

Abstract
G protein‐coupled receptors (GPCRs) constitute the largest family of receptors and membrane proteins in the human genome with ~800 members of which half are olfactory. GPCRs are activated by a very broad range of endogenous signalling molecules and are involved in a plethora of physiological functions. All GPCRs contain a transmembrane domain, consisting of a bundle of seven α‐helices spanning the cell membrane, and forming the majority of the known ortho‐ or allosteric ligand binding sites. Due to their many physiological functions and the accessible and druggable transmembrane pocket, GPCRs constitute the largest family of drug targets mediating the actions of 34% of currently marketed drugs. GPCRs activate one or more of the four G protein families (Gq/11, Gi/o, Gs and G12/13) and/or ß‐arrestin. About a third of the non‐olfactory GPCRs are referred to as orphan receptors which means that their endogenous agonist(s) have not yet been found or firmly established. In this MiniReview, we focus on the orphan GPR139 receptor, for which the aromatic amino acids L‐Trp and L‐Phe as well as ACTH/α‐MSH‐related peptides have been proposed as endogenous agonists. GPR139 has been reported to activate several G protein pathways of which Gq/11 is the primary one. The receptor shows the highest expression in the striatum, thalamus, hypothalamus, pituitary and habenula of the human, rat and mouse CNS. We review the surrogate agonists and antagonists that have been published as well as the agonist pharmacophore and binding site. Finally, the putative physiological functions and therapeutic potential are outlined.
1. G PROTEIN‐COUPLED RECEPTORS
G protein‐coupled receptors (GPCRs) mediate many of the physiological responses to endogenous ligands such as neurotransmitters, hormones, metabolites, ions and sensory signals.1 Although their ligands are structurally very diverse, GPCRs share a common molecular structure of seven transmembrane‐spanning α‐helices connected by three intracellular and three extracellular loops, with an extracellular N terminus and intracellular C terminus.2 Amongst all of the drugs approved by the Food and Drug Administration (FDA), 34% target GPCRs 3 making this the largest class of drug targets. However, despite this, only 27% of all non‐olfactory GPCRs are presently drug targets. Based on previous success with targeting this protein family and strong unexploited disease associations, GPCRs remain highly pursued targets for basic research and drug discovery.3
2. GPCR SIGNAL TRANSDUCTION
In the human genome, there are 16 Gα subunits, 5 Gβ subunits and 12 Gγ subunits, which couple to GPCRs as heterotrimeric Gαβγ proteins.4 The Gα subunits are divided into four classes based on structural and functional similarities termed Gαq/11 (constituted by Gαq, Gα11, Gα14 and Gα15), Gα12/13 (constituted by Gα12 and Gα13), Gαs (constituted by Gαs and Gαolf), and Gαi/o (constituted by Gαi1, Gαi2, Gαi3, Gαo, Gαz, Gαgust, Gαt1 and Gαt2).5
Gαq/11 activate phospholipase C β isoforms (PLCβ) resulting in the hydrolysis of phosphatidylinositol 4,5‐bisphosphate (PIP2) into inositol 1,4,5‐trisphosphate (IP3) and diacylglycerol (DAG). IP3 then induces the release of intracellular Ca2+ from the endoplasmic reticulum, and DAG activates protein kinase C (PKC) (Figure 1A). Both Ca2+ and PKC participate in diverse signalling to induce different cellular events.6, 7
Figure 1.
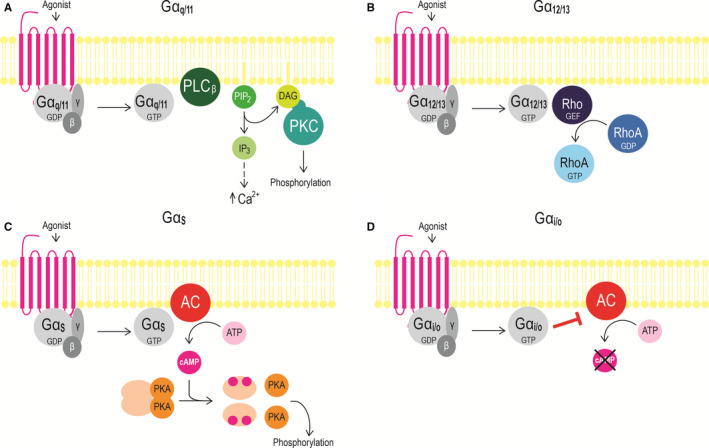
A schematically simplified overview of (A) Gαq/11, (B) Gα12/13, (C) Gαs, and (D) Gαi/o signalling. Abbreviations: AC, adenylyl cyclase; ATP, adenosine 5′‐triphosphate; cAMP, 3′,5′‐cyclic adenosine monophosphate; DAG, diacylglycerol; GDP, guanosine 5′‐diphosphate; GTP, guanosine 5′‐triphosphate; IP3, inositol 1,4,5‐trisphosphate; PIP2, phosphatidylinositol 4,5‐bisphosphate; PKA, protein kinase A; PKC, protein kinase C; PLCβ, phospholipase C‐β; Rho‐GEF, Rho‐guanine nucleotide exchange factor. Adapted from Wettshureck et al5
Activation of Gαs and Gαi/o stimulates and inhibits adenylyl cyclase, respectively,5 which subsequently converts adenosine 5′‐triphosphate (ATP) to 3′,5′‐cyclic adenosine monophosphate (cAMP). cAMP then activates protein kinase A (PKA), which phosphorylates further downstream effectors (Figure 1C, D).5
Gα12/13 is the least characterized G protein signalling pathway. Gα12/13 activates Rho‐guanine nucleotide exchange factors (RhoGEFs) causing a GDP‐GTP exchange in the monomeric GTPase RhoA (Figure 1B).8 No second messenger assay exists for the quantification of the Gα12/13 activation, but this can be measured by monitoring G protein activation by bioluminescence resonance energy transfer (BRET) 9 or by Corning Epic dynamic mass redistribution assay.10
The dimeric Gβγ complex can modulate the activity of various downstream effector molecules, like PLCβ, mitogen‐activated protein kinase (MAPK) and different types of adenylyl cyclase.11
GPCRs also signal through G protein‐independent signalling pathways.12 Until recently, it was assumed that the recruitment of β‐arrestin caused termination of G protein signalling by steric hindrance of G proteins and initiation of receptor internalization into cytosolic endosomes.12 However, it has now been recognized that β‐arrestins can initiate downstream signalling, including activation of extracellular signal‐regulated kinase 1/2 (ERK1/2) 12). Additionally, some GPCRs can continue to signalling after they have been internalized.13, 14, 15, 16
3. ORPHAN GPCRS
An orphan GPCR is a receptor for which an endogenous ligand has not yet been identified. There are presently 121 non‐sensory orphan GPCRs, including GPR139.17 The naming and classification of orphan GPCRs upon pairing with an endogenous ligand is done by the International Union of Basic and Clinical Pharmacology Committee of Receptor Nomenclature and Drug Classification (NC‐IUPHAR), which maintains a database of all non‐sensory human GPCRs (www.guidetopharmacology.org).18 To be considered “deorphanized,” the proposed receptor‐ligand pairing must meet the following criteria (a) have been reported by at least two independent research groups in peer‐reviewed papers and (b) be present in sufficient concentration in the tissue(s) where the receptor is expressed. Additionally, there are two further recommendations: (a) selective antagonists and agonists should block and mimic the action of the putative endogenous ligand on the receptor, respectively, and (b) radioligand binding and functional assays should be performed both in vitro and in native tissue.19
4. DISCOVERY OF GPR139
A partial, one‐exon GPR139 gene, located on chromosome 16 in human beings, was first assigned as a homologue to the thyrotropin‐releasing hormone receptor in 2002.20 In 2005, Gloriam et al21 identified and curated the full‐length GPR139 (mRNA NM_001002911.3 and protein NP_001002911.1) by searches in genome databases. GPR139 has also had a number of aliases, including GPCR12,20 PGR3,22 KOR3L 23 and GPRg1,24 but is now exclusively termed GPR139.17, 19 Basic local alignment search tool (BLAST) searches and phylogenetic analyses showed that GPR139 belongs to the rhodopsin class of GPCRs (also termed class A) and is closest related to another orphan receptor, GPR142.21 Moreover, Gloriam et al found that the full GPR139 gene has two exons and is highly conserved across species, as the amino acid sequence of the human GPR139 protein is 96%, 92% and 70% identical to the mouse, chicken and zebrafish orthologs, respectively.
