

Abstract
Off-target effects continue to impede disease interventions, particularly when targeting a specific protein within a family of similar proteins, such as kinase isoforms that play tumor-subtype-specific roles in cancers. Exploiting the specific electrophilic-metabolite-sensing capability of Akt3, versus moderate or no sensing, respectively, by Akt2 and Akt1, we describe a first-in-class functionally Akt3-selective covalent inhibitor [MK-H(F)NE], wherein the electrophilic core is derived from the native reactive lipid metabolite HNE. Mechanistic profiling and pathway interrogations point to retention of the metabolite’s structure—as opposed to implicit electrophilicity—as being essential for biasing isoform preference, which we found translates to tumor-subtype specificity against pten-null triple-negative breast cancers (TNBCs). MK-H(F)NE further enables novel downstream target identification specific to Akt3-function in disease. In TNBC xenografts, MK-H(F)NE fares better than reversible pan-Akt-inhibitors and does not show commonly observed side-effects associated with Akt1-inhibition. Inhibitors derived from native-metabolite sensing are thus an enabling plan-of-action for unmasking kinase-isoform-biased molecular targets and tumor-subtype-specific interventions.
Short abstract
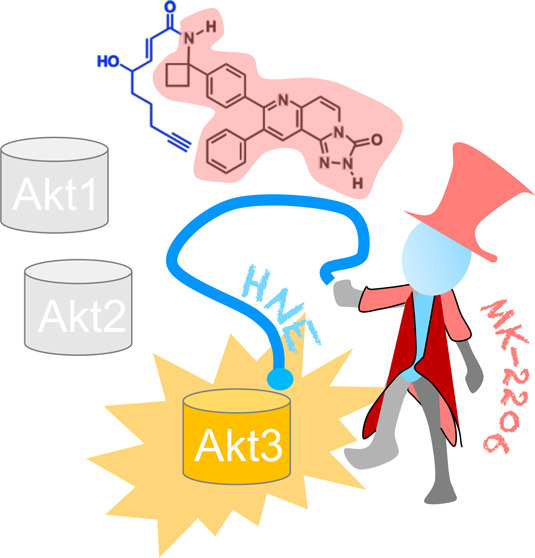
The Ring Master: training of natural electrophiles to target a single protein leads to improved target specificity and drug activity but retains the electrophile’s privileged properties.
Introduction
We have made strides in developing selective drugs, but off-target effects can still severely limit drug efficacy and development. Although widespread, this problem is particularly pertinent to enzymes which use common substrates, such as kinases and ubiquitin-conjugating/deconjugating enzymes. In fact, most of the ∼50 approved small-molecule kinase-targeting drugs have off-target effects that in some way exacerbate efficacy/toxicity. There are continued efforts to identify off-target binding events of kinase drugs for either repurposing or developing general design principles to restrict target spectra.1 Despite some specific successes, novel strategies to engender high selectivity remain limited. This challenge is magnified when targeting specific protein-isoforms. Although some mutant allele-specific anticancer drugs are known, only one isoform-selective kinase inhibitor is currently approved. Because protein-isoforms often show antagonistic functions with discrete regulatory mechanisms depending on disease subtypes and context, this issue remains a significant stumbling block to developing precision therapeutics.
The three Akt-kinase isozymes share 73% sequence-similarity and have overlapping target-protein substrates, as they have a common canonical substrate recognition sequence, (RX)RXXT/S. However, Akt-isozymes also recognize distinct protein–substrates in a complex disease-dependent manner.2−5 Akt1 is associated with tumor invasiveness in some cancers but suppresses metastasis in others; Akt3 is particularly elevated in certain cancer-subtypes. Conversely, Akt2-activation is required for tumor maintenance and metastatic dissemination, particularly when PTEN—an Akt-signaling antagonist—is compromised. Notably, most Akt-inhibitors display affinities highest for Akt1 and lowest for Akt3. Several of these inhibitors have entered Phase-I/II clinical trials. None have been approved.
We recently disclosed that Akt3 is a privileged sensor of the native electrophilic lipid, 4-hydroxynonenal (HNE). Akt3 HNE-sensing is coupled to Akt3-activity downregulation and pathway inhibition.6 HNE also manifests a weak interaction with Akt2, but does not label Akt1. Akt3’s high HNE-ligandability is traceable to a single cysteine, C119 (Figure 1). C119 lies within the linker region bridging the PH- and kinase-domains. Akt3(C119) has no analogue in Akt1. Akt2 possesses a cysteine in the analogous linker region, but the surrounding primary sequence bears no homology to that of Akt3(C119) (Figure 1). Notably, due to HNE-modification of Akt3(C119) eliciting dominant-negative behavior, low occupancy Akt3(C119) HNEylation results in efficient pathway inhibition in both cultured cells and larval fish.6 We thus postulated that HNE could be used as a framework around which to derive Akt-isozyme-selective inhibitors. Such inhibitors would likely have properties useful for drug development, including covalent binding, rapid association kinetics, and dominant-negative behavior.
Figure 1.

