

Abstract
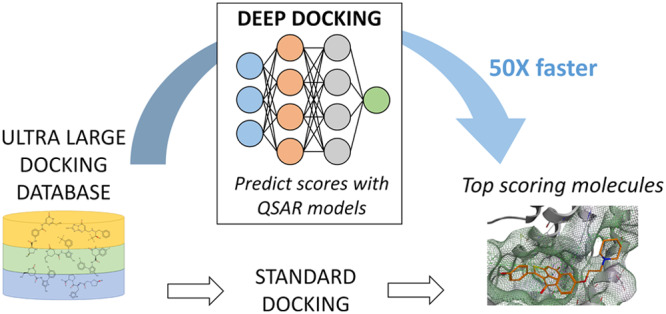
Drug discovery is a rigorous process that requires billion dollars of investments and decades of research to bring a molecule “from bench to a bedside”. While virtual docking can significantly accelerate the process of drug discovery, it ultimately lags the current rate of expansion of chemical databases that already exceed billions of molecular records. This recent surge of small molecules availability presents great drug discovery opportunities, but also demands much faster screening protocols. In order to address this challenge, we herein introduce Deep Docking (DD), a novel deep learning platform that is suitable for docking billions of molecular structures in a rapid, yet accurate fashion. The DD approach utilizes quantitative structure–activity relationship (QSAR) deep models trained on docking scores of subsets of a chemical library to approximate the docking outcome for yet unprocessed entries and, therefore, to remove unfavorable molecules in an iterative manner. The use of DD methodology in conjunction with the FRED docking program allowed rapid and accurate calculation of docking scores for 1.36 billion molecules from the ZINC15 library against 12 prominent target proteins and demonstrated up to 100-fold data reduction and 6000-fold enrichment of high scoring molecules (without notable loss of favorably docked entities). The DD protocol can readily be used in conjunction with any docking program and was made publicly available.
Short abstract
We developed Deep Docking, a deep learning platform that relies on quantitative structure−activity relationship models trained with docking scores of small portions of ultralarge databases to predict scores of remaining entries and, thus, to accelerate virtual screening by 50 times.
Introduction
Drug discovery is an expensive and time-demanding process that faces many challenges, including low hit discovery rates for high-throughput screening, among many others.1,2 Methods of computer-aided drug discovery (CADD) can significantly speed up the pace of such screening and can drastically improve hit rates.3 Molecular docking is routinely used to process virtual libraries containing millions of molecular structures against a variety of drug targets with known three-dimensional structures.
Recent advancements in automated synthesis and surge of available chemicals represent great opportunities for virtual screening (VS) approaches in general and for docking in particular, but also poses entirely novel challenges. For instance, the widely used ZINC library has grown from 700 000 entries in 20054 to over 1.3 billion constituent molecules in 2019,5 representing a remarkable 1000-fold increase. There is still a global lack of experience in screening such libraries, and the advantage of docking them versus smaller collections is still matter of debate.6 However, few recently published works seem to advocate for expanding VS to ultralarge chemical libraries. In a recent groundbreaking study by Lyu et al,7 authors reported docking of 170 million make-on-demand molecular structures, showing that VS of such databases enables the discovery of highly potent inhibitors as well as novel chemical classes that are not present in routinely screened in-stock libraries. Later, other docking studies involving large collections of molecules led to similar conclusions.9,10
Given the current state of docking programs and computational resources available to CADD scientists, one can stipulate that modern docking campaigns can rarely exceed 0.1 billion molecules and that the current chemical space remains largely inaccessible to structure-based drug discovery. One common approach to address this disparity is to filter large chemical collections to manageable drug-, lead-, fragment-, and hit-like subsets (among others) using precomputed physicochemical parameters and drug-like criteria, such as molecular weight, volume, octanol–water partition coefficient, polar surface area, number or rotatable bonds, number of hydrogen bond donors and acceptors, among many others.11 While this approach can effectively reduce an ultralarge docking database to manageable subsets, many potentially useful compounds and novel or unconventional chemotypes (notably emerging from such large collections6) could be lost. In order to take a full advantage of available and emerging “make-on-demand” chemicals, it is essential to maximize the number of database entries tangibly evaluated against a target of interest. It is also important to note that a conventional docking workflow is remarkably neglectful of negative results. A typical docking campaign relies on completing a full docking run and selecting an extremely narrow subset of favorably docked molecules (virtual hits) for future evaluation. Thus, the vast majority of docking data (both favorable and, especially unfavorable) is not being utilized in any way or form, while it could represent a very relevant, well-formatted, and content-rich input for machine learning algorithms.
Previously, the possibility of predicting docking scores through shallow quantitative structure–activity relationship (QSAR) models has been explored by us (using 3D “inductive” descriptors12) and others, using a support vector machine or random forest along with conformal predictors.13,14 None of these methods, however, offer enough speed boost to deal with billions of molecules, and such studies were thus limited to a few millions of compounds at most. Deep learning (DL), on the other hand, is particularly suited for large data set processing,15 and the method is rapidly gaining interest in drug discovery due to its superior performance compared to traditional machine learning techniques.16−18 Thus, we anticipate that the use of DL could unlock a full potential and true synergy between docking and QSAR methodologies and will take a full advantage of ultralarge docking database data.
