

Abstract
Background and Aims
Hepatocellular carcinoma (HCC) is linked to immunosuppression. Relieving immunosuppression has been an attractive strategy to improve the efficacy of cancer immunotherapy. Peptidoglycan recognition protein 2 (PGLYRP2) is a pattern recognition receptor which is specifically expressed in liver and implicated in the regulation of innate immunity and immunosurveillance. However, the role of hepatic PGLYRP2 in modulating immune responses against HCC remains to be investigated.
Approach and Results
In this study, we investigated whether PGLYRP2 is able to influence HCC progression through regulating host antitumor immune responses. We demonstrated that PGLYRP2 was down‐regulated in HCC, which was linked with poor prognosis in patients (P < 0.001). PGLYRP2 overexpression in HCC cells significantly enhanced antitumor immune responses in immune‐competent mice and elevated immune response rates of peripheral blood mononuclear cells against HCC. Mechanistically, DNA methyltransferase 3A–mediated promoter hypermethylation was responsible for the down‐regulation of PGLYRP2 in HCC. PGLYRP2 promoted production of chemokine (C‐C motif) ligand 5 (CCL5) in HCC through binding to the CCL5 promoter, which contributed to the enhanced antitumor immunity.
Conclusions
We provide evidence that tumor‐derived PGLYRP2 acts as a candidate biomarker for adequate immune response against HCC and improved patient outcomes, indicating the importance of hepatic PGLYRP2 in cancer immunosurveillance and in designing immunotherapeutic approaches.
Abbreviations
- 5‐Aza‐CdR
5‐Aza‐2′‐deoxycytidine
- CCL5
chemokine (C‐C motif) ligand 5
- CD
cluster of differentiation
- cDNA
complementary DNA
- ChIP
chromatin immunoprecipitation
- CI
confidence interval
- CXCL
chemokine (C‐X‐C motif) ligand
- DNMT
DNA methyltransferase
- E:T
effector to target ratio
- FoxP3
forkhead box P3
- HCC
hepatocellular carcinoma
- HR
hazard ratio
- IHC
immunohistochemistry
- IL
interleukin
- LIHC
liver HCC
- MDSC
myeloid‐derived suppressor cell
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NF‐κB
nuclear factor kappa B
- NK
natural killer
- OS
overall survival
- PBMC
peripheral blood mononuclear cell
- PGLYRP2
peptidoglycan recognition protein 2
- PRR
pattern recognition receptor
- RNAseq
RNA sequencing
- siRNA
small interfering RNA
- TCGA
The Cancer Genome Atlas
- TIL
tumor‐infiltrating lymphocyte
- Treg
regulatory T cell
The landscape of cancer therapy has been subverted by immunotherapy. Clinical studies have provided substantial evidence that a preexisting antitumor immune response is required for therapeutic benefit from cancer immunotherapy.1, 2, 3, 4 However, due to the comprehensive immunological tumor microenvironment in hepatocellular carcinoma (HCC), the efficiency of the cancer immune response is not satisfied. The desired immunotherapeutic approach for HCC would promote a sustained increase in functional intratumoral immune effector cells through remodeling the tumor microenvironment.5, 6 Understanding the mechanisms that underlie poor intratumoral immune cell infiltration is therefore key to developing rational treatment strategies for cancers that do not respond to immunotherapy.
Pattern recognition receptors (PRRs) function as the initial factors of the innate immune response, and they are closely associated with remodeling of the tumor microenvironment and antitumor immune response.7, 8, 9 PRR‐mediated innate immune responses play important roles in tumor‐infiltrating lymphocyte (TIL) activation, but their potential relevance for prevention and treatment of cancer remains underappreciated.10, 11, 12 Peptidoglycan recognition protein 2 (PGLYRP2) is a bacterial peptidoglycan‐sensing PRR that is primarily expressed at a constitutively high level in the liver but also inducibly expressed in keratinocytes and epithelial cells.13, 14 PGLYRP2 carries peptidoglycan amidase hydrolytic activity (N‐acetylmuramoyl‐l‐alanine amidase activity).15, 16, 17 PGLYRP2‐deficient mice did not display significantly enhanced susceptibility to bacterial infections but exhibited sex‐dependent changes in anxiety‐like behavior.18, 19 Additionally, variants in the PGLYRP2 gene are associated with risk of Parkinson's disease.20 Therefore, the primary function of PGLYRP2 as an innate immune molecule still needs to be further elucidated.
In the present study, we found that a previously unknown fundamental function of PGLYRP2 in hepatocytes is to suppress cancer development by stimulating antitumor immune responses. Here, we investigated the function and mechanism of PGLYRP2 in the regulation of the immune response against HCC. We examined the expression level of PGLYRP2 and its clinical and pathological significance in human hepatoma tissues by immunohistochemical (IHC) staining and real‐time PCR. The correlation among the PGLYRP2 level, activated TILs, and increased chemokine expression in HCC tissues was analyzed by PCR array, chemokine protein array, and immunofluorescence. The tumor suppression function of PGLYRP2 was examined in a tumor mouse model. The effect of PGLYRP2 on the immune response rates of peripheral blood mononuclear cells (PBMCs) was further investigated. Furthermore, the aberrant methylation status of the PGLYRP2 promoter in HCC cells was analyzed by bisulfite DNA sequencing. This report thus provides a direction for improved immunotherapy of hepatoma.
Materials and Methods
Cell Culture and Transfection
The Hep3B, HepG2, C3A, SNU‐387, and Hepa1‐6 cell lines used in this study were originally purchased from the American Type Culture Collection (Manassas, VA). Huh7 was purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China), and NK‐92 was purchased from the China Center for Type Culture Collection of Wuhan University (Wuhan, China). Huh7 was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (Gibco, Life Technologies Inc., Grand Island, NY). Hep3B, HepG2, and C3A were cultured in minimal essential medium supplemented with 10% fetal calf serum. Hepa1‐6 and SNU‐387 were cultured in Roswell Park Memorial Institute‐1640 medium supplemented with 10% fetal calf serum. NK‐92 cells were grown in Eagle's minimal essential medium with Earle's balanced salts supplemented with 10% fetal calf serum and 200 U interleukin 2 (IL‐2). Cells were transiently transfected with the indicated plasmids using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Mycoplasma contamination of cell lines was excluded using a SYBR green–based real‐time PCR assay.21 The identities of all of the cell lines were confirmed by short tandem repeat testing.
Statistics
All of the experiments in our study were independently performed in triplicate. The data are presented as mean ± SEM. All graphs were plotted and analyzed with GraphPad Prism 5 software. P > 0.05 was considered statistically not significant, and the following denotations were used: **P < 0.001 and *P < 0.05.
Full details of these and other methods can be found in the Supporting Information.
Results
Hepatic PGLYRP2 is Significantly Decreased in HCC
To identify cancer immune response–related PRR genes, we hypothesized that any PRR that plays a key regulatory role in the cancer immune response should also show an altered expression pattern in human hepatoma tissues. We first surveyed the expression profile of PRRs in publicly available RNA‐sequencing (RNAseq) data from The Cancer Genome Atlas (TCGA) liver hepatocellular carcinoma (LIHC). At the time of the analyses, the TCGA‐LIHC had RNAseq data available for 421 samples consisting of 371 tumor tissues and 50 tumor‐adjacent tissues. The mRNA expression levels of toll‐like receptors, nonobese diabetic–like receptors, RIG‐I‐like receptors, C‐type lectin receptors, absent in melanoma–like receptors, and peptidoglycan recognition protein (PGRP) family members were quantified; and hierarchical clustering was performed to illustrate the different expression of PRR genes between hepatoma tissues and the adjacent tissues. Using the Student t test and the Mann‐Whitney rank test, we identified three up‐regulated and 18 down‐regulated PRRs (false discovery rate ≤ 0.01, fold‐change ≥ 2) in human hepatoma tissues (Fig. 1A).
Figure 1.
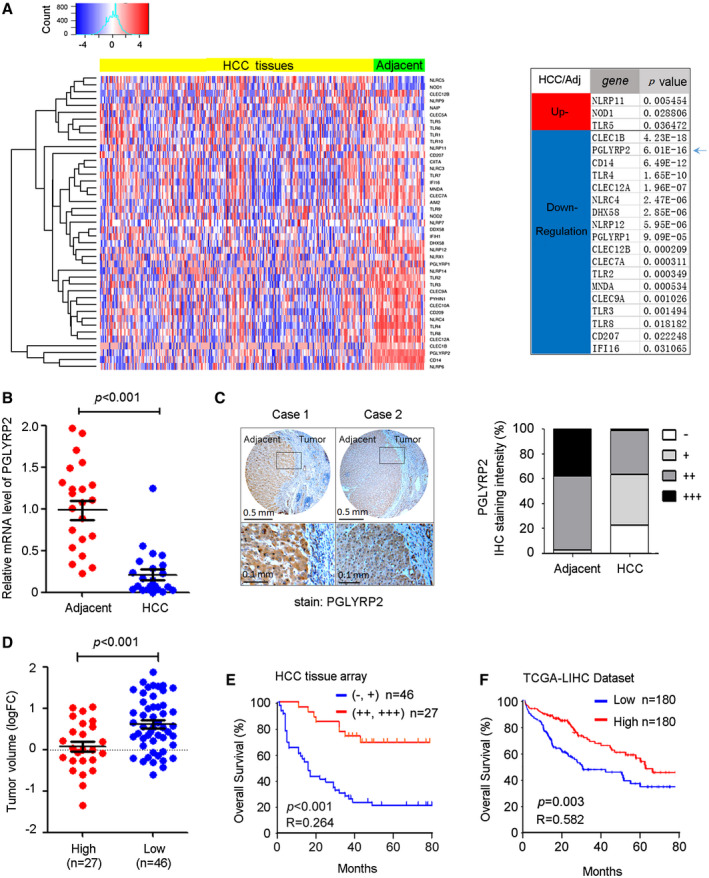
The PGLYRP2 level is down‐regulated in HCC, and the reduced level of PGLYRP2 correlates with poor prognosis in patients with HCC. (A) Left panel, Hierarchical clustering illustrates the different expression of PRR genes in TCGA‐LIHC between hepatoma tissues and the adjacent tissues. Right panel, The most significantly up‐regulated or down‐regulated PRRs between hepatoma tissues and the adjacent tissues are listed. (B) PGLYRP2 mRNA levels in 22 paired HCC tissues and their adjacent noncancerous liver tissues were detected by real‐time PCR assay. The results are reported as mean ± SEM. (C) Left panel, IHC results show PGLYRP2 protein expression in tumor‐adjacent noncancerous liver tissues and HCC tissues. Right panel, Statistical analysis showed PGLYRP2 IHC staining intensity in all of the tested tumor‐adjacent control and HCC tissues. (D) Tumor volume was calculated as 1/2 × tumor length × width2 and compared between PGLYRP2 high–expression and PGLYRP2 low–expression groups. The results are reported as mean ± SEM. (E,F) Kaplan‐Meier survival plots for PGLYRP2 staining were constructed according to the IHC staining intensity (E) and data from TCGA‐LIHC (F). Abbreviation: FC, fold change.
Among these genes, PGLYRP2 is the only PRR that is tissue‐restricted, has constitutively high expression toward the liver, and is significantly down‐regulated in HCC samples (P < 0.001) (Fig. 1A). To confirm the aberrant PGLYRP2 mRNA level in HCC tissues from the TCGA database, real‐time PCR assay was performed in 22 paired HCC tissues and their adjacent noncancerous liver tissues. A significant reduction of PGLYRP2 mRNA levels was observed in HCC tissues (P < 0.001) (Fig. 1B), which was consistent with the data from TCGA‐LIHC (Supporting Fig. S1A,B).
The protein levels of hepatic PGLYRP2 in HCC tissues were further examined by tissue‐array analysis. IHC results also showed that PGLYRP2 protein expression was dramatically reduced in 98 cases of HCC tissues compared with their adjacent nontumor tissues (Fig. 1C; Supporting Fig. S1C,D). PGLYRP2 expression levels in all tested tumor‐adjacent control cases were positive (39%, +++; 58%, ++; 3%, +), while its expression levels in HCC tissues were relative low (1%, +++; 36%, ++; 41%, +, 22%, −) (Fig. 1C; Supporting Fig. S1E). We conclude that PGLYRP2 expression is significantly decreased in HCC.
Association of PGLYRP2 Expression in HCC Tissues With Cancer Progression
In the cohort of HCC patients, the PGLYRP2 protein expression (IHC staining intensity) data set was split to create high‐expression (++, +++) and low‐expression (−, +) groups. Clinical variables were similar in the PGLYRP2 high–expression and PGLYRP2 low–expression groups, with the exception of tumor size (P = 0.008), histological grade (P = 0.011), and tumor–node–metastasis stage (P = 0.008) (Supporting Table S1). Furthermore, compared to that in the PGLYRP2 high–expression group, the tumor volume was significantly increased in the PGLYRP2 low–expression group (P < 0.001) (Fig. 1D). These results were further confirmed by assessing the relationship between neoplasm grades, stages, and expression levels of PGLYRP2 in patients of TCGA‐LIHC, which showed that the reduced levels of PGLYRP2 were associated with high histological grades and advanced stages of HCC (Supporting Fig. S1F).
Long‐term clinical follow‐up data were available in the HCC tissue array. The aforementioned gene expression data were, therefore, correlated with clinical information to determine whether PGLYRP2 expression levels predict the length of overall survival (OS) in HCC. Kaplan‐Meier survival plots for PGLYRP2 staining were constructed according to the IHC staining intensity. Higher expression level of PGLYRP2 was associated with improved OS (P < 0.001; hazard ratio [HR], 0.264; 95% confidence interval [CI], 0.144‐0.485) (Fig. 1E). Next, we used Kaplan‐Meier plotting to generate a validation cohort using LIHC data from TCGA. In this composite analysis of HCC patients, using the median value as the cutoff, we found that high levels of PGLYRP2 were significantly correlated with improved OS (P = 0.003; HR, 0.582; 95% CI, 0.408‐0.831) (Fig. 1F), which supported the observations with the tissue array cohort. These analyses revealed a trend toward a high level of PGLYRP2 and improved patient outcomes. Thus, the relatively high level of PGLYRP2 in HCC tissues correlates with a favorable prognosis in patients.
Aberrant Promoter Methylation and DNA Methyltransferase 3A are Responsible for the Reduced Level of PGLYRP2 in HCC Cells
We next investigated the underlying mechanism of the PGLYRP2 decrease in HCC. Lower expression of PGLYRP2 in HCC tissues was found at the mRNA level, suggesting a pretranslational mechanism. Therefore, we first retrieved methylation and mRNA expression data on PGLYRP2 from HCC tissue samples available in the TCGA database (cBioPortal).22, 23 Linear regression analysis demonstrated a significant negative correlation between PGLYRP2 promoter methylation and mRNA expression (P < 0.001, R = −0.557) (Fig. 2A), suggesting a pivotal regulatory role of promoter methylation on PGLYRP2 expression in HCC.
Figure 2.
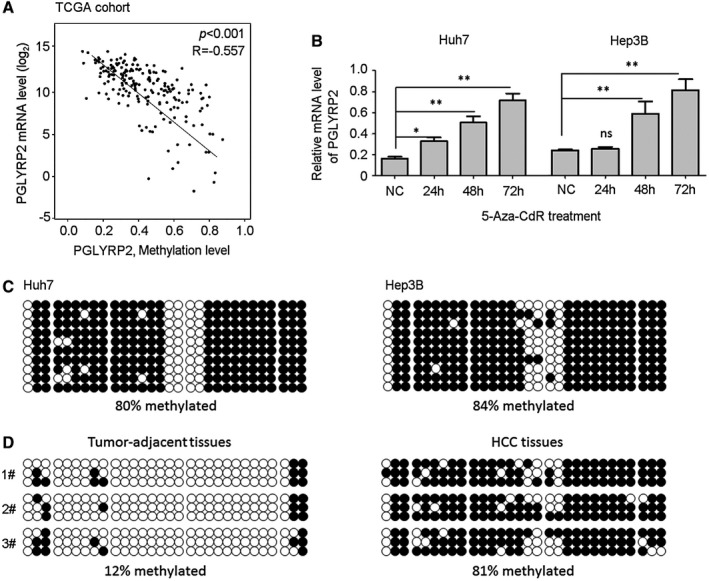
The PGLYRP2 promoter is hypermethylated in HCC cell lines and primary tumors. (A) Linear regression analysis showed a significant negative correlation between methylation level and mRNA expression of the PGLYRP2 gene from HCC tissue samples in the TCGA‐LIHC database. (B) Real‐time PCR results showed that PGLYRP2 levels were significantly elevated when Huh7 and Hep3B cells were treated with 5‐Aza‐CdR at 5 μM for the indicated times. The results represent at least three independent experiments and are reported as mean ± SEM. *P < 0.05, **P < 0.001. (C,D) The methylation status of the PGLYRP2 promoter region was analyzed in Huh7 and Hep3B cells (C) and three paired tumor‐adjacent noncancerous liver tissues and HCC tissues (D) using bisulfate sequencing. Abbreviations: NC, negative control; ns, not significant.