 |
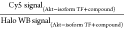 designates the average of signal across all Halo-Akt(n)-expressing
lanes treated with indicated
compound, relative to the corresponding Western blot (WB) signal from
Halo, within the same gel; and
designates the average of signal across all Halo-Akt(n)-expressing
lanes treated with indicated
compound, relative to the corresponding Western blot (WB) signal from
Halo, within the same gel; and 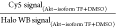 designates the average of signal across all Halo-Akt(n)-expressing lanes that are treated with DMSO,
relative to the corresponding WB signal from Halo, within the same
gel. When signal-to-noise is high (e.g., Figure 1b, left panel), the equation above collapses
to
designates the average of signal across all Halo-Akt(n)-expressing lanes that are treated with DMSO,
relative to the corresponding WB signal from Halo, within the same
gel. When signal-to-noise is high (e.g., Figure 1b, left panel), the equation above collapses
to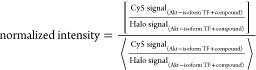 |
Unfortunately, HNE is reactive and toxic. Hence, unlike other natural polypharmacologic compounds, sulforaphane, staurosporine, or quercertin, HNE is not canonical “fodder” for drug discovery. Herein, we disclose a rationally designed Akt3-selective covalent inhibitor that shows moderate selectivity for Akt3 over Akt2 and that labels Akt1 weakly. This inhibitor bears a “tamed” HNE-like appendage fused to a noncovalent, moderately Akt1-selective pan-Akt-inhibitor in Phase-II evaluations, MK-2206. Tagging MK-2206 with generic electrophilic appendages gives hybrid compounds with poor Akt-labeling efficiency in cells; however, HNE-tagging shows potent and selective Akt3-labeling. The resulting Akt3-semiselective inhibitors [MK-H(F)NE] are effective at low ligand occupancy, phenocopying the dominant-negative pathway inhibition incurred upon site-specific native lipid sensing, i.e., Akt3(C119)-hydroxynonenylation. Further interrogations in triple-negative BC (TNBC) cells and TNBC-xenograft models demonstrate that MK-H(F)NE is more efficacious than MK-2206 with an Akt3-biased inhibition profile.
Inhibitor Design and Concept Validation
We examined known Akt inhibitors for a framework around which to develop an Akt3-covalent inhibitor bearing an HNE-like appendage. Akt3 homology modeling using the structure of Akt1 (PDB: 3O96)7 shows that the HNE-reactive residue within Akt3, C119, lies in the flexible linker separating the PH and kinase domains. This linker lies ∼40 Å away from the ATP-binding site (the hinge region), rendering design of HNE-hybrids targeting the ATP-binding site unlikely. However, the ATP-noncompetitive inhibitor, MK-2206, binds ∼25 Å from C1198 (Figure 1), a distance that may render covalent-bond formation feasible since C119 lies on a flexible loop. Unfortunately, MK-2206 has around ∼100-fold higher affinity for Akt1 than Akt3,9,10 and cells resistant to MK-2206 typically overexpress Akt3,11 rendering MK-2206 a challenging framework for Akt3-selective-inhibitor design. However, we reasoned that the proposed benefits of matching the electrophilic moiety to Akt3 could outweigh the negatives of the ligand mismatching the protein. Furthermore, MK-2206 has some desirable chemical attributes, including a pendent amine moiety ripe for functionalization. We thus pursued MK-2206 derivatization and developed MK-HNE, wherein the aldehyde of HNE is converted to an amide, linking the HNE and MK-2206 moieties. Substitution of the aldehyde with an amide lowers electrophilicity of the enone, reducing off-target possibilities. We also created (1) a saturated analogue, MK-HNA, that cannot undergo Michael adduction; and (2) MK-G, derived from a regioisomer of HNE that is more reactive than the parent aldehyde (Figure 1a). An alkyne was installed in all of the above to facilitate target validation.
HEK293T cells ectopically expressing each of the three Akt-isozymes as functional and active HaloTagged-fusions6 (facilitating distinction from endogenous isozymes) were treated (5 μM, 48 h) with the indicated compounds. Lysates were subjected to Click-coupling with azido-fluorophore, reporting inhibitor covalent engagement. Fluorescence was normalized to Halo-expression and background signal from both DMSO-treated transfected and nontransfected cells. As predicted, Akt3 was labeled more efficiently than Akt2. Akt1 and Akt3(C119S), an HNE-sensing-defective-but-otherwise-functional Akt3 (Figure 1b,c), were only weakly labeled under these conditions. Comparative dose-responsive labeling analysis in cells ectopically expressing either Akt3 or Akt2, and fitting the resulting data to a one-site-binding model, showed that MK-HNE labeled Akt3—3.3-fold more efficiently than Akt2 (Figure S2). Given than Akt2 has around 10-fold preference for binding to MK2206 over Akt3,10 this level of selectivity is remarkable (Figure S2). However, the intrinsic labeling preference of MK-HNE for Akt3 is at the lower end of what may be considered as being selective in itself. (We outline other modes-of-action that further promote the Akt3-biasing of MK-HNE and show evidence below for Akt3 being the most relevant isoform functionally targeted by MK-HNE and its derivatives in cells.) MK-HNA-treatment gave no signal intensity above background, supporting the necessity of the enone for covalent association. MK-G showed lower labeling than MK-HNE, and a preference for Akt2 reactivity (Figure 1b,c, Figure S1).
