


Abstract
The identification of new targeted and personalized therapies for cancer requires the fast and accurate assessment of the drug efficacy of potential compounds against a particular biomolecular sample. It has been suggested that the integration of complementary sources of information might strengthen the accuracy of a drug efficacy prediction model. Here, we present a web-based platform for the Prediction of AntiCancer Compound sensitivity with Multimodal Attention-based Neural Networks (PaccMann). PaccMann is trained on public transcriptomic cell line profiles, compound structure information and drug sensitivity screenings, and outperforms state-of-the-art methods on anticancer drug sensitivity prediction. On the open-access web service (https://ibm.biz/paccmann-aas), users can select a known drug compound or design their own compound structure in an interactive editor, perform in-silico drug testing and investigate compound efficacy on publicly available or user-provided transcriptomic profiles. PaccMann leverages methods for model interpretability and outputs confidence scores as well as attention heatmaps that highlight the genes and chemical sub-structures that were more important to make a prediction, hence facilitating the understanding of the model’s decision making and the involved biochemical processes. We hope to serve the community with a toolbox for fast and efficient validation in drug repositioning or lead compound identification regimes.
INTRODUCTION
Despite major investments in drug discovery and drug repurposing, 97% of anticancer candidate drugs fail in clinical trials and never receive food & drug administration (FDA) approval (1). This low success rate has been attributed to either lacking target efficacy or off-target cytotoxicity (2). Indeed, it has been very recently demonstrated that off-target cytotoxicity is not only a frequent, undesired ancillary effect, but also a common mechanism of action of anticancer drugs in clinical trials (3). Furthermore, in cancer precision medicine, high heterogeneity in patients molecular makeup typically results in even higher drug response variability (4). Hence, potent insilico and invitro models that can accurately predict the effect of new candidate drugs on a specific biomolecular profile are necessary to enable personalized therapies.
With the rise of deep learning in drug discovery (5), works that integrate omics information and compound chemical descriptors have been proposed to predict drug sensitivity on specific cell lines (6,7). Many of these works represent compound chemical information using molecular fingerprints descriptors, which have been extensively used in drug discovery, virtual screening and compound similarity search (8). However, the usage of engineered features might constrain the learning ability of machine learning algorithms or not even by available for new compounds. An alternative compound representation is the Simplified Molecular-Input Line-Entry System (SMILES) (9). SMILES allow structure specification by using a small chemical grammar, and are preferred over hand-engineered descriptors such as molecular fingerprints, as they are ubiquitously available, easier to interpret, closer to the actual molecular entity, and furthermore, they enable efficient data augmentation (10).
To address the limitations of previous works and enable the efficient integration of molecular and chemical information in the form of SMILES, we developed in the past the Prediction of AntiCancer Compound sensitivity with Multimodal Attention-based Neural Networks (PaccMann) (11). PaccMann is a multimodal deep learning model for drug sensitivity prediction that integrates three key pillars of information: compounds’ structure in the form of a SMILES sequence, gene expression profiles (GEP) of tumors, and prior knowledge on intracellular interactions from protein–protein interaction networks to predict drug sensitivity as measured by IC50 (half maximal inhibitory concentration) on drug-cell-pairs (Figure 1). The predictions are complemented with two key mechanisms that render the model transparent and interpretable, which is becoming of paramount importance in precision medicine (12). First, PaccMann employs a novel contextual attention mechanism (11) that highlights the most informative genes and compound sub-structures to make a prediction. Second, for each prediction, the model executes a confidence estimate that is computed by measuring aleatoric (or data) uncertainty and epistemic (or model) uncertainty (13).
Figure 1.

PaccMann framework for multimodal prediction of IC50 drug sensitivity. Three key data modalities that influence anticancer drug sensitivity are integrated: biomolecular measurements of cancer cells, e.g. gene expression data, a network of known interactions between the biomolecular entities and the chemical structure of the anticancer compounds (SMILES strings or molecular fingerprints).
Here, we present PaccMann web service (https://ibm.biz/paccmann-aas), an open-access, web-based platform that serves as an access point for the PaccMann model. In the following, we introduce PaccMann’s web application and present a brief evaluation of the model performance, showing that it surpasses state-of-the-art results in anticancer drug sensitivity prediction. At last, we demonstrate in two case studies how PaccMann web service can be applied to drug repurposing and can provide useful insights about a drug mode of action.
METHODOLOGY AND DATA PORTAL
Data
We pooled together the drug sensitivity screening results from two publicly available databases, namely Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Cell Line Encyclopedia (CCLE) (14,15). From GDSC, we retrieved 385 712 IC50 values from 397 compounds (including both target drugs and chemotherapeutics), each of them screened on a subset of 988 pan-cancer cell lines. Since the SMILES representation for 17 of the GDSC compounds was not available, we discarded the associated IC50 values. From CCLE, an additional 325 375 samples from 514 compounds measured on 1038 cell lines were retrieved. The total database consisted of 688 308 samples with drug sensitivity represented as log micromolar IC50. For all investigated cell lines, transcriptomic data (RMA gene expression) of around 20 000 genes were retrieved and favored over genomics data due to its higher predictive power (16). In order to sub-select the most informative genes, a network propagation procedure (17) was applied over the PPI network STRING (18). By restricting to the top 20 neighbors of every drug, an interaction-aware subset of 2128 genes representing ostensible drug targets was assembled (17) (more details about network propagation can be found in the Supplementary Material). The expression values of CCLE and GDSC cell lines were standardized individually prior to applying the ComBat method to remove batch effects (19) (see the Supplementary Materials for more details on the preprocessing).
Model
The implemented neural network architecture is a multiscale convolutional attention (MCA) encoder, as described in (11). This model combines convolutions of varying receptive field sizes over the SMILES sequences with contextual and self-attention mechanisms. The transcriptomics data are processed by a self-attention layer, which highlights the most relevant genes for the current sample, prior to a contextual attention mechanism on the SMILES sequences that aligns the genes with molecular substructures (e.g. toxicophores) with high-predictive power. Moreover, the model is equipped with two simple techniques to assess its own prediction confidence. First, to estimate epistemic uncertainty, we apply Monte Carlo dropout (i.e. dropout during test time) on SMILES sequences, which draws Monte Carlo samples from the approximate predictive posterior (20). Second, to estimate aleatoric uncertainty, we apply test time data augmentation (21). In both cases, 10 forward passes are computed for each drug-cell-pair and the sample standard deviation (i.e. inverse precision) is interpreted as an approximation of the predictive posterior (for details see Supplementary Material).
Data splitting strategy and training regime
Previous work that integrated compound structure information with omics data for drug sensitivity prediction utilized lenient splitting strategies that ensured no drug-cell pair in the test data were seen during training (6,7). However, PaccMann adopts a more stringent splitting strategy. Namely, the PaccMann model was validated following a 25-fold cross validation strategy with completely disjoint sets of both compounds and cell lines in every fold. In this regime, neither any of the drugs nor any of the cell lines in the test set were encountered by the model during training, forcing the model to learn generic relations between transcriptomic information and molecular substructures with anticancer properties and leading to better generalization capabilities. This approach tackles jointly the two complementary goals of drug sensitivity prediction models, namely, the need for a predictive model that generalizes to unseen cancer cell lines (precision medicine regime) and a model that generalizes to unseen compounds (drug discovery regime).
Validation of PaccMann on benchmark datasets
The MCA architecture of PaccMann was thoroughly validated in (11). The method achieves high prediction performance (R2 = 0.86 and RMSE =0.89, coefficient of determination with experimentally determined IC50 values and root mean square error respectively), outperforming several proposed deep learning models as well as previously reported state-of-the-art results for multimodal drug sensitivity prediction. Recently, this has been corroborated independently in an extensive benchmark study (22) that found PaccMann to outperform a stack of 28 deep learning and non-deep learning approaches, and found almost on-par performance on the specific task of protein kinase inhibitor response prediction.
For a more detailed description of the neural network architecture, the data (preprocessing) and the evaluation, please see the associated publications (11,23).
Web application
The PaccMann web application integrates basic functionalities, such as data assembly, upload, processing, inference and interactive visualization using Bokeh (24).
Input and output data
A compound structure can either be provided by selecting a drug from a drop-down menu of existing compounds, by inserting a valid SMILES string in the input field, by interactively drawing a compound in the molecular editor or, for bulk processing, by uploading a file of the format {.smi, .smiles, .json, .rxn, .mol, .sd, .sdf, .cml, .kcj, .kcx} (see Figure 2).
Figure 2.
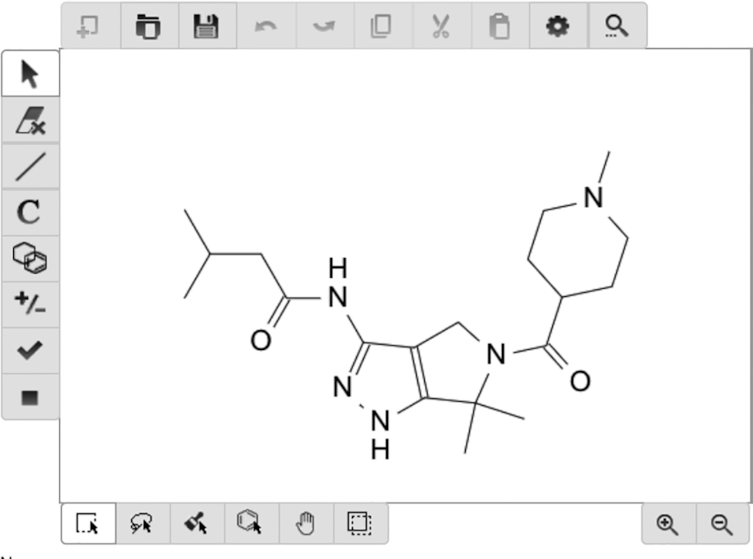
PaccMann molecular editor. Compound structure can be provided in various forms, including single or bulk SMILES or through an interactive molecule editor.
The left panel of the molecule editor contains several tools for editing the molecule, e.g. the erase tool, which erases specific atoms or bonds, the bond tool, which adds a bond from a set of typical bonds, the ring structure tool, which adds a ring from a set of typical rings and the atom and formula tool, which allows to insert new atoms. Upon finishing the molecule design the user can confirm the molecule and run the prediction or simply download and export the molecule into one of the aforementioned formats. Optionally, GEP of a single or multiple cancer cell lines can be uploaded in .csv format. The data matrix is expected to have samples on rows, genes on columns and a header reporting the entity names, as follows:
LAMP3,DDR1,...,GCNT1
-0.48,0.32,...,1.473
-0.89,0.12,...,0.238
The header should specify all 2128 gene names required as model input. The gene list can be downloaded from https://ibm.box.com/v/paccmann-aas-gene-list. Supersets and random permutations of column names are also accepted. Missing genes are mean-imputed and feature-wise standardization is applied. In the absence of a user-defined specification of the transcriptomic profile, the IC50 value for all 2022 cancer cell lines from GDSC and CCLE are predicted simultaneously. Additional column names will be considered as metadata that will be used to complement the prediction results
Following model inference, the IC50 predictions are displayed in an interactive table where columns report: id, the sample identifier (numeric) in the dataset considered; IC50 (min/max scaled), the IC50 score transformed to a unitary scale for each (drug, sample) pair (as seen by the model); IC50 (log(μmol)), the IC50 in logarithmic μmol units for each (drug, sample) pair. In case no omic data are uploaded, the web service provides predictions for a panel of 2022 cell lines (from GDSC and CCLE) and add the following metadata to the table: histology, histological information of the cell line; site, the tissue origin of the cell line; cosmic_id, the COSMIC id of the cell line when available; cell_line, the cell line identifier when available; cell_line_name, the name of the cell line; and dataset, the dataset containing the cell line (GDSC or CCLE). If the user provides custom omic data, the columns of the csv uploaded that are not matching the gene names from the panel considered will be used as metadata in the results table. In case the user enables the prediction confidence estimates, two additional columns will be displayed: epistemic_confidence, the confidence estimated via Monte Carlo dropout; and aleatoric_confidence, the confidence estimated using test time augmentation.
The table can be customized by sorting or filtering individual columns, and results can be exported into csv format.
Upon user request, the service lays over the tested compound the SMILES attention distribution that highlights the molecular sub-structures most relevant for the prediction as shown in (Figure 2). Similarly, the most attended genes and their proportional contribution to the prediction are shown in an interactive barplot. Both attention plots are computed based on an averaging of all samples of the complementary data modality.
User data storage
All entities provided by the users (compound structure and omics profiles) are temporarily stored to run the selected methods and are never persisted in a cloud object storage (COS). Compound structure, predictions and visualizations are stored for one week in the COS and can be accessed for inspection or download by the user that submitted the task.
Asynchronous task system
The service runs asynchronously using a queuing system for the submitted inference tasks. The task queue is dispatched to a set of containerized workers that run the model inference and generate interactive visualizations upon request. The web service will automatically allow the user to open the result visualization tab once the predictions for the uploaded data are completed.
Runtime
The current release offers a selection of methods with different computational complexities. Each forward pass for a compound-cell line pair takes roughly 0.15 s. When no omic data are uploaded, the web service performs a single forward pass of all given compounds for all 2022 cell lines, which can take up to 5 min. On demand, the user can execute the confidence score prediction, which can require several minutes, since multiple forward passes are computed.
CASE STUDIES
Repurposing of Temsirolimus
Temsirolimus is a mTOR and serine/threonine kinases inhibitor that is FDA approved for renal cell carcinoma (an aggressive type of kidney cancer). It was the first discovered clinically effective mTOR inhibitor against mantle cell lymphoma for which it is also approved in Europe. Using PaccMann, Temsirolimus is predicted as effective (IC50 < 1 μm) for a wide range of cell lines, with lowest IC50 on ovary, lung and stomach cancer cell lines Table 1. Furthermore, Temsirolimus is predicted as effective (IC50 < 1 μm) for 74% of the stomach cell lines, 52% of lung and 48% of ovary cell lines—highest proportions across all cancer sites with more than 30 samples, excepting leukemia.
Table 1.
Predictions for Temsirolimus
| Site | Cell line | IC50 [log(μmol)] |
|---|---|---|
| Ovary | A2780 | −3.06 |
| Lung | NCIH1975 | −3.42 |
| Stomach | NCCSTCK140 | −3.12 |
Since mTOR is a key pathway for cell proliferation (25) and mTOR is well known to be dysregulated in many forms of lung cancer, it has been suggested to repurpose mTOR inhibitor novel lung cancer treatments (26–28). However, the only FDA approved mTOR inhibitor for lung cancer is Everolimus, another analog of Rapamycin, that is not part of the GDSC/CCLE database used to train the model. Indeed, Temsirolimus was only recently repurposed for in-vitro and in-vivo studies of lung adenocarcinoma, where it showed desired inhibitory effects, especially when synergistically paired with the chemotherapeutical Cisplatin (29).
mTOR also plays a prominent role in gastric cancer (over-active in 60% of the patients (30)) but to date, no approved mTOR inhibitor for gastric cancer exists. Very recently however, several mTOR inhibitors have been tested in patient-derived xenografts of gastric cancer, which revealed Temsirolimus as the most effective mTOR inhibitor against gastric cancer (31). Since Temsirolimus has already been tested in clinical trials against ovarian cancer, where it did not showed the desired effects (32,33), it may seem reasonable to follow instead the gastric or lung cancer predictions as possible repurposing options for Temsirolimus.
Analysis of attention mechanism
Based on predictions on the entire test samples of the GDSC dataset, we extracted a subset of 371 (out of 2128) highly attended genes (23). A pathway enrichment analysis (34) revealed a significant activation (adjusted P < 0.004) of the apoptosis signaling pathway in PANTHER (35), as expected from anticancer drugs. In Figure 3 we show the gene attention weights for two kidney carcinoma cell lines, ACHN and RCC10RGB. Although the cell lines are genomically distinct, both computed attention weights underline the role of EIF2A, a key gene for tumor initiation (36).
Figure 3.
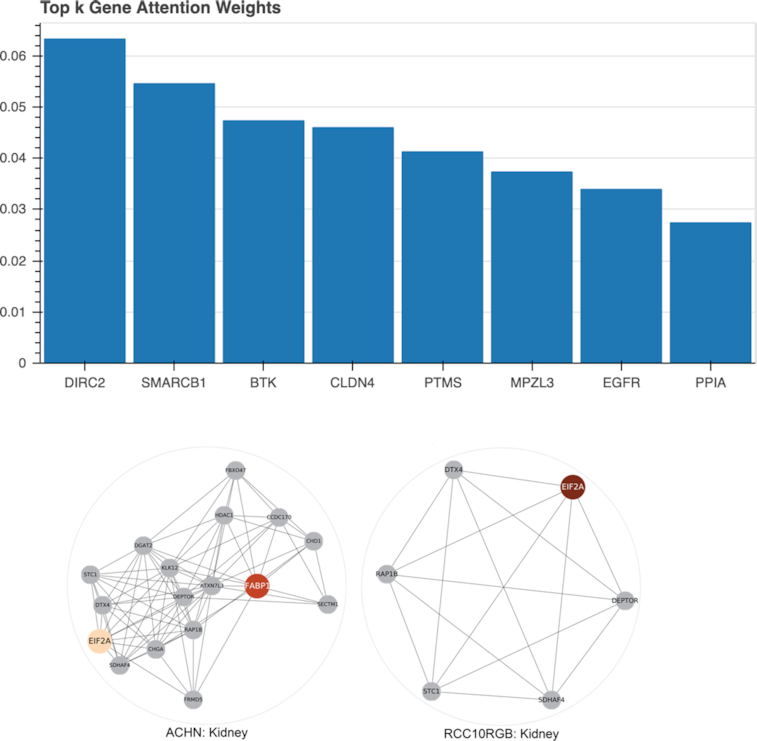
Visualized gene attention weights. Top: The averaged gene attention weights across all cell lines from the panel using the interactive visualization available in the web service. Bottom: Gene attention weights for two kidney cancer cell lines, ACHN (left) and RCC10RGB (right), are displayed using an orange color map. The colored genes received highest attention weights and are displayed with neighboring genes according to the STRING network. This network visualization is not available in the web service.
In Figure 4, we report the molecular attention computed on a chronic myelogenous leukaemia (CML) cell line for two very similar anticancer compounds (Imatinib and Masitinib) (11). The attention weight patterns change in the thiazole and the piperazine group, whereas the remaining regions are unaffected (Pearson’s R = 0.96 and P < 6e − 20 outside functional group and R = 0.29 and P > 0.2 inside functional group). The localized discrepancy in attention centered at the different rings suggests that these substructures drive the sensitivity prediction for the two compounds on the CML cell line. A more quantitative analysis of the attention weights can be found in (11).
Figure 4.

Visualization of the SMILES attention weights. Neural attention on molecules available in the web service. The molecular attention maps demonstrate how the model’s attention is shifted when the thiazole group in Masitinib is replaced by a piperazine group in Imatinib. The change in attention across the two molecules is particularly concentrated around the affected rings, signifying that these functional groups play an important role in the mechanism of action for these tyrosine kinase inhibitors when they act on a CML cell line.
Comparison to existing web servers
There are several existing web servers dedicated related although different drug discovery tasks, such as predicting synergistic effects of cancer drug combination treatment, e.g. DrugComb (37), DeepSynergy (38) or SynergyFinder (39). In addition, DrugMint (40) is a server for predicting drug-likelihood of a compound, although not specifically designed for cancer. Drug ReposER is a database of protein folding structure information that aims to facilitate drug repurposing by sub-structure similarity search (41). At last, way2drug is a web service for predicting cytotoxic effects of chemicals (42). However, their so-called PASS model (based on naive-Bayes) is not specifically dedicated to cancer. Moreover, it does not allow prediction of IC50 scores on the molar scale and can not handle new cell lines (one model per cell-line); and it does not provide any explanation of the results.
DISCUSSION
We have presented here a user-friendly web service called PaccMann. PaccMann provides state-of-the-art performance at the task of predicting drug sensitivity for any pair of compound structure, represented as SMILES and transcriptomic profile, represented by means of 2128 preselected genes. In two case studies, we have shown how PaccMann may be used for drug repositioning while providing interpretable insights about the input features that were more informative to make a prediction.
PaccMann can facilitate in-silico studies of drug efficacy and repurposing of anticancer drugs for novel cancer types. For example, PaccMann can be used for applications in medicinal chemistry that involve comparative analyses such as scaffold hopping, i.e. exploring the chemical space locally around a promising scaffold (43). Furthermore, PaccMann can pave the way for future applications supporting drug discovery, such as small molecule generation using generative models and reinforcement learning to target specific molecular profiles (44). This opens up a scenario where personalized treatments and therapies can become a concrete option for patient care in cancer precision medicine.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the community for using PaccMann and providing us with valuable feedback. We appreciate the comments and suggestions of the anonymous reviewers who helped improve and clarify this manuscript and the PaccMann web service.
Contributor Information
Joris Cadow, Computational Systems Biology Group, IBM Research Europe, Säumerstrasse 4, Rüschlikon, 8803, Switzerland.
Jannis Born, Computational Systems Biology Group, IBM Research Europe, Säumerstrasse 4, Rüschlikon, 8803, Switzerland; Machine Learning & Computational Biology Lab, D-BSSE, ETH Zürich, Mattenstrasse 26, Basel, 4058, Switzerland.
Matteo Manica, Computational Systems Biology Group, IBM Research Europe, Säumerstrasse 4, Rüschlikon, 8803, Switzerland.
Ali Oskooei, Computational Systems Biology Group, IBM Research Europe, Säumerstrasse 4, Rüschlikon, 8803, Switzerland.
María Rodríguez Martínez, Computational Systems Biology Group, IBM Research Europe, Säumerstrasse 4, Rüschlikon, 8803, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
H2020 Societal Challenges [826121]. Funding for open access charge: Own funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wong C.H., Siah K.W., Lo A.W.. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019; 20:273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wehling M. Assessing the translatability of drug projects: what needs to be scored to predict success. Nat. Rev. Drug. Discov. 2009; 8:541–546. [DOI] [PubMed] [Google Scholar]
- 3. Lin A., Giuliano C.J., Palladino A., John K.M., Abramowicz C., Yuan M.L., Sausville E.L., Lukow D.A., Liu L., Chait A.R. et al.. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019; 11:eaaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Geeleher P., Cox N.J., Huang R.S.. Cancer biomarker discovery is improved by accounting for variability in general levels of drug sensitivity in pre-clinical models. Genome Biol. 2016; 17:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen H., Engkvist O., Wang Y., Olivecrona M., Blaschke T.. The rise of deep learning in drug discovery. Drug Discov. Today. 2018; 23:1241–1250. [DOI] [PubMed] [Google Scholar]
- 6. Menden M.P., Iorio F., Garnett M., McDermott U., Benes C.H., Ballester P.J., Saez-Rodriguez J.. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One. 2013; 8:e61318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang Y., Park H., Yang H.-J., Lee S., Lee K.-Y., Kim T.S., Jung J., Shin J.-M.. Cancer drug response profile scan (CDRscan): a deep learning model that predicts drug effectiveness from cancer genomic signature. Sci. Rep. 2018; 8:8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cereto-Massagué A., Ojeda M.J., Valls C., Mulero M., Garcia-Vallvé S., Pujadas G.. Molecular fingerprint similarity search in virtual screening. Methods. 2015; 71:58–63. [DOI] [PubMed] [Google Scholar]
- 9. Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comp. Sci. 1988; 28:31–36. [Google Scholar]
- 10. Bjerrum E.J. SMILES enumeration as data augmentation for neural network modeling of molecules. 2017; arXiv doi:17 May 2017, preprint: not peer reviewedhttps://arxiv.org/abs/1703.07076.
- 11. Manica M., Oskooei A., Born J., Subramanian V., Saez-Rodriguez J., Rodriguez Martinez M.. Toward explainable anticancer compound sensitivity prediction via multimodal attention-based convolutional encoders. Mol. Pharm. 2019; 16:4757–5086. [DOI] [PubMed] [Google Scholar]
- 12. Gilvary C., Madhukar N., Elkhader J., Elemento O.. The missing pieces of artificial intelligence in medicine. Trends Pharmacol. Sci. 2019; 40:555–564. [DOI] [PubMed] [Google Scholar]
- 13. Kendall A., Gal Y.. Guyon I., Luxburg U.V., Bengio S., Wallach H., Fergus R., Vishwanathan S., Garnett R.. What uncertainties do we need in bayesian deep learning for computer vision?. Advances in Neural Information Processing Systems 30. 2017; Red Hook, NY: Curran Associates, Inc; 5574–5584. [Google Scholar]
- 14. Yang W., Soares J., Greninger P., Edelman E.J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J.A., Thompson I.R. et al.. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012; 41:D955–D961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. et al.. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Costello J.C., Heiser L.M., Georgii E., Gönen M., Menden M.P., Wang N.J., Bansal M., Hintsanen P., Khan S.A., Mpindi J.-P. et al.. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014; 32:1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oskooei A., Manica M., Mathis R., Rodríguez Martínez M.. Network-based biased tree ensembles (NetBiTE) for drug sensitivity prediction and drug sensitivity biomarker identification in cancer. Sci. Rep. 2019; 9:15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. et al.. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014; 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson W.E., Li C., Rabinovic A.. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007; 8:118–127. [DOI] [PubMed] [Google Scholar]
- 20. Gal Y., Ghahramani Z.. Balcan F.B., Weinberger K.Q.. Dropout as a bayesian approximation: representing model uncertainty in deep learning. Proceedings of The 33rd International Conference on Machine Learning. 2016; 48:NY: PMLR; 1050–1059. [Google Scholar]
- 21. Ayhan M.S., Berens P.. Test-time data augmentation for estimation of heteroscedastic aleatoric uncertainty in deep neural networks. International conference on Medical Imaging with Deep Learning. 2018; 1–9. [Google Scholar]
- 22. Huang L.-C., Yeung W., Wang Y., Cheng H., Venkat A., Li S., Ma P., Rasheed K., Kannan N.. Quantitative Structure-Mutation-Activity Relationship Tests (QSMART) Model for Protein Kinase Inhibitor Response Prediction. 2019; bioRxiv doi:08 December 2019, preprint: not peer reviewed 10.1101/868067. [DOI] [PMC free article] [PubMed]
- 23. Oskooei A., Born J., Manica M., Subramanian V., Sáez-Rodríguez J., Rodríguez Martínez M.. PaccMann: Prediction of anticancer compound sensitivity with multi-modal attention-based neural networks. 2018; arXiv doi:14 July 2019, Workshop on Machine Learning for Molecules and Materials in NeurIPS 2018https://arxiv.org/abs/1811.06802.
- 24. Bokeh Development Team Bokeh: Python library for interactive visualization. 2019; https://bokeh.org. [Google Scholar]
- 25. Zaytseva Y.Y., Valentino J.D., Gulhati P., Evers B.M.. mTOR inhibitors in cancer therapy. Cancer Lett. 2012; 319:1–7. [DOI] [PubMed] [Google Scholar]
- 26. Ohara T., Takaoka M., Toyooka S., Tomono Y., Nishikawa T., Shirakawa Y., Yamatsuji T., Tanaka N., Fujiwara T., Naomoto Y.. Inhibition of mTOR by temsirolimus contributes to prolonged survival of mice with pleural dissemination of non-small-cell lung cancer cells. Cancer Sci. 2011; 102:1344–1349. [DOI] [PubMed] [Google Scholar]
- 27. Gridelli C., Maione P., Rossi A.. The potential role of mTOR inhibitors in non-small cell lung cancer. Oncologist. 2008; 13:139–147. [DOI] [PubMed] [Google Scholar]
- 28. Vicary G.W., Roman J.. Targeting the mammalian target of rapamycin in lung cancer. Am. J. Med. Sci. 2016; 352:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang H.-W., Wu M.-J., Lin Z.-M., Wang C.-Y., Cheng S.-Y., Lin Y.-K., Chow Y.-H., Ch’ang H.-J., Chang V.H.. Therapeutic effect of repurposed temsirolimus in lung adenocarcinoma model. Front. Pharmacol. 2018; 9:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riquelme I., Tapia O., Espinoza J.A., Leal P., Buchegger K., Sandoval A., Bizama C., Araya J.C., Peek R.M., Roa J.C.. The gene expression status of the PI3K/AKT/mTOR pathway in gastric cancer tissues and cell lines. Pathol. Oncol. Res. 2016; 22:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fukamachi H., Kim S.-K., Koh J., Lee H.S., Sasaki Y., Yamashita K., Nishikawaji T., Shimada S., Akiyama Y., Byeon S.-J. et al.. A subset of diffuse-type gastric cancer is susceptible to mTOR inhibitors and checkpoint inhibitors. J. Exp. Clin. Canc. Res. 2019; 38:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emons G., Kurzeder C., Schmalfeldt B., Reuss A., de Gregorio N., Pfisterer J., Park-Simon T.-W., Mahner S., Schroeder W., Lueck H.-J. et al.. Temsirolimus in women with platinum-resistant ovarian cancer or advanced/recurrent endometrial cancer: a multicenter phase II trial of the AGO Study Group (AGO-GYN 8). J .Clin. Oncol. 2014; 32:5565–5565. [Google Scholar]
- 33. Emons G., Kurzeder C., Schmalfeldt B., Neuser P., De Gregorio N., Pfisterer J., Park-Simon T.-W., Mahner S., Schröder W., Lück H.-J. et al.. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol. Oncol. 2016; 140:450–456. [DOI] [PubMed] [Google Scholar]
- 34. Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. et al.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D.. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2016; 45:D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sendoel A., Dunn J.G., Rodriguez E.H., Naik S., Gomez N.C., Hurwitz B., Levorse J., Dill B.D., Schramek D., Molina H. et al.. Translation from unconventional 5 start sites drives tumour initiation. Nature. 2017; 541:494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zagidullin B., Aldahdooh J., Zheng S., Wang W., Wang Y., Saad J., Malyutina A., Jafari M., Tanoli Z., Pessia A. et al.. DrugComb: an integrative cancer drug combination data portal. Nucleic Acids Res. 2019; 47:W43–W51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Preuer K., Lewis R.P., Hochreiter S., Bender A., Bulusu K.C., Klambauer G.. DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics. 2018; 34:1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ianevski A., He L., Aittokallio T., Tang J.. SynergyFinder: a web application for analyzing drug combination dose–response matrix data. Bioinformatics. 2017; 33:2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dhanda S.K., Singla D., Mondal A.K., Raghava G.P.. DrugMint: a webserver for predicting and designing of drug-like molecules. Biol. Direct. 2013; 8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ab Ghani N.S., Ramlan E.I., Firdaus-Raih M.. Drug ReposER: a web server for predicting similar amino acid arrangements to known drug binding interfaces for potential drug repositioning. Nucleic Acids Res. 2019; 47:W350–W356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lagunin A.A., Dubovskaja V.I., Rudik A.V., Pogodin P.V., Druzhilovskiy D.S., Gloriozova T.A., Filimonov D.A., Sastry N.G., Poroikov V.V.. CLC-Pred: a freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS One. 2018; 13:e0191838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Böhm H.-J., Flohr A., Stahl M.. Scaffold hopping. Drug Discov. Today Technol. 2004; 1:217–224. [DOI] [PubMed] [Google Scholar]
- 44. Born J., Manica M., Oskooei A., Cadow J., Rodríguez Martínez M.. PaccMannRL: Designing anticancer drugs from transcriptomic data via reinforcement learning. Research in Computational Molecular Biology (RECOMB) Proceedings 24. 2020; Springer International Publishing; 231–233. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.