

Abstract
Fatty acid synthases are dynamic ensembles of enzymes that can efficiently biosynthesize long hydrocarbon chains. Here we visualize the interaction between the Escherichia coli acyl carrier protein (AcpP) and β-ketoacyl-ACP-synthase I (FabB) using X-ray crystallography, NMR, and MD simulations. We leveraged this structural information to alter lipid profiles in vivo and provide a molecular basis for how protein-protein interactions can regulate the fatty acid profile in E. coli.
The E. coli fatty acid synthase (FAS) produces fatty acids through an iterative cycle via the activities of 13 discrete proteins that yield both saturated and unsaturated products1,2, with each enzyme carrying out a single, simple transformation. These enzymes are targets for antibiotic inhibition3 and biofuel development4, given their crucial nature and hydrocarbon products. However, the regulation of the discrete steps in this pathway remains poorly understood, and attempts to decipher fundamental phenomena of fatty acid metabolism have been hindered by a lack of information about the molecular interactions between enzymes5.
The critical acyl carrier protein, AcpP, shuttles cargo between partners through iterative biosynthetic cycles (Supplementary Figure 1). As a small, dynamic, monomeric helical bundle6,7, AcpP provides protection to the growing fatty acid intermediates from non-selective reactivity in the cytosol by sequestering them within a central hydrophobic core8. This cargo is covalently tethered to AcpP via thioester linkage to a post-translationally added phosphopantetheine (PPant) arm9. Due to the dynamic nature of both the protein and PPant10, AcpP can sequester diverse cargo lengths and moieties with important cargo-induced structural and regulatory consequences11. Synthetic cargo attachment offers an excellent handle for structural biology studies12,13. During interaction with partner proteins, the cargo completely translocates from the hydrophobic core of AcpP into the partner in a process called “chain flipping”14,15.
β-ketoacyl-ACP-synthase partners, including FabB, facilitate FAS carbon-carbon bond formation in three discrete steps (Supplementary Figure 1). First, an active site cysteine attacks the PPant thioester and releases AcpP. Next, a malonyl-loaded AcpP associates, and, by Claisen-like condensation, extends the chain. Finally, the AcpP dissociates carrying the elongated β-ketoacyl chain. We recently applied a mechanism-based probe to crosslink AcpP with β-hydroxyacyl-AcpP-dehydratase (FabA) and reported the crystal structure of the proteins during interaction13.
To prepare an AcpP-FabB complex, we developed a covalent chloroacryl probe exploiting the nucleophilicity of FabB’s active site cysteine (Supplementary Figure 1)16. This probe was chemoenzymatically appended to AcpP17 followed by incubation with FabB to generate a covalent AcpP2-FabB2 complex (Supplementary Figure 2–3). The 2.4 Å resolution AcpP2-FabB2 crystal structure (Supplementary Table 1) was solved by molecular replacement using a FabB structure as a search model and manual placement of crosslinked AcpPs (Figure 1 and Supplementary Figure 4–7). The AcpP2-FabB2 complex consists of a FabB dimer with each monomer crosslinked to a single AcpP (Figure 1a), similar to the AcpP-FabA complex13. Each AcpP displays a helical bundle fold, with the probe extending from the conserved S36 sidechain at the bottom of AcpP helix II to the active site catalytic cysteine (C163) of FabB (Figure 1b and Supplementary Figure 4a–b).
Figure 1: Crystal structure of E. coli AcpP-FabB complex.
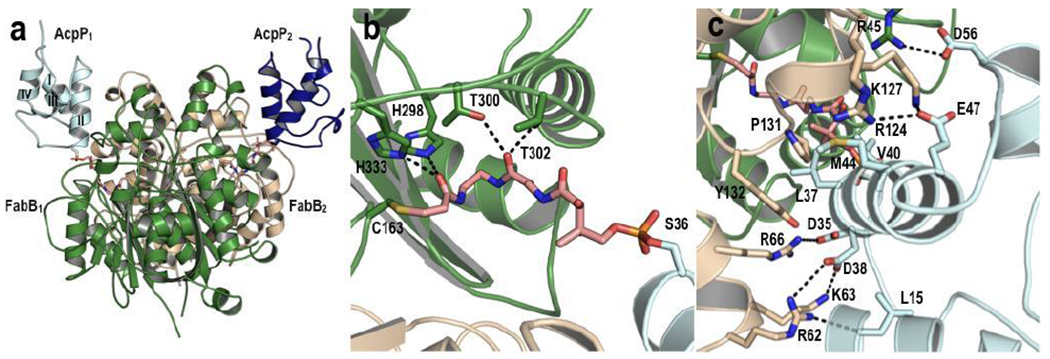
(a) Overall AcpP-FabB complex structure, with the FabB monomers shown in dark green and light tan, the AcpP monomers shown in dark blue and light cyan, and the crosslinker in pink. AcpP helices are labeled I - IV. (b) FabB active site interactions with the crosslinker. (c) AcpP-FabB interface interactions.
Each AcpP makes several interactions primarily through helix II, which is well conserved in carrier proteins18 (Figure 1c and Supplementary Figure 4–5). Both AcpP-FabB interfaces share a set of common interactions. At the bottom of AcpP helix II, D35 and D38 interact with R62, K63, and R66 on FabB, while E47 at the top of helix II interacts with R124 and K127 on FabB. Additionally, each D56 on helix III forms a salt bridge with R45 of its FabB partner, and the backbone of L15 on AcpP forms a hydrogen bond with R62 on FabB. Hydrophobic interactions were observed near the bottom of helix II, primarily between L37, V40, and M44 of AcpP and P131 and Y132 of FabB. A comparison of the AcpP:FabB and AcpP:FabA interfaces reveals distinct AcpP binding motifs for each partner enzyme (Supplementary Figure 8–13); many close contacts with FabB appear near the bottom of helix II, whereas the interactions with FabA are predominantly at the top of helix II. In both structures, salt bridges are observed between helix III and the partner protein (Supplementary Figure 9, 11).
A comparison of the individual AcpP-FabB dimer pairs reveals that the pantetheine binding sites and protein-protein interfaces are generally similar but with key differences. Divergence in the structure is observed in helix III near D56; AcpP1 maintains helical structure at this location, while AcpP2 does not (Supplementary Figure 5e–f). Additionally, high crystallographic B-factors suggest that the residues corresponding to helix III of AcpP2 display more thermal motion or conformational disorder when compared to the ordered helix III on AcpP1 (Supplementary Figure 10). Similar differences were also observed in the AcpP monomers of the AcpP-FabA and AcpP-FabZ structures13,19. Together, these suggest that AcpP interactions with dimeric partner enzymes may be influenced by allosteric regulation or cooperativity. We are currently investigating this using computational and experimental methods to rule out crystal packing as a potential source of these differences, though the fact that this has been observed in all known AcpP-partner structures to date underscores its potential significance.
Solution-state protein NMR experiments characterized the AcpP-FabB interaction in vitro, for comparison with crystallographic observations. 1H,15N-HSQC titration experiments were performed in which labeled octanoyl-AcpP was exposed to increasing molar equivalents of unlabeled FabB (Figure 2a, Supplementary Figure 14–16, Supplementary Table 2–3). Observed peak migrations, quantified as Chemical Shift Perturbations (CSPs), due to the acyl-AcpP FabB interaction matched well with crystallographic observations. D35 and D38 exhibit dramatic CSPs; E47 on helix III demonstrates a moderate CSP. Overall, the NMR CSP plots suggest the transient interaction between AcpP and FabB depends significantly on helix II and, to a lesser extent, on helix III. Additionally, L15, located at the top of helix I at the AcpP:FabB interface, undergoes significant CSP. This is likely due to proximal, transient interaction between the helical backbone and R63 on FabB. Medium-intensity CSPs on the helix III-IV loop as well as helix IV correlate to motion associated with the collapse of the hydrophobic pocket as the acyl substrate is translocated into the FabB core, as seen in our AcpP-FabA work13.
Figure 2: Protein-protein interactions and the fatty acid profile.
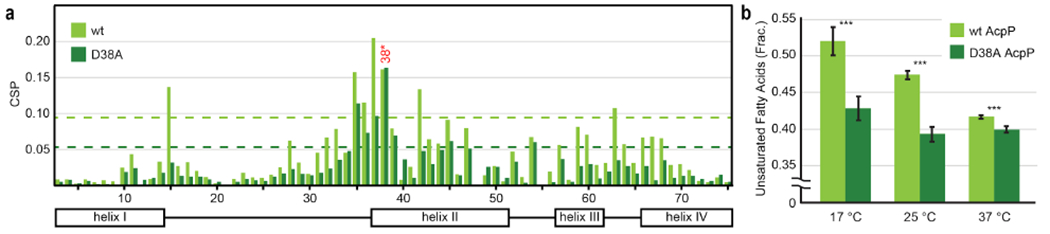
(a) Chemical shift perturbation plot for 1H,15N-HSQC titration experiments of wt C8-AcpP and C8-D38A AcpP against 2.0 molar equivalents FabB. Residue number represented on the lower axis. Dashed lines represent one SD above mean, to highlight significant CSPs. (b) AcpP complementation temperature response. wt AcpP is shown in light green; D38A AcpP is shown in dark green. Four biological replicates were prepared. *** denotes statistical significance with P values below 0.001. Error bars represent SD.
Due to the observed importance of D3820, a D38A mutant was prepared and subjected to the same NMR titration study as the wild type AcpP (Figure 2a, Supplementary Figure 14–16, Supplementary Table 2–3). The D38A AcpP still interacts with FabB; however, the measured CSPs are smaller. The top of helix II appears less perturbed in the D38A mutant than in the wild type, confirming that the D38(AcpP)–K63(FabB), D38–R66, and D38–R62 salt bridges observed crystallographically and computationally are important in stabilizing the complex in solution.
Quantification of this different was achieved by fitting of the CSP curves to calculate the Kd for wt C8-AcpP with FabB and D38A C8-AcpP with FabB, observed to be 37.6 ± 6.6 μM and 167 ± 15 μM respectively. Furthermore, the ΔΔG comparing the wt and D38A interactions was calculated to be 3.84 kJ / mol, confirming the importance of D38 as a hydrogen bonding partner.
Molecular dynamics (MD) simulations were conducted to explore the motions involved in AcpP interactions with FabB. Several long simulations were performed on the complex with AcpP in different states to identify changes in protein-protein dynamics. The AcpPs were modeled as apo (no PPant), holo (empty PPant), C8 acyl-substrate loaded, or C10 acyl-product loaded. Comparisons of the average structures obtained from the various complex states revealed a possible mechanism for communication and recognition between AcpP and FabB (Supplementary Figure 17–19, Supplementary Movie 1–2). In the apo state, AcpP loses its ability to maintain several of the salt bridge contacts observed in the crystal structure. However, upon being activated to its holo state, salt bridge contact times increase on the bottom of helix II. Interestingly, upon actual loading of the PPant with a C10 product (Supplementary Figure 17), AcpP helix III begins to make new interactions with FabB not observed in the crystal structure. A closer look at the secondary structure reveals increased movement in the α6-α7 helix-turn-helix (HTH) motif present in FabB, which is in close proximity to interact with another highly flexible helix (α10) on the same monomer. Both motifs are also able to interact with substrates or products in the active sites; further, α10 is able to interact with helix III of AcpP. Together, these results highlight the importance of anchoring helix II interactions to facilitate correct orientation of the AcpP for productivity. The interactions of helix III change when including the reaction product, suggesting communication between the product-bound state and FabB, and chain translocation may be facilitated by these interactions between FabB and helix III of AcpP (Supplementary Figure 19 and Supplementary Movie 1–2).
Given these observations, we hypothesized that weakening interactions between AcpP and FabB would alter the in vivo fatty acid profile. E. coli increases membrane fluidity in response to temperature reduction by immediate introduction of cis-Δ9 unsaturations into membrane lipids21, via de novo FA synthesis. Regulation appears to occur through coordinated action of multiple dehydratases (FabA and FabZ) and ketosynthases (FabB and FabF)22. Both FabB and FabF perform chain elongation, but only FabB can act on the isomerized 3-cis substrate produced by FabA23. Recent work found that point mutations at the AcpP:Ketosynthase interface were capable of “severely perturbing” substrate shuttling and producing different fatty acid product profiles24.
We performed in vivo studies of the AcpP-FabB interaction via a complementation assay in which a D38A AcpP was used to rescue a wt AcpP knockout. We used a genomic AcpP knockout strain of E. coli developed by Cronan with a wt AcpP on one plasmid and the mutant AcpP on another under orthogonal induction and antibiotic selection25. wt and mutant AcpP cultures were grown to log phase, exposed to a test temperature, and evaluated for fatty acid composition (Figure 2b and Supplementary Figure 20). When complemented with D38A AcpP, E. coli produced smaller fractions of Unsaturated Fatty Acids (UFA) in response to reduced growth temperature than strains complemented with wt AcpP. Although the importance of D38 in other AcpP-partner complexes remains unknown, our structural data and the literature20 demonstrate that D38 is critical to the AcpP-FabB interaction. Weakening this interaction yields reduced CSPs in vitro and diminished UFA production in vivo.
Combined with our previous studies on the AcpP-FabA interaction, this AcpP-FabB structure provides foundational progress towards understanding UFA production in E. coli. Furthermore, the in vitro, in silico, and in vivo results together highlight the importance of AcpP helix II for anchoring and AcpP helix III for chain translocation. Chain flipping may be required for the productive outcome of these protein-protein interactions, but this does not address the subtle differences in each interaction that are critical to pathway regulation, processivity, and cargo communication. The importance of D38 specifically to UFA production provides a glimpse into the subtle differences in specificity that guide whole-organism responses. The differences between the AcpP-FabA and AcpP-FabB interactions reveal the variable interaction networks that can act on the AcpP. Moving forward, these subtle interactions must be thoroughly understood to inform future drug discovery and pathway engineering efforts.
ONLINE METHODS
Protein expression and purification
pET28b 6x His-tagged constructs containing the genes for E. coli AcpP and FabB were separately transformed into E. coli BL21 (DE3) cells by heat shocking at 42 °C for 45 seconds and plated onto LB agar plates supplemented with 50 μg/mL kanamycin. Colonies were transferred to a 10 mL LB starter culture supplemented with 50 μg/mL kanamycin and shaken overnight at 37 °C. The starter culture was then transferred to 1 L of LB supplemented with 50 μg/mL kanamycin and shaken at 37 °C. Once the OD600 reached 0.6, expression was induced by addition of 1 mM IPTG, and the cells were shaken overnight at 18 °C. Cells were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 10 mM imidazole, 10 % glycerol), and flash frozen in liquid nitrogen for storage at −80 °C.
The resuspended cells were lysed by sonication, and the lysate was centrifuged at 21,000 rcf for 60 minutes to remove cellular debris. The lysate was then incubated with 5 mL of Ni-NTA resin (ThermoFisher Scientific) for 1 hour, and the resin was washed with lysis buffer to remove unbound protein. The proteins of interest were eluted in fractions using an imidazole gradient. SDS-PAGE was used to analyze the fractions, and fractions containing the protein of interest were combined and dialyzed into a storage buffer (25 mM Tris pH 7.5, 100 mM NaCl, 1 mM DTT, 5% glycerol). AcpP was further purified using a HiTrap Q FF anion exchange column (GE Healthcare Lifesciences).
Crosslinking, complex purification, and crystallization
Due to endogenous acpS (acyl carrier protein synthase) activity, which attaches phosphopantetheine to a serine residue in a conserved DSL motif, heterologous expression of carrier proteins yields a mixture of apo- and holo- forms. For conversion to homogenous samples, the phosphopantetheine (PPT) prosthetic group was first removed from AcpP by incubation with recombinant MBP-tagged ACP hydrolase (AcpH)26, including 12.5 mM MgCl2 and 2.5 mM MnCl2. After removing the AcpH with amylose resin (New England Biolabs), homogeneous apo-AcpP was chemoenzymatically loaded with the chloroacryl-pantetheine crosslinker16 to form crypto-AcpP using recombinant CoaA, CoaD, CoaE, Sfp, 200 mM ATP, 10 mM MgCl, and a 1.5x molar excess of the crosslinker. The loading was confirmed using MALDI-TOF mass spectrometry. AcpP was purified away from the loading enzymes using a second HiTrap Q FF column.
100 μM FabB was incubated with 200 μM crypto AcpP in 25 mM Tris pH 7.5, 100 mM NaCl, 5 mM DTT, and 5% glycerol overnight at 37 °C. The complex was then purified using a Superdex 200 (GE Healthcare Lifesciences) size exclusion column, concentrated to 6 mg/mL, and flash frozen for storage. The complex was crystallized in 0.1 M sodium acetate pH 5.4, 0.2 M ammonium acetate, and 20% PEG 4000 using the sitting drop vapor diffusion method. The crystals were flash frozen in liquid nitrogen for storage. Diffraction data were measured using beamline 8.2.1 at the Advanced Light Source synchrotron facility and processed using Mosflm27. The structure was solved by molecular replacement using a FabB structure (PDB code: 2VB9) as a search model and refined using the Phenix suite of programs28.
NMR Titration Studies
Isotopically labeled wt and D38A AcpP for NMR studies was prepared as previously reported13,16. In brief, BL21 (DE3) E. coli cells bearing the appropriate construct were first deuterium acclimated then grown at 1 L scale in 15N, 2H M9 minimal media. A 5 mL culture of M9 minimal media prepared with 25% D2O and 75% H2O was inoculated and grown overnight at 37 °C. 100 μL of this dense culture was used to inoculate a 5 mL culture prepared with 50% D2O and 50% H2O, which was grown overnight at 37 °C. In turn, 100 μL of this dense culture was used to inoculate a 5 mL culture prepared with 75% D2O and 25% H2O, which was grown overnight at 37 °C. Finally, 100 μL of this dense culture was used to inoculate a 5 mL culture prepared with 100% D2O and grown overnight at 37 °C. This was used to inoculate a 1 L flask of M9 minimal media prepared with 1 L D2O, 1 g 15NH4Cl, and 4 g glucose. The culture was grown (37 °C, 120 RPM shaking, baffled flask) to an OD600 of 0.8. Expression was induced with the addition of 1 mL of 1M IPTG (in D2O, 0.22 μM sterile filtered), and carried out for 4 hours at 37 °C. Cells were harvested by centrifugation at 600 rcf, 30 min, 6 °C.
Cells were resuspended in 40 mL of lysis buffer (25 mM HEPES pH 7.4, 250 mM NaCl, and 10% glycerol). They were lysed by French pressure cell at ~25,000 PSI, over three presses, with DNAse and RNAse. Lysate was clarified by centrifugation at 12,000 rcf, 45 min, 6 °C and subjected to Ni-NTA (ThermoFisher Scientific) purification. Clarified lysate was tumbled with loose resin at 4 °C for 30 minutes, then washed with 25 mL wash buffer (25 mM HEPES pH 7.4, 250 mM NaCl, and 10% glycerol, 25 mM Imidazole pH 7.4) to remove nonspecific binding. AcpP was eluted with 10 mL elution buffer (25 mM HEPES pH 7.4, 250 mM NaCl, and 10% glycerol, 250 mM Imidazole pH 7.4) then desalted (PD-10 desalting column, GE Healthcare) into 25 mM HEPES pH 7.4, 250 mM NaCl, and 10% glycerol. Preparation of homogeneous apo-AcpP was carried out as reported in the “Crosslinking, complex purification, and crystallization” section. Conversion to C8-AcpP was carried out using the one-pot scheme reported in the “Crosslinking, complex purification, and crystallization” section but using an amide-linked octanoyl fatty acid probe in lieu of the crosslinking probe.
NMR experiments were carried out in the Biomolecular NMR Facility at UCSD, managed by Dr. Xuemei Huang. Titration experiments were performed at 37 °C on a 600 MHz Bruker Avance III system equipped with a cryoprobe. Each 1H-15N HSQC was acquired with uniform sampling, 2048 (R+I) points in the direct dimension, 256 (R+I) points in the 15N dimension, 24 scans, and a 1.5 s recycle delay. Samples were prepared at ~100 μM AcpP in 50 mM potassium phosphate pH 7.4, 0.01% sodium azide, 2.5 mM tris(2-carboxyethyl)phosphine, and 10% D2O. Titration increment points were achieved by preparing a zero molar equivalent and maximum molar equivalent sample, acquiring the first and last points of the titration, then incrementally mixing them to achieve intermediate ratios. This approach was found to yield more accurate ratios and limit protein loss. 1H-15N HSQCs at each equivalent of FabB were collected. Deuteration of the carrier protein was necessary for detection of the backbone amides during titration, because interaction with large FabB dimer produced significant line-broadening effects.
Backbone assignment of the D38A C8-AcpP mutant was achieved by comparison with the published wt C8-AcpP13 and confirmation of the few shifted peaks using short-range amide-amide (i ± 1) NOEs. The 15N NOESY-HSQC was acquired on a 500 MHz Varian VS500 equipped with a room temperature triple-resonance probe, with 4096 (R+I) points in the direct 1H dimension, 128 (R+I) points in the indirect 1H dimension, 96 (R+I) points in the indirect 15N dimension, a 1.5 s recycle delay, and 120 ms mixing time. This sample was prepared at 1.0 mM AcpP in 50 mM potassium phosphate pH 7.4, 0.01% sodium azide, 2.5 mM tris(2-carboxyethyl)phosphine, and 10% D2O.
Data was processed using nmrPipe29 and NMRFAM-SPARKY30. Chemical Shift Perturbations (CSPs) were calculated using the formula31:
CSPs reported in the maintext and figures are from comparing the 0.0 and 2.0 molar equivalent titration points.
CSP values for intermediate titration points were also calculated, and used for Kd determination. The AcpP FabB interaction was observed to be in fast exchange. Kd values of wt C8-AcpP and D38A C8-AcpP against FabB were calculated by fitting the following formula31:
where CSPobs is the observed CSP, CSPmax is the theoretical maximum CSP approached as the interaction is saturated, [P]t is the total protein concentration (AcpP in this case, held constant over the course of the titration), and [L]t is the total ligand concentration (FabB in this case, varied over the course of the titration). The 10 non-aliased residues bearing the largest CSPs were selected for fitting for both wt and D38A AcpP. Curves were fit using the nonlinear least squares implementation in RStudio (RStudio Team, 2015). [P]t CSPmax, and Kd values were fit initially31. The wt and D38A AcpP concentrations were observed to be 0.21 mM and 0.17 mM respectively. The [P]t value was then fixed to the average, and used to locally refit for Kd. The ΔG of the interaction was calculated using the formula: ΔG = RTln(Kd) and reported. The ΔΔG, for comparing wt and D38A AcpP was also calculated and reported. Plots were prepared and reported in Supplementary Figure 15.
Molecular Dynamics (MD) Simulations
The AcpP-FabB complex were modeled using the mechanism-based crosslinked structure described in this paper. Using the software UCSF Chimera, crystal waters were removed and hydrogens added using the Dock Prep tool, and the chloroacryl-based probe covalently linked to S36 was converted in silico into holo, C8-substrate and C10-product representations (Supplementary Figure 18)32. The phosphopantetheinyl serine residues were extracted and capped with N-methyl (NME) and acetyl (ACE) fragments to generate dipeptides for force field preparation and restricted electrostatic potential (RESP) charge fitting. Gaussian 09 Rev C was used for geometry optimization and electrostatic potential calculations using MP2/6-31G(d,p)//MP2/6-31G(d,p). RESP charges were calculated with intramolecular charge restraints and an overall charge of −1 for each non-standard residue and fit using the R.E.D. Server33. When possible, bond parameters were assigned using the main parameter databases in the Amber ff14SB force field. Missing parameters were adopted from the general AMBER force field (GAFF). Four simulation types were setup by configuring S36 on chains C and D of the carrier proteins as: C0D0 (apo:apo), C1D1 (holo:holo), C2D2 (C8-substrate:C8-substrate), and C3D3 (C10-product:C10-product). The crystal structure served as the basis of the initial structure for all simulations. LEaP was used to neutralize the apo systems by adding 52 Na+ ions, and the non-apo systems by adding 54 Na+ ions and solvating the enzyme complexes in 12-Å water buffer TIP3P truncated octahedron boxes. The fully solvated systems contained between 82,554 and 83,278 atoms.
MD was carried out using AMBER 16. -Minimization was carried out in two stages using SANDER from AmberTools 16. The initial stage was carried out over 2,500 steps for the solvent, ions, and post-translationally modified S36 residues of the carrier proteins, with the remaining residues of the proteins restrained by a force constant of 500 kcal/mol/Å2, followed by a second stage carried out over 5,000 steps of the entire system. Heating was performed using SANDER over 100-ps allowing the system to heat up to a temperature of 300K using the Langevin temperature equilibration scheme. During heating the same set of atoms as the initial stage of minimization was restrained, but with a lower force constant of 10 kcal/mol/Å2. PME was used to compute the electrostatic interactions with a real space cutoff of 10 Å, which was also used for the van der Waals interactions. Time steps were set to 2 fs, with hydrogen atoms constrained using the SHAKE algorithm. Equilibration was carried out using SANDER for 10 ns on all simulations. The MD production simulations were carried out using PMEMD.CUDA. The 6 wildtype complex system simulations were run over 1 μs (500,000,000 time steps), and the 18 mutant complex system simulations were run over 150 ns (75,000,000 time steps) for a total of 8.7 μs for all 24 simulations. Simulation speeds of 45 ns/day were observed.
Salt bridge analysis was performed using VMD, where a salt bridge was defined as having a cut-off distance of < 3.2 Å, and is between any oxygen atom on an acidic residue, and a nitrogen atom on a basic residue. Salt bridge contacts were evaluated at every frame, and were used to calculate contact duration as a percentage of present salt bridge over time.
Principal component analysis (PCA) was performed using CPPTraj from AMBER on a 2 μs trajectory file which was generated from the final 500 ns of the apo, holo, C8-substrate, and C8-product simulations. New trajectory files of the principal components were generated to visualize the movements along the major principal components. The starting and end of the principal component trajectories were used to generate porcupine plots using the software PyMol and the modevectors.py script originally authored by Dr. Sean Law (https://pymolwiki.org/index.php/Modevectors).
AcpP Complementation Assay and Fatty Acid Analysis
Codon optimized E. coli AcpP in an Ampr pMAL-c5x (New England Biolabs Inc.) vector was purchased from Genscript. The restriction sites (MfeI, BamHI) were chosen to eliminate the maltose binding protein and express tagless AcpP. Once transformed into DH5ɑ cells, the plasmid was mini prepped (QIAprep Spin Miniprep Kit, QIAGEN) and subjected to site-directed mutagenesis using the Naismith method to produce the D38A construct. The sequence was confirmed by forward and reverse sequencing (Genewiz).
Both wt and D38A tagless pMAL-c5x constructs were transformed into chemically competent CY1877 cells25. CY1877 cells, created by J.E. Cronan’s lab, have no chromosomal AcpP but bear a wt AcpP under arabinose control on a pBAD32220,25 vector with spectinomycin resistance. Chemically competent CY1877 cells were prepared using Rubidium chloride.
Fatty acid complementation growths were carried out in new screw-cap glass test tubes, with 5 mL LB supplemented with 100 μM IPTG, 50 μg/mL Spectinomycin, and 100 μg/mL Ampicillin, in quadruplicate (four separate cell cultures in individual glass tubes). Samples were grown at 37 °C for 3.5 hours, then held at 17 °C, 25 °C, and 37 °C for 1 hour. All growths were grown aerobically and agitated at 100 RPM throughout. Replicates of both wt and D38A were prepared for each temperature. Complementation growths were inoculated with 25 μL of dense overnight cultures (37 °C) grown in LB supplemented with 0.2% Arabinose, 50 μg/mL Spectinomycin, and 100 μg/mL Ampicillin. Complementation growths were harvested by gentle centrifugation in the glass tubes at 500 rcf for 20 minutes and the supernatant was removed.
Fatty acids were converted to the corresponding methyl esters and subjected to GCMS analysis. The wet pellets were resuspended in 1 mL of 1 M HCl in methanol and the tubes were vortexed. Pellets were incubated at 65 C for 30 minutes, with additional vortexing every 10 minutes, then cooled to room temperature. Exactly 1 mL of hexanes was added by syringe to each sample, and they were again vortexed. Samples were briefly spun for 1 minute at 500 rcf for phase separation, and the hexanes layer was transferred by Pasteur pipette into a GCMS sample vial and sealed. Samples were held at 4 °C and subjected to GCMS overnight after extraction.
GCMS was carried out on a 60 meter Db23 column (Agilent) using a 7890A Gas Chromatograph (Agilent) equipped with a 5975C VL MSD Quadrupole Mass Spectrometer (Agilent) and a GC Sampler 80 (Agilent). The inlet temp was set to 250 °C, the MS source was set to 230 °C, and helium was the carrier gas. The method start was at 110 °C followed by a ramp to 200 °C at 15 °C/minute, and a final hold for 21 minutes at 200 °C. Fatty acid identities were compared with reference standards and the NIST database (Wiley Scientific). To limit transfer losses and degradation, the esterification and extraction steps were carried out in the same glass test tubes immediately after harvesting, and GCMS analysis was performed overnight after extraction. Peak volumes were compared and analyzed to generate the fractional plots and statistics reported in the manuscript.
Statistics
NMR titration experiments, as discussed above, were used to calculate Kd values. For each sample (wt AcpP titrated with FabB, and D38A AcpP titrated with FabB) the ten non-aliased residues with the largest CSP values were used in fitting, and the resultant ten Kd values were averaged and the standard deviation calculated. An unpaired t-test (Student’s test), was used to evaluate the statistical significance of the difference in Kd and Cohen’s d for independent groups was calculated using the formula Cohen’s d = (M2 - M1)/SDpooled. Values are reported in Supplementary Table 3.
Fatty acid extractions from the complementation experiments were analyzed by peak volume and divided by the total volume of all peaks. For each FA species, the replicates (separate cultures in separate tubes) were averaged and the standard deviation was calculated. An unpaired t-test (Student’s test) was used to evaluate the statistical significance of the difference in the fraction of unsaturated fatty acids at each temperature. Cohen’s d for independent groups was calculated using the formula Cohen’s d = (M2 - M1)/SDpooled. Values are reported in Supplementary Table 3.
Supplementary Material
ACKNOWLEDGEMENTS
SCT and MDB are supported by GM100305 and GM095970. MDB is also supported by NSF IOS-1516156, and RL is also supported by GM093040 and GM079383. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. The authors thank the staff of beamline 8.2.1 at the Advanced Light Source for support during X-ray diffraction data collection, Dr. Xuemei Huang and Prof. Stan Opella for their guidance and assistance with NMR collection at the UCSD Biomolecular NMR facility, and Dr. Brian Fuglestad for many helpful NMR discussions. The authors thank Prof. John E. Cronan for the CY1877 strain. Additional funding from the institutional Chemical and Structural Biology Training Grant (National Institute of General Medical Sciences Grant T32GM108561) and the National Science Foundation Graduate Research Fellowship is also acknowledged.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
DATA AVAILABILITY
The AcpP-FabB crystal structure coordinates are available through the Protein Data Bank website using the accession code 5KOF. The assignments have been deposited with BMRB, ID# 27872, and are available in Supplementary Table 2.
REFERENCES
- 1.Finzel K, Lee DJ & Burkart MD Using modern tools to probe the structure-function relationship of fatty acid synthases. Chembiochem 16, 528–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng Y & Cronan JE Escherichia coli unsaturated fatty acid synthesis: complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J. Biol. Chem 284, 29526–29535 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright HT & Reynolds KA Antibacterial targets in fatty acid biosynthesis. Curr. Opin. Microbiol 10, 447–453 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu X, Liu T, Zhu F & Khosla C In vitro reconstitution and steady-state analysis of the fatty acid synthase from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 108, 18643–18648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajewski J et al. Engineering fatty acid synthases for directed polyketide production. Nat. Chem. Biol 13, 363–365 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Rock C & Cronan JE Acyl carrier protein from Escherichia coli. Methods Enzymol. 71, 341–351 (1981). [DOI] [PubMed] [Google Scholar]
- 7.Kim Y & Prestegard JH A dynamic model for the structure of acyl carrier protein in solution. Biochemistry 28, 8792–8797 (1989). [DOI] [PubMed] [Google Scholar]
- 8.Roujeinikova A et al. Structural studies of fatty acyl-(acyl carrier protein) thioesters reveal a hydrophobic binding cavity that can expand to fit longer substrates. J. Mol. Biol 365, 135–145 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Majerus PW, Alberts AW & Vagelos PR The Identification Of 4’-Phosphopantetheine as the Prosthetic Group of the Acyl Carrier Protein. Proc. Natl. Acad. Sci. U. S. A 53, 410–417 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan DI, Stockner T, Tieleman DP & Vogel HJ Molecular dynamics simulations of the Apo-, Holo-, and acyl-forms of Escherichia coli acyl carrier protein. J. Biol. Chem 283, 33620–33629 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crosby J & Crump MP The structural role of the carrier protein – active controller or passive carrier. Nat. Prod. Rep 29, 1111 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Thiele GAR et al. Acyl Carrier Protein Cyanylation Delivers a Ketoacyl Synthase–Carrier Protein Cross-Link. Biochemistry 56, 2533–2536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen C et al. Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 505, 427–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cronan JE The chain-flipping mechanism of ACP (acyl carrier protein)-dependent enzymes appears universal. Biochem. J 460, 157–163 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Beld J, Cang H & Burkart MD Visualizing the chain-flipping mechanism in fatty-acid biosynthesis. Angew. Chem. Int. Ed Engl 53, 14456–14461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Worthington AS, Rivera H, Torpey JW, Alexander MD & Burkart MD Mechanism-based protein cross-linking probes to investigate carrier protein-mediated biosynthesis. ACS Chem. Biol 1, 687–691 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Worthington AS & Burkart MD One-pot chemo-enzymatic synthesis of reporter-modified proteins. Org. Biomol. Chem 4, 44–46 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Byers DM & Gong H Acyl carrier protein: structure-function relationships in a conserved multifunctional protein family. Biochem. Cell Biol 85, 649–662 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Zhang L et al. Crystal structure of FabZ-ACP complex reveals a dynamic seesaw-like catalytic mechanism of dehydratase in fatty acid biosynthesis. Cell Res. 26, 1330–1344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Lay NR & Cronan JE Gene-specific random mutagenesis of Escherichia coli in vivo: isolation of temperature-sensitive mutations in the acyl carrier protein of fatty acid synthesis. J. Bacteriol 188, 287–296 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garwin JL & Cronan JE Jr. Thermal modulation of fatty acid synthesis in Escherichia coli does not involve de novo enzyme synthesis. J. Bacteriol 141, 1457–1459 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magnuson K, Jackowski S, Rock CO & Cronan JE Jr. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol. Rev 57, 522–542 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garwin JL, Klages AL & Cronan JE Jr. Beta-ketoacyl-acyl carrier protein synthase II of Escherichia coli. Evidence for function in the thermal regulation of fatty acid synthesis. J. Biol. Chem 255, 3263–3265 (1980). [PubMed] [Google Scholar]
- 24.Rossini E, Gajewski J, Klaus M, Hummer G & Grininger M Analysis and engineering of substrate shuttling by the acyl carrier protein (ACP) in fatty acid synthases (FASs). Chem. Commun 54, 11606–11609 (2018). [DOI] [PubMed] [Google Scholar]
- 25.De Lay NR & Cronan JE In vivo functional analyses of the type II acyl carrier proteins of fatty acid biosynthesis. J. Biol. Chem 282, 20319–20328 (2007). [DOI] [PubMed] [Google Scholar]
ONLINE METHODS REFERENCES
- 26.Kosa NM, Haushalter RW, Smith AR & Burkart MD Reversible labeling of native and fusion-protein motifs. Nat. Methods 9, 981–984 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Battye TGG, Kontogiannis L, Johnson O, Powell HR & Leslie AGW iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr 67, 271–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. in Acta Crystallogr. D Biol. Crystallogr (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Lee W, Tonelli M & Markley JL NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williamson MP Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc 73, 1–16 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Vanquelef E et al. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 39, W511–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.