Click-biotin-enrichment following MK-HNE treatment (5 μM, 12 h) of native HEK293T cells showed that MK-HNE labeled endogenous Akt (Figure S3).
MK-HNE Shows Persistent Inhibition of Akt3
Kinase activity of ectopic Halo-Akt(n) (n = 1—3) can be measured in cells6 by ratiometric-FRET-based assays using Akt-activity reporter (AR) constructs.12 24 h treatment of HEK293T cells expressing Halo-Akt3 and Akt-AR with either MK-2206 (5 μM) or MK-HNE (5 μM) suppressed the ratiometric-FRET signal compared to DMSO-treated cells, implying Akt-activity-downregulation. Reanalysis of the same cells 48 h post-treatment showed preserved inhibition. Media were then switched to media without inhibitor, and cells were incubated for 24 h. Inhibition persisted only in samples originally treated with MK-HNE (Figure 2a). In MK-2206-treated cells, kinase activity was restored. A similar outcome was observed in cells expressing Halo-Akt2 (Figure S4). When Halo-Akt3(C119S) was assayed, both MK-2206 and MK-HNE elicited inhibition prior to compound withdrawal at 24 h; although, at 48 h, MK-HNE treatment inhibited Halo-Akt3(C119S) less effectively than MK-2206 at 48 h (Figure 2b). Inhibitor withdrawal resulted in recovery to normal activity levels for MK-2206 and MK-HNE, indicating that C119 is the principal residue forming a covalent bond to MK-HNE.
Figure 2.

MK-HNE irreversibly inhibits Akt3 in cells by labeling C119. HEK293T cells ectopically expressing AktAR-ratiometric-FRET-reporter and Halo-Akt3 [either wt (a) or C119S (b)] were treated (24–48 h), with MK-HNE (5 μM), MK-2206 (5 μM), or DMSO; followed by rinsing twice with drug-free media, and recovery in drug-free media over another 24 h (denoted as “withdrawal”). Akt-kinase activities were measured at the indicated times, using ratiometric-FRET-based live-cell imaging. Left: representative confocal images. [Cells were excited using an argon laser (458 nm). Emission was observed in cyan (463–498 nm) and yellow (525–620 nm) channels. Scale bar 10 μm.] Right: quantitation of (YFP/CFP)-emission ratio of individual cells. Error bars indicate 5–95% confidence intervals, boxes show upper and lower quartiles, and central bar shows median; dots show outliers. Cell number analyzed = 385, 201, 528, 200, 90, 85, 100, 321, 138 [Akt3(wt)]; and 108, 122, 320, 114, 81, 93, 91, 74, 90 [Akt3(C119S)], from 3 independent sets at different passage numbers. P values are from two-tailed unpaired t-test. Also see Figures S4–S6.
Unlike most covalent drugs currently in clinical use/trials, MK-HNE is a chimera of two different Akt-inhibitors, which have different mechanisms: MK-2206 elicits almost full suppression of Akt3(T305)-phosphorylation; HNE has little effect on phospho-T305-levels.6 We compared how MK-2206 and MK-HNE, respectively, affect Akt3(T305)-phosphorylation to illuminate which component (HNE, or MK-2206) dominates the mode-of-inhibition of Akt3. We analyzed Halo-Akt3 phospho-T305-levels, in cells treated with saturating doses of either MK-2206 or MK-HNE by western blot (Figure S5a) and ELISA (Figure S5b). Akt3(T305)-phosphorylation was not affected by MK-HNE, whereas Akt3(T305) phosphorylation was fully suppressed by MK-2206. Thus, surprisingly the covalent labeling of Akt3 via the HNE-derived component within MK-HNE likely dominates the mechanism of pathway inhibition.
MK-H(F)NE Manifest Privileged kinact Values
To vary oxidative stability and improve reactivity, we replaced the alcohol within MK-HNE with either a fluorine (elevated electron-withdrawing capacity), to give MK-FNE, or a proton (reduced electron-withdrawing capacity), to give MK-NE. Both MK-FNE and MK-NE covalently labeled ectopic Halo-Akt3 in cells (Figure S6). In a coupled assay for Akt3 activity, all 3 inhibitors elicited time-dependent inactivation of recombinant human Akt3. MK-FNE had the best second-order kinetics of the three inhibitors, which was principally due to an effect on Ki, not on kinact (Figure 3a, Figure S7a–e). MK-HNE showed faster second-order inactivation rates than MK-NE, also due to reduction in Ki. These observations indicate that both the OH- and F-substituted inhibitors [MK-H(F)NE] bind Akt3 better than the unsubstituted inhibitor (MK-NE). Such an outcome is consistent with the pendant alkyl chain on the inhibitor promoting inhibition. For all these inhibitors, kinact was ∼12 ms–1, ∼5–10-fold faster than most approved covalent EGFR-kinase inhibitors derived from similar 1,2-disubstituted enone scaffolds (Figure 3c).13 We replicated these experiments with the regioisomer of MK-HNE, MK-G, which showed poor Akt3-labeling potency in the cell-based assay. MK-G emerged to be a poor inhibitor of Akt3 and showed no time dependence over the time course of the assay. This result, especially when taken with the cell-based data, indicates a reversible binding mode. The Ki of MK-G for Akt3 was ∼80 μM (Figure S7g,h).