Results
In the current study, we have introduced the use of fast-computed and target-independent QSAR descriptors (such as 2D molecular fingerprint), the use of iterative and fast random sampling of the docking database, and, principally, the use of DL to predict docking scores of yet unprocessed database entries at each iteration step. As a result, DD achieves up to 100-fold reduction of an ultralarge docking database and up to 6000-fold enrichment for the top-ranked hits, while avoiding significant loss of favorable virtual hits, as it will be discussed below.
DD Pipeline
In its essence, the DD pipeline (Figure 1) relies on the following consecutive steps:
-
(a)
For each entry of an ultralarge docking database (such as ZINC15), the standard set of ligand-based QSAR descriptors (such as molecular fingerprints) is computed;
-
(b)
A reasonably sized training subset is randomly sampled from the database and docked into the target of interest using conventional docking protocol(s);
-
(c)
The generated docking scores of the training compounds are then related to their 2D molecular descriptors through a DL model; a docking score cutoff (typically negative) is then used to divide training compounds in virtual hits (scoring below the cutoff) and nonhits (scoring above the cutoff);
-
(d)
The resulting QSAR deep model (trained on empirical docking scores) is then used to predict docking outcomes of yet unprocessed entries of the database. A predefined number of predicted virtual hits are then randomly sampled and used for the training set augmentation;
-
(e)
Steps b–d are repeated iteratively until a predefined number of iterations is reached, and/or processed entries of an ultralarge docking database are converged.
Figure 1.

Schematic of the DD pipeline. (Top) DD initialization: a small sample of molecules is randomly extracted from an ultralarge docking database and docked to a target under consideration. The generated docking scores are then used to train a QSAR deep model. The created QSAR solution is then used to predict docking outcome for the remainder of a database and to return predicted virtual hits required to start iteration 2. (Bottom) DD screening: from iteration 2 onward, the deep model gets gradually improved by augmenting the training set with randomly sampled QSAR-predicted virtual hits from the previous DD iteration (which also get selected for actual docking). The cycle is repeated for a predefined number of iterations, after which DD returns top scoring molecules from a database. This final library can be postprocessed to remove residual low scoring entities. Alternatively, steps 2–11 can be carried out until the convergence of an ultralarge docking database.
In DD, the virtual hits recall (i.e., the percentage of actual virtual hits that is retrieved from the database) is set implicitly through a probability threshold which is selected to include 90% of the actual virtual hits in the validation set. Then, the same threshold is applied to the independent test set, and the recall of virtual hits is evaluated in order to assess model generalizability. If recalls of the validation and test sets are consistent with each other, the model is applied to all entries of the database (more details can be found in the Methods). Although the recall values could be endorsed explicitly by using, for example, conformal predictors,14,19 we did not observe significant differences in the resulting performance of DD.
The scripts to run DD pipeline are publicly available in GitHub, together with instructions on how to setup runs and a few additional tools to facilitate automation on HPC clusters, at https://github.com/vibudh2209/D2.
Ultra Large Docking Database Sampling
Selection of a representative and balanced training set is a critical step of any modeling workflow. In the context of sampling a chemical space, a proper DD training set should effectively reflect database’s chemical diversity. It could be expected that enlarging the sampling size and preclustering the docking base would ultimately improve or even converge the chemical space coverage. On the other hand, it is currently not feasible to cluster billions of chemical structures in any way or form, and it has also been shown that preclustering large libraries prior to docking can significantly lower the rank of active chemotypes, thus hindering the discovery of new inhibitors or activators.7 Moreover, biasing sampling toward molecules that are highly ranked by DD as potential virtual hits could exclude low ranked, yet true positive molecules from being selected for model training; therefore we selected random sampling for all DD iterations. Finally, the size of DD training set (e.g., the amount of actual docking) would have a pivotal impact on a computational runtime and should be carefully controlled.
To establish an optimal sampling of ZINC15 base, we evaluated the relationship between the size of DD training set and the corresponding means and standard deviations of the test set recall values, reflecting the consistency of model’s performance and its generalizability. For that, we evaluated 12 protein targets from four major drug-target families,20 including nuclear receptors represented by androgen receptor (AR), estrogen receptor-alpha (ERα), and peroxisome proliferator-activated receptor γ (PPARγ). Linases were represented by calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2), cyclin-dependent kinase 6 (CDK6), and vascular endothelial growth factor receptor 2 (VEGFR2). G protein-coupled receptors included adenosine A2A receptor (ADORA2A), thromboxane A2 receptor (TBXA2R), and angiotensin II receptor type 1 (AT1R). Ion channels were represented by Nav1.7 sodium channel (Nav1.7), Gloeobacter ligand-gated ion channel (GLIC), and gamma-aminobutyric acid receptor type A (GABAA) (more details about the selected targets are reported in Table S1). For all 12 studied targets, we investigated the relationships between the sample size and resulting mean test set recall values, which appear to converge to 0.90 when the training set size ranges between 250 000 and 1 million molecules (Figure 2a). We also observed that the standard deviations converge to 0 at about 1 million sample size (Figure 2b). Thus, we have set 1 million molecules as the standard sampling for DD workflow (more details can be found in the Methods).
Figure 2.
