

Abstract
Hidradenitis suppurativa (HS) is a chronic, relapsing, inflammatory skin disease. HS appears to be a primary abnormality in the pilosebaceous-apocrine unit. In this work, we characterized hair follicle stem cells (HFSCs) isolated from HS patients and more precisely the outer root sheath cells (ORSCs). We showed that hair follicle cells from HS patients had an increased number of proliferating progenitor cells and lost quiescent stem cells. Remarkably, we also showed that the progression of replication forks was altered in ORSCs from hair follicles of HS patients, leading to activation of the ATR/CHK1 pathway. These alterations were associated with an increased number of micronuclei and with the presence of cytoplasmic ssDNA, leading to the activation of the IFI16/STING pathway and the production of type I IFNs. This mechanistic analysis of the etiology of HS in the HFSC compartment establishes a formal link between genetic predisposition and skin inflammation observed in HS.
Keywords: Dermatology, Immunology
Keywords: Adult stem cells, Cellular immune response
Introduction
Hidradenitis suppurativa (HS) is a chronic, relapsing, inflammatory skin disease characterized by double comedones; painful, deep, recurrent nodules; and abscesses. It is also known as acne inversa, since it affects the inverse areas, most commonly the axillae, inguinal, perineal, mammary, and inframammary areas (1). This can progress to a chronic inflammatory state with fistula formation, odorous discharge, cribriform scarring, and dermal fibrosis with contractures. HS leads to a high extent of emotional and physical distress, with social embarrassment, isolation, and depression leading to the most severe impairment among common dermatoses of quality of life (2). HS is not a rare disease. The estimated prevalence of HS varies from 0.05% to 4.10%, depending on the methodology used to estimate the prevalence. It affects approximately 1% of the general European population (3) and 98 of 100,000 persons in the United States. Prevalence is highest among women (137 per 100,000), those aged 30 to 39 years (172 per 100,000), and African American (296 per 100,000) and biracial (218 per 100,000) patient groups (4). The effective treatment options are limited. Given that many treatments have been unsatisfactory and there are many recurrences, HS remains difficult to treat. No formal guidelines are currently available for the management of HS. Understanding HS is essential in order to improve treatment and prognosis.
HS appears to be a primary abnormality in the pilosebaceous-apocrine unit, which leads to follicular occlusion, perifollicular cyst development that traps commensal microbes, and rupture into the dermis. This can trigger an exaggerated response of the cutaneous innate immune system (5). Abundant evidence suggests a role for a dysregulated immune response to bacteria and keratin filaments found ectopically in the dermis (6) that may drive a chronic inflammatory phase.
The hair follicle is a complex appendage of the epidermis. It is composed of an infundibulum that opens to the skin surface, sebaceous glands, and the junctional zone between the glands and the bulge, a stem cell niche that acts as a reservoir of multipotent stem cells (7). The cells in the bulge give rise to all epithelial lineages of the skin, including epidermal and follicular keratinocytes, sebocytes, and hair. Quiescent bulge stem cells are located in the outer layer of this niche and contribute to the generation of the outer root sheath (ORS). ORS cells (ORSCs) surround the hair follicle as essentially a stratified epithelium of keratinocytes that is contiguous with the epidermis. The ORS is divided into 4 portions from distal to proximal ends: the infundibulum, the bulge, the sub-bulge, and the lower ORS. These cells display differential expression of stem cell–associated markers and divergent proliferation patterns.
We previously showed that ORSCs isolated from hair follicles of HS patients (HS-ORSCs) spontaneously secrete IP-10 (CXCL10) and RANTES (CCL5) (8). HS-ORSCs secreted significantly more IL-1β on pattern recognition receptor stimulations, and upon IL-1β stimulation, HS-ORSCs further increased production of proinflammatory cytokines IL-6, IL-8, and TNF-α. These results indicated a skewed balance toward a proinflammatory profile of HS-ORSCs that may explain the chronic inflammation without bacterial clearance.
The cGAS/STING pathway is an important cytosolic DNA sensing pathway that activates expression of type I IFNs and other inflammatory cytokines to induce innate immunity for antimicrobial effects in response to viral and bacterial DNA (9). Besides pathogens, endogenous cytosolic DNA activates the cGAS/STING pathway in cancer cells and affects tumor development. The cGAS/STING pathway is constitutively activated in Aicardi-Goutières syndrome, which is caused by germline mutations in genes encoding factors involved in nucleic acid metabolism such as TREX1, RNase H2, and SAMHD1 (10). The characterization of the molecular function of these factors has shed new light on the connections between the DNA damage response and innate immunity in a pathological context. Recent evidence indicates that the processing of stalled forks by DNA repair enzymes leads to the production of small DNA fragments that accumulate in the cytosol and activate the cGAS/STING pathway (11, 12). Stalled forks are detected by the ATR checkpoint kinase, which binds replication protein A–coated ssDNA and activates the effector kinase CHK1 to prevent premature entry into mitosis and promote replication resumption (13).
Other DNA sensors have been identified, such as IFN-inducible protein 16 (IFI16), DDX41, and DNA-dependent protein kinase. IFI16 is expressed in the nucleus in keratinocytes. Under inflammatory conditions, IFI16 could be recruited to STING and induce IP-10 and CCL20 in response to cytoplasmic DNA (14). It has been shown that cGAS and IFI16 cooperate in the sensing of intracellular DNA in human keratinocytes. IFI16 is critical for the full activation of STING, and cooperates with cGAMP in the activation of this key signaling adaptor (15).
In this study, we aim to characterize the molecular mechanisms involved in the proinflammatory phenotype of HS-ORSCs. Transcriptomic analyses of HS-ORSCs highlighted a deregulation of genes involved in cell proliferation and differentiation, and an upregulation of the DNA damage response and type I IFN signature. We observed a perturbation of the hair follicle stem cell (HFSC) compartment in HS patients with the lack of the quiescent bulge stem cell population and the increase of proliferating progenitor cells. Single-molecule analysis of DNA replication revealed that replication fork speed was altered in HS-ORSCs, leading to the activation of the ATR/CHK1 pathway. Accumulation of ssDNA and micronuclei in the cytoplasm of HS-ORSCs contributed to STING activation via the DNA sensor IFI16, inducing type I IFN synthesis. Depletion of STING in ORSCs resulted in modulation of fork progression. These findings support the notion that in HS patients, a perturbation of HFSC homeostasis leading to an increased proliferation induces replication stress and the accumulation of cytoplasmic ssDNA, stimulating type I IFN synthesis through the IFI16/STING pathway.
Results
Transcriptomic analyses of HS-ORSCs revealed an IFN signature and an alteration of cell cycle pathways.