To analyze the methylation status of the PGLYRP2 promoter in HCC cells, Huh7 and Hep3B cells were treated with the DNA demethylating agent 5‐Aza‐2′‐deoxycytidine (5‐Aza‐CdR). PGLYRP2 levels were significantly elevated when both cell lines were treated with 5‐Aza‐CdR at 5 μM for 48 to 72 hours (Fig. 2B). We further characterized the methylation status of the PGLYRP2 promoter region using bisulfate sequencing. The results showed that the PGLYRP2 promoter was hypermethylated in both HCC cells. The methylation level was 80% in Huh7 cells and 84% in Hep3B cells (Fig. 2C). As expected, drastic differences in the overall methylation level were observed between HCC tissues and tumor‐adjacent tissues. The methylation level in HCC tissues was 81%, while in tumor‐adjacent tissues it was reduced to 12% (Fig. 2D). The methylation status of the PGLYRP2 promoter in the three HCC tissues and their corresponding adjacent tissues was negatively associated with the expression levels of PGLYRP2 (Supporting Fig. S2A).
As DNA methylation is catalyzed by members of DNA methyltransferase family including the maintenance enzyme DNA methyltransferase 1 (DNMT 1) and de novo methyltransferases DNMT 3A and DNMT 3B, we examined PGLYRP2 levels in DNMT 1–silenced, DNMT 3A–silenced, or DNMT 3B–silenced Huh7 and Hep3B cells. Real‐time PCR and western blotting results showed that knockdown of DNMT 3A in Huh7 and Hep3B cells significantly reduced the methylation levels of the PGLYRP2 promoter and elevated the expression levels of PGLYRP2 (Fig. 3A,B; Supporting Fig. S2B‐D), suggesting that DNMT 3A was responsible for the aberrant promoter methylation of PGLYRP2 gene in HCC cells. We next assessed whether overexpression of DNMT 3A inhibited PGLYRP2 expression through hypermethylation of its promoter. As the expression levels of PGLYRP2 in both human and mouse HCC cell lines were extremely low (Supporting Fig. S2E‐H), we analyzed the effect of DNMT 3A overexpression on PGLYRP2 promoter activity and methylation status in HEK293/PGLYRP2 promoter luciferase reporter cells. DNMT 3A overexpression in HEK293 cells significantly reduced the PGLYRP2 promoter activity, which was consistent with the elevated promoter activity by DNMT 3A knockdown in Huh7 cells (Supporting Fig. S2I). The methylation levels of PGLYRP2 promoter in control or DNMT 3A–overexpressed HEK293/PGLYRP2 promoter luciferase reporter cells were analyzed using bisulfate sequencing. As expected, DNMT 3A overexpression dramatically increased the methylation level of PGLYRP2 promoter in HEK293/PGLYRP2 promoter cells (Supporting Fig. S2J).
Figure 3.
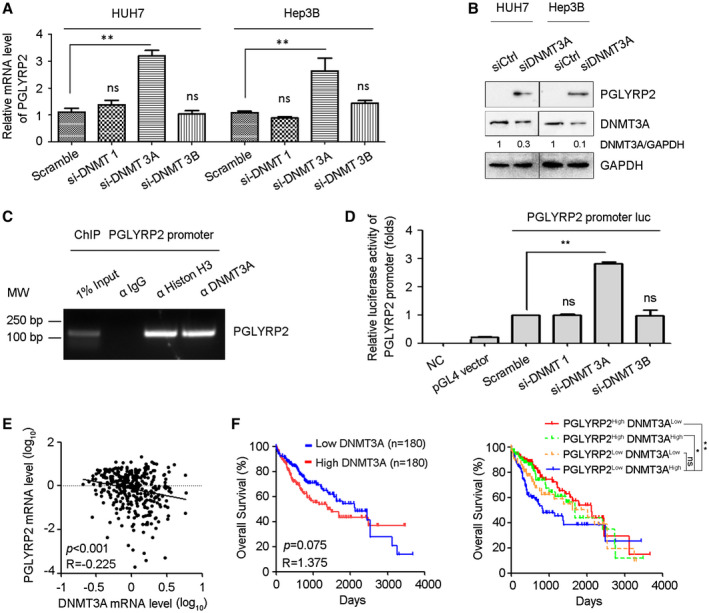
DNMT 3A mediates promoter hypermethylation of PGLYRP2 in HCC. (A) PGLYRP2 levels were examined in DNMT 1–silenced, DNMT 3A–silenced, or DNMT 3B–silenced Huh7 and Hep3B cells by real‐time PCR. The results are reported as mean ± SEM. (B) Western blotting results showed that knockdown of DNMT 3A in Huh7 and Hep3B cells significantly elevated the level of PGLYRP2. (C) The binding activity of DNMT 3A to the PGLYRP2 promoter was assessed by ChIP assay in Huh7 cells. (D) Relative luciferase activity in Huh7 cells transfected with plasmids of pGL4 vector, PGLYRP2 promoter luciferase reporter, scramble siRNA, DNMT 1 siRNA, DNMT 3A siRNA, or DNMT 3B siRNA, alone or in their combinations. The results are reported as mean ± SEM. (E) Confidence ellipse analysis showed that PGLYRP2 levels were inversely correlated with DNMT 3A expression in HCC tissues from TCGA‐LIHC. (F) Kaplan‐Meier analysis showed that the patients from TCGA‐LIHC who expressed low levels of DNMT 3A had a slightly longer OS (left panel), while patients with low DNMT 3A and high PGLYRP2 expression in tumors had a significantly improved OS compared to those in the high–DNMT 3A and low–PGLYRP2 expression group (right panel). *P < 0.05, **P < 0.001. Abbreviations: GAPDH, glyceraldehyde phosphate dehydrogenase; IgG, immunoglobulin G; luc, luciferase; MW, molecular weight; NC, negative control; ns, not significant.
By chromatin immunoprecipitation (ChIP) assay, we analyzed whether DNMT 3A regulated the transcription of PGLYRP2 by binding to its promoter region. DNMT 3A exhibited strong ability to bind to PGLYRP2 promoter in Huh7 cells, while DNMT 1 and DNMT 3B did not show any binding capacity (Fig. 3C; Supporting Fig. S2K). We further investigated whether DNMT 3A regulated the transcription of PGLYRP2 when binding to the promoter regions of PGLYRP2 using a luciferase reporter assay. When DNMT 3A small interfering RNA (siRNA) was transfected, the transcription activity of the PGLYRP2 promoter in Huh7 cells was obviously increased, suggesting that DNMT 3A–mediated promoter methylation regulated the transcription of PGLYRP2 by binding to its promoter regions (Fig. 3D). Then, based on the results of bisulfate sequencing, we chose four regions in the PGLYRP2 promoter to analyze the DNMT 3A binding regions by ChIP assay. Three regions (R1, −1741 to −1491; R2, −1516 to −1362; R4, −440 to −315), but not the R3 region (−736 to −614), from the PGLYRP2 promoter were immunoprecipitated by anti–DNMT 3A (Supporting Fig. S2K). This indicated that the R3 region was not located in the DNMT 3A binding region, which was consistent with the low methylation level of the R3 region as shown in the bisulfite sequencing assay (Fig. 2C).
To further evaluate the physiological relevance of PGLYRP2 and DNMT 3A, we performed in silico analyses of PGLYRP2 and DNMT 3A expression using the TCGA‐LIHC data set. Confidence ellipse analysis results indicated that PGLYRP2 levels were inversely correlated with DNMT 3A expression in HCC tissues (P < 0.001, R = −0.225) (Fig. 3E). Furthermore, Kaplan‐Meier analysis was conducted to determine whether the OS of patients was associated with the DNMT 3A–regulated PGLYRP2 expression in HCC tissues. Expression of PGLYRP2 and DNMT 3A was used to assign patients to the high‐expression (upper 50%) or low‐expression (lower 50%) group. Kaplan‐Meier analysis indicated that patients with tumors that expressed low levels of DNMT 3A had a slightly longer OS (P = 0.075; HR, 1.375; 95% CI, 0.968‐1.954), while patients with low DNMT 3A/high PGLYRP2 expression in tumors had a significantly improved OS compared to those in the high–DNMT 3A/low‐PGLYRP2 expression group (P < 0.001; HR, 0.494; 95% CI, 0.317‐0.769) (Fig. 3F). In addition, high–DNMT 3A/high‐PGLYRP2 and low–DNMT 3A/low‐PGLYRP2 samples were included in our survival analysis. As expected, when compared with the high–DNMT 3A/low‐PGLYRP2 expression group, patients with high DNMT 3A/high PGLYRP2 expression in tumors also showed a significant improvement in OS (P = 0.007; HR, 1.619; 95% CI, 1.015‐2.580) but more modest benefit on OS than patients with low–DNMT 3A/high‐PGLYRP2 expression tumors. Patients with low–DNMT 3A/low‐PGLYRP2‐expressing tumors did not show significantly better OS than the high–DNMT 3A/low‐PGLYRP2 expression group (P = 0.147; HR, 1.325; 95% CI, 0.838‐2.093) (Fig. 3F). Together, these findings suggest that the methylation status of the PGLYRP2 promoter and DNMT 3A levels regulate PGLYRP2 signaling in HCC.
PGLYRP2 Promotes Antitumor Immune Response In Vivo
Tumor progression partially depends on the escape from immunosurveillance. The effect of PGLYRP2 on cell proliferation was excluded using cell colony formation and the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay (Supporting Fig. S3A,B). Considering PGLYRP2 acts as an innate immune molecule, we wondered whether PGLYRP2 silencing in tumor cells would attenuate the host immune response against HCC. To evaluate the relationship between PGLYRP2 and the host immune response, in vivo experiments were performed using two types of mice, BALB/c‐nu/nu and BALB/c mice; and subcutaneous tumor and orthotopic HCC mouse models were established. Human cancer cell lines Huh7/con Huh7/PGLYRP2 and murine cancer cell lines Hepa1‐6/con and Hepa1‐6/PGLYRP2 were subcutaneously inoculated into immunosuppressed BALB/c‐nu/nu mice and immune‐competent BALB/c mice, respectively. PGLYRP2 expression levels in Huh7/PGLYRP2, Hepa1‐6/PGLYRP2, and the corresponding control cells were examined by western blotting (Supporting Fig. S3C). Tumor size was recorded every 5 days, and tumor weight was measured after the mice were sacrificed at the end of the experiment. PGLYRP2 expression exerted no effects on tumor volume and tumor weight in immune‐deficient BALB/c‐nu/nu mice (Fig. 4A,B; Supporting Fig. S3D‐G). Conversely, tumor volume and tumor weight were significantly reduced in immune‐competent BALB/c mice with tumors that stably expressed PGLYRP2 when compared with BALB/c mice with control tumors (Fig. 4C,D; Supporting Fig. S3H,I). Consistently, liver orthotopic xenograft tumor also showed that PGLYRP2 inhibited tumor growth in vivo, whereas PGLYRP2‐deficient tumor cells exhibited aggressive growth (P = 0.004) (Fig. 4E). These results show that PGLYRP2 suppresses HCC progression in immune‐competent mice but not in immune‐deficient mice, suggesting its antitumor role in modulating host immune response.
Figure 4.
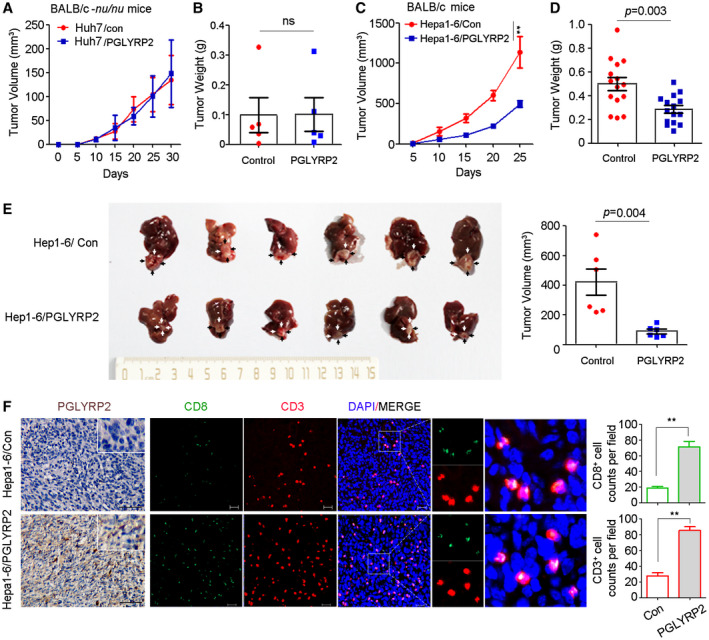
PGLYRP2 suppresses HCC progression in immune‐competent mice but not in immune‐deficient mice. (A‐D) Tumor volume and tumor weight of subcutaneous tumors from immune‐deficient BALB/c‐nu/nu mice (A,B) and immune‐competent BALB/c mice (C,D) are shown. (E) Orthotopic tumors from the indicated groups are shown in the left panel (black or white arrows indicate tumors); tumor volumes are shown in the right panel. (F) IHC staining of PGLYRP2 expression and immunofluorescent IHC staining of CD3 and CD8 were performed on paraffin‐embedded tumor tissue. Scale bar, 100 μm for IHC; 20 μm for immunofluorescence. Statistical analysis showed CD3+ and CD8+ cell counts per field in immunofluorescent IHC staining. All results are reported as mean ± SEM. *P < 0.05, **P < 0.001. Abbreviations: DAPI, 4´,6‐diamidino‐2‐phenylindole; ns, nonsignificant.
Intratumoral cluster of differentiation 3–positive (CD3+), CD8+ T‐cell infiltrates are predictive of response to immunotherapeutic interventions and associate favorably with patient survival.24 To analyze the essential relationship between the antitumor immune response and PGLYRP2 level in tumor tissues, quantitative assessment of CD3 and CD8 in paraffin‐embedded tumor tissue was performed using immunofluorescence assay. When compared with the control tumors in immune‐competent BALB/c mice, there was an expected increment of total CD3+ and CD8+ T cells in tumors with PGLYRP2 expression (Fig. 4F; Supporting Fig. S3J). Overexpression of PGLYRP2 in tumor cells significantly promoted tumor accumulation of CD3+ and CD8+ T cells in tumor tissues (P < 0.001). Therefore, PGLYRP2 contributes to enhancing the antitumor immune response in HCC through turning cold tumors hot.
PGLYRP2 Expression Correlates With Activated TILs in HCC Patients
The presence of activated tumor‐infiltrating immune cells in tumor tissues is known to correlate with better overall outcomes, most likely due to strong antitumor immune responses. We therefore investigated whether PGLYRP2 expression correlated with activated immune cell infiltration using a cohort of 22 primary HCC samples (Fig. 5A). A quantitative RT‐PCR array was used to assess PGLYRP2 mRNA levels of HCC tissues. In addition, the mRNA levels of several established immune markers were quantified, including C2/C3 (complement components), CD3G (total T cells), CD4 (helper T cells), CD8A (cytotoxic T cells), forkhead box P3 (FoxP3; regulatory T cells [Tregs]), CD11b (myeloid‐derived suppressor cells [MDSCs]), CD14/CD68 (monocyte/macrophage markers), CD22 (pan‐B cells; Supporting Fig. S4A, primers in Supporting Table S3).