5. GPR139 EXPRESSION
GPR139 expression studies have mainly been based on mRNA levels and depending on species found to have the highest transcript expression in the striatum, thalamus, hypothalamus, pituitary and habenula of the CNS.24, 25, 26, 27, 28, 29 Profiling of mRNA expression in a broad range of CNS and peripheral tissues have revealed that GPR139 expression is restricted to the CNS in human beings, rat and mouse,29, 30 suggesting that the receptor does not have a peripheral function. An overview of the GPR139 mRNA expression in human, rat and mouse tissue is provided in Table 1. Additionally, GPR139 protein expression has been studied in mice by Liu et al,26 who showed GPR139 antibody immunoreactivity in the habenula in wild‐type mice, but importantly not in knockout mice. Additionally, they confirmed expression of β‐galactosidase in the same brain area in the GPR139 knockout mouse (originating from a LacZ gene replacing the GPR139 gene in the knockout mouse). These findings correlate well with the expression of GPR139 mRNA in the habenula (Table 1). Radiolabelled GPR139 ligands have been developed and used in autoradiography studies on wild‐type vs. GPR139 knockout mouse brains, but no specific binding was detected, most likely due to a too low endogenous receptor expression level or to a too high level of background radioligand binding.26, 31
Table 1.
Regions with highest GPR139 mRNA expression in humans, rat and mouse
| Area of expression | Human beings | Rat | Mouse | |
|---|---|---|---|---|
| CNS | Brainstem | Wang, 2019 | Wang, 2019 | |
| CA1 area of hippocampus | Dvorak, 2014 | Süsen, 2006 | ||
| CA3 area of hippocampus | Dvorak, 2014 | |||
| Dentate gyrus of hippocampus | Dvorak, 2014 | Süsen, 2006 | ||
| Habenula (lateral) | Wagner, 2016 | |||
| Habenula (medial) | Dvorak, 2014; Wagner, 2016 | Matsuo, 2005; Süsen, 2006 | ||
| Hypothalamus |
Liu, 2015; Matsuo, 2005; Wang, 2019 |
Liu, 2015; Wang, 2019 |
Wang, 2019 | |
| Insular cortex | Liu, 2015 | |||
| Medulla | Süsen, 2006; Wang, 2019 | Wang, 2019 | ||
| Olfactory | Wang, 2019 | Wang, 2019 | ||
| Pituitary | Liu, 2015; Matsuo, 2005 | Liu, 2015; Wang, 2019 | ||
| Spinal cord | Wang, 2019 | |||
| Striatum |
Liu, 2015; Matsuo, 2005; Süsen, 2006; Wang 2019 |
Liu, 2015; Dvorak, 2014; Wang, 2019 |
Matsuo, 2005; Wang, 2019 | |
| Substantia nigra | Wang 2019 | Liu, 2015 | ||
| Thalamus | Liu, 2015; Wang, 2019 | Liu, 2015; Wagner, 2016; Wang, 2019 | Wang, 2019 | |
| Zona incerta | Matsuo, 2005 | |||
| Foetus | Matsuo, 2005 | Süsen, 2006a |
mRNA expression was determined using in situ hybridization by Dvorak et al, 201425 and Wagner et al, 2016,28 whereas Liu et al, 201526 used qPCR, Matsuo et al, 200524 used qPCR and in situ hybridization, Süsens et al, 200627 used Northern blot, and Wang et al, 201929 used RNA sequencing. Blank spots in the table indicate that GPR139 expression in these areas has not yet been determined.
Matsuo et al24 were not able to confirm this finding with PCR.
6. PUTATIVE ENDOGENOUS GPR139 RECEPTOR AGONISTS
In 2014, Isberg et al32 developed a pharmacophore model based on the surrogate agonists from Shi et al (see next section) that led to the identification of four aromatic‐containing dipeptides with pharmacological agonist activity at the human GPR139 receptor. The authors continued with a subsequent pharmacological evaluation of each individual amino acid. They assayed all L‐α‐amino acids and found the highest potencies for L‐tryptophan (L‐Trp, EC50 = 220 µmol/L) and L‐phenylalanine (L‐Phe, EC50 = 320 µmol/L) (Figure 2A, B), which were proposed as endogenous agonists for GPR139.32 The activity of L‐Trp and L‐Phe was subsequently confirmed by Liu et al26 at Janssen Research and Development, who also reported that a range of derivatives including tryptamine (Figure 2D), β‐phenylethylamine (Figure 2E) and amphetamine (Figure 2F) activate GPR139.33, 34 Interestingly, Liu et al26 found that the aromatic amino acids were slightly more potent at human GPR139 in their assays (L‐Trp, EC50 = 49 µmol/L; L‐Phe, EC50 = 60 µmol/L) and moreover reported that brain and serum fractions that activated GPR139 contained L‐Trp or L‐Phe. Liu et al26 also reported that rat plasma contains concentrations of L‐Trp (125 µmol/L fasted and 188 µmol/L fed) and L‐Phe (121 µmol/L fasted and 155 µmol/L fed), which is within the dynamic range of the GPR139 receptor. Collectively, these results support that the aromatic amino acids could be the endogenous signalling molecules for GPR139, but evidence of a closer link between the ligands, the receptor and a physiological function is needed to fully prove it.
Figure 2.