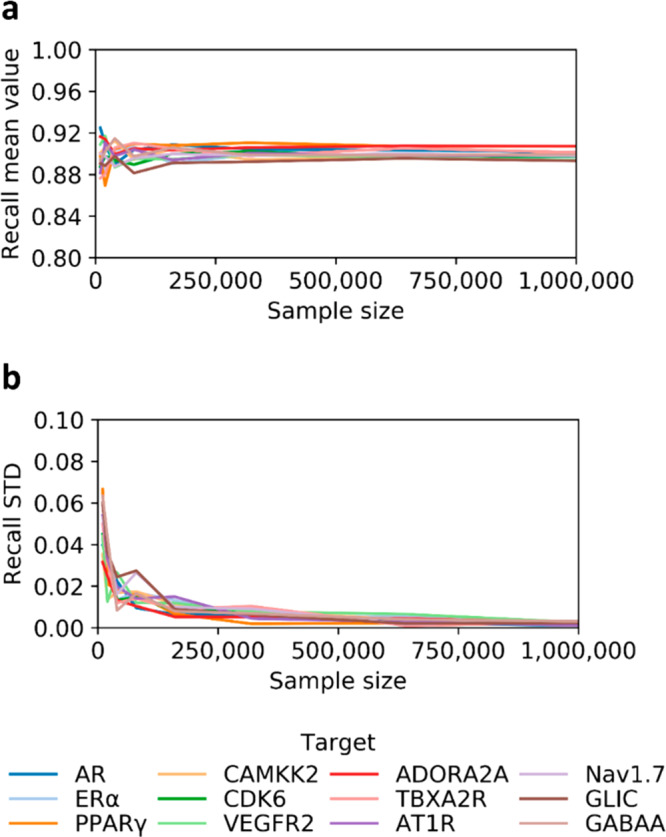
Effect of training set sample size on model generalizability. (a) Mean values for test set recalls computed using different sample sizes. Values approach 0.90 for all targets, when the training set size is within 250 000 and 1 million molecules. (b) Variations of standard deviations (STD) approach 0, for a sample size of 1 million molecules. We ran one iteration for each target and repeated computations five times at each sampling size.
Size Reduction of ZINC15 by DD Virtual Screening
The main goal of DD methodology is to reduce an ultralarge docking database of billions of entries to a manageable few-million-molecules subset which yet encompasses the vast majority of virtual hits. This final molecular subset can then be normally docked into the target using one or several docking programs or can be postprocessed with other VS means. The DD method relies on iterative improvement of the deep neural network (DNN) training by expanding its training set with predicted hit molecules from each previous iteration, while the deciding cutoff also gradually becomes more stringent. We extensively evaluated the performance of this DD protocol by screening all 1.36 billion molecules from ZINC15 against the 12 protein targets introduced above, using docking program FRED.21 Notably, DD itself is not a docking engine, but a DL score predictor to be used in conjunction with any docking program to rapidly eliminate a priori unfavorable, “undockable” molecular entities, and therefore drastically increase the speed of actual docking.
To demonstrate the power of DD, we tested the pipeline with a fixed set of parameters, such as number of iterations, recall values, and others, in order to provide an objective comparison between the 12 investigated systems. It is foreseen that DD users may want to use different simulation parameters than ours, which best suit their time and resource allocations: for example, fewer iterations with more docking per iteration and less DL cycles may be an optimal choice for computing clusters with many CPUs and few GPUs, and vice versa.
For each target, we ran a total of 11 DD iterations—one initial training step (requiring docking of 3 million entries to build training, validation and test sets of 1 million each) and 10 consecutive iterative docking steps, each involving docking of 1 million molecules. Thus, for each target we practically docked only 13 million molecular structures representing less than 1% of the 1.36 billion entries of ZINC15. Figure 3a illustrates the docking score cutoffs used to discern hits and nonhits at each iteration. These values decreased in accordance with the criterion used for defining virtual hits, that becomes more stringent at each iteration (see Methods for details). The majority of nonhits were removed during the first iteration for all targets, while fewer molecules were discarded in successive steps, as expected due to larger portions of unfavorable compounds being present at the beginning of the runs. We observed that the decrease rate and the number of hits identified were target-dependent (Figure 3b). Another notable observation from the analysis of the DD progression is that training sets effectively improve after each iteration, since docking scores of molecules added to training become more negative (favorable) after each round of modeling (Figure 3c). This observation marks progressively more confident performance of DD in recognizing and discarding low scoring molecules, and consequently favorably augmenting of the training set. Consequently, we anticipated that DD is likely to improve the enrichment for virtual hits after each iteration, as visible in Figure 3d, featuring enrichment values for the top 100 molecules ranked by the DNN models in the test sets. As the data indicate, these values increased after each iteration for all targets, also suggesting that model’s performance improves every time the training set is augmented with molecules from each previous DD iteration. Of note, the enrichment values become strongly increased at last iteration, where the models retrieved a very small portion (0.01%) of the top scoring molecules of the database. Further indications of iterative improvement of DNN models are provided by the area under the curve receiver operating characteristics (AUC ROC) values and full predicted database enrichment (FPDE) values, presented in Table S3 for all iterations of the 12 studied targets, as well as the precision of the models to identify high scoring molecules, which improved at each iteration as well, as expected (Figure S2).
Figure 3.

DD performance statistics for 12 drug targets. (a) Variation of score cutoff values used for selecting virtual hits at each iteration. (b) Variation of numbers of molecules predicted as virtual hits after each iteration. (c) Iterative improvement of docking score mean values for randomly selected molecules used for training set augmentation. (d) Enrichment values calculated for 100 top ranked predicted virtual hits in test set after each iteration.