To identify the mechanisms involved in the proinflammatory phenotype of HS-ORSCs, we isolated ORSCs from hair follicles and performed transcriptomic analyses after ORS amplification at passage 3. We compared the gene expression profile in HS patients (n = 6) to that of healthy donors (HD-ORS) (n = 5). Principal component analysis revealed a distinct genomic profile between HS-ORSCs and HD-ORSCs (Figure 1A). The main significant canonical pathways associated with differentially expressed genes between HS-ORSCs and HD-ORSCs are shown in Figure 1B. These revealed a dysregulation of genes involved in cell proliferation and differentiation, and an upregulation of the DNA damage response and cell cycle G2/M checkpoint pathways in HS-ORSCs (Figure 1, C and D). Of note, we observed an induction of genes involved in the IGF1 pathway, which play important roles in hair follicle development and cycling (7). These results also showed the induction of genes strongly associated with type I IFN signature in HS-ORSCs, including an increase in IFN-stimulated genes (ISGs) and IFN regulatory factors (IRFs) (Figure 1C). To confirm this in vitro IFN signature, we assessed expression of ISGs by RT-qPCR in freshly isolated total hair follicle cells. We observed an overexpression of IP-10, IFI27, and OAS1 in HS-ORSCs compared with HD-ORSCs (IP-10: 12.65 ± 15.34 vs. 0.4833 ± 0.37, P = 0.0006; IFI27: 53.45 ± 47.34 vs. 19.71 ± 6.675, P = 0.0048; OAS1: 5.409 ± 2.54 vs. 2.658 ± 1.32, P = 0.019, respectively; Figure 2). MX-1 mRNA expression was increased in HS-ORSCs compared with HD-ORSCs, although the difference was not statistically significant between the HS and HD groups (2.059 ± 1.74 vs. 0.8533 ± 0.75, P = 0.09). These findings indicate that ex vivo total hair follicle cells displayed a type I IFN signature.
Figure 1. Transcriptomic analysis of ORSCs isolated from HS patients revealed a type I IFN signature and an alteration of cell cycle pathways.

(A) Principal component analysis of transcriptomic data (HS n = 6; HD n = 5). (B) Canonical pathways after FDR < 0.2. (C) Main differentially expressed genes between HS-ORSCs and HD-ORSCs associated with cell proliferation and type I IFN signaling. (D) Hierarchical clustering analysis of HS-ORSCs and HD-ORSCs based on the expression of cancer mechanism genes.
Figure 2. IFN signature in hair follicle cells isolated from HS patients.

Relative mRNA levels of MX-1, IP-10, IFI27, and OAS1 in freshly isolated total hair follicle cells from HS patients (n = 14 or 15) and HD controls (n = 5 or 6). Horizontal lines represent the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 by Mann-Whitney U test.
HFSCs from HS patients lacked the quiescent bulge stem cell population.
To confirm transcriptomic data on cell proliferation, immunohistochemical staining for Ki67 was performed on skin lesions. As compared with normal skin, we found in HS lesions an increase of Ki67+ keratinocytes and hair follicle cells (Figure 3A). Ki67+ keratinocytes were especially observed in the epidermis overlying infiltrates and in the sebaceous glands.
Figure 3. HS-ORSCs have an increased proliferation rate.

(A) Immunohistochemical staining of Ki67+ keratinocytes in a chronic HS lesion and in healthy donor. Arrows indicate the sebaceous gland. (B) Colony-forming assay of ORSCs isolated from HS patients (n = 3) and the HD group (n = 3) and evaluation of size and number of colonies formed for each individual. Horizontal lines represent the mean ± SD.
We then focused on ORSC proliferation rates using a colony-forming efficiency assay. At the initial concentration of 5000 cells per well, HS-ORSCs showed a much higher colony-forming ability than HD-ORSCs (Figure 3B). Moreover, there were striking differences in the colony numbers and sizes between HS-ORSCs (n = 3) and HD-ORSCs (n = 3) (Figure 3B). HS-ORSCs gave rise to a larger number of colonies with larger sizes, whereas the HD-ORS population formed a smaller number of colonies with smaller sizes (colonies < 1 mm2, 57 ± 13 vs. 71 ± 18; colonies 1–10 mm2, 80 ± 23 vs. 61 ± 14; colonies 10–100 mm2, 21 ± 5 vs. 3 ± 2; colonies > 100 mm2, 4 ± 4 vs. 0).
HFSCs represent a heterogeneous population and differ strikingly in their cell cycle dynamics. To better characterize HFSC compartments, we performed phenotypic analysis on freshly isolated total hair follicle cells by flow cytometry. We analyzed the CD45–CD117– cells from the starting population in order to exclude CD45+ hematopoietic cells and CD117+ melanocytes. Using the biomarker combinations determined by Inoue et al. (16), we defined cells from the bulge as CD200+CD34–, from the upper sub-bulge as CD200+CD34+ cells, and from the sub-bulge as CD200–CD34+ cells (Figure 4A). We then categorized each population into 2 groups according to cytokeratin 15 (CytoK15) and CD49f expression (Figure 4A). The CytoK15hiCD49fhi cells were in a quiescent state (G0/G1), whereas the CytoK15loCD49flo cells contained a substantial number of S phase cells and G2/M phase cells, indicating dividing cells (Figure 4, A and B). An enrichment of sub-bulge cells was observed in hair follicles isolated from HS patients compared with the HD group, although the difference was not statistically significant (sub-bulge 27.5% ± 16.7% vs. 19% ± 11%, P = 0.27) (Figure 4C). In line with these results, a depletion of bulge cells was noted in hair follicles isolated from HS patients compared with the HD group (9.9% ± 6.2% vs. 20.1% ± 9%, P = 0.018) (Figure 4C). Of note, we found that the well-demarcated quiescent CD200+CD34–CytoK15hiCD49fhi bulge population was significantly decreased in hair follicles isolated from HS patients compared with the HD group (2.2% vs. 6.8%, P = 0.02) (Figure 4D). These findings are consistent with a loss of quiescent stem cells and an increased number of proliferating cells in HS patients.
Figure 4. HFSCs from HS patients lacked the quiescent bulge stem cell population.

(A) Representative data from FACS analyses of freshly isolated total hair follicle cells from the HD group. After gating out CD45+ cells and CD117+ cells, freshly isolated total hair follicle cells were divided into 3 populations, including (a) the bulge cells defined as CD200+CD34–, (b) the upper sub-bulge as CD200+CD34+ cells, and (c) the sub-bulge as CD200–CD34+ cells. Each population was divided into CytoK15hiCD49fhi and CytoK15loCD49flo subsets (red and blue populations, respectively); a histogram demonstrates Ki67 staining in each subpopulation. (B) Representative data from FACS analyses of freshly isolated total hair follicle cells from HS patients. (C) Proportion of each subpopulation in freshly isolated total hair follicle–derived epithelial cells of HS patients (n = 19) and HD group (n = 6). (D) Proportion of CytoK15hiCD49fhi subset in CD200+CD34– cells of HS patients (n = 19) and HD group (n = 6) (HS: first quartile 0.58%; third quartile 3.14%; minimum 0.09%; maximum 9.01% vs. HD: first quartile 1.88%; third quartile 13.41%; minimum 1.2%; maximum 15%). Horizontal lines represent the mean ± SD. *P < 0.05 by Mann-Whitney U test.
Spontaneous activation of the ATR/CHK1 pathway in HS-ORSCs.
The increased number of proliferating ORSCs in HS patients led us to analyze their cell cycle distribution. To this end, ORSCs were amplified in vitro and cell cycle analysis was performed (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI131180DS1). We observed an increased percentage of cells in the S phase in HS-ORSCs compared with HD-ORSCs (22.2% ± 4% vs. 16.7% ± 3.6%, P = 0.0002) (Figure 5A). In line with this result, the percentage of cells in the G0/G1 phase was decreased in HS-ORSCs compared with HD-ORSCs (63% ± 5% vs. 70% ± 5.5%, P = 0.0027). The percentage of cells in the G2/M phase was variable for HS patients from 4.2% to 23.7%, whereas the HD group displayed a more homogeneous pattern of approximately 14% (Figure 5A).
Figure 5. Alteration of fork replication progression in HS-ORSCs.

(A) Cell cycle analysis by FACS of HS-ORSCs (n = 28) and HD-ORSCs (n = 15). (B) DNA fiber spreading analysis of fork progression. ORSCs from the HS and HD groups were labeled with IdU and CldU. Median ldU track lengths are indicated in red. Median ldU track lengths of HD-ORSCs are indicated by the red dotted line. Scale bar: 10 μm. (C) Immunofluorescence analysis of spontaneous γH2AX foci; percentage of HS-ORSCs (n= 11) and HD-ORSCs (n = 5) with γH2AX foci. (D) Immunofluorescence analysis of spontaneous 53BP1 foci. Percentage of HS-ORSCs (n = 9) and HD-ORSCs (n = 5) with 53BP1 foci. Horizontal lines represent the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 by Mann-Whitney U test. NS, not significant.
Because the percentage of cells in the S phase was increased in HS patients, we next asked whether this correlated to increased replication stress, which is defined as a global perturbation of the DNA replication program altering the speed of replication forks and activating the ATR-CHK1 pathway (17). To this end, we labeled ongoing DNA synthesis in HS-ORSCs and HD-ORSCs with 2 successive pulses of the thymidine analogs IdU and CldU and monitored the progression of individual forks by DNA fiber spreading (18). We observed that ldU track length was comparable in the studied ORSCs from 3 HDs, around 5 μm (Figure 5B). Remarkably, HS-ORSCs showed different patterns of ldU track length. ORSCs from 5 out of the 6 HS patients showed a much longer track length compared with HD-ORSCs. Altogether, these results indicated that the progression of replication forks was severely altered in HS-ORSCs.
Next, we asked whether the ATR/CHK1 pathway is spontaneously activated in HS-ORSCs. First, we monitored the phosphorylation of the histone variant H2AX (γH2AX) by immunohistochemistry in ORSCs amplified in vitro. γH2AX was higher in HS-ORSCs compared with HD-ORSCs, but without reaching significance (Figure 5C). Incomplete DNA synthesis during the S phase led to the formation of 53BP1 nuclear bodies in the subsequent G1 phase. Analysis of 53BP1 foci by immunocytochemistry revealed that HS-ORSCs have an increased accumulation of 53BP1 foci after in vitro amplification relative to HD-ORSCs (20.9% ± 9.3% vs. 7.5% ± 4.5%, P = 0.002; Figure 5D). We then monitored the phosphorylation of CHK1 (DNA replication stress checkpoint) and CHK2 (DNA damage checkpoint) by Western blotting after in vitro amplification. CHK1 phosphorylation was statistically increased in HS-ORSCs relative to HD-ORSCs (HS phospho-CHK1/CHK1 0.79 ± 0.84 vs. HD phospho-CHK1/CHK1 0.09 ± 0.13, P = 0.003) (Figure 6A). In contrast, we failed to detect an increased CHK2 phosphorylation in HS-ORSCs relative to HD-ORSCs (Figure 6A and Supplemental Figure 2). These results indicate that HS-ORSCs suffered from a spontaneous replication stress, activating the ATR/CHK1 pathway.
Figure 6. Activation of the DNA damage response in HS-ORSCs.

(A) Western blot analysis of the levels of CHK1. Quantification of phospho-CHK1 and phospho-CHK2 signal intensity in HS-ORSCs (n = 10) and HD-ORSCs (n = 6). (B) Immunofluorescence analysis of spontaneous phospho-CHK1 of total freshly isolated hair follicle cells from HS patients (n = 12) and the HD group (n = 7). (C) Representative images with phospho-CHK1 (green) and cytokeratin 15, an HFSC marker (red); or CD45, a hematopoietic marker (red). Horizontal lines represent the mean ± SD. **P < 0.01 by Mann-Whitney U test. NS, not significant.
To confirm these in vitro data, we analyzed CHK1 phosphorylation in total freshly isolated hair follicle cells. The analysis of CHK1 phosphorylation by immunohistochemistry revealed an increase in phospho-CHK1–positive cells in freshly isolated total hair follicles from HS patients relative to HD controls (17.8% ± 13% vs. 4.6% ± 3.3%, P = 0.0012; Figure 6B). Moreover, CHK1 phosphorylation–positive cells were negative for the leucocyte marker CD45 and positive for the HFSC marker cytokeratin-15 (Figure 6C). We also observed CHK1 phosphorylation–positive cells were negative for CD49f and CD34, both markers expressed by the lower ORS (ref. 16 and Supplemental Figure 3). Together, these data indicate that ex vivo HFSCs from HS patients, but not hematopoietic cells, constitutively exhibit a replication stress profile.
Presence of micronuclei and cytosolic ssDNA and in HS-ORSCs.
Micronuclei formation was increased upon DNA damage, and their rupture exposed DNA to pattern recognition receptors and activated the STING pathway. To detect the presence of micronuclei in ORSCs, we stained cells for lamin B1, which is an integral nuclear envelope protein and thus a reliable marker for micronuclei. Remarkably, HS-ORSCs showed an increased number of micronuclei in comparison with HD-ORSCs (6.54% ± 6.8% vs. 2.29% ± 0.7%, P = 0.04) (Figure 7A).
Figure 7. Presence of micronuclei and cytosolic ssDNA in ORSCs from HS patients.

(A) Representative image with lamin B1 (green) staining the nuclear envelope and DAPI staining DNA (blue). Percentage of HS-ORSCs (n = 9) and HD-ORSCs (n = 7) with micronuclei. White arrow shows micronuclei. Horizontal lines represent the mean ± SD. *P < 0.05 by Mann-Whitney U test. (B) Immunofluorescence analysis of cytoplasmic ssDNA in HS-ORSCs (n = 6) and HD-ORSCs (n = 3). (C) Immunofluorescence analysis of cytoplasmic ssDNA in interfollicular keratinocytes isolated from the same HS patients analyzed in B. Scale bars: 10 μm.
Nuclear DNA damage can also generate ssDNA in the cytoplasm in a less understood process (19). We monitored the presence of cytosolic ssDNA by immunofluorescence microscopy. We detected ssDNA in the cytosol of HS-ORSCs (n = 6; Figure 7B) but not in HD-ORSCs (n = 3). This signal is specific because it decreased after treatment of HS-ORSCs with S1 nuclease (data not shown). Interestingly, no accumulation of ssDNA was detected in the cytoplasm of interfollicular keratinocytes isolated from the same HS patients (n = 3; Figure 7C). These results suggest that in HS patients the accumulation of cytosolic ssDNA is ORSC specific.
Expression of proinflammatory type I IFN via the IFI16/STING pathway in HS-ORSCs.
Cytosolic DNA fragments and micronuclei activate the STING pathway to induce expression of proinflammatory type I IFN. To determine whether the DNA-sensing adaptor STING is involved in type I IFN production observed in ORSCs isolated from HS patients, ORSCs were transfected with STING siRNA (siSTING) and IFN-β transcripts were analyzed because IFN-β is the first target gene of STING pathway activation. In HS patients, ORSCs lacking STING expressed reduced levels of IFN-β transcripts compared with ORSCs transfected with a scramble siRNA (Figure 8A). By contrast, no difference in IFN-β transcript levels was noted in the absence of STING in ORSCs from healthy donors. We also assessed OAS1 and IP-10 as ISGs in ORSCs lacking STING. In HS patients, a decrease in ISG transcripts was observed between ORSCs lacking STING and ORSCs transfected with a scramble siRNA. As expected, no change in ISG transcripts was noted in the absence of STING in ORSCs from the HD group. STING activated transcription factor IRF3: Western blot analysis of IRF3 phosphorylation at Ser396 (p-IRF3) revealed an inhibition of p-IRF3 in HS-ORSCs. In healthy donors, no modification of p-IRF3 was observed between HD-ORSCs lacking STING and HD-ORSCs transfected with a scramble siRNA (Figure 8B).
Figure 8. The IFI16/STING pathway induces type I IFN production in HS-ORSCs.

(A) mRNA levels of IFN-β, IP-10, and OAS1 in ORSCs transfected with siSTING. HS-ORSCs (n = 7) or HD-ORSCs (n = 4) were transfected with siRNA-STING for 2 days before mRNA quantification and Western blot analysis were performed. Red line shows siCtrl level expression for each gene. (B) Western blot analysis of the levels of phospho-IRF3 in ORSCs transfected with siSTING or siCtrl. Quantification of phospho-IRF3 signal intensity in HS-ORSCs (n = 7) and HD group (n = 5). (C) Levels of IFN-β, IP-10, and OAS1 mRNA in ORSCs transfected with siRNA-IFI16. HS-ORSCs (n = 7) were transfected with siRNA-IFI16 for 2 days before mRNA quantification and Western blot analysis were performed. (D) Western blot analysis of the levels of phospho-IRF3 in ORSCs transfected with siIFI16 or siCtrl. Quantification of phospho-IRF3 signal intensity in HS-ORSCs (n = 7). *P < 0.05; **P < 0.01 by Mann-Whitney U test.
Since cGAS cooperates with the DNA binding protein IFI16 during DNA sensing in human keratinocytes, we analyzed IFI16 involvement in type I IFN production of HS-ORSCs. Four out of 7 ORSCs lacking IFI16 expressed reduced IFN-β transcripts compared with ORSCs transfected with a scramble siRNA (Figure 8C). We observed a heterogeneous response for the other tested ISGs, suggesting that other sensors may be involved in STING activation. However, IRF3 phosphorylation in HS-ORSCs depleted of IFI16 was decreased in all cases compared with a scramble siRNA (Figure 8D). The redistribution of a small pool of cellular IFI16 from the nucleus to the cytoplasm is a critical event in stimulation of DNA sensitivity in keratinocytes (14). We observed an increased pool of IFI16 localizing to the cytoplasm in HS-ORSCs compared with HD-ORSCs (5.1% ± 1.4% vs. 3.1% ± 1.7%, P = 0.02), consistent with a role in the detection of cytoplasmic DNA (Figure 9A). Last, we noticed a decrease of cGAS protein expression in ORSCs lacking IFI16 (Figure 9B), suggesting that cGAS and IFI16 cooperate to form a DNA receptor complex as observed in human macrophages (20). Altogether, these data suggested that cytosolic ssDNA in HS-ORSCs induced type I IFN production via the IFI16-STING pathway.
Figure 9. Activation of IFI-16/STING pathway in HS-ORSCs.

(A) Immunofluorescence analysis of cytoplasmic IFI16. Percentage of HS-ORSCs (n = 13) and HD-ORSCs (n = 11) with cytoplasmic IFI16. (B) Western blot analysis of the levels of cGAS in ORSCs transfected with siIFI16 or siCtrl. Quantification of cGAS signal intensity in HS-ORSCs (n = 6). (C) DNA fiber spreading analysis of fork progression in HS-ORSCs transfected with siSTING. HS-ORSCs (n = 2) were transfected with siRNA-STING for 2 days before DNA fiber spreading was performed. *P < 0.05; ****P < 0.0001 by Mann-Whitney U test.
Finally, we asked whether the faster progression of replication forks observed in the majority of HS patients could be due to the constitutive activation of the STING pathway. Remarkably, we observed that ldU track length in the ORSCs from 2 HS patients studied was decreased to the length observed in HD-ORSCs after STING depletion (Figure 9C). These results suggest that STING could modulate replication fork progression.
Discussion
HS is a frequent and nonelucidated disease involving the pilosebaceous-apocrine unit as a primary pathogenic component. Here, for the first time to our knowledge, we identified major homeostatic abnormalities of the HFSC compartment in HS patients. Clonotypic analysis and HFSC characterization demonstrated an increased number of proliferating progenitor cells and a loss of quiescent stem cells associated with a spontaneous replication stress in HS patients. We showed that these cells accumulated cytoplasmic ssDNA and micronuclei and induced IFN synthesis through the IFI16/STING pathway. To our knowledge, such alterations of the HFSC compartment are unique and have not been described in other hair follicle pathologies. In reversible types of alopecia such as alopecia areata, inflammation targets hair follicle progenitor cells but spares HFSCs (7), whereas a defect in conversion of HFSCs to progenitor cells plays a role in the pathogenesis of androgenetic alopecia (21). Our results suggest that the lack of quiescent stem cells is due to increased stem cell differentiation and not to destruction of HFSCs as observed in scarring alopecia associated with permanent hair loss (22).
Another original aspect of our study is that these perturbations of cellular homeostasis were associated with spontaneous replication stress, as illustrated by alterations of replication fork progression, activation of the ATR/CHK1 pathway, and accumulation of cytosolic ssDNA. These alterations were specific to HS-ORSCs since we did not observe CHK1 phosphorylation in hematopoietic cells from patients and ssDNA accumulation in the cytoplasm of interfollicular keratinocytes. These findings suggest a physiopathological process tipping the balance toward cell-intrinsic properties in controlling replication stress and the DNA damage response in skin rather than the local microenvironment. However, a self-perpetuating mechanism secondary to local inflammation, which leads to the production of ROS that may also contribute to genotoxic insults and genomic instability, could not be completely ruled out. Globally, our observation is reminiscent of recent data in mice (23) showing that multipotent HFSCs are more resistant to DNA damage–induced cell death compared with other cells of the epidermis because they express higher levels of the antiapoptotic protein Bcl2 (24). Moreover, murine HFSCs display efficient DNA repair and enhanced nonhomologous end joining (NHEJ) activity (23), which is more error prone than homology-mediated DNA double-strand break repair pathways. We could speculate that a higher resistance of human HFSCs to DNA damage–induced apoptosis and increased NHEJ activity allows for the accumulation of mutations leading to increased genomic instability.
Recent research has provided solid evidence that replication stress and genomic DNA damage trigger inflammatory response through the accumulation of cytosolic DNA fragments and the activation of STING (12, 19). Cytosolic DNA is either generated by aberrant DNA repair activities or by the rupture of micronuclei, which form during mitosis as a consequence of broken or lagging chromosomes (11, 25, 26). Interestingly, both micronuclei and ssDNA fragments were detected in the cytoplasm of HS-ORSCs. The increased frequency of micronuclei in HS-ORSCs could result from their increased replication stress interfering with proper chromosome segregation. Alternatively, it could reflect the fact that HFSCs display increased NHEJ (25), an error-prone DNA repair pathway generating dicentric chromosomes. Cytoplasmic DNA is normally degraded by the TREX1 exonuclease, the loss of which is associated with chronic type I IFN induction (17). Our hypothesis is that, under replication stress conditions, chromosome missegregation induced either by replication completion defects or by NHEJ-mediated chromosome rearrangements increases the accumulation of cytosolic DNA, which overwhelms the buffering capacity of TREX1.
As reported for other pathological mechanisms, our results showed that cytosolic DNA of endogenous origin activates the STING pathway to induce type I IFN production in ORSCs. Indeed, excessive cGAS-STING–dependent type I IFN secretion has been involved in myocardial infarction (27) and in fibrotic interstitial lung disease (28). Genetic deficiencies that compromise DNA damage response functions also induce type I IFN and lead to autoinflammatory disease such as the immunodeficiency syndrome ataxia-telangiectasia due to mutations in the ATM gene (10). Aicardi-Goutières syndrome is driven by chronic type I IFN signaling caused by recessive mutation in one of a few genes involved in nucleic acid metabolism (10).
One of the most striking observations of our study is the fact that replication fork speed was increased by 2- to 3-fold in HS-ORSCs relative to HD-ORSCs. It is generally believed that replication stress is associated with slower replication forks, but a recent study from the laboratory of Jiri Bartek has shown that replication stress can also be caused by faster replication forks (29). Moreover, this study showed that an increase in fork rate above 40% is associated with increased DNA damage, which is the case for most HS-ORSCs. Finally, Lorenza Penengo (University of Zurich – Irchel Campus, Zurich, Switzerland, personal communication and ref. 30) has found that the ubiquitin-like protein ISG15 interacts physically with components of the replisome and accelerates forks. Since ISG15 is the most highly expressed IFN-stimulated gene in HS-ORSCs relative to HD-ORSCs (Figure 1C), these data provide a direct mechanistic link between inflammation, replication stress, and DNA damage in HS-ORSCs.
We identified IFI16 as a DNA sensor critical for the full activation of STING in HS-ORSCs. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes (15). Human keratinocytes normally do not respond to cytoplasmic DNA with an innate immune response. The redistribution of a small pool of cellular IFI16 from the nucleus to the cytoplasm is a critical event in stimulation of DNA sensitivity in keratinocytes (14), and TNF treatment led to accumulation of IFI16 in the cytoplasm. We could argue that IFI16 relocalizes to the cytoplasm because of the inflammation and binds ssDNA to activate the STING pathway.
From a clinical standpoint, our results may have significant implications. First, as discussed above, our results evoke a vicious cycle triggered by HFSC replicative stress leading to chronic inflammation. Second, these results are also reminiscent of the mechanisms involved in Notch signaling pathway defects, the most frequent genetic predisposition reported in familial HS (31). Several manipulated cell line models inactivating the Notch pathway were shown to severely affect the proliferation and differentiation of keratinocyte cell lines (32, 33). Notch signaling is also required for postnatal hair cycle homeostasis because it maintains proper proliferation and differentiation of HFSCs (34). None of the patients enrolled in this study displayed mutations in the γ-secretase complex, suggesting that the homeostatic abnormalities of the HFSC compartment are not associated with familial cases. Third, generation of ORSCs seems a useful and relevant model to understand the pathophysiology of HS. The transcriptomic profile of these cells revealed pathways similar to previous reports from our group and others showing enrichment for keratinocyte differentiation, epidermis development pathways (8, 35), and upregulation of IFN pathways (36, 37) on whole skin isolated from HS lesions. Finally, recent studies have identified a phenotypic heterogeneity in HS patients, suggesting heterogeneous pathogenic pathways. Our results may help to distinguish 2 groups of patients characterized by the presence or absence of a replication stress in HFSCs. We propose here to define patients with a replicative stress as patients with at least 1 of the following criteria: (a) accumulation of cells in the S phase (>25%); (b) an alteration of replication fork progression; (c) an increased proportion of cells with γH2AX foci (>9%). It is noteworthy that 6 out of 8 patients (75%) were men in the group with a replication stress, whereas men represented only 3 out of 10 patients (30%) in the other group. This low number of patients is not sufficient to draw a definitive conclusion. However, if true, this may lead to a new stratification of patients and the possibility of targeted therapeutics. Indeed, patients with replicative stress could benefit from treatment targeting either the cell cycle, such as retinoids, or inhibitors of the JAK/STAT pathway, which could help regulate the expression of inflammatory factors.
Methods
Patient characteristics.
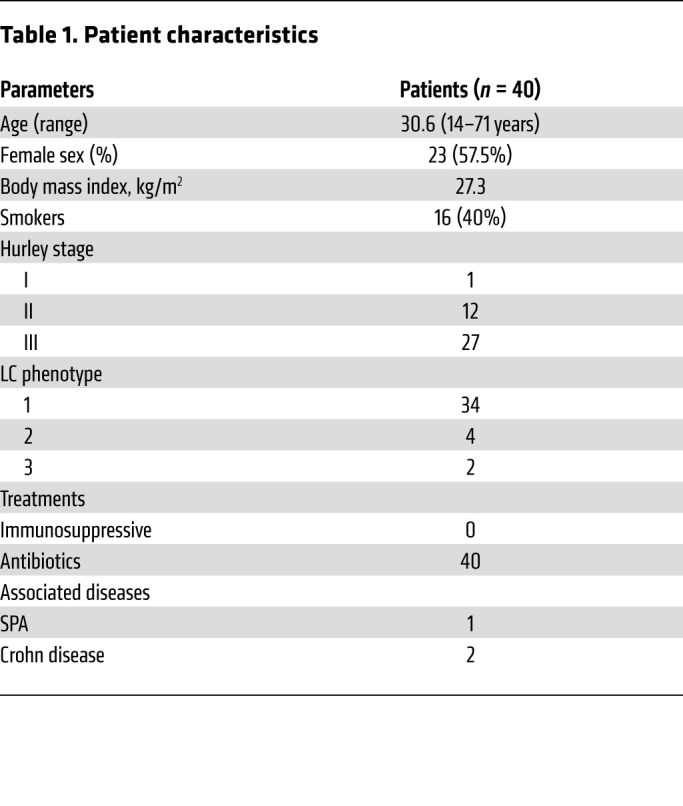
Skin samples were collected from 40 patients with HS who underwent surgery to unroof lesions and from 27 healthy patients after abdominoplasty or brachioplasty at the plastic surgery department of Henri Mondor Hospital. The mean age of the 40 patients with HS was 30.6 years (range: 14–71 years); 23 were women (57.5%) and 16 were smokers (40%). The mean body mass index was 27.3 kg/m2. One, 12, and 27 patients had Hurley stage I, II, and III disease, respectively. No topical treatment was introduced in patients with HS. None of the patients enrolled in this study were under immunosuppressive drug therapy (Table 1). The mean age of the 27 healthy donors was 34.9 years (range: 19–57 years); 22 were women and 5 were men. All healthy individuals had no history of skin disease and no underlying malignancy.
Table 1. Patient characteristics.
Skin dissociation.
Skin samples were obtained after abdominoplasty or brachioplasty for healthy donors and after axillary or perineal surgery for HS patients. Hair-rich skin areas were collected from surgery waste and were processed as described previously (38, 39). Two types of analysis were performed. Cells isolated from hair follicles were directly studied for hair follicle phenotype and phospho-CHK1 staining (total freshly isolated hair follicle cells) or the cells were further expanded on a feeder layer (ORSCs).
ORS cell culture.
When ORSCs were subconfluent, the feeder layer was removed with PBS-EDTA (0.71 mM) and ORSCs were detached after trypsin (TrypLe Express 1X, Life Technologies) incubation. Cells were seeded in defined medium without FCS (Epilife, Life Technologies) or complete DMEM F12 medium on irradiated 3T3 J2 feeder layers (provided by Yann Barrandon, Swiss Federal Institute of Technology Lausanne. Cells were studied at passage 1/2 except for 53BP1 analysis performed at passage 3/4.
Microarray sample preparation.
Total RNA was purified from cells using RNeasy Plus Micro Kit (QIAGEN Inc.), and globin mRNAs were removed by GLOBINclear-Human Kit (Ambion). RNAs were then quantified with Quant-iT RiboGreen RNA Assay Kit (Thermo Fisher Scientific) before quality control on the Bioanalyzer (Agilent). In vitro transcription was generated from RNA using Ambion Illumina TotalPrep RNA Amplification Kits (Applied Biosystems/Ambion). Labelled cRNA was hybridized onto Illumina Human HT-12v4 BeadChips. Quality controls were processed using GenomeStudio software (Illumina). Differentially expressed genes were identified using quantile normalized data as input to gene-specific analysis (GSA) (Partek Flow). Hierarchical clustering was performed using the Euclidean distance method. Only genes with adjusted P values (FDR) of 0.2 or less and a fold change of 1.5 or more were considered as differentially expressed. Functional enrichment analysis of differentially expressed genes was investigated using Ingenuity Pathway software (Ingenuity Systems). All microarray data have been deposited in the MIAME-compliant database Gene Expression Omnibus (GEO accession number: GSE147586).
Clonal expansion.
Clonal expansion was performed at P1, and 5000 ORSCs were seeded on a 0.5 × 106 irradiated 3T3 J2 feeder layer in a 60-mm petri dish and grown for 14 days. ORSCs were fixed with formalin for 10 minutes and immediately colored with rhodamine for 20 minutes. Dishes were washed and left to dry. ORS colonies were analyzed and counted with Image J software.
siRNA.
siRNAs used in this study were the following: siSTING (CUGCAUCCAUCCAUCCCGUdTdT; Sigma-Aldrich, Hs01_00031038) and siIFI16 (L-020004-00-0005, ON-TARGETplus Human IFI16 siRNA SMARTpool; Horizon Discovery). siRNAs were used at a final concentration of 10 μM.
siRNA transfection.
ORSCs were seeded in defined medium without FCS (Epilife, Life Technologies) at P2/P3 and transfected by siRNA at P3/P4. ORSCs were transfected successively at day 0 and day 1 with INTERFERin (PolyPlus Transfection) according to the manufacturer’s protocol. ORSCs were lysed 48 hours after transfection to analyze protein and mRNA expression.
Cell cycle analysis.
ORSCs were grown successively at P2 and P3 in defined medium without FCS (Epilife, Life Technologies). After cells were half-confluent, they were fixed in PBS/70% ethanol on ice. ORSCs were resuspended in FxCycle PI/RNase staining solution according to the manufacturer’s protocol (Invitrogen) and incubated 30 minutes in dark at room temperature before FACS analysis.
Hair follicle phenotype analysis.
Freshly isolated total hair follicle cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Invitrogen) and then fixed with Foxp3/Transcription Factor Staining Buffer (eBioscience). Cells were stained with primary mouse anti–human cytokeratin 15 (LHK15, Thermo Fisher Scientific) and secondary FITC goat anti–mouse Ig (BD Pharmingen) antibodies. Surface staining was performed with a mix of PE mouse anti–human CD34 (8G12, BD Biosciences), APC-H7 mouse anti–human CD45 (2D1, BD Biosciences), Pe-Cy7 mouse anti–human CD117 (104D2, BD Biosciences), BV421 mouse anti–human CD200 (MRC OX-104, BD Biosciences), and PerCp-Cy5.5 rat anti–human CD49f (GoH3, BD Pharmingen) antibodies. Cells were run on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software, version 10.2 (FlowJo LLC).
RNA extraction of total hair follicle cells.
Skin fragments were incubated with Dispase (StemCell Technologies) overnight at 4°C. The next day, 20 hair follicles were collected and put in TRIzol reagent. Then, RNA extraction was performed with Direct-Zol RNA miniPrep KIT (Zymo Research) according to the manufacturer’s protocol.
RNA extraction and reverse transcriptase reaction.
ORSCs were lysed with RLT Plus Buffer (QIAGEN Inc.) and RNA extraction was performed according to the manufacturer’s protocol (RNeasy Plus Mini Kit, QIAGEN Inc.). RNA was converted into cDNA with QuantiTect Reverse Transcription Kit (QIAGEN Inc.) for IFN-β detection and with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) for other genes’ expression.
Quantitative PCR.
Real-time PCR of cDNA samples involved custom-made primer sets with the Brilliant II SYBR Green QPCR master mix (Agilent Technologies) or Quantitect SYBR Green PCR Kit (QIAGEN Inc.) according to the manufacturer’s protocol. Real-time PCR was performed on an MX3000P (Agilent) device. The GAPDH or OAZ1 mRNA level was used for normalization. The mRNA relative expression (ΔCt) of each gene was calculated by comparison with that of GAPDH or OAZ1.
Primers.
Primers used in this study were the following: IFNB (Forward GAATGGGAGGCTTGAATACTGCCT; Reverse TAGCAAAGATGTTCTGGAGCATCTC), IP-10 (Forward GTGTTGAGATCATTGCTACAA; Reverse TCGATTCTGGATTCAGACAT), IFI27 (Forward CTTTGGGCCACGGAATT; Reverse CCACACTGGTCACTGATG), MX-1 (Forward TCCAAGGAGGTGCAGGAGAA; Reverse TTCCTTTGCAAAGTAAGCTCTAA), GAPDH (Forward CTGGCGTCTTCACCATGG; Reverse CATCACGCCACAGTTTCCCGG), OAZ1 (Forward ACTTATTCTACTCCGATGATC; Reverse GAGAATCCTCGTCTTGTC). OAS1 primers were purchased from QIAGEN Inc. (Quantitect Primer Assay, QT00099134).
Western blot.
Lysates were resolved by SDS-PAGE and gels were transferred with an iBlot device (Thermo Fisher Scientific). Membranes were incubated with PBS/0.1% Tween 20 with 5% nonfat milk for 2 hours and then incubated with primary antibody overnight at 4°C according to the manufacturer’s protocol. Then, membranes were incubated with secondary antibodies in PBS-Tween 0.1%-5% nonfat milk for 1 hour at room temperature and revealed with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) by chemiluminescence.
Antibodies.
Western blot antibodies are summarized in Supplemental Table 1.
DNA fiber spreading.
DNA fiber spreading was performed as described previously (40). In brief, subconfluent cells were sequentially labeled, first with 10 μM 5-iodo-2′-deoxyuridine (IdU) and then with 100 μM 5-chloro-2′-deoxyuridine (CldU) for 30 minutes. Cells were loaded onto a glass slide (StarFrost), lysed with spreading buffer (200 mM Tris-HCl pH 7.5, 50 mM EDTA, 0.5% SDS), and run down the slides slowly. Slides were fixed in 3:1 methanol/acetic acid for 10 minutes and allowed to dry. Glass slides were processed for immunostaining with mouse anti-BrdU to detect IdU, rat anti-BrdU to detect CldU, and corresponding secondary antibodies conjugated to various Alexa Fluor dyes. Nascent DNA fibers were visualized by using immunofluorescence microscopy (Zeiss ApoTome). The acquired DNA fiber images were analyzed by using MetaMorph Microscopy Automation and Image Analysis Software (Molecular Devices). Statistical analysis was performed with Prism (GraphPad Software) using a 2-sided Mann-Whitney U test.
Immunohistochemistry.
For the study of proliferation in the epidermis and hair follicles, Ki67 staining was performed on 3-μm-thick skin sections of 5 skin samples with HS lesions and 4 control samples (skin specimens of axillary scars resected from age-matched patients). We used the SP6 clone (Zytomed) with a 1:50 dilution after antigen retrieval at pH 6. The procedure was done using the BOND-III device (Menarini-Leica) with diaminobenzidine chromogen.
Immunocytochemistry.
Total hair follicle cells were cytocentrifuged for 5 minutes at 600 g and dried overnight. Cytospins were fixed in PBS/4% paraformaldehyde (PFA) and kept at –20°C until use. ORS cells were seeded on culture slides and when subconfluence was reached, they were fixed in PBS/4% PFA and kept at –20°C until use. Cells were permeabilized and then blocked as described in Supplemental Table 2. Slides were incubated with the primary antibody in a wet chamber. Slides were then washed 3 times for 5 minutes with PBS/0.1% Tween 20 and incubated with the secondary antibody. Slides were washed 3 times for 5 minutes with PBS/0.1% Tween 20 and mounted with Prolong Gold antifade with DAPI (Molecular Probes).
Statistics.
Groups were compared using the 2-sided nonparametric Mann-Whitney U test. A P value of less than 0.05 was considered significant. All statistical analyses were performed using Prism (version 7.0, GraphPad Software).
Study approval.
This study was approved by the ethics committee of Henri Mondor Hospital (Créteil) in agreement with the Declaration of Helsinki, and written informed consent was received from participants before inclusion in the study.
Author contributions
SH, PP, and YL conceived and supervised the study. SH, PP, YL, CO, YLL, FJL, HH, M Benkirane, NO, YB, and JDL designed the experiments and analyzed the data. CO, YLL, FJL, YB, NO, PT, CL, and M Boniotto performed the experiments. BH, CH, and PW recruited the participants and collected the samples. SH, PP, CO, and YL wrote the manuscript with contributions from all authors.
Supplementary Material
Acknowledgments
This work was supported by the French Society for Dermatology (SFD) and the Labex Vaccine Research Institute (VRI) (Investissements d’Avenir program managed by the ANR under reference ANR-10-LABX-77-01). CO was supported by the DIM One Health Ile de France. Work in the PP lab is supported by grants from INCA (INCA_13934), the Ligue Contre le Cancer (Certified Team), and the MSDAvenir fund. We thank the study volunteers for their participation. We are grateful to Aurélie Wiedemann from the immunomonitoring platform, VRI, for technical assistance with flow cytometry. The platform for FISH analysis of the Department of Pathology, Henri Mondor Hospital, Créteil, France, is using the Pathscan Excilone and is supported by the Lymphoma Academic Research Organisation (LYSARC).
Version 1. 04/02/2020
In-Press Preview
Version 2. 06/08/2020
Electronic publication
Version 3. 07/01/2020
Print issue publication
Version 4. 10/12/2020
Corrected reference 30
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(7):3777–3790.https://doi.org/10.1172/JCI131180.
Contributor Information
Cindy Orvain, Email: cindy.orvain16@gmail.com.
Yea-Lih Lin, Email: Yea-Lih.Lin@igh.cnrs.fr.
Francette Jean-Louis, Email: francette.jean-louis@inserm.fr.
Hakim Hocini, Email: hakim.hocini@inserm.fr.
Barbara Hersant, Email: barbara.hersant@gmail.com.
Yamina Bennasser, Email: yamina.bennasser@igh.cnrs.fr.
Nicolas Ortonne, Email: nicolas.ortonne@hmn.aphp.fr.
Claire Hotz, Email: claire.hotz@gmail.com.
Pierre Wolkenstein, Email: pierre.wolkenstein@hmn.aphp.fr.
Michele Boniotto, Email: michele.boniotto@inserm.fr.
Pascaline Tisserand, Email: pascaline.tisserand@inserm.fr.
Monsef Benkirane, Email: monsef.benkirane@igh.cnrs.fr.
Philippe Pasero, Email: philippe.pasero@igh.cnrs.fr.
References
- 1.Revuz J. Hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2009;23(9):985–998. doi: 10.1111/j.1468-3083.2009.03356.x. [DOI] [PubMed] [Google Scholar]
- 2.Matusiak Ł. Profound consequences of hidradenitis suppurativa: a review. Br J Dermatol. doi: 10.1111/bjd.16603. doi: 10.1111/bjd.16603. [published online May 9, 2018]. [DOI] [PubMed] [Google Scholar]
- 3.Revuz JE, et al. Prevalence and factors associated with hidradenitis suppurativa: results from two case-control studies. J Am Acad Dermatol. 2008;59(4):596–601. doi: 10.1016/j.jaad.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 4.Garg A, Kirby JS, Lavian J, Lin G, Strunk A. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153(8):760–764. doi: 10.1001/jamadermatol.2017.0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen W, Plewig G. Should hidradenitis suppurativa/acne inversa best be renamed as “dissecting terminal hair folliculitis”? Exp Dermatol. 2017;26(6):544–547. doi: 10.1111/exd.13211. [DOI] [PubMed] [Google Scholar]
- 6.van der Zee HH, et al. Alterations in leucocyte subsets and histomorphology in normal-appearing perilesional skin and early and chronic hidradenitis suppurativa lesions. Br J Dermatol. 2012;166(1):98–106. doi: 10.1111/j.1365-2133.2011.10643.x. [DOI] [PubMed] [Google Scholar]
- 7.Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med. 1999;341(7):491–497. doi: 10.1056/NEJM199908123410706. [DOI] [PubMed] [Google Scholar]
- 8.Hotz C, et al. Intrinsic defect in keratinocyte function leads to inflammation in hidradenitis suppurativa. J Invest Dermatol. 2016;136(9):1768–1780. doi: 10.1016/j.jid.2016.04.036. [DOI] [PubMed] [Google Scholar]
- 9.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 11.Coquel F, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature. 2018;557(7703):57–61. doi: 10.1038/s41586-018-0050-1. [DOI] [PubMed] [Google Scholar]
- 12.Coquel F, Neumayer C, Lin YL, Pasero P. SAMHD1 and the innate immune response to cytosolic DNA during DNA replication. Curr Opin Immunol. 2019;56:24–30. doi: 10.1016/j.coi.2018.09.017. [DOI] [PubMed] [Google Scholar]
- 13.Pasero P, Vindigni A. Nucleases acting at stalled forks: how to reboot the replication program with a few shortcuts. Annu Rev Genet. 2017;51:477–499. doi: 10.1146/annurev-genet-120116-024745. [DOI] [PubMed] [Google Scholar]
- 14.Chiliveru S, et al. Inflammatory cytokines break down intrinsic immunological tolerance of human primary keratinocytes to cytosolic DNA. J Immunol. 2014;192(5):2395–2404. doi: 10.4049/jimmunol.1302120. [DOI] [PubMed] [Google Scholar]
- 15.Almine JF, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. 2017;8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue K, et al. Differential expression of stem-cell-associated markers in human hair follicle epithelial cells. Lab Invest. 2009;89(8):844–856. doi: 10.1038/labinvest.2009.48. [DOI] [PubMed] [Google Scholar]
- 17.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A. DNA fiber analysis: mind the gap! Meth Enzymol. 2017;591:55–82. doi: 10.1016/bs.mie.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 19.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299. doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jønsson KL, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. 2017;8:14391. doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garza LA, et al. Bald scalp in men with androgenetic alopecia retains hair follicle stem cells but lacks CD200-rich and CD34-positive hair follicle progenitor cells. J Clin Invest. 2011;121(2):613–622. doi: 10.1172/JCI44478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito M, et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med. 2005;11(12):1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 23.Blanpain C, Mohrin M, Sotiropoulou PA, Passegué E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell. 2011;8(1):16–29. doi: 10.1016/j.stem.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Sotiropoulou PA, et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol. 2010;12(6):572–582. doi: 10.1038/ncb2059. [DOI] [PubMed] [Google Scholar]
- 25.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackenzie KJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King KR, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23(12):1481–1487. doi: 10.1038/nm.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benmerzoug S, et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat Commun. 2018;9(1):5226. doi: 10.1038/s41467-018-07425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018;559(7713):279–284. doi: 10.1038/s41586-018-0261-5. [DOI] [PubMed] [Google Scholar]
- 30.Raso MC, et al. Interferon-stimulated gene 15 accelerates replication fork progression inducing chromosomal breakage. J Cell Biol. 2020;219(8):e202002175. doi: 10.1083/jcb.202002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang B, et al. Gamma-secretase gene mutations in familial acne inversa. Science. 2010;330(6007):1065. doi: 10.1126/science.1196284. [DOI] [PubMed] [Google Scholar]
- 32.Xiao X, He Y, Li C, Zhang X, Xu H, Wang B. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3-kinase/AKT signalling pathways. Br J Dermatol. 2016;174(3):522–532. doi: 10.1111/bjd.14223. [DOI] [PubMed] [Google Scholar]
- 33.Cao L, Morales-Heil DJ, Roberson EDO. Nicastrin haploinsufficiency alters expression of type I interferon-stimulated genes: the relationship to familial hidradenitis suppurativa. Clin Exp Dermatol. 2019;44(4):e118–e125. doi: 10.1111/ced.13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aubin-Houzelstein G. Notch signaling and the developing hair follicle. Adv Exp Med Biol. 2012;727:142–160. doi: 10.1007/978-1-4614-0899-4_11. [DOI] [PubMed] [Google Scholar]
- 35.Gauntner TD. Hormonal, stem cell and Notch signalling as possible mechanisms of disease in hidradenitis suppurativa: a systems-level transcriptomic analysis. Br J Dermatol. 2019;180(1):203–204. doi: 10.1111/bjd.17093. [DOI] [PubMed] [Google Scholar]
- 36.Hoffman LK, et al. Integrating the skin and blood transcriptomes and serum proteome in hidradenitis suppurativa reveals complement dysregulation and a plasma cell signature. PLoS One. 2018;13(9):e0203672. doi: 10.1371/journal.pone.0203672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shanmugam VK, Jones D, McNish S, Bendall ML, Crandall KA. Transcriptome patterns in hidradenitis suppurativa: support for the role of antimicrobial peptides and interferon pathways in disease pathogenesis. Clin Exp Dermatol. 2019;44(8):882–892. doi: 10.1111/ced.13959. [DOI] [PubMed] [Google Scholar]
- 38.Aasen T, Izpisúa Belmonte JC. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat Protoc. 2010;5(2):371–382. doi: 10.1038/nprot.2009.241. [DOI] [PubMed] [Google Scholar]
- 39.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6(3):331–343. doi: 10.1016/S0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 40.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140(6):1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.