Proteins were separated on 10% Zymogram Plus (Gelatin) Protein Gels, (ThermoFisher Scientific). Following electrophoresis, proteins were renatured in 1X renaturing buffer (ThermoFisher Scientific) for 30 min at room temperature, then incubated in 1X developing buffer (ThermoFisher Scientific) overnight at 37°. Gels were rinsed in deionized water and stained with SimplyBlue SafeStain (ThermoFisher Scientific). Gels were imaged with long‐wave ultraviolet light using the Azure C500 (Azure Biosystems; Dublin, CA). Active recombinant human MMP‐9 (Calbiochem, San Diego, CA) was used as a positive control. Densitometry of the digested bands was performed using ImageJ software.
2.12. CRISPR‐Cas9 mediated targeted mutation of Mmp‐9
mmp‐9 mutants were generated according to previously described methods (Hwang et al., 2013). ZiFiT software (available in the public domain at zifit.partners.org) was used to identify a 19 bp sgRNA target sequence for mmp‐9 (GGCTGCTTCATGGCATCAA). Target sequence Oligo1‐ TTGATGCCATGAAGCAGCC and Oligo2‐AAACTTGATGCCATGAAGCAG were annealed and subcloned into the pT7 gRNA vector (Addgene ID: 46759). The pCS2 nCas9n vector (Addgene ID: 46929) was used for Cas9 synthesis. To produce RNAs, the MEGAshortscript T7 kit (Ambion, Foster City, CA: AM1354) and mirVana miRNA Isolation kit (Ambion: AM 1560) were used for the gRNA, whereas, mMesage mMACHINE SP6 kit (Ambion: AM1340) and RNeasy mini Kit from (Qiagen: 74104) were used for the Cas9 mRNA. Single cell‐stage embryos were injected with a 2 nL solution containing 100 pg/nL sgRNA and 150 pg/nL Cas9 mRNA. Founders (F0) were outcrossed to AB wild‐type fish. Mutations were identified using screening primers (F: 5′‐AAGTCTGCAACTACATATCAGC‐3′, R: 5′‐GTACACACTGTAGATGCTGATAAG‐3′) that flanked the mmp‐9 sgRNA target site. Standard protocols for PCR used Platinum Taq‐HF polymerase (Invitrogen) with genomic DNA as the template. The purified PCR product was subcloned into the pGEM‐T Easy vector (Promega, Madison, WI) for Sanger sequencing. Mutant and wild‐type sequences were aligned using pairwise blast (National Center for Biotechnology Information [NCBI], Bethesda, MD). Premature stop codons were identified by comparing predicted amino acid sequences for wild‐type and mutants using the ExPASy translate tool (available in the public domain at (www.expasy.org ). F1 hybrids carrying a mmp‐9 mutation were in‐crossed and homozygous F2 mutants were identified by a combination of Sanger sequencing and T7 endonuclease assays (New England Biolabs, Ipswich, MA) as previously described (www.crisprflydesign.org).
2.13. Imaging
Fluorescence images of retinal sections were captured using the Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). Cell counts were conducted using z‐stack images and analyzed with Imaris software (Bitplane, South Windsor, CT). Regenerated photoreceptors were identified by the colocalization of DAPI, BrdU, and the ISH markers pde6c (cones) or rho (rods).
2.14. Cell counts and area measurement
Cells were counted in both radial sections and retinal wholemounts. In radial sections, cells were counted in three nonadjacent sections in each retina. The number of cells per retina were averaged, and averages were computed for control and experimental groups. In wholemounts stained with an antibody against the tight junction protein, ZO‐1 (Nagashima et al., 2017), cones were identified in optical sections taken at the outer limiting membrane as profiles with perimeters greater than 3.5 μm. Cells were counted in five separate regions in each retina, sampling a totaling 5625μm2 per retina. The average number of cells in control and experimental groups were then averaged.
3. EXPERIMENTAL DESIGN AND STATISTICAL ANALYSES
The statistical significance in mRNA levels between unlesioned and lesioned retinas was calculated using an ANOVA (JMP 9.0; SAS Institute, Inc., Cary, NC). A p‐value ≤.05 was considered significant. The statistical significance in the number of cells between control and treated retinas was calculated using a student's t‐test (GraphPad Prism Software, La Jolla, CA). A p‐value ≤.05 was considered significant. Western and zymogram analyses consisted of three independent biological replicates, and each replicate contained six homogenized retinas from three fish (18 retinas per time point). Statistical significance between optical density values of bands in control and experimental groups was calculated using an ANOVA (JMP 9.0, SAS Institute, Inc., Cary, NC). A p‐value ≤.05 was considered significant.
4. RESULTS
4.1. Photoreceptor injury and death induce the expression of inflammatory genes
To characterize the inflammatory response during photoreceptor ablation and regeneration, qPCR was used to quantify the expression of inflammatory genes, mmp‐9, tnf‐α, tnf‐β, il‐8, nfκb1, and nfκb2, shown previously to regulate various forms of tissue regeneration in zebrafish (de Oliveira et al., 2013; Karra, Knecht, Kikuchi, & Poss, 2015; LeBert et al., 2015; Nelson et al., 2013). The time course of gene expression follows closely the well‐established time course for the injury response in Müller glia and the appearance and amplification of the Müller glia‐derived progenitors and their subsequent differentiation into mature photoreceptors (Figure 1a; see (Gorsuch & Hyde, 2014)). Upregulation of gene expression can be detected by 8 hr post lesion (hpl), and the rising phase of expression corresponds to the period when Müller glia prepare to enter the cell cycle. Expression levels then slowly decline as the Müller glia‐derived photoreceptor progenitors proliferate, migrate to the outer nuclear layer and differentiate (168 hpl; Figure 1a). Interestingly, among all the transcripts examined, mmp‐9 shows the greatest change from the undamaged controls, peaking at 24–48 hpl. Further, by 168 hpl mmp‐9 levels are reduced to only about 70% of the peak value (Figure 1a). The pro‐inflammatory cytokine, Tnf‐α, was shown to be responsible for initiating photoreceptor regeneration in zebrafish (Gorsuch & Hyde, 2014; Nelson et al., 2013), but at all‐time points in this assay, the levels of tnf‐α expression were not significantly different from controls (also see below). Our assay for tnf‐α failed to parallel the previous results (Nelson et al., 2013), and we infer this likely reflects the differences between the qPCR assay used here and the protein analysis used by Nelson et al. (2013). Nonetheless, these data show that in zebrafish photoreceptor death induces rapid expression of inflammatory genes that follows a time course reflecting the well‐described events that underlie photoreceptor regeneration.
Figure 1.
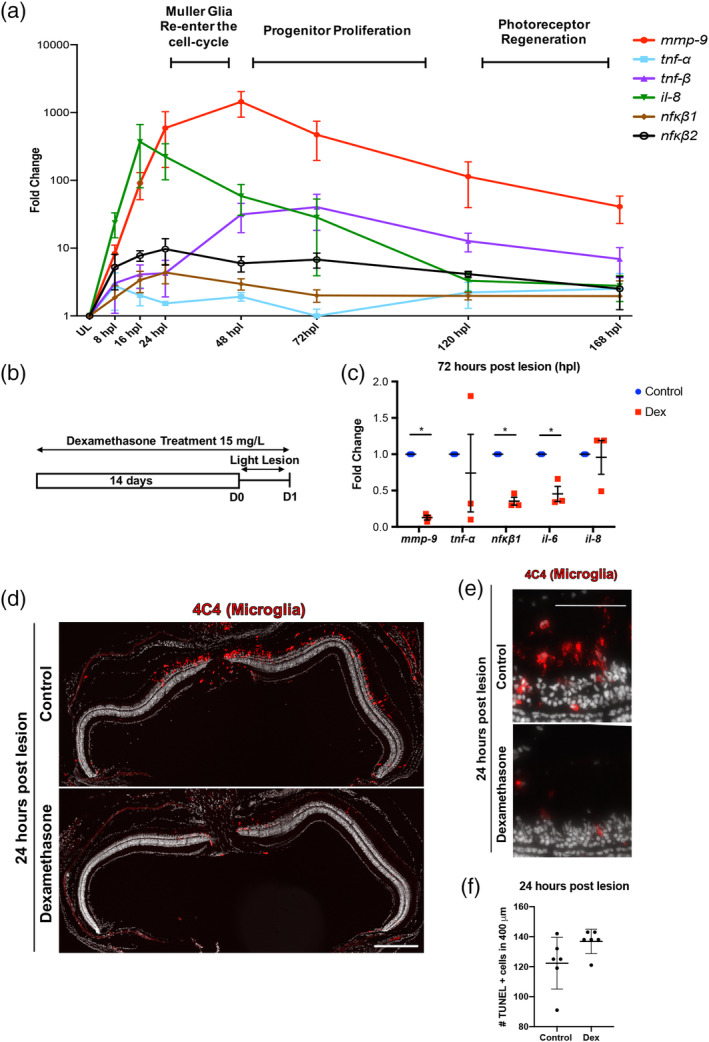
Inflammation is induced following photoreceptor ablation. Dexamethasone treatment suppresses microglia activation and cytokine production, but not photoreceptor death. (a) Time‐course for the expression of inflammatory genes, from 8 to 168 hr post lesion (hpl). Unlesioned retinas served as controls. Expression levels are represented as fold change calculated using DDC T method. (b) Experimental paradigm for the immune suppression. (c) qRT‐PCR for the inflammatory genes mmp‐9 (ANOVA F‐ratio = 22.237, p < .0001), tnf‐α (ANOVA F‐ratio = 1.5507, p = .2209), tnf‐β (ANOVA F‐ratio = 6.5120, p = .001), il‐8 (ANOVA F‐ratio = 8.2296, p = .0003), nfκb1 (ANOVA F‐ratio = 2.5207, p = .0596), and nfκb2 (ANOVA F‐ratio = 3.5591, p = .0168) from control and Dex‐treated retinas at 72 hpl. *p ≤ .05. (d) Immunostaining for microglia using the 4C4 antibody in control (top) and Dex‐treated retinas (bottom). (e) High magnification image of 4C4 staining at 1 dpl control (top) and Dex‐treated retinas (bottom). (f) The number of TUNEL positive cells at 1 dpl in control (122.27 ± 17.03 cells; n = 6) and Dex‐treated animals (136.93 ± 8.29 cells; n = 6). Scale bar equals 50 μm [Color figure can be viewed at wileyonlinelibrary.com]
4.2. Inflammation regulates injury‐induced proliferation and photoreceptor regeneration
To determine if inflammation regulates aspects of photoreceptor regeneration, the glucocorticoid steroid, dexamethasone (Dex), was used to suppress the inflammatory response. A 14‐day pre‐lesion treatment with Dex was chosen (Figure 1b) based on studies performed by Kyritsis et al. (Kyritsis et al., 2012; see also White et al., 2017; Tsarouchas et al., 2018; Caldwell et al., 2019; Bollaerts et al., 2019), and here the dose of Dex, route of administration and sizes and ages of animals were matched with Kyritsis et al. (2012). Whereas this lengthy treatment with Dex may affect homeostatic function in all cells (Juszczak & Stankiewicz, 2018), this treatment in the absence of a lesion does not directly alter constitutive proliferation (Figure S1a; Kyritsis et al., 2012; White et al., 2017; Caldwell et al., 2019), the number of Müller glia or the expression and localization of gfap/Gfap, markers of reactive gliosis (Figure S1b–e). However, pretreatment with Dex does alter the morphology and the number of microglia (Figure S2a,b). Following Dex exposure microglia are less ramified (Figure S2a) and there are significantly fewer transgene/mCherry+ microglia detected in the Dex‐treated retinas (Figure S2b). Further, in the Dex‐treated animals, expression of the inflammatory cytokines, tnfa, tnfb, and the transcriptional regulators of inflammation, nfkb1 and nfkb2, are significantly reduced (Figure S2c). These results indicate that Müller glia do not overtly respond to Dex‐mediated immunosuppression, however pretreatment with Dex suppresses microglial reactivity and the expression of genes that underlie the inflammatory response. To validate the effectiveness of the Dex treatment following light lesion, the expression levels of inflammatory genes at 72 hpl, and the response of microglia and cell death were quantified in control and Dex‐treated groups at 24 hpl. Compared with controls, the expression of mmp‐9, il‐6, and nfκb1 were significantly decreased following Dex treatment (Figure 1c), while, though there was marked variability in the data, the expression of tnf‐α and il‐8 were not significantly different from controls. Comparing the data in Figure S2c and Figure S1f suggest that Dex does not suppress the expression of il‐8. Consistent with reports from larval zebrafish and chicken, Dex treatment suppresses the activation of microglia and their migration to the outer nuclear layer in response to photoreceptor death (Figure 1d,e; White et al., 2017; Gallina, Zelinka, Cebulla, & Fischer, 2015). However, Dex treatment does not alter the extent of photoreceptor cell death (Figure 1f), indicating that in zebrafish Dex is not neuroprotective.
Assays of proliferation and photoreceptor regeneration were performed for control animals and animals treated with Dex. Dividing cells were labeled with BrdU between 48 and 72 hpl to mark progenitors that will give rise to regenerated photoreceptors (Figure 2a). To exclude proliferating microglia, BrdU‐positive cells in the subretinal space were not counted. At 72 hpl, there was significantly fewer BrdU‐labeled cells in Dex‐treated retinas compared to controls (Figure 2b,c). At 7 dpl, there were significantly fewer regenerated rod and cone photoreceptors in Dex‐treated groups compared to controls (Figure 2d–g). These results indicate that, consistent with data showing the requirement for inflammation in neuronal regeneration in the forebrain of zebrafish (Kyritsis et al., 2012), inflammatory mechanisms also promote photoreceptor regeneration in the retina.
Figure 2.
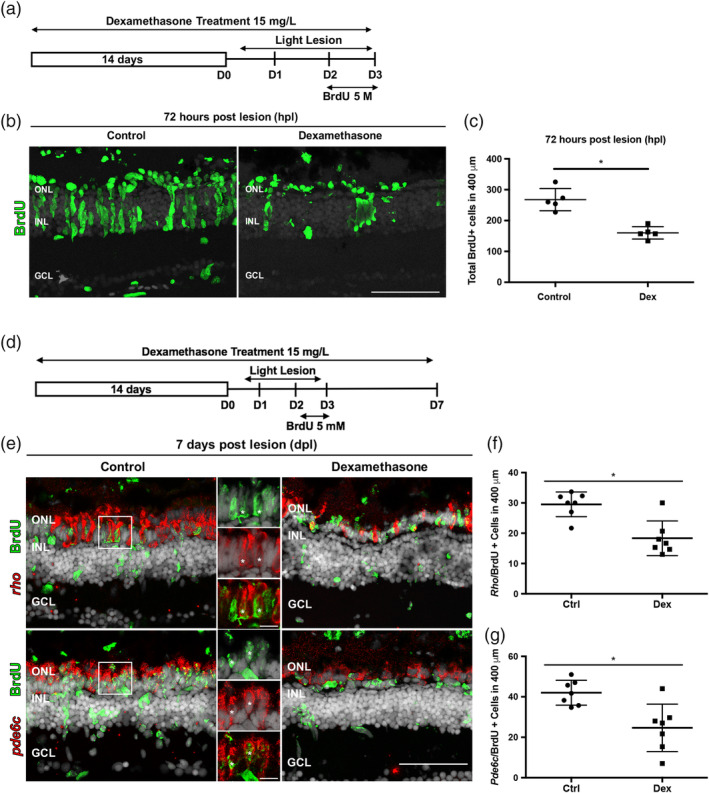
Dexamethasone treatment suppresses proliferation of Müller glia and photoreceptor regeneration. (a) Experimental paradigm of anti‐inflammatory treatment to assay proliferation. (b) BrdU immunostained cells (green) in controls (left) and Dex‐treated animals (right) at 72 hpl. (c) Number of BrdU‐labeled cells in controls (268 ± 36.1 cells; n = 5) and Dex‐treated animals (160.2 ± 20.02 cells; n = 5) at 72 hpl; *p = .0079. (d) Experimental paradigm of anti‐inflammatory treatment. Regenerated photoreceptors were identified as BrdU‐labeled nuclei surrounded by in situ hybridization signal for either rho (rods) or pde6c (cones) at 7 dpl. (e) Double labeled, regenerated photoreceptors using in situ hybridization for rods (rho) and cones (pde6c; red signal) and BrdU (green). The high magnification insets show the colocalization of the two labels (asterisks). (f) Number of regenerated rod photoreceptors in control (29.52 ± 4.1 cells; n = 7) and experimental retinas (18.33 ± 5.71 cells; n = 7); *p = .0047. (g) Number of regenerated cone photoreceptors in control (42 ± 6.1 cells; n = 7) and Dex‐treated animals (25 ± 11.72 cells; n = 7). *p = .0012. Scale bar equals 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer [Color figure can be viewed at wileyonlinelibrary.com]
4.3. Müller glia and Müller glia‐derived progenitors express mmp‐9
MMP‐9 belongs to a large family of Zn2 + −dependent proteases that target a variety of substrates, such as extracellular matrix (ECM), growth factors, chemokines, and cytokines. Mmp‐9 serves diverse functions among vertebrates (Iyer, Nicolle, Gregg, & Merry, 2012; Page‐McCaw, Andrew, & Zena, 2007; Pedersen, Tram, Sissel, & Svein, 2015; Vandooren, Van den Steen, et al., 2013). Mmp‐9 and Mmp‐2 belong to the subfamily of gelatinases, and genes encoding both Mmp‐9 and Mmp‐2 are upregulated by photoreceptor death, though the induction of Mmp‐9 is 10‐times greater than Mmp‐2 (Calinescu et al., 2009; (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13999; data not shown). Due to the magnitude of its induction (Figure 1a) and the varied roles played by mmp‐9 in injured/regenerating tissues and inflammation (Vandooren et al., 2014), the specific function of Mmp‐9 was investigated further. As a first step, in situ hybridization was performed to identify the cellular patterns of expression of mmp‐9. This was undertaken using the Müller glia reporter line, Tg[gfap:EGFP]mi2002 (Bernardos & Raymond, 2006), and BrdU labeling 24 hr prior to sacrifice. By in situ hybridization, mmp‐9 is not detectable in unlesioned retinas. In contrast, by 24 hpl, mmp‐9 is expressed in Müller glia, and this time‐point is prior to their entry into S‐phase of the cell cycle (Figure 3a). By 48 hpl, ~97% of BrdU‐labeled Müller glia express mmp‐9 (Figure 3a,b). At 72 hpl, the expression of mmp‐9 decreases (Figure 3a,c), and the cellular expression shifts to the Müller glia‐derived progenitors, as evidenced by the absence of eGFP in the BrdU‐labeled cells that express mmp‐9 (Figure 3a). The expression of mmp‐9 is also detected in very small number of microglia (data not shown). However, the majority of these cells reside in the subretinal space and were excluded from this analysis. These data indicated that photoreceptor ablation induces the expression of mmp‐9 in Müller glia, and this expression persists in Müller glia‐derived progenitors. Further, these results suggest that mmp‐9 identifies that subset of Müller glia that enter the cell cycle in response to a retinal injury.
Figure 3.
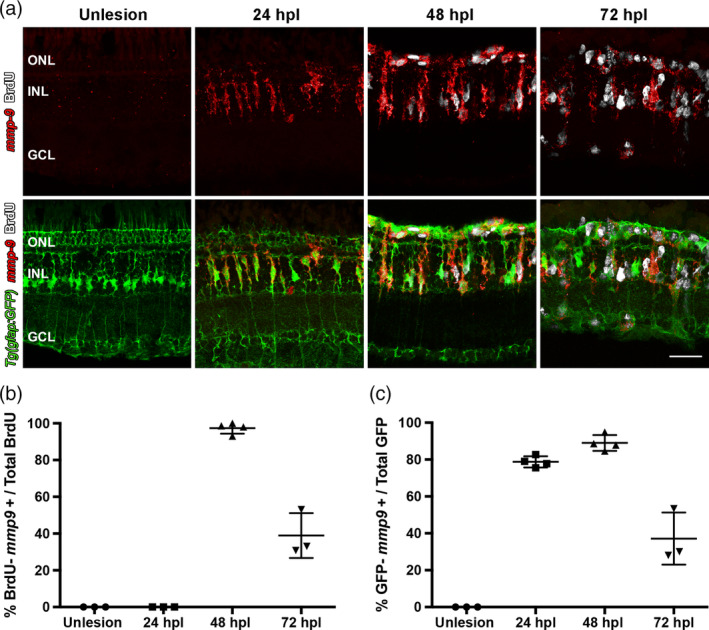
Following a photolytic lesion, Müller glia and Müller glia‐derived progenitors express mmp‐9. These experiments were performed using the transgenic line Tg[gfap:EGFP]mi2002, in which eGFP is expressed in Müller glia. (a) Triple labeling using in situ hybridization for mmp‐9 (red) and immunostaining for BrdU for dividing cells (white), combined with GFP immunostaining (green). (b) Number of BrdU+ cells that express mmp‐9 in unlesioned animals and lesioned animals at 24, 48, and 72 hpl. (c) Number of GFP+ Müller glia that also express mmp‐9 expression at 24, 48, and 72 hpl. Scale bar equals 25 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer [Color figure can be viewed at wileyonlinelibrary.com]
4.4. Mmp‐9 protein is present and catalytically active following photoreceptor death
To characterize protein synthesis and catalytic activity, Western blot analysis and zymogram assays were used, respectively (Vandooren, Geurts, et al., 2013; Vandooren, Van den Steen, et al., 2013; Figure 4). By these assays, Mmp‐9 is not detectable in unlesioned retinas, consistent with the in situ hybridization data (Figures 3a and 4a). In Western blots, at 16 hpl, the first time point sampled, Mmp‐9 is present at very low levels (Figure 4a). Mmp‐9 levels peak by 24 hpl, and then decrease between 24 and 72 hpl (Figure 4a,b). Mmp‐9 was originally named gelatinase B, because its substrate, shared with Mmp‐2, is denatured type‐1 collagen (gelatin). In zymography, the polyacrylamide gel contains gelatin, and after protein renaturation and gelatin staining, areas of protease activity appear as clear, negatively‐stained bands against the stained background (Chattopadhyay & Shubayev, 2009; Vandooren, Geurts, et al., 2013). The same protein samples used for the Western blot analysis were used for zymography. Recombinant, active human MMP‐9 was used as a positive control and shows strong catalytic activity as evidenced by a large negatively‐stained band (Figure 4c). Unlesioned retinas contain no detectable Mmp‐9 catalytic activity. In contrast, the catalytic activity parallels the data from the Western blot analysis (Figure 4c,d). Together, results from the Western blot and zymogram analyses show that catalytically‐active Mmp‐9 is induced by a lesion that selectively kills photoreceptors, and the time course of Mmp‐9 synthesis and catalytic activity parallels the proliferative phase of photoreceptor regeneration.
Figure 4.
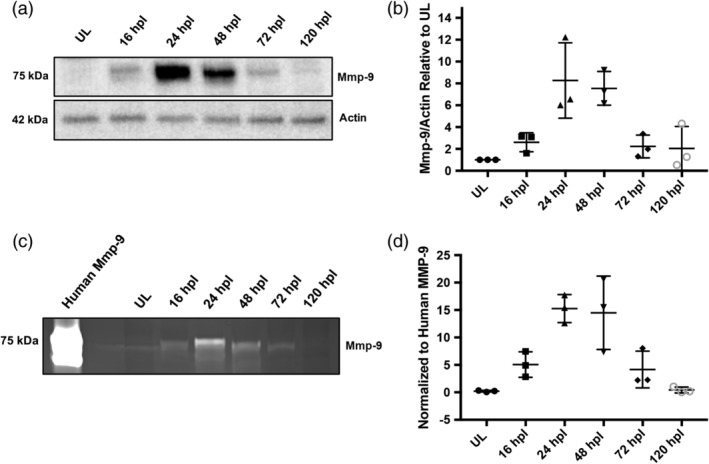
Mmp‐9 is expressed and catalytically active following photoreceptor death. (a) Western blot of retinal proteins from unlesioned retinas (UL) and lesioned retinas at 16, 24, 48, 72, and 120 hpl, stained with anti‐Mmp‐9 and anti‐Actin (loading control) antibodies. (b) Densitometry of Mmp‐9 protein, plotted relative to unlesioned controls. (ANOVA F‐ratio = 8.377, p = .0013) (c) Zymogram of Mmp‐9 catalytic activity from unlesioned retinas (UL) and lesioned retinas at 16, 24, 48, 72, and 120 hpl. Lanes in zymogram correspond to those in the Western blot. Purified human recombinant protein serves as a positive control. (d) Densitometry of Mmp‐9 catalytic activity, plotted relative to the positive control. (ANOVA F‐ratio = 11.870, p = .0003)
4.5. CRISPR mutants lack Mmp‐9 protein and catalytic activity
To investigate the function of mmp‐9 during photoreceptor regeneration, mutants were generated using CRISPR‐Cas9 (Hwang et al., 2013), targeted to the mmp‐9 catalytic domain within exon 2 (Figure 5a). The 19 base sgRNA produced several alleles, two of which were bred to homozygosity and characterized further, a 23 bp deletion (designated mi5003; ZFIN, https://zfin.org/) and an 8 bp insertion (designated mi5004; ZFIN, https://zfin.org/), respectively (Figure 5b). Individuals from the two lines were crossed to create compound heterozygotes to evaluate the potential for off target effects in either of the two independent lines (see next section). Each of the two mmp‐9 lines carries a mutation that results in a frameshift that predicts premature stop codons (Table S1). Retinas from mutants were characterized by Western blot analysis and zymography. Unlesioned retinas had no detectable Mmp‐9 or catalytic activity (Figure 5c,d). As expected, following photoreceptor ablation, Mmp‐9 protein and catalytic activity are present in wild‐type retinas (Figure 5c,d). In contrast, there was trace Mmp‐9 and catalytic activity in the mi5004 line, but no detectable Mmp‐9 or catalytic activity in mi5003 line (Figure 5c,d). Based on these results, subsequent experiments were conducting using the mi5003 line, though the basic observations reported here were confirmed in the mi5004 line (data not shown) and the compound heterozygotes (see below).
Figure 5.
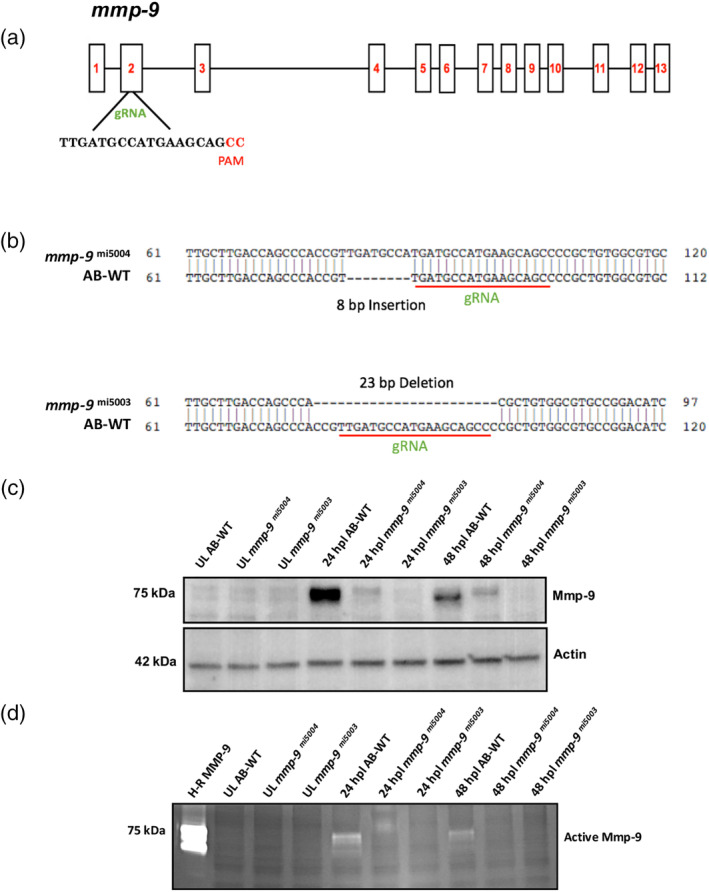
CRISPR mutants lack catalytically‐active Mmp‐9. (a) Genomic structure of mmp‐9 and the gRNA target sequence. (b) Sequence alignment for two indel mutations—8 bp insertion (mmp‐9 mi5004) and 23 bp deletion (mmp‐9 mi5003). Red underline indicates the sequence targeted by the 19 bp gRNA. (c) Western blot of retinal proteins from unlesioned retinas (UL wild‐type; UL mmp‐9 mi5004; UL mmp‐9 mi5003) and lesioned retinas (wild‐type; mmp‐9 mi5004; mmp‐9 mi5003) at 24, and 48 hpl stained with anti‐Mmp‐9 and anti‐Actin (loading control) antibodies. (d) Zymogram of Mmp‐9 catalytic activity. Lanes in zymogram correspond to those in the Western blot. Purified human recombinant protein serves as a positive control [Color figure can be viewed at wileyonlinelibrary.com]
4.6. In Mmp9 mutants, photoreceptor death results in overproduction of photoreceptor progenitors and regenerated photoreceptors
We first sought to determine if there were any differences in retinal development or adult starting points for the injury‐induced proliferation in the mmp‐9 mutants. At 72 hpf, there were no differences between wildtype and the two mutant lines in the size of the eyes or degree of proliferation (Figure S3a,b), indicating early retinal development was not altered by the absence of Mmp9. Similarly, qualitative comparisons of adult retinas showed the appearance of wildtype and mutant retinas did not vary in any identifiable manner (Figure S4a). Finally, comparing the number of Müller glia and the number of mitotic progenitors, which support the persistent neurogenesis in adult retinas (Raymond et al., 2006), showed there were no quantitative differences between wild‐type animals and the mi5003 line in the number of Müller glia or the level of intrinsic cell proliferation (Figure S4b,c).
To determine the consequence of Mmp‐9 loss‐of‐function on retinal regeneration, wild‐type and mutant animals received photolytic lesions. Counts of TUNEL‐labeled cells in retinal sections at 24hpl showed there were no differences in the extent of cell death between WT and mutant animals (131.2 ± 22.64 vs. 137.60 ± 28.98 in 400 μm; p = .6787; n = 6). At 1, 2, and 3 dpl, there were no obvious qualitative or quantitative differences in morphology and the number of microglia (data not shown). To evaluate the loss of Mmp‐9 on proliferation, Müller glia‐derived progenitors were labeled with BrdU between 48–72 hpl, and BrdU‐labeled cells were counted at 72 hpl. Relative to wild‐type animals, mutants had significantly more BrdU‐labeled progenitors at 72 hpl (Figure 6a,b). This over production of the Müller glia‐derived progenitors was also observed in the mi5004 line (data not shown) and compound heterozygotes (Figure S4d), indicating that the hyperproliferation is a consequence of the loss of Mmp9 and is unlikely to result from off‐target effects caused by the CRISPR‐cas9 technique.
Figure 6.
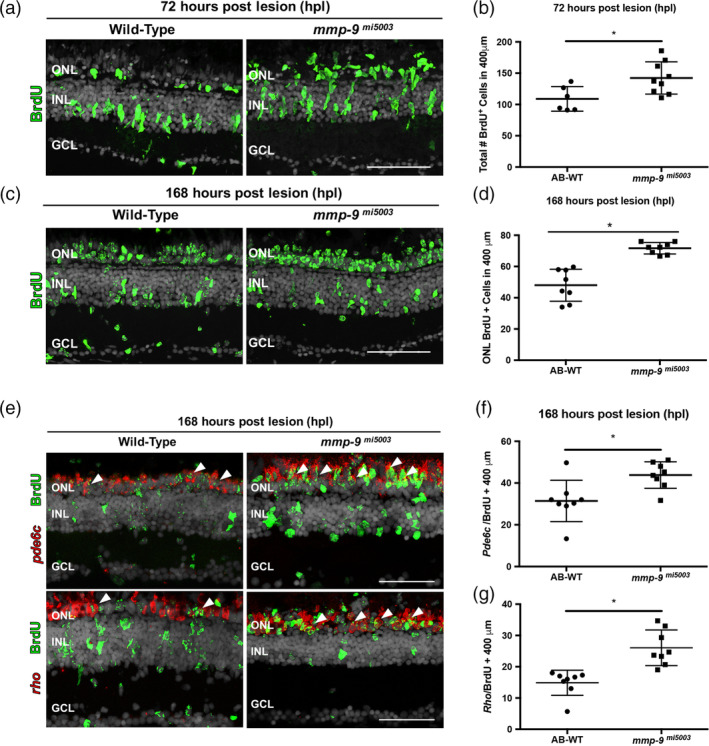
Photolytic lesions in mmp‐9 mutants results in the over production of injury‐induced progenitors and regenerated photoreceptors. (a) BrdU‐labeled cells (green) in wild‐type and mmp‐9 mi5003. (b) Number of BrdU+ cells from wild‐type (109 ± 19.66 cells; n = 6) mutant retinas (142.3 ± 25.72 cells; n = 9) at 72 hpl. *p = .0186. (c) BrdU‐labeled cells (green) in wild‐type and mutant retinas at 168 hpl. (d) Number of BrdU+ cells in the ONL of wild‐type (48 ± 10.24 cells; n = 8) and mutant retinas (71.71 ± 3.7 cells; n = 8). *p = .0001. (e) Double labeled, regenerated photoreceptors using in situ hybridization for rods (rho) and cones (pde6c; red signal) and BrdU (green) at 168hpl. (f) Number of regenerated cone photoreceptors in wild‐type (31.42 ± 9.88 cell; n = 8) and mutant retinas (43.83 ± 10.68 cells; n = 8) at 168 hpl. *p = .0301. (g) Number of regenerated rod photoreceptors in wild‐type (14.88 ± 4.02 cells; n = 8) and mutant retinas (26.04 ± 5.69 cells; n = 8) at 168 hpl. *p = .0005. Scale bars equal 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer [Color figure can be viewed at wileyonlinelibrary.com]
It is well established that Mmp‐9 plays a role in tissue remodeling and cell migration (Page‐McCaw et al., 2007). To determine if Mmp‐9 loss‐of‐function altered the migration of Müller glia‐derived progenitors from the inner nuclear layer to the outer nuclear layer, animals were subjected to photolytic lesions, exposed to BrdU between 48 and 72 hpl and allowed to survive to 7 dpl, a time point where proliferation and migration are complete and photoreceptor differentiation has commenced. Qualitative inspection showed that Mmp‐9 loss‐of‐function does not alter the migration of the Müller glia‐derived progenitors (Figure 6c). Further, cell counts showed that the over production of photoreceptor progenitors observed in mutants at 72 hpl results in significantly more BrdU‐labeled cells within the ONL at 7 dpl (Figure 6b,d). Finally, in the mutants at 7 dpl, there were significantly more regenerated rod and cone photoreceptors (Figure 6e‐g). Together, these data show that, in the absence of an injury, Mmp‐9 loss‐of‐function has no impact retinal development, the proliferative potential of the adult retina or the migration of Müller glia‐derived progenitors following photoreceptor death. The hyperproliferation of the photoreceptor progenitors in mutants indicates that Mmp‐9 functions as a negative regulator of proliferation among Müller glia‐derived progenitors, though the mechanistic details through which this is accomplished are not yet known (see Discussion). Further, though over‐produced, the presence of regenerated photoreceptors at 7 dpl indicates that Mmp‐9 plays no apparent role in governing the timing of cell cycle exit or the onset of photoreceptor differentiation.
4.7. Absence of Mmp‐9 results in elevated levels of tnf‐α
MMP‐9 can cleave inflammatory cytokines, and may modify cytokine signaling during the injury‐induced proliferation (Parks et al., 2004; Ando et al., 2017). To determine if the absence of MMP‐9 results in an altered inflammatory response following photoreceptor death, we compared the expression of inflammatory genes in wildtype fish and mutants. In wildtypes, although the fold increase is less, the expression of tnf‐β (Figure 7a), nfκb1 (Figure 7b), nfκb2 (Figure 7c), il‐8 (Figure 7d) and tnf‐α (Figure 7e) follow similar temporal patterns as observed in Figure 1a. In mutants, the expression of tnf‐β, nfκb1, nfκb2, and il‐8 do not differ from wildtypes, with exception that in mutant retinas the initial induction of il‐8 was significantly reduced (Figure 7a–d). In contrast, in mutants the expression of tnf‐α was significantly greater in both unlesioned retinas and lesioned retinas at all post‐lesion time‐points (Figure 7e). These data indicate that in the absence of Mmp‐9, tnf‐α levels are significantly elevated, and Mmp‐9 negatively regulates the expression of tnf‐α. In zebrafish Tnfα is necessary and sufficient to induce proliferation of Müller glia and Müller glia‐derived progenitors (Nelson et al., 2013; Conner et al., 2014), and we infer that elevated levels of tnf‐α in the Mmp9 mutants may account for the hyperproliferation among Müller glia‐derived progenitors.
Figure 7.
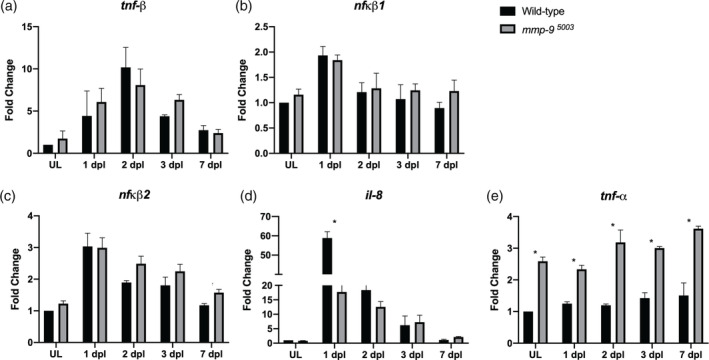
Absence of Mmp‐9 results in an altered inflammatory response following photoreceptor death. Time‐course of the expression of inflammatory genes, tnf‐β (a; ANOVA F‐ratio = 19.2406, p < .0001,), nfκβ1 (b; ANOVA F‐ratio = 10.4104, p < .0001), nfκβ2 (c; ANOVA F‐ratio = 10.4104, p < .0001), il‐8 (d; ANOVA F‐ratio = 185.42, p < .0001), and tnf‐α (e; ANOVA F‐ratio = 54.53, p < .0001) in Wild‐type (black bars) and Mmp‐9 mutant (gray bars) retinas following photoreceptor death. Fold changes relative to unlesioned retinas of Wild‐type are calculated using DDCt method. ANOVA with post‐hoc Tukey, *p < .0001
4.8. Mmp‐9 is required for the survival of regenerated cone photoreceptors
To determine if in the mutants the initial overproduction of photoreceptors persists, regenerated rod and cone photoreceptors were qualitatively and quantitatively compared in wild‐type and mutants at 21 dpl, a time point where regeneration is complete (Powell, Cornblath, Elsaeidi, Wan, & Goldman, 2016). For both groups, regenerated rod and cone photoreceptors were counted in sections and whole mount preparations. These analyses showed there were no qualitative differences between wild‐type and mutants in the appearance of regenerated rod photoreceptors (Figure 8a), and the over production of rod photoreceptors observed at 7 dpl was present at 21 dpl (Figure 8b,c).
Figure 8.
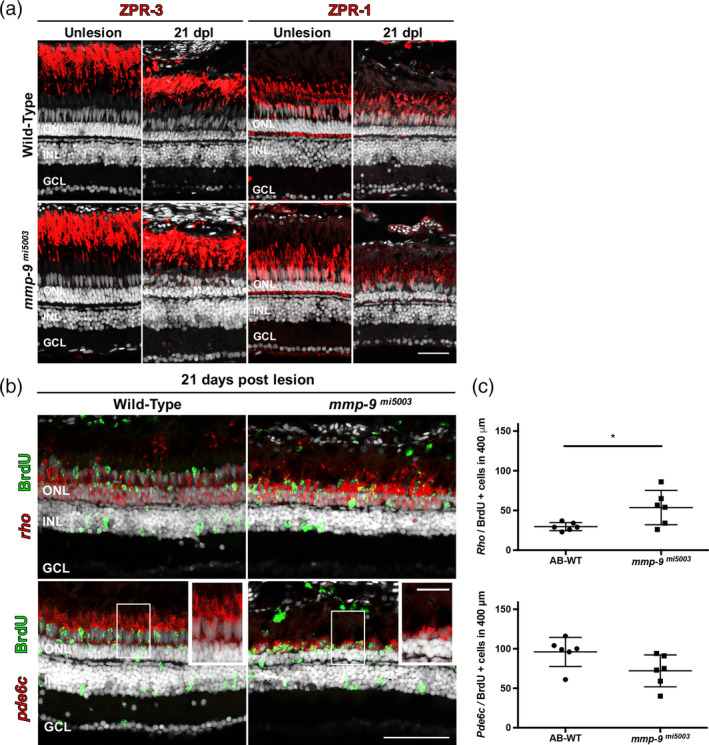
Mmp‐9 regulates the survival of regenerated cone photoreceptors. (a) Immunostaining for red‐green double cones with ZPR‐1 and rods with ZPR‐3. Wild‐type are in the top row; mutants are in the bottom row. Unlesioned retinas are left; lesioned retinas at 21 dpl are right. (b) Double labeled, regenerated photoreceptors at 21dpl using in situ hybridization for rods (rho) and cones (pde6c; red signal) and BrdU (green). Insets detail the morphology of regenerated photoreceptors in wild‐type (left) and mutant retinas (right). (c) Number of regenerated cone photoreceptors (bottom) in wild‐type (95.81 ± 18.49 cells; n = 6) and mutant retinas (71.89 ± 20.37 cells; n = 6) at 21 dpl. p = .059. Number of regenerated rod photoreceptors (top) in wild‐type (29.6 ± 4.94 cells; n = 6) and mutant retinas (53.39 ± 21.62 cells; n = 6) at 21 dpl. *p = .0270. Scale bar equals 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer [Color figure can be viewed at wileyonlinelibrary.com]
In contrast to the regenerated rods, in mutant retinas at 21 dpl, the survival of cones were markedly compromised. In mutants, regenerated cones had shorter outer segments and appeared to be fewer in number (Figure 8a, insets 8B). Counts of regenerated cones in tissues sections showed the initial over production of cones, evident at 7 dpl, was absent at 21 dpl (Figure 8c). Cones were also counted in retinal whole mounts (Figure 9a,b). In both wild‐type and mutant retinas, cone photoreceptors in unlesioned retinas are arranged in a very precise lattice mosaic, a feature of cone photoreceptors in teleost retinas (Nagashima et al., 2017; Figure 9a). There was no significant difference in the number of cone photoreceptors in unlesioned retinas from wild types and the mutants (Figure 9b). Following photoreceptor ablation and regeneration, areas of the retina that contain regenerated photoreceptors are identifiable by the marked spatial degradation of the mosaic, though individual cone photoreceptors remain readily identifiable (Nagashima et al., 2017). Counts of cones in whole mounts show that mutants had significantly fewer regenerated cones than wildtype animals (Figure 9a,b). Finally, as an independent measure of regenerated cones, Western blot analysis was performed using an antibody against Gnat‐2 (transducin), a cone‐specific protein (Figure 9c; Lagman, Callado‐Pérez, Franzén, Larhammar, & Abalo, 2015). Gnat‐2 levels in unlesioned retinas were comparable in wild‐type and mutant animals. In lesioned retinas, Gnat‐2 levels begin to recover in wild‐type animals by 14 dpl, and by 21 dpl values in wild‐type animals were nearly at the levels found in unlesioned retinas (Figure 9c,d). In contrast, Gnat‐2 levels in mutants lagged behind wild‐type levels, and at 21 dpl Gnat‐2 levels in mutant animals were significantly less than in wild‐types (Figure 9c,d). Western blot analysis using rhodopsin antibodies showed there were no differences in rod photoreceptors between wild‐type and mutant animals (data not shown). Together, these results indicate that the absence of Mmp‐9 results in defects specific to regenerated cone photoreceptors, well after injury‐induced proliferation and the initial differentiation of regenerated photoreceptors is complete.
Figure 9.
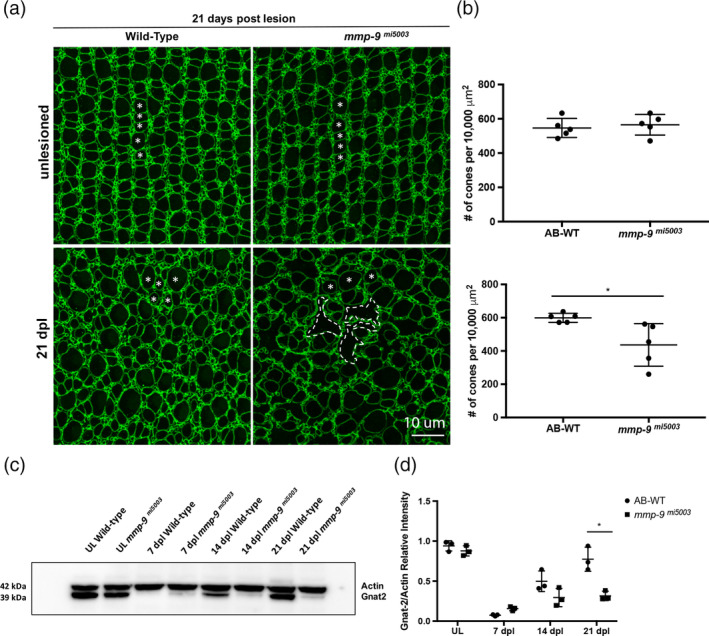
Mmp‐9 is required for the survival of regenerated cone photoreceptors. (a) Wholemounts of wild‐type (left) and mutant retinas (right) immunostained for ZO‐1. Unlesioned retinas are top; lesioned retinas at 21dpl are bottom. Asterisks indicate profiles of cones. In mutants, gaps due to missing cones are replaced by the irregular apical processes of Müller glia (dashed lines). (b) Number of unlesioned cones from wild‐type (546.49 ± 55.69; n = 5) and mmp‐9 mi5003 (565.33 ± 27.42; n = 5) *p = .516; below the number of regenerated cones from wild‐type (599.11 ± 27.42 cones; n = 5) and mutant retinas (436.09 ± 128.04 cones; n = 5) at 21 dpl. *p = .0238. (c) Western blot of retinas stained with antibodies against Gnat‐2 and Actin at 7, 14, and 21 dpl. (d) Densitometry of Gnat‐2 labeling in the Western blot. A significant difference in gnat‐2 levels were observed at 21 dpl. *p = .0014. Scale bar equals 10 μm [Color figure can be viewed at wileyonlinelibrary.com]
4.9. Immune suppression rescues regenerated cones in mmp‐9 mutants
MMP‐9 can modify elements of the inflammatory response (see above; Parks et al., 2004). We hypothesized that the defects in the survival of cone photoreceptors in the Mmp‐9 mutants is a consequence of an altered inflammatory milieu within the outer retina. Activated microglia are a hallmark of inflammation and are associated with retinal degenerative diseases (Ma, Lian Zhao, & Wong, 2012; Madeira, Boia, Santos, Ambrósio, & Santiago, 2015). For example, in rd10 mice, microglia phagocytose living photoreceptors, and genetic ablation of retinal microglia improves photoreceptor survival (Zhao et al., 2015; see also Gallina, Zelinka, & Fischer, 2014). Therefore, we first examined microglia in wildtype and mutant retinas. At 21 dpl, there were significantly more microglia in the mutant retinas (0.1 ± 0.3 cells/400 μm, n = 4 vs. 4 ± 2.2 cells/ 400 μm, n = 4; p = .0148; Figure 10a), suggesting that in the absence of Mmp‐9 there is elevated inflammation following photoreceptor regeneration. To determine if immunosuppression rescues the defects in the survival of regenerated cones, we treated mutants with vehicle solution or Dex between 3 and 13 dpl. This treatment was started after the initial formation of Müller glia‐derived progenitors and lasted throughout the phase of photoreceptor differentiation (Figure 10b). Relative to controls, in the retinas of Dex‐treated mutants there were few microglia (Figure 10c), indicating that Dex treatment in the Mmp‐9 mutant suppresses microglial activation. In the Dex‐treated mutants, regenerated cones were longer and appeared more mature than cones in control retinas (Figure 10d). Finally, there were significantly more regenerated cones in the Dex‐treated retinas than in the vehicle‐treated retinas (Figure 10e,f), and Dex treatment restored the spatial pattern of regenerated cones nearly to that observed in wild‐type animals (Figure 9b; Figure 10e,f). These results suggest that following photoreceptor regeneration the absence of Mmp‐9 results in an altered inflammatory environment within the outer retina, which selectively compromises the survival of cone photoreceptors. Further, our results suggest that in the regenerating retina microglia govern the survival regenerated photoreceptors.
Figure 10.
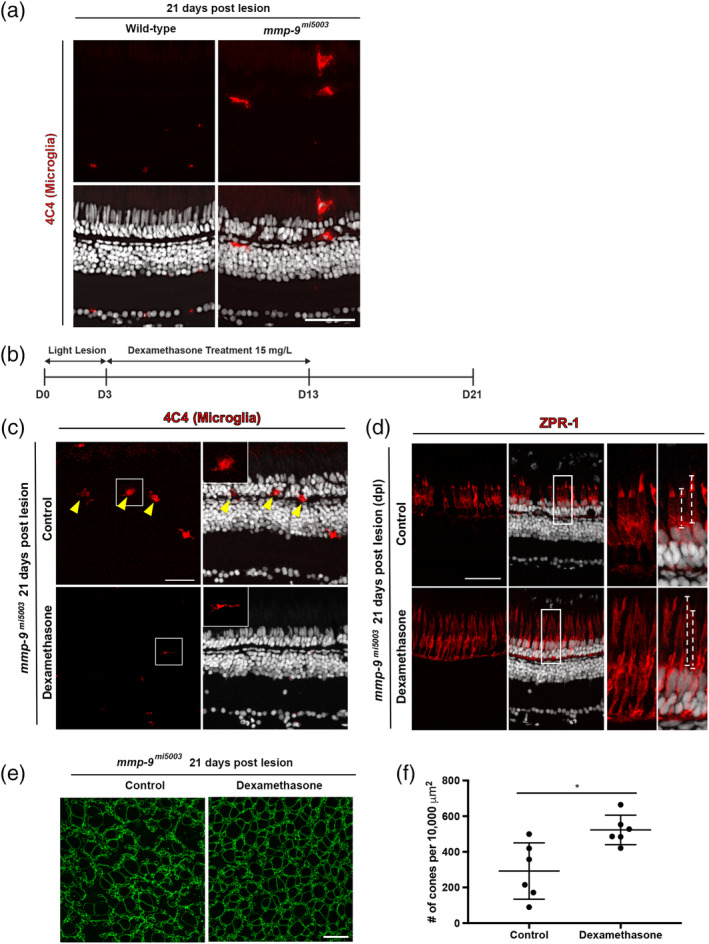
Late anti‐inflammatory treatment rescues defects of regenerated cones in mutants at 21 dpl. (a) Immunostaining for microglia marker, 4C4 in Wild‐type (0.1 ± 0.3 cells per 400 μm, n = 4) and mutant (4.0 ± 2.2 cells per 400 μm, n = 4, p = .0148) at 21 dpl. (b) Experimental paradigm for the photolytic lesions and Dexamethasone treatment. (c) Immunostaining for 4C4 in control (top) and Dex‐treated retinas (bottom). Inserts illustrate ameboid (top) and ramified (bottom) microglia. (d) Immunostaining with ZPR‐1 for red‐green double cones in control (top) and Dex mutants (bottom) 21 dpl. Insets illustrate differences in the lengths of the cone photoreceptors (dashed lines; control, 22.1 ± 7.2 μm, n = 40 cells; Dex‐treated, 29.7 ± 4.8 μm, n = 69 cells; p < .001) (e) Wholemounts of mutant retinas immunostained for ZO‐1. Control retina is left; Dex‐treated retina is right. (f) Number of regenerated cones in control (292.20 ± 158.11 cones; n = 6) and Dex‐treated retinas (522.79 ± 82.55 cones; n = 6) at 21 dpl. Scale bars equal 50 μm in panel (a), 40 μm in panels (c) and (d), and 10 μm in (e) [Color figure can be viewed at wileyonlinelibrary.com]
5. DISCUSSION
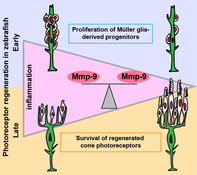
The results of this study are summarized in Figure 11. In response to photoreceptor death, Müller glia undergo a single asymmetric division through interkinetic nuclear migration to produce progenitors, which rapidly proliferate, migrate and differentiate to replace the ablated photoreceptors (Figure 11a,b). Dexamethasone‐induced immunosuppression inhibits proliferation of Müller glia‐derived progenitors, resulting in diminished photoreceptor regeneration (Figure 11c). Loss of Mmp‐9 function results in hyperproliferation and overproduction of regenerated photoreceptors (Figure 11d). However, the absence of Mmp‐9 leads to defects in the survival of cone photoreceptors (Figure 11d). Immunosuppression in mutants following photoreceptor regeneration rescues the survival defects (Figure 11e). Based on these results, we conclude that inflammation is required during the proliferative phase of photoreceptor regeneration, and Mmp‐9 negatively regulates proliferation of Müller glia and their progenitors. Finally, we conclude that Mmp‐9 is required for the survival of regenerated cone photoreceptors.
Figure 11.
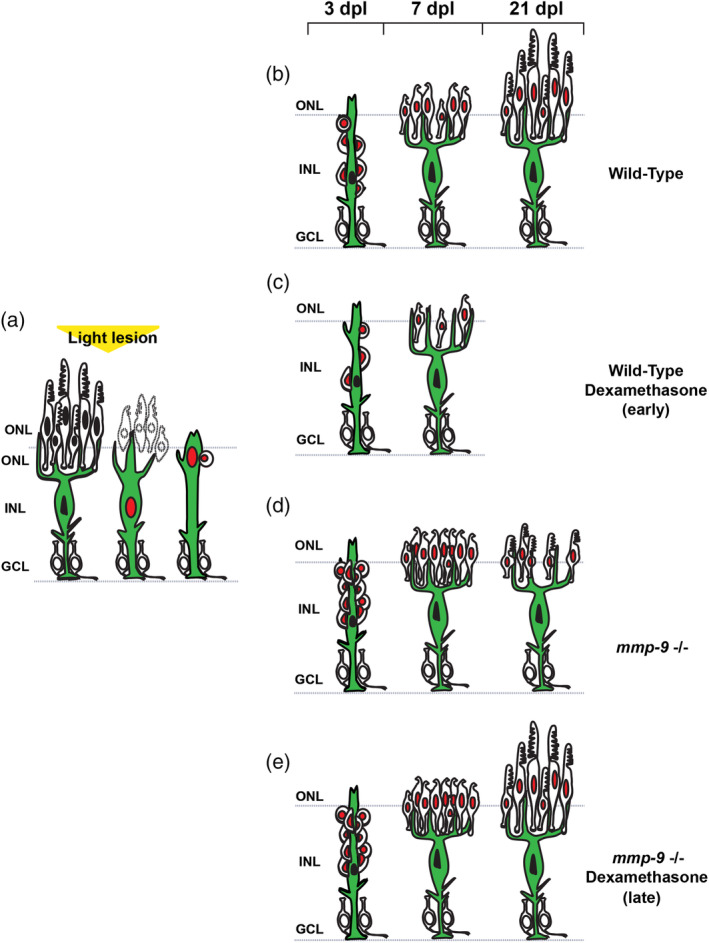
Summary diagram of cone regeneration. (a) Müller glia respond to photoreceptor death by expressing mmp‐9, undergoing interkinetic nuclear migration and a single asymmetric cell division that gives rise to a neuronal progenitor. (b) In wild‐type animals, neuronal progenitors form a neurogenic cluster around the Müller glia (3 dpl), migrate to the ONL, and differentiate into cone photoreceptors (7 dpl) that then mature (21 dpl). (c) Anti‐inflammatory treatment results in fewer Müller glia‐derived progenitors and fewer regenerated photoreceptors. (d) In the absence of Mmp‐9, there is overproduction of Müller glia‐derived progenitors and regenerating photoreceptors. However, at 21 dpl, survival of cone photoreceptors is compromised. (e) In the absence of Mmp‐9, anti‐inflammatory treatment rescues the defects of cone photoreceptors. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer [Color figure can be viewed at wileyonlinelibrary.com]
In the adult zebrafish, immune suppression using Dex inhibits the production of neural progenitors after a lesion (Caldwell et al., 2019; Kyritsis et al., 2012), and we show that treatment with Dex inhibits the proliferation of Müller glia‐derived progenitors. Following a neurotoxic lesion in the retina of the chick, Dex inhibits proliferation of Müller glia‐derived progenitors by activating glucocorticoid receptors on Müller glia (Gallina et al., 2014, 2015; see also Fischer, Zelinka, Gallina, Scott, & Todd, 2014). Our data suggest that Dex inhibits proliferation by altering the microglia response following photoreceptor death, however, we cannot exclude the possibility that in zebrafish Dex also acts directly on Müller glia or Müller glia‐derived progenitors. Regardless of the mechanisms of action, our data showing that immune suppression inhibits photoreceptor regeneration are consistent with previous studies utilizing immune suppression in regeneration paradigms and indicate that acute inflammation is a required component of successful neuronal regeneration in the vertebrate central nervous system.
MMP‐9 is a member of the gelatinases, which includes Mmp‐2. In the chick retina, gelatinase activity is present in unlesioned retinas. Following NMDA damage, global gelatinase activity diminishes slowly as Müller glia‐derived progenitors proliferate (Campbell et al., 2019). Selectively inhibiting Mmp‐9 activity has no effect on the proliferative response, whereas inhibiting MMP‐2 prior to damage or growth factor treatment potentiates proliferation. In the chick Mmp‐2 is produced by oligodendrocytes and non‐astrocyte inner retinal glia, a cell type unique to the chick. In contrast, in zebrafish, in the absence of a lesion, mmp‐2 and mmp‐9 are expressed at very low levels, and neuronal injury very rapidly induces the expression of Mmp‐9 in Müller glia destined to enter the cell cycle. The differences in gelatinase function in the chick and zebrafish, particularly within a tissue that is so highly evolutionarily conserved, is intriguing.
Mmp‐9 loss‐of‐function results in hyperproliferation of Müller glia‐derived progenitors, resulting initially in more regenerated photoreceptors. The overproduction of regenerated photoreceptors is interpreted simply to result from the overproduction of progenitors, and Mmp‐9 plays no role in the timing of cell cycle exit or initial photoreceptor differentiation. There are at least three interpretations that explain the overproduction of photoreceptor progenitors. First, the absence of Mmp‐9 may allow more Müller glia to enter the cell cycle. Previous studies showed that neuronal damage results in only 65–75% of the Müller glia entering the cell cycle (Nagashima et al., 2013). However, we demonstrated that mmp‐9 was expressed only by dividing Müller glia. The absence of proliferation in mmp‐9‐negative Müller glia argues against the possibility that additional Müller glia are recruited to the cell cycle. Second, in the mmp‐9 mutants Müller glia may undergo more than a one round of cell division, thereby increasing the size of the initial pool of Müller glia‐derived progenitors, which then divide at a normal rate. We do not favor this possibility, given a single asymmetric division among Müller glia is a hallmark of regenerative neurogenesis in the zebrafish retina. Third, in the mmp‐9 mutants, proliferation of the Müller glia‐derived progenitors is accelerated. We view this the likeliest of the three possibilities, and propose that, as an extracellular protease, Mmp‐9 regulates the concentration of cytokines and/or growth factors that regulate cell cycle kinetics within the niche of progenitors that cluster around each parental Müller glia, and in the absence of Mmp‐9, mitogenic signals persist, leading to accelerated proliferation (Bosak, Murata, Bludau, & Brand, 2018; Le et al., 2007; Luo et al., 2012; Manicone & Mcguire, 2008; Parks et al., 2004; Vandooren, Van den Steen, et al., 2013). Tnf‐α functions to activate proliferation among Müller glia and Müller glia‐derived progenitors during photoreceptor regeneration (Nelson et al., 2013). Our qPCR data showing elevated tnf‐α in the mutants is consistent with the possibility that in the absence of Mmp‐9 elevated levels of active Tnf‐α accelerate proliferation of Müller glia‐derived progenitors. However, additional studies will be required to determine if Tnf‐α is a direct catalytic substrate of Mmp‐9 and the mechanisms by which the tnf‐α transcript is regulated by this extracellular protease.
We show that immune suppression significantly suppresses mmp‐9 expression. This result is consistent with the literature indicating that Mmp‐9 is a component of the neuroinflammatory response. Following neuronal death, Müller glia secrete the inflammatory cytokines, tnf‐α, leptin, and il‐11 (Nelson et al., 2013; Zhao et al., 2014). Recent transcriptome and gene ontology analysis for Müller glia, isolated during photoreceptor injury and death, identified cytokine signaling and the immune response as activated pathways (Roesch, Stadler, & Cepko, 2012; Sifuentes, Kim, Swaroop, & Raymond, 2016). In the mammalian retina, neuronal death leads to persistent active gliosis in Müller glia and failure of the spontaneous expression of intrinsic reprogramming factors. This limits the ability for neuronal regeneration (Bringmann et al., 2006, 2009; Reichenbach & Bringmann, 2013; see also Ueki et al., 2015). Our data provide additional support that the innate immune response in the retina is required for stem cell‐based neuronal regeneration. It is interesting to note that the activation of cytokine pathways is conserved in the transcriptomes of Müller glia in both zebrafish and mouse models of photoreceptor degeneration (Roesch et al., 2012; Sifuentes et al., 2016), suggesting that photoreceptor death in vertebrates results in a common inflammatory response among Müller glia.
Recently, Kaur et al. (2018) demonstrated a regulatory feedback loop involving Shh and Mmp‐9 during retinal regeneration in zebrafish. They found that mmp‐9 is rapidly induced by a retinal injury, and pharmacological and genetic suppression of Shh signaling caused a significant upregulation in mmp‐9 and inhibited proliferation of Müller glia‐derived progenitors. The hyperproliferation we observed in the mmp‐9 mutants, and the conclusion that Mmp‐9 negatively regulates proliferation of Müller glia‐derived progenitors, is consistent with their data. Kaur et al. (2018) also show there is a positive feedback loop between Shh signaling pathways and Mmp‐9, suggesting that Shh may be a proteolytic substrate for the Mmp‐9. However, Kaur et al. (2018) also report that pharmacological and morpholino‐based inhibition of Mmp‐9 decreases the production of Müller glia‐derived progenitors. This latter observation is inconsistent with our data and raises the possibility that differences in technical approaches or injury paradigms (retinal stab wounds result in bloodborne factors and cells invading the retina (see White et al., 2017)), may account for differences in our respective data.
White et al. (2017) showed that in larval zebrafish the timing of immune suppression can delay or accelerate the regeneration of rod photoreceptors. As a pre‐treatment, dexamethasone suppresses the regeneration of rods. This result aligns with what we report here for adults—inflammatory events are required for the activation of Müller glia and proliferation of Müller glia‐derived progenitors. However, if dexamethasone treatment commences after rod photoreceptor ablation, regeneration kinetics are enhanced. The enhanced regeneration following delayed immune suppression parallels the results from the Mmp‐9 mutants, but it is unlikely a common mechanism can readily account for both results. The molecular milieu is complex in injured tissues, and the potential parallels between delayed suppression of inflammation and the absence of Mmp‐9 await further characterization.
We found that in the Mmp‐9 loss‐of‐function mutants, the survival of regenerated cone photoreceptors is compromised. It is well established that chronic inflammation is a risk factor for photoreceptor dystrophies in humans, such as retinitis pigmentosa and age‐related macular degeneration (AMD; Kauppinen, Paterno, Blasiak, Salminen, & Kaarniranta, 2016; Chen & Xu, 2012; Whitcup, Nussenblatt, Lightman, & Hollander, 2013; Yoshida et al., 2013). A subset of patients with AMD exhibit single nucleotide polymorphism (SNP) variants in the MMP‐9 gene, and elevated inflammation is associated with AMD pathogenesis (Fritsche et al., 2016; Kauppinen et al., 2016; Parks et al., 2004). Mutational variants in the inflammatory regulators, TIMP‐3, TGFB, TNF‐α are also identified in human patients with AMD (Fritsche et al., 2016). We hypothesize that during photoreceptor regeneration in zebrafish, Mmp‐9 function is required to modulate inflammation activated by the death of photoreceptors, and, in the mmp‐9 mutants, there an altered inflammatory environment that is selectively damaging to cones. The mmp‐9 mutants studied here may recapitulate elements of the pathologies that are characteristic of inflammatory photoreceptor diseases in humans, including AMD. Our study begins to shed light on the functional roles of Mmp‐9 in the retina and a potentially important link between intrinsic retinal immunity, human photoreceptor dystrophies and photoreceptor regeneration.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Figure S1 Dexamethasone pretreatment does not alter neurogenesis or induce reactive gliosis of Müller glia. (A) Number of BrdU labeled cells in pretreated unlesioned control (2.1 ± 1.7, n = 4) and dexamethasone (1.3 ± 1.9 n = 4). *p = .5400. (B) Number of Tg[gfap:EGFP]mi2002 + cells (Müller glia) in pretreated controls (65.18 ± 2.58 cells) and dexamethasone (64.88 ± 5.89 cells) groups; p = .9309. (C) A representative image of Tg[gfap:EGFP]mi2002 + cells (Müller glia) from pretreated control and dexamethasone. (D) A representative image of Gfap/Zrf1 + cells labeling Müller glia end feet in pretreated control and dexamethasone. (E) qRT‐PCR for gfap as a readout of gliosis from pretreated control and Dex‐treated retinas. p = .069. Scale bar equals 40 μm
Figure S2. Dexamethasone pretreatment alters the morphology and number of microglia number inflammatory gene expression. (A) A representative image of mpeg:mCherry + cells from pretreated unlesioned control and dexamethasone. (B) Quantification of mpeg:mCherry + cells (Microglia) from pretreated control (111 ± 20.42) and dexamethasone (56.2 ± 15.72). p = .0014. (C‐I) qRT‐PCR gene expression for inflammatory genes, tnf‐α (p = .0075), tnf‐β (p = .0264), nf‐b1 (p = .0287), nf‐b2 (p = .0302), mmp‐9 (p = .2017), il‐6 (p = .3079), and il‐8 (p = .3046) from pretreated control and dexamethasone (n = 3).*p ≤ .05. Scale bar equals 50 μm
Figure S3. Retinal development is unaltered by the absence of Mmp‐9. (A) Sections from 72 hpf larvae immunostained (arrowheads) for ganglion cells (ZN5), rod photoreceptors (ZPR‐3), and amacrine cells (HPC1). (B) Quantification of retinal area in wild‐type (722.36 ± 105.39 μm; n = 12) compared to mmp9 mutants (676.95 ± 75.05 μm; n = 9), p = .286. (C) Quantification of the area of EdU‐labeled cells, as an indicator of proliferation, in wild‐type (30 ± 7.84 μm; n = 12) and to mmp9 mutants (33.5 ± 6.59 μm; n = 9), p = .293. Scale bar equals 25 μm
Figure S4. Anatomy of adult retina in Mmp‐9 mutants. (A) Plastic sections illustrating cellular and synaptic layers in adult retinas from wildtype and Mmp‐9 mutants. These slightly oblique sections also reveal elements of the cone photoreceptor mosaic. (B) Processes of Müller glia (green) stained with the zrf‐1 antibody (top). BrdU‐labeled cells (red, indicated by arrowheads) in wild‐type and mutant retinas (bottom). (B) Immunostained Müller glia (top) and Brdu‐labeled cells (bottom) in wild‐type and mutant retinas. (C) Number of Müller glia in wild‐type (55.83 ± 1.23; n = 4) and mutant retinas (54.58 ± 2.54; n = 4), p = .409 and dividing cells (bottom) in wild‐type (3.66 ± 2.47; n = 4) and mutant retinas (2.75 ± .92; n = 4), p = .515. (D) BrdU‐labeled cells (red) in wild‐type (left) and transheterozygote (TH) retinas, qualitatively illustrating the overproduction of injury‐induced progenitors in transheterozygotes. Scale bars equal 25 μm
Table S1 (A) Amino acid sequence alignments for Mmp‐9 in wild‐type and mmp‐9 mi5003 and mmp‐9 mi5004 mutants. The red amino acids denote the target of the gRNA. Insertion or deletion introduced a frameshift resulting in alteration of the amino acid sequence (mmp‐9 mi5003) or protein truncation (mmp‐9 mi5004)
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health—R01EY07060 (PFH), T32EY013934 (NJS), P30EYO7003 (PFH), R01 EY024519 (DRH), U01EY027267 (DRH), and an unrestricted grant from the Research to Prevent Blindness, New York. The authors thank Dilip Pawar for technical assistance. Fish lines and reagents provided by ZIRC were supported by NIH‐NCRR Grant P40 RR01.
Silva NJ, Nagashima M, Li J, et al. Inflammation and matrix metalloproteinase 9 (Mmp‐9) regulate photoreceptor regeneration in adult zebrafish. Glia. 2020;68:1445–1465. 10.1002/glia.23792
Funding information National Eye Institute, Grant/Award Numbers: P30 EY07003 (PFH), R01 EY024519 (DRH), R01 EY07060 (PHF), U01EY027267 (DRH); Research to Prevent Blindness; NIH‐NCRR, Grant/Award Number: P40 RR01
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Amor, S. , Puentes, F. , Baker, D. , & van der Valk, P. (2010). Inflammation in neurodegenerative diseases. Immunology, 129, 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando, K. , Shibata, E. , Hans, S. , Brand, M. , & Kawakami, A. (2017). Osteoblast production by reserved progenitor cells in zebrafish bone regeneration and maintenance. Developmental Cell, 43, 643–650.e3. [DOI] [PubMed] [Google Scholar]
- Bernardos, R. L. , Barthel, L. K. , Meyers, J. R. , & Raymond, P. A. (2007). Late‐stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. The Journal of Neuroscience, 27, 7028–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardos, R. L. , & Raymond, P. A. (2006). GFAP transgenic zebrafish. Gene Expression Patterns, 6, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Bollaerts I, Van Houcke J, Beckers A, Lemmens K, Vanhunsel S, De Groef L, Moons L. 2019. Prior exposure to immunosuppressors sensitizes retinal microglia and accelerates optic nerve regeneration in zebrafish. Mediators of Inflammation. 6135795, 2019, 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans, C. , Chou, J. , & Werb, Z. (2014). Remodelling the extracellular matrix in development and disease. Nature Reviews. Molecular Cell Biology, 15, 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsini, A. , Zunszain, P. A. , Thuret, S. , & Pariante, C. M. (2015). The role of inflammatory cytokines as key modulators of neurogenesis. Trends in Neurosciences, 38, 145–157. [DOI] [PubMed] [Google Scholar]
- Bosak, V. , Murata, K. , Bludau, O. , & Brand, M. (2018). Role of the immune response in initiating central nervous system regeneration in vertebrates: Learning from the fish. The International Journal of Developmental Biology, 62, 403–417. [DOI] [PubMed] [Google Scholar]
- Bringmann, A. , Pannicke, T. , Grosche, J. , Francke, M. , Wiedemann, P. , Skatchkov, S. N. , … Reichenbach, A. (2006). Müller cells in the healthy and diseased retina. Progress in Retinal and Eye Research, 25, 397–424. [DOI] [PubMed] [Google Scholar]
- Bringmann, A. , Iandiev, I. , Pannicke, T. , Wurm, A. , Hollborn, M. , Wiedemann, P. , … Reichenbach, A. (2009). Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Progress in Retinal and Eye Research, 28, 423–451. [DOI] [PubMed] [Google Scholar]
- Campbell, W. A. , Deshmukh, A. , Blum, S. , Todd, L. , Mendonca, N. , Weist, J. , … Fischer, A. J. (2019). Matrix‐metalloproteinase expression and gelatinase activity in the avian retina and their influence on Müller glia proliferation. Experimental Neurology, 320, 0014–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell, L. J. , Davies, N. O. , Cavone, L. , Mysiak, K. S. , Semenova, S. A. , Panula, P. , … Becker, T. (2019). Regeneration of dopaminergic neurons in adult zebrafish depends on immune system activation and differs for distinct populations. The Journal of Neuroscience, 39, 4694–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calinescu, A.‐A. , Raymond, P. A. , & Hitchcock, P. F. (2009). Midkine expression is regulated by the circadian clock in the retina of the zebrafish. Visual Neuroscience, 26, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay, S. , & Shubayev, V. I. (2009). MMP‐9 controls Schwann cell proliferation and phenotypic remodeling via IGF‐1 and ErbB receptor‐mediated activation of MEK/ERK pathway. Glia, 57, 1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. , & Xu, H. (2012). Inflammation in age‐related macular degeneration—Implications for therapy In Khatami M. (Ed.), Inflammatory diseases—immunopathology, clinical and pharmacological bases (pp. 129–150). London, UK: IntechOpen; 10.5772/25658 [DOI] [Google Scholar]
- Conner, C. , Ackerman, K. M. , Lahne, M. , Hobgood, J. S. , & Hyde, D. R. (2014). Repressing notch signaling and expressing TNF α are sufficient to mimic retinal regeneration by inducing Müller glial proliferation to generate committed progenitor cells. Journal of Neuroscience, 34(43), 14403–14419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, R. , & Wedlich, D. (2001). PCR‐based RNA probes: A quick and sensitive method to improve whole mount embryo in situ hybridizations. BioTechniques, 30, 769–772 74. [PubMed] [Google Scholar]
- De Oliveira, S. , Reyes‐Aldasoro, C. C. , Candel, S. , Renshaw, S. A. , Mulero, V. , & Calado, A. (2013). Cxcl8 (IL‐8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. Journal of Immunology, 190, 4349–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman, B. E. , & Patterson, P. H. (2009). Cytokines and CNS development. Neuron, 64, 61–78. [DOI] [PubMed] [Google Scholar]
- Ekdahl, C. T. , Kokaia, Z. , & Lindvall, O. (2009). Brain inflammation and adult neurogenesis: The dual role of microglia. Neuroscience, 158, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Ellett, F. , Pase, L. , Hayman, J. W. , Andrianopoulos, A. , & Lieschke, G. J. (2011). mpeg1 promoter transgenes direct macrophage‐lineage expression in zebrafish. Blood, 117(4), e49–e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett, B. V. , & Goldman, D. (2006). A role for alpha1 tubulin‐expressing Müller glia in regeneration of the injured zebrafish retina. The Journal of Neuroscience, 26, 6303–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel, S. M. , Montgomery, J. E. , Burket, C. T. , & Hyde, D. R. (2007). Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. The Journal of Neuroscience, 14, 1712–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, A. J. , Zelinka, C. , Gallina, D. , Scott, M. A. , & Todd, L. (2014). Reactive microglia and macrophage facilitate the formation of Müller glia‐derived retinal progenitors. Glia, 62, 1608–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche, L. G. , Igl, W. , Bailey, J. N. C. , Grassmann, F. , Sengupta, S. , Bragg‐Gresham, J. L. , … Heid, I. M. (2016). A large genome‐wide association study of age‐related macular degeneration highlights contributions of rare and common variants. Nature Genetics, 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina, D. , Zelinka, C. , & Fischer, A. (2014). Glucocorticoid receptors in the retina, Müller glia and the formation of Müller glia‐derived progenitors. Development, 141, 3340–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina, D. , Zelinka, C. P. , Cebulla, C. M. , & Fischer, A. J . (2015). Activation of glucocorticoid receptors in Müller glia is protective to retinal neurons and suppresses microglial reactivity. Experimental Neurology, 273, 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemberling, M. , Bailey, T. J. , Hyde, D. R. , & Poss, K. D. (2013). The zebrafish as a model for complex tissue regeneration. Trends in Genetics, 29, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, D. (2014). Müller glial cell reprogramming and retina regeneration. Nature Reviews. Neuroscience, 15, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsuch, R. A. , & Hyde, D. R. (2014). Regulation of Müller glial dependent neuronal regeneration in the damaged adult zebrafish retina. Experimental Eye Research, 123, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramage, E. , D'Cruz, T. , Taylor, S. , Thummel, R. , & Hitchcock, P. F. (2015). Midkine‐a protein localization in the developing and adult retina of the zebrafish and its function during photoreceptor regeneration. PLoS One, 10, e0121789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindi, S. M. , Shin, J. , Ogura, Y. , Li, H. , & Kumar, A. (2013). Matrix Metalloproteinase‐9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS One, 8, e72121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, W. Y. , Fu, Y. , Reyon, D. , Maeder, M. L. , Tsai, S. Q. , Sander, J. D. , … Keith Joung, J. (2013). Efficient genome editing in zebrafish using a CRISPR‐Cas system. Nature Biotechnology, 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, , G. A. , Schmitt, E. A. , Fadool, J. M. , & Dowling, J. E. (1996). Retinoic acid alters photoreceptor development in vivo. Proceedings of the National Academy of Sciences of the United States of America, 93, 13298–13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R. P. , Nicolle, L. P. , Gregg, B. F. , & Merry, L. L. (2012). The history of matrix metalloproteinases: Milestones, myths, and misperceptions. American Journal of Physiology, 303, H919–H930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszczak, G. R. , & Stankiewicz, A. M. (2018). Glucocorticoids, genes and brain function. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 82, 136–168. [DOI] [PubMed] [Google Scholar]
- Karra, R. , Knecht, A. K. , Kikuchi, K. , & Poss, K. D. (2015). Myocardial NF‐κB activation is essential for zebrafish heart regeneration. Proceedings of the National Academy of Sciences of the United States of America, 112, 13255–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen, A. , Paterno, J. J. , Blasiak, J. , Salminen, A. , & Kaarniranta, K. (2016). Inflammation and its role in age‐related macular degeneration. Cellular and Molecular Life Sciences, 73, 1765–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur, S. , Gupta, S. , Chaudhary, M. , Khursheed, M. A. , Mitra, S. , Kurup, A. J. , & Ramachandran, R. (2018). Let‐7 MicroRNA‐mediated regulation of Shh signaling and the gene regulatory network is essential for retina regeneration. Cell Reports, 23, 1409–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizil, C. , Kaslin, J. , Kroehne, V. , & Brand, M. (2012). Adult neurogenesis and brain regeneration in zebrafish. Developmental Neurobiology, 72, 429–461. [DOI] [PubMed] [Google Scholar]
- Kizil, C. , Kyritsis, N. , & Brand, M. (2015). Effects of inflammation on stem cells: Together they strive? EMBO Reports, 16, 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyritsis, N. , Kizil, C. , & Brand, M. (2014). Neuroinflammation and central nervous system regeneration in vertebrates. Trends in Cell Biology, 24, 128–135. [DOI] [PubMed] [Google Scholar]
- Kyritsis, N. , Kizil, C. , Zocher, S. , Kroehne, V. , Kaslin, J. , Freudenreich, D. , … Brand, M. (2012). Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science, 338, 1353–1356. [DOI] [PubMed] [Google Scholar]
- Lagman, D. , Callado‐Pérez, A. , Franzén, I. E. , Larhammar, D. , & Abalo, X. M. (2015). Transducin duplicates in the zebrafish retina and pineal complex: Differential specialisation after the teleost tetraploidisation. PLoS One, 10, e0121330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBert, D. C. , Squirrell, J. M. , Rindy, J. , Broadbridge, E. , Lui, Y. , Zakrzewska, A. , … Huttenlocher, A. (2015). Matrix metalloproteinase 9 modulates collagen matrices and wound repair. Development, 142, 2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski, J. R. , & Raymond, P. A. (2014). Müller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Progress in Retinal and Eye Research, 40, 94–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, N. T. V. , Xue, M. , Castelnoble, L. A. , & Jackson, C. J. (2007). The dual personalities of matrix metalloproteinases in inflammation. Frontiers in Bioscience, 12, 1475–1487. [DOI] [PubMed] [Google Scholar]
- Liddiard, K. , Rosas, M. , Davies, L. C. , Jones, S. A. , & Taylor, P. R. (2011). Macrophage heterogeneity and acute inflammation. European Journal of Immunology, 41, 2503–2508. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Uribe, R. A. , Hayton, S. , Calinescu, A.‐A. , Gross, J. M. , & Hitchcock, P. F. (2012). Midkine‐A functions upstream of Id2a to regulate cell cycle kinetics in the developing vertebrate retina. Neural Development, 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, W. , Zhao, L. , & Wong, W. T. (2012). Microglia in the outer retina and their relevance to pathogenesis of age‐related macular degeneration In: LaVail M., Ash J., Anderson R., Hollyfield J., Grimm C. (eds) Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology, Vol. 723, (pp. 37–42). Boston, MA: Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira, M. H. , Boia, R. , Santos, P. F. , Ambrósio, A. F. , & Santiago, A. R. (2015). Contribution of microglia‐mediated Neuroinflammation to retinal degenerative diseases. Mediators of Inflammation, 2015, 673090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicone, A. , & Mcguire, J. (2008). Matrix metalloproteinases as modulators of inflammation. Seminars in Cell & Developmental Biology, 19, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, J. F. , & Poché, R. A. (2019). Awakening the regenerative potential of the mammalian retina. Development, 146, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masure, S. , Proost, P. , Van Damme, J. , & Opdenakker, G. (1991). Purification and identification of 91‐kDa neutrophil gelatinase. Release by the activating peptide interleukin‐8. European Journal of Biochemistry, 198, 391–398. [DOI] [PubMed] [Google Scholar]
- Nagase, H. , Visse, R. , & Murphy, G. (2006). Structure and function of matrix Metalloproteinases and TIMPs. Cardiovascular Research, 69, 562–573. [DOI] [PubMed] [Google Scholar]
- Nagashima, M. , Barthel, L. K. , & Raymond, P. A. (2013). A self‐renewing division of zebrafish Müller glial cells generates neuronal progenitors that require N‐cadherin to regenerate retinal neurons. Development, 140, 4510–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima, M. , Hadidjojo, J. , Barthel, L. K. , Lubensky, D. K. , & Raymond, P. A. (2017). Anisotropic Müller glial scaffolding supports a multiplex lattice mosaic of photoreceptors in zebrafish retina. Neural Development, 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan, C. (2002). Points of control in inflammation. Nature, 420, 846–852. [DOI] [PubMed] [Google Scholar]
- Nelson, C. M. , Ackerman, K. M. , O'Hayer, P. , Bailey, T. J. , Gorsuch, R. A. , & Hyde, D. R. (2013). Tumor necrosis factor‐alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. The Journal of Neuroscience, 33, 6524–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochocinska, M. J. , & Hitchcock, P. F. (2007). Dynamic expression of the basic helix‐loop‐helix transcription factor neuroD in the rod and cone photoreceptor lineages in the retina of the embryonic and larval zebrafish. The Journal of Comparative Neurology, 501, 1–12. [DOI] [PubMed] [Google Scholar]
- Opdenakker, G. , Masure, S. , Proost, P. , Billiau, A. , & van Damme, J. (1991). Natural human monocyte gelatinase and its inhibitor. FEBS Letters, 284, 73–78. [DOI] [PubMed] [Google Scholar]
- Page‐McCaw, A. , Andrew, J. E. , & Zena, W. (2007). Matrix Metalloproteinases and the regulation of tissue Remodelling. Nature Reviews. Molecular Cell Biology, 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, W. C. , Wilson, C. L. , & López‐Boado, Y. S. (2004). Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature Reviews. Immunology, 4, 617–629. [DOI] [PubMed] [Google Scholar]
- Pedersen, M. E. , Tram, T. V. , Sissel, B. R. , & Svein, O. K. (2015). Matrix metalloproteinases in fish biology and matrix turnover. Journal of International Society for Matrix Biology, 44–46, 86–93. [DOI] [PubMed] [Google Scholar]
- Powell, C. , Cornblath, E. , Elsaeidi, F. , Wan, J. , & Goldman, D. (2016). Zebrafish Müller glia‐derived progenitors are multipotent, exhibit proliferative biases and regenerate excess neurons. Scientific Reports, 6, 24851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond, P. A. , Barthel, L. K. , Bernardos, R. L. , & Perkowski, J. J. (2006). Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Developmental Biology, 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach, A. , & Bringmann, A. (2013). New functions of Müller cells. Glia, 61, 651–678. [DOI] [PubMed] [Google Scholar]
- Roesch, K. , Stadler, M. B. , & Cepko, C. L. (2012). Gene expression changes within Müller glial cells in retinitis pigmentosa. Molecular Vision, 18, 1197–1214. [PMC free article] [PubMed] [Google Scholar]
- Sherpa, T. , Fimbel, S. M. , Mallory, D. E. , Maaswinkel, H. , Spritzer, S. D. , Sand, J. A. , … Stenkamp, D. L. (2008). Ganglion cell regeneration following whole‐retina destruction in zebrafish. Developmental Neurobiology, 68, 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubayev, V. I. , Angert, M. , Dolkas, J. , Marie Campana, W. , Palenscar, K. , & Myers, R. R. (2006). TNFα‐induced MMP‐9 promotes macrophage recruitment into injured peripheral nerve. Molecular and Cellular Neurosciences, 31, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifuentes, C. J. , Kim, J.‐W. , Swaroop, A. , & Raymond, P. A. (2016). Rapid, dynamic activation of Müller glial stem cell responses in zebrafish. Investigative Ophthalmology & Visual Science, 57, 5148–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S. M. , Alvarez‐Delfin, K. , Saade, C. J. , Thomas, J. L. , Thummel, R. , Fadool, J. M. , & Hitchcock, P. F. (2015). The bHLH transcription factor NeuroD governs photoreceptor genesis and regeneration through Delta‐notch signaling. Investigative Ophthalmology & Visual Science, 56, 7496–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsarouchas, T. M. , Wehner, D. , Cavone, L. , Munir, T. , Keatinge, M. , Lamvertus, M. , … Becker, C. G. (2018). Dynamic control of proinflammatory cytokines il‐1β and Tnf‐α by macrophages in zebrafish spinal cord regeneration. Nature Communications, 9, 4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J. L. , Ranski, A. H. , Morgan, G. W. , & Thummel, R. (2016). Reactive gliosis in the adult zebrafish retina. Experimental Eye Research, 143, 98–109. [DOI] [PubMed] [Google Scholar]
- Ueki, Y. , Wilken, M. S. , Cox, K. E. , Chipman, L. , Jorstad, N. , Sternhagen, K. , … Reh, T. A. (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proceedings of National Academy of Sciences of the United States of America, 112, 13717–13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandooren, J. , Geurts, N. , Martens, E. , Van den Steen, P. E. , & Opdenakker, G. (2013). Zymography methods for visualizing hydrolytic enzymes. Nature Methods, 10, 211–220. [DOI] [PubMed] [Google Scholar]
- Vandooren, J. , Van den Steen, P. E. , & Opdenakker, G. (2013). Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase‐9 (MMP‐9): The next decade. Critical Reviews in Biochemistry and Molecular Biology, 48, 222–272. [DOI] [PubMed] [Google Scholar]
- Vandooren, J. , Van Damme, J. , & Opdenakker, G. (2014). On the structure and functions of gelatinase B/matrix metalloproteinase‐9 in neuroinflammation. Progress in Brain Research, 214, 193–206. [DOI] [PubMed] [Google Scholar]
- Vecil, G. G. , Larsen, P. H. , Corley, S. M. , Herx, L. M. , Besson, A. , Goodyer, C. G. , & Yong, V. W. (2000). Interleukin‐1 is a key regulator of matrix metalloproteinase‐9 expression in human neurons in culture and following mouse brain trauma in vivo. Journal of Neuroscience Research, 61, 212–224. [DOI] [PubMed] [Google Scholar]
- Whitcup, S. M. , Nussenblatt, R. B. , Lightman, S. L. , & Hollander, D. A. (2013). Inflammation in retinal disease. International Journal of Inflammation, 2013, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, D. T. , Sengupta, S. , Saxena, M. T. , Xu, Q. , Hanes, J. , Ding, D. , … Mumm, J. S. (2017). Immunomodulation‐accelerated neuronal regeneration following selective rod photoreceptor cell ablation in the zebrafish retina. Proceedings of National Academy of Sciences of the United States of America, 114, E3719–E3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Q. , Cao, L. , Chen, X.‐Y. , Zhao, J. , Gao, L. , Li, S.‐Z. , & Fei, Z. (2017). High expression of MMP9 in glioma affects cell proliferation and is associated with patient survival rates. Oncology Letters, 13, 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, N. , Ikeda, Y. , Notomi, S. , Ishikawa, K. , Murakami, Y. , Hisatomi, T. , … Ishibashi, T. (2013). Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology, 120, e5–e12. [DOI] [PubMed] [Google Scholar]
- Zhao, L. , Zabel, M. , Wang, X. , Ma, W. , Shah, P. , Fariss, R. N. , … Wong, W. T. (2015). Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO, 7(9), 1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X.‐F. , Wan, J. , Powell, C. , Ramachandran, R. , Myers, M. G. , & Goldman, D. (2014). Leptin and IL‐6 family cytokines synergize to stimulate Müller glia reprogramming and retina regeneration. Cell Reports, 9, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Dexamethasone pretreatment does not alter neurogenesis or induce reactive gliosis of Müller glia. (A) Number of BrdU labeled cells in pretreated unlesioned control (2.1 ± 1.7, n = 4) and dexamethasone (1.3 ± 1.9 n = 4). *p = .5400. (B) Number of Tg[gfap:EGFP]mi2002 + cells (Müller glia) in pretreated controls (65.18 ± 2.58 cells) and dexamethasone (64.88 ± 5.89 cells) groups; p = .9309. (C) A representative image of Tg[gfap:EGFP]mi2002 + cells (Müller glia) from pretreated control and dexamethasone. (D) A representative image of Gfap/Zrf1 + cells labeling Müller glia end feet in pretreated control and dexamethasone. (E) qRT‐PCR for gfap as a readout of gliosis from pretreated control and Dex‐treated retinas. p = .069. Scale bar equals 40 μm
Figure S2. Dexamethasone pretreatment alters the morphology and number of microglia number inflammatory gene expression. (A) A representative image of mpeg:mCherry + cells from pretreated unlesioned control and dexamethasone. (B) Quantification of mpeg:mCherry + cells (Microglia) from pretreated control (111 ± 20.42) and dexamethasone (56.2 ± 15.72). p = .0014. (C‐I) qRT‐PCR gene expression for inflammatory genes, tnf‐α (p = .0075), tnf‐β (p = .0264), nf‐b1 (p = .0287), nf‐b2 (p = .0302), mmp‐9 (p = .2017), il‐6 (p = .3079), and il‐8 (p = .3046) from pretreated control and dexamethasone (n = 3).*p ≤ .05. Scale bar equals 50 μm
Figure S3. Retinal development is unaltered by the absence of Mmp‐9. (A) Sections from 72 hpf larvae immunostained (arrowheads) for ganglion cells (ZN5), rod photoreceptors (ZPR‐3), and amacrine cells (HPC1). (B) Quantification of retinal area in wild‐type (722.36 ± 105.39 μm; n = 12) compared to mmp9 mutants (676.95 ± 75.05 μm; n = 9), p = .286. (C) Quantification of the area of EdU‐labeled cells, as an indicator of proliferation, in wild‐type (30 ± 7.84 μm; n = 12) and to mmp9 mutants (33.5 ± 6.59 μm; n = 9), p = .293. Scale bar equals 25 μm
Figure S4. Anatomy of adult retina in Mmp‐9 mutants. (A) Plastic sections illustrating cellular and synaptic layers in adult retinas from wildtype and Mmp‐9 mutants. These slightly oblique sections also reveal elements of the cone photoreceptor mosaic. (B) Processes of Müller glia (green) stained with the zrf‐1 antibody (top). BrdU‐labeled cells (red, indicated by arrowheads) in wild‐type and mutant retinas (bottom). (B) Immunostained Müller glia (top) and Brdu‐labeled cells (bottom) in wild‐type and mutant retinas. (C) Number of Müller glia in wild‐type (55.83 ± 1.23; n = 4) and mutant retinas (54.58 ± 2.54; n = 4), p = .409 and dividing cells (bottom) in wild‐type (3.66 ± 2.47; n = 4) and mutant retinas (2.75 ± .92; n = 4), p = .515. (D) BrdU‐labeled cells (red) in wild‐type (left) and transheterozygote (TH) retinas, qualitatively illustrating the overproduction of injury‐induced progenitors in transheterozygotes. Scale bars equal 25 μm
Table S1 (A) Amino acid sequence alignments for Mmp‐9 in wild‐type and mmp‐9 mi5003 and mmp‐9 mi5004 mutants. The red amino acids denote the target of the gRNA. Insertion or deletion introduced a frameshift resulting in alteration of the amino acid sequence (mmp‐9 mi5003) or protein truncation (mmp‐9 mi5004)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.