Analysis of DD Performance
As indicated earlier, the main objective of the DD methodology is to reduce an ultralarge docking database to a highly enriched library of molecules that can be processed using conventional docking programs and computational resources. While studying 12 selected targets, we observed that the sizes of final, remaining subsets ranged between 1% and 12% of the original ultralarge docking database (Figure 4a). It is foreseen that these remaining enriched and DNN-ranked libraries can then be postprocessed to remove residual low scoring molecules. Alternatively, DD can be carried out until the convergence of an ultralarge docking database.
Figure 4.

Final data set sizes and enrichment values resulting from DD runs. (a) Total number of molecules predicted as virtual hits remaining after the 11th DD iteration. Values are also reported in terms of percentage of ZINC15 entries that were retained (right vertical axis). (b) FPDE values resulting from the last iterations of DD experiments. (c) Enrichment values for top 10, top 100, and top 1000 selected virtual hits (in the test sets).
DD demonstrated its best performance for PPARγ protein, where the database was reduced to 1% of its size. Thus, considering docking required to train the model as well as to postprocess the final subset, DD screening of ZINC15 against this target requires docking of 50 times fewer molecules than conventional VS. On the other hand, DD was least effective on the GABAA target, where 12% of ZINC15 molecules were left after the last iteration due to low precision of the model. These results clearly suggest that, like any other computational tool, DD shows performances that are target-dependent. Encouragingly, the recall values were consistently transferred to the test sets in all cases (see Table S3). We also compared the number of molecules expected to be returned by DD at each iteration based on the test sets with the number that was actually returned when DD models were applied to ZINC15, observing no significant differences (Figure S3). Taken together, these results suggest that all underlying DL models were generalizable in a consistent way. To further assess the overall DD performance, we evaluated final FPDE values, which ranged from 8 to 89, indicating that DD enriched final subsets with high scoring molecules in a target-dependent way. As expected, FPDE values matched the trends observed for the number of DD predicted virtual hits (Figure 4b).
We also evaluated enrichment values for the top 10, 100, and 1000 predicted virtual hits identified in the test sets after the final iteration, observing values ranging from 240 to 6000 as demonstrated on Figure 4c. Such enrichments decreased consistently in all cases when evaluating larger portions of top ranked structures, thus suggesting that true hits are highly concentrated at the top of the DD rank, and molecules at the bottom of the rank are mostly false positives. It is important to note that we have set stringent 0.90 recall values for our DD runs to preserve the vast majority of virtual hits in the final subset. However, one can lower such recall cutoff to sacrifice the retention of virtual hits in a DD workflow, but to significantly reduce the number of unprocessed molecules retained at the end, and to shorten the runtime.
To investigate the effect of decreasing the recall of virtual hits, we ran DD for two systems, namely, PPARγ and GABAA (the best and worst targets in terms of number of molecules remained unprocessed after 11 DD iterations as described above), using recall values of 0.75 and 0.60. Encouragingly, the number of remaining entries significantly decreased in both cases compared to DD runs with 0.90 recall (Figure 5a). For the PPARγ system, the size of ZINC15 was scaled down 800 times at 0.75 recall, and 2300 times at 0.60. Similarly, we observed a 66- and 654-fold size reduction using recalls of 0.75 and 0.60 respectively, for the GABAA target. Importantly, the enrichment of the molecular subsets increased noticeably when lower recalls were used, reaching up to 1450 for FPDE values and again clearly indicating that density of virtual hits rapidly decreases as we move away from the top of the DNN rank (Figure 5b). Thus, the recall value can be chosen according to the needed speed boost or computational resources available to the user in order to further reduce the amount of docking and/or postprocessing required at the end of DD runs.
Figure 5.

DD size reduction power depends on recall values. (a) Final number of molecule (in logarithmic scale) predicted as virtual hits by DD and (b) FPDE values obtained for PPARγ and GABAA systems by varying the virtual hit recall values.
Overall, the above analysis indicates that the DD procedure can effectively discard most of unqualified molecules in a ultra large docking database, without losing more than a predefined percentage of virtual hits. In our opinion, this makes DD methodology an efficient mean for conducting large-scale VS campaigns involving billions of small molecule structures, and a valid alternative to brute force approaches demanding large amounts of computational resources.
DD Virtual Screening and Active Ligands
We also investigated how DD method deals with active ligands that are present in ultralarge docking databases. For five of the investigated systems, namely, AR, ERα, PPARγ, VEGFR2, and ADORA2A, the Database of Useful Decoys: Enhanced22 (DUD-E) provided sets of confirmed active ligands (details about the data sets are reported in Table S2) that we docked to their respective targets together with 1 million of random compounds from ZINC15 (considered as inactive molecules). Docking performances of FRED were variable across targets, with AUC ROC values ranging from 0.52 to 0.91 (Figure 6a). In parallel, we evaluated how DD ranks of compounds (evaluated at 0.90 recall) correlated with their docking scores, in particular, with their distance from the score cutoff used to define virtual hits and nonhits in the last iteration, using random samples of 1 million ZINC15 molecules docked to each target (Figure 6b). DD appears to clearly bias final sets toward high docking scores, and since active ligands score better than inactives based on AUC values, we expected active ligands to be discarded at lower rates than inactive molecules. Thus, we plotted the rank of active ligands against their score distance from the virtual hit cutoff (Figure 6c). As expected, DD worked particularly well for systems with many high scoring actives and good AUC values, such as AR and ERα, for which all active, top scoring ligands were retained. Interestingly, DD retained also a significantly large amount of active ligands with nonhits scores for all targets, at about 10-fold higher rates than remaining ZINC15 nonhits.
Figure 6.

(a) ROC curves for FRED docking of active ligands to five targets. Actives were mixed with 1 million of randomly sampled ZINC15 molecules. AUC value for each target is reported in brackets. (b) DD rank (logarithmic scale) versus distance of scores from cutoff values of random samples of 1 million ZINC15 compounds docked to the five targets. (c) DD rank (logarithmic scale) versus distance from score cutoff for active ligands. Kept (recalled) ligands are represented with yellow dots, discarded ligands are represented with blue dots. (d) Enrichment factor (EF, logarithmic scale) of active ligands when top 100 000, 1 000 000, 10 000 000, and all DD hits are considered.
Then, we calculated the enrichment factors (EFs) for active compounds on top 100 000, 1 000 000, and 10 000 000 ranked molecules, for the fivr systems. Not surprisingly, the highest values were observed for AR and ERα, for which EFs for the top 100 000 molecules were encouragingly high, 660 for AR and 477 for ERα, and consistently decreased by evaluating larger portion of top scoring molecules (Figure 6d). For the other three systems, a clear trend for enrichments was not observed when different sizes were considered. Nevertheless, all the considered subsets of molecules were enriched with active ligands, showing EFs of at least six. Thus, selecting a subset of top ranked DD molecules seems to be a valid alternative to lowering the recall value in order to limit the amount of final docking, when limited computational resources are available. Furthermore, EFs calculated over all DD hits ranged between 11 and 14 for all systems. Thus, DD not only enables extraction of controlled portions of virtual hits from an ultralarge docking database, but also enriches final subsets with active hit molecules. Remarkably, this unexpected DD feature of retaining actives regardless of their scores resulted in enrichment also for targets showing near-to-random docking performance, such as the ADORA2A system.
Discussion
With the increasing automation of synthetic procedures, the focus of modern drug discovery campaigns will be shifting toward screening of increasingly larger molecular libraries consisting of billions of chemical structures. To reinforce such opportunity, docking protocols demonstrate improved performance on larger make-on-demand databases, effectively yielding novel, diverse, and nontraditional chemotypes for drug discovery endeavors.7 It could be noted, however, that most of time and resources invested in modern docking campaigns are spent on processing unfavorable molecular structures, while the emerging “negative” data are also not being utilized in any way or form.
Hence, to keep the pace with an ever-expanding chemical Big Data space and to fully utilize results generated by docking programs “on a fly”, we have developed the Deep Docking protocol DD, a DNN-based method for processing large chemical libraries with conventional computational and software resources. The method relies on iterative docking of a small portion of a parental ultralarge docking database (such as ZINC15) using a docking program of choice and utilizes generated scores (both favorable and unfavorable) to train ligand-based QSAR models. These models then enable approximation of the docking outcome for unprocessed database entries. Importantly, DL allows the use of simple 2D protein-independent descriptors such as Morgan fingerprints to capture the docking scores. We have demonstrated that such approach can yield a manageably small subset of a database, highly enriched with favorably “dockable” molecular structures.
We proved the power of DD by screening all 1.36 billion entries of ZINC15 against 12 prominent drug targets, where the original ultralarge docking database was significantly reduced while retrieving a controlled, high portion of favorably docked molecules. At the same time, most of low scoring molecules were removed without investing time and resources in them, and the generated ZINC subsets were highly enriched in potential virtual hits. Notably, screening an ultralarge docking database using DD requires docking up to 50 times fewer molecules compared to conventional docking, while losing only about 10% of virtual hits. We also showed that DD can further shrink an ultralarge docking database to a few hundreds of thousands of molecules while still retrieving a significant part (60%) of top scoring hits. Moreover, DD appears to enrich final subsets with active ligands, even when only small portions of top ranked molecules are considered. This unexpected result suggests that true binders carry on certain chemical features that are complementary to the binding pocket and that the model is able to capture such features through the QSAR descriptors.
Conclusion
In the current work, we introduced the use of deep learning in structure-based drug discovery in a novel way. The developed Deep Docking approach utilizes QSAR models trained on actual docking scores of a small subset of a molecular database to predict docking scores for the rest. Such approach, being used in an iterative manner (with predefined recall parameters) allows significant savings of docking runtime, without notable loss of potentially “dockable” entities or active ligands. The use of Deep Docking circumvents computational limits of large-scale docking campaigns and makes billion-entries molecular databases accessible even with limited computational resources.
Collectively, our results strongly advocate the use of deep learning for exploration of continuously expanding chemical space in search for new therapeutics.
Methods
QSAR Descriptors
SMILES of 1.36 billion molecular structures were downloaded from ZINC15.5 Morgan fingerprints with a size of 1024 bits and a radius of 2 were generated using the RDKit package.23
Protein Targets
The X-ray structures of AR,24 ERα,25 PPARγ,26 CAMKK2,27 CDK6,28 VEGFR2,29 ADORA2A,30 TBXA2R,31 AT1R,32 Nav1.7,33 GLIC,34 and GABAA35 containing cocrystallized ligands were extracted from the Protein Data Bank (PDB).36 Details about the selected target structures are summarized in Table S1.