Figure 3.

MK-FNE manifests an efficient covalent-inactivation rate in vitro and dominant pathway-suppression in cells through Akt3(C119)-labeling. (a–c) Akt3-covalent inhibition analyzed by NADH-coupled kinase-activity assays. (Error bars indicate s.e.m.; n ≥ 3 replicates from two independent sets of Akt3 kinase.) Also see Figure S7. (a) Progress curves for inhibition of recombinant-human-Akt3 (0.15 μM) by MK-FNE. Solid curves: best nonlinear fits using eq i. Inset: (top) residual analysis of the fit; (right) eqs i—iii. (For individual terms, see Figure S7.) (b) kobs [fit to eq ii] against [MK-FNE]. (c) Akt3-covalent-inactivation parameters for the indicated compounds under identical conditions from eqs ii and iii. (d, e) Dominant loss of Akt3-kinase activity measured by AktAR-ratiometric-FRET reporter assays (see Figure 2, Figures S4 and S8). HEK293T cells were cotransfected with AktAR-reporter-plasmid and an equivalent amount of a plasmid mix containing Halo-Akt3(wt:C119S) in the indicated ratios. After 12 h, cells were treated (48 h) with MK-FNE (5 μM) or DMSO; washed twice with drug-free media; and allowed to recover over another 24 h (indicated as “withdrawal”). (d) Representative images. (e) Image-J quantitation of YFP/CFP-emission ratios after 24 h recovery in drug-free media. Data were normalized to the respective DMSO-treated samples subjected to otherwise identical conditions. Error bars indicate 5–95% percentile of data with n (cell no.) = 497, 326, 375, 312, 337, from 3 independent sets of cells at different passage numbers. P values are from two-tailed unpaired t-test.
MK-FNE Engenders Dominant-Negative Inhibition of Akt3, but Not Akt2
MK-FNE also showed irreversible Akt3 inhibition in HEK293T cells expressing Halo-Akt3(wt). Inhibition was reversible in cells expressing Akt3(C119S) (Figure 3d,e, Figure S8). When a 50:50 mixture of Akt3:Akt3(C119S) was expressed, complete inhibition persisted postwithdrawal. Thus, MK-FNE retains dominant-negative effects similar to Akt3(C119)-specific hydroxynonenylation.6 When we repeated this experiment in cells expressing 50:50 Halo-Akt2(wt):Halo-Akt3(C119S), no similar dominant-negative effects were observed; i.e., significant release from inhibition was observed postremoval of MK-FNE (Figure S9e,f). The difference in the outcomes observed in the two heterozygote-mimicking backgrounds further shows that HNE-like motifs possess additional modes of inhibition when interacting with Akt3, as opposed to other Akt isoforms. These modes could synergize with the improved kinetic selectivity shown by MK-FNE for Akt3. We investigated these effects further below.
MK-FNE Manifests Increased Selectivity and Greater Sustained Efficacy than MK-2206 in pten–/– Triple-Negative BC-Cells
Because Akt3-knockdown in MDA-MB-468 attenuates the tumor-growth rate,14 we explored the correlation between cellular Akt-isoform dependency and MK-H(F)NE potency in BCs. We chose 6 BC lines not believed to rely on Akt2 and Akt3 for survival, two of which, T47D and Hs578T, require Akt1 for growth. We also chose 2 BC lines highly dependent on Akt2 and Akt3 for survival (MDA-MB-468 and BT-549). MK-FNE showed similar toxicity to MK-2206 against the latter two cell lines (Figure 4a). Consistent with the regain in ectopic-Akt3(wt)-activity upon MK-2206-withdrawal (Figure 2a), when MK-2206 was withdrawn from MDA-MB-468 line, statistically significant regrowth was observed: regrowth was not observed in MK-H(F)NE-treated cells (Figure S10). Noting that dominant-negative inhibition was observed specifically during Akt3-targeting, and Akt1-targeting was not covalent, these data provide further evidence for an Akt3-functionally selective mechanism in MDA-MB-468. In this line, HNE-amide-induced proliferation inhibition EC50 was >200 μM (Figure S11). HNE-amide also did not show any synergism with MK2206 (%Figure S11), indicating that chemical fusion of the twain is required to observe the effects we see with MK-H(F)NE.
Figure 4.

 |
Importantly, in non-Akt2/3-reliant lines (T47D and Hs578T), MK-FNE induced significantly less toxicity than MK-2206 (Figure 4a). Since many lines become Akt3-dependent upon Akt1-selective inhibitor exposure, we tested if MK-FNE exhibits better efficacy against MK-2206-resistant lines than MK-2206. We used a previously developed T47D (Akt1-dependent) line exhibiting a 5-fold increase in resistance to MK-2206 treatment, due to upregulation of Akt3-expression.11 Consistent with our proposed mode of toxicity, the MK-2206-resistant-T47D cells showed only a 1.6-fold increase in resistance to MK-FNE (Figure S12).
MDA-MB-468 Exhibits Selective Sensitivity to MK-FNE upon Akt3-Specific Knockdown
To show that endogenous Akt3 is a key functionally relevant target of our compounds, we used isoform-selective Akt-knockdown (Figure S13) combined with inhibitor treatment. Partial knockdown of the principal protein target of the compound should sensitize cells to the compound. Comparing the EC50-values of proliferation suppression between MK-2206 and MK-FNE, we found that Akt3-knockdown sensitized cells to MK-FNE, whereas knockdown of Akt1 sensitized cells to MK-2206 (Figure 4b, Table S1). Although one siRNA targeting Akt2 showed moderate synergy with MK-FNE, the overwhelming majority of siRNAs (4 out of 5, showing 50–75% knockdown efficiency, Figure S13) showed no synergy. These outputs are strongly consistent with the proposed selectivity profiles, namely, that MK2206 targets Akt1, and MK-FNE targets Akt3.
MK-FNE and MK-2206 Regulate Different Akt-Dependent Phosphorylations
Although this property was not shared by all Akt-targeting drugs, e.g., GDC0068 and GSK690693, MK2206 and MK-F(H)NE elicited similar cell cycle changes (G1/G0-stall) (Table S2). Nevertheless, based on the divergent toxicity profiles and isoform-selective sensitization displayed by MK-FNE and MK-2206, we hypothesized that these inhibitors may differentially affect the phosphorylation of Akt targets. We used SILAC (Figure 5a) to map the specific phosphorylated targets altered in response to MK-FNE vs MK-2206 in MDA-MB-468 cells. Using an antibody recognizing the canonical Akt-phosphorylation-site sequence (RXXT/S; where the italicized site indicates a phosphorylated residue), coprecipitated proteins were quantitatively profiled. Each inhibitor-treated sample (heavy-Arg-/Lys-labeled cells) was compared to DMSO (light).
Figure 5.

MK-FNE modulates novel downstream targets distinct from those affected by MK-2206. (a) SILAC-proteomics analysis identifies Akt3-specific substrates. 1:1 mixing of “Heavy” MDA-MB-468-cells treated (36 h) with either MK-FNE or MK-2206 (at EC60-concentrations) and DMSO-treated “Light” cells were lysed and phosphorylated target-proteins were enriched using an immobilized phospho-Akt-substrate antibody (110b7E). Enriched targets were resolved by SDS-PAGE, trypsin-digested, and analyzed by LC-MS/MS. (b–e) Selection criteria and validations deployed. (b) Among the 47 hits suppressed exclusively upon MK-FNE-treatment, 11 were previously not clearly linked to Akt. Similar selection criteria discovered 3 hits from MK-2206-subset. (c) Cytotoxicity of MK-HNE and MK-2206 analyzed by alamarBlue in indicated knockdown lines: inset (top-right). Data show ± s.d. (n ≥ 8). Frequency-distribution analysis selects knockdown lines deviating by ±1σ from group mean. (d) Knockdown of selected hits, KIFC1, Grsf1, and JUP, assessed by western blot: representative data from three independent shKIFC1-lines against two shControl-lines. See also Figure S16. (e) Proliferation inhibition of MDA-MB-468 cells by either MK-FNE or MK-2206 was compared under either DMSO- or AZ82-treatment. Two-tailed unpaired t-test was applied (independent biological replicates: MK-FNE, n = 3, MK-2206, n = 15). See also Figures S12–S21.
There was substantial overlap between the immunoprecipitated proteins profiled in the two sets (i.e., MK-2206 vs DMSO; and MK-FNE vs DMSO) irrespective of Heavy:Light ratios. Twenty-five total proteins profiled within these two data sets were known Akt-substrates reported in PhosphoSitePlus (PSP) (Figure S14). Analyzing the depleted protein targets (i.e., suppression of substrate phosphorylation following inhibitor treatment; targets with low Heavy:Light ratios) (Figure 5b) revealed several nonoverlapping hits, i.e., protein substrates whose phosphorylation was differentially affected upon MK-2206- vs MK-FNE-treatment. From these nonoverlapping hits, we further imposed selection rules (Figure 5b), giving 14 hits for further investigations. These 14 hits all deviated by −3σ from the mean Heavy:Light ratio of the two respective data sets (Figure 5b).
KIFC1 Is a Novel Downstream Target Underpinning MK-HNE Sensitivity
For these 14 genes, three independent shRNA-knockdown MDA-MB-468 lines were generated. The sensitivity of each knockdown line to MK2206 vs MK-HNE was profiled relative to shControl lines (Figure 5b, inset). For 3 of the 14 proteins knocked down, 2 of the 3 knockdown lines showed significantly different sensitivity to MK2206 vs MK-HNE, compared to shControl cells and other knockdown lines. These 3 proteins, Grsf1, KIFC1, and JUP, all house potential Akt-phosphorylation sites (Figure S15). Western-blot-based knockdown validation ruled out JUP but allowed retention of Grsf1 and KIFC1, which were both in the MK-FNE-exclusively depleted data sets.
Grsf1 is not known to be associated with the Akt-pathway, and only one report has to date implicated KIFC1 as an Akt-pathway regulator, albeit with an unclear mechanism.15 The selectivity EC50(MK-HNE)/EC50(MK-2206) both increased upon knocking down either KIFC1 or Grsf1 in MDA-MB-468 cells [75 and 15% knockdown efficiencies, respectively (Figure S16, Figure 5d)], indicating that the depletion of either of these targets renders the cells less sensitive to MK-HNE with respect to MK-2206. We further investigated potential synergy with the KIFC1-inhibitor, AZ82. Consistent with our shRNA data, AZ82 selectively promoted resistance to MK-FNE, but not MK-2206 (Figure 5e).
We examined the correlation between the expression level of individual Akt isoforms and that of KIFC1 and Grsf1, in breast, lung, gastric, colorectal cancers, and melanomas in the patient database. A negative correlation was found in all patients between KIFC1 and Akt3, and also between Grsf1 and Akt3, in all but melanoma. Notably, Akt1 and Akt2 expressions were not consistently correlated with either KIFC1 or Grsf1 (Figures S17–S21). Furthermore, Akt1-expression was also negatively correlated with Akt3-expression in all except gastric and colorectal cancers. These data overall underscore that MK-FNE functions are distinct from MK-2206 and showed that these inhibitors can be exploited to identify hitherto-unknown pathway intersections functionally selective to Akt3.
MK-FNE and MK-HNE Are More Efficacious than MK-2206 in Mouse Xenograft Models
In mouse microsome stability assays, MK-2206 had t1/2 = 86 min, whereas our inhibitors had significantly shorter half-lives (t1/2: 15 and 29 min for MK-FNE and MK-HNE, respectively) (Figure 6a). Nevertheless, for covalent drugs, metabolic stability is often less important than achieving target engagement, and further, we had evidence above that only fractional occupancy of Akt3 by MK-FNE is required for blocking Akt3-signaling.
Figure 6.

We thus evaluated these compounds in xenograft tumor models (Figure 6b–d, Figure S22). Polyclonal MDA-MB-468 cells were injected into each flank of female nude mice, and after 7 days, each inhibitor or DMSO was administered by oral gavage. To analyze the overall growth rates, tumor volume normalized to the zero point was plotted as a function of time (Figure 6c), and growth rates were calculated by linear regression, which best fit the data. MK-2206 showed moderate, but significant (14%) tumor-growth suppression overall. However, our hybrid inhibitors, MK-HNE and MK-FNE, showed 31% and 26% suppression, indicating that our molecules are significantly more potent than MK-2206 in this assay. Animals showed no weight loss despite repeated dosing with compound for 3 weeks. Importantly blood glucose levels in MK-2206-treated mice were elevated relative to MK-FNE-treated mice (Figure 6d), indicating that MK-FNE does not show one of the most common side effects of MK-2206 treatment.16 Excision of the tumors and staining of the compound using Click-assays showed that our hybrid-compounds were able to label tumors covalently (Figure 6e). Finally, H&E staining of the excised tumors showed overall less microvessel formation in tumors treated with MK-H(F)NE than MK-2206 and DMSO (Figure 6f).
Discussion
Covalent drug design has become a significant weapon in the armory of medicinal chemists. However, of the ∼50 kinase inhibitors approved, only 6 are definitively covalent inhibitors, of which most were approved over the past 10 years. Given our interest in the electrophile signaling field,17 we were struck that the common methods to fashion electrophile drugs consist of attachment of an ostensibly unfunctionalized enone moiety to a pre-existing noncovalent drug scaffold.5 Building on our recent data, we were compelled to test the hypothesis that a druglike molecule could effectively deliver HNE to a target protein, eschewing the known off-target effects of HNE, which render HNE an unlikely drug candidate, but retaining the protein-specific beneficial effects of HNEylation. In this paper, we provide evidence that such a strategy is possible and gives predictable, and overall improved, outputs compared to parent ligands/equivalent electrophiles. Indeed, our analyses of labeling, mechanism, pathway modulation (e.g., T305-phosphorylation), and cell-line specific toxicities offer a consistent vein of logic attesting to the fact that MK-HNE and MK-FNE have properties more similar to the covalent moiety HNE, over the parent noncovalent ligand MK-2206. These data imply that drugs designed from natural lipid-derived electrophiles likely have different pharmacological properties to their parent noncovalent ligands. Such an ability to engender a diverse series of physiological outcomes from a single “ligandable” interaction is very useful. Furthermore, since the effects of the covalent moiety are predictable from methods such as T-REX, this makes a powerful combination for rational design that we will investigate further.
Perhaps the most striking aspect of these data is the semiselective targeting of Akt3 by MK-H(F)NE. Indeed, Akt has proven a difficult family of proteins to target selectively18 with numerous drugs reaching early phase clinical trials, but none progressing to approval often due to off-target effects. Among the isozymes of Akt, Akt3-specific inhibition has proven particularly challenging, and most Akt-targeting drugs target Akt1 selectively over the other Akt-isozymes.19 Thus, our first thought was to examine the selectivity of these compounds for the different isoforms of Akt. Our compounds did not readily form covalent bonds to Akt1. However, our molecules showed some ability to label Akt2. This event reached 50% saturation at a concentration 3.5-fold higher than Akt3 labeling. Furthermore, Akt2 labeling did not elicit a dominant-negative effect, like it did for Akt3. Because of these synergizing factors, MK-HNE/FNE appear to be functionally quite selective Akt3-inhibitors, certainly with respect to Akt1, and, to a moderate extent, Akt2 as well. In order to evaluate how Akt3-functional targeting was involved in the mode-of-action of these molecules, we first turned to measuring synergy with knockdown of specific Akt-isoforms. Consistent with endogenous Akt3 being a significant component of the pharmaceutical activity of our compounds, MK-FNE showed synergy only with knockdown of Akt3, whereas MK2206, an Akt1-semiselective drug, showed synergy with only siAkt1. siAkt-2 showed no synergy with either molecule. Indeed, MK-FNE showed a higher extent of synergy with Akt3 than the synergy shown by MK-2206 for Akt1. This trend was reflected in the fact that MK2206-resistant lines,11 which are Akt3-dependent, were sensitized to MK-FNE. Thus, Akt3 is a pharmaceutically important target of MK-FNE: Akt3 is not a particularly important target of MK-2206, as expected. Clearly these observations do not rule out that MK-H(F)NE has other targets. However, the data implicate Akt3 targeting, as opposed to other Akt-isoforms, as a significant event in the pharmaceutical behavior of MK-H(F)NE.
We progressed to examine the utility of our molecules, as there is little data available to show how well tolerated or efficacious semiselective Akt3-inhibition could be, particularly in advanced models. Our data attest that Akt3-selective targeting can be efficacious. In cells and in mouse models, Akt3-targeting exhibits lower side-effects and higher efficacy than a compound presently in clinical trials. Finally, by comparing the effects of MK-2206 and MK-FNE on Akt-dependent phosphorylations, we found novel allele-specific drug intersections, demonstrating that our inhibitors are sufficiently selective and informative to provide new biologically relevant insight. The most interesting is KIFC1, which we believe deserves more extensive follow up.
Although we are aware that one single success does not prove generality,5 we conclude that our data are most encouraging for developing drugs based on reactive lipids. Critically, these natural electrophiles20 have broad target spectra that we show here for the first time can be biased through ligandability. We will continue to investigate these effects by modifying other inhibitors.
Safety Statement and Animal Model Research
All chemical and biological studies in vitro and cultured cells were conducted in accordance with institutional chemical and biosafety protocols. Studies involving mice models were performed by Washington Biotech. Inc., following ethical standards for animal studies of the Office for Laboratory Animal Welfare (OLAW), division of the US Public Health Service as administered by the US National Institutes of Health.
Acknowledgments
Miss Sanjna Surya (a former undergraduate researcher in Y.A. laboratory, and now an MD-student at Perelman School of Medicine, University of Pennsylvania) is acknowledged for her assistance in initial studies. Mr. Jesse R. Poganik (PhD student in Y.A. laboratory) is acknowledged for assistance with viability and knockdown assays, and Ms. Bella Aprilia, Ms. Jolyn Pan, Mr. Tiger Li, and Ms. Ivy Guan (summer students in X.L. laboratory) are acknowledged for assistance with blind quantification of imaging data. Dr. Sheng Zhang and staff members at Cornell University proteomics facility are acknowledged for assistance with SILAC-data processing and analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b00893.
Biochemical assays involving purified proteins, and chemical biological methods performed in cells and in mice; chemical synthesis procedures; figure legends for Figures S21–S22; and compound characterization data (PDF)
Additional data and figures (PDF)
SILAC proteomics data file related to MK-2206 (XLSX)
SILAC proteomics data file related to MK-FNE (XLSX)
Author Contributions
X.L.: investigation, data analysis, writing (compilation of data, method details, references, and manuscript editing). M.J.C.L.: conceptualization, initial toxicity assays, data analysis, writing (manuscript draft and critical review); B.D.H.: cell-line panel screening, scientific discussions; C.L.: HNE-amide synthesis, and toxicity assays, data analysis; L.W.: cell-based dose–response assays, and data analysis; Y.A.: conceptualization, data analysis, writing (manuscript critical review and writing), supervision, project administration, funding acquisition. All authors assisted with the preparation of corresponding Supporting Information sections and final proofing of the manuscript.
Swiss National Science Funding (SNSF) Project Funding (310030_184729); NCCR Chemical Biology (SNSF); NIH Director’s New Innovator (1DP2GM114850); Novartis Medical-Biological Research Foundation (Switzerland); Swiss Federal Institute of Technology Lausanne (EPFL) (to Y.A.).
The authors declare the following competing financial interest(s): Akt-inhibitors discussed in this work was filed for US Patent application by the former institution of X.L., M.J.C.L., and Y.A., Cornell University.
Supplementary Material
References
- Ferguson F. M.; Gray N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discovery 2018, 17 (5), 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Manning B. D.; Toker A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169 (3), 381–405. 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E.; McGraw T. E. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 2009, 8 (16), 2502–2508. 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E. C.; Lyssiotis C. A.; Cantley L. C.. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. In Metabolism in Cancer; Cramer T., A. Schmitt C., Eds.; Springer International Publishing: Cham, 2016; pp 39–72. [DOI] [PubMed] [Google Scholar]
- Long M. J. C.; Aye Y. Privileged Electrophile Sensors: A Resource for Covalent Drug Development. Cell Chem. Biol. 2017, 24 (7), 787–800. 10.1016/j.chembiol.2017.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J. C.; Parvez S.; Zhao Y.; Surya S. L.; Wang Y.; Zhang S.; Aye Y. Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat. Chem. Biol. 2017, 13 (3), 333–338. 10.1038/nchembio.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Voegtli W. C.; Sturgis H. L.; Dizon F. P.; Vigers G. P. A.; Brandhuber B. J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One 2010, 5 (9), e12913 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan M.; Beg M. A.; Parveen S.; Damanhouri G. A.; Zaher G. F. Computational Insights into the Inhibitory Mechanism of Human AKT1 by an Orally Active Inhibitor, MK-2206. PLoS One 2014, 9 (10), e109705 10.1371/journal.pone.0109705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H.; Sootome H.; Nakatsuru Y.; Miyama K.; Taguchi S.; Tsujioka K.; Ueno Y.; Hatch H.; Majumder P. K.; Pan B.-S.; Kotani H. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9 (7), 1956. 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- Quambusch L.; Landel I.; Depta L.; Weisner J.; Uhlenbrock N.; Müller M. P.; Glanemann F.; Althoff K.; Siveke J. T.; Rauh D. Covalent-Allosteric Inhibitors to Achieve Akt Isoform-Selectivity. Angew. Chem., Int. Ed. 2019, 58 (52), 18823–18829. 10.1002/anie.201909857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottrup C.; Tsang T.; Chin Y. R. Upregulation of AKT3 Confers Resistance to the AKT Inhibitor MK2206 in Breast Cancer. Mol. Cancer Ther. 2016, 15 (8), 1964–1974. 10.1158/1535-7163.MCT-15-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Zhang J. Spatiotemporal Analysis of Differential Akt Regulation in Plasma Membrane Microdomains. Mol. Biol. Cell 2008, 19 (10), 4366–4373. 10.1091/mbc.e08-05-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. A.; Kuzmic P.; Solowiej J.; Bergqvist S.; Bolanos B.; Almaden C.; Nagata A.; Ryan K.; Feng J.; Dalvie D.; Kath J. C.; Xu M.; Wani R.; Murray B. W. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (1), 173–178. 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin Y. R.; Yoshida T.; Marusyk A.; Beck A. H.; Polyak K.; Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014, 74 (3), 964–973. 10.1158/0008-5472.CAN-13-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Chong T.; Yang J.; Li H.; Chen H. Kinesin Motor Protein KIFC1 Is a Target Protein of miR-338–3p and Is Associated With Poor Prognosis and Progression of Renal Cell Carcinoma. Oncol. Res. 2018, 27 (1), 125–137. 10.3727/096504018X15213115046567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinsky K.; Sparano J. A.; Andreopoulou E.; Taback B.; Wiechmann L. S.; Feldman S. M.; Ananthakrishnan P.; Hibshoosh H.; Manavalan J.; Crew K. D.; Maurer M. A.; Hershman D. L. Presurgical evaluation of the AKT inhibitor MK-2206 in patients with operable invasive breast cancer. J. Clin. Oncol. 2014, 32 (15s), 2613–2613. 10.1200/jco.2014.32.15_suppl.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Long M. J. C.; Poganik J. R.; Aye Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118 (18), 8798–8888. 10.1021/acs.chemrev.7b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi R.; Thompson C. B. The survival kinases Akt and Pim as potential pharmacological targets. J. Clin. Invest. 2005, 115 (10), 2618–2624. 10.1172/JCI26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitulescu G. M.; Margina D.; Juzenas P.; Peng Q.; Olaru O. T.; Saloustros E.; Fenga C.; Spandidos D. A.; Libra M.; Tsatsakis A. M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48 (3), 869–885. 10.3892/ijo.2015.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J. C.; Liu X.; Aye Y. Genie in a bottle: controlled release helps tame natural polypharmacology?. Curr. Opin. Chem. Biol. 2019, 51, 48–56. 10.1016/j.cbpa.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.