

Abstract
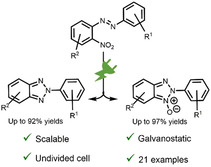
Electrosynthesis of 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles and their N‐oxides from 2‐nitroazobenzene derivatives is reported. The electrolysis is conducted in a very simple undivided cell under constant current conditions with a leaded bronze cathode and a glassy carbon anode. The product distribution between 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles and their N‐oxides can be guided by simply controlling the current density and the amount of the charge applied. The reaction tolerates several sensitive functional groups in reductive electrochemistry. The usefulness and the applicability of the synthetic method is demonstrated by a formal synthesis of an antiviral compound.
Keywords: azo compounds, electrochemistry, nitrogen heterocycles, reduction, sustainable chemistry
Electrosynthesis takes the lead: Electrosynthesis enables green and atom‐economic access to 2H‐2‐(aryl)‐benzo[d]‐1,2,3‐triazoles or their N‐oxides from 2‐nitroazobenzenes. This operationally very simple protocol is conducted in an undivided cell by constant current electrolysis. Optionally, inexpensive NaOH can be used as both a supporting electrolyte and additive.
The intriguing and versatile chemical and physical properties of 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles have made them a frequently used substructure in many different functional molecules and commercial products. Just to name a few examples, 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazole containing molecules have been studied thus far as novel ligands1 (Scheme 1), as potential treatment to Duchenne muscular dystrophy2 and as antivirals with either a broad inhibitory spectrum or a high selectivity towards human poliovirus type 1 Sabin strain (HPV‐1, Sb‐1).3 In technical products, the motif has found use in UV absorbers (BASF, Tinuvin©) and its use as fluorescent light‐emitting element has been patented.4 Furthermore, also 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazole‐1‐oxides are versatile synthetic intermediates as the oxygen can be exploited to tune the reactivity in C−H functionalizations.5 Hence, developing new sustainable synthetic methods for both 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles and to their N‐oxides preferably from a single starting material is of very high importance.
Scheme 1.
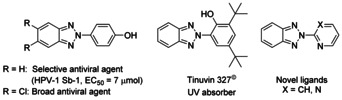
Various applications of the 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazole motif.
Broadly, 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles are synthesized either with an arylation of N‐unsubstituted benzo[d]‐1,2,3‐triazoles or by cyclizing 2‐substituted azobenzenes either oxidatively or reductively (Scheme 2). In the former, benzo[d]‐1,2,3‐triazoles are N2‐arylated with aryl halogens catalyzed by Pd,6 Cu7 or Fe/Cu,8 with iodine(III) compounds with9 or without10 transition metal catalysts, with silyl aryl triflates,11 and in SNAr reactions with electron‐deficient fluoroaryls.12
Scheme 2.
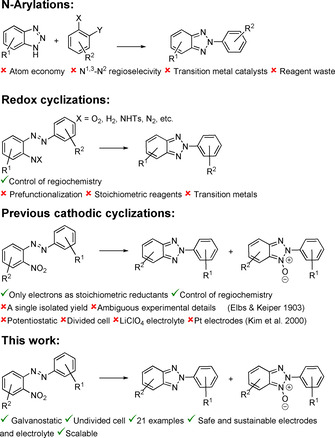
Synthetic approaches to 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles and 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazole‐1‐oxides.
In the second strategy, 2‐substituted azobenzenes or azoxybenzenes are either oxidatively or reductively cyclized. Oxidatively, this sequence has been realized with (2‐tosylamino)azobenzenes13 and 2‐aminoazoxybenzenes14 using PhI(OAc)2 as oxidant, or by oxidizing 2‐aminoazobenzenes with CuII,15 Cu/Py/O2,16 PhI(OAc)2 17 or NaOCl.18 Substrates with ortho‐N3‐moiety, that can also be prepared in situ from suitable starting materials, are cyclized thermally19 or by various different reagents such as BCl3,20 BF3,20 Pd/TBHP,21 and CuI/TMEDA.22 In addition, reduction of 2‐nitroazobenzenes can also give access to 2‐substituted benzo[d]‐1,2,3‐triazoles and this has been realized using P(III/V)/PhSiH3,23 Zn,24 SmI2,25 hydroxylamine,26 thiourea S,S‐dioxide,27 baker's yeast,28 Ru3(CO)12/CO29 or Ag.30, 31 Furthermore, also galvanostatic32 and potentiostatic33 electroreductive conditions have been reported in 1903 and 2000, respectively. Notably, many of these reductive protocols focus only on the synthesis of 2H‐2‐(2‐hydroxyphenyl)benzo[d]‐1,2,3‐triazole derivatives and synthetic methods with a broader scope and a more diverse variation in the aryl rings have not been elaborated. Beyond these described strategies, also intramolecular cyclization of 2‐nitrophenyl(phenyl)carbodiimide can give access to 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazole with a loss of CO2.34
Unfortunately, one or more drawbacks hamper the synthetic utility of these strategies (Scheme 2). For example, regioselectivity between N1, 3‐N2 sites is a common and severe challenge in the arylations of the N‐unsubstituted benzo[d]‐1,2,3‐triazoles. Although there have been recently significant advances to address this shortcoming, most of these methods also suffer from a poor atom economy. The regioselectivity issue is averted when 2‐substituted azobenzenes or azoxybenzenes are cyclized oxidatively or reductively, but again, these approaches are hindered either by the use of transition‐metal catalysts, highly sophisticated organocatalytic systems or enzymes, the necessity to install N‐substituents, or by the required (over)stoichiometric amounts of oxidants or reductants. The published electrochemical procedures elegantly eliminate the need of redox reagents and also address other important issues, but they as well have limitations. In the first communication, only a single isolated yield was reported, whereas the latter report focuses mainly onto the synthesis of 2H‐2‐(2‐hydroxyphenyl)benzo[d]‐1,2,3‐triazoles.32, 33 The yields obtained with them were excellent, but a significant drop was realized when the 2‐hydroxyphenyl moiety was substituted (40–63 %). Furthermore, the use of LiClO4 as the supporting electrolyte, the three‐electrode arrangement, and the platinum electrodes combined with the high amount of required charge present safety, scalability, and economic issues, respectively. This makes these methods very limited useful for synthetic applications and not exploitable for technical processes.
Synthetic organic electrochemistry has in recent years experienced a renaissance and it can be regarded as a pinnacle of sustainability in many terms. For instance, electrons or holes can replace chemical redox reagents that reduces both the waste and the cost of the synthesis. The green aspects of electrochemistry are evident in numerous different types of reactions,35 particularly in arylations.36 Encouraged by our recent success in the electroreductive functionalization of nitroarenes37 and the discovery of leaded bronzes as safe and efficient alternatives for lead,38 we set to develop new general electrochemical conditions for the synthesis of 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles. The most efficient way to identify suitable electrolytic operational conditions is achieved by electrosynthetic screening.39 We report that an operationally very simple galvanostatic electrolysis of 2‐nitroazobenzene derivatives in an undivided cell can be used to access both 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles as well as their N‐oxides in high yields from single starting materials. The control of product distribution is achieved easily by controlling only the current density and the amount of applied charge.
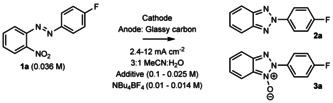
We initiated the study by choosing the cyclization of 4’‐fluoro‐2‐nitroazobenzene (1 a) to 2H‐(4‐fluorophenyl)‐2‐benzo[d]‐1,2,3‐triazole (2 a) and 2H‐(4‐fluorophenyl)‐2‐benzo[d]‐1,2,3‐triazole 1‐oxide (3 a) as the test reaction for the screening together with modified reaction conditions, which we have previously found highly suitable for nitroarenes.37 We first explored different cathode materials such as lead, boron‐doped diamond (BDD), glassy carbon (GC), CuSn10Pb10 and CuSn5Pb20 leaded bronzes in a mixture of MeCN/H2O (3:1) with AcONa/AcOH (pH 3.7, 0.1 m) buffer additive by using GC as an anode with a 12 mA cm−2 current density (Table 1, entries 1–5). Out of these materials, CuSn5Pb20 gave the best result (entry 5).
Table 1.
Screening of the reaction conditions.
|
| |||||
|---|---|---|---|---|---|
|
Entry |
Cathode |
Additive[a,b] |
Current density [mA cm−2] |
Applied charge [F] |
Yield[c] [%] of 2 a/3 a (2 a+3 a) |
|
1 |
BDD |
AcONa/AcOH |
12 |
5 |
18/27 45 |
|
2 |
Pb |
AcONa/AcOH |
12 |
5 |
27/15 42 |
|
3 |
GC |
AcONa/AcOH |
12 |
5 |
16/6 22 |
|
4 |
CuSn10Pb10 |
AcONa/ AcOH |
12 |
5 |
36/16 52 |
|
5 |
CuSn5Pb20 |
AcONa/AcOH |
12 |
5 |
40/21 61 |
|
6 |
CuSn5Pb20 |
HCOONa/HCOOH |
12 |
5 |
40/21 61 |
|
7 |
CuSn5Pb20 |
Na2CO3 |
12 |
5 |
43/16 59 |
|
8 |
CuSn5Pb20 |
– |
12 |
5 |
40/19 59 |
|
9 |
CuSn5Pb20 |
NaOH |
12 |
5 |
62/24 86 |
|
10 |
CuSn5Pb20 |
KOH |
12 |
5 |
48/19 67 |
|
11 |
CuSn5Pb20 |
LiOH*2H2O |
12 |
5 |
37/37 74 |
|
12[d] |
CuSn7Pb15 |
NaOH |
4.1 |
8.4 |
87/9 96 |
|
13[d] |
CuSn7Pb15 |
NaOH |
2.4 |
8.4 |
0/94 94 |
[a] In entries 1–11, NBu4BF4 was used at the concentration of 0.01 m and in entries 12—13 at the concentration of 0.014 m. [b] Added as a 0.1 m solution in H2O (0.025 m) (entries 1–6) or as a solid (entries 9–13) (0.1 m). [c] Conversion‐based yield was determined with 1H or 19F NMR spectroscopy by using 1,3,5‐trimethoxybenzene or NBu4BF4 as the internal standard. [d] MeOH was used as a solvent instead of MeCN/H2O (3:1).
We then continued by screening several different buffers and additives. Formate buffer (pH 3.7, 0.1 m), Na2CO3 (2.67 equiv), and electrolysis in the absence of any additive gave comparable results to AcONa/AcOH buffer (entries 6–8), whereas the use NaOH‐additive increased the yield notably (entry 9). We then explored the use of 2.67 equiv of KOH, and LiOH*2H2O but these modifications did not improve the obtained yields (Entries 10–11). Changing the anode to stainless steel (SS, 1.4301) undergoes severe corrosion, whereas another SS (VA 1.4571) and nickel furnished slightly lower yields than GC. Finally, after lowering the current density from 12 to 4.1 mA cm−2, exchanging the cathode from CuSn5Pb20 to CuSn7Pb15, and replacing the solvent to MeOH due to the poor solubility of some of the starting materials as well as the slight immiscibility of MeCN and H2O caused by the added NaOH, we were able to obtain 2 a in 87 % NMR yield (entry 12). In general, the reaction mixtures were clearer in MeOH than in MeCN/H2O (3:1). Interestingly, 3 a could be accumulated as the main product when the current density was even further attenuated to 2.4 mA cm−2 (entry 13).
With this set of reaction conditions, we then started to explore the scope of the reaction. We first attempted the synthesis of different benzo[d]‐1,2,3‐triazoles with a 4.1 mA cm−2 current density. In addition to 2 a, which could be isolated in an 86 % yield (Scheme 3), also unsubstituted substrate 2 b was obtained in a high 89 % yield. To our delight, also electron‐rich 2‐(4‐MePh) (2 d) and 2‐(4‐OMePh) (2 e) substituted compounds were obtained in excellent 92 % and 88 % yields, respectively. The latter example is in particular important, as the Sb‐1 inhibitor (Scheme 1) can be easily accessed from 2 e in 82 % yield through demethylation by following a literature‐known protocol.40 We then explored the halogen‐series as they are excellent synthetic handles for further functionalization and they have easy accessible LUMOs that can accept electrons instead of NO2 or its subsequently reduced reaction intermediates. This tendency could lead to side‐reactions, such as dehalogenations, in the employed protic media. Interestingly, 2‐(4‐ClPh) (2 c) was obtained in an 83 % yield whereas 2‐(4‐BrPh) (2 g) was isolated in a 62 % yield, together with 6 % of the dehalogenated compound 2 b (Scheme 3). Also, 5‐Br‐2‐Ph (2 f) substituted benzo[d]‐1,2,3‐triazole could be obtained in a moderate 57 % yield. In this case, dehalogenated side product was also observed in the reaction mixture. Unfortunately, we were unable to isolate the 2‐(4‐IPh) derivative 2 k as the major component (23:77 I/H) with our reaction conditions. Furthermore, we obtained 2 f and 2 g more selectively, when the current density was lowered from 4.1 to 2.4 mA cm−2. Moreover, 2‐(3,5‐bis‐(CF3)Ph) derivative 1 i converted readily to 2 i with an excellent yield of 89 %. The 2‐(4‐CNPh) was first obtained in a 29 % yield, but when we changed the additive from NaOH to Et3N (12 equiv), 45 % yield was received. Surprisingly, 2‐(2‐COOMePh) (2 h) derivative was obtained in a 12 % yield. For 2 c, 2 f–2 h, 2 j,k, we used 3:2 THF/MeOH mixture as the reaction solvent, because MeOH alone did not sufficiently solubilize their starting materials.
Scheme 3.
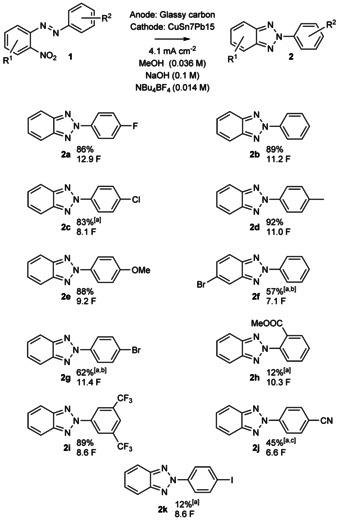
Scope of the 2H‐2‐(aryl)‐benzo[d]‐1,2,3‐triazole synthesiswith isolated yields. [a] 3:2 THF/MeOH instead of MeOH. [b] Current density of 2.4 mA cm−2 was used. [c] 12 equiv of Et3N instead of NaOH.
The reductive cyclization of 2‐nitroazobenzenes to 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles is a 4 e−/4H+ process. Therefore, 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles N‐oxides should become analogously accessible, if the starting materials are only reduced by 2 e−/2 H+. Based on this assumption, we then attempted synthesis of benzo[d]‐1,2,3‐triazole N‐oxides. In all cases, we lowered the current density to 2.4 mA cm−2 to make reactions more selective (Scheme 4). Again, the electron‐rich 2‐(4‐MePh) (3 d) and 2‐(4‐OMePh) (3 e) were obtained in excellent yields of 91 % and 82 %, respectively. Also, unsubstituted 2‐(Ph) (3 b), 2‐(4‐FPh) (3 a), and 2‐(4‐ClPh) (3 c) derivatives were obtained in 83 %, 97 % and 81 % yields, respectively. In addition, 2‐(3,5‐bis‐(CF3)Ph)‐derivative 1 i furnished 3 i in an 88 % yield, and 1 h yielded 31 % of 2‐(2‐COOMePh) derivative 3 h. Moreover, we were able to isolate 5‐Br‐2‐Ph (3 f) and 2‐(4‐BrPh) (3 g) with good yields of 83 % and 86 % and, surprisingly, also 2‐(4‐IPh) (3 k) could be obtained in a 74 % yield. Although we were unable to obtain 2H‐2‐(4‐iodophenyl)benzo[d]‐1,2,3‐triazole (2 k) directly, the relatively high yield of its N‐oxide 3 k indicates that the utilization of iodine‐derivatives is also possible in a multistep synthesis. It is conceivable that after stopping the reductive cyclization at the N‐oxide state, the iodo substituent can be utilized as a synthetic handle for further functionalization that is followed by electrochemical deoxygenation to functionalized benzo[d]‐1,2,3‐triazoles. As with the synthesis of 2H‐2‐(aryl)benzo[d]‐1,2,3‐triazoles, 3 c, 3 f–3 h, 3 k were obtained in 3:2 THF/MeOH mixture.
Scheme 4.
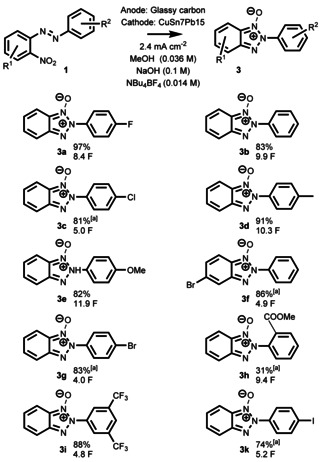
Scope of the 2H‐2‐(aryl)‐benzo[d]‐1,2,3‐triazole N‐oxide synthesis with isolated yields. [a] 3:2 THF/MeOH instead of MeOH.
Regarding the anodic counter reaction, during reaction monitoring in MeOH, we observed evolution of a 1H NMR resonance at 8.49 ppm that we confirmed to originate from a sodium formate by comparing to an authentic sample. Thus, it is plausible that sodium hydroxide additive has multiple roles in the reaction. On the one hand, it might contribute to the decrease of the oxidation potential of the methanol in the anodic counter oxidation41 and, on the other hand, it is an extra electrolyte that lowers the terminal voltage. At this point, we became interested whether NBu4BF4 could be omitted altogether from the reaction mixture as this would simplify any downstream processes. Gratifyingly, 2 a and 2 d were obtained in a 92 % and a 91 % yield (NMR) in the absence of NBu4BF4 after passing 11.7 F and 10.6 F of charge, respectively (Scheme 5). Furthermore, reaction of 1 d to 2 d was scaled‐up from 0.18 mmol to 3.4 mmol (19×) in the latter experiment, which confirms the excellent robustness and scalability of the studied reaction.
Scheme 5.
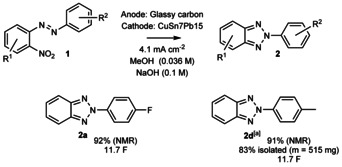
Experiments without NBu4BF4 supporting electrolyte. [a] At 3.4 mmol (m=814 mg).
A plausible mechanistic rationale from preliminary studies is proposed in Scheme 6. At the beginning, 1 a is reduced to nitroso intermediate (Int‐2) at the cathode either by addition of 2 e−/2 H+, or alternatively, by 4 e−/4 H+, which generates first the hydroxylamine intermediate (Int‐1) that is further reversibly oxidized at the anode with a loss of 2 e−/2H+ to Int‐2.42 Cyclic voltammetry (CV) of 1 a shows two broad reductive waves at −0.71 V and −1.17 V (vs. Ag/Ag+, see Supporting Information for further details). Furthermore, from CV of 2 a, we deduce that the latter reductive wave of 1 a is associated to 2 a. Therefore, 2 a can accumulate already during the reductive phase of the CV of 1 a and the oxidation of Int‐1 to Int‐2 is not a prerequisite for the generation of 2 a. Next, 5‐atom 6π‐electrocyclization converts Int‐2 to 2 a. Calculations at DLPNO‐CCSD(T)/def2‐QZVPP/SMD(MeOH)//PBE0‐D3/def2‐TZVP/CPCM(MeOH) level of theory reveal that the electrocyclization of Int‐2(b) to 2 b has a ΔG ≠ of 8.2 kcal mol−1 and the cyclization is overall exothermic by −17.1 kcal mol−1 (Supporting Information). After electrocyclization, 2 e−/2 H+ reduction of N‐oxide furnishes the final product. At the anode, methanol oxidation to sodium formate supplies electrons for the reduction.
Scheme 6.
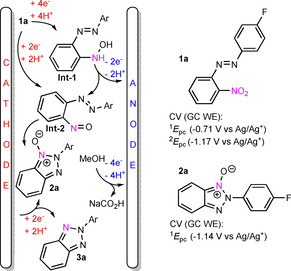
Plausible mechanistic rationale (left) and cyclovoltammetry (CV) E pc potentials of 1 a–2 a (right). CV measurements with glassy carbon (GC) working, GC counter, and Ag/Ag+ reference electrode in saturated LiCl/EtOH by using 150 mVs−1 scan rate in 0.1 m NaOH in MeOH.
To conclude, we have realized an efficient synthetic protocol for accessing both 2H‐2‐(aryl)‐benzo[d]‐1,2,3‐triazoles and their N‐oxides from the same starting materials. The developed electrochemical conditions have several elements that make it highly interesting over its precedents in academic and industrial settings. The reactions are set up under operationally very simple galvanostatic conditions in an undivided cell and the reductive cyclizations can be optionally performed by using inexpensive NaOH as both the supporting electrolyte and additive, which simplifies possible downstream processes. Furthermore, leaded bronze cathodes and glassy carbon anodes are safe and sustainable materials, and the scalable and robust reaction proceeds in general with high yields and tolerates several challenging functional groups.
Experimental Section
General protocol for the synthesis of 2H ‐2‐(aryl)benzo[d]‐1,2,3‐triazoles and 2H ‐2‐(aryl)benzo[d]‐1,2,3‐triazole N‐oxide derivatives: Starting material (0.18 mmol), NBu4BF4 (0.07 mmol), and NaOH (0.5 mmol) or Et3N (2.16 mmol) were dissolved to either MeOH or 3:2 THF/MeOH (5 mL). The reaction mixture was then stirred for about 30 min at 33–37 °C (RT) and during that time the CuSn7Pb15 cathode was gently polished and glassy carbon anode was cleaned with H2O and acetone before constant current electrolysis (either 2.4 or 4.1 mA cm−2) was started. The reaction was controlled by analyzing the crude reaction mixture periodically by 1H or 19F NMR. Once the reaction was complete, the reaction mixture was evaporated. Purification of the crude material by flash column chromatography (SiO2) by using mixtures of cyclohexane/ethyl acetate as eluents afforded the title compounds.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support by the DFG (Wa1276/17‐2) is highly appreciated. T.W. gratefully acknowledges the fellowship by the Oskar Huttunen Foundation.
T. Wirtanen, E. Rodrigo, S. R. Waldvogel, Chem. Eur. J. 2020, 26, 5592.
In memory of Jun‐ichi Yoshida.
Contributor Information
Dr. Tom Wirtanen, http://www.chemie.uni‐mainz.de/OC/AK‐Waldvogel/
Prof. Dr. Siegfried R. Waldvogel, Email: waldvogel@uni-mainz.de.
References
- 1. Obijalska E., Kaszynski P., Jankowiak A., Young V. G., Polyhedron 2011, 30, 1339–1348. [Google Scholar]
- 2. de Moor O., Dorgan C. R., Johnson P. D., Lambert A. G., Lecci C., Maillol C., Nugent G., Poignant S. D., Price P. D., Pye R. J., Storer R., Tinsley J. M., Vickers R., van Well R., Wilkes F. J., Wilson F. X., Wren S. P., Wynne G. M., Bioorg. Med. Chem. Lett. 2011, 21, 4828–4831. [DOI] [PubMed] [Google Scholar]
- 3. Loddo R., Novelli F., Sparatore A., Tasso B., Tonelli M., Boido V., Sparatore F., Collu G., Delogu I., Giliberti G., La Colla P., Bioorg. Med. Chem. 2015, 23, 7024–7034. [DOI] [PubMed] [Google Scholar]
- 4. Tanabe J., Lennartz C., Fluoroscent Organic Light Emitting Elements Having High Effiency (US 9853224 B2), 2017.
- 5. Tian Q., Chen X., Liu W., Wang Z., Shi S., Kuang C., Org. Biomol. Chem. 2013, 11, 7830–7833. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Ueda S., Su M., Buchwald S. L., Angew. Chem. Int. Ed. 2011, 50, 8944–8947; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9106–9109; [Google Scholar]
- 6b. Beletskaya I. P., Davydov D. V., Moreno-Mañas M., Tetrahedron Lett. 1998, 39, 5617–5620. [Google Scholar]
- 7. Garnier T., Danel M., Magné V., Pujol A., Bénéteau V., Pale P., Chassaing S., J. Org. Chem. 2018, 83, 6408–6422. [DOI] [PubMed] [Google Scholar]
- 8. Shi S., Kuang C., J. Org. Chem. 2014, 79, 6105–6112. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Beletskaya I. P., Davydov D. V., Moreno-Mañas M., Tetrahedron Lett. 1998, 39, 5621–5622; [Google Scholar]
- 9b. Davydov D. V., Chernyshev V. V., Rybakov V. B., Oprunenko Y. F., Beletskaya I. P., Mendeleev Commun. 2018, 28, 287–289. [Google Scholar]
- 10.
- 10a. Riedmüller S., Nachtsheim B., Synlett 2015, 26, 651–655; [Google Scholar]
- 10b. Roshandel S., Lunn M. J., Rasul G., Ravinson D. S. M., Suri S. C., Prakash G. K. S., Org. Lett. 2019, 21, 6255–6258. [DOI] [PubMed] [Google Scholar]
- 11. Liu Z., Larock R. C., J. Org. Chem. 2006, 71, 3198–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y., Yan W., Chen Y., Petersen J. L., Shi X., Org. Lett. 2008, 10, 5389–5392. [DOI] [PubMed] [Google Scholar]
- 13. Ryu T., Min J., Choi W., Jeon W. H., Lee P. H., Org. Lett. 2014, 16, 2810–2813. [DOI] [PubMed] [Google Scholar]
- 14. Li H., Deng H., Synthesis 2017, 49, 2711–2720. [Google Scholar]
- 15. Jo J., Lee H. Y., Liu W., Olasz A., Chen C.-H., Lee D., J. Am. Chem. Soc. 2012, 134, 16000–16007. [DOI] [PubMed] [Google Scholar]
- 16. Terpugova M. P., Amosov Y. I., Kotlyarevskii I. L., Russ. Chem. Bull. 1982, 31, 1040–1042. [Google Scholar]
- 17. Dyall L. K., Harvey J. J., Jarman T. B., Aust. J. Chem. 1992, 45, 371–384. [Google Scholar]
- 18. Dyall L. K., Aust. J. Chem. 1984, 37, 2013–2026. [Google Scholar]
- 19.
- 19a. Hall J. H., J. Org. Chem. 1968, 33, 2954–2956; [Google Scholar]
- 19b. Zincke T., Jaenke H., Ber. Dtsch. Chem. Ges. 1888, 21, 540–548; [Google Scholar]
- 19c. Zincke T., Lawson A. Th., Ber. Dtsch. Chem. Ges. 1887, 20, 1176–1183; [Google Scholar]
- 19d. Carboni R. A., Castle J. E., J. Am. Chem. Soc. 1962, 84, 2453–2454; [Google Scholar]
- 19e. Carboni R. A., Kauer J. C., Castle J. E., Simmons H. E., J. Am. Chem. Soc. 1967, 89, 2618–2625. [Google Scholar]
- 20. Spagnolo P., Zanirato P., J. Chem. Soc. Perkin Trans. 1 1988, 2615–2620. [Google Scholar]
- 21. Khatun N., Modi A., Ali W., Patel B. K., J. Org. Chem. 2015, 80, 9662–9670. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Shang X., Zhao S., Chen W., Chen C., Qiu H., Chem. Eur. J. 2014, 20, 1825–1828; [DOI] [PubMed] [Google Scholar]
- 22b. Li J., Cong W., Gao Z., Zhang J., Yang H., Jiang G., Org. Biomol. Chem. 2018, 16, 3479–3486. [DOI] [PubMed] [Google Scholar]
- 23. Nykaza T. V., Harrison T. S., Ghosh A., Putnik R. A., Radosevich A. T., J. Am. Chem. Soc. 2017, 139, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Dong J., Jin B., Sun P., Org. Lett. 2014, 16, 4540–4542; [DOI] [PubMed] [Google Scholar]
- 24b. Liu G.-B., Zhao H.-Y., Yang H.-J., Gao X., Li M.-K., Thiemann T., Adv. Synth. Catal. 2007, 349, 1637–1640; [Google Scholar]
- 24c. Recca A., Libertini E., Finocchiaro P., Munro H. S., Clark D. T., Macromolecules 1988, 21, 2641–2642. [Google Scholar]
- 25. Kim B. H., Kim S. K., Lee Y. S., Jun Y. M., Baik W., Lee B. M., Tetrahedron Lett. 1997, 38, 8303–8306. [Google Scholar]
- 26. Wilshire J. F. K., Aust. J. Chem. 1988, 41, 617–622. [Google Scholar]
- 27.
- 27a. Rosevear J., Wilshire J. F. K., Aust. J. Chem. 1984, 37, 2489–2497; [Google Scholar]
- 27b. Tanimoto S., Kamano T., Synthesis 1986, 1986, 647–649. [Google Scholar]
- 28. Baik W., Park T. H., Kim B. H., Jun Y. M., J. Org. Chem. 1995, 60, 5683–5685. [Google Scholar]
- 29. Pizzotti M., Cenini S., Psaro R., Costanzi S., J. Mol. Catal. 1990, 63, 299–304. [Google Scholar]
- 30. Li J., Zhou H., Zhang J., Yang H., Jiang G., Chem. Commun. 2016, 52, 9589–9592. [DOI] [PubMed] [Google Scholar]
- 31.For extensive list of methods in the patent literature, please refer to Ref. 24 b.
- 32. Elbs K., Keiper W., J. Prakt. Chem. 1902, 67, 580–584. [Google Scholar]
- 33. Hyo Kim B., Byung Lee D., Ho Kim D., Han R., Moo Jun Y., Baik W., Heterocycles 2000, 53, 841–850. [Google Scholar]
- 34. Houghton P. G., Pipe D. F., Rees C. W., J. Chem. Soc. Perkin Trans. 1 1985, 1471–1479. [Google Scholar]
- 35.
- 35a. Frontana-Uribe B. A., Little R. D., Ibanez J. G., Palma A., Vasquez-Medrano R., Green Chem. 2010, 12, 2099–2119; [Google Scholar]
- 35b. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35c. Jiang Y., Xu K., Zeng C., Chem. Rev. 2018, 118, 4485–4540; [DOI] [PubMed] [Google Scholar]
- 35d. Kärkäs M. D., Chem. Soc. Rev. 2018, 47, 5786–5865; [DOI] [PubMed] [Google Scholar]
- 35e. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149; [Google Scholar]
- 35f. Wiebe A., Gieshoff T., Möhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594–5619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694–5721. [Google Scholar]
- 36. Waldvogel S. R., Lips S., Selt M., Riehl B., Kampf C. J., Chem. Rev. 2018, 118, 6706–6765. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Rodrigo E., Waldvogel S. R., Green Chem. 2018, 20, 2013–2017; [Google Scholar]
- 37b. Rodrigo E., Waldvogel S. R., Chem. Sci. 2019, 10, 2044–2047; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37c. Rodrigo E., Baunis H., Suna E., Waldvogel S. R., Chem. Commun. 2019, 55, 12255–12258. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Gütz C., Grimaudo V., Holtkamp M., Hartmer M., Werra J., Frensemeier L., Kehl A., Karst U., Broekmann P., Waldvogel S. R., ChemElectroChem 2018, 5, 247–252; [Google Scholar]
- 38b. Gütz C., Bänziger M., Bucher C., Galvão T. R., Waldvogel S. R., Org. Process Res. Dev. 2015, 19, 1428–1433; [Google Scholar]
- 38c. Gütz C., Selt M., Bänziger M., Bucher C., Römelt C., Hecken N., Gallou F., Galvão T. R., Waldvogel S. R., Chem. Eur. J. 2015, 21, 13878–13882; [DOI] [PubMed] [Google Scholar]
- 38d. Gálvez-Vázquez M. d. J., Moreno-García P., Guo H., Hou Y., Dutta A., Waldvogel S. R., Broekmann P., ChemElectroChem 2019, 6, 2324–2330. [Google Scholar]
- 39.
- 39a. Gütz C., Klöckner B., Waldvogel S. R., Org. Process Res. Dev. 2016, 20, 26–32; [Google Scholar]
- 39b. Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 13325–13329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13509–13513; [Google Scholar]
- 39c. Lips S., Frontana-Uribe B. A., Dörr M., Schollmeyer D., Franke R., Waldvogel S. R., Chem. Eur. J. 2018, 24, 6057–6061; [DOI] [PubMed] [Google Scholar]
- 39d. Wiebe A., Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 14727–14731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14920–14925; [Google Scholar]
- 39e. Lips S., Wiebe A., Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 10872–10876; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11031–11035; [Google Scholar]
- 39f. Wiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 11801–11805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11979–11983. [Google Scholar]
- 40. Rosevear J., Wilshire J. F. K., Aust. J. Chem. 1987, 40, 1663–1673. [Google Scholar]
- 41. Kwon Y., Lai S. C. S., Rodriguez P., Koper M. T. M., J. Am. Chem. Soc. 2011, 133, 6914–6917. [DOI] [PubMed] [Google Scholar]
- 42.Some electroreductive intramolecular cyclizations of nitro derivatives have been reported to proceed through phenylhydroxylamine intermediates that are oxidized to nitroso-derivatives. See for example: Frontana-Uribe B. A., Moinet C., Tetrahedron 1998, 54, 3197–3206. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary