

Abstract
Pathologic intracellular inclusions formed from polymers of misfolded α-synuclein (αsyn) protein define a group of neurodegenerative diseases termed synucleinopathies which includes Parkinson’s disease (PD). Prion-like recruitment of endogenous cellular αsyn has been demonstrated to occur in animal models of synucleinopathy, whereby misfolded αsyn can induce further pathologic αsyn inclusions to form through a prion-like mechanism. It has been suggested that misfolded αsyn may assume differing conformations which lead to varied clinical and pathological manifestations of disease; this phenomenon bears similarities to that of prion strains whereby the same misfolded protein can produce unique diseases. It is unclear what factors influence the development of unique αsyn strains, however post-translational modifications (PTMs) such as phosphorylation and truncation that are present in misfolded αsyn in disease may play a role due to their modulation of biochemical and structural αsyn properties. Herein, we investigate the prion-like properties of misfolded αsyn polymers containing either phosphomimetic (S129E) αsyn, 5 different major carboxy (C)-truncated forms of αsyn (1-115, 1-119, 1-122, 1-125, and 1-129 αsyn), ora mixture of these PTM containing αsyn forms compared to full-length (FL) αsyn in HEK293T cells and M83 transgenic mice overexpressing A53T αsyn. It is demonstrated that upon peripheral intramuscular injection of these C-truncated or S129E αsyn polymers into M83 mice, prion-like progression and time to disease onset in this mouse model is elongated when any of these PTMs are present, demonstrating that common modifications to the C-terminus of αsyn present in disease modulates the prion-like seeding properties of αsyn.
Keywords: α-synuclein, Parkinson’s disease, truncation, fibril, inclusion formation, amyloid, prion, neurodegeneration, Lewy body
Introduction
Synucleinopathies including Parkinson’s disease (PD), Lewy body dementia (LBD), and Multiple system atrophy (MSA) are a heterogeneous group of diseases both clinically and pathologically, however all of these diseases are characterized by the presence of neuronal or glial inclusions comprised of misfolded alpha-synuclein (αsyn) aggregated into beta-sheet rich fibrils [1–3]. Pathologic αsyn fibrils have been shown to harbor prion-like properties, as insoluble αsyn fibrils isolated from Lewy bodies (LBs), glial cytoplasmic inclusions (GCIs), and even those generated in-vitro as pre-formed fibrils (PFFs) recruit endogenous physiologic αsyn to misfold and form additional fibrils in a process termed conformational templating [3,4]. The prion-like nature of pathologic αsyn fibrils has been demonstrated in a variety of in-vivo experiments whereby injection of PFFs intracerebrally or even intramuscularly through invasion of peripheral nerves is sufficient to induce the progressive formation of pathologic aggregates throughout the nervous system resulting in neuronal loss and subsequent behavioral and motor deficits [4–6]. Observed patterns in the clinical and pathologic progression of PD is congruent with a molecular and cellular prion-like mechanism of disease in a significant percentage of patients [7–9]. Particularly, staging schemes for PD may imply peripheral initiation of αsyn fibrils in the gastrointestinal tract followed by retrograde vagal spread of pathology to the brainstem and subsequently mesencephalic and telencephalic regions although this concept is still under investigation [10–12]. αsyn prion-like activities may also explain the heterogeneity amongst the synucleinopathies, whereby it has been postulated that molecularly diverse polymer seeds amongst these disorders results in strain-like variations in pathological initiation resulting in varied pathologic and clinical presentations of disease [3,13].
Post-translational modifications (PTMs) of αsyn, including phosphorylation and truncation, that are commonly associated with PD, LBD, and MSA [14–16] can potentially yield diverse forms of pathologic strain-like αsyn variant polymers. Structural characterization of αsyn fibrils suggests that modifications to the carboxy (C)-terminus such as truncation can greatly alter the fibril structure [17–19]; consequent alterations in prion-like seeding as a result of these modifications may be important in explaining strain-like diversity in fibril structure and seeding capacity isolated from LBs and GCIs [13,20–24]. Indeed, it has been demonstrated that αsyn fibrils are generally trafficked for lysosomal processing intracellularly, where extensive proteolytic truncation of exposed C-terminal regions occurs [4,25–27], thus warranting further investigation of the impact of these modifications on prion-like activity of αsyn fibrils in disease models.
Inoculation studies using the M83 human A53T αsyn transgenic mouse model [5,28] have been successfully utilized to demonstrate varied strain-like seeding activities including PD versus MSA lysate with significant differences in time to disease onset [21,24,29,30]. In this mouse model, PFFs can be injected peripherally or intra-cerebrally resulting in induction of αsyn inclusion pathology in the CNS and subsequent development of a fatal movement disorder [5,6,31]. Herein, using the hemizygous M83 (M83+/−) intramuscular injection (IM) model [5,6], significant variation in functional prion-like seeding activity is demonstrated for PFFs comprised of C-truncated or phosphomimetic αsyn which has implications for disease mechanisms of induction and progression in synucleinopathies.
Materials and Methods
Antibodies
Mouse monoclonal antibody 81A recognize αsyn but only when phosphorylated at Ser129 (pSer129) [32]. Antibodies 2H6 and 94-3A10 are mouse monoclonal antibodies that were raised against N-terminal (2-21) and C-terminal (130-140) regions of human αsyn, respectively, and demonstrate conformational specificity for aggregated αsyn [33]. A rabbit polyclonal antibody specific for p62/sequestosome-1 was utilized as a general inclusion marker (ProteinTech, Rosemont, IL). SNL-4 is a rabbit polyclonal antibody raised against residues 2-12 of αsyn [34].
Expression and purification of recombinant αsyn proteins
Recombinant FL human or mouse αsyn (residues 1-140), N-terminal truncated human αsyn (residues 21-140; 21-140 αsyn), Δ71-82 αsyn (deleted residues 71-82), and various C-terminal truncated forms of human αsyn (residues 1-115, 1-119, 1-122, 1-125, 1-129) were expressed from the pRK172 plasmids containing the cDNA for the SNCA gene with appropriately located stop codons as described previously [35–37]. QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA) using mutant-specific oligonucleotides was used to generate the phosphomimetic mutation S129E in cDNA encoding FL human αsyn and 21-140 human αsyn in the pRK172 plasmid as previously described [38]. The presence of the mutation and the absence of errors throughout the length of the αsyn cDNA was confirmed by Sanger sequencing. All recombinant forms of αsyn were expressed in E.coli BL21 (DE3) and purified as previously described utilizing size exclusion and Mono Q anion exchange chromatography [35,36]. All recombinant proteins were diluted in pH 7.4 sterile phosphate buffered saline (PBS), and concentrations were determined using the bicinchoninic acid (BCA) assay (Pierce, Waltham, MA) with bovine serum albumin (BSA) as the standard.
Mice
All animal experimental procedures were performed in accordance to University of Florida Institutional Animal Care and Use Committee regulatory policies following approval. Mice were housed in a stable environment with a 12-hour light/dark cycle and access to food and water ad libitum. M83+/− transgenic mice hemizygous for human αsyn with the A53T mutation (M83+/−) under control of the mouse prion protein promoter were utilized for all animal experiments described [28].
αsyn fibril preparation and intramuscular injection
FL, S129E, N-truncated, and C-truncated forms of αsyn were individually assembled into PFFs by incubation at 37°C at 5 mg/ml in sterile PBS with continuous shaking at 1,050 rpm. When combinations of FL with C-truncated forms of αsyn were co-fibrillized, a 1:1 molar ratio was used at a 5 mg/ml total concentration, αsyn fibril formation was validated with K114 fluorometry as previously described [36]. PFFs were diluted in sterile PBS and fragmented into an array of shortened fibrils by mild water bath sonication for one hour prior to injection [6].
For comparison of prion-like seeding properties between FL and C-truncated forms of αsyn, 6 cohorts of 10-13 mice (Table 1) were injected unilaterally into the gastrocnemius muscle as previously described [6] with 5μL of solution (sterile PBS) containing 5 μg of FL αsyn PFFs or the molar equivalent (based off the molecular mass of monomers) of each of the C-truncated forms of αsyn used (1-115, 1-119, 1-122, 1-125 or 1-129 αsyn [36]). 5 μg of PFFs comprised of S129E αsyn were intramuscularly injected into a cohort of 10 mice. To determine the seeding activity of co-fibrils containing both FL αsyn and C-truncated forms of αsyn, 5 cohorts of 9-10 mice were similarly injected with 5μL of solution (sterile PBS) containing 4-5 μg of mixed PFFs that are molar equivalent to 5 μg of FL αsyn which were generated in a 1:1 molar ratio (1-115/FL, 1-119/FL, 1-122/FL, 1-125/FL or 1-129/FL αsyn). Following inoculations, animals were regularly assessed for motor deficits indicative of a diseased phenotype and were sacrificed upon onset of fatal limb paralysis.
Table 1. Summary of varied PFFs used in M83+/− IM injection experiments.
Each cohort is shown along with composition and dosage of injected PFFs, age of animals at injection, mean ± standard deviation and median days post-injection until motor phenotype onset. Dosages of C-truncated αsyn PFFs or C-truncated/FL αsyn mixed PFFs were chosen to be molar equivalent to the control 5 μg FL αsyn PFFs.
| Cohort | Inoculum | N | Age at Injection (months) | Mean Survival Post-Injection ± std (days) | Median Survival Post-Injection (days) |
|---|---|---|---|---|---|
| Dosage | |||||
| 0.1 μg | 0.10 μg mPFFs | 11 | 2 | 164±31 | 159 |
| 0.5 μg | 0.50 μg mPFFs | 13 | 2 | 146±37 | 136 |
| 1 μg | 1.00 μg mPFFs | 11 | 2 | 149±31 | 134 |
| 2 μg | 2.00 μg mPFFs | 8 | 2 | 142±26 | 128 |
| 5 μg | 5.00 μg mPFFs | 8 | 2 | 133±11 | 135 |
| 10 μg | 10.00 μg mPFFs | 8 | 2 | 126±13 | 127 |
| Sex | |||||
| Male | 0.50-10.00 μg mPFFs | 24 | 2 | 138±32 | 131 |
| Female | 0.50-10.00 μg mPFFs | 24 | 2 | 143±25 | 135 |
| C-terminal modification | |||||
| 1-115 | 4.00 μg 1-115 hPFFs | 11 | 2 | 211±37 | 205 |
| 1-119 | 4.15 μg 1-119 hPFFs | 10 | 2 | 241±58 | 213 |
| 1-122 | 4.26 μg 1-122 hPFFs | 10 | 2 | 176±38 | 186 |
| 1-125 | 4.39 μg 1-125 hPFFs | 10 | 2 | 170±37 | 171 |
| 1-129 | 4.55 μg 1-129 hPFFs | 10 | 2 | 212±46 | 199 |
| 1-140 (FL) | 5.00 μg FL hPFFs | 12 | 2 | 121±23 | 115 |
| S129E | 5.00 μg S129E hPFFs | 10 | 2 | 153±22 | 146 |
| 1-115/FL | 4.50 μg 1-115/FL hPFFs | 11 | 2 | 144±35 | 142 |
| 1-119/FL | 4.58 μg 1-119/FL hPFFs | 10 | 2 | 173±58 | 147 |
| 1-122/FL | 4.63 μg 1-122/FL hPFFs | 10 | 2 | 149±23 | 148 |
| 1-125/FL | 4.70 μg 1-125/FL hPFFs | 10 | 2 | 131±22 | 134 |
| 1-129/FL | 4.78 μg 1-129/FL hPFFs | 9 | 2 | 124±14 | 125 |
For dosage studies, 3 cohorts of 11-13 mice (Table 1) were injected unilaterally into the gastrocnemius muscle with 5 μL of solution (sterile PBS) containing either 0.1, 0.5, or 1.0 μg of FL mouse αsyn PFFs (mPFFs). A control group of 14 mice was similarly injected unilaterally into the gastrocnemius with 20 μg total (twice 5 μl containing 10 ug) of Δ71-82 αsyn (Table 1) that is impaired in aggregation and prion-like seeding [35,39,40]; mice were sacrificed at the predetermine time of 200 days post injection if motor impairment/paralysis did not develop. Mice were monitored and sacrificed upon onset of limb paralysis; survival curves were integrated with previously published cohorts of 8 mice each injected in the same location and volume but containing 2.0, 5.0, or 10.0 μg of mPFFs [6]. Survival curves containing the 0.5, 1.0, 2.0, 5.0, and 10.0 μg mPFF cohorts were additionally stratified based on sex of each mouse.
Tissue preparation and immunohistochemistry
Mice were euthanized with CO2 and perfused with a heparin/PBS solution as previously described [6]. Brain and spinal tissue were fixed in 70% ethanol/150 mM NaCl, embedded in paraffin, and cut into 5 μm sections as described [6]. For immunohistochemical analysis, sections were stained utilizing established methods [41]. Sections were deparaffinized and rehydrated, followed by antigen retrieval in a steam bath of 0.05% Tween 20 for 30 minutes. Endogenous peroxidases were inactivated in a solution of 2% peroxide/PBS and sections were blocked in 2% fetal bovine serum (FBS)/0.1M Tris. Antibodies were diluted in block solution and applied overnight to sections at 4°C. Species specific secondary antibodies (Vector Laboratories, Burlingame, CA) were applied for 1 hour at room temperature, followed by incubation with avidin-biotin complex (ABC) reagent (Vectastain ABC Elite kit; Vector Laboratories, Burlingame, CA) in similar conditions. Immunocomplexes were detected upon exposure to chromogen 3,3′-diaminobenzidine (DAB kit; KPL, Gaithersburg, MD). Sections were counterstained with hematoxylin. All slides were digitally scanned using an Aperio ScanScope CS instrument (40× magnification; Aperio Technologies Inc., Vista, CA), and images of representative areas of pathology were captured using the ImageScope software (40× magnification; Aperio Technologies Inc.).
Mammalian expression plasmids
The cDNA encoding FL human αsyn was previously cloned in the mammalian expression vector pcDNA3.1(+) [36]. Site directed mutagenesis was used to introduce the S129E mutation in the FL human αsyn pcDNA3.1 plasmid. The mutation and the absence of errors throughout the entire length of the αsyn cDNA was confirmed by Sanger sequencing.
HEK293T cell culture and transfection
HEK293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (Invitrogen, Carlsbad, CA) supplemented to contain 2 mM L-glutamine, 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin, at 37°C and 5% CO2. Cells were plated into 4 cm2 wells, and allowed to reach ~30% confluency at which time cells were transfected using a modified calcium phosphate protocol [36]. For each of 3 replicate wells per construct, 1.5 μg of pcDNA3.1 vector expressing FL or S129E human αsyn was used for transfection. One hour after transfection, PFFs comprised of 21-140 αsyn or 21-140 αsyn with the S129E mutation were added to 1 μM in each well, with the concentration being based on that of the monomeric subunits. Cells were harvested for biochemical fractionation at a final time point of 64 hours post transfection.
HEK293T cell biochemical fractionation and western blot analysis
HEK293T cells were washed once in PBS, and subsequently lysed in 200 μL/well detergent extraction buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 20 mM NaF) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl and 1 mg/ml each of pepstatin, leupeptin, N-tosyl-L-phenylalanyl chloromethyl ketone, N-tosyl-lysine chloromethyl ketone and soybean trypsin inhibitor). Lysate was separated into triton soluble and insoluble fractions and prepared for western blot analysis as previously described [36]. Equal volumes of Triton-soluble and Triton-insoluble fractions were loaded onto 15% polyacrylamide gels and resolved by SDS-PAGE, followed by electrophoretic transfer onto 0.2 μm pore size nitrocellulose membranes (Bio-Rad, Hercules, CA), in carbonate transfer buffer (10 mM NaHCO3, 3 mM Na2CO3, pH 9.9) [42]. Membranes were blocked in 5% dry milk/Tris buffered saline (TBS) and incubated overnight at 4°C with primary antibody SNL4 (residues 2-12) diluted in block solution. After washing in TBS, membranes were incubated in goat anti-secondary antibody conjugated to horseradish peroxidase (Jackson Immuno Research Labs, Westgrove, PA) diluted in 5% dry milk/TBS for 1 hour; immunocomplexes were detected using Western Lightning-Plus ECL reagents (PerkinElmer, Waltham, MA) followed by chemiluminescence imaging (PXi, Syngene, Frederick, MD). Densitometry was performed using ImageJ software to quantify the ratio of detergent insoluble to total αsyn.
Quantitative and Semi-quantitative analysis
Survival curves were generated in GraphPad Prism software, and both the Log-rank test and Gehan-Breslow-Wilcoxon were used to individually compare various cohorts to detect significant differences in survival (Tables 1, 2). For immunohistochemical data from mice injected with human FL PFFs or C-truncated PFFs, semi-quantitative grading of density of pathology on a 4-point scale was performed for the spinal cord and brainstem from all mice as these are the regions with the heaviest pathology in this mouse model [6]. The analysis was performed by 3 independent observers for the main 4 antibodies utilized (81A, 2H6, 94-3A10, and p62-sequestome 1). For western blot data, soluble and insoluble bands were quantified using ImageJ software (NIH, Bethesda, MD). The insoluble αsyn fraction was reported as the ratio of the intensity of the αsyn band in the insoluble fraction divided by the summed intensities of αsyn in the soluble and insoluble fractions. Densitometric comparisons were performed in GraphPad Prism software using one-way analysis of variance (ANOVA), with post hoc analysis using Dunnett’s test to compare combination of aggregation conditions to the FL + 21-140 αsyn control.
Table 2. Statistical summary of survival curves for each varied PFF injection experiment.
All mPFF dosage experiments were compared to the 5 μg mPFFs cohort as the control. All comparisons for the PFFs containing C-truncated or phosphomimetic S129E αsyn were made with FL αsyn PFFs as the control. Shown are p-value summaries for survival curve comparisons using the log-rank test or Gehan-Breslow-Wilcoxon (GBW) test.
| Comparison | Log rank p value | GBW test p value |
|---|---|---|
| Modified hPFFs | ||
| 1-115 vs FL | **** | **** |
| 1-119 vs FL | **** | **** |
| 1-122 vs FL | *** | *** |
| 1-125 vs FL | ** | ** |
| 1-129 vs FL | **** | **** |
| S129E vs FL (WT) | * | ** |
| Mixed hPFFs | ||
| 1-115/FL vs FL | NS | NS |
| 1-119/FL vs FL | ** | ** |
| 1-122/FL vs FL | * | ** |
| 1-125/FL vs FL | NS | NS |
| 1-129/FL vs FL | NS | NS |
| Dosage | ||
| 10ug vs 5ug | NS | NS |
| 2ug vs 5ug | NS | NS |
| 1ug vs 5ug | NS | NS |
| 0.5ug vs 5ug | NS | NS |
| 0.1ug vs 5ug | ** | ** |
| Sex | ||
| male vs female | NS | NS |
NS = no significance,
= p ≤ 0.05,
= p ≤ 0.01,
= p ≤ 0.001,
= p ≤ 0.0001.
Results
Onset of disease in M83+/− mice IM injected with mPFFs is consistent over a wide dosage range and is not altered by sex
It was previously shown that unilateral hind limb IM injection of PFFs into M83+/− mice results in a progressive accumulating of αsyn inclusion pathology in an ascending pattern from the spinal cord to the brain over the course of ~4 months post injection associated with motor neuron demise and severe motor impairment/paralysis [5,6]. To assess if this model was suitable for detecting differences in prion-like seeding of varying PFFs, robustness of the model to phenotype development across a range of PFF dosages was tested. Cohorts of M83+/− Tg mice (n=11-13 per cohort) were unilaterally IM injected with 0.1, 0.5, or 1 μg of mPFFs; time to the severe motor impairments/paralysis phenotype was measured and compared with our previously published cohorts of similarly injected M83+/−Tg mice (n=8 per cohort) in which 2, 5, or 10 μg of mPFFs were utilized [6]; Kaplan-Meier survival curves were generated for each cohort and compared to the 5 μg PFF group as a control under the log rank and Gehan-Beslow-Wilcoxon (GBW) tests (Figure 1, Tables 1, 2). The 5 μg mPFF IM injected cohort had a highly consistent mean time to death at 133± 11 (std) days post-injection (dpi) and median 135 dpi; survival curves for dosages of 10, 2, 1, and 0.5 μg were not significantly different from the 5 μg mPFF cohort and the median survival for each cohort ranged from 127 to 136 dpi (Figure 1, Tables 1, 2). Only the 0.1 μg mPFF IM injected cohort was significantly different in survival time from the 5 μg cohort, with a median survival time of 159 dpi compared to 135 dpi for 5 μg (Figure 1, Tables 1, 2). Overall, the model is extremely robust to even tenfold variations in PFF concentration suggesting that differing survival times for PFF variants are unlikely to be explained by dissimilarity in dosage.
Figure 1. The M83+/− IM seeding model is consistent in motor impairments and paralysis presentation across a range of mPFF dosages and is not affected by sex of animal.
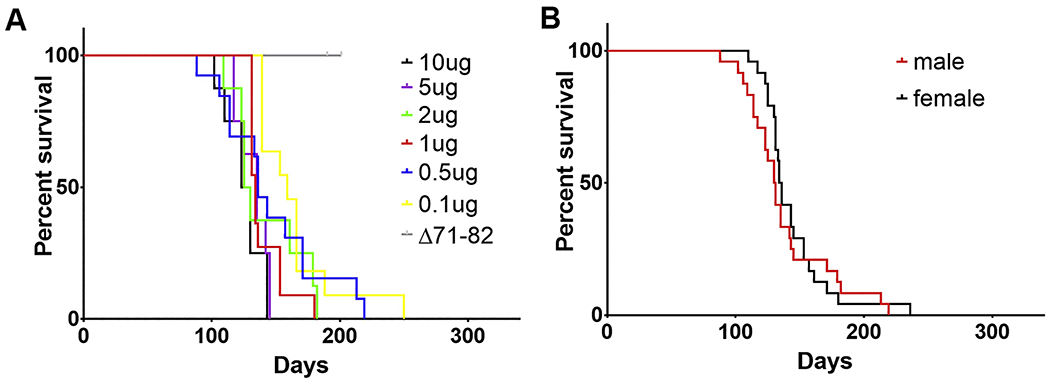
(A) Varied doses of mPFFs (see also Table 1) were unilaterally injected into the gastrocnemius of 2-month-old M83+/− αsyn transgenic mice (n=8-13 per dosage) and time to fatal motor phenotype development was monitored. Varying the dosage of IM injected mPFFs from 10 μg to 0.5 μg did not significantly affect time to onset of fatal motor impairment; only injection of 0.1 μg resulted in a modest, but statistically significant, delay in disease onset time (statistical details see Table 2). Unilateral injection (20 ug) of the aggregation-deficient deletion protein Δ71–82 αsyn as a control did not result in this motor impairment phenotype. (B) Utilizing the mice injected with 0.5, 1, 2, 5, or 10 μg of mPFFs from (A), cohorts were pooled and time to disease onset was compared between male (n=24) and female (n=24) injected mice; no significant difference in time to motor disease was detected (statistical details see Table 2). In both graphs, the x-axis is days post IM injection of mPFF until fatal motor phenotype onset.
Many mouse models of neurodegeneration have varied phenotypes depending on the sex of the animal [43]. In order to determine whether sex alters the survival time in the M83+/− injection model, survival curves containing the 0.5, 1.0, 2.0, 5.0, and 10.0 μg mPFF cohorts were stratified based on sex of each mouse (n=24 male, n=24 female) and compared as described for the dosage studies (Figure 1, Tables 1, 2). No difference in survival time was imparted due to sex of the animals; the median survival times were 131 dpi for males and 135 dpi for females.
To demonstrate that this mouse model progresses due to prion-like seeding, M83+/−Tg mice (n=14) were intramuscularly injected with 20 μg of Δ71-82 αsyn which has previously been shown to be deficient in amyloid aggregation and seeding potency due to removal of a critical hydrophobic motif [35,39,40]. Of the Δ71-82 injected mice, none developed a phenotype by 200 dpi (Figure 1, Table 1) at which point the mice were sacrificed for histologic analysis. These findings demonstrate that survival differences in the M83+/− injection mouse model imparted by differing PFF types are unlikely to be confounded due to dosage used or the sex of animal.
Onset of disease in M83+/− mice is significantly delayed when injected with PFFs comprised of C-terminally modified human αsyn suggesting functional alterations in prion-like seeding due to these PTMs
Pathologic αsyn inclusions contain αsyn harboring extensive PTMs, notably C-terminal phosphorylation (pSer129) and truncation at multiple sites [14,44]. In order to assess how the presence of these common PTMs of αsyn may alter prion-like seeding, human PFFs (hPFFs) were generated either fully comprised of 6 different C-truncated or full-length (FL) forms of human αsyn (1-115, 1-119, 1-122, 1-125, 1-129, 1-140 (FL)) or as co-fibrils generated in a 1:1 molar ratio with each C-truncated αsyn and FL αsyn (1-115/FL, 1-119/FL, 1-122/FL, 1-125/FL, 1-129/FL). Additionally, phosphomimetic S129E αsyn hPFFs were generated (1-140 αsyn with a S129E mutation). All fibrils were verified to be amyloid fibrils through K114 fluorometry [45], and IM injections in cohorts of M83+/− mice (12 cohorts of n=9-12 each) were performed unilaterally with dosages of 5 μg for FL hPFFs or the molar equivalent for hPFFs containing C-truncated αsyn (Table 1).
Survival curves were significantly prolonged for M83+/− mice injected with hPFFs comprised solely of C-truncated (1-115, 1-119, 1-122, 1-125, or 1-129) or S129E αsyn relative to FL hPFFs (Figure 2, Tables 1, 2), and the 1-119/FL and 1-122/FL hPFF injected cohorts also demonstrated a significant increase in survival time post injection (Figure 2, Tables 1, 2). All hPFFs comprised fully of C-truncated αsyn resulted in elongated survival times relative to FL hPFFs for injected M83+/− mice and all were moreso than even the 0.1 μg mPFF cohort, demonstrating that significant alterations in prion-like properties may be imparted upon C-terminal truncation of αsyn.
Figure 2. Time to motor impairments and paralysis in the M83+/− IM seeding model is significantly delayed in the context of hPFFs comprised of C-terminally truncated or Ser129 phosphomimetic αsyn indicative of strain-like differences.
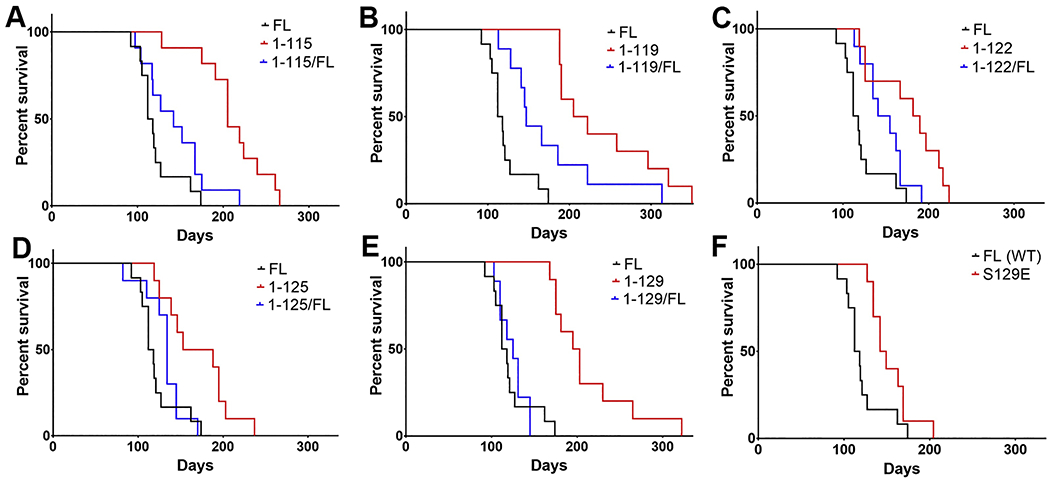
(A-F) Cohorts of M83+/− αsyn transgenic mice (n=9-12 per hPFF type; also see Table 1) were unilaterally injected with 5 μg of hPFFs (or molar equivalent for truncated forms) into the gastrocnemius at 2 months old and time to disease onset was monitored; survival curves for all hPFFs were compared with FL αsyn hPFFs (statistical details see Table 2). Mice were injected with hPFFs comprised of (A) 1-115 αsyn or 1-115 and FL αsyn in a 1:1 ratio, (B) 1-119 αsyn or 1-119 and FL αsyn in a 1:1 ratio, (C) 1-122 αsyn or 1-122 and FL αsyn in a 1:1 ratio, (D) 1-125 αsyn or 1-125 and FL αsyn in a 1:1 ratio, (E) 1-129 αsyn or 1-129 and FL αsyn in a 1:1 ratio, or (F) S129E αsyn. Details of injections and statistical summary of presented data see Tables 1 and 2.
The finding that cohorts inoculated with copolymerized 1-119/FL and 1-122/FL hPFFs [36] increase in survival time post injection relative to FL hPFFs (Figure 2, Tables 1, 2) indicates that even partial truncation of hPFFs could results in strain-like differences in seeding activities. Although not statistically significant, the 115/FL, 125/FL, and 129/FL hPFFs all displayed greater median survival times compared with FL hPFFs and were close to significance. hPFFs comprised of 1-119 αsyn demonstrated the largest effect on motor phenotype development, as median survival was 213 dpi for 1-119 hPFFs, 147 dpi for the 1-119/FL hPFFs, and 115 dpi for the FL hPFFs; a summary of mean and median survival times (dpi) along with statistical summaries for survival comparisons are provided for all other C-truncated hPFFs (Tables 1,2).
Phosphopmimetic S129E αsyn hPFFs also resulted in what may be altered prion-like properties evidenced by a significant difference in survival curves compared with FL wild-type (WT) hPFF injected mice (median survival 146 dpi for S129E and 115 dpi for FL) (Figure 2F, Table 1); the effect size on survival for S129E hPFFs was lesser than any of the fully C-truncated hPFFs which shows that a single amino acid modification is unlikely to impact prion-like properties as severely as multiple amino acid truncation.
Density, distribution, and immunohistochemical profile of induced pathologic αsyn inclusions in end stage IM seeded M83+/− mice are similar regardless of C-terminal modified forms of αsyn within PFFs
Multiple cellular and animal models of neurodegeneration have demonstrated that inoculation with varied PFF strains may result in either similar or differing pathologies as the propagation of strain may also depend on cell autonomous factors and not only the αsyn seed fibril [20,24,46]. As the various cohorts of M83+/− IM injected PFF mice displayed significant differences in survival curves, all mice injected with PFFs containing C-truncated or phosphomimetic αsyn (1-115, 1-119, 1-122, 1-125, 1-129, 1-115/FL, 1-119/FL, 1-122/FL, 1-125/FL, 1-129/FL, and S129E) were sacrificed upon end stage paralysis and immunohistochemically profiled for comparison of αsyn pathology with the FL αsyn PFF injected cohort (Figures 3–5). For all cohorts, sections of the spinal cord and brain were stained with antibodies including those specific for pSer129 αsyn (81A), p62-sequestosome-1 as a general inclusion marker (p62), an N-terminal αsyn epitope (residues 2-21; 2H6), and a C-terminal αsyn epitope (residues 130-140; 94-3A10).
Figure 3. Induced CNS pSer129 αsyn and p62-sequestosome-1 inclusion pathology is similar at motor impairment end stage of IM seeded M83+/− mice regardless of the αsyn C- truncated hPFFs used for seeding.
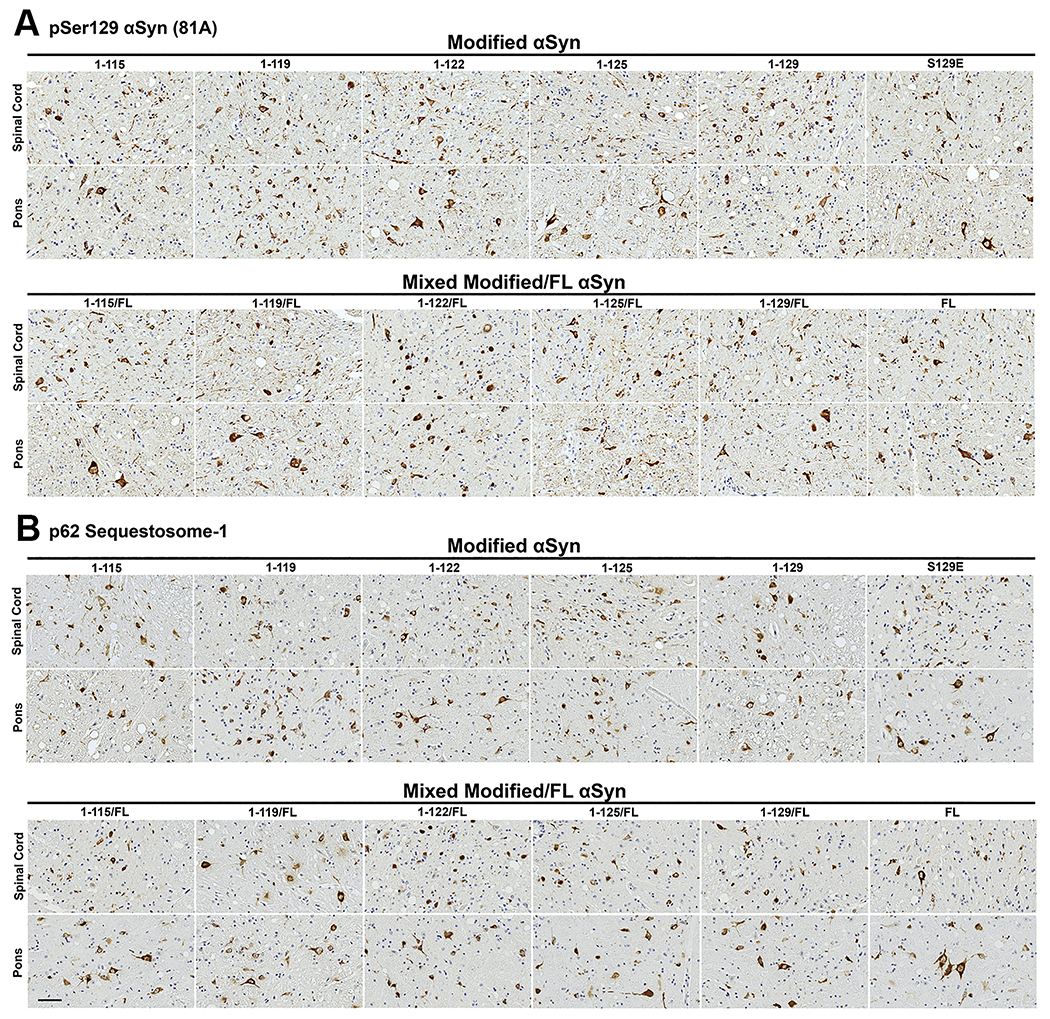
Representative immunohistochemical sections from the spinal cord and pons are shown from cohorts IM injected with hPFFs of varied compositions (1-115, 1-119, 1-122, 1-125, 1-129, S129E, and FL human αsyn or 1:1 mixed fibrils with each C-truncated form of αsyn and FL human αsyn). (A) Immunohistochemical staining with pSer129 αsyn antibody 81A. (B) Immunohistochemical staining with anti-p62-sequestosome-1 antibody. Similar densities of neuritic and cellular inclusions are seen for all cohorts in the spinal cord and pons. Scale bar 50 μm.
Figure 5. Semiquantitative comparisons of inclusion pathology with various antibodies demonstrate equivalent pathologic αsyn profiles at motor impairment end stage of IM seeded M83+/− mice irrespective of the αsyn C-truncated hPFFs used as inoculum.
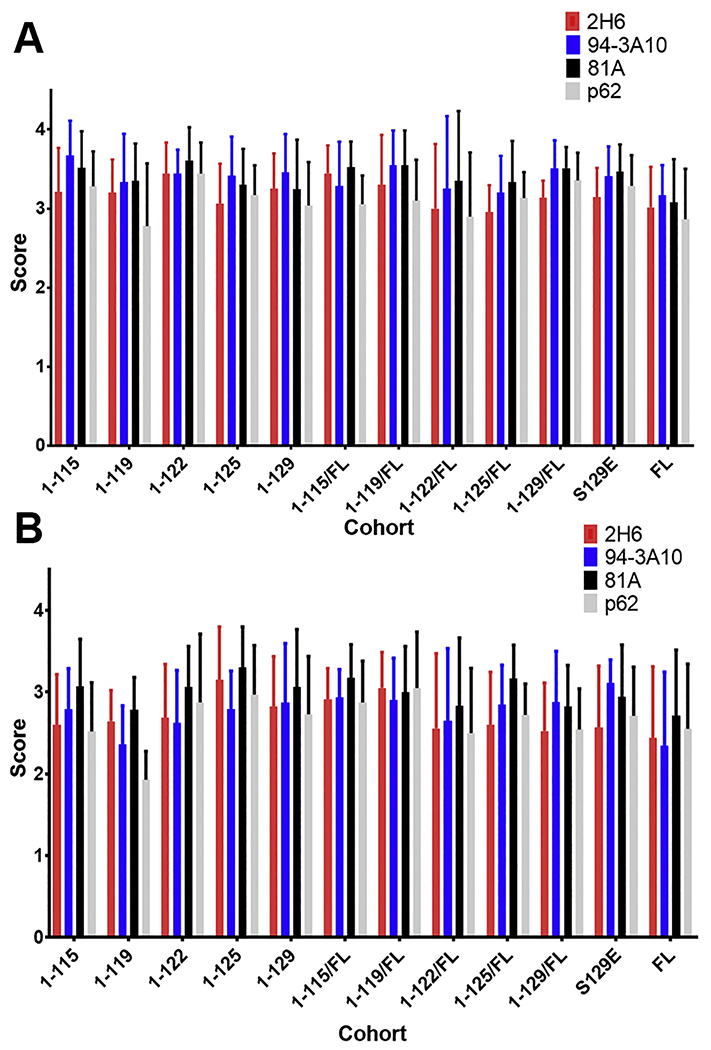
. For cohorts of M83+/− αsyn transgenic mice IM injected with hPFFs of varied compositions (1-115, 1-119, 1-122, 1-125, 1-129, S129E FL, and FL human αsyn or 1:1 mixed fibrils with each C-truncated form of αsyn and FL human αsyn), semiquantitative grading of pathology detected with immunohistochemistry in the (A) spinal cord and (B) pons was performed by 3 independent observers on a 4 point scale for all mice of each cohort using a panel of 4 antibodies with varying epitopes (pSer129 αsyn; 81A, p62-sequestosome-1, 2-21 αsyn; 2H6, 130-140 αsyn; 94-3A10). In general, burden of pathologic αsyn inclusions was increased in the spine relative to the pons for all cohorts of varied PFFs, however no difference was detected between cohorts or between the 4 antibodies within a cohort.
For all 4 of these antibodies and all mice, pathology density in the spinal cord and pons was graded on a 4-point scale by 3 independent observers in order to detect differences in end-stage pathology (Figure 5). The spinal cord and pons were the focus of examination as these contain the earliest and most dense αsyn pathology in this model, however the distribution of pathology throughout the rest of the neuroaxis for all cohorts was similar to that previously described as regions of selective vulnerability at end stage for this model [5,6]. In density and distribution of pathology, all aforementioned C-truncated and phosphomimetic PFF injected cohorts displayed similarity in the number of inclusions per 20x visual field in the spinal cord compared with the FL PFF cohort, with most spinal pathology concentrated in ventral motor regions; density of pathology is similar regardless of which of the 4 antibodies were used suggesting that epitope exposure due to altered αsyn conformations is not occurring (Figures 3–5). In the pons, all C-truncated and phosphomimetic PFF injected cohorts displayed qualitatively less pathology compared with the spinal cord, however between the cohorts within the pons there is again similarity in the number of inclusions per 20x visual field regardless of antibody used (Figures 3–5). Morphologically, αsyn pathology in both the spinal cord and pons across all cohorts is similar to that previously described for PFF injected M83+/− mice [5,6], with a mixture of LB-like neuronal inclusions and a meshwork of neuropil pathology (Figures 3–4).
Figure 4. αsyn inclusion pathology assessed with N- and C-terminal specific αsyn antibodies is similar at motor impairment end stage of IM seeded M83+/− mice irrespective of the αsyn C-truncated hPFFs used for seeding.
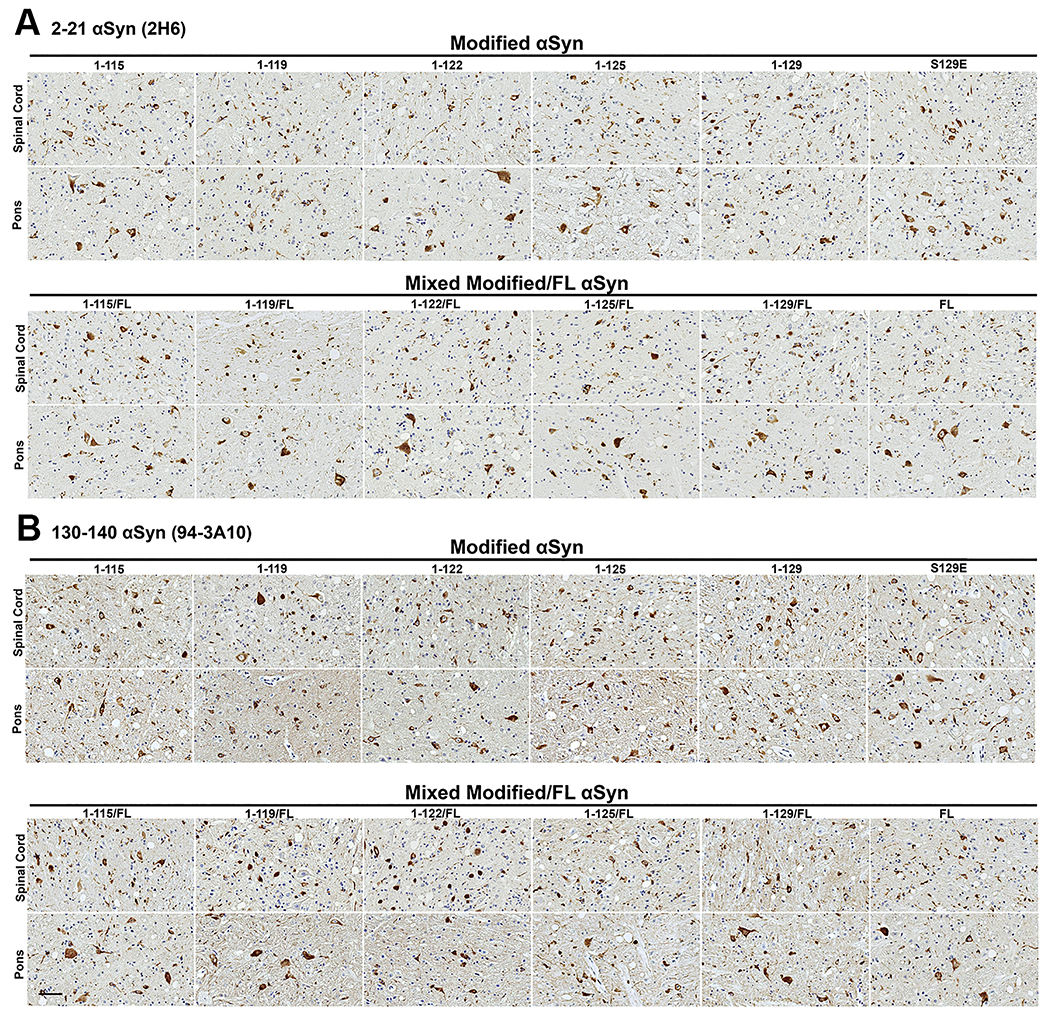
Representative immunohistochemical sections from the spinal cord and pons are shown from cohorts IM injected with hPFFs of varied compositions (1-115, 1-119, 1-122, 1-125, 1-129, S129E, and FL human αsyn or 1:1 mixed fibrils with each C-truncated form of αsyn and FL human αsyn). (A) Immunohistochemical staining with monoclonal antibody 2H6 (N-terminal epitope residues 2-21 αsyn) or (B) with monoclonal antibody 94-3A10 (C-terminal epitope residues 130-140 αsyn). Similar densities of neuritic and cellular αsyn inclusions are seen for all cohorts in the spinal cord and pons. Scale bar 50 μm.
Immunohistochemical staining for the varied mPFF dosage and Δ71-82 injected cohorts was performed identically to that of the C-truncated and phosphomimetic PFF injected cohorts (Supplementary figure 1). No differences in inclusion density, distribution, or antibody staining profile was detected in the spinal cord and pons for varied PFF dosage mice, however Δ71-82 injected mice demonstrated no pathology with any antibody which is congruent with the lack of a phenotype in these mice.
Cumulatively, these results show that in the M83+/− prion-like seeding IM model there is highly stereotypic αsyn pathology at end-stage paralysis regardless of the nature of PFF used here and that survival curve differences rather than histologic analysis at endpoint may be a more sensitive parameter for comparing strain-like variants of PFFs.
The αsyn S129E phosphomimetic results in both decreased prion-like seeding and aggregation in both homotypic and heterotypic paradigms in HEK293T cells
Phosphorylation at Ser129 is the most common PTM of αsyn in synucleinopathies, however experiments have provided mixed results regarding the effect of this PTM on toxicity, prion-like seeding, and aggregation which are of importance in disease progression [47]. Additionally, it is unclear whether this Ser129 hyper phosphorylation that is present in αsyn pathologic inclusions is occurring to monomeric αsyn leading to aggregation or after αsyn fibrillizes and coalescence to form inclusions [47]. In order to further investigate the findings that S129E hPFFs in the M83+/− injected cohort displayed decreased seeding of endogenous FL αsyn, we extend these studies in an HEK293T seeding paradigm. HEK293T cells were transfected to overexpress FL αsyn with or without the S129E phosphomimetic, and likewise hPFFs comprised of 21-140 αsyn with or without the S129E mutation were added to induce intracellular seeding (n=6 wells per combination; 2 replicates of n=3); control transfections with no hPFFs were also performed. The hPFFs used for these studies lack the first 20 amino acids that allows to specifically detect αsyn expressed in cells with N-terminal specific antibody SNL4.
At time 48 hours after hPFF addition, cells were biochemically fractionated into Triton X-100 detergent soluble (monomeric) and insoluble (fibrils) fractions. In this experimental paradigm the accumulation of Triton X-100 insoluble intracellularly expressed αsyn is due to prion-like amyloid seeding of the exogenously added αsyn fibrils [38]. The fraction of cellular Triton X-100 insoluble αsyn that formed was determined through western blotting with antibody SNL4. Without exogenous seeds, FL WT and S129E αsyn were completely soluble (Figure 6). As previously described [36], the treatment with exogenous 21-140 WT PFFs to cells that express FL αsyn induced the formation of insoluble, aggregated αsyn with 37.3±6.3% accumulating on this fraction (Figure 6). The presence of the S129E phosphomimetic only to the 21-140 PFFs or only to the expressed FL αsyn resulted in a significant reduction of insoluble αsyn fraction with 21.5±4.2% and 17.6±6.0% respectively; this trend was accentuated when both the 21-140 PFFs and expressed FL αsyn carried the S129E substitution at which point the insoluble αsyn fraction was reduced to 9.4±5.1% (Figure 6). These results indicate that the presence of S129E phosphomemetic reduces aggregation and prion-like seeding of αsyn.
Figure 6. Phosphomimetic S129E αsyn alters both heterotypic and homotypic seeding and aggregation in cultured HEK293T cells.
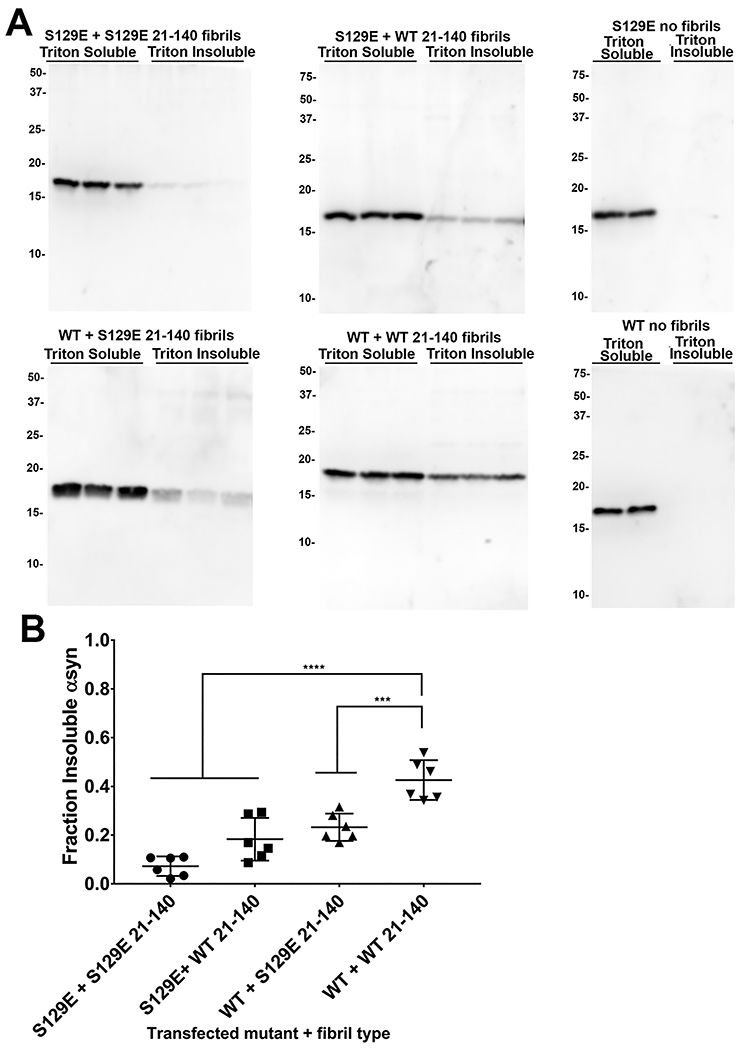
(A) Western blots displaying Triton X-100 soluble and insoluble fractions for HEK293T cells transfected to express either WT αsyn or S129E αsyn (n=6) followed by treatment with WT 21-140 αsyn or S129E 21-140 αsyn hPFFs as indicated; control transfections with no added hPFFs are also displayed. Antibody SNL4 against residues 2-12 was utilized for detection of transfected αsyn. The presence of S129E in the PFFs or cellularly expressed human αsyn decreases prion-like seeding and aggregation propensity; S129E homotypic seeding and aggregation is further attenuated. The mobility of molecular mass markers (kDa) are displayed on the left. (B) Densitometric analysis of the blots in A (n=6, error bars = s.d.); one-way ANOVA with Dunnet’s test determined the seeding and aggregation efficiency for each PFF and transfection combination relative to FL αsyn treated with 21-140 αsyn PFFs. *** = p ≤ 0.001, **** = p ≤ 0.0001.
Discussion
It is becoming increasingly clear that different conformers or “strains” of misfolded αsyn [13] may in part govern the progression of initial αsyn pathology into different synucleinopathies ranging from the relatively innocuous incidental Lewy body disease [48] to rapidly progressive MSA [1]. Even within the same disease, subtypes of PD and MSA with differential symptomatic and pathologic progression [1] may be explained in part by this strain concept. Indeed, missense mutations in SNCA αsyn gene resulting in pathologic αsyn fibrils with unique structural properties [19,49] present with varied clinical symptoms and pathology in terms of whether the familial human disease is most similar to PD, LBD, or MSA [50]. Furthermore, unique αsyn pathologic conformers with altered prion-like seeding properties may presumably appear due to different cellular biochemical environments [20,24,51], or through direct PTMs such as phosphorylation and truncation [17,36,46]. Herein, we demonstrated that αsyn C-terminal PTMs within PFFs impact the overall pace of prion-like peripheral to CNS synucleinopathy in the IM seeding M83 transgenic model with quantifiable alteration in the time of disease presentation.
In the course of disease progression in PD and LBD, it is hypothesized that αsyn aggregates are able to traverse the synaptic cleft from an afflicted neuron to a naive neuron where prion-like seeds are able to induce pathology through conformational templating in the recipient neuron [4]. Upon uptake of αsyn fibrils into the naive cell, it has been repeatedly demonstrated that fibrils are quickly localized to lysosomes where truncation occurs [4,25–27] which may alter the fibril structure compared to non-truncated fibrils [17–19]. How the presence of truncated αsyn within a pathologic fibril impacts prion-like seeding of αsyn in vivo has not been extensively studied, but it has been reported that prion-like seeding of endogenous FL αsyn within mice may be potentiated or attenuated depending on the exact truncation present [46,52,53] or even if other proteins such as tau are recruited into the seeding fibril [53]. Our laboratory has previously demonstrated that αsyn fibrils comprised of progressively more C-truncated αsyn fibrils display reduced cross-seeding of monomeric FL αsyn in cultured cells and in non-transgenic mice [36,52], however these later studies along with others [46,53] have relied on histologic examination of tissue at preset time-points which may not fully capture how PTMs can effect disease progression compared to a survival based model. Additionally, stereotactic surgical approaches to investigating prion-like activity introduces complicating variables including direct brain injury relating to the surgery and associated neuroinflammatory responses [54], uptake of PFFs by glial cells instead of neurons due to direct parenchymal injection [55], and outcomes dependent on effective PFF dosage as increased sonication to produce smaller fibrils results in exacerbated seeding of pathology [56]. The intramuscular injection model used herein does not involve any direct brain lesions, is presumably due predominantly to interneuronal spreading as injected fibrils invade the CNS through the sciatic nerve [5,29], and is robust in phenotype development across a wide range of PFF dosages suggesting that only very few neurons are involved in initial αsyn pathology from which further neuronal spread occurs. The features of this model are optimal for determining whether strain-like differences in C-truncated forms of αsyn impact prion-like properties in vivo.
Although the C-terminus of αsyn remains largely unstructured and does not contribute to the core of pathologic fibrils, C-terminal truncation has been recently demonstrated to alter the features of the overall fibril structure [17–19] which may have functional consequences for prion-like seeding. It was demonstrated in vitro that truncation at certain sites leads to increased templating ability [17,26,46,57] of FL αsyn by truncated fibrils and while others observed a decrease [18,58,59] compared to FL αsyn fibrils. One study that demonstrated decreased seeding with fibrils comprised of 1-108 αsyn demonstrated that the high degree of helical periodicity, or “twisting”, in these fibrils create a steric barrier to the addition of FL αsyn monomers as the fibril structure may be less able to accommodate the intact C-terminus of FL αsyn [18]; in comparison, a study that observed increased seeding of FL αsyn with 1-103 αsyn fibrils noted the increased twist in the fibrils but in contrast to the previous study found that FL αsyn readily added on to these fibrils and were templated to form fibrils with a similar twisted structure [17]. In the aforementioned animals studies, increased or decreased seeding of endogenous FL αsyn as measured by histologic formation of inclusions is again seemingly dependent on the exact truncation studied [46,52,53]. Thus, the exact structure(s) of αsyn fibrils comprised of C-truncated αsyn seems to be dependent on the exact truncations present (or other PTMs) and likely dictates whether monomeric FL αsyn can efficiently add to these fibrils, resulting in altered cross-seed efficiency, and propagation of unique strain.
Herein, we examined 6 different C-truncations previously shown to form in cultured cells or human brain pathological inclusions [25,36] and demonstrated that cross-seeding of the expressed FL αsyn in the mice is generally decreased by C-truncation, however the effect size is strongly dependent on the exact truncation where truncation at residues 115, 119, and 129 had a much larger effect on time to disease presentation than C-truncation at residues 122 or 125. Additionally, even fibrils that are comprised of only partially C-truncated αsyn as studied here are similarly affected, with the mixed 119/FL αsyn fibrils demonstrating increased survival time compared with 115/FL αsyn or 122/FL αsyn fibrils. The elongated survival time observed with certain C-truncated PFFs is likely due to structural incompatibility between the PFFs and FL αsyn, resulting in a longer time for templating of FL αsyn to occur. After the initial templating of endogenous αsyn, it is likely that a normal course of prion-like spread occurs resulting in the observed stereotypical pathology seen for M83 mice that we observed for all PFFs used in terms of location, cell type affected, and morphology [5,6]. Other studies noting increased seeding propensity or formation of unique pathologies in M83 mice [21,24] may have utilized αsyn fibril strains that are uniquely able to impart their conformation onto monomeric αsyn in a true prion-like fashion as observed in vitro for certain truncations [17].
Modification of the C-terminus through introduction of the S129E phosphomimetic mutation also resulted in what may be significantly altered prion-like seeding in the M83 injection model when S129E PFFs were used, although the effect size was not as large as for PFFs comprised of most of the C-truncated forms of αsyn assessed. This suggests that even slight alteration of the C-terminus through phosphorylation of a single residue is sufficient to affect the fibril structure and prion-like seeding activity. Additionally, the heterotypic and homotypic seeding and aggregation of S129E αsyn was examined in cultured cells which demonstrated that pSer129 may in general impair both aggregation and prion-like seeding, as the combination of S129E fibrils with S129E αsyn monomers produced the least aggregation. In comparison, with C-terminal truncations the negative charges are eliminated by this process which is implicated in the increased aggregation propensity from this PTM [36], and it would be expected that the addition of negativity through phosphorylation would decrease aggregation which is what we observed. Interpretation of this data may be complicated as phosphomimetic mutations do not fully replicate the effects of phosphorylation [47], however these results again demonstrate that αsyn C-terminal PTMs can govern prion-like properties.
The ramifications of C-truncation are particularly important in PD, as this PTM has been demonstrated to promote initial αsyn aggregation [36,52] and also be prominent within intestinal neuronal plexi [53,60] where αsyn pathology is postulated to initiate under standard staging schema [7]. Our results demonstrate that C-truncation that occurs in disease modulates prion-like seeding activity in vivo. In human disease whether this type of modification results in slowed prion-like spread due to these altered αsyn fibrils being less efficient at seeding endogenous αsyn or the formation of new strains with impartible conformations resulting in context-dependent potentiated seeding is unclear. It is possible that C-terminal truncation of αsyn is both harmful in initial pathology formation while subsequently dampening the rate of prion-like spread when fibrils undergo lysosomal processing following neuronal uptake, which may be a factor in the decades long time-course of human pathology progression [8] compared with transgenic mice. Nonetheless, these results demonstrate that C-truncation of αsyn can have strong functional consequences on prion-like seeding, and this PTM in combination with other PTMs or unknown factors may be a component of the unique strains that arise in disease.
Supplementary Material
Figure S1. αsyn inclusion pathology is similar at motor impairment end stage regardless of PFF dosage used for IM seeded M83+/− mice. Representative immunohistochemical sections from the spinal cord and pons are shown for various antibodies from cohorts injected with mPFFs of varied dosage (0.1, 0.5, or 1 μg) or Δ71-82 αsyn (20 μg). Immunohistochemical staining with anti-pSer129 αsyn monoclonal antibody 81A (A), anti-p62-sequestosome-1 antibody (B), monoclonal antibody 2H6 (N-terminal residues 2-21 αsyn) (C), or monoclonal antibody 94-3A10 (C-terminal residues 130-140 αsyn) (D). Similar densities of neuritic and cellular αsyn inclusions are seen for all cohorts in the spinal cord and pons, except for Δ71-82 αsyn where no pathology was observed. Scale bar 50 μm.
Highlights.
Carboxy-terminal modification of αsyn through truncation or introduction of a phosphomimetic functionally changes prion-like seeding as evidenced by elongated time to death in a prion-like mouse model of synucleinopathy
All mice injected with each carboxy-modified form of αsyn developed similar pathology and end stage phenotype despite the elongated time to death
In cultured cells, the presence of the S129E phosphomimetic form of αsyn decreases prion-like seeding and aggregation
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01NS089022, R01NS100876 and F30AG063446).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare they have no conflict of interest with this article.
References
- [1].McCann H, Stevens CH, Cartwright H, Halliday GM, α-Synucleinopathy phenotypes, Parkinsonism Relat. Disord 20 (2014) S62–S67. doi: 10.1016/S1353-8020(13)70017-8. [DOI] [PubMed] [Google Scholar]
- [2].Spillantini MG, Goedert M, Neurodegeneration and the ordered assembly of α-synuclein, Cell Tissue Res. 373 (2018) 137–148. doi: 10.1007/s00441-017-2706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Uchihara T, Giasson BI, Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies., Acta Neuropathol. 131 (2016) 49–73. doi: 10.1007/s00401-015-1485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Karpowicz RJ, Trojanowski JQ, Lee VM-Y, Transmission of α-synuclein seeds in neurodegenerative disease: recent developments, Lab. Investig 99 (2019) 971–981. doi: 10.1038/s41374-019-0195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI, Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice, Proc. Natl. Acad. Sci 111 (2014) 10732–10737. doi: 10.1073/PNAS.1321785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sorrentino ZA, Xia Y, Funk C, Riffe CJ, Rutherford NJ, Ceballos Diaz C, Sacino AN, Price ND, Golde TE, Giasson BI, Chakrabarty P, Motor neuron loss and neuroinflammation in a model of α-synuclein-induced neurodegeneration, Neurobiol. Dis 120 (2018) 98–106. doi: 10.1016/j.nbd.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Del Tredici K, Braak H, Review: Sporadic Parkinson’s disease: development and distribution of α-synuclein pathology, Neuropathol. Appl. Neurobiol 42 (2016) 33–50. doi: 10.1111/nan.12298. [DOI] [PubMed] [Google Scholar]
- [8].Kalia LV, Lang AE, Parkinson’s disease., Lancet (London, England). 386 (2015) 896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- [9].Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJA, Kraneveld AD, Exploring Braak’s Hypothesis of Parkinson’s Disease., Front. Neurol 8 (2017) 37. doi: 10.3389/fneur.2017.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lionnet A, Leclair-Visonneau L, Neunlist M, Murayama S, Takao M, Adler CH, Derkinderen P, Beach TG, Does Parkinson’s disease start in the gut?, Acta Neuropathol. 135 (2018) 1–12. doi: 10.1007/s00401-017-1777-8. [DOI] [PubMed] [Google Scholar]
- [11].Liddle RA, Parkinson’s disease from the gut, Brain Res. 1693 (2018) 201–206. doi: 10.1016/j.brainres.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Klingelhoefer L, Reichmann H, Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors., Nat. Rev. Neurol 11 (2015) 625–36. doi: 10.1038/nrneurol.2015.197. [DOI] [PubMed] [Google Scholar]
- [13].Peelaerts W, Bousset L, Baekelandt V, Melki R, α-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences, Cell Tissue Res. 373 (2018) 195–212. doi: 10.1007/s00441-018-2839-5. [DOI] [PubMed] [Google Scholar]
- [14].Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ, Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease, J. Biol. Chem 281 (2006) 29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- [15].Liu C-W, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ, A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease., J. Biol. Chem 280 (2005) 22670–8. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- [16].Zhang J, Li X, Da Li J, The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases, Front. Neurosci 13 (2019). doi: 10.3389/fnins.2019.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ni X, McGlinchey RP, Jiang J, Lee JC, Structural Insights into α-Synuclein Fibril Polymorphism: Effects of Parkinson’s Disease-Related C-Terminal Truncations, J. Mol. Biol 431 (2019) 3913–3919. doi: 10.1016/j.jmb.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Iyer A, Roeters SJ, Kogan V, Woutersen S, Claessens MMAE, Subramaniam V, C-Terminal Truncated α-Synuclein Fibrils Contain Strongly Twisted β-Sheets, J. Am. Chem. Soc 139 (2017) 15392–15400. doi: 10.1021/jacs.7b07403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Meade RM, Fairlie DP, Mason JM, Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles, Mol. Neurodegener 14 (2019) 29. doi: 10.1186/s13024-019-0329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, Trojanowski JQ, Lee VM-Y, Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies, Nature. 557 (2018) 558–563. doi: 10.1038/s41586-018-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K, Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism., Proc. Natl. Acad. Sci. U. S. A 112 (2015) E5308–17. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lavenir I, Passarella D, Masuda-Suzukake M, Curry A, Holton JL, Ghetti B, Goedert M, Silver staining (Campbell-Switzer) of neuronal α-synuclein assemblies induced by multiple system atrophy and Parkinson’s disease brain extracts in transgenic mice, Acta Neuropathol. Commun 7 (2019) 148. doi: 10.1186/s40478-019-0804-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song E-S, Cairns NJ, Kotzbauer PT, Diamond MI, Parkinson’s disease and multiple system atrophy have distinct α-synuclein seed characteristics, J. Biol. Chem 294 (2019) 1045–1058.doi: 10.1074/jbc.RA118.004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lau A, So RWL, Lau HHC, Sang JC, Ruiz-Riquelme A, Fleck SC, Stuart E, Menon S, Visanji NP, Meisl G, Faidi R, Marano MM, Schmitt-Ulms C, Wang Z, Fraser PE, Tandon A, Hyman BT, Wille H, Ingelsson M, Klenerman D, Watts JC, α-Synuclein strains target distinct brain regions and cell types., Nat. Neurosci 23 (2020) 21–31. doi: 10.1038/s41593-019-0541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pieri L, Chafey P, Le Gall M, Clary G, Melki R, Redeker V, Cellular response of human neuroblastoma cells to α-synuclein fibrils, the main constituent of Lewy bodies., Biochim. Biophys. Acta 1860 (2016) 8–19. doi: 10.1016/j.bbagen.2015.10.007. [DOI] [PubMed] [Google Scholar]
- [26].McGlinchey RP, Lacy SM, Huffer KE, Tayebi N, Sidransky E, Lee JC, C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson’s disease, J. Biol. Chem 294 (2019) 9973–9984. doi: 10.1074/jbc.RA119.008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sacino AN, Brooks MM, Chakrabarty P, Saha K, Khoshbouei H, Golde TE, Giasson BI, Proteolysis of α-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology., J. Neurochem 140 (2017) 662–678. doi: 10.1111/jnc.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y, Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein., Neuron. 34 (2002) 521–33. [DOI] [PubMed] [Google Scholar]
- [29].Ayers J.l, Riffe CJ, Sorrentino ZA, Diamond J, Fagerli E, Brooks M, Galaleldeen A, Hart PJ, Giasson BI, Localized induction of wild-type and mutant alpha-synuclein aggregation reveals propagation along neuroanatomical tracts, J. Virol 92 (2018) JVI.00586-18. doi: 10.1128/JVI.00586-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rutherford NJ, Dhillon J-KS, Riffe CJ, Howard JK, Brooks M, Giasson BI, Comparison of the in vivo induction and transmission of α-synuclein pathology by mutant α-synuclein fibril seeds in transgenic mice., Hum. Mol. Genet 26 (2017) 4906–4915. doi: 10.1093/hmg/ddx371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VMY, Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice., J. Exp. Med 209 (2012) 975–86. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rutherford NJ, Brooks M, Giasson BI, Novel antibodies to phosphorylated α-synuclein serine 129 and NFL serine 473 demonstrate the close molecular homology of these epitopes., Acta Neuropathol. Commun 4 (2016) 80. doi: 10.1186/s40478-016-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dhillon J-KS, Riffe C, Moore BD, Ran Y, Chakrabarty P, Golde TE, Giasson BI, A novel panel of α-synuclein antibodies reveal distinctive staining profiles in synucleinopathies, PLoS One. 12 (2017) e0184731. doi: 10.1371/journal.pone.0184731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY, A panel of epitope-specific antibodies detects protein domains distributed throughout human α-synuclein in lewy bodies of Parkinson’s disease, J. Neurosci. Res 59 (2000) 528–533. doi:. [DOI] [PubMed] [Google Scholar]
- [35].Giasson BI, Murray IVJ, Trojanowski JQ, Lee VM-Y, A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly, J. Biol. Chem 276 (2001) 2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- [36].Sorrentino ZA, Vijayaraghavan N, Gorion K-M, Riffe CJ, Strang KH, Caldwell J, Giasson BI, Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions, J. Biol. Chem 293 (2018) 18914–18932. doi: 10.1074/jbc.RA118.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ayers JI, Brooks MM, Rutherford NJ, Howard JK, Sorrentino ZA, Riffe CJ, Giasson BI, Robust Central Nervous System Pathology in Transgenic Mice following Peripheral Injection of α-Synuclein Fibrils., J. Virol 91 (2017) e02095–16. doi: 10.1128/JVI.02095-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Waxman EA, Giasson BI, A novel, high-efficiency cellular model of fibrillar α-synuclein inclusions and the examination of mutations that inhibit amyloid formation, J. Neurochem 113 (2010) 374–388. doi: 10.1111/j.1471-4159.2010.06592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sacino AN, Thomas MA, Ceballos-Diaz C, Cruz PE, Rosario AM, Lewis J, Giasson BI Golde TE, Conformational templating of α-synuclein aggregates in neuronal-glial cultures., Mol. Neurodegener 8 (2013) 17. doi: 10.1186/1750-1326-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM-Y, Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells., Proc. Natl. Acad. Sci. U. S. A 106 (2009) 20051–6. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VM, Trojanowski JQ, Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy., J. Neuropathol. Exp. Neurol 59 (2000) 830–41. [DOI] [PubMed] [Google Scholar]
- [42].Dunn SD, Effects of the modification of transfer buffer composition and the renaturation of proteins in gels on the recognition of proteins on Western blots by monoclonal antibodies., Anal. Biochem 157 (1986) 144–53. [DOI] [PubMed] [Google Scholar]
- [43].Jankowsky JL, Zheng H, Practical considerations for choosing a mouse model of Alzheimer’s disease, Mol. Neurodegener 12 (2017) 89. doi: 10.1186/s13024-017-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK, Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations., Proc. Natl. Acad. Sci. U. S. A 102 (2005) 2162–7. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Crystal AS, Giasson BI, Crowe A, Kung M-P, Zhuang Z-P, Trojanowski JQ, Lee VM-Y, A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114., J. Neurochem 86 (2003) 1359–68. [DOI] [PubMed] [Google Scholar]
- [46].Terada M, Suzuki G, Nonaka T, Kametani F, Tamaoka A, Hasegawa M, The effect of truncation on prion-like properties of α-synuclein, J. Biol. Chem 293 (2018) 13910–13920. doi: 10.1074/jbc.RA118.001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Oueslati A, Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade?, J. Parkinsons. Dis 6 (2016) 39–51. doi: 10.3233/JPD-160779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Markesbery WR, Jicha GA, Liu H, Schmitt FA, Lewy Body Pathology in Normal Elderly Subjects, J. Neuropathol. Exp. Neurol 68 (2009) 816–822. doi: 10.1097/NEN.0b013e3181ac10a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sahay S, Ghosh D, Singh PK, Maji SK, Alteration of Structure and Aggregation of α-Synuclein by Familial Parkinson’s Disease Associated Mutations, Curr. Protein Pept. Sci 18 (2016) 656–676. doi: 10.2174/1389203717666160314151706. [DOI] [PubMed] [Google Scholar]
- [50].Rosborough K, Patel N, Kalia LV, α-Synuclein and Parkinsonism: Updates and Future Perspectives, Curr. Neurol. Neurosci. Rep 17 (2017) 31. doi: 10.1007/s11910-017-0737-y. [DOI] [PubMed] [Google Scholar]
- [51].Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R, Structural and functional characterization of two alpha-synuclein strains, Nat. Commun 4 (2013) 2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sorrentino ZA, Xia Y, Gorion K, Hass E, Giasson BI, Carboxy-terminal truncations of mouse alpha-synuclein alter aggregation and prion-like seeding, FEBS Lett. (2020) 1873–3468.13728. doi: 10.1002/1873-3468.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ahn EH, Kang SS, Liu X, Chen G, Zhang Z, Chandrasekharan B, Alam AM, Neish AS, Cao X, Ye K, Initiation of Parkinson’s disease from gut to brain by δ-secretase., Cell Res. 30 (2020) 70–87. doi: 10.1038/s41422-019-0241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Koller EJ, Brooks MMT, Golde TE, Giasson BI, Chakrabarty P, Inflammatory preconditioning restricts the seeded induction of α-synuclein pathology in wild type mice., Mol. Neurodegener 12 (2017) 1. doi: 10.1186/s13024-016-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sorrentino ZA, Giasson BI, Chakrabarty P, α-Synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease, Acta Neuropathol. 138 (2019) 1–21. doi: 10.1007/s00401-019-01977-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Froula JM, Castellana-Cruz M, Anabtawi NM, Camino JD, Chen SW, Thrasher DR, Freire J, Yazdi AA, Fleming S, Dobson CM, Kumita JR, Cremades N, Volpicelli-Daley LA, Defining α-synuclein species responsible for Parkinson’s disease phenotypes in mice, J. Biol. Chem 294 (2019) 10392–10406. doi: 10.1074/jbc.RA119.007743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang Z, Kang SS, Liu X, Ahn EH, Zhang Z, He L, Iuvone PM, Duong DM, Seyfried NT, Benskey MJ, Manfredsson FP, Jin L, Sun YE, Wang J-Z, Ye K, Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson’s disease., Nat. Struct. Mol. Biol 24 (2017) 632–642. doi: 10.1038/nsmb.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mishizen-Eberz AJ, Norris EH, Giasson BI, Hodara R, Ischiropoulos H, Lee VM-Y, Trojanowski JQ, Lynch DR, Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein., Biochemistry. 44 (2005) 7818–29. doi: 10.1021/bi047846q. [DOI] [PubMed] [Google Scholar]
- [59].van der Wateren IM, Knowles TPJ, Buell AK, Dobson CM, Galvagnion C, C-terminal truncation of α-synuclein promotes amyloid fibril amplification at physiological pH., Chem. Sci 9 (2018) 5506–5516. doi: 10.1039/c8sc01109e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Killinger BA, Madaj Z, Sikora JW, Rey N, Haas AJ, Vepa Y, Lindqvist D, Chen H, Thomas PM, Brundin P, Brundin L, Labrie V, The vermiform appendix impacts the risk of developing Parkinson’s disease, Sci. Transl. Med 10 (2018) eaar5280.doi: 10.1126/scitranslmed.aar5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. αsyn inclusion pathology is similar at motor impairment end stage regardless of PFF dosage used for IM seeded M83+/− mice. Representative immunohistochemical sections from the spinal cord and pons are shown for various antibodies from cohorts injected with mPFFs of varied dosage (0.1, 0.5, or 1 μg) or Δ71-82 αsyn (20 μg). Immunohistochemical staining with anti-pSer129 αsyn monoclonal antibody 81A (A), anti-p62-sequestosome-1 antibody (B), monoclonal antibody 2H6 (N-terminal residues 2-21 αsyn) (C), or monoclonal antibody 94-3A10 (C-terminal residues 130-140 αsyn) (D). Similar densities of neuritic and cellular αsyn inclusions are seen for all cohorts in the spinal cord and pons, except for Δ71-82 αsyn where no pathology was observed. Scale bar 50 μm.