Molecular Docking
PDB structures were optimized using the Protein Preparation Wizard module from the Schrödinger suite,37 and docking grids were prepared using the MakeReceptor utility from OpenEye.38 SMILES were processed using QUACPAC8 in order to generate dominant tautomer and ionization states at pH 7.4. The OMEGA’s pose module39,40 was used to generate multiple 3D optimal conformers for FRED docking. Docking simulations were carried out using FRED program and Chemgauss4 scoring function from OpenEye.21
Database Sampling
The optimal number of molecules required for the training set was determined by running one DD iteration for each target, using different sizes for training, validation, and test set (10 000, 20 000, 40 000, 80 000, 160 000, 320 000, 640 000, and 1 million molecules). For each sample size, computations were repeated five independent times. The optimal sampling size was then chosen by evaluating means and standard deviations of recall values in the test sets for all targets.
DD Workflow
Initial training, validation, and test sets used for the DL model consisted of 1 million molecules each that were randomly sampled from ZINC15 during the first DD iteration. Each set was docked to the target of interest and then divided into virtual hits and nonhits based on the generated docking scores. The score cutoff used to determine the class of molecules was determined in order to split the validation in 1% top scoring molecules (virtual hits) and 99% nonhits. Molecules of each set with docking scores equal or more favorable than the cutoff value were assigned to the virtual hit class, while remaining molecules were assigned to the nonhit class. The DNN model was trained using classes and molecular descriptors of the processed entries and then used to predict virtual hits and unqualified molecules from the whole ZINC15 based on molecular descriptors. From the second iteration onward, the training set was expanded with 1 million molecules randomly sampled from hits predicted in the previous iteration, while validation and test sets remained unchanged for all the length of the DD run. The score cutoff was gradually decreased (corresponding to higher predicted target affinity) after each iteration to keep selecting better compounds. This reduction was done by linearly lowering the percentage of top scoring molecules in the validation set assigned to the virtual hit class from 1% in the first iteration to 0.01% in the last one. A linear variation of the score cutoff was chosen in order to avoid large variations in initial iterations, which could restrict the model to comprehensively explore chemical classes. Thus, the cutoff value in iteration 2 corresponded to the highest (worst) docking score of the top 0.9% ranked compounds, in iteration 3 it corresponded to the highest docking score of top 0.8% compounds, and so on.
Evaluation Metrics
All evaluation metrics were calculated on the test sets. Precision was calculated as
| 1 |
where TP (true positives) were virtual hits correctly predicted by the DNN, and FP (false positives) were actual nonhits that were incorrectly classified as virtual hits by the DNN.
Recall was calculated as
| 2 |
where FN (false negatives) were virtual hits incorrectly discarded by the DNN.
Enrichment values were calculated as
| 3 |
with TP|top N as the number of TP found within the top N ranked molecules by the DNN, and TP|random N as the number of TP found within N randomly sampled molecules. N was set to values equal to 10, 100, and 1000.
FPDE was calculated as the ratio between precision (eq 1) and random precision:
| 4 |
Deep Learning
The Keras Python library41 was used for building and training feed-forward DNN models.42 Model hyperparameters were set as the number of hidden layers and neurons, dropout frequency, as well as oversampling of the minority class and class weights, in order to deal with highly imbalanced data sets (1–0.01% of virtual hits). A lower threshold value was established for the DNN probabilities (indicating the likelihood of molecules of being virtual hits) and used as criterion to assign molecules to the virtual hit class upon prediction. The threshold was chosen each time in order to retrieve 90% of the actual virtual hits (i.e., top scoring molecules) of the validation set. Model selection was performed by running a basic grid search to identify the set of hyperparameters providing the highest FPDE value in the test set. The best model was then applied to all ZINC15 entries in order to predict virtual hits and nonhits.
Active Ligands
Active compounds were obtained from the DUD-E repository for available targets.22 If not already present in ZINC15, SMILES and relative Morgan fingerprints of compounds were calculated and added to it. The molecules were prepared and docked to the respective targets as previously described. Enrichment factors after top N molecules,43 ranked by DD probability of virtual hit-likeness, were calculated as
| 5 |
with actives|N being the number of active ligands found in top N molecules, actives|database the total number of active ligands, and mol|database the database size.
Hardware
We used a 6 Intel(R) Xeon(R) Silver 4116 CPU @ 2.10 GHz (a total of 60 cores) for docking and 4 Nvidia Tesla V100 GPUs with 32 GB memory for DNN model training and inference.
Acknowledgments
This work has been supported by the Canadian Cancer Society Research Institute (CCSRI) Impact Grant 2019 No. 706145. F.G. would like to thank the Ermenegildo Zegna Founder’s scholarship program for financial support.
Glossary
Abbreviations
- CADD
computer-aided drug discovery
- VS
virtual screening
- QSAR
quantitative structure–activity relationship
- DL
deep learning
- DD
Deep Docking
- AR
androgen receptor
- ERα
estrogen receptor-alpha
- PPARγ
peroxisome proliferator-activated receptor γ
- CAMKK2
calcium/calmodulin-dependent protein kinase kinase 2
- CDK6
cyclin-dependent kinase 6
- VEGFR2
vascular endothelial growth factor receptor 2
- ADORA2A
adenosine A2A receptor
- TBXA2R
thromboxane A2 receptor
- AT1R
angiotensin II receptor type 1
- Nav1.7
Nav1.7 sodium channel
- GLIC
Gloeobacter ligand-gated ion channel
- GABAA
gamma-aminobutyric acid receptor type A
- DNN
deep neural network
- AUC ROC
area under the curve receiver operating characteristics
- FPDE
full predicted database enrichment
- PDB
Protein Data Bank
- TP
true positives
- FP
false positives
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00229.
Docking results of cocrystallized ligands, additional performance analysis, details about protein targets, details of DUD-E ligands, supplementary references (PDF)
DD code is publicly available in GitHub at https://github.com/vibudh2209/D2. Docking data sets used for building models reported in the manuscript are freely available at https://drive.google.com/drive/folders/1UNGfkGiL_8heQWwxaaQPfIoU4AAqoUoA.
AUC ROC and FPDE values for all iterations (XLSX)
Author Contributions
A.C., F.G., and V.A. designed research. F.G., V.A., and M.H. performed research. F.G., V.A., M.H., A.-T.T, F.B., and U.N. analyzed data. F.G., M.H., and A.C. drafted the manuscript, with all authors contributing. A.C. and M.E.G. supervised the research.
Author Contributions
# F.G. and V.A. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- DiMasi J. A.; Grabowski H. G.; Hansen R. W. Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of Health Economics 2016, 47, 20–33. 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Hughes J. P.; Rees S.; Kalindjian S. B.; Philpott K. L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162 (6), 1239–49. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban F.; Dalal K.; Li H.; LeBlanc E.; Rennie P. S.; Cherkasov A. Best Practices of Computer-Aided Drug Discovery: Lessons Learned from the Development of a Preclinical Candidate for Prostate Cancer with a New Mechanism of Action. J. Chem. Inf. Model. 2017, 57 (5), 1018–1028. 10.1021/acs.jcim.7b00137. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45 (1), 177–82. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T.; Irwin J. J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55 (11), 2324–37. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpfe D.; Bajorath J.. Current Trends, Overlooked Issues, and Unmet Challenges in Virtual Screening. J. Chem. Inf. Model. 2020, 10.1021/acs.jcim.9b01101 [DOI] [PubMed] [Google Scholar]
- Lyu J.; Wang S.; Balius T. E.; Singh I.; Levit A.; Moroz Y. S.; O’Meara M. J.; Che T.; Algaa E.; Tolmachova K.; Tolmachev A. A.; Shoichet B. K.; Roth B. L.; Irwin J. J. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566 (7743), 224. 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUACPAC 2.0.1.2; OpenEye Scientific Software, Santa Fe, NM, USA, 2019.
- Stein R. M.; Kang H. J.; McCorvy J. D.; Glatfelter G. C.; Jones A. J.; Che T.; Slocum S.; Huang X. P.; Savych O.; Moroz Y. S.; Stauch B.; Johansson L. C.; Cherezov V.; Kenakin T.; Irwin J. J.; Shoichet B. K.; Roth B. L.; Dubocovich M. L. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579 (7800), 609–614. 10.1038/s41586-020-2027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgulla C.; Boeszoermenyi A.; Wang Z. F.; Fischer P. D.; Coote P.; Padmanabha Das K. M.; Malets Y. S.; Radchenko D. S.; Moroz Y. S.; Scott D. A.; Fackeldey K.; Hoffmann M.; Iavniuk I.; Wagner G.; Arthanari H. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663. 10.1038/s41586-020-2117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004, 1 (4), 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Cherkasov A. R.; Galkin V. I.; Cherkasov R. A. A new approach to the theoretical estimation of inductive constants. J. Phys. Org. Chem. 1998, 11 (7), 437–447. . [DOI] [Google Scholar]
- Ahmed L.; Georgiev V.; Capuccini M.; Toor S.; Schaal W.; Laure E.; Spjuth O. Efficient iterative virtual screening with Apache Spark and conformal prediction. J. Cheminf. 2018, 10 (1), 8. 10.1186/s13321-018-0265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson F.; Norinder U.; Bender A. Improving screening efficiency through iterative screening using docking and conformal prediction. J. Chem. Inf. Model. 2017, 57 (3), 439–444. 10.1021/acs.jcim.6b00532. [DOI] [PubMed] [Google Scholar]
- Chen H.; Engkvist O.; Wang Y.; Olivecrona M.; Blaschke T. The rise of deep learning in drug discovery. Drug Discovery Today 2018, 23 (6), 1241–1250. 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- Mayr A.; Klambauer G.; Unterthiner T.; Hochreiter S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3, 80. 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- Fernandez M.; Ban F.; Woo G.; Hsing M.; Yamazaki T.; LeBlanc E.; Rennie P. S.; Welch W. J.; Cherkasov A. Toxic Colors: The Use of Deep Learning for Predicting Toxicity of Compounds Merely from Their Graphic Images. J. Chem. Inf. Model. 2018, 58 (8), 1533–1543. 10.1021/acs.jcim.8b00338. [DOI] [PubMed] [Google Scholar]
- Playe B.; Stoven V. Evaluation of deep and shallow learning methods in chemogenomics for the prediction of drugs specificity. J. Cheminf. 2020, 12 (1), 11. 10.1186/s13321-020-0413-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer G.; Vovk V. A tutorial on conformal prediction. J. Machine Learning Res. 2008, 9 (Mar), 371–421. [Google Scholar]
- Santos R.; Ursu O.; Gaulton A.; Bento A. P.; Donadi R. S.; Bologa C. G.; Karlsson A.; Al-Lazikani B.; Hersey A.; Oprea T. I.; Overington J. P. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 2017, 16 (1), 19. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26 (8), 897–906. 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55 (14), 6582–94. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum G.Rdkit documentation, Release 2013; 1, 1–79.
- Hur E.; Pfaff S. J.; Payne E. S.; Grøn H.; Buehrer B. M.; Fletterick R. J. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol. 2004, 2 (9), e274 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowski A. M.; Pike A. C.; Dauter Z.; Hubbard R. E.; Bonn T.; Engström O.; Öhman L.; Greene G. L.; Gustafsson J.-Å.; Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389 (6652), 753. 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- van Marrewijk L. M.; Polyak S. W.; Hijnen M.; Kuruvilla D.; Chang M. R.; Shin Y.; Kamenecka T. M.; Griffin P. R.; Bruning J. B. SR2067 Reveals a Unique Kinetic and Structural Signature for PPARgamma Partial Agonism. ACS Chem. Biol. 2016, 11 (1), 273–83. 10.1021/acschembio.5b00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukimoto-Niino M.; Yoshikawa S.; Takagi T.; Ohsawa N.; Tomabechi Y.; Terada T.; Shirouzu M.; Suzuki A.; Lee S.; Yamauchi T.; Okada-Iwabu M.; Iwabu M.; Kadowaki T.; Minokoshi Y.; Yokoyama S. Crystal structure of the Ca2+/calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. J. Biol. Chem. 2011, 286 (25), 22570–22579. 10.1074/jbc.M111.251710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Lee N. V.; Hu W.; Xu M.; Ferre R. A.; Lam H.; Bergqvist S.; Solowiej J.; Diehl W.; He Y.-A.; Yu X.; Nagata A.; VanArsdale T.; Murray B. W. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther. 2016, 15 (10), 2273–2281. 10.1158/1535-7163.MCT-16-0300. [DOI] [PubMed] [Google Scholar]
- McTigue M.; Murray B. W.; Chen J. H.; Deng Y.-L.; Solowiej J.; Kania R. S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (45), 18281–18289. 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R. K.; Segala E.; Robertson N.; Deflorian F.; Doré A. S.; Errey J. C.; Fiez-Vandal C.; Marshall F. H.; Cooke R. M. Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 2017, 25 (8), 1275–1285. 10.1016/j.str.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Fan H.; Chen S.; Yuan X.; Han S.; Zhang H.; Xia W.; Xu Y.; Zhao Q.; Wu B. Structural basis for ligand recognition of the human thromboxane A 2 receptor. Nat. Chem. Biol. 2019, 15 (1), 27. 10.1038/s41589-018-0170-9. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Unal H.; Gati C.; Han G. W.; Liu W.; Zatsepin N. A.; James D.; Wang D.; Nelson G.; Weierstall U.; Sawaya M. R.; Xu Q.; Messerschmidt M.; Williams G. J.; Boutet S.; Yefanov O. M.; White T. A.; Wang C.; Ishchenko A.; Tirupula K. C.; Desnoyer R.; Coe J.; Conrad C. E.; Fromme P.; Stevens R. C.; Katritch V.; Karnik S. S.; Cherezov V. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161 (4), 833–44. 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S.; Mukund S.; Deng L.; Khakh K.; Chang E.; Ho H.; Shriver S.; Young C.; Lin S.; Johnson J. Structural basis of Nav1. 7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350 (6267), aac5464 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- Pan J.; Chen Q.; Willenbring D.; Mowrey D.; Kong X.-P.; Cohen A.; Divito C. B.; Xu Y.; Tang P. Structure of the pentameric ligand-gated ion channel GLIC bound with anesthetic ketamine. Structure 2012, 20 (9), 1463–1469. 10.1016/j.str.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Noviello C. M.; Teng J.; Walsh R. M. jr; Kim J. J.; Hibbs R. E. Structure of a human synaptic GABAA receptor. Nature 2018, 559 (7712), 67–72. 10.1038/s41586-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman J. L.; Lin D.; Jiang J.; Manning N. O.; Prilusky J.; Ritter O.; Abola E. E. Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54 (6), 1078–1084. 10.1107/S0907444998009378. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2019-3: Maestro; Schrödinger, LLC: New York, NY, USA: ,2019.
- OEDOCKING 3.3.1.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50 (4), 572–84. 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P.; Skillman A.; Warren G.; Ellingson B.; Stahl M.. OMEGA 3.1.0.3, 2019.
- Chollet F.Keras, 2015.
- Svozil D.; Kvasnicka V.; Pospichal J. Introduction to multi-layer feed-forward neural networks. Chemom. Intell. Lab. Syst. 1997, 39 (1), 43–62. 10.1016/S0169-7439(97)00061-0. [DOI] [Google Scholar]
- Bender A.; Glen R. C. A discussion of measures of enrichment in virtual screening: comparing the information content of descriptors with increasing levels of sophistication. J. Chem. Inf. Model. 2005, 45 (5), 1369–75. 10.1021/ci0500177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.