

Abstract
Autism spectrum disorders (ASDs) are increasingly being diagnosed. Hypotheses link ASD to genetic, epigenetic, or environmental factors. The role of oxidative stress and the imbalance between excitatory and inhibitory neurotransmission in the pathogenesis of ASD has been suggested. Rats in which ASD symptoms are induced by valproate (VPA) or thalidomide (THAL) application in utero are useful models in ASD studies. Our study investigated whether rats in ASD models show changes in metabolite levels in the brain consistent with the hypothetical pathomechanisms of ASD. Female rats were fed one dose of 800 mg/kg VPA or 500 mg/kg THAL orally on the 11th day of gestation, and 1-month offspring were used for the experiments. Metabolic profiles from proton nuclear magnetic resonance spectroscopy of hydrophilic and hydrophobic extracts of rat hippocampi were subjected to OPLS-DA statistical analysis. Large differences between both models in the content of several metabolites in the rat hippocampus were noticed. The following metabolic pathways were identified as being disturbed in both ASD models: steroid hormone biosynthesis; fatty acid biosynthesis; the synthesis and degradation of ketone bodies; glycerophospholipid metabolism; cholesterol metabolism; purine metabolism; arginine and proline metabolism; valine, leucine, and isoleucine biosynthesis and degradation. These results indicate disorders of energy metabolism, altered structure of cell membranes, changes in neurotransmission, and the induction of oxidative stress in the hippocampus. Our data, consistent with hypotheses of ASD pathomechanisms, may be useful in future ASD studies, especially for the interpretation of the results of metabolomics analysis of body fluids in rat ASD models.
Keywords: Autism, Metabolomics, NMR spectroscopy
Introduction
Autism spectrum disorder (ASD) is an increasingly emerging disease that appears worldwide, and the prevalence of ASD ranges from 25 to 110 cases per 10,000 children, depending on the country [1]. It seems that ASD occurs 2–3 times more often in boys than that in girls [2]. The etiology of autism disease is complex and not yet fully explained. There are hypotheses linking symptoms of ASD with genetic [3–5], epigenetic, or environmental factors [1]. Epigenetic factors could modify the expression of mRNA or miRNA, which could affect the conformation and content of proteins involved in body functions [6]. Environmental factors, due to their toxicity, could affect not only young subjects but also fetuses through toxic influences on pregnant mothers [7–9]. Drugs [1], as well as disturbances in the levels of metal ions (i.e., zinc) [10, 11], in hormonal systems [12] and in brain amino acid–mediated neurotransmission [13] could also be implicated in ASD etiology and/or pathogenesis. According to this interpretation, behavioral disorders observed in ASD may result from changes at the level of gene expression and in the level and activity of specific proteins, which may consequently be reflected in changes in the level of certain metabolites in the brain.
There are several analytical methods that allow us to study the abovementioned factors. One of them is nuclear magnetic resonance (NMR). Magnetic resonance spectroscopy (MRS) can be applied in vivo to examine the content of various metabolites in the brains of autistic children, while NMR can be used ex vivo to study biofluids such as serum, urine, or saliva [14, 15]. MRS is a low-resolution study and does not allow the recognition of all amino acids and lipids. One of the most complex methods for the study of small molecules is metabolomics based on NMR spectroscopy. A critical advantage of NMR spectroscopy is its ability to detect many compounds present in the examined sample in a single experiment. Additionally, this study method makes it possible to perform quantitative analysis with the use of a single reference compound. NMR spectroscopy is most useful if no single biomarker could be identified for differential diagnosis. It could also be applied to study concentrations of neuroactive amino acids, such as glutamate, GABA, and glutamine, as well as taurine, which, according to the glutamatergic hypothesis of ASD, could be candidates for biomarkers [13, 16]. Previous pioneering studies of autism and schizophrenia using NMR spectroscopy–based metabolomics led to the disclosure of disturbances in glutamate and taurine concentrations in the urine of children with ASD [16–18]. However, detailed tissue analysis, in particular of ex vivo brain extracts using NMR spectroscopy in practice, is limited to studies using animal models of various diseases.
There are numerous animal models of autism used in laboratory studies, including genetically modified animals [19] or those based on the use of specific substances, e.g., teratogenic drugs valproic acid (VPA) or thalidomide (THAL). Drugs in these animal models of ASD are administered to mothers during the critical period of gestation [20, 21]. Similar neurodevelopmental effects in offspring can be achieved by causing thyroid hormone [22] or zinc [23] deficiency in pregnant mothers, as well as by inducing specific inflammation [24, 25]. In all these models, offspring exhibit behavior similar to ASD, validated using animal behavioral tests [26–29]. The advantage of these models compared with transgenic animals is the similarity of behavioral disorders induced in this way to the idiopathic symptoms of autism [20, 21]. It seems that these animal models of autism could be useful for the study of the content of biochemical compounds in the brain to test the compliance of observed changes with hypotheses regarding the pathogenesis of ASD. Such studies are impossible to carry out on humans.
In our previous study [30], in which NMR spectroscopy was used in addition to HPLC, we showed changes in glutamate, glutamine, and GABA levels in the rat hippocampus in VPA- and THAL-induced models of autism. However, the results obtained differed depending on the analytical method used, the experimental group, and the animal sex. This made it difficult to draw useful conclusions for the determination of the role of changes in excitatory and inhibitory neurotransmission in the pathogenesis of autistic-like behaviors in the animals studied. Therefore, based on this previous experience, we decided to focus our further study on only NMR spectroscopy and to try to identify metabolites other than amino acid neurotransmitters, both hydrophilic and hydrophobic, that can be determined using this method, the content of which in the hippocampus is changed in rat ASD models.
The aim of the current study was to better characterize metabolic changes in the rat brain in chemically induced ASD models, which would be useful for the validation of these models for further studies on the pathogenesis of ASD. We intended to check if by using these models we could detect changes in substance concentrations that could indicate the contribution of postulated mechanisms associated with the pathogenesis of autism, such as oxidative stress or an imbalance between excitatory and inhibitory neurotransmission. Another goal was to attempt to identify metabolites whose levels undergo similar changes in two selected experimental models. In the future, this may help in the identification of potential ASD biomarkers. In this study, we used two established rat ASD models prenatally induced by the application of VPA and THAL. NMR spectrometry was used to determine the ex vivo content of a number of detectable hydrophilic and hydrophobic metabolites in hippocampal extracts of juvenile 1-month-old rats.
Methods
Animal Models of Autism
Experiments were performed using Wistar rats of both sexes. The animals were bred in the Animal Colony of the Mossakowski Medical Research Centre, Polish Academy of Sciences, in Warsaw. The animals were provided water and fed ad libitum and kept on a 12-h dark/light cycle at room temperature with a constant humidity of approximately 60%. All procedures involving animals were in accordance with the EC Directive for the use of experimental animals 2010/63/EU from 2010, with further modifications, and the national law.
They were approved by the Fourth Local Ethical Committee in Warsaw (resolution no. 43/2015 of May 22, 2015).
In this project, two chemical teratogenic models of autism were used. The procedure was performed exactly as previously described [30]. Female rats on the 11th day of gestation were fed one dose of 800 mg/kg b.w. VPA or 500 mg/kg b.w. THAL. VPA was mixed with 1 ml of saline, THAL was mixed with vegetable oil, and both were administered orally via an intragastric tube. Control animals were fed 1 ml of a mixture of oil and saline, 1:1 v/v [20, 21]. The 31 (± 2)-day-old Wistar rats of both sexes (F: female and M: male) were used for NMR experiments. The use and distribution in groups of all animals are shown in Table 1. The rats of each group came from two litters. Each group had a different number of female and male rats. There were also animals used in other studies not described in this manuscript. The total number of pups in the two litter of each group was as follows: in the control group, 27 (16F:11M); in the VPA group, 22 (11F:11M); and in THAL group, 19 (7F:12M). Finally, in these studies, there were 11 (6F + 5M) control animals, 11 (5F + 6M) VPA-treated animals, and 13 (4F+9M) THAL-treated animals.
Table 1.
The use and distribution in groups of all animals from the two litters used in this study
| Control | VPA | THAL | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ∑ | M | F | ∑ | M | F | ∑ | M | F | ||
| I litter | Total animals | 13 | 4 | 9 | 13 | 6 | 7 | 12 | 6 | 6 |
| This study | 4 | 3 | 1 | 7 | 3 | 4 | 6 | 3 | 3 | |
| Other study | 9 | 1 | 8 | 6 | 3 | 3 | 6 | 3 | 3 | |
| II litter | Total animals | 14 | 7 | 7 | 9 | 5 | 4 | 7 | 6 | 1 |
| This study | 7 | 5 | 2 | 4 | 3 | 1 | 7 | 6 | 1 | |
| Other study | 7 | 2 | 5 | 5 | 2 | 3 | 0 | 0 | 0 | |
VPA valproate treated group, THAL thalidomide treated group, M male, F female animals
Sample Preparation
After decapitation, one hippocampus was removed and homogenized for 2 min by hand in 500 μl of ice-cold saline using a plastic/Teflon homogenizer. Then, 400-μl aliquots of fresh hippocampal homogenates were extracted for NMR studies using the Bligh and Dyer method [31], with slight modification, exactly according to the procedure described previously [30]. Briefly, the homogenates were vortexed for 1 min with 1875 μl of a mixture of 99% methanol, 98% chloroform, and 36% HCl, 40:20:1 (v/v). As the next step, 625 μl chloroform was added, and the mixture was again vortexed for 1 min. After that, 625 μl of water was added and vortexed for 1 min. Then, the mixture was centrifuged at 2000×g for 15 min using a swing-out rotor to obtain three phases: upper, water/methanol containing substances diluted in water; lower, containing lipids; and middle, containing proteins. Upper and lower phases were extracted for the NMR examination. Middle phases were collected to assess total protein content in samples (using the Lowry test) to normalize the concentration of compounds obtained in the hippocampus. The water/methanol phase of the sample was dried using nitrogen. Dry residues were then diluted in 700 μl of D2O and immediately tested. Additionally, the lipid phase was dried using nitrogen, dissolved in 700 μl of CDCl3, and immediately tested.
Spectra Acquisition
The pH of the samples was adjusted to 7.5 ± 0.2 using HCl. 3-Trimethylsilyl propionic acid (TSP) at a final concentration of 1 mM was used as an internal reference for the normalization of all spectra and quantitative statistical analysis. All NMR spectra of hydrophilic compounds were acquired at 25 °C using an Avance III HD 500 MHz (Bruker, Germany) spectrometer. Excitation sculpting [32] was used to suppress the water signal while minimizing phase distortion of the spectrum and utilized a 2-ms square inversion pulse in a double pulse field gradient spin echo. Line broadening of 0.5 and baseline and phase corrections were applied to each spectrum using software implemented in the spectrometer. Hydrophobic compounds were measured using a single pulse sequence at 20 °C and 128 transients with a 5-s repetition time.
All spectra were first both baseline and phase corrected and analyzed. There were 98 signals of hydrophilic and 45 hydrophobic functional groups of compounds. Signal assignments were performed using our own database of spectra of reference compounds and literature data, considering correction for the modified extraction method [33]. For the confirmation of signal assignment, other NMR experiments were performed as follows: 1H-1H COSY, 31P, 1H-31P HSQC. For further statistical analyses, we selected the 55 hydrophilic and 24 hydrophobic most isolated NMR signals that represent all assigned and unassigned compounds, and their magnitudes were measured and normalized to the TSP or CDCl3 rest signal prior to statistical analyses.
Statistical Analysis
Univariate statistical analyses, one-way ANOVA, and two-way ANOVA tests followed by Dunn’s corrections were performed using the SigmaPlot 12.5 software package (Systat Software, Inc.). Two-way ANOVA was carried out for group and sex factors. A p value lower than 0.05 was considered significant. Statistical multivariate analyses (MVAs) of principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) were described in detail in our previous publication [30]. In the analysis, the X-matrix (independent variables) represents all data obtained from NMR spectral analysis, and the Y-matrix (dependent variable) represents all groups [34]. Models were validated using an analysis of variance of cross-validation estimation (CV-ANOVA). The variable importance in the projection (VIP) value of each variable in the model was calculated. MVA was performed using the SIMCA software package, ver. 15, Sartorius Stedim Data Analytics AB, Sweden [35].
Results
The content of metabolites in the samples was expressed as the magnitude of their NMR signals, which is known to correspond to the concentration of the compound, normalized to the protein concentration in the sample. Fifty-five hydrophilic NMR signals representing various compounds were statistically analyzed, of which ten signals were not assigned to the compounds. In turn, 24 hydrophobic NMR signals representing various compounds or compound complexes were statistically analyzed, of which three signals were not assigned to the compounds. PCA was used to identify outliers. Two outliers were identified: one sample in the VPA group and one sample in the THAL group, and their data were removed from further analyses. Finally, in statistical analyses, the number of samples in each group was as follows: control, 11; VPA, 10; and THAL, 12. Because it is difficult to identify a single biomarker that, with high sensitivity and specificity, distinguishes a patient from the healthy population, the entire dataset of compounds was subjected to multivariate OPLS-DA analysis. When added to the analysis of the group parameter (Y-matrix), we did not observe any influence of the sex parameter on the metabolic profile MVA. The result of the MVA was the same, regardless of whether we considered sex and the group or just the group.
Hydrophilic Compounds
Considering hydrophilic substances, statistically significant differences between the VPA-treated and control groups (Table 2) were observed for hypoxanthine, 3-OH-butyrate, and glutathione. The concentrations of 3-OH-butyrate and glutathione decreased in the VPA group, while the concentration of hypoxanthine increased. Statistically significant differences in the concentrations of hydrophilic compounds between the control and THAL groups (Table 1) were observed for 8-hydroxyadenine, hypoxanthine, GMP, guanine, and xanthine complex signals; thymine, allantoin, myo-inositol, taurine, taurine, and phosphoethanolamine complex signals; glycerophosphorylcholine, phosphorylcholine, choline, creatinine, L-cysteic acid, creatine, aspartate, L-glutamine, L-glutamate, acetate, NAA, GABA, L-alanine, lactate, L-threonine, and L-valine; overlapping signals from leucine and isoleucine; and overlapping signals from 3-OH-valerate and methylmalonate, and unassigned signals Nos. 2 and 4. All signal magnitudes except guanine, xanthine, and overlapping signals from 3-OH-valerate and methylmalonate were higher in the THAL group than those in the control group.
Table 2.
Hydrophilic compounds found in NMR analysis of hippocampus extracts
| Chemical shift (ppm) | Compound | VPA | THAL | ||||
|---|---|---|---|---|---|---|---|
| % of control | ANOVA | VIP value | % of control | ANOVA | VIP value | ||
| 9.29 | Unassigned 1 | 93 | F(1,20) = 0.0504 | 1.25 | 133 | H(1) = 0.750 | 1.0 |
| p = 0.387 | |||||||
| p = 0.825 | |||||||
| 9.24 | Unassigned 2 | 84 | H(1) = 2.623 | 124 | F(1,22) = 4.369 | ||
| p = 0.105 | p = 0.049* | ||||||
| 8.96 | (–C(4)H–) N-Methylnicotinamide | 104 | F(1,20) = 0.0735 | 122 | H(1) = 2.182 | ||
| p = 0.140 | |||||||
| p = 0.789 | |||||||
| 8.70 | (=C(2)H–) 8-Hydroxyadenine | 94 | H(1) = 0.0198 | 1.32 | 160 | F(1,22) = 4.571 | 1.44 |
| p = 0.888 | p = 0.044* | ||||||
| 8.68 | (–C(2)H–) Inosine | 105 | F(1,20) = 0.0827 | 110 | F(1,22) = 0.195 | ||
| p = 0.663 | |||||||
| p = 0.777 | |||||||
| 8.59 | (–C(8)H–) IMP | 84 | F(1,20) = 0.824 | 1.08 | 119 | F(1,22) = 0.708 | |
| p = 0.375 | p = 0.410 | ||||||
| 8.45 | (–C(8)H–) Hypoxanthine | 141 | H(1) = 4.170 | 1.84 | 243 | F(1,22) = 14.808 | 1.55 |
| p = 0.041* | |||||||
| p < 0.001* | |||||||
| 8.29 | (–C(8)H–) GMP | 72 | H(1) = 0.838 | 1.04 | 155 | H(1) = 4.125 | |
| p = 0.360 | p = 0.042* | ||||||
| 8.24 | Unassigned 3 | 55 | H(1) = 3.354 | 1.39 | 87 | H(1) = 0.136 | |
| p = 0.067 | p = 0.712 | ||||||
| 7.96 | (–C(4)H–) Cytosine | 102 | F(1,20) = 0.0148 | 118 | F(1,22) = 1.067 | ||
| p = 0.313 | |||||||
| p = 0.904 | |||||||
| 7.67 | (–C(8)H–) Guanine/(–C(2)H–) xanthine | 107 | H(1) = 0.317 | 36 | H(1) = 9.470 | 1.69 | |
| p = 0.573 | p = 0.002* | ||||||
| 7.43 | Unassigned 4 | 84 | H(1) = 1.607 | 131 | H(1) = 4.640 | ||
| p = 0.205 | p = 0.031* | ||||||
| 7.33 | (–C(6)H–) Thymine | 94 | H(1) = 0.124 | 144 | F(1,22) = 10.350 | ||
| p = 0.725 | |||||||
| p = 0.004* | |||||||
| 7.10 | Unassigned 5 | 83 | H(1) = 1.790 | 108 | F(1,22) = 0.397 | ||
| p = 0.181 | p = 0.536 | ||||||
| 6.92 | Unassigned 6 | 86 | H(1) = 0.972 | 116 | F(1,22) = 0.690 | ||
| p = 0.324 | p = 0.415 | ||||||
| 6.85 | (–CH=CH–) Fumarate | 96 | F(1,20) = 0.0547 | 125 | F(1,22) = 1.858 | ||
| p = 0.187 | |||||||
| p = 0.818 | |||||||
| 6.80 | (–C(2,6)H–) Tyrosine | 87 | H(1) = 0.496 | 112 | H(1) = 0.640 | ||
| p = 0.424 | |||||||
| p = 0.481 | |||||||
| 6.22 | (–C(1′)H–) AMP | 90 | F(1,20) = 0.429 | 130 | F(1,22) = 2.551 | ||
| p = 0.520 | p = 0.125 | ||||||
| 5.98 | Unassigned 7 | 103 | F(1,20) = 0.0721 | 113 | F(1,22) = 0.933 | ||
| p = 0.345 | |||||||
| p = 0.791 | |||||||
| 5.39 | (–C(4)H–) Allantoin | 60 | H(1) = 0.526 | 340 | H(1) = 7.542 | 1.56 | |
| p = 0.468 | p = 0.006* | ||||||
| 4.19 | (–C(1)H2–) Phosphorylcholine | 90 | H(1) = 0.0198 | 142 | H(1) = 3.186 | 1.10 | |
| p = 0.888 | p = 0.074 | ||||||
| 4.04 | (–C(2)H2–) Glycolic acid | 67 | H(1) = 0.124 | 1.21 | 117 | F(1,22) = 0.249 | |
| p = 0.725 | p = 0.623 | ||||||
| 3.86 | Unassigned 8 | 92 | H(1) = 0.972 | 110 | F(1,22) = 0.527 | ||
| p = 0.324 | p = 0.477 | ||||||
| 3.79 | (–C(1)H2–) Guanidinoacetate | 102 | H(1) = 0.317 | 144 | H(1) = 1.515 | ||
| p = 0.573 | p = 0.218 | ||||||
| 3.71 | Unassigned 9 | 136 | F(1,20) = 1.775 | 1.72 | 754 | H(1) = 3.409 | 1.20 |
| p = 0.198 | p = 0.065 | ||||||
| 3.66 | Unassigned 10 | 80 | F(1,20) = 2.513 | 1.21 | 94 | F(1,22) = 0.224 | |
| p = 0.129 | p = 0.641 | ||||||
| 3.55 | (–C(2)H2–) Glycine | 91 | H(1) = 1.790 | 1.36 | 133 | F(1,21) = 3.384 | |
| p = 0.181 | p = 0.081 | ||||||
| 3.52 | (–C(6,4)H–) Myo-inositol | 110 | F(1,20) = 0.467 | 148 | F(1,22) = 11.197 | 1.07 | |
| p = 0.503 | |||||||
| p = 0.003* | |||||||
| 3.42 | (–C(3)H2–) Taurine | 85 | H(1) = 0.972 | 141 | H(1) = 6.367 | 1.06 | |
| p = 0.324 | p = 0.012* | ||||||
| 3.35 | ((–CH–)6) Scyllo-inositol | 87 | H(1) = 1.433 | 65 | F(1,22) = 2.020 | 1.06 | |
| p = 0.231 | p = 0.170 | ||||||
| 3.25 | (–C(3)H2–) Taurine/(–C(2)H2–) Phosphoethanolamine | 90 | H(1) = 179 | 163 | H(1) = 8.015 | 1.28 | |
| p = 0.673 | p = 0.005* | ||||||
| 3.23 | (–N–(CH3)3) Phosphorylcholine/glycerophosphorylcholine | 90 | F(1,20) = 0.562 | 177 | F(1,22) = 21.144 | 1.33 | |
| p = 0.463 | |||||||
| p < 0.001* | |||||||
| 3.20 | (–N–(CH3)3) Choline | 72 | H(1) = 3.352 | 196 | F(1,22) = 13.111 | 1.17 | |
| p = 0.067 | |||||||
| p = 0.002* | |||||||
| 3.14 | (–N–(CH3)3) Creatinine | 117 | F(1,20) = 0.596 | 1.21 | 212 | F(1,22) = 16.402 | 1.51 |
| p = 0.450 | |||||||
| p < 0.001* | |||||||
| 3.09 | (–C(2)H–) L-cysteic acid | 94 | F(1,20) = 0.205 | 167 | F(1,22) = 14.240 | 1.17 | |
| p = 0.656 | |||||||
| p = 0.001* | |||||||
| 3.07 | (–C(6)H3–) Creatine | 87 | F(1,20) = 0.774 | 141 | F(1,22) = 5.349 | 1.03 | |
| p = 0.390 | p = 0.031* | ||||||
| 2.97 | (–C(3)H2–) Aspartate | 95 | H(1) = 0.0 | 145 | F(1,22) = 9.080 | 1.09 | |
| p = 1.000 | p = 0.007* | ||||||
| 2.88 | (N–(CH3)2) Dimethylamine | 93 | H(1) = 0.0198 | 124 | F(1,22) = 2.563 | ||
| p = 0.888 | p = 0.124 | ||||||
| 2.68 | (–C(2)–CH3) Pyruvate/(–C(2,3)H2–) succinate | 93 | F(1,20) = 0.329 | 115 | F(1,22) = 1.244 | ||
| p = 0.573 | p = 0.277 | ||||||
| 2.63 | (–C(4)H2–) L-Glutamate | 113 | F(1,20) = 0.609 | 154 | F(1,22) = 8.600 | ||
| p = 0.445 | p = 0.008* | ||||||
| 2.49 | (–C(4)H2–) L-Glutamine | 88 | F(1,20) = 1.129 | 138 | F(1,22) = 10.721 | ||
| p = 0.301 | |||||||
| p = 0.004* | |||||||
| 2.38 | (–C(10)H2–) Glutathione | 74 | H(1) = 7.934 | 1.34 | 91 | F(1,22) = 1.624 | |
| p = 0.217 | |||||||
| p = 0.005* | |||||||
| 2.22 | (–C(15)H3) NAAG | 81 | H(1) = 0.496 | 21 | H(1) = 2.562 | 1.69 | |
| p = 0.481 | p = 0.109 | ||||||
| 2.10 | (–C(2)H3) Acetate | 92 | F(1,20) = 0.313 | 168 | F(1,22) = 15.804 | 1.27 | |
| p = 0.582 | |||||||
| p < 0.001* | |||||||
| 2.04 | (–C(7)H3) NAA | 101 | F(1,20) = 0.0741 | 157 | F(1,22) = 12.394 | 1.18 | |
| p = 0.932 | p < 0.002* | ||||||
| 1.96 | (–C(3)H2–) GABA | 94 | H(1) = 0.124 | 155 | F(1,22) = 13.302 | 1.12 | |
| p = 0.725 | |||||||
| p = 0.002* | |||||||
| 1.78 | (–C(5)H2–) Lysine | 91 | H(1) = 0.0446 | 108 | H(1) = 0.545 | ||
| p = 0.833 | p = 0.460 | ||||||
| 1.55 | (–C(3)H3) L-Alanine | 97 | F(1,20) = 0.044 | 132 | F(1,22) = 5.281 | ||
| p = 0.836 | p = 0.032* | ||||||
| 1.42 | (–C(3)H3) Lactate | 76 | F(1,20) = 2.150 | 1.07 | 149 | H(1) = 5.186 | 1.02 |
| p = 0.159 | p = 0.023* | ||||||
| 1.35 | (–C(3)H3) L-Threonine | 88 | F(1,20) = 0.818 | 145 | F(1,22) = 7.909 | ||
| p = 0.377 | p = 0.010* | ||||||
| 1.20 | (C(3)–(CH3)2) 3-hydroxyisovalerate/(–C(2)–CH3) methylmalonate | 99 | H(1) = 0 | 0 | H(1) = 6.540 | 1.11 | |
| p = 1.000 | p = 0.011* | ||||||
| 1.18 | (–C(2)H2–) 3-Hydroxybutyrate | 41 | H(1) = 4.765 | 2.50 | 334 | H(1) = 2.004 | |
| p = 0.029* | p = 0.157 | ||||||
| 1.14 |
(–C(3)–(CH3)2) 2- oxoisovalerate |
50 | H(1) = 3.615 | 2.05 | 308 | H(1) = 2.761 | |
| p = 0.057 | p = 0.097 | ||||||
| 1.08 | (–C(4)H3) L-Valine | 98 | H(1) = 0.00496 | 144 | H(1) = 6.367 | 1.05 | |
| p = 0.944 | p = 0.012* | ||||||
| 0.96 | (–C(5.6)H3) L-Leucine/(–C(3)–CH3) isoleucine | 102 | H(1) = 0.124 | 148 | F(1,22) = 7.383 | 1.12 | |
| p = 0.725 | p = 0.013* | ||||||
Only VIP values > 1 obtained in MVA analysis are presented
*Statistically significant
MVA of the hydrophilic compounds detected in the hippocampal homogenates collected from the VPA-treated group allowed us to build a valid model (p = 0.036) (Fig. 1a). The model consisted of two components: one predictive and one orthogonal to the data. R2 for this model was 0.733, and Q2 was 0.455. The model fits the data well and was good for prediction. Samples were classified correctly in 95.2% of their groups (p < 0.001): 90.91% in the control group (10 out of 11) and 100% in the VPA group. MVA indicated significant group differentiation (VIP > 1) of the following compounds: 3-hydroxybutyrate, 2-oxoisovalerate, lactate, glutathione, creatinine, glycine, glycolic acid, GMP, hypoxanthine, IMP, 8-hydroxyadenine, and unassigned compound signals Nos. 1, 3, 9, and 10 (Table 2).
Fig. 1.
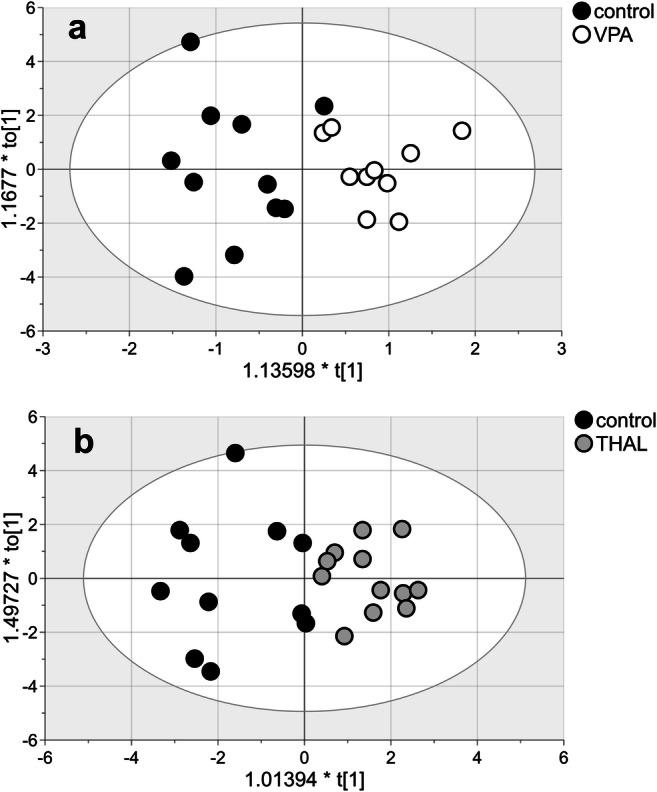
The score plot of the two-component OPLS-DA model for hydrophilic compounds of NMR data for VPA vs control (a) and THAL vs control group (b); to[1] represents within-class variation in the first orthogonal component, whereas t[1] represents between-class variation in the first predictive component. Ellipse represents Hotelling’s T2 with 95% confidence in score plots
MVA of the hydrophilic compounds for the THAL group also allowed us to build a valid model (p = 0.007). The model consisted of two components: one predictive and one orthogonal to the data (Fig. 1b). R2 for this model was 0.721, and Q2 was 0.527. Samples were classified correctly to their groups in 87% (p < 0.001): 72.7% (8 out of 11) in the control group and 100% in the THAL group. The following substances were found: L-valine, leucine/isoleucine, GABA, NAA, acetate, lactate, 3-hydroxyisovalerate and methylmalonate, NAAG, aspartate, choline, creatine, L-cysteic acid, creatinine, taurine, myo-inositol, allantoin, hypoxanthine, taurine/phosphoethanolamine, scyllo-inositol, phosphocholine/glycerophosphocholine, 8-hydroxyadenine, phosphorylcholine, guanine and xanthine overlapped signal, and unassigned compound signals Nos. 1 and 9 (Table 2).
Hydrophobic Compounds
The analysis of the content of hydrophobic substances showed statistically significant differences between the VPA group and the control group (Table 3) in the common signal of the saturated/monounsaturated/polyunsaturated fatty acids (FAs/MUFAs/PUFAs) complex, estriol, and 24-hydroxycholesterol. The concentrations of all these substances decreased in the VPA group compared with those in the control group.
Table 3.
Hydrophobic compounds found in NMR analysis of hippocampus extracts
| Chemical shift (ppm) | Compound/functional group | VPA | THAL | ||||
|---|---|---|---|---|---|---|---|
| % of control | ANOVA | VIP value | % of control | ANOVA | VIP value | ||
| 6.87 | Unassigned 1 | 85 | F(1,21) = 2.920 | 1.06 | 86 | F(1,23) = 1.665 | |
| p = 0.103 | p = 0.210 | ||||||
| 6.09 | (–HC(2,4)=) Estriol | 78 | F(1,21) = 5.222 | 1.35 | 71 | F(1,23) = 9.735 | 1.37 |
| p = 0.033* | p = 0.005* | ||||||
| 5.87 | (–C(4)H=C(5)H–) Testosterone | 165 | H(1) = 0.683 | 2.27 | 53 | H(1) = 1.137 | 1.52 |
| p = 0.407 | p = 0.286 | ||||||
| 5.14 | (–HC=CH–) Olefinic group in MUFA | 93 | F(1,21) = 0.228 | 96 | H(1) = 4.720 | 1.35 | |
| p = 0.638 | p = 0.030* | ||||||
| 5.03 | Unassigned 2 | 82 | F(1,21) = 0.690 | 1.67 | 117 | F(1,23) = 0.582 | 1.16 |
| p = 0.416 | p = 0.454 | ||||||
| 4.70 | Unassigned 3 | 68 | H(1) = 2.804 | 1.54 | 112 | H(1) = 0.0680 | |
| p = 0.794 | |||||||
| p = 0.094 | |||||||
| 4.15 | (–C(2)H–) Sphingomyelin (d18:1/16:0) | 103 | F(1,21) = 0.0719 | 119 | F(1,23) = 2.359 | ||
| p = 0.139 | |||||||
| p = 0.791 | |||||||
| 3.88 | (–CH2–CH2–N(–CH3)3) L-α-Phosphatidylcholine (16:0/18:2) | 104 | F(1,21) = 0.145 | 124 | H(1) = 3.123 | ||
| p = 0.708 | p = 0.077 | ||||||
| 3.68 | (ROCH2–CHOH–CH2OH) in glyceryl group in 1-MG | 112 | F(1,21) = 0.556 | 145 | F(1,23) = 7.328 | 1.10 | |
| p = 0.465 | p = 0.013* | ||||||
| 3.24 | (–O–CH2–CH2–NH3) L-α-Phosphatidylethanolamine (18:0/18:0) | 91 | F(1,21) = 0.515 | 101 | F(1,23) = 0.00762 | ||
| p = 0.483 | |||||||
| p = 0.932 | |||||||
| 2.30 | (–OCO–CH2–), (–COOH–CH2–) Acyl groups in 1,3-DG, 1-MG, and FA | 107 | F(1,21) = 0.262 | 93 | F(1,23) = 0.225 | ||
| p = 0.614 | p = 0.643 | ||||||
| 2.22 | (–C(α)H2–) in saturated FA, MUFA, and PUFA | 64 | F(1,21) = 11.580 | 1.60 | 61 | F(1,23) = 10.494 | 1.37 |
| p = 0.005* | |||||||
| p = 0.003* | |||||||
| 2.00 | (–CH2–CH=CH–) in acyl groups of FA and phospholipids | 88 | F(1,21) = 1.093 | 103 | F(1,23) = 0.0717 | ||
| p = 0.310 | p = 0.792 | ||||||
| 1.70 | (–C(15)H2–) Palmitic acid in L-α- lysophosphatidylcholine and FA | 84 | H(1) = 2.381 | 1.17 | 140 | H(1) = 3.545 | 1.07 |
| p = 0.123 | p = 0.060 | ||||||
| 1.60 | (–OCO–CH2–CH2–) in acyl groups in 1,3-DG, 1-MG, and FA | 89 | F(1,21) = 1.516 | 106 | F(1,23) = 0.359 | ||
| p = 0.233 | p = 0.555 | ||||||
| 1.28 | (−(CH2)n–) in acyl groups in FA | 95 | F(1,21) = 0.365 | 106 | F(1,23) = 0.509 | ||
| p = 0.552 | p = 0.483 | ||||||
| 1.08 | (–C(21)H3) Cholestenol | 101 | F(1,21) = 0.0148 | 124 | p = 0.029* | ||
| p = 0.904 | |||||||
| 0.98 | (–C(ω)H3) in PUFA/MUFA | 108 | F(1,21) = 0.385 | 131 | F(1,23) = 6.927 | 1.21 | |
| p = 0.542 | p = 0.015* | ||||||
| 0.93 | (–C(21)H3) Free cholesterol and 25-hydroxycholesterol | 104 | F(1,21) = 0.118 | 125 | F(1,23) = 6.076 | 1.10 | |
| p = 0.734 | p = 0.022* | ||||||
| 0.88 | (–CH3) in acyl groups of FA and sterols | 93 | F(1,21) = 1.000 | 107 | F(1,23) = 0.805 | ||
| p = 0.329 | p = 0.379 | ||||||
| 0.72 | (–C(18)H3) 24-Hydroxycholesterol | 70 | H(1) = 7.788 | 1.34 | 83 | F(1,23) = 2.228 | |
| p = 0.005* | p = 0.150 | ||||||
| 0.69 | (–C(18)H3) Free cholesterol and cholesterol esters | 103 | F(1,21) = 0.0971 | 121 | F(1,23) = 5.099 | 1.00 | |
| p = 0.034* | |||||||
| p = 0.759 | |||||||
| 0.62 | (–C(18)H3) Progesterone | 87 | F(1,21) = 0.233 | 1.03 | 113 | F(1,23) = 0.240 | 1.28 |
| p = 0.635 | p = 0.629 | ||||||
| 0.53 | (–C(18)H3) Lathosterol | 108 | F(1,21) = 0.950 | 115 | F(1,23) = 2.759 | ||
| p = 0.341 | p = 0.111 | ||||||
Only VIP values > 1 obtained in MVA analysis are presented
*Statistically significant
Statistically significant differences between the control and the THAL group (Table 3) were observed for estriol, the olefinic group in MUFAs, the FAs/MUFAs/PUFAs complex signal, cholestenol, free cholesterol and 25-hydroxycholesterol, free cholesterol and cholesterol esters, and the glyceryl group in 1-MG and 1,2-DG. All compound concentrations increased in the THAL group compared with those in the control group except for the FAs/MUFAs/PUFAs complex signal and estriol, whose concentrations decreased.
MVA of the lipid data for the VPA group allowed us to build a valid model (p = 0.04). The model consisted of two components: one predictive and one orthogonal to the data (Fig. 2a). R2 for this model was 0.833, and Q2 was 0.546. The model fits the data well and was good for prediction. Samples were classified correctly (p < 0.001) in 100% of their groups. MVA indicated significant group differentiation (VIP > 1) of the following substances: estriol, testosterone, progesterone, saturated FAs/MUFAs/PUFAs complex signal, palmitic acid in LPtdC and FAs, 24-hydroxycholesterol, and unassigned compound signals Nos. 1–3. Among substances with altered concentrations in the VPA-treated group, only testosterone concentration was higher, while concentrations of all other compounds were lower compared with those in the control group (Table 3).
Fig. 2.
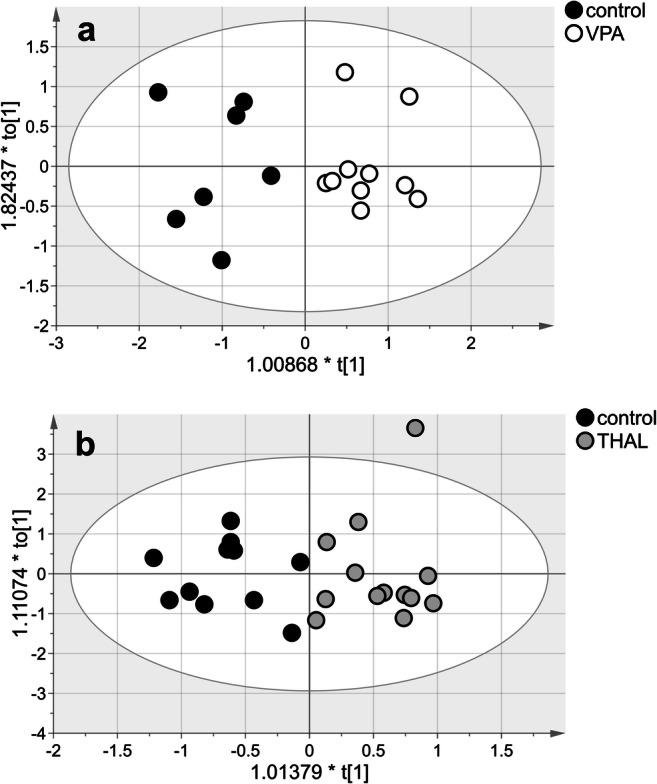
The score plot of the two-component OPLS-DA model for hydrophobic compounds of NMR data for VPA vs control (a) and THAL vs control group (b); to[1] represents within-class variation in the first orthogonal component, whereas t[1] represents between-class variation in the first predictive component. Ellipse represents Hotelling’s T2 with 95% confidence in score plots
MVA of the lipid data for the THAL group also allowed us to build a valid model (p = 0.04). The model consisted of three components: one predictive and two orthogonal to the data (Fig. 2b). R2 for this model was 0.778, and Q2 was 0.518. Samples were classified correctly (p < 0.001) in 100% of their groups. MVA analysis indicated significant group differentiation (VIP > 1) signals from estriol, testosterone, progesterone, MUFAs, 1-MG, palmitic acid in LPtdC and FAs/MUFAs/PUFAs complex signal, free cholesterol/cholesterol ester complex signal, free cholesterol/25-hydroxycholesterol complex signal, and unassigned signal No. 2 (Table 2). Among substances with changed concentrations in the THAL-treated group, only testosterone, estriol, and olefinic acid in MUFAs and FAs/MUFAs/PUFAs concentrations were decreased, while concentrations of all other compounds were increased compared with those in the control group (Table 3).
Discussion
In our studies, instead of genetic models, only pharmacological models of ASD on rats were used. This is due to our priority interest in the role of environmental factors, especially neurotoxic ones, in the etiopathogenesis of autism. Both VPA and THAL are used in therapy, so it is important to better understand their adverse effects on the fetus in the early stages of pregnancy, which may result in ASD in the offspring. In addition, we expected that a comparison of metabolomic changes in both pharmacological models could help clarify whether both models can be used alternatively, e.g., for testing new therapies. In this work, research focused on the hippocampus, one of the brain regions whose developmental disorders are particularly strongly associated with the pathogenesis of behavioral disorders in ASD [36]. This is a continuation and extension of the scope of our previous studies [30], in which only changes in the concentration of neuroactive amino acids in the hippocampus in rat ASD models were analyzed.
Our discussion of the results obtained in this study mainly focuses on the role of the substances identified in the NMR spectra whose content in rat hippocampus homogenates from experimental groups proved to be significantly (VIP > 1) different from that in the control group in the MVA and whose changes can be related to hypothetical mechanisms implicated in the pathogenesis of ASD. When necessary, we also discuss the results for some other substances.
Hydrophobic and Hydrophilic Compounds in Rat ASD Models and Their Potential Role in Excitation/Inhibition Imbalance in the Hippocampus
In both experimental groups, MVA showed a statistically significant decrease in the levels of FAs/MUFAs/PUFAs. Fatty acids (FAs), carboxylic acids with aliphatic chains, are the main components of lipids that form the cell membrane and thus play an important role in the structure and function of the nervous system. FA composition affects the activity of ion channels and receptors. High levels of n-6 PUFAs are generally pro-inflammatory, while those of n-3 PUFAs are beneficial for neuronal functions. High levels of n-6 PUFAs are generally pro-inflammatory, while those of n-3 PUFAs are beneficial for neuronal functions [37, 38]. Deficiencies of n-3 PUFAs and an increased n-6/n-3 PUFA ratio in erythrocytes have been reported in humans with mental disorders including ASD [39–41]. The reduction in unsaturated fatty acid content that we observed in this work in both rat ASD models can therefore be an element of the pathomechanisms of behavioral changes and a candidate ASD biomarker.
In the present study, we noticed that in the THAL group, estriol and testosterone levels were decreased by 30% and 47%, respectively, whereas progesterone levels were increased. A slightly different pattern of changes was found in the VPA group, where estriol levels were reduced by 22%, while testosterone levels were increased by 65%. Estriol is produced from estradiol by the cytochrome P450 family 1 subfamily A polypeptide (data from KEGG - Kyoto Encyclopedia of Genes and Genomes). Estriol is the only form of estrogen we could identify as an isolated NMR signal, and therefore, it serves as an indicator of the content of estrogen in the brain. Estrogen is one of the main regulators of brain energy metabolism [42] and coordinates functional interactions among organs, cells, and genes [43]. Estrogen is synthesized in the ovaries and adrenal glands and in the brain, where it can also be synthesized from cholesterol [44, 45]. Regardless of gender, estrogen receptors are present in many parts of the brain, mostly in the hippocampus and cerebral cortex, both in neurons and in glial cells [46].
We hypothesize that in the THAL group, there are disorders of enzyme activity involved in converting progesterone to testosterone and estradiol, while the changes observed in the VPA group could be explained by the dysfunction of aromatase, an enzyme converting testosterone into estradiol. There have been reports of impaired functioning of this enzyme in patients with ASD [47]. Male and female sex hormones differentially regulate the expression of a novel autism candidate gene, retinoic acid–related orphan receptor-alpha (RORA), which transcriptionally regulates aromatase [48]. Hypothetically, sex steroids modulate the E/I balance, and changes in their concentration may sensitize the male brain to ASD-inducing factors [5]. Estrogens modulate GABA signaling by regulating the expression of glutamic acid decarboxylase [49] or the potassium-chloride cotransporter KCC2 [50], while androgens lead to GABAA-mediated excitotoxicity in the developing hippocampus of male rats [51].
Our study also showed elevated cholesterol levels and various changes in the content of its derivatives in both ASD models. Cholesterol is a precursor to steroid hormones, suggesting a hypothetical explanation that altered cholesterol metabolism can cause disturbances in sex hormone levels. The role of cholesterol metabolism disorders and the participation of their metabolites, such as testosterone, estrogen, cortisol, and vitamin D, in the pathogenesis of ASD have been suggested [52]. Cholesterol participates in numerous functions of the cell membrane, regulating, among others, permeability and fluidity [53]. The cellular level of free cholesterol is strictly regulated by a network of transcriptional and post-translational mechanisms sensitive to levels of free cholesterol and oxysterols (oxidized cholesterol derivatives), which can lower cholesterol through a negative feedback mechanism and prevent its toxic effects [54, 55]. 25-Hydroxycholesterol (25-HC), which was detected in our study, reduces free cholesterol by increasing cholesterol esterification by acyl-CoA-cholesterol acyltransferase in the endoplasmic reticulum [56]. We also detected 24(S)-hydroxycholesterol (24(S)-HC), which is the major brain cholesterol metabolite produced by cholesterol 24-hydroxylase. This pathway is crucial for brain cholesterol metabolism [57]. Unlike free cholesterol, 24(S)-HC is membrane permeable and thus could be metabolized in the periphery [58]. These dependencies were reflected in the decrease in the level of 24 (S) -HC in both experimental groups with an increase in the level of free cholesterol, which indicates a decrease in cholesterol metabolism via 24-hydroxylase cholesterol. Moreover, when considering the mechanisms of a hypothetical excitation/inhibition (E/I) imbalance in autism, modulation of NMDAR activity by oxysterols should be taken into account. 24(S)-HC is a selective and strong positive allosteric modulator of NMDARs, while 25-HC antagonizes this effect [59], providing neuroprotection also an NMDAR-independent mechanism [60]. A reduced level of 24(S)-HC, which we noticed in both experimental groups, is not consistent with the hypothesis being tested.
Many of the changes in the content of hydrophilic substances in the hippocampus shown in this work can also be referred to the hypothesis about the role of E/I imbalance in the pathogenesis of ASD, which suggests that, in autism, brain stimulatory glutaminergic neurotransmission prevails over inhibitory GABAergic neurotransmission [61–63]. This has been partly supported by the results of our previous studies using the same rat models of autism [30]. Abnormalities in the gene encoding the glycine receptor α2 subunit have been observed in a boy with autism [64]. In the current study, an increase in the content of glycine, taurine, and alanine in the hippocampus in the THAL model was noticed, while in the VPA model, it was slightly reduced, but the level of GABA increased (Table 2). GABA, as well as glycine, taurine, and alanine, are tonic agonists of GABAA and glycine receptors, respectively, which are coupled to chloride channels and in the adult brain play the role of major inhibitory neurotransmitters [65, 66]. However, this is a more complex issue because, in the early stages of development, GABA and glycine depolarize neurons due to the relatively high intracellular concentration of Cl− ions, but during development, GABA and glycine function shifts from excitatory to inhibitory neurotransmitters [67–69]. It has been suggested that a delay in this shift may result in neurodevelopmental disorders, including ASD [70]. This may also apply to animal, pharmacological ASD models.
In this study, we observed a decrease in guanine content and an increase in GMP/IMP concentration in the hippocampus in the THAL model. These results may be related to the pathogenesis of autism because urine metabolomics studies of autistic children indicated impaired purine transformation [71], and may indirectly point to neurotransmission disorders and E/I imbalance in this ASD model. The guanine-based purinergic system (GBP) has many functions in nerve cells, including the modulation of NMDA receptor activity important in protecting against excitotoxicity. Moreover, GMP reduces glutamate binding to receptors (for review, see [72]), guanosine, a guanine substrate in the purine metabolic cycle (KEGG), prevents glutamate release in hippocampal slices [73], and GBP interacts with the activity of the glutamate transporter [74]. Chronic administration of GMP reduces the expression of NMDA and AMPA receptor subunits and the glutamate transporters EAAC1 and GLT-1 in the rat cerebral cortex [75].
The abovementioned disturbances in brain amino acid concentrations in both rat ASD models also concern the neuropeptide N-acetylaspartylglutamate (NAAG), an endogenous agonist of presynaptic metabotropic glutamate receptor 3 (mGluR3) which inhibits glutamate release. In the VPA model, the NAAG level and the levels of aspartate and NAA, which are NAAG precursors, were lower than those in the control group. In the THAL model, however, the NAA level was increased, while NAAG was decreased. These results could indicate a deficiency in the activity of N-acetylaspartylglutamate/N-acetylaspartylglutamylglutamate synthase or the hyperactivity of glutamate carboxypeptidase II (KEGG). Although there is no data in the literature linking disturbances in NAA/NAAG levels with ASD, they were observed in schizophrenia [76, 77].
Hypotheses of Impaired Energy Metabolism and Oxidative Stress in the Brain in ASD
In our studies using proton and phosphorus NMR spectroscopy, we did not observe changes in the spectra of the ATP, ADP, and AMP signals due to the low concentrations of these substances, below the detection limit of the method. Also, the acetyl-CoA molecule is not detectable. However, increased lactate levels observed in the VPA group may be a consequence and an indicator of energy deficit. Lactate is an important intermediary in numerous metabolic processes [78, 79] and a preferred neuronal fuel [80, 81]. In the VPA model, reduced levels of methylmalonate and acetate and a deficiency in pyruvate, 2-oxo-3-hydroxyisovalerate, 2-oxoisovalerate, L-leucine, and L-valine were also noted, which can be hypothetically explained by acetyl-CoA deficiency (KEGG). Another circumstantial evidence indicating a possible energy deficit is the elevated levels of ketone bodies in the THAL model. Typically, ketone bodies are converted in the brain to acetyl-CoA, which can then enter the citric acid cycle to produce ATP (KEGG). The ketone body β-hydroxybutyrate has been shown to depolarize the plasma membrane and interfere with the synaptic vesicle cycle [82], which shows the possible functional consequences of this phenomenon.
An increase in the level of hypoxanthine in the hippocampus in both rat ASD models can lead to disorders in adenosine transport and imbalances in the activity of adenosine, dopamine, and serotonin receptors, as well as to abnormalities in the development of neurons, resulting from altered expression of some genes responsible for early neuronal differentiation [83, 84]. Moreover, this effect is indirectly indicative of energy deficit. Hypoxanthine has been shown to induce energy disorders in the brain [85]; its intrastriatal administration altered neuroenergetic parameters and caused mitochondrial dysfunction and apoptotic cell death [86]. These disorders and a decrease in ATP levels appear to be associated with oxidative stress because they may be prevented by pretreatment with free radical scavengers [87].
The results of several studies indicate a possible contribution of oxidative stress to the pathomechanisms of ASD [88–90]. A number of our results, such as the increase in hypoxanthine level in the THAL model discussed above, are consistent with this hypothesis. It is known that increased levels of hypoxanthine may lead to an increase in the production of reactive oxygen species and an exacerbation of the oxidative stress response [91, 92], leading to impairment of brain energy metabolism [93]. Oxidative stress is indicated by a decrease in the concentration of glutathione (GSH), which is a natural free radical scavenger in the cell, in the VPA model, and a similar trend in the THAL model. Moreover, in the THAL model, we found an increase in the level of 8-hydroxyadenine, which is a marker of DNA damage that may be a consequence of GSH deficiency [94, 95].
In our studies, hypoxanthine and allantoin which are markers of free radical production [96] were elevated in the THAL model.
Other Hydrophilic Compounds and Their Possible Role in ASD Pathomechanisms
In both animal models, we observed changes in the levels of many other hydrophilic compounds that can affect neuronal function and contribute to the development of autism. In the THAL model, we noticed increased levels of choline (Cho) and creatine (Cr) and the Cho/Cr ratio, while in the VPA model, they were lowered. The results of clinical studies also differ; both a significant increase in the Cho/Cr ratio [97] and a decreased Cho/Cr levels in the brains of ASD children [98] were reported.
In the brains of rats from the THAL group, an increased level of myo-inositol and a reduced level of scyllo-inositol were found, which indicate disturbances in the metabolism of inositol, an important carbocyclic sugar that is involved in cellular signal transduction and osmoregulation. Preclinical studies showed positive effects of scyllo-inositol supplementation in in vivo and in vitro models of Alzheimer’s disease (AD) [99], while inositol supplementation in individuals with ASD yielded negative results [100]. We also noticed increased valine, leucine, and isoleucine levels in both ASD models, which may be due to the dysfunction of the enzyme amino acid transferase (KEGG). A reduction in the levels of 3-hydroxyisovalerate and methyl malonate confirms this interpretation. Similar disturbances in the levels of these amino acids in the urine of autistic children have been reported [101]. Reduced blood valine levels have been observed in children with propionic acidosis and autistic features [102].
Conclusions
Although it is known that very similar behavioral symptoms that correspond to symptoms in autistic patients are observed in the two ASD rat models used in these studies, our NMR spectroscopic analysis showed large differences between these models in the content of several metabolites in the hippocampi. Table 4 provides a summary of the abovementioned compounds whose levels have been changed, directions of change, and metabolic pathways in which they are involved. We believe that these differences between VPA and THAL models could be a reflection of the spectrum of phenotypes observed in patients with ASD. Despite these differences, the following metabolic pathways were identified that were disrupted in both ASD models: steroid hormone biosynthesis; FAs biosynthesis; the synthesis and degradation of ketone bodies; glycerophospholipid metabolism; cholesterol metabolism; purine metabolism; arginine and proline metabolism; and valine, leucine, and isoleucine biosynthesis and degradation. These results may indicate disturbances in energy production, altered cell membrane structure, disturbances in excitatory and/or inhibitory neurotransmission, and the induction of oxidative stress in the hippocampus. The causal relationship between these disorders, the primary triggering mechanism(s), or their role in behavioral disorders similar to autism remains unclear. However, the obtained results may be useful in the future in the choice of the optimal animal model for ASD studies. In addition, these results may be advantageous for interpreting the results of our ongoing metabolomics studies using body fluids collected from rats from both ASD models.
Table 4.
Functions and metabolic pathways of metabolites (VIP > 1) in two models of autism; directions of changes found in each of the models compared with the control are presented
| Metabolites | Metabolic pathway | Probably pathomechanism | VPA model | THAL model |
|---|---|---|---|---|
| Progesterone | Steroid hormone biosynthesis | Energy and neurotransmission disturbances | ↓ | ↑ |
| Testosterone | ↑ | ↓ | ||
| Estriol | ↓ | ↓ | ||
| FA, MUFA, PUFA | FA biosynthesis | Energy production disturbances, cell membrane functional disturbances | ↓ | ↓ |
| Lysophosphatidylcholine | Glycerophospholipid metabolism | Cell membrane functional disturbances | ↓ | ↑ |
| Cholesterol | Cholesterol metabolism | Neurotransmission disturbances | ↑ | |
| Cholesterol ester | ↑ | |||
| 25-Hydroxycholesterol | ↑ | |||
| 24-Hydroxycholesterol | ↓ | |||
| 8-Hydroxyadenine | Purine metabolism | Oxidative stress | ↓ | ↑ |
| Hypoxanthine | ↑ | ↑ | ||
| GMP/IMP | ↓ | ↑ | ||
| Allantoine | ↓ | ↑ | ||
| Guanine, xanthine | ↓ | |||
| Glycine | Glycine, serine metabolism | ↓ | ||
| Threonine | Threonine metabolism | Neurotransmission disturbances | ↑ | |
| Glutathione | Glutathione/cysteine and methionine metabolism | Oxidative stress | ↓ | |
| Phosphorylcholine | Glycerophospholipid metabolism | Neurotransmission disturbances | ||
| ↑ | ||||
| Phosphoethanolamine | ↑ | |||
| Glycerophosphorylcholine | ↑ | |||
| Choline | ↑ | |||
| Creatine | Arginine and proline metabolism | Neurotransmission disturbances Oxidative stress | ↑ | |
| Creatinine | ↑ | ↑ | ||
| GABA | ↑ | |||
| Taurine | Taurine, alanine and glutamate, pyruvate metabolism | Neurotransmission disturbances Oxidative stress | ↑ | |
| Aspartate | ↑ | |||
| Cysteic acid | ↑ | |||
| Lactate | ↑ | |||
| NAA | ↑ | |||
| NAAG | ↓ | ↓ | ||
| Glycolic acid | Glyoxylate and dicarboxylate metabolism | Oxidative stress | ↓ | |
| Acetate | Synthesis and degradation of ketone bodies | Energy production disturbances | ↑ | |
| 3-Hydroxybutyrate | ↓ | |||
| Myo-inositol | Inositol metabolism | Energy production disturbances | ↑ | |
| Scyllo-inositol | ↓ | |||
| 3-Hydroxyisovalerate | Valine, leucine, isoleucine biosynthesis Pyrimidine metabolism | Neurotransmission disturbances Oxidative stress | ↓ | |
| Thymine | ↑ | |||
| Methylmalonate | ↓ | |||
| 2-Oxoisovalerate | Valine, leucine, isoleucine degradation | Neurotransmission disturbances Oxidative stress | ↑ | |
| Valine, leucine, isoleucine | ↑ |
Author Contributions
EZ: originated the research concept and contributed to the design of the study, prepared animal models for experiments, prepared samples for NMR measurements, contributed to interpreting the results and preparation of the figures, and participated in the draft of the manuscript. BT: contributed to the design of the study, measured and analyzed NMR spectra, performed the statistical analysis of experimental data, contributed to interpreting the results and preparation of the figures, and participated in the draft of the manuscript. PS: analyzed NMR spectra and participated in the draft of the manuscript. JWL: PI of the grant, contributed to the design of the study, interpreted the results reviewing them critically, and participated in the draft of the manuscript. All authors revised and approved the final version of the manuscript.
Funding Information
This study was supported by the Polish National Science Centre (grant number: 2014/15/B/NZ4/04490). The founding sponsors had no role in the design of this study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; and in the decision to publish the results.
Compliance with Ethical Standards
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Competing Interests
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Almandil NB, Alkuroud DN, AbdulAzeez S, AlSulaiman A, Elaissari A, Borgio JF (2019) Environmental and genetic factors in autism spectrum disorders: special emphasis on data from Arabian studies. Int J Environ Res Public Health 16(4). 10.3390/ijerph16040658 [DOI] [PMC free article] [PubMed]
- 2.Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383(9920):896–910. doi: 10.1016/S0140-6736(13)61539-1. [DOI] [PubMed] [Google Scholar]
- 3.Fernandes D, Santos SD, Coutinho E, Whitt JL, Beltrao N, Rondao T, Leite MI, Buckley C, Lee HK, Carvalho AL. Disrupted AMPA receptor function upon genetic- or antibody-mediated loss of autism-associated CASPR2. Cereb Cortex. 2019;29:4919–4931. doi: 10.1093/cercor/bhz032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Yin F, Gao J, Fan X. Association between 5-HTTLPR polymorphism and the risk of autism: a meta-analysis based on case-control studies. Front Psychiatry. 2019;10:51. doi: 10.3389/fpsyt.2019.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubenstein JL. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr Opin Neurol. 2010;23(2):118–123. doi: 10.1097/WCO.0b013e328336eb13. [DOI] [PubMed] [Google Scholar]
- 6.Vaccaro TDS, Sorrentino JM, Salvador S, Veit T, Souza DO, de Almeida RF (2018) Alterations in the microRNA of the blood of autism spectrum disorder patients: effects on epigenetic regulation and potential biomarkers. Behav Sci (Basel) 8(8). 10.3390/bs8080075 [DOI] [PMC free article] [PubMed]
- 7.Grova N, Schroeder H, Olivier JL, Turner JD. Epigenetic and neurological impairments associated with early life exposure to persistent organic pollutants. Int J Genomics. 2019;2019:2085496–2085419. doi: 10.1155/2019/2085496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry. 2015;77(1):66–74. doi: 10.1016/j.biopsych.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landrigan PJ, Lambertini L, Birnbaum LS. A research strategy to discover the environmental causes of autism and neurodevelopmental disabilities. Environ Health Perspect. 2012;120(7):a258–a260. doi: 10.1289/ehp.1104285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ha HTT, Leal-Ortiz S, Lalwani K, Kiyonaka S, Hamachi I, Mysore SP, Montgomery JM, Garner CC, Huguenard JR, Kim SA. Shank and zinc mediate an AMPA receptor subunit switch in developing neurons. Front Mol Neurosci. 2018;11:405. doi: 10.3389/fnmol.2018.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora M, Reichenberg A, Willfors C, Austin C, Gennings C, Berggren S, Lichtenstein P, Anckarsater H, Tammimies K, Bolte S. Fetal and postnatal metal dysregulation in autism. Nat Commun. 2017;8:15493. doi: 10.1038/ncomms15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherskov A, Pohl A, Allison C, Zhang H, Payne RA, Baron-Cohen S. Polycystic ovary syndrome and autism: a test of the prenatal sex steroid theory. Transl Psychiatry. 2018;8(1):136. doi: 10.1038/s41398-018-0186-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rojas DC. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J Neural Transm (Vienna) 2014;121(8):891–905. doi: 10.1007/s00702-014-1216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, Spooren W, Barker GJ, Kunnecke B, Murphy DG. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry. 2018;8(1):106. doi: 10.1038/s41398-018-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lussu M, Noto A, Masili A, Rinaldi AC, Dessi A, De Angelis M, De Giacomo A, Fanos V, Atzori L, Francavilla R. The urinary (1) H-NMR metabolomics profile of an italian autistic children population and their unaffected siblings. Autism Res. 2017;10(6):1058–1066. doi: 10.1002/aur.1748. [DOI] [PubMed] [Google Scholar]
- 16.Yap IK, Angley M, Veselkov KA, Holmes E, Lindon JC, Nicholson JK. Urinary metabolic phenotyping differentiates children with autism from their unaffected siblings and age-matched controls. J Proteome Res. 2010;9(6):2996–3004. doi: 10.1021/pr901188e. [DOI] [PubMed] [Google Scholar]
- 17.Holmes E, Tsang TM, Tabrizi SJ. The application of NMR-based metabonomics in neurological disorders. NeuroRx. 2006;3(3):358–372. doi: 10.1016/j.nurx.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsang TM, Woodman B, McLoughlin GA, Griffin JL, Tabrizi SJ, Bates GP, Holmes E. Metabolic characterization of the R6/2 transgenic mouse model of Huntington’s disease by high-resolution MAS 1H NMR spectroscopy. J Proteome Res. 2006;5(3):483–492. doi: 10.1021/pr050244o. [DOI] [PubMed] [Google Scholar]
- 19.Patel J, Lukkes JL, Shekhar A. Overview of genetic models of autism spectrum disorders. Prog Brain Res. 2018;241:1–36. doi: 10.1016/bs.pbr.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Kolozsi E, Mackenzie RN, Roullet FI, deCatanzaro D, Foster JA. Prenatal exposure to valproic acid leads to reduced expression of synaptic adhesion molecule neuroligin 3 in mice. Neuroscience. 2009;163(4):1201–1210. doi: 10.1016/j.neuroscience.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 21.Narita N, Kato M, Tazoe M, Miyazaki K, Narita M, Okado N. Increased monoamine concentration in the brain and blood of fetal thalidomide- and valproic acid-exposed rat: putative animal models for autism. Pediatr Res. 2002;52(4):576–579. doi: 10.1203/00006450-200210000-00018. [DOI] [PubMed] [Google Scholar]
- 22.Sadamatsu M, Kanai H, Xu X, Liu Y, Kato N. Review of animal models for autism: implication of thyroid hormone. Congenit Anom (Kyoto) 2006;46(1):1–9. doi: 10.1111/j.1741-4520.2006.00094.x. [DOI] [PubMed] [Google Scholar]
- 23.Grabrucker S, Haderspeck JC, Sauer AK, Kittelberger N, Asoglu H, Abaei A, Rasche V, Schon M, Boeckers TM, Grabrucker AM. Brain lateralization in mice is associated with zinc signaling and altered in prenatal zinc deficient mice that display features of autism spectrum disorder. Front Mol Neurosci. 2017;10:450. doi: 10.3389/fnmol.2017.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pardo CA, Meffert MK (2018, 299) Animal models in autism research: the legacy of Paul H. Patterson. Exp Neurol (Pt A):197–198. 10.1016/j.expneurol.2017.11.004 [DOI] [PMC free article] [PubMed]
- 25.Kazdoba TM, Leach PT, Yang M, Silverman JL, Solomon M, Crawley JN. Translational mouse models of autism: advancing toward pharmacological therapeutics. Curr Top Behav Neurosci. 2016;28:1–52. doi: 10.1007/7854_2015_5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazdoba TM, Leach PT, Crawley JN. Behavioral phenotypes of genetic mouse models of autism. Genes Brain Behav. 2016;15(1):7–26. doi: 10.1111/gbb.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30(1):80–89. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- 28.Schneider T, Roman A, Basta-Kaim A, Kubera M, Budziszewska B, Schneider K, Przewlocki R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology. 2008;33(6):728–740. doi: 10.1016/j.psyneuen.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Schneider T, Turczak J, Przewlocki R. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology. 2006;31(1):36–46. doi: 10.1038/sj.npp.1300767. [DOI] [PubMed] [Google Scholar]
- 30.Zieminska E, Toczylowska B, Diamandakis D, Hilgier W, Filipkowski RK, Polowy R, Orzel J, Gorka M, Lazarewicz JW. Glutamate, glutamine and GABA levels in rat brain measured using MRS, HPLC and NMR methods in study of two models of autism. Front Mol Neurosci. 2018;11:418. doi: 10.3389/fnmol.2018.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 32.Hwang TL, Shaka AJ. Water suppression that works - excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J Magn Reson Ser A. 1995;112(2):275–279. doi: 10.1006/jmra.1995.1047. [DOI] [Google Scholar]
- 33.Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13(3):129–153. doi: 10.1002/1099-1492(200005)13:3<129::AID-NBM619>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 34.Weigl W, Milej D, Gerega A, Toczylowska B, Sawosz P, Kacprzak M, Janusek D, Wojtkiewicz S, Maniewski R, Liebert A. Confirmation of brain death using optical methods based on tracking of an optical contrast agent: assessment of diagnostic feasibility. Sci Rep. 2018;8(1):7332. doi: 10.1038/s41598-018-25351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis DI, Dunn WB, Griffin JL, Allwood JW, Goodacre R. Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics. 2007;8(9):1243–1266. doi: 10.2217/14622416.8.9.1243. [DOI] [PubMed] [Google Scholar]
- 36.Varghese M, Keshav N, Jacot-Descombes S, Warda T, Wicinski B, Dickstein DL, Harony-Nicolas H, De Rubeis S, Drapeau E, Buxbaum JD, Hof PR. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 2017;134(4):537–566. doi: 10.1007/s00401-017-1736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer’s disease. PLoS One. 2014;9(6):e100519. doi: 10.1371/journal.pone.0100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mischoulon D, Freeman MP. Omega-3 fatty acids in psychiatry. Psychiatr Clin North Am. 2013;36(1):15–23. doi: 10.1016/j.psc.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Agostoni C, Nobile M, Ciappolino V, Delvecchio G, Tesei A, Turolo S, Crippa A, Mazzocchi A et al (2017) The role of omega-3 fatty acids in developmental psychopathology: a systematic review on early psychosis, autism, and ADHD. Int J Mol Sci 18(12). 10.3390/ijms18122608 [DOI] [PMC free article] [PubMed]
- 40.Bell JG, Miller D, MacDonald DJ, MacKinlay EE, Dick JR, Cheseldine S, Boyle RM, Graham C, O’Hare AE. The fatty acid compositions of erythrocyte and plasma polar lipids in children with autism, developmental delay or typically developing controls and the effect of fish oil intake. Br J Nutr. 2010;103(8):1160–1167. doi: 10.1017/S0007114509992881. [DOI] [PubMed] [Google Scholar]
- 41.Parletta N, Niyonsenga T, Duff J. Omega-3 and Omega-6 polyunsaturated fatty acid levels and correlations with symptoms in children with attention deficit hyperactivity disorder, autistic spectrum disorder and typically developing controls. PLoS One. 2016;11(5):e0156432. doi: 10.1371/journal.pone.0156432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brinton RD. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008;31(10):529–537. doi: 10.1016/j.tins.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rettberg JR, Yao J, Brinton RD. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol. 2014;35(1):8–30. doi: 10.1016/j.yfrne.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balthazart J, Ball GF. Is brain estradiol a hormone or a neurotransmitter? Trends Neurosci. 2006;29(5):241–249. doi: 10.1016/j.tins.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Ovejero D, Azcoitia I, Doncarlos LL, Melcangi RC, Garcia-Segura LM. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res Brain Res Rev. 2005;48(2):273–286. doi: 10.1016/j.brainresrev.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 46.Mitterling KL, Spencer JL, Dziedzic N, Shenoy S, McCarthy K, Waters EM, McEwen BS, Milner TA. Cellular and subcellular localization of estrogen and progestin receptor immunoreactivities in the mouse hippocampus. J Comp Neurol. 2010;518(14):2729–2743. doi: 10.1002/cne.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crider A, Thakkar R, Ahmed AO, Pillai A. Dysregulation of estrogen receptor beta (ERbeta), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol Autism. 2014;5(1):46. doi: 10.1186/2040-2392-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarachana T, Xu M, Wu RC, Hu VW. Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PLoS One. 2011;6(2):e17116. doi: 10.1371/journal.pone.0017116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88(1):91–124. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galanopoulou AS. GABA receptors as broadcasters of sexually differentiating signals in the brain. Epilepsia. 2005;46(Suppl 5):107–112. doi: 10.1111/j.1528-1167.2005.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nunez JL, McCarthy MM. Androgens predispose males to GABAA-mediated excitotoxicity in the developing hippocampus. Exp Neurol. 2008;210(2):699–708. doi: 10.1016/j.expneurol.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gillberg C, Fernell E, Kocovska E, Minnis H, Bourgeron T, Thompson L, Allely CS. The role of cholesterol metabolism and various steroid abnormalities in autism spectrum disorders: a hypothesis paper. Autism Res. 2017;10(6):1022–1044. doi: 10.1002/aur.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 54.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002;110(7):905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gill S, Chow R, Brown AJ. Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res. 2008;47(6):391–404. doi: 10.1016/j.plipres.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 56.Du X, Pham YH, Brown AJ. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J Biol Chem. 2004;279(45):47010–47016. doi: 10.1074/jbc.M408690200. [DOI] [PubMed] [Google Scholar]
- 57.Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278(25):22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- 59.Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, Crawford DC, Linsenbardt AJ, Shu HJ, Izumi Y, Mennerick SJ, Zorumski CF. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci. 2013;33(44):17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun MY, Taylor A, Zorumski CF, Mennerick S. 24S-hydroxycholesterol and 25-hydroxycholesterol differentially impact hippocampal neuronal survival following oxygen-glucose deprivation. PLoS One. 2017;12(3):e0174416. doi: 10.1371/journal.pone.0174416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dickinson A, Jones M, Milne E. Measuring neural excitation and inhibition in autism: Different approaches, different findings and different interpretations. Brain Res. 2016;1648(Pt A):277–289. doi: 10.1016/j.brainres.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 62.Carlson GC. Glutamate receptor dysfunction and drug targets across models of autism spectrum disorders. Pharmacol Biochem Behav. 2012;100(4):850–854. doi: 10.1016/j.pbb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cellot G, Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr. 2014;2:70. doi: 10.3389/fped.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pilorge M, Fassier C, Le Corronc H, Potey A, Bai J, De Gois S, Delaby E, Assouline B, Guinchat V, Devillard F, Delorme R, Nygren G, Rastam M, Meier JC, Otani S, Cheval H, James VM, Topf M, Dear TN, Gillberg C, Leboyer M, Giros B, Gautron S, Hazan J, Harvey RJ, Legendre P, Betancur C. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol Psychiatry. 2016;21(7):936–945. doi: 10.1038/mp.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sipila ST, Spoljaric A, Virtanen MA, Hiironniemi I, Kaila K. Glycine transporter-1 controls nonsynaptic inhibitory actions of glycine receptors in the neonatal rat hippocampus. J Neurosci. 2014;34(30):10003–10009. doi: 10.1523/JNEUROSCI.0075-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mori M, Gahwiler BH, Gerber U. Beta-alanine and taurine as endogenous agonists at glycine receptors in rat hippocampus in vitro. J Physiol. 2002;539(Pt 1):191–200. doi: 10.1113/jphysiol.2001.013147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 2014;15(10):637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang C, Shimizu-Okabe C, Watanabe K, Okabe A, Matsuzaki H, Ogawa T, Mori N, Fukuda A, Sato K. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 2002;139(1):59–66. doi: 10.1016/s0165-3806(02)00536-9. [DOI] [PubMed] [Google Scholar]
- 69.Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557(Pt 3):829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito S. GABA and glycine in the developing brain. J Physiol Sci. 2016;66(5):375–379. doi: 10.1007/s12576-016-0442-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bitar T, Mavel S, Emond P, Nadal-Desbarats L, Lefevre A, Mattar H, Soufia M, Blasco H, Vourc’h P, Hleihel W, Andres CR. Identification of metabolic pathway disturbances using multimodal metabolomics in autistic disorders in a Middle Eastern population. J Pharm Biomed Anal. 2018;152:57–65. doi: 10.1016/j.jpba.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 72.Tasca CI, Lanznaster D, Oliveira KA, Fernandez-Duenas V, Ciruela F. Neuromodulatory effects of guanine-based purines in health and disease. Front Cell Neurosci. 2018;12:376. doi: 10.3389/fncel.2018.00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Molz S, Dal-Cim T, Tasca CI. Guanosine-5′-monophosphate induces cell death in rat hippocampal slices via ionotropic glutamate receptors activation and glutamate uptake inhibition. Neurochem Int. 2009;55(7):703–709. doi: 10.1016/j.neuint.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 74.Dal-Cim T, Martins WC, Thomaz DT, Coelho V, Poluceno GG, Lanznaster D, Vandresen-Filho S, Tasca CI. Neuroprotection promoted by guanosine depends on glutamine synthetase and glutamate transporters activity in hippocampal slices subjected to oxygen/glucose deprivation. Neurotox Res. 2016;29(4):460–468. doi: 10.1007/s12640-015-9595-z. [DOI] [PubMed] [Google Scholar]
- 75.Ganzella M, de Oliveira ED, Comassetto DD, Cechetti F, Cereser VH, Jr, Moreira JD, Hansel G, Almeida RF, Ramos DB, Figueredo YN, Souza DG, Oses JP, Worm PV, Achaval M, Netto CA, Souza DO. Effects of chronic guanosine treatment on hippocampal damage and cognitive impairment of rats submitted to chronic cerebral hypoperfusion. Neurol Sci. 2012;33(5):985–997. doi: 10.1007/s10072-011-0872-1. [DOI] [PubMed] [Google Scholar]
- 76.Bergeron R, Coyle JT. NAAG, NMDA receptor and psychosis. Curr Med Chem. 2012;19(9):1360–1364. doi: 10.2174/092986712799462685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jessen F, Fingerhut N, Sprinkart AM, Kuhn KU, Petrovsky N, Maier W, Schild HH, Block W, Wagner M, Traber F. N-acetylaspartylglutamate (NAAG) and N-acetylaspartate (NAA) in patients with schizophrenia. Schizophr Bull. 2013;39(1):197–205. doi: 10.1093/schbul/sbr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. 2004;558(Pt 1):5–30. doi: 10.1113/jphysiol.2003.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Owen L, Sunram-Lea SI. Metabolic agents that enhance ATP can improve cognitive functioning: a review of the evidence for glucose, oxygen, pyruvate, creatine, and L-carnitine. Nutrients. 2011;3(8):735–755. doi: 10.3390/nu3080735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown AM, Wender R, Ransom BR. Metabolic substrates other than glucose support axon function in central white matter. J Neurosci Res. 2001;66(5):839–843. doi: 10.1002/jnr.10081. [DOI] [PubMed] [Google Scholar]
- 81.Hassel B, Brathe A. Cerebral metabolism of lactate in vivo: evidence for neuronal pyruvate carboxylation. J Cereb Blood Flow Metab. 2000;20(2):327–336. doi: 10.1097/00004647-200002000-00014. [DOI] [PubMed] [Google Scholar]
- 82.Voronina PP, Adamovich KV, Adamovich TV, Dubouskaya TG, Hrynevich SV, Waseem TV, Fedorovich SV. High concentration of ketone body beta-hydroxybutyrate modifies synaptic vesicle cycle and depolarizes plasma membrane of rat brain synaptosomes. J Mol Neurosci. 2020;70(1):112–119. doi: 10.1007/s12031-019-01406-9. [DOI] [PubMed] [Google Scholar]
- 83.Torres RJ, Puig JG. Hypoxanthine deregulates genes involved in early neuronal development. Implications in Lesch-Nyhan disease pathogenesis. J Inherit Metab Dis. 2015;38(6):1109–1118. doi: 10.1007/s10545-015-9854-4. [DOI] [PubMed] [Google Scholar]
- 84.Ceballos-Picot I, Mockel L, Potier MC, Dauphinot L, Shirley TL, Torero-Ibad R, Fuchs J, Jinnah HA. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum Mol Genet. 2009;18(13):2317–2327. doi: 10.1093/hmg/ddp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harkness RA. Hypoxanthine, xanthine and uridine in body fluids, indicators of ATP depletion. J Chromatogr. 1988;429:255–278. doi: 10.1016/s0378-4347(00)83873-6. [DOI] [PubMed] [Google Scholar]
- 86.Biasibetti-Brendler H, Schmitz F, Pierozan P, Zanotto BS, Prezzi CA, de Andrade RB, Wannmacher CMD, Wyse ATS. Hypoxanthine induces neuroenergetic impairment and cell death in striatum of young adult Wistar rats. Mol Neurobiol. 2018;55(5):4098–4106. doi: 10.1007/s12035-017-0634-z. [DOI] [PubMed] [Google Scholar]
- 87.Bavaresco CS, Chiarani F, Kolling J, Netto CA, de Souza Wyse AT. Biochemical effects of pretreatment with vitamins E and C in rats submitted to intrastriatal hypoxanthine administration. Neurochem Int. 2008;52(6):1276–1283. doi: 10.1016/j.neuint.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 88.El-Ansary A, Bjorklund G, Chirumbolo S, Alnakhli OM. Predictive value of selected biomarkers related to metabolism and oxidative stress in children with autism spectrum disorder. Metab Brain Dis. 2017;32(4):1209–1221. doi: 10.1007/s11011-017-0029-x. [DOI] [PubMed] [Google Scholar]
- 89.Parker W, Hornik CD, Bilbo S, Holzknecht ZE, Gentry L, Rao R, Lin SS, Herbert MR, Nevison CD. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J Int Med Res. 2017;45(2):407–438. doi: 10.1177/0300060517693423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17(4):389–401. doi: 10.1038/mp.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bavaresco CS, Chiarani F, Wannmacher CM, Netto CA, Wyse AT. Intrastriatal hypoxanthine reduces Na(+),K (+)-ATPase activity and induces oxidative stress in the rats. Metab Brain Dis. 2007;22(1):1–11. doi: 10.1007/s11011-006-9037-y. [DOI] [PubMed] [Google Scholar]
- 92.Biasibetti H, Pierozan P, Rodrigues AF, Manfredini V, Wyse ATS. Hypoxanthine intrastriatal administration alters neuroinflammatory profile and redox status in striatum of infant and young adult rats. Mol Neurobiol. 2017;54(4):2790–2800. doi: 10.1007/s12035-016-9866-6. [DOI] [PubMed] [Google Scholar]
- 93.Ishii T, Miyazawa M, Onouchi H, Yasuda K, Hartman PS, Ishii N. Model animals for the study of oxidative stress from complex II. Biochim Biophys Acta. 2013;1827(5):588–597. doi: 10.1016/j.bbabio.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 94.Weidner AM, Bradley MA, Beckett TL, Niedowicz DM, Dowling AL, Matveev SV, LeVine H, 3rd, Lovell MA, Murphy MP. RNA oxidation adducts 8-OHG and 8-OHA change with Abeta42 levels in late-stage Alzheimer’s disease. PLoS One. 2011;6(9):e24930. doi: 10.1371/journal.pone.0024930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96(3):825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- 96.Marklund N, Ostman B, Nalmo L, Persson L, Hillered L. Hypoxanthine, uric acid and allantoin as indicators of in vivo free radical reactions. Description of a HPLC method and human brain microdialysis data. Acta Neurochir. 2000;142(10):1135–1141. doi: 10.1007/s007010070042. [DOI] [PubMed] [Google Scholar]
- 97.Margari L, De Giacomo A, Craig F, Palumbi R, Peschechera A, Margari M, Picardi F, Caldarola M, Maghenzani MA, Dicuonzo F. Frontal lobe metabolic alterations in autism spectrum disorder: a (1)H-magnetic resonance spectroscopy study. Neuropsychiatr Dis Treat. 2018;14:1871–1876. doi: 10.2147/NDT.S165375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baruth JM, Wall CA, Patterson MC, Port JD. Proton magnetic resonance spectroscopy as a probe into the pathophysiology of autism spectrum disorders (ASD): a review. Autism Res. 2013;6(2):119–133. doi: 10.1002/aur.1273. [DOI] [PubMed] [Google Scholar]
- 99.Ma K, Thomason LA, McLaurin J. Scyllo-inositol, preclinical, and clinical data for Alzheimer’s disease. Adv Pharmacol. 2012;64:177–212. doi: 10.1016/B978-0-12-394816-8.00006-4. [DOI] [PubMed] [Google Scholar]
- 100.Levine J, Aviram A, Holan A, Ring A, Barak Y, Belmaker RH. Inositol treatment of autism. J Neural Transm (Vienna) 1997;104(2–3):307–310. doi: 10.1007/BF01273191. [DOI] [PubMed] [Google Scholar]
- 101.Li C, Shen K, Chu L, Liu P, Song Y, Kang X. Decreased levels of urinary free amino acids in children with autism spectrum disorder. J Clin Neurosci. 2018;54:45–49. doi: 10.1016/j.jocn.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 102.Witters P, Debbold E, Crivelly K, Vande Kerckhove K, Corthouts K, Debbold B, Andersson H, Vannieuwenborg L, Geuens S, Baumgartner M, Kozicz T, Settles L, Morava E. Autism in patients with propionic acidemia. Mol Genet Metab. 2016;119(4):317–321. doi: 10.1016/j.ymgme.2016.10.009. [DOI] [PubMed] [Google Scholar]