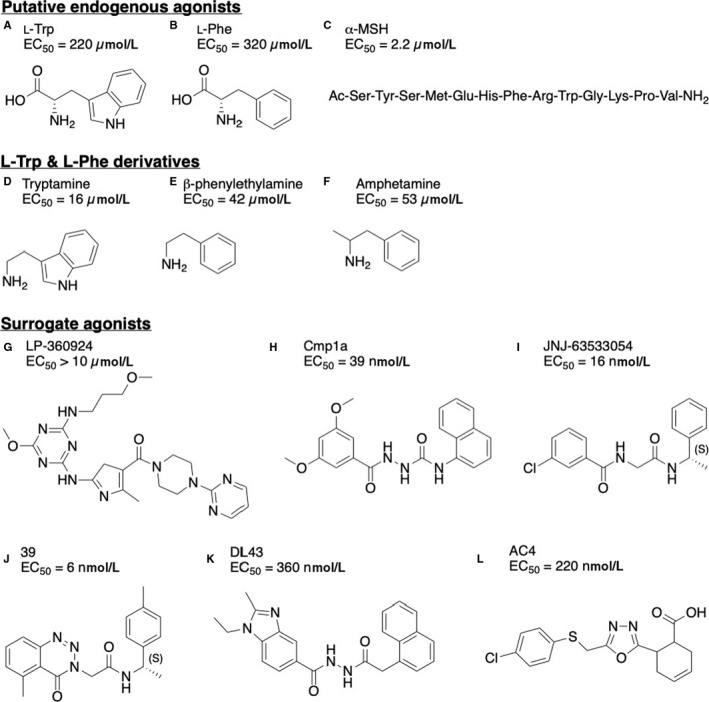
Structures of selected GPR139 agonists (A) L‐tryptophan,26, 32 (B) L‐phenylalanine,26, 32 (C) α‐MSH,35 (D) tryptamine,25, 26 (E) β‐phenylethylamine,25, 26 (F) amphetamine,25, 26 (G) LP‐360924 by Lexicon Pharmaceuticals,37 (H) cmp1a by H. Lundbeck A/S,38 (I) JNJ‐63533054 (also known as 7c) by Janssen Research and Development,26, 39 (J) 39 by Takeda Pharmaceutical Company Limited,40 (K) DL43 a bioisostere of cmp1a 41 and (L) AC4 43
Recently, Nøhr et al reported similarities between the ligand binding pocket residues of GPR139 and the melanocortin 4 receptor (MC4R). The authors subsequently demonstrated GPR139 activity of three known endogenous MC4R agonists: neuropeptides adrenocorticotropic hormone (ACTH), α‐melanocyte‐stimulating hormone (α‐MSH) (Figure 2C) and β‐melanocyte‐stimulating hormone (β‐MSH), as well as their conserved common motif HFRW.35 All three peptides activated GPR139 in the high nanomolar to low micromolar range.35 Another noteworthy finding was that a predicted cleavage product (α‐MSH1‐10) of the pre‐pro‐protein pro‐opiomelanocortin (POMC) activated the receptor in the submicromolar range (EC50 = 318 nmol/L), thus qualifying the peptide as the most potent and selective endogenous agonist of GPR139 identified to date.35 Given that the potency of the peptides is higher than L‐Trp and L‐Phe, the authors proposed that GPR139 is a peptide‐binding GPCR, albeit the endogenous peptide agonist might be another more potent peptide (derivative) than tested in the study.35 However, as detailed below, Nepomuceno et al (Janssen Research and Development) found the ACTH, α‐MSH and β‐MSH peptides to be less potent, but interestingly, the peptide agonists shifted the signalling pathway of the cognate melanocortin receptors when co‐expressed with GPR139.36 Collectively, these results suggest that GPR139 could also play a role as peptide receptor, either directly or indirectly via heterodimerization. However, more studies are required to elucidate the physiological endogenous agonist(s) for GPR139.
7. SURROGATE GPR139 RECEPTOR AGONIST LIGANDS, PHARMACOPHORE AND STRUCTURE‐ACTIVITY RELATIONSHIPS
Shortly after the discovery of GPR139, the first two series of small‐molecule surrogate agonists were reported by Hu et al (exemplified by LP‐360924, Figure 2G)37 and Shi et al (exemplified by cmp1a, Figure 2H).38 The compounds were selective towards GPR139 compared to β2AR and GPR142 and a selectivity panel consisting of 90 different receptors, ion channels and transporters. Furthermore, Janssen Research and Development identified a potent and selective (tested against a panel consisting of 50 different GPCRs, ion channels and transporters, including GPR142) surrogate small‐molecule agonist (JNJ‐63533054, also known as compound 7c) (Figure 2I) that crosses the rat blood‐brain barrier, reaching brain levels in the micromolar range after oral administration.26, 39 Subsequently, several groups have reported GPR139 surrogate agonist for GPR139, including Hitchcock et al (compound 39, Figure 2J 40), Shehata et al (DL43, Figure 2K 41) and Nøhr et al (AC4, Figure 2L).42
Shehata et al reported an updated agonist pharmacophore model (Figure 3) highlighting ligand features important for GPR139 agonism.41 The model spans six features: Two terminal aromatic systems (R1 and R2) separated by a linker consisting of two hydrogen bond acceptors (A3 and A4); one hydrogen bond donor (D5); and a dual hydrophobic/halogen or hydrogen bond acceptor element (H6/A2) placed at position 3 of R1. The model matches known GPR139 agonists and thus represents a typical GPR139 agonist. The authors also represented a combined structure‐activity relationship (SAR) of published GPR139 agonists 41 and reported that a reduction of the linker length results in loss of activity, while compounds with linkers longer than 6‐atoms only had reduced activity. The 1‐carbonyl oxygen atom represents a critical hydrogen bond acceptor, and removal leads to loss of activity. Methylation or carbon substitution of the 2‐nitrogen atom leads to a reduced potency, indicating the importance of a hydrogen bond donor on this position. Contrary to this, carbon substitution of the 3‐nitrogen has minor effect on potency. The 4‐carbonyl oxygen atom represents another important hydrogen bond acceptor, and removal results in loss of activity. Nitrogen to carbon substitution on the 5‐position only causes a minor shift in potency. Locking the linker 1‐2 atoms and/or the aromatic ring in a favourable conformation generally improves potency, as opposed to 5‐6‐cyclization. The R1 aromatic ring is present in all active surrogate agonists and most likely contributes to ligand potency. Both L‐Trp and L‐Phe overlay with R1 and R2, respectively, as well as with several linker characteristics, indicating that they also accommodate the pharmacophore and the same binding site. Interestingly, the most recently published surrogate agonist, AC4 (Figure 2L), contains a novel scaffold, which does not fully fit the pharmacophore as it lacks the second terminal ring. However, AC4 still possesses nanomolar potency and binds to the receptor in the same binding site as other GPR139 agonists (cf. section “GPR139 receptor agonist binding site” below).42
Figure 3.
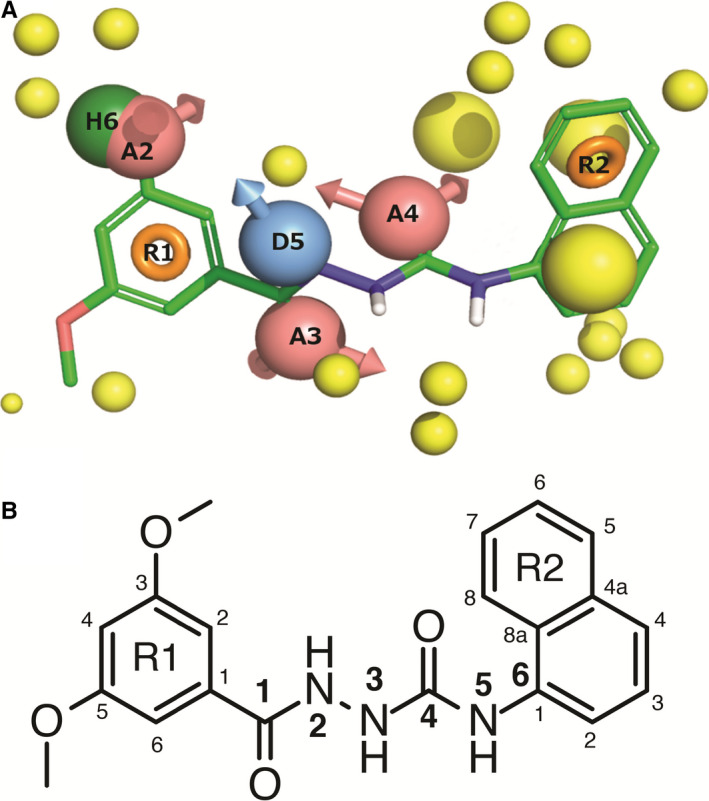
A, Agonist pharmacophore model showing cmp1a with green carbons and the following pharmacophore features: red: hydrogen bond acceptor, blue: hydrogen bond donor, green: hydrophobic element, orange: aromatic system and yellow: exclusion volume. B, Chemical structure of cmp1a with atom numbering used to discuss the structure‐activity‐relationship. From 41 with permission under Creative Commons Attribution 4.0 International License
8. GPR139 RECEPTOR AGONIST BINDING SITE
Nøhr et al elegantly confirmed that the endogenous L‐Trp and L‐Phe share a common binding site with the surrogate agonists cmp1a, JNJ‐63533054 and AC4.42, 43 By combining in silico and in vitro mutagenesis, they identified a common binding pocket consisting of a hydrophobic region deeply buried between F1093x33, H1875x43 and W2416x48, and a proximal polar region enclosed by N2717x38, R2446x51 and E1083x32 (Figure 4).43 The binding site was confirmed by Wang et al, who also identified a less buried hydrophobic binding site defined by V762x73, F792x56, I1043x28, V832x60 and I802x57 shared by larger GPR139 agonists.44 They furthermore identified several series of human GPR139 mutations resulting in gain, reduction or loss of function. The two first sets of mutations were located in the common binding pocket and near the G protein binding site on helix 5 and 6, which are known to move upon G protein binding. Another set of mutations impact the transmembrane helices structure and thus might change the common ligand binding site or the ability to bind G proteins. The authors speculate that the last two set of mutations have a structural impact on GPR139 function.44
Figure 4.
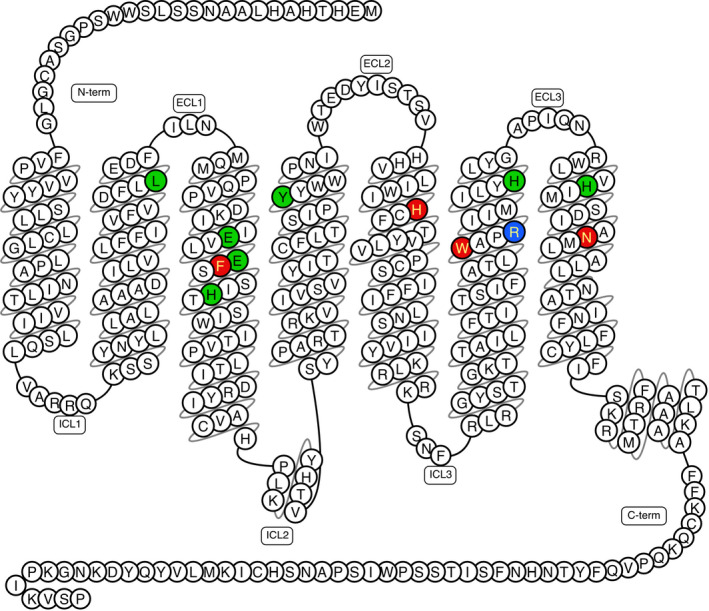
Snake plot diagram of GPR139 agonist binding site showing 12 mutated residues in red (hot spot), green (no effect) and blue (very low expression). Drawn in GPCRdb 61 based on data from.43 N‐term: amino‐terminal, C‐term: carboxy‐terminal, ECL: extracellular loop and ICL: intracellular loop
9. GPR139 RECEPTOR ANTAGONISTS
Finding antagonists for GPR139 has proven to be more challenging. However, Hu et al have reported two antagonists LP‐471756 and LP‐114958 (Figure 5A, B),37 that are selective towards the GPR139 receptor, compared to vector and β2AR cells. Wang et al identified five small‐molecule antagonists (NCRW0001‐C02, NCRW0005‐F05, NCRW0008‐C04, NCRW0095‐F03 and NCRW0105‐E06) representing four different scaffolds but reported no selectivity data (Figure 5C‐G).45 Lastly, Andersen et al report a GPR139 antagonist compound 4 (Figure 5H).46 Overall, these antagonists have relatively low potency and unknown receptor selectivity severely limiting their usability as pharmacological tools to study function and physiology of the GPR139 receptor. However, Janssen Research and Development recently reported the discovery of the GPR139 antagonist JNJ‐3792165 (Figure 5I), which is fairly potent (pKb = 7.4 in [35S]GTPγS assay and pKb = 6.9 in calcium mobilization assay) and selective compared to 50 neurotransmitter and neuropeptide receptors.36
Figure 5.
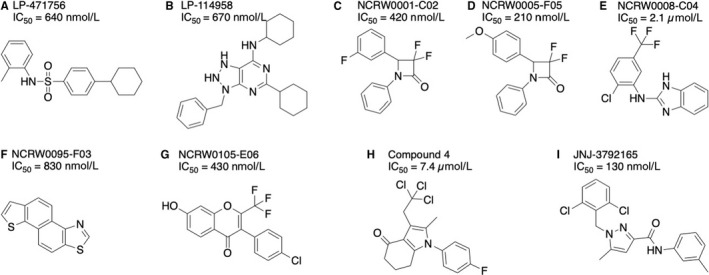
GPR139 antagonist structures. A, LP‐471756, (B) LP‐114958 by Lexicon Pharmaceuticals,37 (C) NCRW0001‐C02, (D) NCRW0005‐F05, (E) NCRW0008‐C04, (F) NCRW0095‐F03, (G) NCRW0105‐E06 by the National Center for Drug Screening, CAS, Shanghai,45 (H) compound 4 by H. Lundbeck A/S 46 and (I) JNJ‐3792165 by Janssen Research and Development 36
10. GPR139 SIGNALLING PATHWAYS
There is some discrepancy in the literature reports of GPR139 signalling pathways. Of note, the signalling pathways have only been studied in transfected cells, because a cell line or primary cells endogenously expressing GPR139 have not yet been identified. All the reported results on GPR139 signalling pathways are summarized in Figure 6 and described briefly below.
Figure 6.
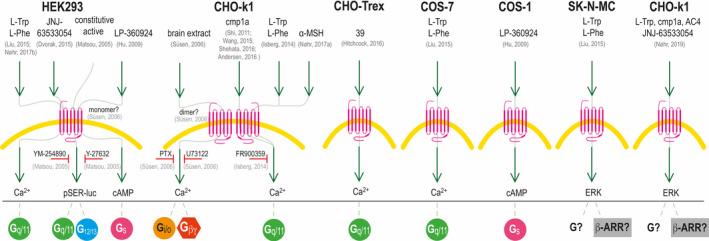
Schematic drawing of the reported GPR139 signalling in different cell types. GPR139 signalling has been reported in HEK293, CHO‐k1, COS‐1, COS‐7, SK‐N‐MC and CHO‐Trex cells. Green arrows indicate an effect on GPR139, and red horizontal T‐bars indicate inhibition of the signalling pathways by different pathway inhibitors: YM‐254890 is a Gq/11 inhibitor; Y‐27632 is a ROCK inhibitor that inhibits the G12/13 signalling pathway; PTX, pertussis toxin, is a Gi/o inhibitor; U73122 is a PLC inhibitor, and FR900359 is a Gq/11 inhibitor. At the bottom of the drawing, the association with the different G proteins and ß‐arrestin (ß‐ARR) is given. The references are listed in light grey: Andersen et al, 201646; Dvorak et al, 201539; Hitchcock et al, 201639; Hu et al, 200937; Isberg et al, 201437; Liu et al, 201526; Matsuo et al, 200526; Nøhr et al, 2017a,35 Nøhr et al 2017b35; Nøhr et al 201942; Shehata et al, 201643; Shi et al, 201138; Süsens et al, 200638; Wang et al, 201545
Matsuo et al report constitutive activity of GPR139 expressed in 293‐EBNA cells (HEK293 cell line expressing Epstein‐Barr virus). They were able to block this constitutive activity with a Gq/11 inhibitor (YM‐254890) and partly inhibit it with a G12/13 pathway inhibitor (Y‐27632 binding to Rho‐associated protein kinase), suggesting coupling to Gq/11 and G12/13. 24 Shi et al, Wang et al and Isberg et al have all used the same stable transfected CHO‐GPR139 cell line and have consistently shown that cmp1a activates a Ca2+ response 32, 38, 45 that can be inhibited by the Gq/11 inhibitor FR900359, suggesting Gq/11 coupling.32 Furthermore, Nøhr et al and Liu et al show that HEK293 cells transiently transfected with GPR139 give a Ca2+ response upon stimulation with L‐Trp and L‐Phe.26, 43 Additionally, Hitchcock et al show Ca2+ signalling in CHO‐TREx cells expressing GPR139 and Dvorak et al show a Ca2+ response in stably expressing HEK293‐GPR139 cells upon stimulation with the agonist JNJ‐63533054.39, 40 Altogether, there is strong evidence of Gq/11 as the primary transducer of GPR139 signalling.
A few studies have also suggested other signalling pathways. Hu et al observed a cAMP stimulation response in HEK293 and COS‐1 cells expressing GPR139 upon stimulation with the agonist LP‐360924 indicative of Gs coupling,37 whereas Süsen et al via indirect measurements suggested that GPR139 expressed in CHO‐k1 cells couples to Gi/o upon stimulation with brain extracts.27 However, Shehata et al have been unable to detect Gs or Gi/o‐mediated responses from cmp1a, JNJ‐63533054, DL43, L‐Trp or L‐Phe in CHO‐GPR139 cells.41 This does not rule out the possibility that GPR139 might be able to activate non‐Gq/11‐mediated signalling, as the cellular background could influence the preferred signalling pathway depending on G protein and β‐arrestin subtype expression. Another possibility is that distinct ligands could induce signalling bias.47, 48 More studies are thus needed to delineate the role of the non‐Gq/11 pathways in GPR139 receptor signalling.
Interestingly, Nøhr et al recently demonstrated a robust GPR139‐mediated ERK phosphorylation response in stably transfected CHO‐GPR139 cells,42 which further elaborated initial observations by Liu et al.26 All the tested compounds (incl. L‐Trp, cmp1a, JNJ‐63533054 and AC4) displayed a concentration‐dependent response, with potencies and potency rank order similar to what had previously been observed in the calcium mobilization assay.42 More studies are needed to elucidate whether the ERK is mediated via Gq/11, ß‐arrestin or a third signalling pathway.
11. PUTATIVE GPR139 HETERODIMERIZATION
As described above, Nøhr et al have reported that high nanomolar to low micromolar concentrations of the peptides ACTH, α‐MSH and variants hereof activated the GPR139 receptor stably expressed in a CHO cell line, albeit with generally relatively lower potencies than reported at the canonical targets; melanocortin receptors 1‐5 (MC1‐5).35 When reproducing this, Nepomuceno et al 36 observed GPR139 receptor activity of these peptides at the highest tested concentrations in some cell lines and pharmacological assays, but obtained lower potencies.35 The reason for this discrepancy is unclear but it is well known that GPCR and/or G protein expression levels can vary between studies and affect agonist potencies.47 However, in both studies, the potencies of the peptides were likely too low to be physiologically relevant albeit peptide variants of different length or with post‐translational modifications might be more potent and thus endogenous agonists.35
Interestingly, Nepomuceno et al 36 observed that co‐expression of GPR139 with MC3 or MC5 enabled robust and potent intracellular calcium mobilization by ACTH, α‐MSH and ß‐MSH. MC3 and MC5 are Gs coupled receptors which usually do not couple to intracellular calcium mobilization when expressed alone, so this study suggests that the GPR139 receptor can heterodimerize with other GPCRs and modulate their signalling. Wang et al subsequently performed a study where they observed that mRNA expression of the GPR139 and dopamine D2 receptor correlated in a broad range of CNS regions, which led the authors to hypothesize that they might functionally interact.29 Interestingly, co‐expression of the two receptors in HEK293 cells enabled the dopamine D2 receptor to increase intracellular calcium, while the D2 receptor did not respond in this pathway when expressed alone. Moreover, the calcium response from the co‐expressed receptors could be antagonized by either a D2 or GPR139 receptor antagonist. Collectively, these results suggest that the receptors interact functionally and that GPR139 modulate D2 receptor signalling.29 Further studies on cells or tissues with native GPR139 receptor expression are required to fully elucidate the role of the GPR139 receptor on ACTH/α‐MSH/ß‐MSH pharmacology and function and on the role of GPR139 in modulating melanocortin and dopamine D2 receptor signalling.
12. PHYSIOLOGICAL FUNCTION AND THERAPEUTIC POTENTIAL OF GPR139
GPR139 is still classified as an orphan receptor, as its endogenous ligand and function remain to be firmly established according to the NC‐IUPHAR guidelines (cf. section “Orphan GPCRs” above). However, based on the expression pattern, putative endogenous ligands and preliminary animal studies, four major hypotheses about the physiological function and therapeutic potential of the receptor have been developed.
12.1. Hypothesis I: Locomotor activity
The consistent cross‐species expression of GPR139 mRNA in the striatum (Table 1) suggests that GPR139 may have a role in the control of locomotor activity (movement). Parkinson’s disease (PD) is a major movement disorder. Its aetiology/cause is still largely unknown; however, patients with PD suffer from degeneration of dopaminergic (DA) neurons in substantia nigra, which gives input to the striatum. The striatum consists of caudate nucleus and putamen and is part of the basal ganglia.33 There is a direct motor loop through the basal ganglia that facilitates the initiation of willed movements, which is disrupted in patients with PD. More than 6 million people worldwide have PD 49 and presently only the symptoms can be treated; hence, novel treatment strategies are acutely needed.50 Besides GPR139 mRNA expression in the striatum, there are three studies pointing towards a role in locomotor activity for the receptor:
A patent from Regeneron Pharmaceuticals Inc in 2004 describes GPR139 knockout mouse phenotypes.23 Interestingly, they showed that in a rotarod performance test, which is a test known to evaluate balance and motor co‐ordination, the mean latency time to fall off the rod was significantly lower for GPR139 knockout than wild‐type mice. Furthermore, when their GPR139 knockout mice were placed on a 2‐cm‐thick beam, they fell off at rates that were significantly faster than their wild‐type littermates. However, other tests for balance and motor control like a gait analysis and a thin 5‐mm beam balance test showed no difference between wild‐type mice and their GPR139 knockout littermates.23 Nonetheless, all together these tests suggest impairment in balance and in sensory motor integration in their GPR139 knockout mice, but further behavioural studies are needed to clarify this.
Recently, Liu et al26 published a potent GPR139 agonist JNJ‐63533054 (Figure 2I). They dosed male Sprague Dawley rats with this compound and observed significantly decreased locomotor activity compared to vehicle and to rats dosed with the less active enantiomer of JNJ‐63533054. However, future work is needed to elucidate whether the mechanism behind this locomotor effect was mediated by GPR139, as the compound could potentially have sedative effects that could partly explain the observed decrease in locomotor activity.26
Lastly, Andersen et al46 showed that GPR139 agonists (including cmp1a) protected primary dopaminergic (DA) neurons against the toxin 1‐methyl‐4‐phenylpyridinium (MPP+) in vitro. MPP+produces similar neuropathological defects as observed in PD patients by degenerating DA neurons 51 and is therefore often used in animal models for PD.52 However, GPR139 agonists did not show any neuroprotective activity upon treatment with rotenone,46 which is another toxin used in animal models of PD.52 Despite the inclusive results, Andersen and coworkers suggest that GPR139 could be a potential target for neuroprotection in PD.46
12.2. Hypothesis II: Metabolism
GPR139 mRNA is expressed in the hypothalamus and pituitary in humans and rat, and both GPR139 mRNA and proteon have been found in the habenula in rodents (Table 1). On this basis, GPR139 has been postulated to regulate food consumption and/or energy expenditure.28
The close GPR139 homologue GPR142 shares the activation by L‐Trp and L‐Phe, but is mainly expressed in the pancreas and gut, where it is involved in the regulation of insulin and incretin secretion, respectively, making GPR142 a potential target in type II diabetes.53, 54 As L‐Trp and L‐Phe are both also putative endogenous ligands of GPR139,26, 32 it has been suggested that GPR139 is a nutrient‐sensing receptor.26 Thus, based on the expression pattern and nature of the putative endogenous agonists, GPR139 could be involved in metabolism‐related disorders like type II diabetes.
Furthermore, in the non‐peer‐reviewed patent from Regeneron Pharmaceuticals, it is stated that the GPR139 knockout mice had increased lean body mass and decreased body fat compared to wild‐type mice.23 Hence, the patent supports the hypothesis that GPR139 plays a role in energy homeostasis. Additionally, as described above, it has been shown that the appetite‐regulating hormones ACTH, α‐MSH and β‐MSH 55 can activate GPR139 at relatively high concentrations 35 or that GPR139 modulate the signalling the canonical melanocortin receptors,36 which further support a role of GPR139 in energy homeostasis. In conclusion, GPR139 could be a potential target for the treatment of metabolism‐related disorders like obesity and type II diabetes.
12.3. Hypothesis III: Alcohol addiction and hyperalgesia
The habenula is also known as a brain region involved in addiction.56 Interestingly, a recent study has shown that systemic or habenula‐specific injection of the GPR139 selective agonist JNJ‐63533054 reduced alcohol self‐administration in a subgroup of alcohol‐dependent rats that exhibited a compulsive‐like phenotype. Moreover, the GPR139 agonist also reduced withdrawal‐induced hyperalgesia.34 Importantly, these effects were not observed in non‐dependent rats and the agonist did not affect water or saccharin intake. It was thus concluded that GPR139 is a potential new drug target for treatment of alcohol misuse disorders and potentially also other drugs of abuse.
12.4. Hypothesis IV: Phenylketonuria (PKU)
Phenylketonuria (PKU) is an autosomal recessive genetic disorder, which is associated with intellectual disabilities when untreated. Patients suffering from PKU have a non‐functional phenylalanine hydroxylase enzyme, which is a hepatic enzyme that metabolizes L‐Phe to L‐Tyr. In the absence of this enzyme, L‐Phe accumulates in the body to a neurotoxic level (reviewed in 57). Since it has been shown that L‐Phe is an agonist for GPR139,26, 32 it has been suggested that the abnormally high levels of L‐Phe in PKU patients cause overstimulation of GPR139 in the brain which could be one of the reasons for the intellectual disabilities associated with PKU.25 Inhibiting GPR139 in PKU patients might therefore limit the intellectual disabilities seen in these patients.
12.5. Other potential (patho) physiological roles of GPR139
Genetic variations in the GPR139 locus have been linked to schizophrenia58 and symptoms of inattention in attention deficit hyperactivity disorder (ADHD).59 GPR139 has also been proposed to have a role in depression40 and foetal development.27 Recently, Janssen Research and Development showed that the brain penetrant agonist JNJ‐63533054 had no or only minor effects in a range of rodent mood/anxiety models questioning the GPR139 receptor role in mood/anxiety.60
13. CONCLUSION AND PERSPECTIVES
Recent years have brought a number of significant tools in the studies of the GPR139 receptor including a range of potent and selective GPR139 agonists of which some have been shown to penetrate the blood‐brain barrier and be active in animal models. The amino acids L‐Trp and L‐Phe as well as derivatives of the peptide hormones ACTH and α‐MSH have been suggested as potential endogenous GPR139 receptor agonists, but remain to be fully validated. A specific radioligand and antibody have been published but are not commercially available limiting application. Likewise, only limited information has been drawn from published GPR139 knockout mice. The main tool remaining to be developed to study the GPR139 receptor is an even more potent and selective antagonist, preferably blood‐brain barrier permeable and metabolic stable. The lower number of reported antagonists is striking, given the many potent and selective agonists that have been published. It would also be of great interest to investigate the full range of signalling pathways employed by GPR139 in cells with native receptor expression and whether some of the many agonists developed show signalling bias, which could translate into different physiological/therapeutic effects. Finally, it would also be very valuable to develop the first allosteric modulators, which might have therapeutic benefits compared to the orthosteric ligand. Nevertheless, the tools available will undoubtedly become very valuable in future years to further characterize the pharmacology, physiological function and therapeutic potential of the GPR139 receptor.
ACKNOWLEDGEMENTS
Our work on the GPR139 receptor has been supported by grants to H.B.‐O from the Lundbeck Foundation (R151‐2013‐14399) and the Carlsberg Foundation (CF17‐0132) and to D.E.G. from the Lundbeck Foundation (R163‐2013‐16327), the Danish Council for Independent Research (DFF‐1331‐00180) and the European Research Council (DE‐ORPHAN 639125).
Vedel L, Nøhr, AC , Gloriam DE, Bräuner‐Osborne H. Pharmacology and function of the orphan GPR139 G protein‐coupled receptor. Basic Clin Pharmacol Toxicol. 2020;126(Suppl. 6):35–46. 10.1111/bcpt.13263
Line Vedel and Anne C. Nøhr equal contributions as shared first authors.
David E. Gloriam and Hans Bräuner‐Osborne equal contributions as shared senior authors.
Parts of the present MiniReview have been published previously in the PhD thesis of Dr. Anne C. Nøhr.
REFERENCES
- 1. Bockaert J, Pin JP. Molecular tinkering of G protein‐coupled receptors: an evolutionary success. EMBO J 1999;18:1723‐1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: a G protein‐coupled receptor. Science 2000;289:739‐745. [DOI] [PubMed] [Google Scholar]
- 3. Hauser AS, Attwood MM, Rask‐Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 2017;16:829‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heldin CH, Lu B, Evans R, Gutkind JS. Signals and receptors. Cold Spring Harb Perspect Biol 2016;8:a005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev 2005;85:1159‐1204. [DOI] [PubMed] [Google Scholar]
- 6. Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev 2016;96:1261‐1296. [DOI] [PubMed] [Google Scholar]
- 7. Wu‐Zhang AX, Newton AC. Protein kinase C pharmacology: refining the toolbox. Biochem J 2013;452:195‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siehler S. Regulation of RhoGEF proteins by G12/13‐coupled receptors. Br J Pharmacol 2009;158:41‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, Martemyanov KA. Distinct profiles of functional discrimination among G proteins determine the actions of G protein‐coupled receptors. Sci Signal 2015;8:ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schröder R, Janssen N, Schmidt J, et al. Deconvolution of complex G protein‐coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotech 2010;28:943‐949. [DOI] [PubMed] [Google Scholar]
- 11. Dupre DJ, Robitaille M, Rebois RV, Hebert TE. The role of Gβγ subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol 2009;49:31‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β‐arrestin‐biased agonism at seven‐transmembrane receptors. Annu Rev Pharmacol Toxicol 2012;52:179‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomsen AR, Plouffe B, Cahill TJ 3rd, et al. GPCR‐G protein‐ß‐arrestin super‐complex mediates sustained G protein signaling. Cell 2016;166:907‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calebiro D, Nikolaev VO, Gagliani MC, et al. Persistent cAMP‐signals triggered by internalized G‐protein‐coupled receptors. PLoS Biol 2009;7:e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrandon S, Feinstein TN, Castro M, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 2009;5:734‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irannejad R, Tomshine JC, Tomshine JR, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013;495:534‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander S, Christopoulos A, Davenport AP, et al. The concise guide to pharmacology 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017;174(Suppl 1):S17‐s129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davenport AP, Alexander S, Sharman JL, et al. International union of basic and clinical pharmacology. LXXXVIII. G protein‐coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013;65(3):967‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein‐coupled receptor genes from the human genome sequence. FEBS Lett 2002;520:97‐101. [DOI] [PubMed] [Google Scholar]
- 21. Gloriam DE, Schiöth HB, Fredriksson R. Nine new human Rhodopsin family G‐protein coupled receptors: identification, sequence characterisation and evolutionary relationship. Biochim Biophys Acta 2005;1722:235‐246. [DOI] [PubMed] [Google Scholar]
- 22. Vassilatis DK, Hohmann JG, Zeng H, et al. The G protein‐coupled receptor repertoires of human and mouse. Proc Natl Acad Sci USA 2003;100:4903‐4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murphy AJ, Croll‐Kalish S KOR3 like‐proteins and methods of modulating KOR3L‐mediated activity. WO2004/074841 A2, Regeneron Pharmaceuticals Inc., 2004. https://patents.google.com/patent/US20040161799.
- 24. Matsuo A, Matsumoto S‐I, Nagano M, et al. Molecular cloning and characterization of a novel Gq‐coupled orphan receptor GPRg1 exclusively expressed in the central nervous system. Biochem Biophys Res Commun 2005;331:363‐369. [DOI] [PubMed] [Google Scholar]
- 25. Dvorak CA, Liu C, Kuei C. Physiological ligands for GPR139. WO2014/152917 A2, Janssen Pharmaceuticals, 2014. https://patents.google.com/patent/WO2014152917A2.
- 26. Liu C, Bonaventure P, Lee G, et al. GPR139, an orphan receptor highly enriched in the habenula and septum, is activated by the essential amino acids L‐tryptophan and L‐phenylalanine. Mol Pharmacol 2015;88:911‐925. [DOI] [PubMed] [Google Scholar]
- 27. Süsens U, Hermans‐Borgmeyer I, Urny J, Schaller HC. Characterisation and differential expression of two very closely related G‐protein‐coupled receptors, GPR139 and GPR142, in mouse tissue and during mouse development. Neuropharmacology 2006;50:512‐520. [DOI] [PubMed] [Google Scholar]
- 28. Wagner F, Bernard R, Derst C, French L, Veh RW. Microarray analysis of transcripts with elevated expressions in the rat medial or lateral habenula suggest fast GABAergic excitation in the medial habenula and habenular involvement in the regulation of feeding and energy balance. Brain Struct Funct 2016;221:4663‐4689. [DOI] [PubMed] [Google Scholar]
- 29. Wang L, Lee G, Kuei C, et al. GPR139 and dopamine D2 Receptor co‐express in the same cells of the brain and may functionally interact. Front Neurosci 2019;13:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein‐coupled receptor expression. Cell 2008;135:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuhne S, Nøhr AC, Marek A, et al. Radiosynthesis and characterisation of a potent and selective GPR139 agonist radioligand. RSC Adv 2016;6:947‐952. [Google Scholar]
- 32. Isberg V, Andersen KB, Bisig C, Dietz G, Bräuner‐Osborne H, Gloriam DE. Computer‐aided discovery of aromatic L‐alpha‐amino acids as agonists of the orphan G protein‐coupled receptor GPR139. J Chem Inf Model 2014;54:1553‐1557. [DOI] [PubMed] [Google Scholar]
- 33. Bear MF, Connors BW, Paradiso MA. Neuroscience Exploring the Brain. 3rd. edition ed Baltimore, MD: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 34. Kononoff J, Kallupi M, Kimbrough A, Conlisk D, de Guglielmo G, George O. Systemic and intra‐habenular activation of the orphan G protein‐coupled receptor GPR139 decreases compulsive‐like alcohol drinking and hyperalgesia in alcohol‐dependent rats. eNeuro. 2018;5:ENEURO.0153‐18.2018. 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nøhr AC, Shehata MA, Hauser AS, et al. The orphan G protein‐coupled receptor GPR139 is activated by the peptides: Adrenocorticotropic hormone (ACTH), α‐, and β‐melanocyte stimulating hormone (α‐MSH, and β‐MSH), and the conserved core motif HFRW. Neurochem Int 2017;102:105‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nepomuceno D, Kuei C, Dvorak C, Lovenberg T, Liu C, Bonaventure P. Re‐evaluation of adrenocorticotropic hormone and melanocyte stimulating hormone activation of GPR139 in vitro. Front Pharmacol 2018;9:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu LA, Tang PM, Eslahi NK, Zhou T, Barbosa J, Liu Q. Identification of surrogate agonists and antagonists for orphan G‐protein‐coupled receptor GPR139. J Biomol Screen 2009;14:789‐797. [DOI] [PubMed] [Google Scholar]
- 38. Shi F, Shen JK, Chen D, et al. Discovery and SAR of a series of agonists at orphan G protein‐coupled receptor 139. ACS Med Chem Lett 2011;2:303‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dvorak CA, Coate H, Nepomuceno D, et al. Identification and SAR of glycine benzamides as potent agonists for the GPR139 receptor. ACS Med Chem Lett 2015;6:1015‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hitchcock S, Lam B, Monenschein H, Reichard H. 4‐oxo‐3,4‐dihyrol‐1,2,3‐benzotriazine modulators of GPR139. US Patent US2016/0145218 A1, Takeda Pharmaceutical Company Limited, 2016. https://patents.google.com/patent/US20160145218A1/.
- 41. Shehata MA, Nøhr AC, Lissa D, et al. Novel agonist bioisosteres and common structure‐activity relationships for the orphan G protein‐coupled receptor GPR139. Sci Rep 2016;6:36681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nøhr AC, Shehata MA, Palmer D, et al. Identification of a novel scaffold for a small molecule GPR139 receptor agonist. Sci Rep 2019;9:3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nøhr AC, Jespers W, Shehata MA, et al. The GPR139 reference agonists 1a and 7c, and tryptophan and phenylalanine share a common binding site. Sci Rep 2017;7:1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang L, Lee G, Shih A, et al. Mutagenesis of GPR139 reveals ways to create gain or loss of function receptors. Pharmacol Res Perspect 2019;7:e00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J, Zhu LY, Liu Q, Hentzer M, Smith GP, Wang MW. High‐throughput screening of antagonists for the orphan G‐protein coupled receptor GPR139. Acta Pharmacol Sin 2015;36:874‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Andersen KB, Johansen JL, Hentzer M, Smith GP, Dietz G. Protection of primary dopaminergic midbrain neurons by GPR139 agonists supports different mechanisms of MPP+ and rotenone toxicity. Front Cell Neurosci 2016;10:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bräuner‐Osborne H, Ebert B, Brann MR, Falch E, Krogsgaard‐Larsen P. Functional partial agonism at cloned muscarinic acetylcholine receptors. Eur J Pharmacol 1996;313:145‐150. [DOI] [PubMed] [Google Scholar]
- 48. Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 2011;336:296‐302. [DOI] [PubMed] [Google Scholar]
- 49. Rocca WA. The burden of Parkinson’s disease: a worldwide perspective. Lancet Neurol 2018;17:928‐929. [DOI] [PubMed] [Google Scholar]
- 50. Oertel W, Schulz JB. Current and experimental treatments of Parkinson disease: a guide for neuroscientists. J Neurochem 2016;139(Suppl 1):325‐337. [DOI] [PubMed] [Google Scholar]
- 51. Langston JW, Langston EB, Irwin I. MPTP‐induced Parkinsonism in human and non‐human primates–clinical and experimental aspects. Acta Neurol Scand 1984;100:49‐54. [PubMed] [Google Scholar]
- 52. Duty S, Jenner P. Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 2011;164:1357‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rudenko O, Shang J, Munk A, et al. The aromatic amino acid sensor GPR142 controls metabolism through balanced regulation of pancreatic and gut hormones. Mol Metab 2019;19:49‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin HV, Efanov AM, Fang X, et al. GPR142 controls tryptophan‐induced insulin and incretin hormone secretion to improve glucose metabolism. PLoS ONE 2016;11:e0157298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Austin J, Marks D. Hormonal regulators of appetite. Int J Pediatr Endocrinol 2009;2009:141753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Batalla A, Homberg JR, Lipina TV, et al. The role of the habenula in the transition from reward to misery in substance use and mood disorders. Neurosci Biobehav Rev 2017;80:276‐285. [DOI] [PubMed] [Google Scholar]
- 57. Al Hafid N, Christodoulou J. Phenylketonuria: a review of current and future treatments. Transl Pediatr 2015;4:304‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Castellani CA, Awamleh Z, Melka MG, O’Reilly RL, Singh SM. Copy number variation distribution in six monozygotic twin pairs discordant for schizophrenia. Twin Res Hum Genet 2014;17:108‐120. [DOI] [PubMed] [Google Scholar]
- 59. Ebejer JL, Duffy DL, van der Werf J, et al. Genome‐wide association study of inattention and hyperactivity‐impulsivity measured as quantitative traits. Twin Res Hum Genet 2013;16:560‐574. [DOI] [PubMed] [Google Scholar]
- 60. Shoblock JR, Welty N, Fraser I, et al. In vivo characterization of a selective, orally available, and brain penetrant small molecule GPR139 agonist. Front Pharmacol 2019;10:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Isberg V, Mordalski S, Munk C, et al. GPCRdb: an information system for G protein‐coupled receptors. Nucleic Acids Res 2016;44:D356‐D364. [DOI] [PMC free article] [PubMed] [Google Scholar]