Figure 5.
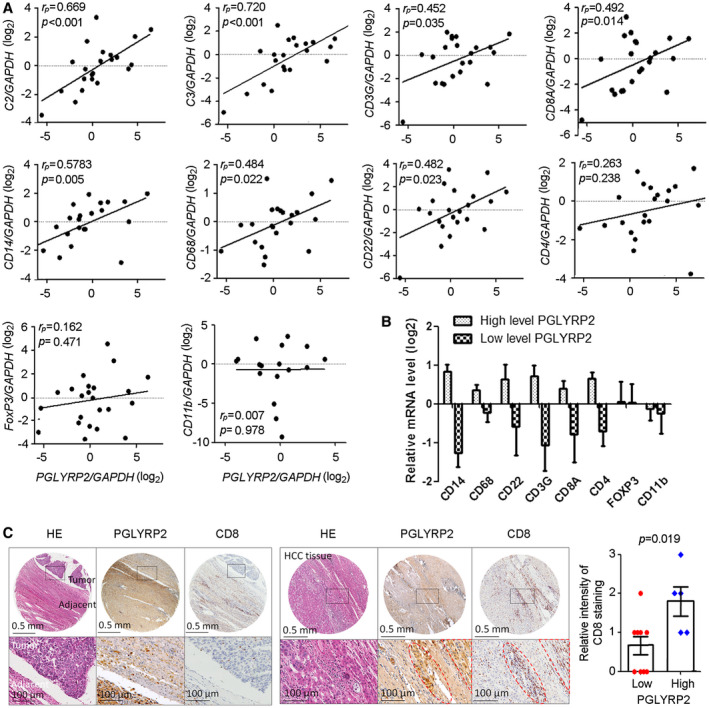
PGLYRP2 mRNA expression level correlates with activated immune cell infiltration in HCC. (A) PGLYRP2 expression in 22 primary HCC samples was detected using a quantitative RT‐PCR assay. The mRNA levels of several established immune markers were quantified, and their association with PGLYRP2 level was analyzed. The markers included complement components markers C2 and C3, total or cytotoxic T‐cell markers CD3G and CD8A, monocyte/macrophage markers CD14 and CD68, pan‐B‐cell marker CD22, helper T‐cell marker CD4, Treg‐cell marker FoxP3, and MDSC marker CD11b. (B) PGLYRP2 mRNA levels in the real‐time PCR data set were split based on a median value, and the mRNA levels of immune cells were analyzed in the high–PGLYRP2 expression or low–PGLYRP2 expression group. (C) IHC assays showed PGLYRP2 and CD8 protein expression in HCC tissues. Statistical analysis showed the relative intensity of CD8 IHC staining in high–PGLYRP2 expression (++, +++; n = 5) and low–PGLYRP2 expression (−, +; n = 9) HCC tissues. Results are expressed as the mean ± SEM. P > 0.05 indicates that there is no significant difference. Abbreviations: GAPDH, glyceraldehyde phosphate dehydrogenase; HE, hematoxylin and eosin.
Strong positive correlations were observed between PGLYRP2 mRNA expression levels and C2 (P < 0.001, rP = 0.669), C3 (P < 0.001, rP = 0.720), CD14 (P = 0.005, rP = 0.578), CD3G (P = 0.035, rP = 0.452), CD8A (P = 0.014, rP = 0.492), CD68 (P = 0.022, rP = 0.484), and CD22 (P = 0.023, rP = 0.482). The exceptions were CD11b (P = 0.978, rP = 0.007), CD4 (P = 0.238, rP = 0.263), and FoxP3 (P = 0.471, rP = 0.162) (Fig. 5A), which linked with suppressive function of MDSCs and CD4+ Tregs. In the real‐time PCR data set, based on a median split, most markers of immune effectors were in the PGLYRP2 high–expression group, except the MDSC marker CD11b and the Treg marker FoxP3 (Fig. 5B), further suggesting that the positive signals emanate from bona fide activated antitumor immunity. The correlation between PGLYRP2 mRNA level and immune cell markers was further confirmed in the TCGA cohort (Supporting Fig. S4B). Thus, these data indicated that PGLYRP2 expression was associated with tumor accumulation of antitumoral TILs but not recruitment of immune‐suppressive MDSCs and Tregs.
To confirm the correlation between PGLYRP2 expression and the accumulation of TILs in HCC tissues, IHC staining of PGLYRP2 and CD8 was carried out in 14 HCC tissues. The HCC tissues were split into groups with high (++, +++; n = 5) and low (−, +; n = 9) PGLYRP2 expression. Compared with that in high‐PGLYRP2‐expressing HCC tissues, the intensity of CD8 staining was much weaker in low‐PGLYRP2‐expressing tissues (P = 0.019) (Fig. 5C; Supporting Fig. S4C).
PGLYRP2 Induces Expression of Chemokine (C‐C MOTIF) Ligand 5 in HCC
Considering that chemokines were responsible for recruitment of TILs, the relationship between PGLYRP2 and chemokines expression was analyzed. In a chemokine protein array, chemokine expression levels were detected in HCC tissue lysates and cell culture supernatants. Analysis of multiple analytes in tissue lysates by chemokine array demonstrated elevated chemokine (C‐C motif) ligand 5 (CCL5) levels as well as decreased levels of chemokine (C‐X‐C motif) ligand 4 (CXCL4), CXCL7, CXCL12, CCL21, Midkine, IL‐16, and Chemerin in lysate of high‐PGLYRP2‐expressing HCC tissue compared with those in lysate of low‐PGLYRP2‐expressing HCC tissue (Fig. 6A). The expression level of PGLYRP2 in HCC tissues was detected by IHC staining (Fig. 6A). In addition to the lysis of PGLYRP2‐sufficient HCC tissues, the supernatant of the stable cell line Huh7/PGLYRP2 displayed a higher amount of secreted CCL5 than that of Huh7/Con cells. The high level of CCL5 expression in Huh7/PGLYRP2 cells was further confirmed by real‐time PCR. A significant difference in CCL5 level was observed between Huh7/Con and Huh7/PGLYRP2 cells cultured in vitro, suggesting that tumor cells themselves rather than tumor‐infiltrating cells are the main source of this chemokine in vivo. Thus, these results indicated that tumor‐derived PGLYRP2 induced expression of CCL5 in HCC cells.
Figure 6.
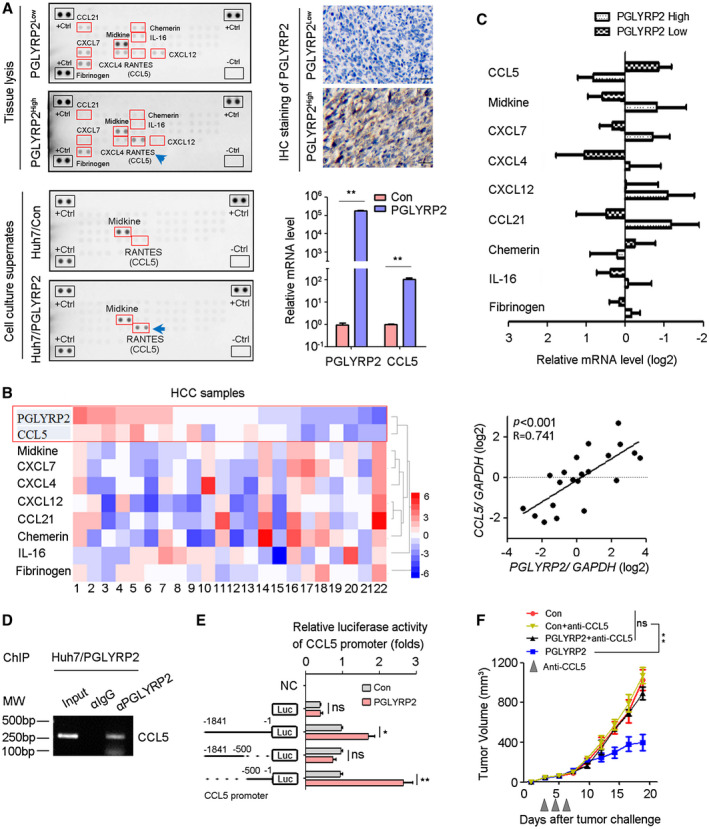
PGLYRP2 promotes chemokine CCL5 expression in HCC. (A) Chemokine levels in HCC tissue lysate and supernatants of Huh7 cells were detected using a human chemokine protein array. The PGLYRP2 expression level in HCC tissues was measured by IHC. Scale bars, 100 μm. Expression levels of PGLYRP2 and CCL5 in Huh7/Con and Huh7/PGLYRP2 cells were detected by real‐time PCR. (B,C) Expression of PGLYRP2 and chemokine was detected in 19 HCC samples using a quantitative RT‐PCR assay. Heat map showed different expression of chemokines in 19 HCC tissues (B, left panel). The association between PGLYRP2 and CCL5 level in HCC samples was analyzed (B, right panel). The mRNA levels of chemokines were analyzed in the high–PGLYRP2 expression and low–PGLYRP2 expression groups (C). (D) The binding activity of PGLYRP2 to the CCL5 promoter was assessed by ChIP assay in Huh7/PGLYRP2 cells. (E) Relative luciferase activity in Huh7/Con and Huh7/PGLYRP2 cells transfected with plasmids of pGL4 vector, PGLYRP2 promoter (–1841 to –1 bp) luciferase reporter, PGLYRP2 promoter (–1841 to –500 bp) luciferase reporter, or PGLYRP2 promoter (–500 to –1 bp) luciferase reporter alone. (F) Tumor volume of Hepa 1‐6/control or Hepa 1‐6/PGLYRP2 tumors in immunocompetent BALB/c mice and anti‐CCL5‐treated BALB/c mice. Mice that did not receive anti‐CCL5 treatment were treated with an equivalent concentration of immunoglobulin G isotype control antibody. All results are reported as mean ± SEM. *P < 0.05, **P < 0.001. Abbreviations: Ctrl, control; IgG, immunoglobulin G; Luc, luciferase; MW, molecular weight; NC, negative control; ns, not significant; RANTES, regulated upon activation, normal T cell expressed, and secreted.
The correlation between PGLYRP2 and chemokine expression was further confirmed in 22 HCC samples using a complementary DNA (cDNA) array. The data showed that there was a higher mRNA level of CCL5 in high‐PGLYRP2‐expressing tumor tissues than in low‐PGLYRP2‐expressing tumor tissues (Fig. 6B,C) and that CCL5 expression in HCC tissues showed a significantly positive correlation with PGLYRP2 level (P < 0.001, R = 0.741, 95% CI, 0.454‐0.888) (Fig. 6B). We next analyzed the correlation between CCL5 and PGLYRP2 mRNA levels in the TCGA‐LIHC cohort. The data showed that there was only a moderate correlation between CCL5 and PGLYRP2 in the TCGA cohort (P = 0.05, R = 0.1; Supporting Fig. S5A), which was not consistent with our cDNA array result. The main and initial difference between the two experiments is the source of HCC samples. Patients with HCC and viral hepatitis accounted for >68% (15/22) in all of the samples of the cDNA array, while there were few hepatitis virus–infected patients with HCC in the TCGA data set. Considering that virus‐activated nuclear factor kappa B (NF‐κB) p65/RELA is the major upstream transcription factor of CCL5, we split the TCGA‐LIHC samples into high‐RELA and low‐RELA expression groups (50%:50%). There was no correlation between CCL5 and RELA levels in the TCGA cohort (P = 0.307, R = 0.05; Supporting Fig. S5B). In the low‐RELA group, there was no correlation between CCL5 and PGLYRP2 (P = 0.166, R = −0.104; Supporting Fig. S5C), while a significant correlation was observed in the high‐RELA group (P = 0.029, R = 0.162; Supporting Fig. S5D). These results indicated that PGLYRP2‐regulated CCL5 expression partially relied on NF‐κB p65 (RELA).
An immunofluorescence assay showed that PGLYRP2 partly localized in the nucleus (Supporting Fig. S5 E). Therefore, we analyzed whether PGLYRP2 regulated the transcription of CCL5 by binding to its promoter region by ChIP assay. PGLYRP2 exhibited its ability to bind to the CCL5 promoter in Huh7/PGLYRP2 cells (Fig. 6D; Supporting Fig. S5F). We further investigated whether PGLYRP2 regulated the transcriptional activity of CCL5 using a luciferase reporter assay. The data showed that transfection of the luciferase reporter containing CCL5 promoter sequences from −1,841 to −1 bp or −500 to −1 bp led to higher promoter activity in Huh7/PGLYRP2 cells than that in Huh7/control. Meanwhile, transfection of the luciferase reporter containing CCL5 promoter sequences from −1,841 to −500 bp did not result in significantly higher activity in Huh7/PGLYRP2 cells (Fig. 6E).
Next, we assessed if PGLYRP2‐mediated CCL5 expression was responsible for tumor regression observed in immunocompetent BALB/c mice with PGLYRP2‐sufficient tumors. The volume of Hepa1‐6/control or Hepa1‐6/PGLYRP2 tumors in immunocompetent BALB/c mice and anti‐CCL5‐treated BALB/c mice was analyzed. PGLYRP2 expression and immune infiltration were examined using IHC staining (Supporting Fig. S5G). When CCL5 was blocked by intratumoral administration of anti‐CCL5 blocking antibody, tumor regression was abrogated in mice with PGLYRP2‐overexpressing tumors (P < 0.001) (Fig. 6F).
PGLYRP2 Improves In Vitro and In Vivo Antitumor Immune Response
PBMCs contain a mixture of natural killer (NK) cells, monocytes, T cells, and B cells. To investigate the effect of PGLYRP2 on the antitumor immune response rates of PBMC cells, an ex vivo study was carried out in PBMC tumor cell coculture system. PBMCs were isolated from healthy donors and then incubated with target cells (stably transfected Huh7/Con or Huh7/PGLYRP2) at an effector to target (E:T) ratio of 5:1 (Fig. 7A). After 12 or 24 hours of incubation, PBMCs were removed from the coculture system, and the tumor‐killing activity of PBMC cells on tumor cells was determined using cell counts and the MTT assay. The data showed that the number of tumor cells killed by PBMCs in the PGLYRP2 group was much higher than that in the control group (P < 0.001) (Fig. 7B), suggesting that overexpression of PGLYRP2 significantly enhanced the antitumor immune response rates of PBMCs.
Figure 7.
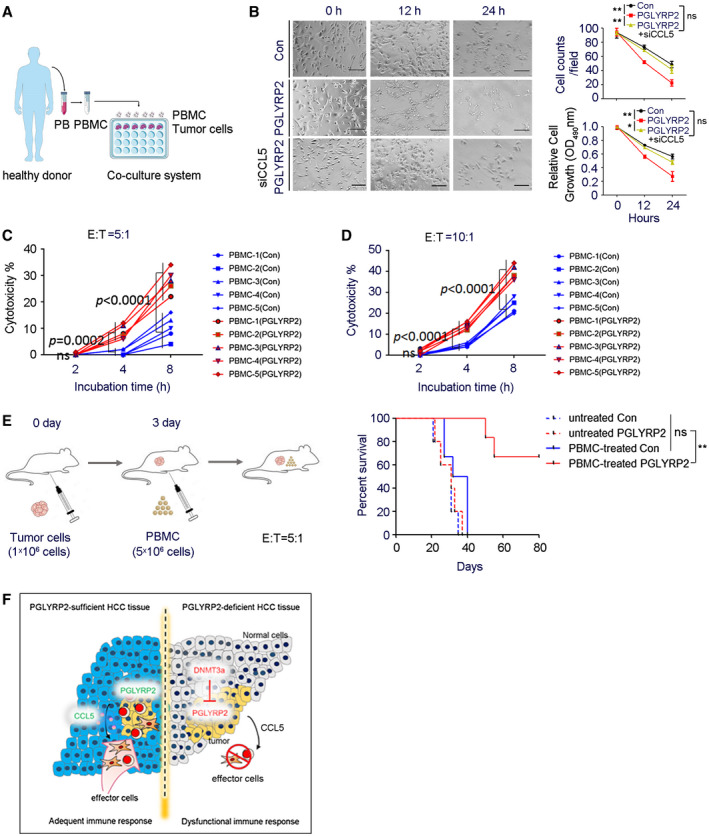
PGLYRP2 elevates antitumor activity of PBMC. (A) Model of PBMC–tumor cell coculture system. (B) PBMCs were incubated with stable Huh7/Con, Huh7/PGLYRP2, or Huh7/PGLYRP2/siCCL5 cells at an E:T ratio of 5:1 for the indicated incubation times. Scale bar, 300 μm. After 12 or 24 hours of incubation, PBMCs were removed from the cell culture plates, and the antitumor activity of PBMCs on tumor cells was determined using cell counts and MTT assay. **P < 0.001. (C,D) The effect of PGLYRP2 expression on the antitumor activity of PBMCs was analyzed by incubating target Huh7/Con or Huh7/PGLYRP2 cells with five independent PBMC donors for 2, 4, and 8 hours at an E:T ratio of 5:1 (C) or 10:1 (D). (E) Left, Evaluation model of antitumor activity of PBMCs in Huh7 xenografted BALB/c‐nu/nu mice. Right, Survival of mice in untreated or PBMC‐treated Huh7/Con or Huh7/PGLYRP2 group (n = 5) is shown. (F) Working model of PGLYRP2‐mediated antitumor immune response against HCC. All results are expressed as mean ± SEM. *P < 0.05, **P < 0.001. Abbreviations: ns, not significant; OD, optical density; PB, peripheral blood.
The expression levels of PGLYRP2 were extremely low in HCC cell lines, including human HCC cell lines C3A, Huh7, HepG2, Hep3B, and SNU387 and mouse HCC cell line Hepa 1‐6 (Supporting Fig. S2E‐H). CCL5 levels were increased in stable cell lines Huh7/PGLYRP2 and Hepa 1‐6/PGLYRP2, while knockdown of PGLYRP2 inhibited CCL5 expression in stable cell lines Huh7/PGLYRP2 and Hepa 1‐6/PGLYRP2 (Supporting Fig. S6A‐C). The increased tumor‐killing activity and antitumor immune response could further be abrogated by knockdown of CCL5 or PGLYRP2 in stable Huh7/PGLYRP2 cells (Fig. 7B; Supporting Fig. S6D). This indicated that the PGLYRP2‐elevated tumor‐killing activity of immune cells was dependent on the expression of CCL5.
To identify the immune effector cells of PGLYRP2‐mediated antitumor immunity, depletion of immune cells in immunocompetent BALB/c mice was performed by intraperitoneally administering depleting antibody prior to tumor injection. The data showed that depletion of NK and CD8+ T cells significantly decreased tumor growth compared to the undepleted Hepa 1‐6/PGLYRP2 group, while depletion of CD4+ T cells did not have an obvious impact on tumor growth. The depletion of immune cells was confirmed using flow‐cytometric analysis (Supporting Fig. S7A).
A cytotoxicity assay25 was performed by incubating target cells with PBMCs for 2, 4, and 8 hours at indicated E:T ratios. The PBMCs were from five independent healthy donors, and the tumor‐killing activity of PBMCs was verified in a PBMC–K562 coculture system (Supporting Fig. S7B). Assessment with the five independent PBMCs showed significant enhancement of immune response rates by PGLYRP2 after 4 hours of incubation (P < 0.001) and that PGLYRP2 enhanced PBMC‐mediated killing of tumor cells in an E:T ratio–dependent manner (Fig. 7C,D). The cytotoxicity assay was further confirmed by incubating tumor cells with the NK cell line NK‐92 or cultured NK cells from PBMCs of healthy donors (Supporting Fig. S7C‐E). Subsets of PBMCs and cultured NK cells from healthy donors were identified by flow cytometry (Supporting Fig. S7F,G). The PGLYRP2‐enhanced cytotoxicity could be significantly decreased when CCL5 blocking antibody was added into the coculture system (Supporting Fig. S6E), which further confirmed the role of the PGLYRP2/CCL5 axis in regulating the tumor‐killing activity of immune cells.
Finally, the PGLYRP2‐elevated antitumor immune response of PBMCs was assessed in Huh7 xenografted BALB/c‐nu/nu mice. A total of 1 × 106 Huh7/Con or Huh7/PGLYRP2 cells were injected intraperitoneally into nude mice, and 3 days later 5 × 106 isolated PBMCs from healthy donors were intraperitoneally injected (n = 5). The survival time of mice was monitored, and the status until day 80 is shown in Fig. 7E. All mice in the Huh7/Con+PBMC group died between day 27 and day 40 following tumor transplantation; in contrast, mice in the Huh7/PGLYRP2+PBMC group died after 50 days post–tumor inoculation, and 3/5 mice survived throughout the study and were censored at the end. In mice with PGLYRP2‐overexpressing tumor, a significantly prolonged survival rate was achieved (P < 0.001), while PGLYRP2 knockdown in stable Huh7/PGLYRP2 cells led to a poor survival rate similar to that of the Huh7/Con group (Supporting Fig. S6F). Therefore, these data suggest that PGLYRP2 has the potential to inhibit tumor growth by improving the response rates of immune effectors.
Discussion
Harnessing the immune system against cancer has been an effective therapy option. The identification of clinical features and biomarkers would allow better patient selection for individual immunotherapy.26 Our studies have led to the surprising identification of innate immune receptor PGLYRP2 as a candidate biomarker for adequate immune response against HCC and improved patient outcomes. Unlike other PGRP family members, prior work has shown that PGLYRP2 is restrictively expressed constitutively in liver tissue.15 Analysis of a cohort of 98 HCC patients revealed that the PGLYRP2 level was dramatically reduced in HCC tissues compared to adjacent noncancerous tissues. These results were validated at the mRNA level by real‐time PCR and at the protein level by IHC (Fig. 1B,C), which was also confirmed by the RNAseq data from TCGA‐LIHC and proteomic data from the Chinese Human Proteome Project27 (Supporting Fig. S1A,B,G). Strong positive correlations were observed between PGLYRP2 level and several established immune markers, including complement components and TILs (Fig. 5A). Clinical data further showed that PGLYRP2 was also associated with improved outcomes of patients with HCC (Fig. 1E,F).
Substantial evidence has indicated that certain chemokines and complement components contribute to the recruitment and activation of lymphocytes.28, 29, 30, 31, 32, 33 Our data indicate that the increased level of CCL5 is induced by PGLYRP2 (Fig. 6A), while the elevated amount of complement components is not mediated by PGLYRP2 (data not shown), which might be from the chemokines–responsive lymphocytes in the tumor microenvironment. CCL5 has been found to recruit immune effectors into the tumor microenvironment and thus promote cancer immune control32 (Supporting Fig. S5I). Given that PGLYRP2 is restrictively expressed toward the liver, the fact that during a local tumor immune response PGLYRP2 deficiency in HCC tissues mediated local, but not systemic, reduction in CCL5 production may consequentially alter the TILs population in HCC (Fig. 7F). Therefore, it is possible that the early innate immune responses mediated, at least in part, by PGLYRP2 orchestrate the generation of adaptive immune responses by regulating the chemoattractant signals.
PGLYRP2 is a nonclassic soluble PRR secreted in plasma, which belongs to a unique family of antimicrobial proteins.34 PGLYRP2 has N‐acetyl‐muramoyl‐l‐alanine amidase activity that hydrolyzes peptidoglycan from gram‐positive bacteria, but PGLYRP2 did not show direct bactericidal activity in host defense to bacterial infection.16 In addition to the soluble, secreted form of PGLYRP2, we discovered that parts of PGLYRP2 are localized in the nucleus (Supporting Fig. S5E) and that the nuclear PGLYRP2 induces CCL5 expression (Fig. 6A,D,E), suggesting that PGLYRP2 plays a certain role in nucleus beyond the classical immune functions. Considering that the liver maintains a state of immune homeostasis in normal physiological conditions, based on previous reports and our data, we deduce that normally PGLYRP2 is secreted into the blood from the liver to perform the function of immune surveillance but that under certain stresses, such as the cancer microenvironment, parts of them are translocated from the cytoplasm into the nucleus, and thus regulate the immune response.
DNMT 3A–mediated de novo DNA methylation is essential for genome regulation. Our data indicated that DNMT 3A indirectly regulated CCL5 expression, but no association was found between DNMT 3A and CCL5 mRNA level in the TCGA cohort (Supporting Fig. S5H). DNMT 3A is crucial for global methylation; that is to say, DNMT 3A regulates global gene expression, including that of the PGLYRP2 gene, while CCL5 gene expression is regulated by PGLYRP2 but also by multiple transcription factors, such as NF‐κB. Therefore, we deduced that DNMT 3A was required for regulation of PGLYRP2 expression in HCC cells but not necessary for regulation of CCL5 expression.
In this study, PGLYRP2 was identified as an antitumor innate immune molecule. We found that PGLYRP2 was epigenetically altered and transcriptionally down‐regulated in HCC cells. Chi‐squared tests and Cochran‐Armitage trend were used to identify associations between PGLYRP2 and TILs (Fig. 5A). PGLYRP2 further showed promising induction of PBMC‐mediated killing of tumor cells. But PGLYRP2 did not directly inhibit cancer cell proliferation or induce HCC cell apoptosis in vitro (Supporting Figs. S3A and S8). This evidence indicated the importance of PGLYRP2 in tumor immune surveillance and in designing immunotherapeutic approaches. Therefore, we identified a relationship between PGLYRP2 and the antitumor immune response, in which PGLYRP2 acted as a functional bridge between innate and adaptive immune responses.
Supporting information
Acknowledgment
We thank Dr. Shan Wang (University of Texas Southwestern Medical Center) and Professor Lanfen Chen (Xiamen University) for useful discussion. We also thank Professor Yu Li, Mr. Peng Dou, Mr. Yao Zhang, and Ms. Yue Song (Harbin Institute of Technology) for technical help.
Supported by the National Natural Science Foundation of China (31870891 and 81502781), the China Scholarship Council (CSC 201806125051), and China's Manned Space Preresearch Project (17420205).
Potential conflict of interest: Nothing to report.
Contributor Information
Hongjin Wu, Email: wuhongjin@hit.edu.cn.
Ming Shi, Email: shiming@hit.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Gandhi L, Rodriguez‐Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non‐small‐cell lung cancer. N Engl J Med 2018;378:2078‐2092. [DOI] [PubMed] [Google Scholar]
- 2. Gettinger SN, Choi J, Mani N, Sanmamed MF, Datar I, Sowell R, et al. A dormant TIL phenotype defines non‐small cell lung carcinomas sensitive to immune checkpoint blockers. Nat Commun 2018;9:3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res 2017;27:96‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 2013;39:74‐88. [DOI] [PubMed] [Google Scholar]
- 5. Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, Slingluff CL Jr. Immune cell infiltration and tertiary lymphoid structures as determinants of antitumor immunity. J Immunol 2018;200:432‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune checkpoint blockade. Clin Cancer Res 2019;25:1612‐1623. [DOI] [PubMed] [Google Scholar]
- 7. Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales‐Puchalt A, et al. Microbially driven TLR5‐dependent signaling governs distal malignant progression through tumor‐promoting inflammation. Cancer Cell 2015;27:27‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41:830‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klein JC, Moses K, Zelinskyy G, Sody S, Buer J, Lang S, et al. Combined toll‐like receptor 3/7/9 deficiency on host cells results in T‐cell‐dependent control of tumour growth. Nat Commun 2017;8:14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corrales L, McWhirter SM, Dubensky TW Jr., Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest 2016;126:2404‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565‐1570. [DOI] [PubMed] [Google Scholar]
- 12. Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang RF. Tumor‐infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll‐like receptor signaling pathway. Immunity 2007;27:334‐348. [DOI] [PubMed] [Google Scholar]
- 13. Li X, Wang S, Wang H, Gupta D. Differential expression of peptidoglycan recognition protein 2 in the skin and liver requires different transcription factors. J Biol Chem 2006;281:20738‐20748. [DOI] [PubMed] [Google Scholar]
- 14. Wang H, Gupta D, Li X, Dziarski R. Peptidoglycan recognition protein 2 (N‐acetylmuramoyl‐L‐Ala amidase) is induced in keratinocytes by bacteria through the p38 kinase pathway. Infect Immun 2005;73:7216‐7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, van der Fits L, Voerman JS, Melief MJ, Laman JD, Wang M, et al. Identification of serum N‐acetylmuramoyl‐l‐alanine amidase as liver peptidoglycan recognition protein 2. Biochim Biophys Acta 2005;1752:34‐46. [DOI] [PubMed] [Google Scholar]
- 16. Wang ZM, Li X, Cocklin RR, Wang M, Fukase K, Inamura S, et al. Human peptidoglycan recognition protein‐L is an N‐acetylmuramoyl‐L‐alanine amidase. J Biol Chem 2003;278:49044‐49052. [DOI] [PubMed] [Google Scholar]
- 17. De Pauw P, Neyt C, Vanderwinkel E, Wattiez R, Falmagne P. Characterization of human serum N‐acetylmuramyl‐L‐alanine amidase purified by affinity chromatography. Protein Expr Purif 1995;6:371‐378. [DOI] [PubMed] [Google Scholar]
- 18. Arentsen T, Khalid R, Qian Y, Diaz Heijtz R. Sex‐dependent alterations in motor and anxiety‐like behavior of aged bacterial peptidoglycan sensing molecule 2 knockout mice. Brain Behav Immun 2018;67:345‐354. [DOI] [PubMed] [Google Scholar]
- 19. Xu M, Wang Z, Locksley RM. Innate immune responses in peptidoglycan recognition protein L‐deficient mice. Mol Cell Biol 2004;24:7949‐7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldman SM, Kamel F, Ross GW, Jewell SA, Marras C, Hoppin JA, et al. Peptidoglycan recognition protein genes and risk of Parkinson's disease. Mov Disord 2014;29:1171‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishikawa Y, Kozakai T, Morita H, Saida K, Oka S, Masuo Y. Rapid detection of mycoplasma contamination in cell cultures using SYBR green‐based real‐time polymerase chain reaction. Vitro Cell Dev Biol Anim 2006;42:63‐69. [DOI] [PubMed] [Google Scholar]
- 22. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Egelston CA, Avalos C, Tu TY, Simons DL, Jimenez G, Jung JY, et al. Human breast tumor‐infiltrating CD8+ T cells retain polyfunctionality despite PD‐1 expression. Nat Commun 2018;9:4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshikawa T, Nakatsugawa M, Suzuki S, Shirakawa H, Nobuoka D, Sakemura N, et al. HLA‐A2‐restricted glypican‐3 peptide‐specific CTL clones induced by peptide vaccine show high avidity and antigen‐specific killing activity against tumor cells. Cancer Sci 2011;102:918‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inarrairaegui M, Melero I, Sangro B. Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin Cancer Res 2018;24:1518‐1524. [DOI] [PubMed] [Google Scholar]
- 27. Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics identifies new therapeutic targets of early‐stage hepatocellular carcinoma. Nature 2019;567:257‐261. [DOI] [PubMed] [Google Scholar]
- 28. Wu F, Zou Q, Ding X, Shi D, Zhu X, Hu W, et al. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J Neuroinflammation 2016;13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol 2018;40:37‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kang S, Xie J, Ma S, Liao W, Zhang J, Luo R. Targeted knock down of CCL22 and CCL17 by siRNA during DC differentiation and maturation affects the recruitment of T subsets. Immunobiology 2010;215:153‐162. [DOI] [PubMed] [Google Scholar]
- 31. Lim K, Hyun YM, Lambert‐Emo K, Capece T, Bae S, Miller R, et al. Neutrophil trails guide influenza‐specific CD8+ T cells in the airways. Science 2015;349:aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza‐Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018;172:1022‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Araujo JM, Gomez AC, Aguilar A, Salgado R, Balko JM, Bravo L, et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci Rep 2018;8:4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Royet J, Gupta D, Dziarski R. Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nat Rev Immunol 2011;11:837‐851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials