

Abstract
Background and Aims
Acetaminophen (APAP) overdose induces severe liver injury and hepatic failure. While the activation of c‐Jun NH2‐terminal kinase (JNK) has been implicated as a mechanism in APAP‐induced liver injury, the hepatic defense system controlled by nuclear factor erythroid 2–related factor 2 (Nrf2) plays a central role in the mitigation of APAP toxicity. However, the link between the two signaling pathways in APAP‐induced liver injury (AILI) remains unclear.
Approach and Results
In this study, we demonstrated that the activation of JNK in mouse liver following exposure to APAP was correlated with the phosphorylation of Nrf2 and down‐regulation of the antioxidant response element (ARE)‐driven genes, NAD(P)H:quinone dehydrogenase 1, glutathione S‐transferase α3, glutathione S‐transferase M1, glutathione S‐transferase M5, and aldo‐keto reductase 1C. The JNK inhibitor, SP600125, or knockdown of JNK by infection of adenovirus expressing JNK small interfering RNA, ameliorated the APAP induced liver toxicity, and inhibited the phosphorylation of Nrf2 and down‐regulation of detoxifying enzymes by stabilizing the transcription factor. Mechanistically, JNK antagonized Nrf2‐ and ARE‐driven gene expression in a Kelch‐like ECH‐associated protein 1–independent manner. Biochemical analysis revealed that phosphorylated JNK (P‐JNK) directly interacted with the Nrf2‐ECH homology (Neh) 1 domain of Nrf2 and phosphorylated the serine‐aspartate‐serine motif 1 (SDS1) region in the Neh6 domain of Nrf2.
Conclusions
Mass spectrometric analysis identified serine 335 in the SDS1 region of mNrf2 as the major phosphorylation site for modulation of Nrf2 ubiquitylation by P‐JNK. This study demonstrates that Nrf2 is a target of P‐JNK in AILI. Our finding may provide a strategy for the treatment of AILI.
Abbreviations
- AAV
adeno‐associated virus
- AILI
APAP‐induced liver injury
- AKR1C
aldo‐keto reductases 1C
- ALT
alanine aminotransferase
- APAP
acetaminophen
- ARE
antioxidant response element
- Bcl‐2
B‐cell lymphoma 2
- β‐TrCP
beta‐transducin repeat‐containing protein
- BW
body weight
- ChIP
chromatin immunoprecipitation
- GCLC
glutamate‐cysteine ligase catalytic subunit
- GSH
reduced glutathione
- GSK‐3
glycogen synthase kinase‐3
- GST
glutathione S‐transferase
- Gstα3
glutathione S‐transferase α3
- Gstm1
glutathione S‐transferase M1
- Gstm5
glutathione S‐transferase M5
- HO‐1
heme oxygenase 1
- JNK
c‐Jun NH2‐terminal kinase
- Keap1
Kelch‐like ECH‐associated protein 1
- MEFs
mouse embryo fibroblasts
- MKP‐1
mitogen‐activated protein kinase phosphatase 1
- mNrf2
mouse Nrf2
- MS
mass spectrometry
- NAPQI
N‐acetyl‐p‐benzoquinoneimine
- Neh
Nrf2‐ECH homology
- NQO1
NAD(P)H:quinone oxidoreductase 1
- Nrf2
nuclear factor erythroid 2–related factor 2
- PBS
phosphate‐buffered saline
- P‐JNK
phosphorylated JNK
- P‐Nrf2
phosphorylated Nrf2
- SDS1
serine‐aspartate‐serine motif 1
- SDS2
serine‐aspartate‐serine motif 2
- Ser
serine
- siRNA
small interfering RNA
- tBHQ
tert‐butylhydroquinone
- Thr
threonine
- Tyr
tyrosine
- WT
wild type
N‐acetyl‐p‐aminophenol (APAP; acetaminophen) is a commonly used non‐narcotic analgesic drug used to reduce fever and relieve pain. It is generally considered to be safe at normal therapeutic levels, but is a major cause of liver failure and causes death when taken in excess.1 Under normal conditions, APAP is primarily metabolized in the liver by glucuronidation catalyzed by uridine diphosphate glucuronosyltransferases (UGTs) and sulfation by sulfotransferases. A small amount of the drug is metabolized by several of the cytochrome p450 enzymes into the reactive intermediate, N‐acetyl‐p‐benzoquinoneimine (NAPQI), which is normally detoxified by reduced glutathione (GSH) both nonenzymatically and enzymatically in a reaction catalyzed by glutathione S‐transferases (GSTs). With an overdose, sulfation and glucuronidation become saturated, and GSH is depleted by NAPQI. Excess NAPQI causes oxidative stress and binds covalently to liver proteins, leading to hepatic necrosis.2, 3, 4 Although the precise mechanism by which APAP or its metabolites cause cellular injury is still unknown, it has been speculated that cell death and organ failure most likely result from the cumulative and additive effects of oxidative damage, depressed mitochondrial function, and disruption of Ca2+ homeostasis with redox imbalance.3
c‐Jun N‐terminal protein kinases (JNKs) are a family of serine (Ser)/threonine (Thr) kinases important in responding to environmental stresses as well as growth factors and cytokines.5, 6 JNK activation is an important component of the stress response in cells, but when the activation is sustained, it is believed to promote cell injury and death. JNK has been shown to play a central role in APAP‐induced liver injury (AILI).7 APAP treatment causes JNK activation and its translocation to mitochondria, where JNK induces the mitochondrial permeability transition and inhibits mitochondrial bioenergetics.8 Mice injected with a JNK inhibitor (SP600125) or lacking JNK (after antisense treatment or in knockout mice) are significantly protected against AILI.7, 9, 10 Previous studies have suggested that NAPQI‐mediated damage alone is insufficient to cause hepatocyte death, and that activation of specific signal‐transduction pathways involving JNK is necessary for hepatocyte death with APAP treatment.3 The downstream targets of JNK that mediate APAP‐induced injury remain elusive.
The transcription factor, Nrf2 (nuclear factor erythroid 2–related factor 2), is a master regulator of redox homeostasis, controlling expression of a battery of genes involved in protecting cells against oxidative damage. Several lines of evidence support the fact that Nrf2‐mediated gene regulation is efficacious in protection against APAP‐induced hepatotoxicity. These Nrf2‐mediated genes are required for regulation of hepatic functions related to GSH synthesis, conjugation, detoxification, and transport. Nrf2 mediates basal and inducible expression of detoxifying genes by binding the antioxidant response element (ARE) in their promoters. Nrf2‐regulated genes include NAD(P)H:quinone oxidoreductase 1 (NQO1), GSTs, catalase, superoxide dismutase 1, and UGT. The greater susceptibility of Nrf2‐null mice and extreme resistance of hepatocyte keap1‐null mice to APAP toxicity support the hypothesis that the Nrf2‐mediated gene battery serves as a target for hepatoprotection.11, 12, 13 However, regulation of Nrf2 during APAP treatment is still not entirely clear.
Nrf2 is principally controlled through regulation of protein turnover by the ubiquitin‐proteasome system. Stability of Nrf2 protein is regulated by two E3 ubiquitin ligase adaptors, Keap1 (Kelch‐like ECH‐associated protein 1) and β‐TrCP (beta‐transducin repeat‐containing protein). In normal cells, Nrf2 is constitutively recognized by the substrate adaptor, Keap1, by the DIDLID and ETGE motifs in its N‐terminal Nrf2‐ECH homology (Neh) 2 domain and is presented for cullin‐3–based E3 ligase ubiquitination and proteasomal degradation. Under normal redox conditions, Keap1 mediates ubiquitination of Nrf2 and its proteasomal degradation. Upon exposure to oxidants or electrophiles, Keap1 becomes modified, leading to a disturbance of its interaction with Nrf2. Nrf2 therefore accumulates in the nucleus, where it activates transcription of a large battery of cytoprotective genes by binding to their AREs.14, 15, 16 In contrast, β‐TrCP regulates Nrf2 protein stability in a redox‐independent manner. Glycogen synthase kinase 3β (GSK‐3β) phosphorylates the two serines in DSGIS338 (residues 334‐338), a β‐TrCP binding motif, leading to Skp1‐Cullin1‐F‐box–containing protein/β‐TrCP–dependent degradation of Nrf2.17 However, it is not clear whether other kinases phosphorylate Nrf2 for its degradation.
Despite the critical roles of Nrf2 and JNK in AILI, the relationship between the two signaling pathways during such injury remains elusive. In this study, we present evidence that phosphorylated JNK (P‐JNK) impairs the cytoprotective system controlled by Nrf2/ARE through phosphorylating Nrf2. The crosstalk between P‐JNK and Nrf2 is important for promoting AILI.
Materials and Methods
Details of chemicals, antibodies, cell cultures, and plasmids are provided in the Supporting Information. The antibody defined as anti‐P‐Nrf23S, which specifically recognizes the phosphorylated cluster of serine (Ser)‐335, Ser‐338, and Ser‐342 of Nrf2, was generated in this laboratory. Procedures for the preparation and characterization of anti‐P‐Nrf23S are provided in the Supporting Information and Supporting Fig. S1. The single‐colony cell lines, 293T‐mNrf2ΔE and 293T‐mNrf2ΔEΔSDS1, which stably express Flag‐tagged mNrf2ΔETGE and Flag‐tagged mNrf2ΔETGEΔSDS1, respectively, were generated in this laboratory as described in the Supporting Information. Details for constructing plasmids pET41a‐mNrf2Neh1, pET41a‐mNrf2Neh6, pET41a‐mNrf2Neh3, PETDuet‐1‐His‐mNrf2ΔETGE, pcDNA3.1B/V5/his‐mNrf2ΔETGE,S335A, pEGFP‐mNrf2ΔETGE,S335A, pcDNA3.1/V5‐mNrf2S335A, pFuipw‐mNrf2∆ETGE, pEGFP‐C1‐mNrf2∆ETGE∆SDS1, pFuipw‐mNrf2∆ETGE∆SDS1, pETDuet‐1‐JNK1, pETDuet‐1‐JNK1Y185A, pETDuet‐1‐JNK1T183E/Y185E, AAV019‐mNrf2, and AAV019‐mNrf2S335A are described in the Supporting Information.
C57BL/6J wild‐type (WT) mice were purchased from the Shanghai Laboratory Animal Center (Chinese Academy of Sciences, Shanghai, China). C57BL/6J Nrf2−/− mice were kindly provided by Prof. Masayuki Yamamoto (University of Tsukuba, Tsukuba, Japan). Male mice (6‐8 weeks of age) were randomly assigned to the different treatment groups (n = 8‐15). Animals were fasted overnight, but allowed access to water, before experiments. APAP (50, 100, 200, or 300 mg/kg body weight [BW] IP) was dissolved in warm phosphate‐buffered saline (PBS; 55°C) that was cooled to 37°C before injection. SP600125 was dissolved in polyethylene glycol 400 and diluted with PBS (40% in PBS). In pretreatment experiments, SP600125 (10 mg/kg IP) was given 1 hour before injection of APAP. The dose of SP600125 chosen in this study was the same as those used in previous studies.7, 9, 10 Mice were sacrificed at 1.5, 3, 6, and 24 hours after administration of APAP. Blood and livers were harvested as described.18 Serum alanine aminotransferase (ALT) analysis was as described.19 All animal procedures were approved by the Laboratory Animals Ethics Committee of Zhejiang University (Hangzhou, China).
Experimental procedures for transfections, luciferase reporter gene activity, GST pulldown, immunoprecipitation, recombinant protein purification, western blotting analysis, immunohistochemical (IHC) analysis, in vitro phosphorylation assay, chromatin immunoprecipitation (ChIP) assay, RT‐qPCR, mass spectrometric analysis, mouse primary hepatocyte isolation and culture, and adenoviruses and adeno‐associated virus (AAV) infection of mice are provided in the Supporting Information.
Statistical Analysis
Statistical comparisons were made using an unpaired Student t test. A value of P < 0.05 was considered statistically significant.
Results
APAP Down‐regulates the Expression of Nqo1, glutathione S‐transferase α3, glutathione S‐transferase M1, glutathione S‐transferase M5, and aldo‐keto reductase 1C in Mouse Liver
Administration of a nonlethal dose of APAP (300 mg/kg IP) to male C57BL/6J mice resulted in hepatic injury as measured by serum ALT levels, which were markedly increased to 2,550 U/L at 6 hours (Fig. 1A). Centrilobular hepatocellular necrosis was observed in livers from APAP‐treated mice (Fig. 1B, e). As expected, phosphorylation of JNK was induced at 6 hours after APAP treatment (Supporting Fig. S2A, lane 2). In agreement with previous reports,13, 20, 21 an increased expression of Nrf2 was detected at 6 hours post‐APAP. However, despite this increased accumulation of Nrf2, mRNA levels of the Nrf2 target genes, Nqo1, glutathione S‐transferase α3 (Gstα3), glutathione S‐transferase M1 (Gstm1), glutathione S‐transferase M5 (Gstm5), and aldo‐keto reductase 1C (AKR1C), dropped rapidly at 6 hours post‐APAP (Supporting Fig. S2B), and their protein expressions were changed significantly at 6 hours (Fig. 1C, lanes 7 and 8), but markedly decreased at 24 hours post‐APAP (Fig. 1C, lanes 11 and 12). These data indicate that for certain ARE‐driven genes, the Nrf2/ARE transcriptional program is impaired in hepatocytes during AILI.
Figure 1.
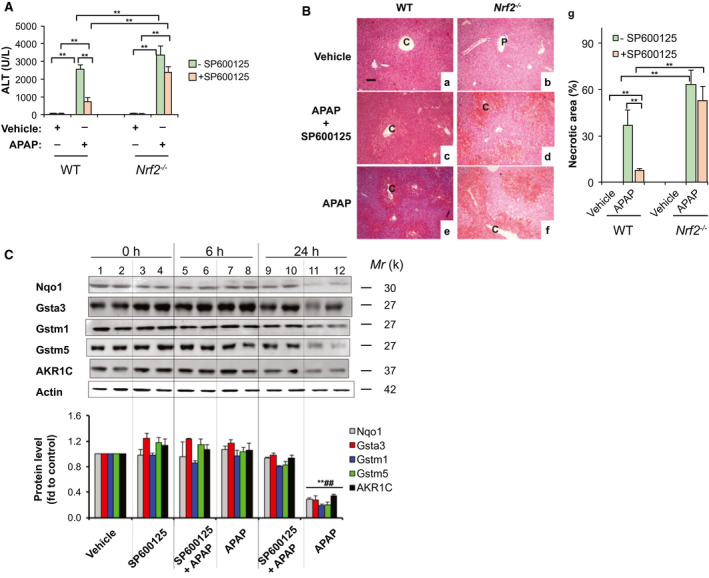
The JNK inhibitor, SP600125, blocks down‐regulation of ARE‐driven genes induced by APAP in mouse liver. SP600125 (10 mg/kg IP) was given to WT and Nrf2−/− mice 1 hour before injection of APAP (300 mg/kg IP). Livers were harvested 6 and 24 hours after administration of APAP. (A) Serum ALT levels at 6 hours (n = 3‐6). (B) Hematoxylin and eosin staining of liver sections at 6 hours (original magnification, ×200; scale bars, 50 μm; P, portal venules; C, central venules). g, the percentage of necrotic area by semiquantification (mean ± SD; n = 3‐5). (C) SP600125 blocked the decrease of Nqo1, AKR1C, Gstα3, Gstm1, and Gstm5 expression in APAP‐treated liver. Protein extracts were prepared from livers of WT mice harvested at 6 and 24 hours after administration of APAP. Western immunoblottings were probed with the indicated antibodies. Each lane contains a sample from a single mouse. Lower panel, semiquantitative results of blottings. The value from the WT group treated with vehicle was set at 1. Values are mean ± SD (n = 3). *P < 0.05; **P < 0.01 versus WT vehicle. ## P < 0.01 versus WT treated with SP600125 and APAP at 24 hours.
JNK Inhibitor SP600125 Blocks Down‐regulation of Nqo1, Gstα3, Gstm1, Gstm5, and AKR1C in APAP‐Treated Liver of WT Mice
JNK activation is a critical early event in AILI.7 To evaluate whether down‐regulation of ARE‐driven genes is linked to JNK activation, mice were subjected to intraperitoneal injection of the JNK inhibitor, SP600125, before APAP treatment. As expected, WT mice with SP600125 (10 mg/kg) pretreatment had only a slight elevation of serum ALT, 748 U/L at 6 hours after APAP treatment (Fig. 1A), and nearly normal liver histology (Fig. 1B,C). Remarkably, reductions of Nqo1, AKR1C, Gstα3, Gstm1, and Gstm5 mRNA levels at 6 hours (Supporting Fig. S2B) were markedly attenuated, whereas decreases of protein levels at 24 hours post‐APAP were almost completely blocked in livers from WT mice pretreated with SP600125 (Fig. 1C, lanes 9 and 10), compared to those treated with APAP alone (Fig. 1C, lanes 11 and 12). However, the JNK inhibitor failed to provide protective effects similar to Nrf2−/− mice. At 6 hours post‐APAP, serum ALT levels (Fig. 1A) and grade of centrilobular necrosis in Nrf2−/− mice (Fig. 1B,D,F,G) were not significantly changed by SP600125 pretreatment. Our results indicate that P‐JNK is implicated in down‐regulation of ARE genes in AILI, and that the protective effect of the JNK inhibitor, SP600125, is Nrf2/ARE dependent.
P‐JNK Increases Nrf2 Turnover In Vitro Through a Keap1‐Independent Mechanism
We next treated mice with SP600125 (10 mg/kg IP) alone. Interestingly, Nrf2 protein level was increased in liver (Fig. 2A, lanes 3 and 4). After 24 hours, liver extracts (Fig. 1C, lanes 3 and 4) exhibited marginal increases of AKR1C and Gstα3 protein levels comparable to those of WT mice treated with vehicle (Fig. 1C, lanes 1 and 2), presumably by inhibiting basal P‐JNK. Given that Keap1 is a well‐known key repressor of Nrf2, to assess any possibility of the involvement of Keap1 in the effect of SP600125 on Nrf2, we treated Keap1 −/− mouse embryonic fibroblasts (MEFs) with SP600125. Western blottings showed marked inhibition of the phosphorylation of JNK1/2 by SP600125 (10 μM; Fig. 2B). When WT MEFs were exposed to SP600125 (10 μM) for 16 hours, Nrf2 steady‐state level increased 1.5‐fold (Fig. 2B, lane 2). Moreover, SP600125 (10 μM) enhanced the Nrf2 level induced by the Nrf2 activator, tert‐butylhydroquinone (tBHQ), from 2.4‐ to 5‐fold (Fig. 2B, lanes 3 and 4). As expected, baseline Nrf2 protein level in Keap1−/− MEFs was 2.5‐fold than in WT counterparts, attributable to knockout of Keap1 (Fig. 2B, lane 5). Importantly, SP600125 (10 μM) further increased Nrf2 protein level in Keap1 −/− MEFs to 4.5‐fold (Fig. 2B, lane 6). These data indicate that Keap1 is not required for the action of SP600125.
Figure 2.
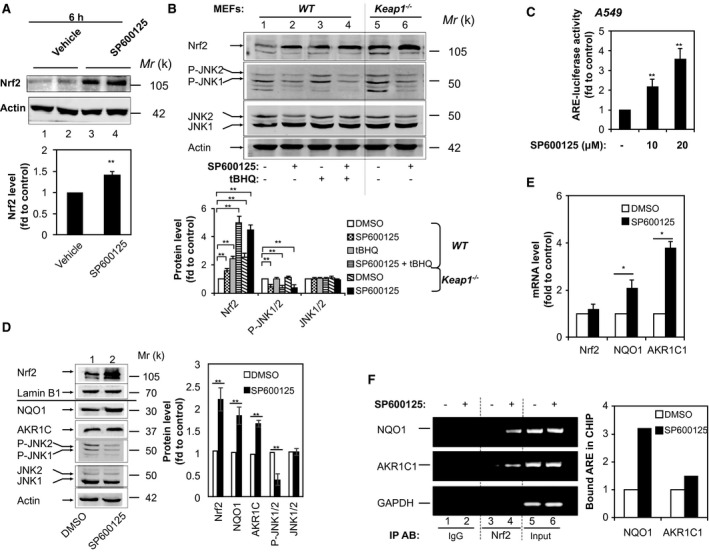
The JNK inhibitor, SP600125, up‐regulates expression of Nrf2‐ and ARE‐driven genes. (A) SP600125 increases expression of Nrf2 in liver. Mice were given SP600125 (10 mg/kg IP). Livers were harvested 6 hours after administration. Western immunoblottings of protein extracts from livers probed with anti‐Nrf2 or anti‐actin. Each lane contains a sample from a single mouse. Lower panel, semiquantitative results of the blottings. The value from the WT group treated with vehicle was set at 1. Values are mean ± SD (n = 3). (B) SP600125‐induced Nrf2 expression is independent of Keap1. WT and Keap1 −/− MEFs were treated with SP600125 (10 μM) in the presence or absence of tBHQ (20 μM) for 16 hours. Cell lysates were probed by immunoblotting with anti‐Nrf2, anti‐P‐JNK, anti‐JNK, or anti‐actin. Lower panel, semiquantitative results of the blottings. The value for DMSO treatment was set at 1. Values are mean ± SD (n = 3). (C) SP600125 activates ARE‐luciferase activity dose dependently in A549 cells. A549 cells were transfected with pGL‐GSTA2‐41bp‐ARE in combination with pRL‐TK. Twenty‐four hours later, cells were treated with 10 or 20 μM of SP600125 for 16 hours and the dual luciferase activity was determined. The value for DMSO treatment was set at 1 (control). Values are mean ± SD (n = 3). (D,E) SP600125 increases the expression of Nrf2‐ and ARE‐driven genes in A549 cells. Immunoblotting with anti‐Nrf2 and anti‐lamin B1 of nuclear extracts from A549 cells subjected to serum depletion for 16 hours before treatment with 10 μM of SP600125 for 16 hours. Whole‐cell extracts were probed by immunoblotting with anti‐NQO1, anti‐AKR1C, anti‐P‐JNK, and anti‐JNK. Right panel, semiquantitative results of the blottings. Relative levels of Nrf2 normalized to lamin B1. Relative levels of NQO1, AKR1C, P‐JNK, and JNK normalized to actin. The value for DMSO treatment was set at 1. Values are mean ± SD (n = 3). (E) Statistics for mRNA levels of NQO1 and AKR1C as determined by RT‐PCR. 18S rRNA was used as an internal control. The value for DMSO treatment was set at 1. Data are presented as the mean ± SD of triplicate experiments. (F) SP600125 increases Nrf2 binding to ARE sites in the promoters of NQO1 and AKR1C1. A549 cells were treated with 10 μM of SP600125 for 6 hours and used for ChIP analysis. GAPDH served as a negative control. PCR reactions were not saturated. Results are representative of three separate experiments. The relative value of NRF2 binding to ARE sites was determined as described in Materials and Methods. *P < 0.05; **P < 0.01. Abbreviations: DMSO, dimethyl sulfoxide; fd, fold; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; IgG, immunoglobulin G; IP AB, immunoprecipitation antibody; rRNA, ribosomal RNA; tBHQ, tert‐butylhydroquinone.
We next carried out further mechanistic studies in non‐small‐cell lung cancer A549 cells, which carry a dysfunctional somatic mutation of Keap1.22 To evaluate whether SP600125 has a direct effect on the Nrf2/ARE system, an ARE‐driven luciferase reporter construct, pGL‐GSTA2.41bp‐ARE,23 was transfected into A549 cells. We found that SP600125 induced ARE‐luciferase activity dose dependently in A549 cells. After cells were stimulated for 16 hours with 10 μM of SP600125, ARE‐luciferase activity doubled, whereas 20 μM of SP600125 triggered >3‐fold induction (Fig. 2C). Western blotting showed an >2‐fold increase in the Nrf2 protein level after 16 hours of exposure to 10 μM of SP600125 (Fig. 2D, lanes 1 and 2), along with marked elevation of NQO1 and AKR1C1 mRNA and protein levels (Fig. 2D,E). Moreover, ChIP assays showed increased Nrf2 binding to ARE sequences in the promoters of NQO1 and AKR1C1 after 6 hours of exposure to SP600125 (20 μM; Fig. 2F, lane 4). Taken together, our results demonstrate that the P‐JNK–mediated down‐regulation of Nrf2/ARE signaling is not dependent on Keap1‐mediated degradation of Nrf2.
Inverse Relationship Between Nrf2 Steady‐State Level and P‐JNK
To further evaluate the ability of JNK to antagonize the Nrf2/ARE system, we used small interfering RNA (siRNA) specific to JNK1/2 to knock down JNK in A549 cells. Western immunoblotting demonstrated the successful knockdown of both P‐JNK and JNK (Fig. 3A, lane 2). Importantly, JNK1/2 knockdown increased the Nrf2 protein level, leading to elevation of mRNA and protein levels of NQO1 and AKR1C (Fig. 3A,B) and 3‐fold higher ARE‐luciferase activity (Fig. 3C). Conversely, overexpression of JNK by transient transfection of pSG5‐JNK1 into A549 cells markedly reduced the amount of Nrf2 (Supporting Fig. S3A), along with significant reduction of AKR1C1 and NQO1 at mRNA and protein levels (Supporting Fig. S3A,B), as well as ARE‐luciferase activity (Supporting Fig. S3C). Moreover, mRNA levels of Nrf2 remained unchanged by SP600125 (Fig. 2E), JNK knockdown (Fig. 3B), or JNK overexpression (Supporting Fig. S3B), indicating that P‐JNK exerts its anti‐Nrf2 effect by acting on Nrf2 protein, not its mRNA.
Figure 3.
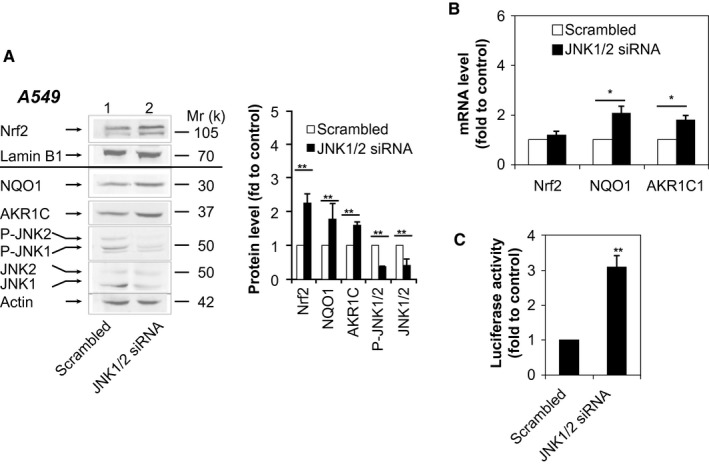
Knockdown of JNK increases expression of Nrf2‐ and ARE‐driven genes. (A,B) A549 cells were transfected with JNK1/2 siRNA or scrambled siRNA. Forty‐eight hours later, nuclear extracts were probed by immunoblotting with anti‐Nrf2 and anti‐lamin B1, and whole‐cell lysates were probed by immunoblotting with anti‐NQO1, anti‐AKR1C, anti‐P‐JNK, anti‐JNK, and anti‐actin (A). Right panel (A), semiquantitative result of blotting. Relative levels of Nrf2 normalized to lamin B1. Relative levels of NQO1, AKR1C, P‐JNK, and JNK normalized to actin. The value for scrambled siRNA was set at 1. Values are mean ± SD (n = 3). mRNA levels of NQO1 and AKR1C1 were determined by RT‐PCR (B). The level of 18S rRNA was used as an internal control. The value for scrambled siRNA was set at 1. (C) Luciferase activity 48 hours after transfection of A549 cells with JNK1/2 siRNA or scrambled siRNA plus pGL‐GSTA2.41bp‐ARE reporter vector and pRL‐TK. Data are presented as the mean ± SD of triplicate experiments. *P < 0.05; **P < 0.01. Abbreviation: rRNA, ribosomal RNA.
Active JNK Interacts Directly With Neh1 of Nrf2
JNKs are activated by dual phosphorylation on a specific Thr and a specific tyrosine (Tyr) in a typical Thr‐X‐Tyr motif within their “activation/phosphorylation loop” sequences.24 To determine whether activated JNK can interact with Nrf2, we generated an expression vector encoding His‐tagged JNK1T183E‐Y185E, which mimics P‐JNK with mutations of the Thr‐183 and Tyr‐185 residues to glutamic acid. An expression vector encoding the kinase‐dead form of His‐JNK1Y185A, with the Tyr‐185 residue mutated to alanine, was also constructed as a control. To determine the region of Nrf2 that is required to interact with JNK, a series of Nrf2‐truncated proteins tagged with GST (see Supporting Fig. S4A) were expressed and their ability to interact with purified recombinant His‐JNK1 or mutants was tested by GST‐pulldown assay. We found that JNK1T183E‐Y185E interacted with hNrf2339‐605 protein and mNrf2Neh1 (Supporting Fig. S4B, lanes 6 and 8), indicating that the Neh1 domain is the site that interacts with the kinase‐active P‐JNK. In contrast, the kinase‐dead form of JNK or JNKY185A failed to interact with the C‐terminal Nrf2 proteins (Supporting Fig. S4B, lanes 2 and 4). These results led us to speculate that Nrf2 is a P‐JNK substrate.
Active JNK Phosphorylates Ser Residues in the Neh6 Domain of Nrf2
To determine whether P‐JNK phosphorylates Nrf2 directly and identify the phosphorylation site(s), we performed in vitro kinase assays. His‐tagged mouse Nrf2 (mNrf2) was produced in Escherichia coli and purified as a substrate for kinase assays. Kinase‐active JNK was prepared by immunoprecipitation with anti‐JNK from lysates of HEK293 cells that had been treated with the JNK activator, anisomycin. The phosphorylated His‐tagged Nrf2 band on the gel was then excised and subjected to mass spectrometry (MS). MS‐MS and MS3 spectra indicated that Ser‐335 (major; Supporting Fig. S5; Supporting Table S2) and Ser‐347 (major; Supporting Table S2) were phosphorylated by P‐JNK.
It has been shown that Nrf2 is repressed by β‐TrCP through DSGIS and DSAPGS motifs in its Neh6 domain (Fig. 4A).17, 25 Although each of these motifs is sufficient to enable ubiquitylation of Nrf2, only phosphorylation of the DSGIS motif in serine‐aspartate‐serine motif 1 (SDS1) increases its degron activity. The DSAPGS motif is not influenced by GSK‐3 activity.17, 25 Given that Ser‐335 resides within the DSGIS motif whereas Ser‐347 is located outside the SDS1 and serine‐aspartate‐serine motif 2 (SDS2) regions, we speculated that Ser‐335 phosphorylation could be crucial for the anti‐Nrf2 effect of P‐JNK and focused our subsequent validation of the liquid chromatography/MS‐MS results on Ser‐335. We made an antibody named phosphorylated Nrf2 (P‐Nrf2)3S, which specifically recognizes the phosphorylated Ser cluster (Ser‐335, Ser‐338, and Ser‐342; Supporting Fig. S1). We also created an expression vector encoding His‐tagged mNrf2ΔETGE,S335A, in which the Ser‐335 residue was replaced with nonphosphorylatable alanine and the ETGE motif was deleted. We performed the in vitro kinase assay to phosphorylate His‐mNrf2 mutants as described above. As expected, P‐JNK directly phosphorylated His‐mNrf2ΔETGE (Fig. 4B, lane 2) in vitro. In contrast, anti‐P‐Nrf23S failed to react with His‐mNrf2ΔETGE in the absence of P‐JNK immunoprecipitation (Fig. 4B, lane 1). Importantly, mutation of the Ser‐335 site abolished the phosphorylation by P‐JNK (Fig. 4B, lane 3), confirming the Ser‐335 site for P‐JNK. To analyze the role of Ser‐335 in the regulation of Nrf2 by JNK, A549 cells were transfected with an ARE‐driven luciferase reporter construct (pGL‐GSTA2.41bp‐ARE). In addition, cells were cotransfected with vector pcDNA3.1/V5/His, pcDNA3.1/V5‐mNrf2 encoding WT mNrf2, or pcDNA3.1/V5‐mNrf2S335A encoding mutant mNrf2, in which the Ser‐335 residue was replaced with alanine. As expected, mNrf2 or mNrf2S335A overexpression induced ARE‐luciferase activity at a similar level of 2 to 3‐fold (Fig. 4C). In cells overexpressing WT mNrf2, SP600125 (10 μM) enhanced ARE‐luciferase activity further to 3.8‐fold, attributable to inhibition of endogenous P‐JNK. In contrast, cells overexpressing mNrf2S335A were insensitive to the JNK inhibitor (Fig. 4C), indicating that phosphorylation of Ser‐335 is required for the anti‐Nrf2 action of JNK.
Figure 4.
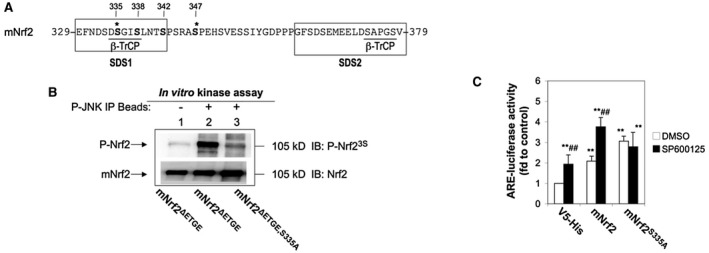
Identification of the site at which Nrf2 is phosphorylated by P‐JNK. (A) Amino‐acid sequences of mNrf2 between residues 329 and 379. (B) Mutation of Ser‐335 to Ala in the Neh6 domain of Nrf2 abolishes its phosphorylation by P‐JNK. HEK293 cells were treated with 5 μg/mL of anisomysin for 30 minutes. The cell lysate was immunoprecipitated with anti‐P‐JNK. Immunoprecipitates were subjected to in vitro kinase assays with purified recombinant His‐mNrf2ΔETGE or His‐mNrf2ΔETGE,S335A proteins, followed by western immunoblotting. Upper blottings, immunodetection of P‐Nrf2 with anti‐P‐Nrf23S. Lower blottings, immunodetection of Nrf2 mutant proteins to ensure similar quantity per reaction. Lane 1, kinase assay in the absence of P‐JNK immunoprecipitates. The blottings presented are typical examples of at least three independent experiments. (C) Mutation of Ser‐335 to Ala in the Neh6 domain of Nrf2 abolishes SP600125‐enhanced ARE‐luciferase activity. A549 cells were transfected with pcDNA3.1/V5/His, pcDNA3.1/V5‐mNrf2, or pcDNA3.1/V5‐mNrf2S335A plus pGL‐GSTA2.41bp‐ARE reporter vector and pRL‐TK. Cells were treated with 10 μM of SP600125 for 16 hours before dual luciferase activity was analyzed. The value for cells transfected with pcDNA3.1/V5, pGL‐GSTA2.41bp‐ARE reporter vector, and pRL‐TK and treated with DMSO was set at 1. Data are presented as the mean ± SD of triplicate experiments. *Significantly different from cells transfected with pcDNA3.1/V5/His, pGL‐GSTA2.41bp‐ARE reporter vector, and pRL‐TK and treated with DMSO. #Significantly different from cells transfected with the same plasmids and treated with DMSO. **P < 0.01; ## P < 0.01. Abbreviations: Ala, alanine; DMSO, dimethyl sulfoxide; fd, fold; IB, immunoblotting; IP, immunoprecipitation; Ser‐335, serine 335; V5‐His, V5 peptide with polyhistidine tag.
P‐JNK Mediates Ubiquitylation of Nrf2
To test whether P‐JNK phosphorylates endogenous Nrf2, A549 cells were exposed to the JNK activator, anisomycin, for 30 minutes, followed by immunoprecipitation with anti‐Nrf2 that recognized both the phosphorylated and nonphosphorylated forms. Immunoblotting analysis of the precipitates showed that the steady‐state level of Nrf2 was markedly reduced by anisomycin (Supporting Fig. S6, lane 2). In contrast, analysis with anti‐P‐Nrf23S revealed a dramatic increase in P‐Nrf2, indicating that the Ser cluster was phosphorylated (Supporting Fig. S6, lane 2). To examine the degradation of Nrf2 by the ubiquitin‐proteasome system, 549 cells were exposed to anisomycin with or without MG132 treatment. Whole‐cell lysates were immunoprecipitated with anti‐Nrf2. Analysis of precipitates showed that increased amounts of ubiquitinated molecules were effectively conjugated to Nrf2 to form polyubiquitinated species in response to anisomycin treatment (Fig. 5A, upper blot, lane 3), correlated with the dramatic reduction of Nrf2 protein level (Fig. 5A, lower blot, lane 3). MG132 cotreatment led to a significant further increase of ubiquitin‐conjugated species (Fig. 5A, lanes 2 and 4). To define the role of SDS1 in Nrf2 degradation mediated by P‐JNK, we generated the stable cell lines, 293T‐mNrf2ΔE and 293T‐mNrf2ΔEΔSDS1, which expressed Flag‐tagged mNrf2ΔETGE (lacking the ETGE motif) and mNrf2ΔETGEΔSDS1 (lacking the ETGE and SDS1 regions). These cells were treated with anisomycin and analyzed by western immunoblotting. Flag‐mNrf2ΔETGE had a half‐life of 22 minutes (Fig. 5B, upper panels (lanes 1‐4) and lower histogram). In contrast, Flag‐mNrf2ΔETGEΔSDS1 exhibited a higher steady‐state level on anisomycin treatment, with a half‐life of 45 minutes (Fig. 5B, lanes 5‐8 and lower histogram). Furthermore, to determine whether Nrf2 protein accumulation by SP600125 was attributed to an increase in its stability, we measured the half‐life of Nrf2 in A549 cells. Cells cotreated with SP600125 (10 μM) and cycloheximide (CHX; 20 μM) presented a delayed Nrf2 degradation curve compared to cells treated with CHX alone (Supporting Fig. S7). That is, the half‐life of Nrf2 increased from 45 to 80 minutes in the presence of SP600125 (Supporting Fig. S7, lower panel). These results together showed that JNK‐mediated degradation of Nrf2 is through polyubiquitination at SDS1.
Figure 5.
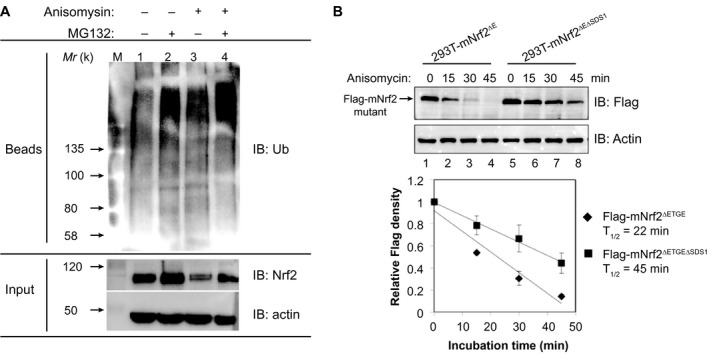
P‐JNK regulates Nrf2 stability. (A) Activated JNK increases the polyubiquitination of endogenous Nrf2 in A549 cells. A549 cells were pretreated with 10 μM of MG132 for 3 hours, before 1‐hour treatment with 5 μg/mL of anisomysin in the continued presence or absence of 10 μM of MG132. Whole‐cell lysates were subjected to immunoprecipitation with anti‐Nrf2. Immunoprecipitates were analyzed by immunoblotting using antibody against Ub. Input, 10% of the cell lysate used for immunoprecipitation. The results presented are typical examples from at least three independent experiments. (B) Deletion of SDS1 in the Neh6 domain blocks degradation of Nrf2 induced by anisomycin. 293T‐mNrf2ΔE and 293T‐mNrf2ΔEΔSDS1 cells expressing Flag‐tagged mNrf2ΔETGE and Flag‐tagged mNrf2ΔETGEΔSDS1, respectively, were treated with 5 μg/mL of anisomysin for 15‐45 minutes. Whole‐cell lysates were immunoblotted with anti‐Flag or anti‐actin. Lower panel, relative levels of Flag‐mNrf2 mutants normalized to actin. The value at time 0 for the same protein was set at 1. Results are from three separate experiments. Values are mean ± SD. Abbreviations: IB, immunoblotting; Ub, ubiquitin.
APAP Induces Phosphorylation of Nrf2 by P‐JNK in Mouse Liver
To assess the relationship of the phosphorylation status of Nrf2 and P‐JNK in AILI, WT mice received 0‐300 mg/kg BW of APAP. IHC analysis with anti‐P‐Nrf23S revealed that in livers from mice treated with vehicle or nontoxic 100 mg/kg BW at 6 hours post‐APAP, P‐Nrf2 staining was very weak (Supporting Fig. S8A,B, a and b). With an >200‐mg/kg BW dose, P‐Nrf2 expression was markedly increased (Supporting Fig. S8B, c‐e), and correlated with the increase of APAP toxicity (Supporting Fig. S8A). We next carried out early time‐course studies after 300 mg/kg of APAP. To define the association between the extent of Nrf2 phosphorylation and P‐JNK specifically, we performed immunoprecipitation with protein extracts from livers with anti‐Nrf2 antibodies, which recognized both the phosphorylated and nonphosphorylated forms, followed by immunoblotting with anti‐P‐Nrf23S and anti‐Nrf2 antibodies, respectively. The treatment elicited dramatically increased P‐Nrf2 at 6 hours (Fig. 6A, a, lane 4 and Supporting Fig. S8C, d), in line with activation of P‐JNK (Fig. 6A, b, lane 4). In contrast, Nrf2 level was markedly reduced at 6 hours (Fig. 6A, a, lane 4), inversely associated with activation of P‐JNK (Fig. 6A, b, lane 4). Moreover, exposing mouse primary hepatocytes to APAP (10 mM), P‐Nrf2 was induced within 2 hours (Fig. 6B, lane 4), again associated with activation and phosphorylation of JNK (Fig. 6B, lane 4). Intriguingly, pretreatment with SP600125 resulted in significantly weaker staining of P‐Nrf2 in liver (Fig. 6C, b and d). Alternatively, we used adenoviruses expressing JNK1/2 siRNA to down‐regulate JNK expression in mouse liver. Adeno‐JNK1/2 siRNA ameliorated APAP‐induced (300 mg/kg) liver toxicity (Fig. 7A), following knockdown of both basal JNK and APAP‐induced P‐JNK in mouse liver (Fig. 7B, a [lanes 2 and 4], b, and c). Importantly, adeno‐JNK1/2 siRNA inhibited APAP‐induced phosphorylation of Nrf2 (Fig. 7C, lane 4) and enhanced both basal and APAP‐induced Nrf2 levels (Fig. 7C, lanes 2 and 4). Moreover, inhibition of Nqo1, Gstα, Gstm1, Gstm5, and AKR1C levels at 24 hours by APAP was impaired by adeno‐JNK siRNA (Fig. 7D, lane 4). Next, Nrf2−/− mice were injected with AAV019‐mNrf2 expressing Flag‐mNrf2, AAV019‐mNrf2S335A expressing Flag‐mNrf2S335A, or HBAAV2/9‐GFP, a control virus. Immunoblottings showed comparable expression levels of Flag‐mNrf2 and Flag‐mNrf2S335A in Nrf2−/− mouse liver (Supporting Fig. S9A). APAP (300 mg/kg BW) caused 71.4% (5/7) lethality in the control AAV virus group within the first 6 hours. In contrast, no such lethality was observed in either the AAV mNrf2 group or AAV mNrf2S335A group. Notably, at 6 hours post‐APAP, the AAV mNrf2S335A group showed a significantly lower ALT level and markedly decreased histological necrosis, compared to the AAV mNrf2 group (Supporting Fig. S9B). Taken together, these data suggest that P‐JNK directs phosphorylation of the serine cluster in the Neh6 domain of Nrf2 in APAP‐induced liver toxicity in the early hours, leading to its dysfunction and hence the silencing of ARE‐driven genes.
Figure 6.
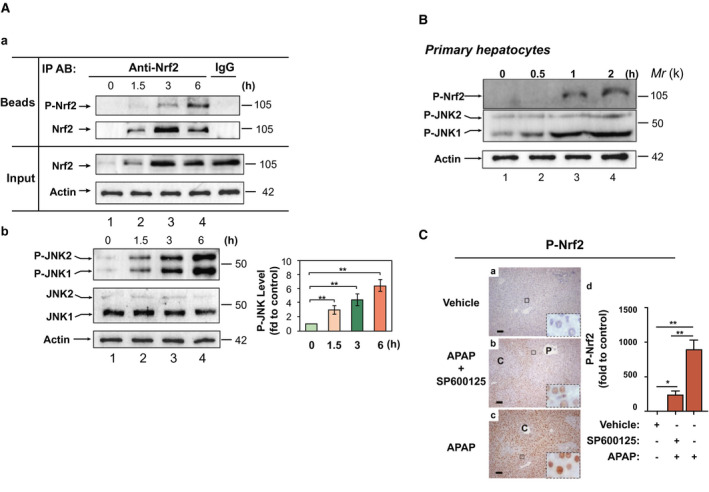
APAP induces phosphorylation of Nrf2 by P‐JNK in mouse liver. (A) WT mice were given APAP (300 mg/kg BW IP). Livers were harvested 1.5, 3, or 6 hours later. (a) Soluble extracts (100 μg) from livers in 3 randomly selected mice each group were subjected to immunoprecipitation with anti‐Nrf2. After washing, each half of the immunoprecipitates was then subjected to immunoblotting with anti‐Nrf2 or anti‐P‐Nrf23S. IgG with extracts from livers in the 6‐hour APAP group was used as a negative control. Input, 10% of the lysate used for immunoprecipitation. The blottings shown represent results from three independent experiments. (b) Western blotting of P‐JNK and JNK in 3 randomly selected mice each group. Right panel, semiquantitative results of the blottings. The control (vehicle) was set at 1. Values are mean ± SD (n = 3). **P < 0.01 versus vehicle. (B) APAP induces phosphorylation of Nrf2 in mouse primary hepatocytes. Freshly isolated mouse hepatocytes were exposed to 10 mM of APAP for 30 minutes, 1 hour, and 2 hours. Cell lysates were subjected to immunoprecipitation with anti‐Nrf2. After washing, each of the immunoprecipitates was then subjected to western blotting with anti‐P‐Nrf23S; the flowthroughes blotting with anti‐P‐JNK, and anti‐actin, respectively. Blottings are representative of two independent experiments. (C) SP600125 inhibits APAP‐induced phosphorylation of Nrf2 in mouse liver. SP600125 (10 mg/kg IP) was given to mice 1 hour before injection of APAP (300 mg/kg IP). Livers were harvested 6 hours after APAP. (a‐c) Liver sections were probed with anti‐P‐Nrf23S (original magnification, ×200; scale bars, 50 μm; insets, original magnification, ×1000; P, portal venules; C, central venules). (d) Statistics from experiments as in (a–c). The control (vehicle at 6 h) was set at 1. Values are mean ± SD (n = 3). *P < 0.05; **P < 0.01. Abbreviations: fd, fold; IgG, immunoglobulin G; IP AB, immunoprecipitation antibody.
Figure 7.
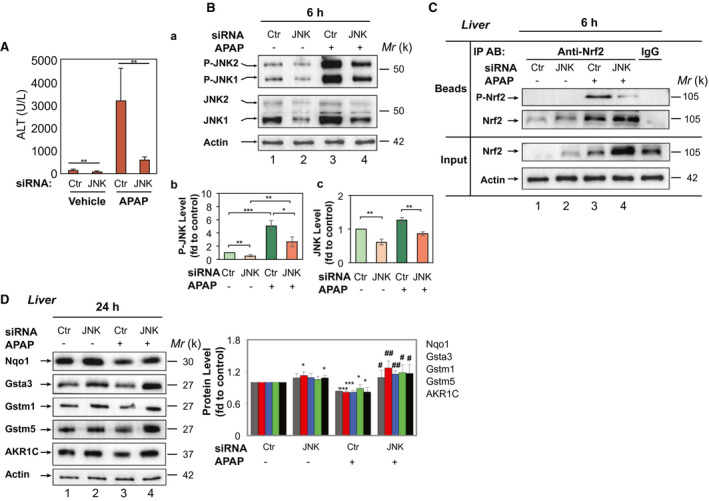
Knockdown of JNK in mouse liver attenuates phosphorylation of Nrf2 and down‐regulation of Nqo1, AKR1C, Gstα3, Gstm1, and Gstm5 induced by APAP. WT mice were treated with an adenovirus‐expressing control or JNK1/2 siRNA (5 × 109 PFU per mouse IV) for 10 days, then by APAP (300 mg/kg IP) for 6 or 24 hours. (A) Serum ALT levels at 24 hours post‐APAP (n = 5 each, means ± SD). (B) Western blotting of P‐JNK1/2 and JNK1/2 in 3 randomly selected mice each group at 6 hours post‐APAP. Lower panel, semiquantitative results of the blottings. (C) Soluble extracts (100 μg) from livers in 3 randomly selected mice each group at 6 hours post‐APAP were first subjected to immunoprecipitation with anti‐Nrf2. After washing, each half of the immunoprecipitates was then subjected to immunoblotting with anti‐Nrf2 or anti‐P‐Nrf23S antibodies. IgG with extracts from livers in the Ctr siRNA‐APAP group was used as a negative control. Input, 10% of the lysate used for immunoprecipitation. Blottings shown represent results from three independent experiments. (D) Western blotting of Nqo1, AKR1C, Gstα3, Gstm1, and Gstm5 in 3 randomly selected mice each group at 24 hours post‐APAP. Right panel, semiquantitative results of blottings. (B‐D) Each lane represents a different mouse. The control (Ctr siRNA and treated with PBS at 6 hours) was set at 1. Values are mean ± SD (n = 3). *P < 0.05; **P < 0.01 versus Ctr siRNA + vehicle; # P < 0.05; ## P < 0.01 versus Ctr siRNA + APAP (300 mg/kg). Abbreviations: Ctr, control; fd, fold; IgG, immunoglobulin G; IP AB, immunoprecipitation antibody.
Discussion
APAP is a widely used antipyretic drug, but can cause significant morbidity and mortality in cases of toxic‐dose ingestion or improper use. Although JNK activation is known to play a pathological role in AILI,7 the downstream signaling pathways that are important in mediating such injury are not well understood. JNK has many targets in the early stages of APAP toxicity, including B‐cell lymphoma 2 (Bcl‐2) proteins such as Bcl‐2 interacting mediator of cell death, p53 up‐regulated modulator of apoptosis, BCL2‐associated X apoptosis regulator, and, most important, SH3‐domain binding protein that preferentially associates with Btk protein.26 In this study, we identified Nrf2 as a target of JNK signaling in AILI.
The gene battery regulated by Nrf2 plays a critical role in the multiple steps associated with mitigation of APAP toxicity. The Nrf2‐dependent antioxidant defense system, composed of GSH synthesis, phase II detoxifying enzymes, and reactive oxygen species–inactivating enzymes, protects cells upon oxidative damage by NAPQI.27 In addition, Nrf2 controls hepatic gene expression of multidrug‐resistance–associated protein transport, by which xenobiotics and their conjugates are excreted into bile or urine rather than accumulating in the liver.28, 29 It has been documented that NAPQI, the hepatotoxic metabolite of APAP, activates Nrf2 by modifying Keap1.21 APAP treatment elicits Nrf2 nuclear translocation in mouse liver,20 and mRNA levels of its target genes increase transiently in the first few hours after APAP.13, 30 In this study, although we observed a similar change of certain ARE‐driven genes at 3 hours post‐APAP (data not shown), mRNA levels of Nqo1, Gstα3, Gstm1, Gstm5, and AKR1C6 dropped markedly at 6 hours (Supporting Fig. S2B), correlated with JNK activation and Nrf2 phosphorylation. Our results indicate that the Nrf2/ARE transcriptional programs for certain ARE‐driven genes are disrupted at an early time point during AILI. Taken together, our data suggest that after nuclear translocation of Nrf2 upon APAP treatment, whereas the nonphosphorylated Nrf2 switches on the transcription of ARE genes, a certain amount of Nrf2 is targeted and phosphorylated by P‐JNK, which triggers Nrf2 degradation, consequently down‐regulating ARE‐driven genes. The balance between nonphosphorylated and phosphorylated Nrf2 determines the activation or inhibition of Nrf2 target genes. At a relatively low dose of APAP, when less Nrf2 is phosphorylated by P‐JNK, up‐regulation of ARE‐driven genes predominates, protecting against liver injury. However, a higher dose of APAP induces the dramatic activation of JNK and phosphorylation of Nrf2, down‐regulation of Nrf2, and blocking the transcription of ARE‐driven genes, so impairment of the Nrf2/ARE system contributes to APAP hepatotoxicity. In agreement with previous studies,20, 21 at the toxic 300‐mg/kg dose, we also found that the treatment elicited dramatic inductions of heme oxygenase‐1 (Ho‐1) and glutamate‐cysteine ligase catalytic subunit (Gclc) mRNAs during the early 6 hours post‐APAP (data not shown). Marked induction of Ho‐1 and a slight increase of Gclc abundance at 24 hours were also observed (Supporting Fig. S10). The distinctly different expression profiles of Ho‐1 and Gclc from those of Nqo1, Gstα3, Gstm1, Gstm5, and AKR1C may be attributed to the fact that other transcription factors in addition to Nrf2 are also involved in the regulation of Ho‐131 and Gclc.32
Nrf2 is principally controlled by protein ubiquitylation, which targets it for proteasomal degradation. Through distinct mechanisms, Keap1 and β‐TrCP regulate the turnover of Nrf2 by separate protein domains, the redox‐sensitive Neh2 degron and the redox‐insensitive Neh6 degron.16, 17, 33, 34, 35 Whereas Keap1 mediates degradation of Nrf2 primarily in the cytoplasm, the redox‐insensitive Neh6 degron predominantly mediates turnover of Nrf2 in the nucleus. The Neh6 domain contains two distinct destruction motifs, DSGIS and DSAPGS, which recruit β‐TrCP,17, 25, 36 a substrate adaptor for the Skp1‐Cul1‐Rbx1 core E3 complex. Phosphorylation of DSGIS increases its degron activity, and this is positively regulated by GSK‐3. We showed here that P‐JNK phosphorylates Ser‐335 in the DSGIS motif (residues 334‐338) of Nrf2, leading to its degradation in a Keap1‐indedendent manner. In a manner similar to Nrf2 regulation, Lee et al. reported that P‐JNK–mediated degradation by the ubiquitin–proteasome system of p45/nuclear factor erythroid 2, another member of the cap‘n’collar/basic leucine zipper family, occurs during differentiation of mouse erythroleukemia cells.37 Keum et al. have shown that JNKs are involved in activation of Nrf2 by the chemopreventive agent, phenethylisothiocyanate.38 Given that JNKs regulate diverse cellular programs, it remains elusive whether the observed effects occur through direct phosphorylation of Nrf2 or through indirect mechanisms. Previously, Sun et al. used MS to identify phosphorylated sites of Nrf2 by overexpression of mitogen‐activated protein kinases.39 Although several phosphorylated sites of Nrf2 were detected, neither Ser‐335 nor Ser‐347 phosphorylation was reported in their study.39 This discrepancy may be attributable to the different cell lines and culture conditions and the fact that we used the JNK activator, anisomycin, to activate JNK. Nevertheless, the present study clearly showed that Nrf2 is a substrate of P‐JNK. Together, our data demonstrated that Nrf2 is destabilized as a consequence of its phosphorylation by JNK and subsequent ubiquitination through the DSGIS motif.17
Mitogen‐activated protein kinase phosphatase 1 (MKP‐1), also referred to as dual‐specificity phosphatase 1, is a critical negative regulator of JNK. Wancket et al. reported that Mkp‐1 protects mice against APAP‐induced hepatic injury, possibly by inhibiting JNK activity.40 Previous studies from our laboratory have shown that MKP‐1 and Nrf2 form a forward‐feedback loop. On one hand, MKP‐1 enhances Nrf2/ARE signaling by directly interacting with the Neh2 domain of Nrf2 and increases its stability; on the other hand, MKP‐1 is an Nrf2 target gene. Nrf2 induces MKP‐1 transcription by binding to the ARE site in the promoter of MKP‐1.18, 41 The finding in this study implies that phosphorylation and degradation of Nrf2 by P‐JNK not only causes dysfunction of the ARE‐cytoprotective system, but may also repress MKP‐1 signaling, leading to prolonged activation of JNK. Attenuation of both Nrf2 and MKP‐1 signaling pathways causes liver injury.
Taken together, we discovered a signaling pathway during AILI. When APAP treatment activates JNK, the electrophilic metabolite, NAQPI, modifies Keap1, leading to nuclear accumulation of Nrf2 (Supporting Fig. S11). P‐JNK phosphorylates the Neh6 domain of Nrf2 and triggers its degradation. As a result, the Nrf2‐directed transcriptional program is inhibited, and, consequently, the detoxification and cytoprotection system is impaired. Limitation of Nrf2 phosphorylation provides a strategy for protection against APAP hepatotoxicity.
Author Contributions
Y.C., K.L., J.Z., Y.H., P.W., H.W., Q.L., J.Y., Y.G., and Y.L. performed the experiments and analyzed the data. C.C.L. performed Mass Spectrometry and analyzed the data. X.J.W. and X.T. designed the experiments, analyzed the data. X.J.W. and X.T. drafted and revised the manuscripts.
Supporting information
Acknowledgments
We thank Prof. John Hayes (University of Dundee, Dundee, UK) for kindly providing antisera against Gstα3, Gstm1, Gstm5, and AKR1C and Prof. Masayuki Yamamoto (University of Tsukuba, Tsukuba, Japan) for providing the Nrf2 −/− mice and Keap1 −/− MEFs. This work was supported by the National Natural Science Foundation of China (31571476, 91643110, and 31971188). We thank Yanwei Li (Core facilities, Zhejiang University School of Medicine) for help with qRT‐PCR.
See Editorial on Page 1530
Potential conflict of interest: Nothing to report.
Supported by NSFC (31571476, 91643110, and 31971188).
Contributor Information
Xiuwen Tang, Email: xiuwentang@zju.edu.cn.
Xiu Jun Wang, Email: xjwang@zju.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Wallace CI, Dargan PI, Jones AL. Paracetamol overdose: an evidence based flowchart to guide management. Emerg Med J 2002;19:202‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dahlin DC, Miwa GT, Lu AY, Nelson SD. N‐acetyl‐p‐benzoquinone imine: a cytochrome P‐450‐mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A 1984;81:1327‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov 2005;4:489‐499. [DOI] [PubMed] [Google Scholar]
- 4. Pumford NR, Halmes NC. Protein targets of xenobiotic reactive intermediates. Annu Rev Pharmacol Toxicol 1997;37:91‐117. [DOI] [PubMed] [Google Scholar]
- 5. Johnson GL, Nakamura K. The c‐jun kinase/stress‐activated pathway: regulation, function and role in human disease. Biochim Biophys Acta 2007;1773:1341‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol 2007;19:142‐149. [DOI] [PubMed] [Google Scholar]
- 7. Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c‐Jun N‐terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006;131:165‐178. [DOI] [PubMed] [Google Scholar]
- 8. Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen‐induced liver injury. J Biol Chem 2008;283:13565‐13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, et al. Critical role of c‐jun (NH2) terminal kinase in paracetamol‐induced acute liver failure. Gut 2007;56:982‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen‐induced liver injury. Hepatology 2007;45:412‐421. [DOI] [PubMed] [Google Scholar]
- 11. Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. Hepatocyte‐specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun 2006;339:79‐88. [DOI] [PubMed] [Google Scholar]
- 12. Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O'Connor T, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE‐regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 2001;59:169‐177. [DOI] [PubMed] [Google Scholar]
- 13. Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A 2001;98:4611‐4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dinkova‐Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 2002;99:11908‐11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino‐terminal Neh2 domain. Genes Dev 1999;13:76‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin‐mediated ubiquitylation of proteins by a “tethering” mechanism: a two‐site interaction model for the Nrf2‐Keap1 complex. J Biol Chem 2006;281:24756‐24768. [DOI] [PubMed] [Google Scholar]
- 17. Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta‐TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK‐3 activity. Oncogene 2013;32:3765‐3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo L, Chen Y, Wang H, Wang S, Liu K, Li X, et al. Mkp‐1 protects mice against toxin‐induced liver damage by promoting the Nrf2 cytoprotective response. Free Radic Biol Med 2018;115:361‐370. [DOI] [PubMed] [Google Scholar]
- 19. Wang XJ, Li Y, Luo L, Wang H, Chi Z, Xin A, et al. Oxaliplatin activates the Keap1/Nrf2 antioxidant system conferring protection against the cytotoxicity of anticancer drugs. Free Radic Biol Med 2014;70:68‐77. [DOI] [PubMed] [Google Scholar]
- 20. Goldring CE, Kitteringham NR, Elsby R, Randle LE, Clement YN, Williams DP, et al. Activation of hepatic Nrf2 in vivo by acetaminophen in CD‐1 mice. Hepatology 2004;39:1267‐1276. [DOI] [PubMed] [Google Scholar]
- 21. Copple IM, Goldring CE, Jenkins RE, Chia AJ, Randle LE, Hayes JD, et al. The hepatotoxic metabolite of acetaminophen directly activates the Keap1‐Nrf2 cell defense system. Hepatology 2008;48:1292‐1301. [DOI] [PubMed] [Google Scholar]
- 22. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1‐NRF2 interaction in non‐small‐cell lung cancer. PLoS Med 2006;3:e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, et al. RXRalpha inhibits the NRF2‐ARE signalling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res 2013;73:3097‐3108. [DOI] [PubMed] [Google Scholar]
- 24. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:239‐252. [DOI] [PubMed] [Google Scholar]
- 25. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/β‐TrCP promotes glycogen synthase kinase 3‐dependent degradation of the Nrf2 transcription factor in a Keap1‐independent manner. Mol Cell Biol 2011;31:1121‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han D, Dara L, Win S, Than TA, Yuan L, Abbasi SQ, et al. Regulation of drug‐induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol Sci. 2013;34:243‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reisman SA, Csanaky IL, Aleksunes LM, Klaassen CD. Altered disposition of acetaminophen in Nrf2‐null and Keap1‐knockdown mice. Toxicol Sci 2009;109:31‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aleksunes LM, Slitt AL, Maher JM, Augustine LM, Goedken MJ, Chan JY, et al. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol 2008;226:74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maher JM, Dieter MZ, Aleksunes LM, Slitt AL, Guo G, Tanaka Y, et al. Oxidative and electrophilic stress induces multidrug resistance‐associated protein transporters via the nuclear factor‐E2‐related factor‐2 transcriptional pathway. Hepatology 2007;46:1597‐1610. [DOI] [PubMed] [Google Scholar]
- 30. Aleksunes LM, Slitt AM, Cherrington NJ, Thibodeau MS, Klaassen CD, Manautou JE. Differential expression of mouse hepatic transporter genes in response to acetaminophen and carbon tetrachloride. Toxicol Sci 2005;83:44‐52. [DOI] [PubMed] [Google Scholar]
- 31. Yeligar SM, Machida K, Kalra VK. Ethanol‐induced HO‐1 and NQO1 are differentially regulated by HIF‐1alpha and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem 2010;285:35359‐35373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang H, Wang J, Huang ZZ, Ou X, Lu SC. Cloning and characterization of the 5′‐flanking region of the rat glutamate‐cysteine ligase catalytic subunit. Biochem J 2001;357:447‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2‐DLGex degron and Keap1. Mol Cell Biol 2014;34:832‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox‐regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox‐sensitive Neh2 degron and the redox‐insensitive Neh6 degron. J Biol Chem 2004;279:31556‐31567. [DOI] [PubMed] [Google Scholar]
- 35. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two‐site molecular recognition model. Mol Cell Biol 2006;26:2887‐2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rada P, Rojo AI, Evrard‐Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta‐TrCP axis. Mol Cell Biol 2012;32:3486‐3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee TL, Shyu YC, Hsu PH, Chang CW, Wen SC, Hsiao WY, et al. JNK‐mediated turnover and stabilization of the transcription factor p45/NF‐E2 during differentiation of murine erythroleukemia cells. Proc Natl Acad Sci U S A 2009;107:52‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keum YS, Owuor ED, Kim BR, Hu R, Kong AN. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC). Pharm Res 2003;20:1351‐1356. [DOI] [PubMed] [Google Scholar]
- 39. Sun Z, Huang Z, Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2‐dependent antioxidant response. PLoS One 2009;4:e6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wancket LM, Meng X, Rogers LK, Liu Y. Mitogen‐activated protein kinase phosphatase (Mkp)‐1 protects mice against acetaminophen‐induced hepatic injury. Toxicol Pathol 2012;40:1095‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li J, Wang H, Zheng Z, Luo L, Wang P, Liu K, et al. Mkp‐1 cross‐talks with Nrf2/Ho‐1 pathway protecting against intestinal inflammation. Free Radic Biol Med 2018;124:541‐549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials