

Summary
Coronavirus disease 2019 (COVID‐19) is frequently associated with severe systemic consequences, including vasculitis, a hyperinflammatory state and hypercoagulation. The mechanisms leading to these life‐threatening abnormalities are multifactorial. Based on the analysis of publicly available interactomes, we propose that severe acute respiratory syndrome coronavirus‐2 infection directly causes a deficiency in C1 esterase inhibitor, a pathogen‐specific mechanism that may help explain significant systemic abnormalities in patients with COVID‐19.
Keywords: contact system, serpins, virology, complement, SARS‐CoV‐2
Severe manifestations of coronavirus disease 2019 (COVID‐19) include acute respiratory distress, cardiovascular affectation, multi‐organ involvement 1 and coagulopathy, 2 , 3 , 4 which may be compatible with disseminated intravascular coagulation (DIC). 5 , 6 The causal mechanisms of the systemic manifestations associated with COVID‐19 are attributed to the cytokine storm accompanying severe inflammatory syndrome, 7 , 8 , 9 direct viral disruption of endothelial integrity, 10 release of coagulation factors by inflammasome‐activated macrophages, 11 liver dysfunction, 12 anti‐phospholipid antibodies, 13 hypertension, hypoxia, stress from mechanical ventilation, limited mobility of the patients, or a combination of factors.
While examining the published interactomes of severe acute respiratory syndrome coronavirus (SARS‐CoV)14 and SARS‐CoV‐215 proteins [hereafter, CoV‐1 and CoV‐2, respectively) with human proteins, we noted that C1 esterase inhibitor [C1‐INH, encoded by the serine proteinase inhibitor (serpin) family G member 1 (SERPING1) gene] is an interactor for seven distinct CoV‐1 proteins and polypeptides, encoded by open reading frame (ORF) 3b, ORF7b, ORF14, non‐structural protein (nsp)2ab, nsp13ab, nsp14ab and nsp8ab (Fig. 1A). These CoV‐1 proteins are highly similar to their orthologous CoV‐2 proteins (Figure S1B). Along with interferon and innate immune signalling components, such as interferon regulatory factor 3 (IRF3), transmembrane protein 173 (TMEM173), TANK binding kinase 1 (TBK1), inhibitor of nuclear factor kappa B kinase subunit epsilon (IKBKE), tripartite motif containing 25 (TRIM25), mitochondrial antiviral signalling protein (MAVS) or DExD/H‐box helicase 58 (DDX58), C1‐INH is one of the proteins with the highest connectivity in the merged CoV‐1 and CoV‐2 interactomes (Fig. 1C), suggesting a relevant role for these interactions in the life cycle of CoV‐1 and, by similarity, CoV‐2.
Fig 1.
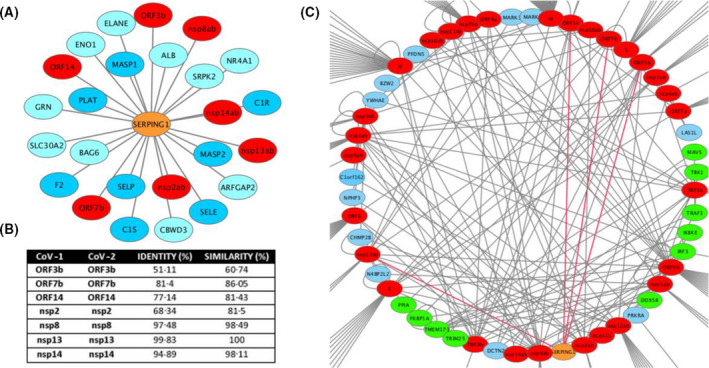
(A) Cytoscape rendering of SERPING1 interactions retrieved from BioGrid. Viral proteins are in red, contact system and coagulation factors in dark blue. (B) Similarities between CoV‐1 and CoV‐2 proteins, estimated from pairwise alignments (Supporting Information). (C) Cytoscape rendering of the nodes with the highest connectivity of human proteins with CoV‐1 and CoV‐2 proteins, merged from BioGrid interaction networks (https://thebiogrid.org). SERPING1 is shown in orange and edges of its interactions with CoV‐1 proteins are in red. Viral proteins are in red; innate immunity and interferon pathway components are in yellow.
Of the SERPING1 CoV interactors, nsp8, together with nsp7, functions as a primase that forms part of the RNA polymerase complex, 16 nsp13 is a helicase, 17 nsp14 is a proofreading exoribonuclease 18 and an S‐adenosyl methionine (SAM)‐dependent (guanine‐N7) methyl transferase. 19 The protein encoded by ORF3b antagonises interferon responses through as yet uncharacterised molecular mechanisms, 20 , 21 the protein encoded by ORF7b bears a transmembrane domain through which it localises to the Golgi complex and whose expression upon viral infection can lead to aberrant localisation of cell‐surface glycoproteins, 22 and ORF14 encodes a 70 (CoV‐1) or 73 (CoV‐2) amino acid protein for which no function has been described. We speculate that the likelihood of a direct interaction of with SERPING1 is lower for nsp7, nsp13 and nsp14, given their above‐described functions, although the formation of multimolecular complexes remains a possibility.
The 105‐KDa glycoprotein C1‐INH, a serine protease inhibitor bearing a conserved serpin domain, is the main inhibitor of the classical complement enzymes C1r and C1 esterase (C1s). 23 It is also the primary inhibitor of the activated Factors XII (FXIIa) and XI (FXIa) and activated plasma kallikrein 23 and, more modestly, of plasmin, tissue‐type plasminogen activator (tPA) and thrombin (Fig. 2). C1‐INH is the sole natural inhibitor of C1r and C1s, is an inhibitor of the lectin pathway of complement activation via inactivation of mannan‐binding lectin‐associated serine protease‐1 and ‐2 (MASP1 and MASP2), and inhibits the alternative pathway of activation by binding to C3b. Thus, C1‐INH is a major regulator of all three pathways of complement activation. 24 C1‐INH is the most heavily glycosylated plasma protein, and bears a sialyl Lewisx‐related moiety through which it binds to endothelial cell‐surface selectins E and P, in competition with the binding of leucocytes, 25 exerting, as such, an anti‐inflammatory function.
Fig 2.
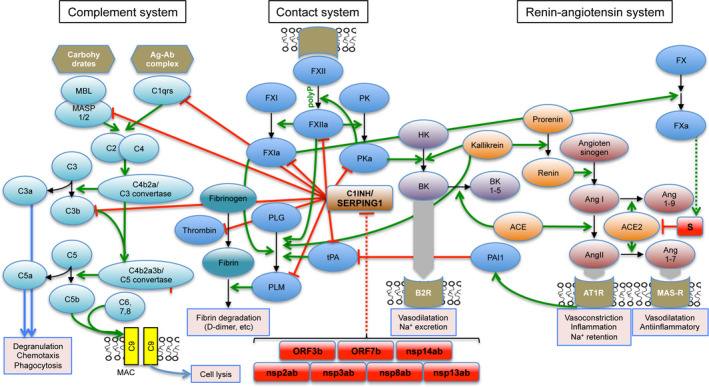
Schematic illustration of the contact, complement and renin–angiotensin systems. Green arrows represent activating functions and red stop rods represent inhibitory functions. Only selected reactions are represented, relevant to the discussion. Contact system: FXII, FXI: Factors XII and XI; PK, prekallikrein; PKa, activated kallikrein; HK, high‐molecular weight kininogen; B2R, bradykinin receptor B2; BK, bradykinin; BK1‐5, bradykinin 1‐5; PLG, plasminogen; PLM, plasmin; tPA, tissue‐type plasminogen activator. Renin–angiotensin system: ACE, angiotensin‐converting enzyme; ACE2, angiotensin‐converting enzyme 2; AngI, angiotensin I; AngII, angiotensin II; Ang1‐7, angiotensin 1‐7; AT1R, angiotensin type 1 receptor; MAS‐R, MAS receptor. Complement system: MBL, mannose‐binding lectin; MASP1/2, mannan‐binding lectin‐associated serine protease‐1/2. Other factors and components of the three systems are shown with conventional designations. SARS‐CoV and SARS‐CoV‐2 predicted proteins are shown as red‐filled rectangles. Also represented is the negative regulation of cell‐surface ACE2 through internalisation upon interaction and entry of the CoV‐1 and CoV‐2 spike proteins.
Through covalent bond formation with the complement components C1s, C1r, MASP1 and MASP2 and reversible binding to C3a24, C1‐INH attenuates the consequences of complement activation, including the generation of pro‐inflammatory anaphylatoxins, especially C5a, and the formation of a membrane attack complex (MAC) that leads to cell lysis. Of note, severe acute respiratory distress syndrome (ARDS) in SARS was found to be associated with excessive activation of C3.26. Further, MASP2 has been found to be a target of the nucleocapsid (N) protein of Middle East respiratory syndrome (MERS)‐CoV, SARS‐CoV and SARS‐CoV‐2,27 and a blocking antibody to C5a has shown benefit in patients with COVID‐19 with severe lung injury.
The presence of pulmonary oedema is another feature in patients with COVID‐19 with ARDS, and aberrant activation of the kallikrein–bradykinin (BK) system has been proposed as an explanatory mechanism. 28 A further connection with aberrant activation of the contact system in COVID‐19 is provided by the emergence of cases of Kawasaki disease (KD)‐like syndrome in SARS‐CoV‐2‐positive children, 29 characterised by skin rash, gastrointestinal affectation, myocarditis and shock syndrome. Laboratory tests indicated ongoing fibrinolysis, with elevated blood D‐dimer levels. KD is the most frequent childhood vasculitis 30 and is frequently accompanied with excessive thrombocytosis, 31 and evidence of activation of the classical complement pathway. 32
The plasma contact system is a pro‐coagulant and pro‐inflammatory protease cascade that occurs on the surface of endothelial cells. 33 Upon contact with surface‐bound negatively charged polymers such as polyphosphate (polyP), proteoglycans or RNA, FXII is activated to catalyse the proteolytic activation of plasma kininogen to kallikrein which, in turn, converts high‐molecular‐weight kininogen to BK 23 (Fig. 1D). FXII also binds to the endothelial cell surface through interactions with the urokinase receptor or integrins (through fibronectin‐like domain), exerting signalling functions independent of its catalytic activity. BK is liganded to the constitutively expressed cell‐surface G‐protein‐coupled receptor, B2R, causing vasodilatation and increased vascular permeability. BK is degraded by carboxypeptidase N (in plasma) or carboxypeptidase M (on endothelial cells), yielding des‐arg‐9 BK, which interacts with a second, cytokine‐induced receptor, B1R, through which it may prolong the vascular response until it is fully inactivated by angiotensin‐converting enzyme (ACE), aminopeptidase P, or neutral endopeptidase. 34 A deficit in C1‐INH, found in the rare diseases types I and II hereditary angioedema (HAE), acquired angioedema and age‐related macular degeneration, results in excessive activation of FXII and unchecked production of BK, leading to angioedema. 35
Upon contact activation, FXIa initiates the coagulation cascade through sequential activation of Factors X, IX and prothrombin (Factor II), resulting in the polymerisation of fibrin from fibrinogen. Incidentally, Factor Xa cleaves the spike protein (S) of CoV‐1 at the S1–S2 boundary, enhancing the fusion of viral particles to cell membranes. 36 It has not yet been determined if the CoV‐2 spike protein, which is cleaved at the S1–S2 boundary by transmembrane protease, serine 2 (TMPRSS2)37 and furin 38 is also cleaved by FXa. At the endothelial cell surface, C1‐INH regulates the intrinsic coagulation pathway by inhibiting multiple enzymes: the pro‐coagulant enzymes FXIIa, FXIa and thrombin, and the pro‐fibrinolytic enzymes tPA and plasmin (Fig. 2). As such, loss of expression or function of C1‐INH would be expected to result in augmented coagulation and fibrinolysis and, indeed, blood D‐dimer levels can be elevated during angioedema attacks in HAE. 39 However, except in anecdotal cases with concurring pro‐coagulant events, 40 thromboembolism is generally not observed in HAE. In this regard, because patients severely deficient in FXII, high‐molecular weight kininogen or prekallikrein display normal haemostasis, the intrinsic coagulation pathway was considered not to have a function in physiological haemostasis. This seeming paradox was resolved by the finding that FXII is potently activated by activated platelets, platelet‐shed vesicles and solid‐phase bound (membrane‐associated) negatively charged molecules such as polyP (stored in platelets in complex with Ca2+) or RNA. 41
The interaction of several CoV‐1 (and, by similarity, CoV‐2) proteins with C1‐INH suggests that this essential regulator of the contact system is inhibited during viral infection, leading to a propensity to activate the complement cascade, the BK pathway and the intrinsic coagulation cascade. For the latter to promote thromboembolism and other clinical manifestations of pathological coagulation, a concomitant activation or destruction of platelets, or local release of negatively charged polymers may be required. SARS‐CoV‐2 can infect multiple human cell types through the engagement of ACE2, expressed on the surface of alveolar type 2 pneumocytes, enterocytes, kidney epithelial cells and endothelial cells, 42 causing extensive cell death and tissue destruction. Viral destruction of endothelial cells, 10 either through direct cell disruption or indirectly through inflammatory mechanisms, activates the extrinsic coagulation pathway, initiated by interaction of platelets with collagen and tissue factor and initial production of thrombin. This is followed by amplified production of thrombin through the intrinsic pathway. 43 As such, a deficit in available C1‐INH caused by interacting CoV‐2 or CoV‐1 proteins would be expected to prime the intrinsic coagulation pathway, leading to a pro‐coagulant state that can overcome physiological anticoagulant activities.
Depletion of C1‐INH below certain thresholds increases the risk of angioedema attacks in HAE. 44 C1‐INH replacement therapy has long been used in acute and prophylactic treatment of HAE, 45 and could be useful in the management of COVID‐19‐associated coagulopathy. Further, heparin and sulphated glycans amplify the inhibitory functions of C1‐INH on the contact system, 46 which, in addition to the activities of heparin on coagulation factors, could help explain the beneficial effects of heparin in the management of patients with COVID‐19.
Relevantly, a recent limited observational study in five patients with COVID‐19 with severe pneumonia reported a rapid improvement in their clinical and laboratory parameters after treatment with C1‐INH (Ruconest, Pharming, Leiden, The Netherlands). The reported outcomes were resolution of fever within 48 h in four of the patients, accompanied with decreased blood C‐reactive protein and interleukin 6, followed by rapid full recovery and discharge. The remaining patient had a slightly delayed recovery (https://www.pharming.com/news/pharming‐reports‐encouraging‐results‐use‐ruconestr‐covid‐19‐patients). The fact that replenishment of C1‐INH prompts the recovery of patients with severe COVID‐19 not responding to a given standard‐of‐care (hydroxychloroquine and lopinavir/ritonavir) strongly suggests that such patients have C1‐INH depletion. To our knowledge, our hypothesis provides the first mechanistic explanation for such a deficiency. On these bases, we propose C1‐INH replenishment as a useful adjunct in the management of COVID‐19.
Competing interests
The authors declare no competing interests.
Author contributions
Timothy M. Thomson conceived and designed the study; Timothy M. Thomson and Emily Toscano‐Guerra performed analyses and prepared the figure; Timothy M. Thomson wrote the manuscript; Rosanna Paciucci and Ernesto Casis provided critical information and assessment.
Supporting information
Fig S1. Supplementary Figure 1B.
Acknowledgements
This study was supported by grant COVID‐006 from the Spanish Science Council (CSIC) (to T.M.T.). Research by R.P. is funded by the Spanish Ministry of Science and Innovation (RTI 2018‐096055‐B‐100).
References
- 1. Wu Z, McGoogan JM. Characteristics of and important lessons from the Coronavirus Disease 2019 (COVID‐19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–42. DOI: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 2. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–7. DOI: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62. DOI: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091. DOI: 10.1136/bmj.m1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18:1094–9. DOI: 10.1111/jth.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID‐19: a prospective cohort study. Ann Intern Med. 2020. [Epub ahead of print]. DOI: 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borges AH, O'Connor JL, Phillips AN, Baker JV, Vjecha MJ, Losso MH, et al. Factors associated with D‐dimer levels in HIV‐infected individuals. PLoS One. 2014;9:e90978. DOI: 10.1371/journal.pone.0090978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramacciotti E, Agati LB, Aguiar VC, Wolosker N, Guerra JC, de Almeida RP, et al. Zika and chikungunya virus and risk for venous thromboembolism. Clin Appl Thromb Hemost. 2019;25:1076029618821184. DOI: 10.1177/1076029618821184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smither SJ, O'Brien L, Eastaugh L, Woolley T, Lever M, Fletcher T, et al. Haemostatic changes in five patients infected with Ebola virus. Viruses. 2019;11:647. DOI: 10.3390/v11070647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395:1417–8. DOI: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe 2020;27:556–570.e6. DOI: 10.1016/j.chom.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang C, Shi L, Wang FS. Liver injury in COVID‐19: management and challenges. Lancet Gastroenterol Hepatol. 2020;5:428–30. DOI: 10.1016/S2468-1253(20)30057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid‐19. N Engl J Med. 2020;382:e38. DOI: 10.1056/NEJMc2007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pfefferle S, Schöpf J, Kögl M, Friedel CC, Müller MA, Carbajo‐Lozoya J, et al. The SARS‐coronavirus‐host interactome: identification of cyclophilins as target for pan‐coronavirus inhibitors. PLoS Pathog. 2011;7:e1002331. DOI: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020. [Online ahead of print]. DOI: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhai Y, Sun F, Li X, Pang H, Xu X, Bartlam M, et al. Insights into SARS‐CoV transcription and replication from the structure of the nsp7‐nsp8 hexadecamer. Nat Struct Mol Biol. 2005;12:980–6. DOI: 10.1038/nsmb999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ivanov KA, Thiel V, Dobbe JC, van der Meer Y, Snijder EJ, Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J Virol. 2004;78:5619–32. DOI: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011;8:270–9. DOI: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y, Cai H, Pan J, Xiang N, Tien P, Ahola T, et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci USA. 2009;106:3484–9. DOI: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou P, Li H, Wang H, Wang LF, Shi Z. Bat severe acute respiratory syndrome‐like coronavirus ORF3b homologues display different interferon antagonist activities. J Gen Virol. 2012;93:275–81. DOI: 10.1099/vir.0.033589-0. [DOI] [PubMed] [Google Scholar]
- 21. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS‐CoV‐2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant. bioRxiv, 2020. [Online ahead of print]. DOI: 10.1101/2020.05.11.088179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schaecher SR, Diamond MS, Pekosz A. The transmembrane domain of the severe acute respiratory syndrome coronavirus ORF7b protein is necessary and sufficient for its retention in the Golgi complex. J Virol. 2008;82:9477–91. DOI: 10.1128/JVI.00784-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maas C, Oschatz C, Renne T. The plasma contact system 2.0. Semin Thromb Hemost 2011:37:375–81. DOI: 10.1055/s-0031-1276586. [DOI] [PubMed] [Google Scholar]
- 24. Jiang H, Wagner E, Zhang H, Frank MM. Complement 1 inhibitor is a regulator of the alternative complement pathway. J Exp Med. 2001;194:1609–16. DOI: 10.1084/jem.194.11.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai S, Davis AE 3rd. Complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial‐leukocyte adhesion. J Immunol. 2003;171:4786–91. DOI: 10.4049/jimmunol.171.9.4786. [DOI] [PubMed] [Google Scholar]
- 26. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio 2018;9:e01753–18. DOI: 10.1128/mBio.01753-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP‐2‐mediated complement over‐activation. medRxiv. 2020. [Online ahead of print]. DOI: 10.1101/2020.03.29.20041962. [DOI] [Google Scholar]
- 28. van de Veerdonk Frank L, Netea MG, van Deuren M, van der Meer Jos WM, de Mast Q, Brüggemann RJ, et al. Kallikrein‐kinin blockade in patients with COVID‐19 to prevent acute respiratory distress syndrome. Elife. 2020;9:e57555. DOI: 10.7554/eLife.57555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toubiana J, Poirault C, Corsia A, Bajolle F, Fourgeaud J, Angoulvant F, et al. Outbreak of Kawasaki disease in children during COVID‐19 pandemic: a prospective observational study in Paris, France. medRxiv. 2020. [Online ahead of print]. DOI: 10.1101/2020.05.10.20097394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schnabel A, Hedrich CM. Childhood vasculitis. Front Pediatr. 2018;6:421. DOI: 10.3389/fped.2018.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nigrovic LE, Nigrovic PA, Harper MB, Chiang VW. Extreme thrombocytosis predicts Kawasaki disease in infants. Clin Pediatr (Phila). 2006;45:446–52. DOI: 10.1177/0009922806289621. [DOI] [PubMed] [Google Scholar]
- 32. Kohsaka T, Abe J, Asahina T, Kobayashi N. Classical pathway complement activation in Kawasaki syndrome. J Allergy Clin Immunol. 1994;93:520–5. DOI: 10.1016/0091-6749(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 33. Long AT, Kenne E, Jung R, Fuchs TA, Renne T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. 2016;14:427–37. DOI: 10.1111/jth.13235. [DOI] [PubMed] [Google Scholar]
- 34. Kaplan AP, Ghebrehiwet B. The plasma bradykinin‐forming pathways and its interrelationships with complement. Mol Immunol. 2010;47:2161–9. DOI: 10.1016/j.molimm.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 35. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382:1136–48. DOI: 10.1056/NEJMra1808012. [DOI] [PubMed] [Google Scholar]
- 36. Du L, Kao RY, Zhou Y, He Y, Zhao G, Wong C, et al. Cleavage of spike protein of SARS coronavirus by protease factor Xa is associated with viral infectivity. Biochem Biophys Res Commun. 2007;359:174–9. DOI: 10.1016/j.bbrc.2007.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. DOI: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoffmann M, Kleine‐Weber H, Pohlmann S. A multibasic cleavage site in the spike protein of sARS‐CoV‐2 Is essential for infection of human lung cells. Mol Cell. 2020;78:779–784.e5. DOI: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reshef A, Zanichelli A, Longhurst H, Relan A, Hack CE. Elevated D‐dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy. 2015;70:506–13. DOI: 10.1111/all.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oguma K, Suzuki T, Mano S, Takeuchi S, Takeda J, Maruyama Y, et al. Hereditary angioedema with deep vein thrombosis and pulmonary thromboembolism during pregnancy. Taiwan J Obstet Gynecol. 2019;58:895–6. DOI: 10.1016/j.tjog.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 41. Maas C, Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131:1903–9. DOI: 10.1182/blood-2017-04-569111. [DOI] [PubMed] [Google Scholar]
- 42. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS‐CoV‐2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9:45. DOI: 10.1186/s40249-020-00662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoffman M. Remodeling the blood coagulation cascade. J Thromb Thrombolysis. 2003;16:17–20. DOI: 10.1023/B:THRO.0000014588.95061.28. [DOI] [PubMed] [Google Scholar]
- 44. Spath PJ, Wuthrich B, Butler R. Quantification of C1‐inhibitor functional activities by immunodiffusion assay in plasma of patients with hereditary angioedema–evidence of a functionally critical level of C1‐inhibitor concentration. Complement. 1984;1:147–59. DOI: 10.1159/000467830. [DOI] [PubMed] [Google Scholar]
- 45. Henry Li H, Riedl M, Kashkin J. Update on the use of C1‐esterase inhibitor replacement therapy in the acute and prophylactic treatment of hereditary angioedema. Clin Rev Allergy Immunol. 2019;56:207–18. DOI: 10.1007/s12016-018-8684-1. [DOI] [PubMed] [Google Scholar]
- 46. Schoenfeld AK, Lahrsen E, Alban S. Regulation of complement and contact system activation via C1 inhibitor potentiation and factor XIIa activity modulation by sulfated glycans – structure‐activity relationships. PLoS One. 2016;11:e0165493. DOI: 10.1371/journal.pone.0165493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Supplementary Figure 1B.