

Abstract
Background and Aims
Cholestatic liver disease is characterized by gut dysbiosis and excessive toxic hepatic bile acids (BAs). Modification of gut microbiota and repression of BA synthesis are potential strategies for the treatment of cholestatic liver disease. The purpose of this study was to examine the effects and to understand the mechanisms of the probiotic Lactobacillus rhamnosus GG (LGG) on hepatic BA synthesis, liver injury, and fibrosis in bile duct ligation (BDL) and multidrug resistance protein 2 knockout (Mdr2−/−) mice.
Approach and Results
Global and intestine‐specific farnesoid X receptor (FXR) inhibitors were used to dissect the role of FXR. LGG treatment significantly attenuated liver inflammation, injury, and fibrosis with a significant reduction of hepatic BAs in BDL mice. Hepatic concentration of taurine‐β‐muricholic acid (T‐βMCA), an FXR antagonist, was markedly increased in BDL mice and reduced in LGG‐treated mice, while chenodeoxycholic acid, an FXR agonist, was decreased in BDL mice and normalized in LGG‐treated mice. LGG treatment significantly increased the expression of serum and ileum fibroblast growth factor 15 (FGF‐15) and subsequently reduced hepatic cholesterol 7α‐hydroxylase and BA synthesis in BDL and Mdr2−/− mice. At the molecular level, these changes were reversed by global and intestine‐specific FXR inhibitors in BDL mice. In addition, LGG treatment altered gut microbiota, which was associated with increased BA deconjugation and increased fecal and urine BA excretion in both BDL and Mdr2−/− mice. In vitro studies showed that LGG suppressed the inhibitory effect of T‐βMCA on FXR and FGF‐19 expression in Caco‐2 cells.
Conclusion
LGG supplementation decreases hepatic BA by increasing intestinal FXR–FGF‐15 signaling pathway–mediated suppression of BA de novo synthesis and enhances BA excretion, which prevents excessive BA‐induced liver injury and fibrosis in mice.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BA
bile acid
- BDL
bile duct ligation
- BSEP
bile salt export pump
- BSH
bile salt hydrolase
- C4
7‐alpha‐hydroxy‐4‐cholesten‐3‐one
- CDCA
chenodeoxycholic acid
- CFU
colony‐forming units
- CK‐19
cytokeratin 19
- Cyp2c70
cytochrome P450 family 2, subfamily c, polypeptide 70
- CYP7A1
cholesterol 7α‐hydroxylase
- CYP8B1
sterol 12α‐hydroxylase
- CYP7B1
oxysterol 7α‐hydroxylase
- CYP27A1
sterol 27‐hydroxylase
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- Gly‐MCA
glycine‐β‐muricholic acid
- IL‐6
interleukin‐6
- LCA
lithocholic acid
- LGG
Lactobacillus rhamnosus GG
- α/β‐MCA
α‐/β‐muricholic acids
- Mdr2
multidrug resistance protein 2
- MRP
multidrug resistance–associated protein
- OST‐α/‐β
organic solute transporter subunits alpha/beta
- PBC
primary biliary cholangitis
- SHP
small heterodimer partner
- α‐SMA
alpha‐smooth muscle actin
- Timp‐1
tissue inhibitor of metalloproteinase 1
- T‐α‐/T‐β‐MCA
taurine‐α‐/β‐MCAs
- TNF‐α
tumor necrosis factor alpha
- Z‐Gu
(Z)‐guggulsterone
Cholestatic liver disease is characterized by an impairment of bile flow to the gallbladder and duodenum.1 Hepatic accumulation of bile acids (BAs) is central to the pathogenesis of cholestasis‐induced liver injury, and an excess of cytotoxic BAs in the liver can lead to liver fibrosis and cirrhosis and even liver carcinogenesis.2 During cholestasis, repression of BA synthesis is a protective mechanism against hepatic BA accumulation and cytotoxicity.
BAs are synthesized and conjugated in the hepatocytes and then secreted into the intestine. BA homeostasis is tightly regulated through enterohepatic signaling. Farnesoid X receptor (FXR) is a member of the steroid/thyroid hormone receptor family of ligand‐activated transcription factors, and certain BAs are its endogenous ligands.3 In the liver, FXR plays a prominent role in the feedback repression of BA synthesis through small heterodimer partner (SHP) by reducing the expression of the cytochrome p450 enzymes cholesterol 7α‐hydroxylase (CYP7A1) and sterol 12α‐hydroxylase (CYP8B1) to suppress BA synthesis. In the ileum, FXR is critically involved in the BA reabsorption process through regulation of BA transporter expression. Activation of FXR in the ileum also induces the expression of fibroblast growth factor‐19/15 (human FGF‐19, mouse FGF‐15), a hormone that is secreted into the portal blood and transported to the liver to repress the expression of CYP7A1, a rate‐limiting enzyme for BA synthesis.4 Thus, hepatic FXR and intestinal FXR coordinate to regulate BA synthesis through CYP7A1 regulation. Systemic activation of FXR was shown to protect against cholestasis through a putative hepatic FXR‐mediated effect.5 Interestingly, research using tissue‐specific FXR knockout mice demonstrated that the intestinal FXR–FGF‐15 pathway plays a prominent role, with respect to the hepatic FXR–SHP pathway, in repressing CYP7A1 expression.6 In addition, gut microbiota have a profound systemic effect on BA metabolism.7 Gut microbiota regulate BA metabolism by reducing the level of taurine‐β‐muricholic acid (T‐β‐MCA) and then increasing FXR‐15‐dependent production and reducing BA synthesis.8 Therefore, modification of gut microbiota is a potential strategy in the treatment of liver diseases caused by excessive BA exposure. Lactobacillus rhamnosus GG (LGG), the best‐characterized probiotic strain, has been widely used in the prevention and treatment of liver diseases in patients and animal models, with one mechanism of action being gut microbiota modification.9, 10 A recent study showed that LGG could reduce hepatic fibrosis in rats.11 However, the mechanism underlying this protection is unclear.
The current study aimed to examine the preventive effect of LGG in cholestatic liver disease. We demonstrated that probiotic LGG prevents liver fibrosis by inhibiting hepatic BA synthesis through increasing intestinal FXR–FGF‐15 signaling and enhancing BA excretion in bile duct ligated (BDL) mice and multidrug resistance protein 2 knockout (Mdr2−/−) mice. Our study further showed that intestine‐specific inhibition of FXR blunted the beneficial effect of LGG in BDL mice. In addition, LGG enhanced gut microbiota that have bile salt hydrolase (BSH) activity to enhance BA excretion.
Materials and Methods
Animal Study
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Louisville. Male C57BL/6J mice (8 weeks of age) and Mdr2−/− (FVB.129P2‐Abcb4tm1Bor/J) mice (7 weeks of age) were obtained from Jackson Laboratory (Bar Harbor, ME). They were maintained at 22°C with a 12‐hour:12‐hour light/dark cycle and had free access to normal chow diet and sterile water.
The BDL procedure was conducted as described.12 Briefly, mice were anesthetized with avertin intraperitoneally. After laparotomy, the bile duct was double‐ligated with nonresorbable surgical sutures. Sham‐operated mice underwent the same surgical procedure with the exception of BDL and resection. After surgery, 42 male mice were randomly divided into six groups: (A) sham, (B) BDL, (C) BDL + LGG, (D) LGG, (E) BDL + LGG + (Z)‐Guggulsterone (Z‐Gu), and (F) BDL + LGG + glycine‐β‐muricholic acid (Gly‐MCA). Live LGG (suspended in physiological saline) was given to mice by oral gavage at a dose of 109 colony‐forming units (CFU)/day for 11 days. Mice in groups A and B were gavaged with the same volume of physiological saline. Z‐Gu, a global FXR inhibitor, or Gly‐MCA, an intestine‐specific FXR inhibitor, were given to respective groups of mice at a dose of 10 mg/kg body weight/day by oral gavage. For Mdr2−/− mice, LGG or saline was administered by oral gavage at the same dose as for BDL mice for 2 weeks.
Statistical Analysis
Statistical analyses were performed using the statistical computer package GraphPad Prism, version 6 (GraphPad Software Inc., San Diego, CA). Results are expressed as means ± SEM. Statistical comparisons were made using two‐way analysis of variance (ANOVA) with Bonferroni’s post hoc test or one‐way ANOVA with Tukey’s post hoc test or Student t test where appropriate. Differences were considered significant at P < 0.05.
Please see additional methods in the Supporting Information.
Results
LGG Treatment Improved Liver Injury and Fibrosis in BDL Mice
We hypothesized that probiotic LGG may be protective against BA‐induced cholestatic liver disease. BDL was performed on mice that were then followed for 11 days, and LGG was supplemented at the beginning of the time period and daily thereafter. Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) activity and total bilirubin and direct bilirubin levels in BDL mice were significantly increased compared to mice in the sham group. LGG treatment significantly reduced serum levels of AST, ALP, and direct bilirubin and tended to reduce ALT and total bilirubin levels in BDL mice (Fig. 1A). Compared to sham mice, BDL mice showed severe changes in liver morphology, including necrosis of liver cells, diffuse severe/high bile duct hyperplasia, portal edema, and mild portal infiltrates. These changes in liver morphology were markedly improved by LGG treatment (Fig. 1B). In BDL mice, a marked fibrosis was observed in portal tracts, which was significantly decreased by LGG treatment (Fig. 1B,C).
Figure 1.
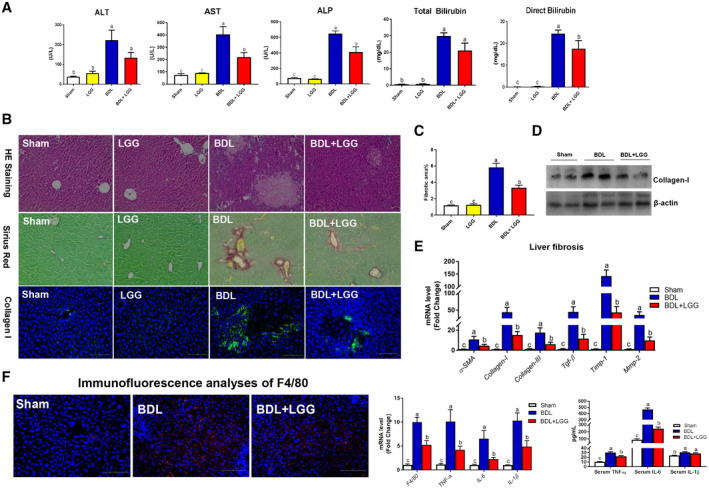
LGG treatment improved liver injury and fibrosis in BDL mice. (A) Serum ALT, AST, and ALP levels. Serum total bilirubin and direct bilirubin levels. (B) Representative images of liver specimens stained with hematoxylin and eosin (original magnification ×200), sirius red (×200), and immunofluorescence analyses of collagen I (×200). (C) Morphometric quantification of sirius red staining–positive area. (D) Hepatic protein expression of collagen I. (E) Hepatic mRNA expression of liver fibrosis–related genes. (F) Immunofluorescence analyses of F4/80 (×200); hepatic mRNA expression of liver inflammation‐related genes; and serum levels of TNF‐α, IL‐6, and IL‐1β. Data are expressed as mean ± SEM (n = 5‐8). Columns with different letters differ significantly (P < 0.05). Abbreviations: HE, hematoxylin and eosin; Mmp‐2, matrix metallopeptidase 2.
The results showed that BDL caused an elevation of hepatic collagen I, the most abundant extracellular matrix protein in the fibrotic liver, which was significantly reduced by LGG treatment (Fig. 1B,D). mRNA expression of the liver fibrosis markers alpha‐smooth muscle actin (α‐SMA), collagen I, collagen III, transforming growth factor beta (Tgf‐β), tissue inhibitor of metalloproteinase 1 (Timp‐1), and matrix metallopeptidase 2 were significantly increased in BDL mice and reduced by LGG treatment (Fig. 1E).
We evaluated the inflammatory response in hepatic and intestinal tissues and system inflammation. BDL caused a significant elevation of hepatic F4/80 protein and mRNA levels of F4/80, tumor necrosis factor alpha (TNF‐α), interleukin‐6 (IL‐6), and IL‐1β, which were significantly reduced by LGG treatment (Fig. 1F). Serum levels TNF‐α and IL‐6 were significantly reduced by LGG in BDL mice (Fig. 1F). In addition, LGG treatment significantly decreased ileum mRNA levels of proinflammatory markers and mediators, including F4/80, TNF‐α, IL‐6, and IL‐1β (Supporting Fig. S6). These results indicated that LGG treatment suppressed the BDL‐induced proinflammatory response.
BDL caused an elevation of hepatic cytokeratin 19 (CK‐19) expression, a bile duct proliferation marker, which was significantly reduced by LGG treatment (Supporting Fig. S9).
LGG Treatment Decreased Hepatic BA Level
Massive BA accumulation is a hallmark of cholestatic liver disease. Compared to sham mice, the total serum BA level in BDL mice was significantly increased, which was not altered by LGG treatment (Fig. 2A). Hepatic BA level and total BA pool size in BDL mice were significantly increased, which were significantly reduced by LGG treatment (Fig. 2A). Notably, we found that hepatic chenodeoxycholic acid (CDCA) levels were significantly decreased but that T‐αMCA and T‐βMCA concentrations were significantly increased in BDL mice. Interestingly, the hepatic mRNA expression level of cytochrome P450 family 2, subfamily c, polypeptide 70 (Cpy2c70), which is responsible for the conversion of CDCA to MCAs in mice,13 was increased by BDL. These changes were reversed by LGG treatment (Fig. 2B). It is known that CDCA is a potent endogenous FXR agonist and that T‐βMCA is an FXR antagonist. Thus, LGG treatment likely increases FXR activity in BDL mice. Detailed hepatic and serum BA alterations are shown in Supporting Fig. S1A,B.
Figure 2.
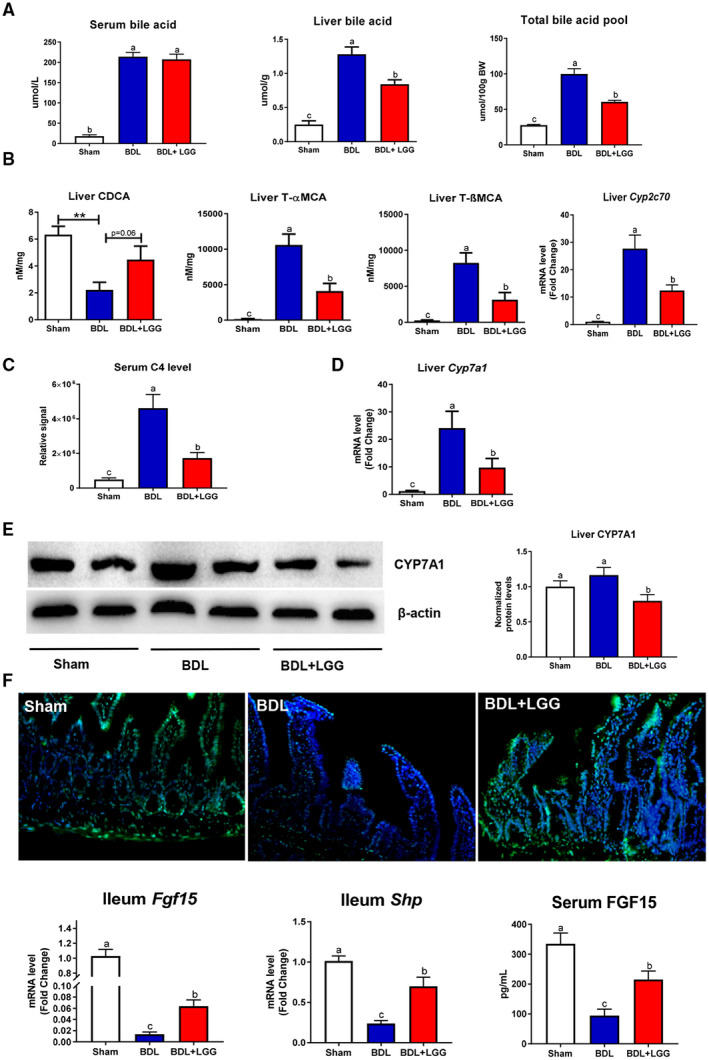
LGG treatment changed hepatic BA metabolism and activated intestinal FXR signaling. (A) Serum BA levels, liver total BA levels, and total BA pool. (B) Hepatic levels of CDCA, T‐αMCA, T‐βMCA, and mRNA expression of Cyp2c70. (C) Serum C4 level. (D) Hepatic mRNA expression of Cyp7a1. (E) Hepatic protein expression and quantification of CYP7A1. (F) Immunofluorescence analyses of ileum FGF‐15 (×200), ileum mRNA expression of Fgf‐15 and Shp, and serum FGF‐15 levels. Data are expressed as mean ± SEM (n = 5‐8). Columns with different letters differ significantly (P < 0.05).
Importantly, BDL caused a robust elevation of the serum 7α‐hydroxy‐4‐cholesten‐3‐one (C4) level, a surrogate of BA synthesis, which was significantly reduced by LGG treatment (Fig. 2C). BDL mice had elavated hepatic mRNA expression levels of Cyp7a1, the major enzyme in the classic BA synthesis pathway, which was significantly reduced by LGG (Fig. 2D). mRNA levels of Cyp8b1, sterol 27‐hydroxylase (Cyp27a1), and oxysterol 7α‐hydroxylase (Cyp7b1) were not altered by BDL; and LGG treatment significantly decreased Cyp27a1 and Cyp7b1 mRNA levels (Supporting Fig. S2A). The CYP7A1 protein level was unchanged in BDL mice but significantly reduced by LGG (Fig. 2E). In addition, the protein expression of CYP8B1 and CYP7B1 was decreased by BDL and further reduced by LGG treatment, while CYP27A1 protein was not altered (Supporting Fig. S2). Hepatic mRNA expression of Fxr and Shp was significantly increased by BDL and significantly reduced by LGG (Supporting Fig. S3). These data suggest that LGG treatment suppressed BA synthesis in BDL mice.
In addition to the regulated synthesis of BAs, a variety of hepatic and intestinal membrane transporters play critical roles in maintaining BA homeostasis. Hepatic mRNA expression levels of the bile salt export pump (Bsep), multidrug resistance–associated protein 3 (Mrp3), Mrp4, organic solute transporter subunit beta (OST‐β), Na+‐taurocholic acid cotransporting polypeptide (Ntcp), and organic anion transporting polypeptide 4 were significantly increased in BDL mice and significantly reduced by LGG (Supporting Fig. S4A). Western blot results showed that the expression of BSEP, MRP4, and OST‐β were significantly increased in BDL mice and reduced by LGG. The NTCP level was increased in BDL mice but not altered by LGG (Supporting Fig. S4B). In the intestine, LGG had no significant effect on mRNA expression of ileum apical sodium‐dependent bile acid transporter but significantly decreased mRNA levels of Ost‐α/Ost‐β, Mrp2, and Mrp3 (Supporting Fig. S5).
Together, these data suggest that, under cholestatic conditions, LGG functions to restore hepatic BA homeostasis by reducing BA synthesis in the liver. Although expression levels of BA transporters are selectively up‐regulated as part of the adaptive response to cholestasis, they were normalized by LGG treatment due to normalized BA levels.
LGG Treatment Activated Intestinal FXR–FGF‐15 Signaling
In the intestine, BAs activate FXR to induce FGF‐15 expression. Secreted FGF‐15 binds to FGF receptor/β‐Klotho complex on hepatocytes and represses BA synthesis.14 In sham mice, we observed a strong expression of FGF‐15 protein. BDL significantly reduced FGF‐15 expression, which was restored by LGG. Similarly, mRNA expression levels of Fgf‐15 in ileal tissues were decreased by BDL and increased by LGG (Fig. 2F). In addition, ileal mRNA expression of an FXR target gene, Shp, was significantly reduced by BDL and normalized by LGG, indicating an activation of intestinal FXR by LGG treatment in BDL mice (Fig. 2F), with no significant effect on mRNA expression (Supporting Fig. S5). Finally, BDL caused a significant reduction of circulating FGF‐15 protein levels, which was prevented by LGG (Fig. 2F). These data suggest that LGG treatment–mediated reduction of BA synthesis in BDL mice is through up‐regulation of intestinal FXR–FGF‐15 signaling.
Inhibition of Intestinal FXR Activation Attenuated the Protective Effects of LGG
The data presented above suggest a critical involvement of FXR in LGG‐mediated reduction of BA synthesis. We hypothesized that inhibition of FXR activation may blunt the beneficial effects of LGG. To test this hypothesis, mice undergoing 11‐day BDL were gavaged with LGG and/or Z‐Gu, a global FXR inhibitor.15 The reduction of serum AST and ALP levels by LGG was attenuated by Z‐Gu treatment (Fig. 3A). Global inhibition of FXR by Z‐Gu significantly attenuated the protective effects on liver injury and fibrosis by LGG in BDL mice (Fig. 3B,C). Consistently, Z‐Gu treatment markedly suppressed the LGG‐reduced mRNA expression levels of α‐SMA and collagen I (Fig. 3D). Serum BA levels were not changed by either LGG or Z‐Gu (Fig. 3E). However, Z‐Gu treatment significantly blunted the reducing effect of LGG on liver BA levels (Fig. 3E). Intestinal BA levels were not changed by LGG or Z‐Gu (Fig. 3E). As expected, increased intestinal mRNA expression of Fgf‐15 and Shp (Fig. 3F) and decreased hepatic Cyp7a1 mRNA level by LGG were almost completely suppressed by Z‐Gu treatment (Fig. 3F). Similarly, liver Shp and Bsep mRNA expression was also significantly inhibited by Z‐Gu treatment (Fig. 3F), confirming the global inhibition of FXR by Z‐Gu. In addition, the suppressive effects of LGG on hepatic mRNA levels of F4/80, TNF‐α, IL‐6, and IL‐1β were markedly reduced by Z‐Gu treatment (Supporting Fig. S7). Z‐Gu treatment also attenuated the inhibitory effect of LGG on CK‐19 expression (Supporting Fig. S9).
Figure 3.
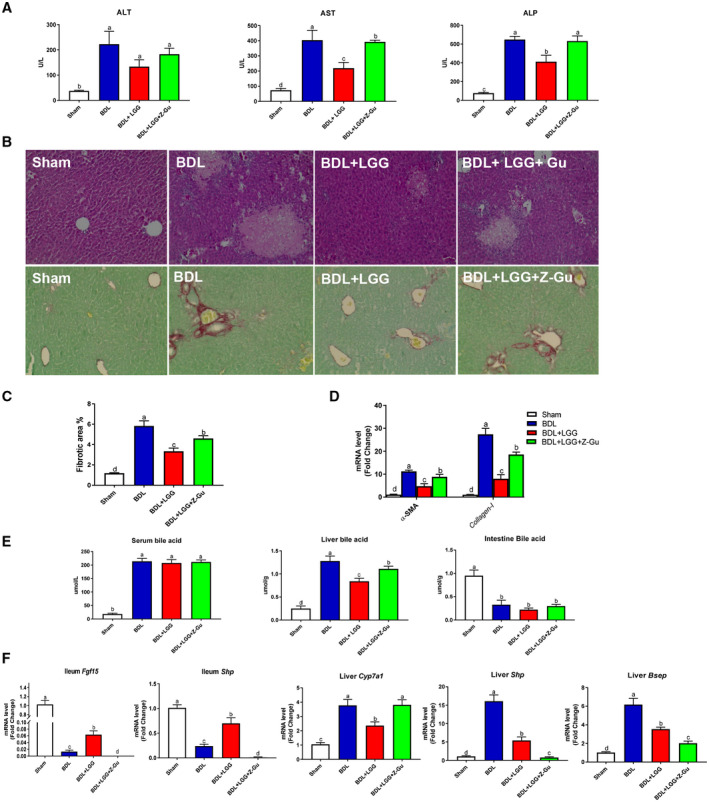
Global inhibition of FXR activation abolished the protective effects of LGG. (A) Serum ALT, AST, and ALP activities. (B) Representative images of liver specimens stained with hematoxylin and eosin (×200) and sirius red (×200). (C) Morphometric quantification of sirius red staining–positive area. (D) Hepatic mRNA expression of α‐SMA and collagen I. (E) Serum BA levels, Hepatic total BA levels, and intestinal BA levels. (F) Ileum mRNA expression of Fgf‐15 and Shp and hepatic mRNA expression of Cyp7a1, Shp, and Bsep. Data are expressed as mean ± SEM (n = 5‐8). Columns with different letters differ significantly (P < 0.05).
We further hypothesized that inhibition of intestine‐specific FXR is responsible for the beneficial effects of LGG. To test this hypothesis, mice were treated with an intestine‐specific FXR inhibitor, Gly‐MCA, which has been identified as an orally available, small molecule, intestine‐selective FXR inhibitor and is not hydrolyzed by gut bacterial BSH.16 As expected, Gly‐MCA did not suppress liver Shp expression, but the intestinal Shp mRNA level was completely inhibited (Fig. 4A), indicating the intestinal specificity of FXR inhibition by Gly‐MCA. Ileal mRNA and serum FGF‐15 protein level were significantly increased by LGG and completely inhibited by Gly‐MCA in BDL mice (Fig. 4A). As a result, the reduction of serum C4 level, hepatic Cyp7a1 mRNA expression, hepatic BA levels, and total BA pool size by LGG treatment in BDL mice were all significantly increased by Gly‐MCA (Fig. 4B). Gly‐MCA significantly reduced the protective effects on liver injury and fibrosis by LGG (Fig. 4C,D). Moreover, mRNA expression α‐SMA and collagen I was increased in Gly‐MCA compared to LGG‐treated mice (Fig. 4E). LGG‐mediated reduction of serum levels of AST and ALP in BDL mice was also reduced by Gly‐MCA (Fig. 4F). In addition, decreased hepatic protein levels of CK‐19 by LGG were markedly increased by Gly‐MCA (Supporting Fig. S9) and decreased hepatic mRNA levels of F4/80, TNF‐α, IL‐6, and IL‐1β by LGG were markedly increased by Gly‐MCA (Supporting Fig. S8). These data unambiguously demonstrate that inhibition of intestinal FXR was sufficient to reduce the protective effects of LGG on BA‐associated liver fibrosis and injury in BDL mice.
Figure 4.

Inhibition of intestinal FXR activation abolished the protective effects of LGG. (A) Hepatic mRNA expression of Shp, ileum mRNA expression of Shp, ileum mRNA expression of Fgf‐15, and serum FGF‐15 levels. (B) Serum C4 level, hepatic mRNA expression of Cyp7a1, hepatic total BA levels, and total BA pool. (C) Representative images of liver specimens stained with hematoxylin and eosin (×200) and sirius red (×200). (D) Morphometric quantification of sirius red staining–positive area. (E) Hepatic mRNA expression of α‐SMA and collagen I. (F) Serum ALT, AST, and ALP activities. Data are expressed as mean ± SEM (n = 5‐8). Columns with different letters differ significantly (P < 0.05). Abbreviation: BW, body weight.
LGG Treatment Attenuated T‐βMCA Suppression of FXR Activities in Intestinal Epithelial Cells
To further determine the effect of LGG on FXR activity/functions on intestinal epithelial cells, we treated human intestinal epithelial Caco‐2 cells with the FXR agonist CDCA and concurrently with the FXR antagonist T‐βMCA in the presence or absence of LGG. CDCA treatment markedly increased mRNA expression of FGF‐19 and SHP, which was blocked by T‐βMCA and recovered by LGG (Fig. 5A). Next, we determined whether LGG directly activates FXR using a luciferase activity assay in transfected cells. LGG restored T‐βMCA‐suppressed FXR‐luciferase activity in both Caco‐2 and HEK‐293 cells (Fig. 5B). Previous studies suggest that LGG may exert its function through its fermentation products. To test whether LGG culture supernatant (LGGs) has a similar effect on FXR activation, Caco‐2 and HEK293 cells were treated with LGGs. Reporter assays showed that LGGs treatment similarly restored CDCA‐stimulated and T‐βMCA‐suppressed FXR‐luciferase activity (data not shown).
Figure 5.
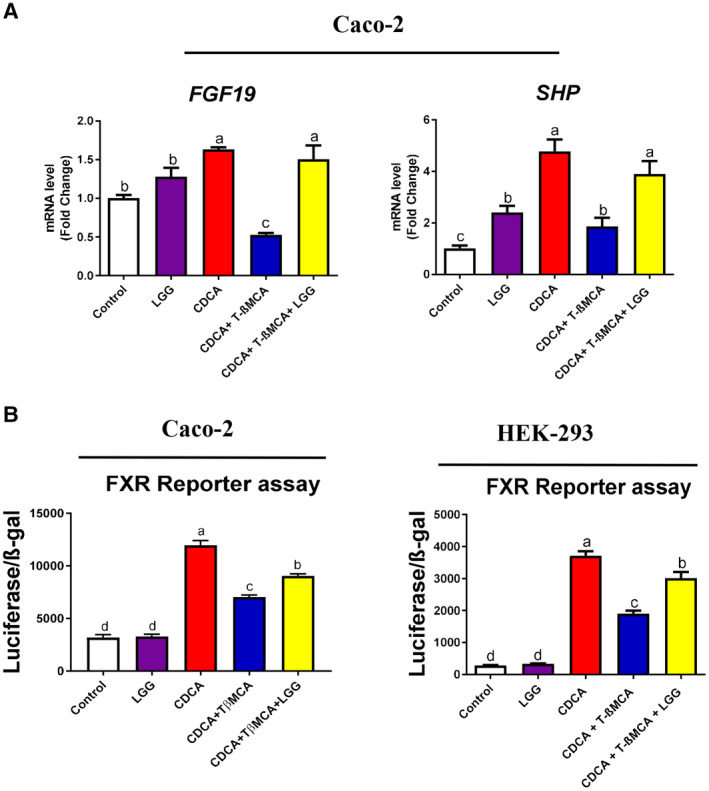
LGG treatment attenuated T‐βMCA suppression of FXR activities in intestinal epithelial cells. (A) Caco‐2 cells were treated with CDCA (100 μM) alone or in combination with T‐βMCA (100 μM), with or without live LGG (104 CFU/well) for 6 hours, then cells were lysed, and total RNA was extracted for real‐time PCR analysis of FXR target genes FGF‐19 and SHP. (B) Luciferase reporter assay was performed in Caco‐2 and HEK293 cells transfected with FXR expression plasmid, FXR reporter plasmid, and β‐galactosidase expression plasmid. Cells were stimulated with CDCA (20 μM) alone or in combination with T‐βMCA (60 μM), with or without live LGG (104 CFU/well) for 6 hours. Data are expressed as mean ± SEM (n = 3). Columns with different letters differ significantly (P < 0.05).
LGG Treatment Changed Gut Microbiota and Increased BA Excretion
Gut bacteria play a key role in intestinal BA deconjugation and excretion.8 Deconjugated BAs are more hydrophobic and more easily excreted into feces. Bacteria having high BSH activity promote BA deconjugation.17 Gut microbiota analysis revealed that phylum levels of Firmicutes and Actinobacteria, known phyla that harbor bacteria with high BSH activity,18 were significantly increased by LGG treatment (Fig. 6A). In contrast, Bacteroidetes, a major bacterial phylum that harbors bacteria with low BSH activity, was not altered by LGG treatment. Interestingly, both Z‐Gu and Gly‐MCA did not inhibit the effect of LGG on Firmicutes, Actinobacteria, and Bacteroidetes levels (Fig. 6A). These results suggest that the inhibitory effects of Z‐Gu and Gly‐MCA on intestinal FXR are mainly due to the antagonism by the drugs rather than microbiota changes.
Figure 6.

LGG treatment changed gut microbiota and increased BA excretion. (A) Fecal microbiota changes. (B) Fecal BSH activity. (C) Fecal total BA levels. (D) Fecal LCA levels. (E) Fecal T‐βMCA/β‐MCA ratio. (F) Urine BA levels and kidney mRNA expression of Mrp‐2 and Mrp‐4. Data are expressed as mean ± SEM (n = 5‐8). *P < 0.05, **P < 0.01. Abbreviation: OD, optical density.
Enzymatic assay of BSH activity in fecal samples confirmed the results (Fig. 6B). Fecal total BAs were significantly increased by LGG treatment (Fig. 6C). In an ex vivo study, we showed that the intestinal basolateral side of exposure to taurocholic acid increased BA levels on the lumen side (Supporting Fig. S10), suggesting that BA could transport to intestine and gut lumen through blood circulation. Interestingly, a deconjugated BA, lithocholic acid (LCA), which has high cytotoxicity, was significantly increased in fecal samples of LGG‐treated mice (Fig. 6D). The fecal T‐βMC/β‐MCA ratios was significantly decreased by LGG (Fig. 6E), indicating a likely elevated intestinal FXR activity due to the decreased antagonism. Detailed relative BA alterations in feces are shown in Supporting Fig. S1C.
To further determine the BA excretion, urine BAs were also analyzed. BDL increased urine BA level due to high circulating levels. Importantly, LGG treatment further increased urine BAs (Fig. 6F). BDL significantly decreased the kidney BA transporter Mrp‐2, but not Mrp‐4, while LGG treatment significantly increased both Mrp‐2 and Mrp‐4 mRNA expression (Fig. 6F). Because MRP‐4 is not an FXR target, the results suggest an LGG‐FXR‐independent mechanism in renal BA excretion. This notion was confirmed by treatments with the FXR antagonists Z‐Gu and Gly‐MCA (Supporting Fig. S11). Both Z‐Gu and Gly‐MCA did not inhibit the effect of LGG on elevated urine BA excretion.
LGG Prevented Liver Injury and Fibrosis Through Inhibiting Hepatic BA Synthesis and Enhancing BA Excretion in Mdr2−/− Mice
The effects of LGG on liver injury and fibrosis were examined in Mdr2−/− mice, which develop spontaneous cholangitis and liver fibrosis with high levels of BA. LGG treatment significantly reduced the serum ALP level and the direct bilirubin level and tended to reduce ALT, AST, and total bilirubin levels in Mdr2−/− mice (Fig. 7A). Mdr2−/− mice showed a marked liver injury and fibrosis, which was significantly decreased by LGG treatment (Fig. 7B,C). To further determine the effect of LGG on hepatic fibrosis, we measured hepatic fibrosis–related gene expression. mRNA expression of the liver fibrosis markers α‐SMA, collagen I, Tgf‐β, and Timp‐1 was significantly reduced by LGG treatment in Mdr2−/− mice (Fig. 7D).
Figure 7.
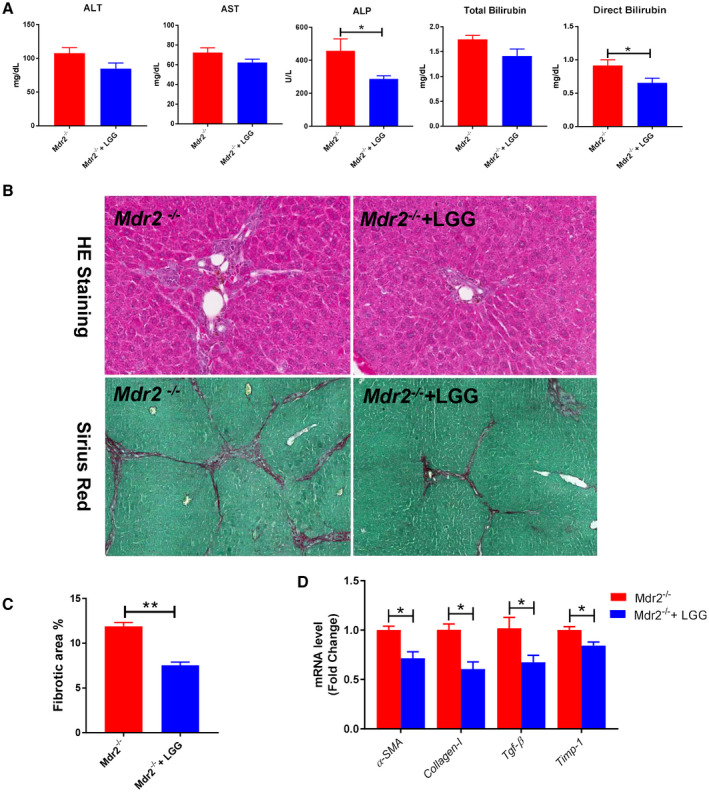
LGG treatment improved liver injury and fibrosis in Mdr2−/− mice. (A) Serum levels of ALT, AST, ALP, total bilirubin, and direct bilirubin. (B) Representative images of liver specimens stained with hematoxylin and eosin (×100) and sirius red (×40). (C) Morphometric quantification of sirius red staining–positive area. (D) Hepatic mRNA expression of liver fibrosis–related genes. Data are expressed as mean ± SEM (n = 4‐6). *P < 0.05, **P < 0.01. Abbreviation: HE, hematoxylin and eosin.
LGG treatment significantly decreased BA levels in serum, liver, and intestine tissues in Mdr2−/− mice. Total BA pool size was also reduced by LGG treatment (Fig. 8A). Hepatic mRNA expression of Cyp7a1 and CYP7A1 protein expression were significantly reduced by LGG (Fig. 8B). Importantly, serum C4 level was markedly reduced by LGG (Fig. 8C), further suggesting a decrease of BA synthesis by LGG. LGG treatment significantly decreased liver Fxr and Shp but significantly increased ileum Fxr and Shp mRNA expression (Fig. 8D). In addition, ileum Fgf‐15 mRNA and serum FGF‐15 protein expression were significantly increased by LGG treatment (Fig. 8D). These results demonstrate that LGG treatment decreases hepatic BA levels through intestinal FXR–FGF‐15 up‐regulation in Mdr2−/− mice, which agrees with the findings in BDL mice. Furthermore, fecal and urine BA levels were significantly elevated by LGG in Mdr2−/− mice, suggesting that LGG treatment enhances BA excretion (Fig. 8E).
Figure 8.
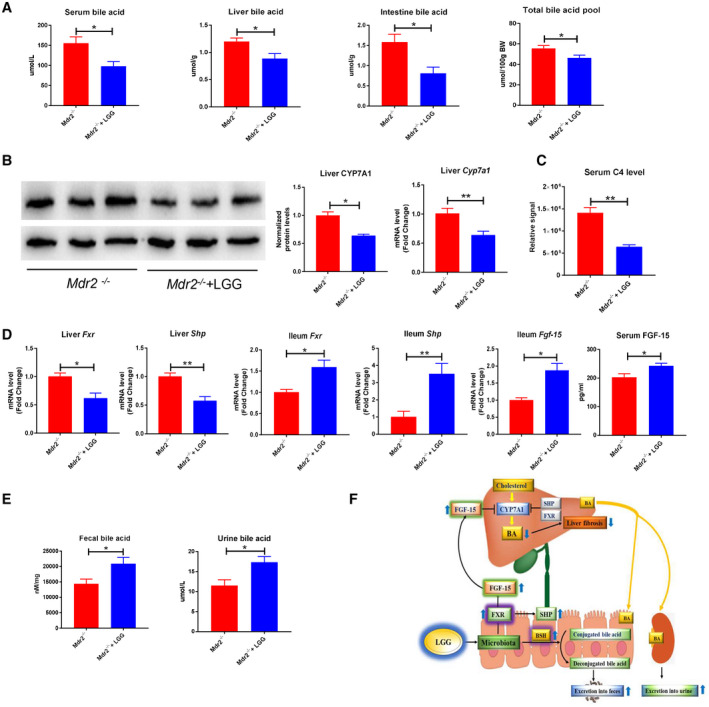
LGG treatment decreased hepatic BA synthesis and increased BA excretion in Mdr2−/− mice. (A) Serum BA levels, liver total BA levels, intestine BA levels, and total BA pool. (B) Hepatic protein expression and quantification of CYP7A1 and hepatic mRNA expression of Cyp7a1. (C) Serum C4 levels. (D) Hepatic mRNA expression of Fxr and Shp; ileum mRNA expression of Fxr, Shp, and Fgf‐15; and serum FGF‐15 levels. (E) Fecal total BA levels and urine BA levels. (F) A proposed model of LGG prevention against liver injury and fibrosis through inhibiting hepatic BA synthesis and enhancing BA excretion in BDL and Mdr2−/− mice. Data are expressed as mean ± SEM (n = 4‐6). *P < 0.05, **P < 0.01.
Discussion
Cholestasis is a pathologic condition characterized by impairment or cessation of the bile flow with consequent liver damage.19 Hepatic accumulation of toxic BAs plays a pivotal role in cholestatic liver disease and is associated with multiple other types of liver diseases.20 Reducing hepatic BA overload is a therapeutic goal for the management of BA‐associated liver diseases. Current treatments of cholestasis include several BAs and their derivatives. Ursodeoxycholic acid (UDCA), which works through inhibiting intestinal BA absorption and eliminating toxic BA in hepatocytes, is a standard treatment for primary biliary cholangitis (PBC) and has been used for the treatment of primary sclerosing cholangitis (PSC).21 Obeticholic acid, a derivative of CDCA with high FXR activation potency, has been used in several clinical trials for the treatment of PBC and PSC,22 and it is used clinically for patients with PBC who are intolerant or poorly responsive to UDCA. Recent studies suggest that intestinal microbiota play an important role in the pathophysiology of cholestatic liver disease.7, 23 Targeting microbiota may offer treatment options for cholestatic liver disease. Earlier studies have evaluated the therapeutic function of probiotics in cholestatic liver disease in humans and animal models.24 However, the mechanisms by which probiotics improve liver injury and fibrosis have not been fully studied. In the current study, we evaluated the effectiveness and the potential mechanisms of probiotic LGG in the treatment of BDL‐induced and Mdr2 knockout–induced liver injury/fibrosis, and we found that the beneficial effects of LGG were mediated by an intestinal FXR–FGF‐15 signaling pathway to inhibit BA de novo synthesis and by increasing BA excretion.
Our study demonstrated that LGG treatment improved liver injury and fibrosis in BDL mice by a notable reduction in hepatic BA levels. Reducing hepatic BAs could be achieved by strategies such as increasing BA flow, inhibition of BA reabsorption from intestine, and inhibition of BA synthesis in the liver.25 CYP7A1 is the rate‐limiting enzyme for the conversion of cholesterol to BA. Hepatic activation of FXR induces elevation of SHP, which inhibits CYP7A1 expression.26, 27 Previous studies have shown that systemic activation of FXR by specific synthetic FXR ligands such as GW4064 and 6α‐ethyl‐CDCA (INT‐747) protected animals from BDL‐induced, α‐naphthyl isothiocyanate–induced, and ethinyl estradiol–induced cholestasis.28, 29 The observed protective effects were ascribed, in part, to the induction of BSEP, MDR2, and MRP2 to enhance excretion of BAs and phospholipids from the liver into the small intestine and, in part, to the repression of CYP7A1 through hepatic SHP to reduce BA synthesis.30 One of the major findings in the current study was that LGG treatment significantly reduced hepatic CYP7A1 expression, which was associated with increased hepatic concentration of CDCA, an FXR agonist, and decreased the concentration of T‐β‐MCA, an FXR antagonist, in BDL mice, implying that LGG treatment may elevate hepatic FXR activity. Interestingly, BDL increased both CYP7A1 and SHP expression, suggesting that BA synthesis increases even under conditions of BA overloading. The increase of hepatic SHP expression may be an adaptive response to elevated BA due to BDL operation. However, hepatic SHP was decreased by LGG in BDL mice. A possible explanation is that the reductive effect of LGG on CYP7A1 is mediated by a mechanism independent of the hepatic FXR–SHP axis, and this regulatory axis of BA is not required to rebalance BA homeostasis in these 11‐day BDL‐operated mice. In fact, previous studies showed that SHP regulation is ligation time–dependent in BDL mice.31
Recent studies showed that hepatic BA synthesis is also regulated by intestine‐derived FGF‐15.32 Under sham conditions, BAs activate intestinal FXR and subsequently elevate the endocrine hormone FGF‐15, which binds the FGF receptor and β‐Klotho complex on the surface of hepatocytes to inhibit CYP7A1 expression.33 Obstruction of bile flow reduces intestinal BA concentration, leading to a significant reduction of FXR activity and FGF‐15 expression. However, we found that BAs exist in the intestine of BDL mice. This phenomenon can be explained by the BDL procedure with possibly loose ligation and a likely reverse transport of BA into the intestine from the circulation where BA concentration is high. In fact, our ex vivo study showed that taurocholic acid can enter the intestinal lumen from the basolateral side (Supporting Fig. S10). Importantly, LGG treatment significantly decreased the T‐β‐MCA/β‐MCA ratio in feces, implying a reduction of FXR antagonism in the intestine. This notion is confirmed by the increased ileal expression and serum levels of FGF‐15 in LGG‐treated mice and by an in vitro study using human intestinal epithelial Caco‐2 cells. The suppressive effect of LGG on T‐β‐MCA‐inhibited FXR activity was further demonstrated in a reporter assay. It is known that FGF‐15 is solely expressed in the intestine in rodents, while humans also express FGF‐19 in the liver.34 Interestingly, one study showed that livers of cholestatic patients ectopically expressed FGF‐19.35 This increase might be a protective mechanism against BA synthesis and the progression of cholestasis.
Previous studies demonstrated that activation of intestinal FXR in BDL mice could reduce hepatic BA level.5 We further confirmed that activation of intestinal FXR by LGG is sufficient to inhibit hepatic BA synthesis in BDL mice. Treatment with a global FXR inhibitor, Z‐Gu, eliminated the protective effects of LGG on liver injury and fibrosis and on the reduction of hepatic BAs. Notably, treatment with an intestine‐specific FXR inhibitor, Gly‐MCA, also eliminated the protective effect of LGG. It is thus clear that LGG exerts its function through intestinal FXR activation and FGF‐15 production to inhibit hepatic BA synthesis in BDL mice.
LGG regulation of BA metabolism seems to be BA concentration–dependent. When applied to sham operated control mice, which have balanced BAs in the liver and a significant amount of BAs in the gut, LGG increased hepatic CYP7A1 and decreased intestinal SHP and FGF‐15 expression. As a result, the hepatic BAs remained unchanged (Supporting Fig. S12). Therefore, the function of LGG is likely to regulate cholesterol homeostasis when hepatic BAs are not significantly altered. When applied to BDL mice, which have high levels of hepatic BAs and low intestinal BAs, LGG treatment increased intestinal SHP and FGF‐15 and inhibited hepatic CYP7A1 leading to an inhibition of BA synthesis. Therefore, LGG may play an important role in maintaining BA homeostasis.
Our findings in BDL mice were confirmed in Mdr2−/− mice. LGG treatment prevents spontaneous fibrosis and liver injury. Importantly, LGG treatment decreased the hepatic BA level through intestinal FXR–FGF‐15 signaling–mediated hepatic BA synthesis inhibition in Mdr2−/− mice, implying that the effect of BA lowering by LGG might be mediated by the common mechanism under conditions of disorders with hepatic BA accumulation.
Gut microbiota and BA metabolism are mutually regulated. Changes in BA signatures as well as modulation of the enterohepatic FXR/FGF‐15/CYP7A1 axis were found in germ‐free, gnotobiotic, and antibiotic‐treated animals.36 Bacterial metabolism–mediated primary BA deconjugation and subsequent conversion determine the amount of secondary BAs that might be detrimental to intestinal epithelium architecture.37 Gut bacteria–derived BSH is a major enzyme that catalyzes the “gateway” reaction in the bacterial metabolism of conjugated BAs to produce deconjugated BAs.38 We showed here that LGG treatment altered gut microbiota with an enrichment of the gut flora with BSH‐containing phyla. LGG itself has high BSH activity. The deconjugation increases BA fecal excretion because deconjugated BAs are less hydrophilic and therefore less likely to be reabsorbed.39 Furthermore, we showed that enhanced urine BA excretion by LGG is FXR‐independent because inhibiting FXR did not block the increase of urine BA excretion by LGG.
It should be noted that although gut bacteria can metabolize BAs that contribute to the balance of BA‐based FXR activators and inhibitors, gut bacteria might produce FXR ligands that are not derived from BAs. Although we observed a change in BAs that activate or inhibit FXR, the decreased FXR activity during BDL might be the result of low intestinal BA level, and increased FXR activity by LGG might be a result of direct stimulation of FXR with unknown metabolites and mechanism. Therefore, screening FXR ligands from LGG culture supernatant and gut lumen content of mice treated by LGG deserves further study to enhance our understanding of the protective effects of LGG in cholestatic liver disease. In addition, LGG might be effective on BA metabolism in other forms of liver diseases in which excessive BA accumulation exists. Previous studies have also shown that some metabolic disorders are associated with hepatic accumulation of BAs.40 Therefore, LGG may provide protection against these metabolic disorders.
Inflammation plays a critical role in the initiation and progression of liver fibrosis. LGG has been reported to decrease systemic inflammation in patients with cirrhosis.41 The transcriptional activity of FXR has been reported to be regulated by BA ligands, but enhanced inflammation can also decrease FXR activation, even in the presence of BA ligands.42 In our study, LGG treatment significantly decreased liver and ileum inflammation in BDL mice, suggesting that LGG‐increased FXR activity may also be mediated by reduced inflammation in BDL mice.
In conclusion, the current study demonstrates that LGG treatment prevents BDL‐induced and Mdr2−/−‐induced liver injury and fibrosis by inhibition of liver BA synthesis through intestinal FXR–FGF‐15 signaling. LGG treatment increases fecal BA excretion through increasing BSH‐containing gut bacteria. In addition, LGG treatment promotes urine BA excretion through an FXR‐independent pathway (Fig. 8F). Together, these results suggest that LGG may be used to prevent/treat liver disease associated with excessive hepatic BA levels.
Author Contributions
Y.L. designed and performed the experiments, analyzed the data and drafted the manuscript. K.C., F.L., Z.G., L.H., T.S., Q.S., F.Z., L.Z., M.J., Y.Z., and X.Z. provided technical support. S.B. provided critical discussion on manuscript revision. C.M. contributed to the conceptual review of the study and critical discussion on manuscript revision. W.F. conceived, designed and supervised the study and wrote the manuscript.
Supporting information
Acknowledgment
We thank Dr. Andrew D. Patterson from The Pennsylvania State University for providing intestine‐specific FXR inhibitor, Drs. Matthew Cave and K. Cameron Falkner from the University of Louisville for the plasmids, and Dr. Wei Jia from the University of Hawaii for BA analysis. We thank Marion McClain for proofreading the manuscript.
Supported by the National Institutes of Health (R21AA020848 and R01AA023190, to W.F.; U01AA021901, U01AA021893‐01, U01AA022489‐01A1, R01AA023681, P20GM113226, and P50AA024337, to C.J.M.) and by the US Department of Veterans Affairs (1I01BX002996, C.J.M).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Jansen PL, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017;65:722‐738. [DOI] [PubMed] [Google Scholar]
- 2. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11:55‐67. [DOI] [PubMed] [Google Scholar]
- 3. Gomez‐Ospina N, Potter CJ, Xiao R, Manickam K, Kim MS, Kim KH, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun 2016;7:10713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 5. Modica S, Petruzzelli M, Bellafante E, Murzilli S, Salvatore L, Celli N, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012;142:355‐365. [DOI] [PubMed] [Google Scholar]
- 6. Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue‐specific farnesoid X receptor in suppressing the expression of genes in bile‐acid synthesis in mice. Hepatology 2012;56:1034‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wahlström A, Sayin SI, Marschall H‐U, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 2016;24:41‐50. [DOI] [PubMed] [Google Scholar]
- 8. Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H‐U, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐beta‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225‐235. [DOI] [PubMed] [Google Scholar]
- 9. Mantegazza C, Molinari P, D'Auria E, Sonnino M, Morelli L, Zuccotti GV. Probiotics and antibiotic‐associated diarrhea in children: a review and new evidence on Lactobacillus rhamnosus GG during and after antibiotic treatment. Pharmacol Res 2018;128:63‐72. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Liu Y, Kirpich I, Ma Z, Wang C, Zhang M, et al. Lactobacillus rhamnosus GG reduces hepatic TNFα production and inflammation in chronic alcohol‐induced liver injury. J Nutr Biochem 2013;24:1609‐1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hammes TO, Leke R, Escobar TDC, Fracasso LB, Meyer FS, Andrades ME, et al. Lactobacillus rhamnosus GG reduces hepatic fibrosis in a model of chronic liver disease in rats. Nutr Hosp 2017;34:702‐709. [DOI] [PubMed] [Google Scholar]
- 12. Miyoshi H, Rust C, Roberts PJ, Burgart LJ, Gores GJ. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999;117:669‐677. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 2016;57:2130‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile‐acid signalling for metabolic diseases. Nat Rev Drug Discov 2008;7:678‐693. [DOI] [PubMed] [Google Scholar]
- 15. Urizar NL, Liverman AB, Dodds DT, Silva FV, Ordentlich P, Yan Y, et al. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 2002;296:1703‐1706. [DOI] [PubMed] [Google Scholar]
- 16. Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, et al. Intestine‐selective farnesoid X receptor inhibition improves obesity‐related metabolic dysfunction. Nat Commun 2015;6:10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geng W, Lin J. Bacterial bile salt hydrolase: an intestinal microbiome target for enhanced animal health. Anim Health Res Rev 2016;17:148‐158. [DOI] [PubMed] [Google Scholar]
- 18. Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA 2008;105:13580‐13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trauner M, Fuchs CD, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 2017;65:1393‐1404. [DOI] [PubMed] [Google Scholar]
- 20. Fickert P, Wagner M. Biliary bile acids in hepatobiliary injury—what is the link? J Hepatol 2017;67:619‐631. [DOI] [PubMed] [Google Scholar]
- 21. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet 2015;386:1565‐1575. [DOI] [PubMed] [Google Scholar]
- 22. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol 2015;62(1 Suppl.):S25‐S37. [DOI] [PubMed] [Google Scholar]
- 23. Fuchs CD, Paumgartner G, Mlitz V, Kunczer V, Halilbasic E, Leditznig N, et al. Colesevelam attenuates cholestatic liver and bile duct injury in Mdr2−/− mice by modulating composition, signalling and excretion of faecal bile acids. Gut 2018;67:1683‐1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orso G, Mandato C, Veropalumbo C, Cecchi N, Garzi A, Vajro P. Pediatric parenteral nutrition–associated liver disease and cholestasis: novel advances in pathomechanisms‐based prevention and treatment. Dig Liver Dis 2016;48:215‐222. [DOI] [PubMed] [Google Scholar]
- 25. Pathak P, Liu H, Boehme S, Xie C, Krausz KW, Gonzalez F, et al. Farnesoid X receptor induces Takeda G‐protein receptor 5 cross‐talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem 2017;292:11055‐11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 2009;50:1955‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol 2013;58:155‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu L, Liu X, Yuan Z, Li X, Yang H, Yuan Z, et al. SRT1720 alleviates ANIT‐induced cholestasis in a mouse model. Front Pharmacol 2017;8:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang CL, Xu YJ, Xiang D, Yang JY, Lei K, Liu D. Pharmacokinetic characteristics of baicalin in rats with 17alpha‐ethynyl‐estradiol‐induced intrahepatic cholestasis. Curr Med Sci 2018;38:167‐173. [DOI] [PubMed] [Google Scholar]
- 30. Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra‐ and extrahepatic cholestasis. J Clin Invest 2003;112:1678‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zollner G, Fickert P, Silbert D, Fuchsbichler A, Stumptner C, Zatloukal K, et al. Induction of short heterodimer partner 1 precedes downregulation of Ntcp in bile duct‐ligated mice. Am J Physiol Gastrointest Liver Physiol 2002;282:G184‐G191. [DOI] [PubMed] [Google Scholar]
- 32. Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 2016;24:41‐50. [DOI] [PubMed] [Google Scholar]
- 33. Lee S, Choi J, Mohanty J, Sousa LP, Tome F, Pardon E, et al. Structures of beta‐klotho reveal a “zip code”‐like mechanism for endocrine FGF signalling. Nature 2018;553:501‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7α‐hydroxylase gene expression. Hepatology 2009;49:297‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schaap FG, van der Gaag NA, Gouma DJ, Jansen PL. High expression of the bile salt‐homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology 2009;49:1228‐1235. [DOI] [PubMed] [Google Scholar]
- 36. Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150‐2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wahlstrom A, Kovatcheva‐Datchary P, Stahlman M, Backhed F, Marschall HU. Crosstalk between bile acids and gut microbiota and its impact on farnesoid X receptor signalling. Dig Dis 2017;35:246‐250. [DOI] [PubMed] [Google Scholar]
- 38. Begley M, Hill C, Gahan CG. Bile salt hydrolase activity in probiotics. Appl Environ Microbiol 2006;72:1729‐1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr–Fgf15 axis in mice. Cell Rep 2014;7:12‐18. [DOI] [PubMed] [Google Scholar]
- 40. Just S, Mondot S, Ecker J, Wegner K, Rath E, Gau L, et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018;6:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, Puri P, Sterling RK, et al. Randomised clinical trial: Lactobacillus GG modulates gut microbiome, metabolome and endotoxemia in patients with cirrhosis. Aliment Pharmacol Ther 2014;39:1113‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frankenberg T, Miloh T, Chen FY, Ananthanarayanan M, Sun AQ, Balasubramaniyan N, et al. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 2008;48:1896‐1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials