

Abstract
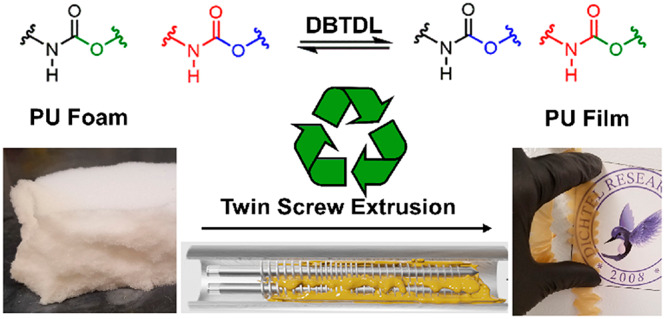
Cross-linked polyurethane (PU) is extensively used as thermoset foam; however, methods to directly reprocess PU foam waste derived from commercial sources into similar value materials have not been developed. We demonstrate that introducing dibutyltin dilaurate (DBTDL) into cross-linked PU foams and films enables their reprocessing at elevated temperatures via dynamic carbamate exchange reactions. Both model and commercial cross-linked PU foams were continuously reprocessed using twin-screw extrusion to remove gaseous filler and produce PU filaments or films with elastomeric or rigid thermoset mechanical properties. The properties of microcompounded model PU foam were in excellent agreement with PU film synthesized using the same monomers, indicating that this process occurs efficiently. These findings will enable the bulk reprocessing of commercial thermoset PU waste and inspire the further development of reprocessing methods for other thermosets and the compatibilization of chemically distinct cross-linked materials.
Short abstract
A method for recycling cross-linked polyurethane foam by embedding carbamate exchange catalyst and continuously reprocessing which could potentially reduce polyurethane waste generation is presented.
Introduction
Polyurethanes (PUs) are commonly synthesized as thermoset polymer networks used as foams, elastomers, coatings, sealants, adhesives, and rubbers.1 PUs represent 31% of the thermoset materials market and are widely incorporated into mattresses and furniture, thermal and sound insulation, automobiles, footwear, and construction materials.2,3 Although many of these applications represent durable goods, inevitable wear and replacement of PU-containing products generates a vast waste stream and necessitates the synthesis of new PUs, largely from toxic isocyanate-containing precursors. Methods to reprocess PUs, especially PU foams, are therefore attractive, but thermosetting foams are by definition chemically cross-linked, which precludes melt reprocessing and severely restricts their practical reuse. Current approaches for repurposing and recycling PUs are limited to mechanical methods, including rebonding for carpeting, or chemically recycling via catalyzed glycolysis, which results in the formation of reactive oligomeric polyols.4 Both of these approaches are forms of downcycling rather than direct recycling of the PU waste into similar value products.5 These limitations motivate alternative strategies to reprocess PU foams into similar or even higher-value products, which will improve the sustainability and circularity of their manufacture and use.
These sustainability goals might be achieved if processes to controllably reconfigure the cross-links of commercial PU foams are introduced. One promising approach is to formulate these systems as covalent adaptable networks (CANs),6−11 whose cross-links undergo exchange in response to a stimulus, such as increased temperature or photoexcitation. An ideal CAN combines the robust mechanical properties associated with traditional thermosets along with the processability of thermoplastics.6−11 Reports of CANs based on reversible Diels–Alder adducts12−14 and alkene addition/fragmentation15 demonstrated the ability to impart malleable character into cross-linked networks. Following this observation, Leibler and co-workers demonstrated bulk reprocessability of cross-linked polyester networks using Zn2+-catalyzed transesterification chemistry.16 This seminal work directly related rapid stress relaxation to efficient reprocessability of cross-linked polymers. Subsequently, stress relaxation and bulk reprocessing have been demonstrated in CANs using many dynamic linkages, including boronate esters,17 disulfides,18,19 carbonates,20 and vinylogous urethanes.21 Many of the synthesized CANs are amenable to typical plastic processing methods such as injection molding,16 compression molding,22 melt blowing,23,24 and twin-screw extrusion.25 Despite these advances, CANs rely on dynamic linkages that are not generally present in established commercial thermosets, which will require new materials to be developed and formulated within a commoditized marketplace that does not yet fully value circularity and sustainability. Recently, Manas-Zloczower and co-workers demonstrated that introducing dynamic cross-links into networks is possible by postsynthetically introducing transesterification catalysts into polyester networks via solution swelling methods.26 Most reported CANs rely on catalyzed dynamic exchange and are directly synthesized in the presence of these catalysts. Extending this processability to already synthesized static PUs remains largely unexplored and will enable repurposing of vast amounts of already-deployed materials into equal- or even higher-value products.
The direct reprocessing of PUs using the dynamics of urethane bonds was demonstrated in polyhydroxyurethanes, which reprocess in the absence of external catalysts likely through hydroxyl-carbamate exchange reactions.27,28 More recently, Lewis acid and tertiary amine activated carbamate exchange reactions have been employed in cross-linked PU networks that exhibit a rapid stress relaxation behavior.29−34 Although very promising, all of these examples of PU CANs involve rigid or elastomeric films, despite the fact that 67% of commercial PU materials are synthesized as foams.35 Consideration of PU foam gas content and cell morphology, additional commercial additives such as halogenated flame-retardants and surfactants, and various polymer backbone chemical functionalities, such as urea and isocyanurates, add more processing complexities and potential side reactions that may hinder the breakage and reformation of carbamate cross-links.36 Due to the prevalence of cross-linked PU foam materials in daily life, coupled with the significant space polyurethane foams take up in landfills,37 developing bulk reprocessing methods for the recycling of cross-linked PU foam is essential to ensure the sustainable use of these materials.
Here, we reprocess cross-linked PU foams by postsynthetically introducing dibutyltin dilaurate (DBTDL) as a carbamate exchange catalyst (Figure 1A) followed by melt reprocessing at elevated temperatures. This process is first demonstrated for a noncommercial rigid polyester PU foam without additives, and then for a flexible, open-cell commercial PU foam used in furniture. While catalyst-loaded model foams showed efficient dynamics during stress relaxation experiments, poor efficiency was observed in reprocessing by compression molding alone, which we attribute to poor mixing and the presence of trapped air in reprocessed materials. Therefore, we developed a twin-screw extrusion methodology that enables continuous reprocessing of PU foams (Figure 1B) into films with mechanical properties similar to other films made from the same monomers. We anticipate that these findings will enable the recycling of postconsumer PUs and will empower the use of cross-linked PU systems in emerging additive manufacturing techniques.
Figure 1.
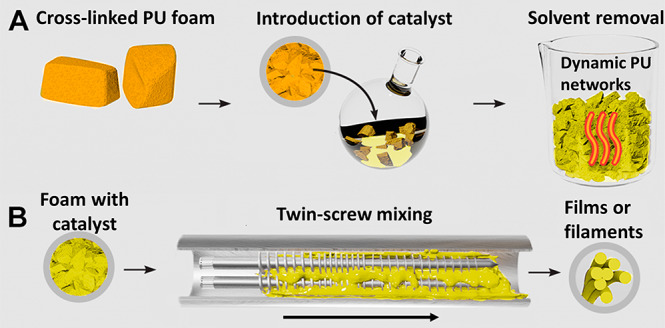
(A) Method of introduction of catalyst into static PU foams postsynthetically using solvent swelling in dichloromethane catalyst solution. (B) Scheme of microcompounding catalyst-loaded PU foam into filaments or films with removal of trapped air.
Results and Discussion
To determine if our proposed methodology of postsynthetically introducing catalyst and directly reprocessing foams would be practical, we first developed a model system to better understand how carbamate exchange occurs in both PU foams and films. Using methylene diphenyl diisocyanate and a commercially available polyester polyol (f = 2.5), cross-linked PU films were first cast from toluene at 60 °C with no added catalyst to serve as comparatives to model foams (Scheme 1). The stress relaxation and thermomechanical properties of both catalyst-free and catalyst-loaded films were compared to foams synthesized using the same monomers to understand how the foam morphology influences reprocessability. Rigid, cross-linked PU foams were synthesized by reacting the same monomers used in film synthesis in the presence of either isopentane or water as physical blowing (PB) or chemical blowing (CB) agents, respectively. CB foams lose cross-links through generation of CO2 and contain urea functional groups as byproducts. Although mechanical properties will differ in CB foams, it is important to probe their stress relaxation because most foams are chemically blown with water.38 The synthesis of foams required low concentrations of a DBTDL catalyst (0.35 mol %) to appropriately balance the rates of polymerization and gas generation. These model PU foams were evaluated prior to testing commercial PU foam formulations because the commercial products contain proprietary additives that might complicate carbamate exchange stress relaxation. Both foams and films showed complete disappearance of the isocyanate stretch at 2285 cm–1 and appearance of a carbamate stretch at 1708–1724 cm–1 by Fourier-transform infrared spectroscopy (FT-IR), indicating complete curing of the polymer networks (Figure S1). The films and foams both degraded to volatile byproducts between Td = 260 and 284 °C, as characterized by thermogravimetric analysis (TGA) (Figure S2), which is typical of cross-linked PUs. Differential scanning calorimetry (DSC) showed that the PB foams had higher glass transition temperatures (Tg = 61 °C) compared to control films (Tg = 45 °C Figure S3), which was attributed to the air in foam. Gel fractions of model PU films and PB foams were 87% and 89%, respectively, which is consistent with a well-cured polymer network. Both PB and CB foams exhibit closed cell morphologies with many cells collapsed, likely because silicone surfactants typically used to prevent this behavior were omitted (Figure S4).39 However, the exclusion of common foam additives means that the synthesized PU films and PB foams are chemically similar, enabling a more direct comparison of the differences in reprocessing between film and foam morphologies.
Scheme 1. Synthesis of PU Film with No DBTDL (Green), Physically Blown Foam with 0.35 mol % DBTDL (Red), and Chemically Blown Foam with 0.35 mol % DBTDL (Blue).

To determine if diffusing DBTDL into cross-linked PUs by postsynthetically swelling in a concentrated catalyst solution would provide processable PU networks, we first evaluated this method using catalyst-free PU films. PU film was ground and stirred (100 mg PU/mL) in a solution of DBTDL in CH2Cl2 (30 mg/mL), after which the PU was filtered, collected, and dried under vacuum. Inductively coupled plasma–optical emission spectrometry (ICP–OES) was used to determine that the PU film contained 0.56 wt % of Sn after swelling in catalyst solution (Table S1). The catalyst-loaded, powdered PU sample was subjected to compression molding at 160 °C for 12 min, during which the sample fused into a homogeneous solid film. Stress relaxation analysis (SRA) was performed to compare the viscoelastic properties of the as-synthesized PU film with no catalyst and compression molded catalyst-containing PU films. Films treated with catalyst solution relaxed stress rapidly (τ* = 28 s at 160 °C), suggesting that the material undergoes rapid dynamic exchange under these conditions. Films lacking the catalyst relaxed stress much more slowly and showed poor reprocessability, suggesting that carbamate exchange only occurs rapidly in catalyst-treated PU (Figure 2A). DBTDL was next introduced to both physically blown and chemically blown foams, and the foams were compression molded using the previous procedure. ICP–OES confirmed the presence of Sn at a concentration of 0.92 wt % in PB PU foam which is notably higher than the PU film results (0.56 wt %). Despite being chemically identical to PU control films and containing higher Sn by weight, PB foams relaxed stress more slowly than PU films (71 s at 160 °C, Figure 2B). CB foams relaxed more rapidly (53 s at 160 °C) than the PB foams, indicating that dynamic carbamate exchange is not hindered by other functional groups formed in PU foams blown with water (e.g., ureas). At all temperatures measured, stress relaxation times were significantly slower in compression molded PU foams compared to compression molded PU films. Activation energies of relaxation in both compression molded PB PU foam (159 ± 6 kJ/mol) and CB PU foam (188 ± 16 kJ/mol) are higher than that in PU film (143 ± 4 kJ/mol) suggesting that foam morphology increases the temperature dependence of stress relaxation in reprocessed PU material. Overall, the stress relaxation differences between compression molded foams and films could be due to the fact that the PUs’ initial foam morphology restricts the viscous flow of the resulting compression molded material.
Figure 2.

(A) Stress relaxation analysis of PU films without catalyst treatment (black) and with catalyst treatment (red) at 160 °C. (B) DMTA of compression molded postsynthetically treated PU films (red) and PB foams (blue).
In addition to influencing reprocessability, the initial foam morphology of the PU negatively impacts the thermomechanical properties of the materials reprocessed by compression molding. The Tg and rubbery plateaus of the reprocessed materials were determined by dynamic mechanical thermal analysis (DMTA) (Figure 2B). Compression molded PB foams exhibited broader peaks in tan(δ), with only 61% integrated area relative to compression molded PU films. Area under the tan(δ) curve is thought to correspond to the chain mobility, damping, and impact strength of the network.40 Even when compression molding was performed for longer time (1 h), the resulting films exhibited broad glass transitions (Figure S5). To determine if catalyst loading was responsible for the broad glass transitions, PU film and PB PU foam were synthesized with 1 mol % DBTDL and subsequently compression molded. The glass transition remained broad in compression molded foam suggesting that foam morphology is the major contributing factor (Figure S6). We attributed the broader glass transitions of the compression molded foams to inhomogeneity caused by trapped air still present within the resulting films (Figure 3A). Indeed, scanning electron microscopy (SEM) revealed voids in films derived from compression molded PU foams (Figure 3B). Optical microscopy of the molded foam samples showed cracking and greater inhomogeneity compared to samples derived from film-to-film reprocessing (Figure S7). These findings suggest that compression molding is not an effective reprocessing method for reprocessing PU foams, although it is effective for film-to-film PU reprocessing.
Figure 3.

(A) Image of compression molded PU film (left) and compression molded physically blown (PB) PU foam (right). (B) SEM of reprocessed PB foam using compression molding. (C) Image of continuously extruded PB foam into film. (D) SEM of reprocessed PB foam using twin-screw microcompounding.
Although the compression molded foam-to-film PU samples have inferior thermomechanical properties and stress relaxation rates compared to their film-to-film counterparts, foam-to-film samples nevertheless exhibit relatively rapid stress relaxation at elevated temperatures associated with dynamic carbamate exchange, suggesting that PU foam is still reprocessable. The broad glass transitions, higher rubbery plateaus, and higher glass transition temperatures are similar to those of polymer networks containing fillers.41−43 In these systems, fillers occupy free volume of the polymer network and interfere with segmental motion, which broadens the glass transition and increases the rubbery plateau modulus.44−46 Foams are formally gas–solid composite materials,47−49 and their processing into films therefore requires the removal of air from the network. Therefore, we turned our attention to alternatives to compression molding that would provide improved mixing and gas removal to achieve more homogeneous films.
Microcompounding refers to mixing of polymer formulations in the melt state on a small scale and is commonly accomplished with continuous mixing in a twin-screw extruder. We hypothesized that distributive and dispersive mixing mechanisms involved in microcompounding would cause the PU to homogenize more effectively and more efficiently expel air from the extrudate.50,51 Therefore, catalyst-loaded PB foam was microcompounded in a twin-screw batch mixer and extruded as a film, whose rheological properties and microscopic morphology were directly tested (Figure 3C). The extrusion was performed at 200 °C under nitrogen with a residence time of ∼1 min. The voids observed in the SEM images of compression molded PB foams were notably absent in images of the microcompounded PB foams (Figure 3D), which appear uniform.
Given the microscopic evidence of air removal in microcompounded PB foam, DMTA was used to quantitatively evaluate the reprocessing efficiency. Compared to compression molded PB foam, microcompounded PB foams exhibited drastically sharper tan(δ) responses more similar to compression molded PU films (Figure 4A). The area under the tan(δ) curve exceeds that of the compression molded PU film (1.27 normalized to that of compression molded films), demonstrating that the inherent damping behavior and molecular motion of the PU are largely recovered. We speculate that the excess area under the tan(δ) curve is related to the increased density of microcompounded PU foam compared to cast PU films (1.33 vs 1.13 g/cm3), but more work is required to determine the cause of the increase in area. Microscopic evidence coupled with rheology strongly suggests that microcompounding results in superior reprocessing of PU foams into films, compared with compression molding methods.
Figure 4.

(A) DMTA of compression molded PU films (red), compression molded physically blown (PB) PU foams (blue), and microcompounded PB PU foams (green). (B) Tensile testing of synthesized PU films (red), compression molded PB PU foams, and microcompounded PB PU foams into continuous films.
To determine the practical implications of this improved mixing methodology, tensile testing was performed on a continuously extruded film and compared to both as-synthesized PU films and compression molded PB foam-to-film samples (Figure 4B). The tensile strengths and yield points of both early and late portions of microcompounded PB foams were within error of those of as-synthesized PU films (Table S2), demonstrating excellent recovery of these material mechanical properties throughout the extrudate. In contrast, compression molded PB foams exhibited drastically lower tensile strengths and elongations at break, which supports our hypothesis that improved mixing and air removal are essential for obtaining desirable mechanical properties in foam-to-film reprocessed samples. Despite its excellent reprocessability, we observed that the extrudate does not relax stress significantly after microcompounding (Figure S8). To determine the major source of degradation of catalyst, PU films with catalyst were annealed at 200 °C for 2 min in air, which mimic the temperature of screw extrusion conditions but lack the mechanical agitation. SRA of these samples indicated only mild reduction of the stress relaxation, such that we speculate that high shear followed by subsequent air cooling may be the source of catalyst degradation (Figure S9).52 This degradation can be a desirable feature if films with minimal creep and maximum shape stability are desired, at the expense of multiple reprocessing cycles. However, these results suggest that other, more stable carbamate exchange catalysts should be investigated if materials capable of many reprocessing operations are desired. Based on our previous study, DBTDL was replaced with the less toxic and greener catalyst, bismuth neodecanoate (Bi(neo)3). Microcompounding led to noncontinuous extrusion and discoloration of the resulting materials (Figure S10). Interestingly, the materials retained stress relaxation in both light and dark portions of the PU film (Figure S11). Nevertheless, despite the loss of dynamic behavior after extrusion, the DBTDL-catalyzed films extrude continuously while maintaining excellent mechanical properties.
Having established reprocessing conditions for model PB PU foams, we applied this method to PU foams taken from consumer products, envisioning that this methodology could be applied to reprocess the vast amounts of PU waste created annually (1.3 million tons of PU waste in the US alone).53 Commercial PU foam (Air Lite) was purchased, infused with DBTDL catalyst, and microcompounded under similar conditions. ICP–OES confirmed that the Sn catalyst was present in treated commercial PU foams at similar weight percentages (0.64 wt %). As with the model PB PU foam, compression molding the commercial PU foam yielded inhomogeneous films with broad glass transitions, as well as obvious air voids observed by SEM (Figure S12). The catalyst-loaded commercial PU foam, despite containing a proprietary mixture of additives, such as surfactants, flame retardants, and stabilizers, underwent microcompounding and extrusion similar to the model PB PU foams (Figure 5A), suggesting that this methodology is tolerant of commercial PU formulations. Efforts to microcompound foams without added DBTDL failed, demonstrating the importance of introducing the catalyst to facilitate dynamic exchange and malleability into commercial PU foam (Figure S13). Optical microscopy and SEM of the extruded film indicated relatively homogeneous materials that were free of trapped air (Figure S14). DMTA of microcompounded films indicates much more homogeneous films with a near 50% increase in the area under the tan(δ) curve relative to compression molded samples (Figure 5B). Microcompounded foams exhibited a 5 °C lower glass transition by DSC compared to as-supplied foams, which is similar to the glass transition decrease in microcompounded PB PU foams (Figure S15). The tensile strength of microcompounded commercial PU foam was 3.4 MPa with average elongation of break of 168%, values typical of loosely cross-linked elastomeric materials (Figure S16). Interestingly, the microcompounded commercial foams relax stress after microcompounding (Figure S17) which is in stark contrast to model PB PU foam, suggesting that these commercial materials may be capable of further reprocessing cycles and demonstrating the promise of this methodology for continuous recycling of commercial PU foam waste.
Figure 5.

(A) Images of the PU foam reprocessed into films by microcompounding. (B) DMTA of compression molded commercial PU foam (blue) and microcompounded PU foams (green).
Conclusions
We have demonstrated that PU thermosets can be made continuously processable via postsynthetic introduction of DBTDL as a carbamate exchange catalyst. This process imparts malleability into cross-linked PU materials traditionally considered as thermosets. DMTA and SEM imaging indicate that reprocessing via compression molding is inefficient at removing air from foam materials. Microscopy and thermomechanical experiments show that air can be efficiently removed from PU foam using industrially relevant twin-screw extrusion which allows for continuous quantitative reprocessing of cross-linked PU foam into film. Additionally, we demonstrate the efficacy of this reprocessing method with commercial PU foams, suggesting its applicability to continuous recycling of the large amounts of PU waste currently produced. We expect that postsynthetic introduction of catalyst combined with twin-screw extrusion mixing will enable recycling of a wide variety of PU waste into high-value materials and will inspire the development of a similar methodology for the recycling of other commodity thermosets. These findings should also motivate the further development of carbamate exchange catalysts that withstand multiple reprocessing cycles and are both green and nontoxic.
Acknowledgments
This research was supported by the National Science Foundation (NSF) through the Center for Sustainable Polymers (CHE-1413862). This research made use of the Materials Characterization and Imaging Facility, which receives support from the MRSEC Program (NSF DMR-1121262) of the Materials Research Center at Northwestern University, and the Integrated Molecular Structure Education and Research Center at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology. This work made use of the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. We thank John Beumer for the artistic design of Figure 1.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00083.
Synthesis and characterization of PU foams, methods of imaging reprocessed materials, and thermal and mechanical characterization of synthesized and reprocessed films (PDF)
The authors declare the following competing financial interest(s): Northwestern University, the University of Minnesota, and Cornell University have filed a patent application related to the findings described in this manuscript.
Supplementary Material
References
- Szycher M.Szycher’s Handbook of Polyurethanes, 2nd ed.; Taylor & Francis, 2012; pp 6–12. [Google Scholar]
- Sonnenschein M. F.Polyurethanes: Science, Technology, Markets, and Trends. Wiley: Hoboken, NJ, 2015; pp 1–10. [Google Scholar]
- Polyurethanes (PUR) and Isocyanates (MDI & TDI); Ceresana Market Study, January 2016.
- Al-Salem S. M.; Lettieri P.; Baeyens J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manage. 2009, 29 (10), 2625–2643. 10.1016/j.wasman.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Thomas S.; Rane A. V.; Kanny K.; VK A.; Thomas M. G.. Recycling of Polyurethane Foams; Elsevier Science, 2018; pp 1–18. [Google Scholar]
- Zou W.; Dong J.; Luo Y.; Zhao Q.; Xie T. Dynamic Covalent Polymer Networks: from Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29 (14), 1606100. 10.1002/adma.201606100. [DOI] [PubMed] [Google Scholar]
- Fortman D. J.; Brutman J. P.; De Hoe G. X.; Snyder R. L.; Dichtel W. R.; Hillmyer M. A. Approaches to Sustainable and Continually Recyclable Cross-Linked Polymers. ACS Sustainable Chem. Eng. 2018, 6 (9), 11145–11159. 10.1021/acssuschemeng.8b02355. [DOI] [Google Scholar]
- Kloxin C. J.; Bowman C. N. Covalent adaptable networks: smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42 (17), 7161–7173. 10.1039/C3CS60046G. [DOI] [PubMed] [Google Scholar]
- Denissen W.; Winne J. M.; Du Prez F. E. Vitrimers: permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7 (1), 30–38. 10.1039/C5SC02223A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride M. K.; Worrell B. T.; Brown T.; Cox L. M.; Sowan N.; Wang C.; Podgorski M.; Martinez A. M.; Bowman C. N. Enabling Applications of Covalent Adaptable Networks. Annu. Rev. Chem. Biomol. Eng. 2019, 10 (1), 175–198. 10.1146/annurev-chembioeng-060718-030217. [DOI] [PubMed] [Google Scholar]
- Rowan S. J.; Cantrill S. J.; Cousins G. R. L.; Sanders J. K. M.; Stoddart J. F. Dynamic Covalent Chemistry. Angew. Chem., Int. Ed. 2002, 41 (6), 898–952. . [DOI] [PubMed] [Google Scholar]
- Kloxin C. J.; Scott T. F.; Adzima B. J.; Bowman C. N. Covalent Adaptable Networks (CANs): A Unique Paradigm in Crosslinked Polymers. Macromolecules 2010, 43 (6), 2643–2653. 10.1021/ma902596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzima B. J.; Aguirre H. A.; Kloxin C. J.; Scott T. F.; Bowman C. N. Rheological and chemical analysis of reverse gelation in a covalently crosslinked Diels-Alder polymer network. Macromolecules 2008, 41 (23), 9112–9117. 10.1021/ma801863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Dam M. A.; Ono K.; Mal A.; Shen H.; Nutt S. R.; Sheran K.; Wudl F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295 (5560), 1698–1702. 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- Kloxin C. J.; Scott T. F.; Bowman C. N. Stress relaxation via addition-fragmentation chain transfer in a thiol-ene photopolymerization. Macromolecules 2009, 42 (7), 2551–2556. 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarnal D.; Capelot M.; Tournilhac F.; Leibler L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334 (6058), 965–968. 10.1126/science.1212648. [DOI] [PubMed] [Google Scholar]
- Rottger M.; Domenech T.; van der Weegen R.; Breuillac A.; Nicolay R.; Leibler L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356 (6333), 62–65. 10.1126/science.aah5281. [DOI] [PubMed] [Google Scholar]
- Fortman D. J.; Snyder R. L.; Sheppard D. T.; Dichtel W. R. Rapidly Reprocessable Cross-Linked Polyhydroxyurethanes Based on Disulfide Exchange. ACS Macro Lett. 2018, 7 (10), 1226–1231. 10.1021/acsmacrolett.8b00667. [DOI] [PubMed] [Google Scholar]
- Rekondo A.; Martin R.; Ruiz de Luzuriaga A.; Cabañero G.; Grande H. J.; Odriozola I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 2014, 1 (2), 237–240. 10.1039/C3MH00061C. [DOI] [Google Scholar]
- Snyder R. L.; Fortman D. J.; De Hoe G. X.; Hillmyer M. A.; Dichtel W. R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51 (2), 389–397. 10.1021/acs.macromol.7b02299. [DOI] [Google Scholar]
- Denissen W.; Rivero G.; Nicolaÿ R.; Leibler L.; Winne J. M.; Du Prez F. E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25 (16), 2451–2457. 10.1002/adfm.201404553. [DOI] [Google Scholar]
- Delahaye M.; Winne J. M.; Du Prez F. E. Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry. J. Am. Chem. Soc. 2019, 141 (38), 15277–15287. 10.1021/jacs.9b07269. [DOI] [PubMed] [Google Scholar]
- Jin K.; Kim S.-s.; Xu J.; Bates F. S.; Ellison C. J. Melt-Blown Cross-Linked Fibers from Thermally Reversible Diels–Alder Polymer Networks. ACS Macro Lett. 2018, 7 (11), 1339–1345. 10.1021/acsmacrolett.8b00685. [DOI] [PubMed] [Google Scholar]
- Jin K.; Banerji A.; Kitto D.; Bates F. S.; Ellison C. J. Mechanically Robust and Recyclable Cross-Linked Fibers from Melt Blown Anthracene-Functionalized Commodity Polymers. ACS Appl. Mater. Interfaces 2019, 11 (13), 12863–12870. 10.1021/acsami.9b00209. [DOI] [PubMed] [Google Scholar]
- Taplan C.; Guerre M.; Winne J. M.; Du Prez F. E. Fast processing of highly crosslinked, low-viscosity vitrimers. Mater. Horiz. 2020, 7 (1), 104–110. 10.1039/C9MH01062A. [DOI] [Google Scholar]
- Yue L.; Bonab V. S.; Yuan D.; Patel A.; Karimkhani V.; Manas-Zloczower I. Vitrimerization: A Novel Concept to Reprocess and Recycle Thermoset Waste via Dynamic Chemistry. Global Challenges 2019, 1800076. 10.1002/gch2.201800076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortman D. J.; Brutman J. P.; Cramer C. J.; Hillmyer M. A.; Dichtel W. R. Mechanically activated, catalyst-free polyhydroxyurethane vitrimers. J. Am. Chem. Soc. 2015, 137 (44), 14019–14022. 10.1021/jacs.5b08084. [DOI] [PubMed] [Google Scholar]
- Fortman D. J.; Brutman J. P.; Hillmyer M. A.; Dichtel W. R. Structural effects on the reprocessability and stress relaxation of crosslinked polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134 (45), 44984. 10.1002/app.44984. [DOI] [Google Scholar]
- Fortman D. J.; Sheppard D. T.; Dichtel W. R. Reprocessing Cross-Linked Polyurethanes by Catalyzing Carbamate Exchange. Macromolecules 2019, 52 (16), 6330–6335. 10.1021/acs.macromol.9b01134. [DOI] [Google Scholar]
- Yan P.; Zhao W.; Fu X.; Liu Z.; Kong W.; Zhou C.; Lei J. Multifunctional polyurethane-vitrimers completely based on transcarbamoylation of carbamates: thermally-induced dual-shape memory effect and self-welding. RSC Adv. 2017, 7 (43), 26858–26866. 10.1039/C7RA01711A. [DOI] [Google Scholar]
- Yan P.; Zhao W.; Jiang L.; Wu B.; Hu K.; Yuan Y.; Lei J. Reconfiguration and shape memory triggered by heat and light of carbon nanotube-polyurethane vitrimer composites. J. Appl. Polym. Sci. 2018, 135 (5), 45784. 10.1002/app.45784. [DOI] [Google Scholar]
- Yan P.; Zhao W.; Wang Y.; Jiang Y.; Zhou C.; Lei J. Carbon Nanotubes-Polyurethane Vitrimer Nanocomposites with the Ability of Surface Welding Controlled by Heat and Near-Infrared Light. Macromol. Chem. Phys. 2017, 218 (20), 1700265. 10.1002/macp.201700265. [DOI] [Google Scholar]
- Schneiderman D. K.; Vanderlaan M. E.; Mannion A. M.; Panthani T. R.; Batiste D. C.; Wang J. Z.; Bates F. S.; Macosko C. W.; Hillmyer M. A. Chemically Recyclable Biobased Polyurethanes. ACS Macro Lett. 2016, 5 (4), 515–518. 10.1021/acsmacrolett.6b00193. [DOI] [PubMed] [Google Scholar]
- Brutman J. P.; Fortman D. J.; De Hoe G. X.; Dichtel W. R.; Hillmyer M. A. Mechanistic Study of Stress Relaxation in Urethane-Containing Polymer Networks. J. Phys. Chem. B 2019, 123 (6), 1432–1441. 10.1021/acs.jpcb.8b11489. [DOI] [PubMed] [Google Scholar]
- Gama N. V.; Ferreira A.; Barros-Timmons A. Polyurethane Foams: Past, Present, and Future. Materials 2018, 11 (10), 1841. 10.3390/ma11101841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesczewski B.Polymerization Chemistry of Polyurethanes. In Encyclopedia of Materials: Science and Technology; Buschow K. H. J., Cahn R. W., Flemings M. C., Ilschner B., Kramer E. J., Mahajan S., Veyssière P., Eds.; Elsevier: Oxford, 2001; pp 7632–7634. [Google Scholar]
- Kylili A.; Seduikyte L.; Fokaides P. A.. Life Cycle Analysis of Polyurethane Foam Wastes. In Recycling of Polyurethane Foams; Thomas S., Rane A. V., Kanny K., V.K A., Thomas M. G., Eds.; William Andrew Publishing, 2018; Chapter 9, pp 97–113. [Google Scholar]
- Frisch K. C.; Klempner D.. Polyurethanes. In Comprehensive Polymer Science and Supplements; Allen G., Bevington J. C., Eds.; Pergamon: Amsterdam, 1989; Chapter 24, pp 413–426. [Google Scholar]
- Zhang X. D.; Macosko C. W.; Davis H. T.; Nikolov A. D.; Wasan D. T. Role of Silicone Surfactant in Flexible Polyurethane Foam. J. Colloid Interface Sci. 1999, 215 (2), 270–279. 10.1006/jcis.1999.6233. [DOI] [PubMed] [Google Scholar]
- Menard K. P.; Menard N. R. Dynamic Mechanical Analysis in the Analysis of Polymers and Rubbers. Encyclopedia of Polymer Science and Technology 2015, 1–33. 10.1002/0471440264.pst102.pub2. [DOI] [Google Scholar]
- Yim A.; Chahal R. S.; Pierre L. E. S. The effect of polymer—filler interaction energy on the T′g of filled polymers. J. Colloid Interface Sci. 1973, 43 (3), 583–590. 10.1016/0021-9797(73)90406-2. [DOI] [Google Scholar]
- Reid C. G.; Greenberg A. R. Influence of silica reinforcement upon the glass transition behavior of acrylic polymers. J. Appl. Polym. Sci. 1990, 39 (4), 995–1014. 10.1002/app.1990.070390417. [DOI] [Google Scholar]
- Manson J. A.Polymer Blends and Composites; Springer US, 2012; pp 51–75. [Google Scholar]
- Toussaint A. Influence of pigmentation on the mechanical properties of paint films. Prog. Org. Coat. 1974, 2 (3), 237–267. 10.1016/0300-9440(74)80004-4. [DOI] [Google Scholar]
- Iisaka K.; Shibayama K. Effect of filler particle size on dynamic mechanical properties of poly(methyl methacrylate). J. Appl. Polym. Sci. 1978, 22 (5), 1321–1330. 10.1002/app.1978.070220513. [DOI] [Google Scholar]
- Howard G. J.; Shanks R. A. The Influence of Filler Particles on the Mobility of Polymer Molecules. J. Macromol. Sci., Chem. 1982, 17 (2), 287–295. 10.1080/00222338208063261. [DOI] [Google Scholar]
- Lee S. T.; Ramesh N. S.. Polymeric foams: mechanisms and materials; CRC Press: Boca Raton, 2004; pp 1–19. [Google Scholar]
- Lee L. J.; Zeng C.; Cao X.; Han X.; Shen J.; Xu G. Polymer nanocomposite foams. Compos. Sci. Technol. 2005, 65 (15), 2344–2363. 10.1016/j.compscitech.2005.06.016. [DOI] [Google Scholar]
- Eaves D.Handbook of Polymer Foams; Rapra Technology, 2004; pp 1–8. [Google Scholar]
- Martin C. Twin Screw Extruders as Continuous Mixers for Thermal Processing: a Technical and Historical Perspective. AAPS PharmSciTech 2016, 17 (1), 3–19. 10.1208/s12249-016-0485-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur. J. Pharm. Biopharm. 2002, 54 (2), 107–117. 10.1016/S0939-6411(02)00061-9. [DOI] [PubMed] [Google Scholar]
- Ghazi D.; Rasheed Z.; Yousif E. J. d. Review of organotin compounds: chemistry and applications. AOICS 2018, 3, 344–352. 10.32474/AOICS.2018.03.000161. [DOI] [Google Scholar]
- Nikje M. M. A.; Garmarudi A. B.; Idris A. B. Polyurethane Waste Reduction and Recycling: From Bench to Pilot Scales. Des. Monomers Polym. 2011, 14 (5), 395–421. 10.1163/138577211X587618. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.