

Abstract
Pediatric cancers, particularly high-risk solid tumors, urgently need effective and specific therapies. Their outlook has not appreciably improved in decades. Immunotherapies such as immune checkpoint inhibitors offer much promise, but most are only approved for use in adults. Though several hundred clinical trials have tested immune-based approaches in childhood cancers, few have been guided by biomarkers or clinical-grade assays developed to predict patient response and, ultimately, to help select those most likely to benefit. There is extensive evidence in adults to show that immune profiling has substantial predictive value, but few studies focus on childhood tumors, because of the relatively small disease population and restricted use of immune-based therapies. For instance, only one published study has retrospectively examined the immune profiles of pediatric brain tumors after immunotherapy. Furthermore, application and integration of advanced multiplex techniques has been extremely limited. Here, we review the current status of immune profiling of pediatric solid tumors, with emphasis on tumor types that represent enormous unmet clinical need, primarily in the context of immune checkpoint inhibitor therapy. Translating optimized and informative immune profiling into standard practice and access to personalized combination therapy will be critical if childhood cancers are to be treated effectively and affordably.
Introduction to immune profiling
The immune system is critical for detection and elimination of transformed cells. However, it can promote tumor development through selection of less immunogenic, unstable variants and suppression of antitumor responses (1, 2). The tumor microenvironment is a complex and dynamic network of tumor cells, fibroblasts, endothelial cells, and immune cells, the last of which include T and B lymphocytes, monocytes and macrophages, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), and natural killer (NK) cells (3). These tumor-infiltrating leukocytes (TILs) play diverse roles in tumorigenesis. Different subsets support or suppress growth and metastasis, through direct interactions and production of soluble factors including cytokines, chemokines, and growth factors (4, 5).
Quantification of type, number, and location of TILs within the tumor has considerable prognostic value (6). Infiltration of CD8+ T cells or NK cells is associated with improved outcomes in many adult cancers, while myeloid cells such as MDSCs have a negative correlation with survival (7, 8). These analyses form the basis of immune profiling, i.e., the identification of profiles or signatures that predict patient outcomes (9, 10). With recent developments in technology, immunoprofiling has evolved from interrogating one marker by immunohistochemistry (IHC) to investigating many subsets and phenotypes by multiplex IHC and flow cytometry, and hundreds of immune-related genes by sequencing (11–13). Novel techniques that interrogate gene and protein expression at the single-cell level, including single-cell RNA sequencing (14–16), T cell receptor (TCR) sequencing (17, 18), and mass cytometry (19–22), have revealed important insights into the diversity, specificity, and complexity of, and spatial relationships between, TILs in adult cancers, which could be applied to children (Figure 1).
Figure 1. Techniques used for immunoprofiling.

IHC, immunohistochemistry; mIHC, multiplex IHC; RNA-Seq, RNA sequencing; TCR-Seq, T cell receptor sequencing.
Adult tumors can be classified into three basic immune profiles (23). Immune-inflamed, or “hot,” tumors have considerable infiltration of cytotoxic T cells and other TILs into the tumor parenchyma and express proinflammatory cytokines, chemokines, and immune checkpoints. Immune-altered “intermediate” tumors are characterized by restriction of T cells to the surrounding stroma and production of chemokines and vascular and tumor-derived inhibitory factors (ref. 24 and Figure 2). For example, TGF-β is a key mediator of immune suppression and is associated with the immune-altered/excluded phenotype (25). Another immunosuppressive mediator, indoleamine 2,3-dioxygenase (IDO), is expressed by DCs and has a role in controlling inflammation (26); increased IDO expression is associated with poor prognosis in multiple cancer types (27).
Figure 2. Multiplex IHC in melanoma using two different antibody panels to immunoclassify solid tumors.

(A and B) Immuno-infiltrated tumors. (C and D) Immuno-altered tumors. A and C include CD4+, CD8+, FoxP3+CD4+ Treg, SOX10+, PD-L1, and DAPI. B and D include CD3+, CD20+, CD11c+, PD-L1+, SOX10+, CD68+, and DAPI. Original magnification of OPAL images, ×200.
Immune-desert “cold” tumors lack substantial CD8+ T cell infiltration altogether and are populated by antiinflammatory myeloid cells and regulatory T cells, which express cytokines associated with immune suppression, tolerance, and homeostasis (23). Even this basic level of tumor analysis can help predict which adult patients are most likely to respond to immunotherapies (24). A hot tumor is more likely to respond to immune checkpoint inhibitors than a cold tumor (23).
Immune checkpoint inhibitors are one of the most important therapeutic breakthroughs for cancer treatment. By blocking inhibitory receptor/ligand interactions, these can restore innate or adaptive immune responses against tumor cells. Here, we focus primarily on checkpoint inhibitors that restore the adaptive T cell response. Several antibodies are currently in the clinic, including those specific for programmed death-1 (PD-1) and its ligand (PD-L1) and cytotoxic T lymphocyte–associated protein-4 (CTLA-4); or in development, including lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin- and mucin domain–containing-3 (TIM-3), and B7-H3 and T cell immunoreceptor with Ig and ITIM domains (TIGIT) (28). Anti–PD-1 has shown the greatest clinical efficacy across a range of adult cancer types, and response data are now available for large cohorts (29). Retrospective analyses suggest it may be possible to prospectively use immune profiling to identify patients who are likely to respond to anti–PD-1. For example, in melanoma, anti–PD-1 is more effective in tumors with high CD8+ T cell infiltration, a T cell–inflamed gene expression profile (GEP), and PD-L1 expression (30, 31). This suggests that a preexisting immune response in the tumor is required for anti–PD-1 therapy to work (32). In addition to the PD-1/PD-L1 axis, B7-H3 is an immune checkpoint that inhibits cytotoxic T cell function (33). B7-H3 is expressed at higher levels in adult cancers and is associated with a worse prognosis (34, 35).
Although immune profiling can predict adult patient responses to immunotherapy, application to the clinic is limited. The Immunoscore is a clinical IHC assay that enumerates CD8+ and CD45RO+ T cells in the tumor. It is highly prognostic in colon cancer; however, its applicability to other cancers and utility as a predictive biomarker remain unclear (36, 37). Several IHC assays have been developed to quantify PD-L1 expression, to provide rationale for anti–PD-1/PD-L1 treatment (38). However, PD-L1 as a single biomarker is not optimal in certain tumor subtypes. In the CheckMate 067 study (ClinicalTrials.gov NCT01844505), the level of PD-L1 expression alone was a poor predictive biomarker of response to PD-1/PD-L1 inhibition in patients with melanoma (39). In contrast, in the KEYNOTE-042 trial (NCT02220894), in which patients with non–small cell lung cancer were treated with anti–PD-1 or anti–CTLA-4 alone or in combination, the PD-L1 tumor proportion score was considered a reliable predictive biomarker of response (40). However, in the KEYNOTE-051 trial, in which PD-L1 expression was a criterion for pediatric patient selection, only 5.9% of patients with solid tumors or lymphomas achieved an objective response (41).
The approval of multiple PD-L1 diagnostic assays presents a substantial challenge. Although PD-L1 testing is now established as routine clinical practice, there is still considerable variability in the techniques used and the staining cutoffs used to stratify patients in different clinical trials. There are four available commercial assays, each of which uses a different antibody clone (SP142, SP263, 22C3, and 28-8) and either of two IHC platforms (Dako and Ventana) to estimate PD-L1 expression in formalin-fixed, paraffin-embedded specimens. Each of these assays has a different scoring system and different thresholds to evaluate PD-L1 expression. Therefore, interpretation of PD-L1 IHC assays remains a clinical challenge. The various assays may produce conflicting results on the same tissue specimen, and their performance is heavily dependent on tumor type. To address this issue, several multicenter studies are under way to directly compare performance (42, 43).
Microsatellite instability (MSI) status and DNA mismatch repair deficiencies (dMMRs) are used as biomarkers to predict response to anti–PD-1 (44); however, only 30%–60% of adult patients with high MSI and/or dMMR benefit from therapy (39, 45, 46). High tumor mutation burden (TMB), a common feature of dMMR, also predicts response, but in only 20%–60% of patients (47–50). Although high TMB, MSI, and/or dMMR are currently used to select for patients in some clinical trials, these are not ideal stand-alone biomarkers. Other biomarkers that predict response to checkpoint inhibitors, such as high neoantigen load and high TCR diversity, are emerging in the literature, but as yet have not been translated into the clinic (51–54). Several assays based on analysis of gene expression are being developed to identify immune-inflamed and noninflamed tumors from RNA sequencing data and to define predictive signatures (11). The T cell–inflamed GEP is an 18-gene signature that predicts response to anti–PD-1 in melanoma (30). It has been validated in several pan-cancer adult cohorts (55).
The T cell–inflamed GEP has been combined with other biomarkers to enrich for responders. Patients with high GEP or high TMB benefited from anti–PD-1, but those with both were the best responders (56). In another large pan-cancer study, patients with high TMB in combination with high T cell–inflamed GEP or expression of PD-L1 showed the best responses (57). These studies highlight the benefit of applying a multibiomarker immune profiling platform, using complementary technologies that examine different aspects of the immune response, to enrich for responders. In 2017, OmniSeq released a comprehensive pan-cancer Immune Report Card that measures multiple immune and molecular biomarkers, using IHC and sequencing analysis. This test is available for use in children; however, as it is a relatively new platform, its utility for making immunotherapy recommendations is yet to be determined.
Predictive immune profile assays will be critical to achieve maximum clinical benefit from immunotherapy (58–60). Most clinical trials for immunotherapy in pediatric cancers to date have not enriched for potential responders. Entry criteria often simply require a specified cancer type and resistance to standard therapies. This means that many enrolled children are unlikely to benefit, and this will contribute to the low overall response rates reported so far. It is also important to remember that cancers arising in children are biologically and molecularly distinct from those in adults (56, 61). Hypermutated cancers are uncommon in childhood, and are most commonly cancers of the central nervous system associated with germline mutations in mismatch repair genes, with or without somatic mutations in replication/repair-associated DNA polymerases (61). On average, the single-nucleotide mutation burden is an order of magnitude lower than in adult tumors, as childhood cancers are far more frequently driven by structural gene variants and copy number aberrations. The rate of germline alterations in cancer predisposition genes is higher (62, 63). Thus, while some clinical trials include pediatric patients with highly mutated tumors (NCT02992964, NCT03668119), there are further patients, for example those with highly inflamed tumors but a low mutational burden, who could potentially respond. The impact of genomic features such as copy number aberrations, structural variants such as fusion genes, and RNA-splicing variants on the generation of neoantigens and the possibility of immune-mediated tumor clearance may be more important in pediatric cancer than absolute mutation burden.
Another important consideration is that the immune system of a child is still developing. It is naive and relatively impaired, with weak Th1 inflammatory and antibody responses compared with those of older children and adults (64). However, as the thymus is highly active in childhood and continually produces new T cells (65), it could potentially generate new tumor-specific clones. A few studies have shown that pediatric cancers are generally infiltrated by macrophages, have few DCs (66), and express low levels of immune checkpoints compared with adult tumors (67, 68). Thus, immunoprofiling assays developed for adult cancers may need modification to better predict outcomes in children. There are a limited number of studies to inform the development of pediatric cancer–specific assays. Here, we summarize the current literature that utilizes single or multiple immune biomarkers for stratification, prognosis, or prediction of responses to immunotherapy in specific classes of pediatric solid tumors.
Immune profiling of pediatric solid tumors
Brain tumors.
Brain tumors are the most common solid tumor of children, accounting for about 25% of cancers and the leading cause of cancer-related death (69). Glioma and medulloblastoma are the most common subtypes (70). Gliomas can arise in any part of the brain and spinal cord (71), and account for approximately 50% of pediatric brain tumors (70, 72). Most are slow-growing, low-grade gliomas that respond well to treatment. However, a large proportion are high-grade gliomas (HGGs), such as diffuse intrinsic pontine glioma (DIPG) (73, 74). Despite treatment, these tumors invariably progress rapidly, with a median survival of less than 1 year (75). Medulloblastomas are aggressive embryonal tumors that account for approximately 10%–15% of pediatric brain tumors (70, 72). Although survival rates have increased in recent years, the prognosis remains very poor for patients who relapse (76, 77).
Immune profiling studies in pediatric brain cancers are summarized in Table 1. Low-grade glioma trends toward a more inflamed microenvironment than HGG (78). Pilocytic astrocytoma and ependymoma tend to be more inflamed when compared with medulloblastoma and glioblastoma (79). There are differences between different molecular subtypes of the same tumor. The sonic hedgehog (SHH) subgroup of medulloblastoma had increased numbers of macrophages compared with other subtypes (80, 81). Expression of immune response–related genes and myeloid cell genes was enriched in the mesenchymal subtype of pediatric HGG (82). In a recent trial, hypermutated HGGs and those resembling pleomorphic xanthoastrocytoma were typically characterized as immune hot and responded better to anti-VEGF (bevacizumab) in conjunction with chemotherapy, while histone H3 subgroups were immune cold and had poorer outcomes (83).
Table 1. Immune profiling studies in brain tumors.

Expression of PD-L1 is generally low in brain cancers, with some exceptions. The supratentorial RELA fusion (ST-RELA) subtype of ependymoma expressed higher PD-L1 levels than other ependymoma subtypes (84), as did HGG in comparison with DIPG (85). Conflicting results were reported for PD-L1 expression in medulloblastoma (86, 87), possibly as a result of the PD-L1 IHC assays used. There were also conflicting data regarding whether CD8+ T cell infiltration correlated with improved or poorer outcomes in medulloblastoma (86, 87). Several studies have identified potential targets for immunotherapy in brain tumors. DIPG expressed high levels of B7-H3, while HGG expressed high levels of PD-L1, B7-H3, and TGF-β (85, 88). CD1d is a nonclassical antigen presentation molecule that presents phospholipid-derived antigen to invariant natural killer T (iNKT) cells (89). The expression of CD1d was high in medulloblastoma, particularly in infants, which suggests that iNKT cell–based therapy could be effective in these tumors (81).
Overall, most brain tumors are immune cold, with high myeloid signatures and low T cell infiltration, particularly aggressive subtypes such as DIPG and medulloblastoma (83, 85). However, there is evidence that some subtypes are more inflamed than others and may respond to immune checkpoint inhibitors. Advanced multiplex technologies such as multiplex IHC and RNA sequencing analyses are yet to be broadly applied to pediatric brain cancers to examine their immune profiles. The resolution that can be achieved by comprehensive analyses is clear in studies that combine extensive sequencing with IHC (83). Further studies in brain tumors will be key to providing a deeper understanding of the complexities of the immune microenvironment to inform therapeutic decisions and improve outcomes.
Neuroblastoma.
Neuroblastomas arise from neural crest progenitors and are the most commonly diagnosed extracranial solid tumor in children, accounting for approximately 10% of childhood cancers (90). Neuroblastoma is heterogeneous in biology and behavior. Spontaneous regression can be seen in neonates with either local or systemic disease, while metastatic disease in patients older than 18 months is usually fatal despite intensive treatment (91). It is plausible that spontaneous regression may be due to a host-mediated immune response. Interestingly, the paraneoplastic opsoclonus-myoclonus syndrome is associated with antineuronal antibodies and a more favorable outcome (92). If this hypothesis is valid, a better understanding of the immunology of tumor regression could potentially result in new strategies to induce regression in a greater proportion of cases.
Distinct immune profiles have been used to stratify high- and low-risk neuroblastomas, as summarized in Table 2. However, there are many conflicting reports. In some studies, low-risk tumors are reported as being more inflamed and having an improved outcome compared with high-risk tumors (93, 94), whereas other studies suggest that high-risk disease is associated with increased T cell infiltration and activation (95, 96). Some inconsistencies have also been reported in the correlation between amplification of the MYCN oncogene and immune infiltration. One study has shown that TIL density is independent of MYCN amplification (93), while another found that MYCN is associated with reduced infiltration (94).
Table 2. Immune profiling studies in neuroblastoma.

In a recent comprehensive study, high-risk neuroblastomas could be stratified into three groups based on their immune profiles (97). High-risk, non–MYCN-amplified tumors (clusters 3 and 4) had higher T cell signatures and TCR heterogeneity and increased expression of immune checkpoints compared with high-risk, MYCN-amplified tumors (cluster 1). This study highlights the complexity of the different microenvironments that can exist for high-risk neuroblastomas and may explain why different phenotypes have been reported in other studies. Further application and integration of advanced multiplex techniques is critical to resolve the immune context of neuroblastomas and to identify subsets that are most likely to respond to immunotherapy.
There are also conflicting data for PD-L1 expression in neuroblastomas. PD-L1 was expressed by all low/intermediate-risk samples, most high-risk samples, and about 50% of metastatic neuroblastomas in one study (95). In another study, only 14% of neuroblastomas were PD-L1+, which was associated with inferior overall survival (OS) (98). A separate study found that PD-L1 expression was associated with better OS, but increased risk of relapse (99). These differences may be explained by the substantial variation seen in PD-L1 staining using different assays. Further studies in larger cohorts of neuroblastoma are clearly needed to clarify these discrepancies.
Overall, the immune microenvironment of neuroblastoma has been studied more than that of any other pediatric tumor. While some tumors clearly can have an immune-inflamed phenotype, express PD-L1, and thus may appear likelier to respond to checkpoint blockade, a consistent association with a particular neuroblastoma subtype or subtypes has not emerged. Recent studies have described multiple cell-type states of neuroblastoma cells within the same tumor, termed undifferentiated mesenchymal and committed adrenergic. These cells display a divergent GEP (100). Other studies have also highlighted the genetic intratumor heterogeneity of neuroblastoma (101, 102). Giving consideration to the potential interplay between intratumor and microenvironmental heterogeneity will be of utmost importance to developing new immunotherapy strategies. So far, only a few studies have integrated genomic, transcriptomic, and protein-based analyses to provide a comprehensive insight into the complex immune microenvironments of neuroblastomas; such studies are critical for the identification of patients for whom checkpoint blockade will provide therapeutic benefit.
Sarcoma.
Sarcomas are a heterogeneous group of tumors that can arise in bone or soft tissues anywhere in the body. Together, they account for about 10% of pediatric cancers (90). Only 20%–30% of children with recurrent or metastatic disease survive, despite aggressive chemoradiotherapy (103). Osteosarcomas are the most prevalent primary malignancy of bone that occurs in children and young adults (104). Alveolar rhabdomyosarcomas (ARMS) and embryonal rhabdomyosarcomas (ERMS) are the most common soft tissue sarcomas, accounting for about 5% of childhood cancers (105). Malignant peripheral nerve sheath tumors (MPNSTs), synovial sarcomas, and desmoplastic small round cell tumors (DSRCTs) are other soft tissue sarcomas that occur in children. Although these subtypes are rare, they are associated with high recurrence and poor outcomes (106–108). Immune profiling studies conducted in pediatric sarcomas are overviewed in Table 3. Osteosarcomas are more frequently immune inflamed than other subtypes, with high CD8+ T cell infiltrate (109) and higher levels of PD-L2, B7-H3, and the immune suppressive molecule indoleamine 2,3 dioxygenase-1 (IDO1) than many other cancers (110). Approximately 25% of osteosarcomas expressed PD-L1 and were more likely to be infiltrated by PD-1+ TILs (111). ERMS had higher numbers of T cells compared with ARMS (112). Furthermore, low-risk ERMS had increased T cell infiltration compared with high-risk. However, T cell numbers were relatively low in rhabdomyosarcomas (RMS) compared with other cancer types. There are some conflicting data on PD-L1 expression in RMS. One study reports that PD-L1 was absent in these tumors (112), while another found that PD-L1 was primarily expressed in RMS compared with other sarcomas (109). PD-L1 expression correlated with improved event-free survival, OS, and metastasis-free survival in ARMS. Coexpression of PD-1 and CD8 (with or without PD-L1) trended toward a better metastasis-free survival. In another study, PD-L1 was highly expressed by ARMS (86%), followed by Ewing sarcomas (57%), ERMS (50%), and osteosarcomas (47%) (95). However, those with the highest proportion of PD-L1 expression had the worst outcomes. But PD-L1+ patients with high numbers of CD8+ TILs had better survival. In a study in Ewing sarcoma, patients with high numbers of CD8+ TILs also had better OS (113). Some sarcoma subtypes have substantial NK cell infiltrate, which represents a potential immunotherapeutic approach for these cancers. In a recent study, HLA-A/B/C score, β2-microglobulin protein levels, and CD56+ NK cells were highest in MPNSTs compared with other NF1-associated tumors (114). PD-L1 expression was highly variable in these tumors. In another study, higher expression of HLA-A/B/C and β2-microglobulin was observed in synovial sarcoma and DSRCT samples compared with those taken at diagnosis. Increased CD56+ NK cells were observed in DSRCT samples from diagnosis to recurrence. PD-L1 was minimally expressed in these tumors (58). In summary, the immune profiles of pediatric sarcomas have not been comprehensively explored, and there are no profiling studies published for some subtypes. The expression of PD-L1 is highly variable in sarcomas, with some conflicting data.
Table 3. Immune profiling studies in sarcomas.

Translating immune profiling to guide pediatric tumor immunotherapy
Barriers to clinical translation of immune profiling.
Considerable progress in the appreciation of the immune microenvironment of pediatric cancers is needed before immunoprofiling can be reliably used to guide clinical decisions in children. There are a considerable number of studies in some cancers, such as neuroblastoma, but few in others, particularly sarcomas. Tissue sample size often limits multiple analyses. Small biopsies are taken to minimize the impact of invasive procedures, unless sampling occurs during scheduled surgery. This limits the application of techniques such as flow cytometry and mass cytometry. This also makes it difficult to see the whole picture with respect to immunoprofiling, especially in tumors that display high intratumor heterogeneity like neuroblastoma (101). Many current studies rely on standard IHC to assay individual markers. While these analyses undoubtedly yield useful information, they do not resolve the complexities of immune subsets, phenotypes, and activation status, nor can they address intricacies in the spatial relationships between different immune cells and tumor cells. In the future, multifaceted approaches should provide a deeper understanding of the immune microenvironments of pediatric cancers, and offer new insights into combinations of molecular markers that best predict outcome or point to the most appropriate therapy (83, 97).
Conflicting immunoprofiling data have been reported for some tumors, which presents another roadblock to translation. In neuroblastoma, T cells are associated with favorable outcomes in some studies (93, 94, 97) but with high-risk disease in others (95, 96). In medulloblastoma, CD8+ T cells correlate with improved outcomes in one study (87) but poorer outcomes in another (86). The lack of resolution achieved using standard techniques may contribute to these inconsistencies. The relatively small size of pediatric cohorts may also contribute to this issue. Counterintuitive results described for some pediatric cancers also complicate clinical translation; for example, the presence of CD8+ T cells is associated with worse outcomes in some tumor types (86, 95, 109). This highlights the need for further interrogation of the mechanisms driving these observations. Another consideration is that many studies, particularly with brain cancer, use samples taken after immunosuppressive treatments such as corticosteroids, or at autopsy. These limitations are for the most part unavoidable. However, it is critical that, when possible, immunoprofiling studies report these details to ensure that these factors are incorporated into the interpretation of the data. Pediatric tumors are generally characterized by high macrophage infiltrate, lack DCs, and have low proportions of T cells. Based on adult data, this would constitute a cold immune profile that is unlikely to respond to checkpoint inhibitor monotherapy. However, it is not clear whether the same “cutoff” for an immune-inflamed or noninflamed tumor should be directly applied to children, and whether these profiles have the same clinical relevance in predicting response to immunotherapies.
This leads to perhaps one of the largest barriers to the application of immune profiling to childhood cancers: the near-complete lack of data from pediatric patients treated with immunotherapies. Only one study has been reported to date (83). Although immune checkpoint inhibitors have been tested in children in recent trials and have shown clinical benefit in some (41, 115, 116), the small patient numbers do not permit us to predict which cancer types may show the greatest benefit. Furthermore, no attempts have been made to understand which characteristics of each patient may be predictive of response or resistance. It is critical that these data be retrospectively analyzed in future trials to determine whether the immune-inflamed phenotype in pediatric cancers predicts response to immunotherapies, in combination with other biomarkers such as PD-L1 expression, high TMB, MSI, and dMMR. It is unclear whether these biomarkers, derived from adult cancers, translate directly to pediatric malignancies. Owing to profound differences in the biological, molecular, and immunological profiles of pediatric and adult cancers, it is likely that immune profiling would need to be optimized specifically for pediatric tumors.
Although hundreds of clinical trials are testing immunotherapies in children across the globe, it is important to remain cautious of the potential side effects of these approaches. Recent trials have reported similar pharmacokinetics and short-term toxicity of checkpoint inhibitors in children and adults (117). In the KEYNOTE-051 trial, in which pediatric patients with advanced melanoma or a PD-L1+ advanced/relapsed or refractory tumor were treated with pembrolizumab, treatment did not appear to have any major effect on the developing immune system (41). In contrast, addition of bevacizumab to chemotherapy and radiotherapy in pediatric patients with HGG was associated with more serious adverse events (118). Within the next decade, we should see reports of retrospective studies in pediatric malignancies, which will provide the long-term safety data and inform the translation of immunoprofiling into the clinic.
Approaches to treating immune-hot and -cold tumors.
It would seem intuitive that immune-hot tumors, particularly those that also have a high TMB, would be most likely to benefit from immunotherapy and could be treated with one or multiple immune checkpoint inhibitors. However, this does not mean that cold tumors should be excluded. Combination strategies that activate the intrinsic antitumor response may be the key to unlocking the therapeutic potential of immunotherapy for all pediatric tumors (Figure 3). Several drugs with immunomodulatory properties are being tested in clinical trials with checkpoint inhibitors in adults, including antibodies directed against immunosuppressive molecules, histone deacetylase inhibitors, and tyrosine kinase inhibitors; but few trials are open to pediatric patients (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI137181DS1). There is also substantial evidence to show that low-dose pulses of cytotoxic chemotherapy can promote an antitumor response, eliminate suppressive cells from the tumor microenvironment, and potentiate immunotherapy (119). The same applies to radiotherapy, in which low doses can promote immune infiltration and enhance immunotherapy (120). Several trials in pediatric patients that combine checkpoint inhibitors with low-dose radiotherapy or chemotherapy are currently recruiting (e.g., NCT02961101, NCT03585465, NCT03690869, NCT02989636). There are also several trials combining checkpoint inhibitors with other types of immunotherapy in children, such as chimeric antigen receptor T cells and cancer vaccines, to improve responses (e.g., NCT02775292, NCT04239040). These combination therapy approaches are not only relevant in the context of treatment with immune checkpoint inhibitors, but could be applied more broadly to improve the efficacy of other immunotherapy and immunomodulatory drugs in immune-cold pediatric tumors that have not been discussed within the scope of this Review.
Figure 3. New approach using immune context classification of tumors to guide anticancer therapy and increase the response to immune checkpoint inhibitors.
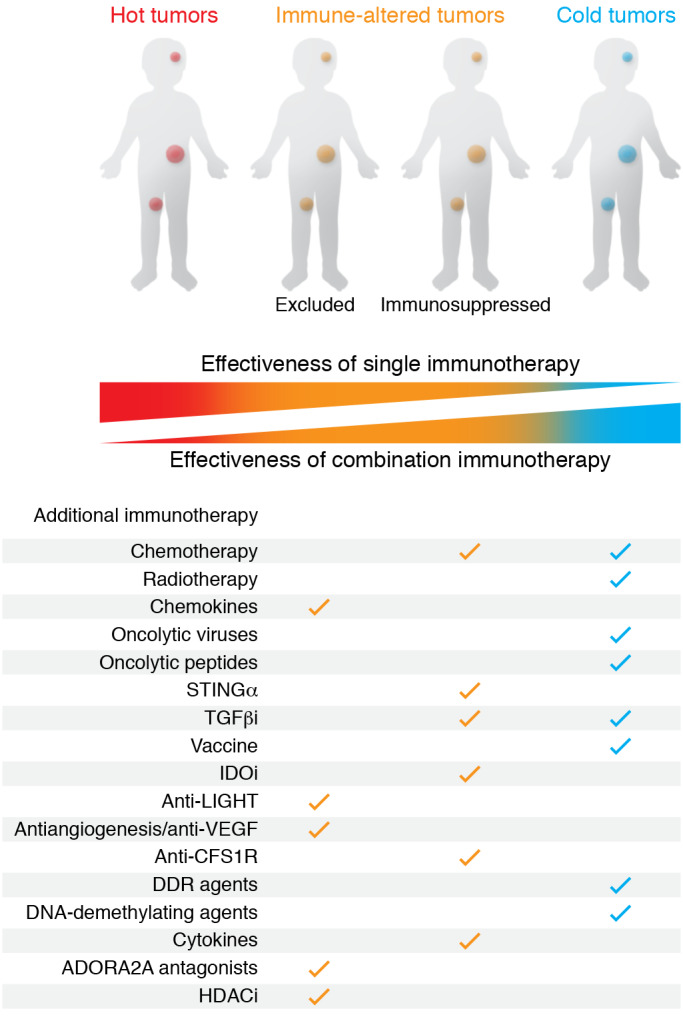
ADORA2A, A2a adenosine receptor; CSF1R, colony-stimulating factor 1 receptor; DDR, DNA damage response; HDAC, histone deacetylase; IDO, indoleamine 2,3-dioxygenase; LIGHT, tumor necrosis factor superfamily member 14; STING, stimulator of IFN genes.
A personalized medicine approach will likely yield the best results in children, by identifying the optimal immune checkpoint to target and the best immunomodulatory agent to combine it with. As an example, the checkpoint B7-H3 is highly expressed in DIPG (85, 88). Anti–B7-H3 has recently been tested in clinical trials in children with solid tumors (NCT02982941). Unfortunately, this study did not include brain tumors. Nevertheless, B7-H3 could be a good target in DIPG. For an appropriate cotherapy, DIPG is reported to express VEGF (85), and there is evidence to suggest that anti-VEGF has some clinical activity in adult and pediatric brainstem gliomas (121). Although anti-VEGF is not optimal as a single agent, it is currently being tested in combination with anti–PD-1 in several clinical trials (Supplemental Table 1). However, as PD-L1 expression is reported to be very low in DIPG, and anti–PD-1 has not shown efficacy as a single agent in DIPG, B7-H3 may represent a better checkpoint than PD-1/PD-L1 to use in combination with anti-VEGF. A second example, anti-RANKL, could be used in osteosarcoma to enhance T cell responses to anti–PD-1. Osteosarcomas express PD-L1, and anti–PD-1 is currently being tested in pediatric trials (NCT03359018, NCT02301039, NCT02304458, NCT03628209). In separate trials, anti-RANKL is also being tested in children with osteosarcomas (NCT02470091). Anti-RANKL is used to limit tumor growth in osteosarcoma, through inhibition of osteoclastogenesis (122). However, recent studies have shown that the RANK/RANKL pathway is critical for maintaining central tolerance in the thymus, by eliminating self-reactive T cells (123). In the context of cancer, this may mean that tumor-specific T cell clones are sequestered. This has been shown in preclinical studies in melanoma, in which anti-RANKL rescued melanoma-specific T cells and enhanced anti–PD-1 (124). There are several clinical trials testing anti-RANKL plus anti–PD-1 in melanoma and non–small cell lung carcinoma (Supplemental Table 1), but no pediatric trials. This combination could potentially be very effective in pediatric osteosarcoma.
Overall, many clinical trials are combining immune checkpoint inhibitors with immunomodulatory agents to improve response in adult cancers, but few trials in children. Once the safety and efficacy of these combinations are established, they will likely be extended to pediatric malignancies. This will result in new treatment combinations for childhood cancers that otherwise would be nonresponsive to immunotherapies, and will meaningfully improve patient outcomes.
Summary
Recent developments in immunotherapy give new hope for children with high-risk cancers (59, 125). However, because of the lack of reliable biomarkers and clinical assays to inform treatment decisions (126), many children who receive immunotherapies do not respond. There is ample evidence to show that immunoprofiling can predict which adult patients are more likely to respond to immune checkpoint inhibitors (127), but these data do not yet exist for children (7, 8). However, there are hundreds of clinical trials under way that are testing immunotherapies in childhood cancers. These studies will generate data and samples that can be retrospectively analyzed to identify immune signatures predictive of responses. The development of comprehensive clinical-grade assays, specific for pediatric cancers, will be key to improved treatment. Furthermore, immunoprofiling may identify new immune-based treatment opportunities for these cancers. Finally, the opportunity for children to access personalized combination treatments tailored to their specific tumor type, molecular characteristics, and immune profile will be critical to improving long-term survival and quality of life.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the Medical Research Future Fund Australian Brain Cancer Mission supporting the ZERO Childhood Cancer initiative to ensure that all Australian children with high-risk brain cancer may access precision medicine to improve survival, quality of life, and treatment options. DM has scholarship funding from Plan Cancer, l’Institut Servier, and Fondation Nuovo-Soldati, and gratefully acknowledges Tour de Cure for their support. JAT is supported by Investigator and Program grants from the National Health and Medical Research Council (NHMRC) of Australia, and is a Rosie Lew Fellow of the Peter MacCallum Cancer Foundation. PJN is supported by a program grant from the NHMRC of Australia.
Version 1. 06/15/2020
Electronic publication
Version 2. 07/01/2020
Print issue publication
Footnotes
Conflict of interest: PN received research funding from BMS, Roche Genentech, Allergan, Juno-Celgene, and Compugen.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(7):3391–3402.https://doi.org/10.1172/JCI137181.
Contributor Information
Deborah Meyran, Email: deborah.meyran@petermac.org.
David S. Ziegler, Email: d.ziegler@unsw.edu.au.
Michelle Haber, Email: mhaber@ccia.unsw.edu.au.
Paul G. Ekert, Email: PEkert@ccia.org.au.
References
- 1.Fridman WH. From cancer immune surveillance to cancer immunoediting: birth of modern immuno-oncology. J Immunol. 2018;201(3):825–826. doi: 10.4049/jimmunol.1800827. [DOI] [PubMed] [Google Scholar]
- 2.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 3.Binnewies M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 5.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 7.Becht E, et al. Immune contexture, Immunoscore, and malignant cell molecular subgroups for prognostic and theranostic classifications of cancers. Adv Immunol. 2016;130:95–190. doi: 10.1016/bs.ai.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Bethmann D, Feng Z, Fox BA. Immunoprofiling as a predictor of patient’s response to cancer therapy—promises and challenges. Curr Opin Immunol. 2017;45:60–72. doi: 10.1016/j.coi.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Ascierto PA, et al. The additional facet of immunoscore: immunoprofiling as a possible predictive tool for cancer treatment. J Transl Med. 2013;11:54. doi: 10.1186/1479-5876-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fridman WH, et al. The immune microenvironment of human tumors: general significance and clinical impact. Cancer Microenviron. 2013;6(2):117–122. doi: 10.1007/s12307-012-0124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finotello F, Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother. 2018;67(7):1031–1040. doi: 10.1007/s00262-018-2150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyons YA, Wu SY, Overwijk WW, Baggerly KA, Sood AK. Immune cell profiling in cancer: molecular approaches to cell-specific identification. NPJ Precis Oncol. 2017;1(1):26. doi: 10.1038/s41698-017-0031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danaher P, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer. 2017;5:18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng C, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 15.Zavidij O, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer. 2020;1(5):493–506. doi: 10.1038/s43018-020-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sade-Feldman M, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175(4):998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564(7735):268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 18.Azizi E, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174(5):1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chevrier S, et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169(4):736–749.e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krieg C, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24(2):144–153. doi: 10.1038/nm.4466. [DOI] [PubMed] [Google Scholar]
- 21.de Vries NL, et al. High-dimensional cytometric analysis of colorectal cancer reveals novel mediators of antitumour immunity. Gut. 2019;69(4):691–703. doi: 10.1136/gutjnl-2019-318672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner J, et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. 2019;177(5):1330–1345.e18. doi: 10.1016/j.cell.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 24.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 25.Mariathasan S, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) 2012;4:734–745. doi: 10.2741/e414. [DOI] [PubMed] [Google Scholar]
- 28.Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11(1):39. doi: 10.1186/s13045-018-0582-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8. doi: 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayers M, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daud AI, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34(34):4102–4109. doi: 10.1200/JCO.2016.67.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halse H, et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci Rep. 2018;8(1):11158. doi: 10.1038/s41598-018-28944-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun J, et al. Clinical significance and regulation of the costimulatory molecule B7-H3 in human colorectal carcinoma. Cancer Immunol Immunother. 2010;59(8):1163–1171. doi: 10.1007/s00262-010-0841-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zang X, et al. B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc Natl Acad Sci U S A. 2007;104(49):19458–19463. doi: 10.1073/pnas.0709802104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galon J, et al. Immunoscore and immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge 2015. J Transl Med. 2016;14:273. doi: 10.1186/s12967-016-1029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pages F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–2139. doi: 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 38.Liu D, Wang S, Bindeman W. Clinical applications of PD-L1 bioassays for cancer immunotherapy. J Hematol Oncol. 2017;10(1):110. doi: 10.1186/s13045-017-0479-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolchok JD, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mok TSK, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–1830. doi: 10.1016/S0140-6736(18)32409-7. [DOI] [PubMed] [Google Scholar]
- 41.Geoerger B, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020;21(1):121–133. doi: 10.1016/S1470-2045(19)30671-0. [DOI] [PubMed] [Google Scholar]
- 42.Adam J, et al. Multicenter harmonization study for PD-L1 IHC testing in non-small-cell lung cancer. Ann Oncol. 2018;29(4):953–958. doi: 10.1093/annonc/mdy014. [DOI] [PubMed] [Google Scholar]
- 43.Tsao MS, et al. PD-L1 Immunohistochemistry comparability study in real-life clinical samples: results of Blueprint phase 2 project. J Thorac Oncol. 2018;13(9):1302–1311. doi: 10.1016/j.jtho.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diaz LA, et al. Pembrolizumab therapy for microsatellite instability high (MSI-H) colorectal cancer (CRC) and non-CRC. J Clin Oncol. 2017;35(15 suppl):3071 [Google Scholar]
- 45.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Overman MJ, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodman AM, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luksza M, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551(7681):517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li B, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet. 2016;48(7):725–732. doi: 10.1038/ng.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hogan SA, et al. Peripheral blood TCR repertoire profiling may facilitate patient stratification for immunotherapy against melanoma. Cancer Immunol Res. 2019;7(1):77–85. doi: 10.1158/2326-6066.CIR-18-0136. [DOI] [PubMed] [Google Scholar]
- 54.Yusko E, et al. Association of tumor microenvironment T-cell repertoire and mutational load with clinical outcome after sequential checkpoint blockade in melanoma. Cancer Immunol Res. 2019;7(3):458–465. doi: 10.1158/2326-6066.CIR-18-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Danaher P, et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA) J Immunother Cancer. 2018;6(1):63. doi: 10.1186/s40425-018-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cristescu R, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362(6411):eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ott PA, et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol. 2019;37(4):318–327. doi: 10.1200/JCO.2018.78.2276. [DOI] [PubMed] [Google Scholar]
- 58.Wedekind MF, Haworth KB, Arnold M, Stanek JR, Lee D, Cripe TP. Immune profiles of desmoplastic small round cell tumor and synovial sarcoma suggest different immunotherapeutic susceptibility upfront compared to relapse specimens. Pediatr Blood Cancer. 2018;65(11):e27313. doi: 10.1002/pbc.27313. [DOI] [PubMed] [Google Scholar]
- 59.Park JA, Cheung NV. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat Rev. 2017;58:22–33. doi: 10.1016/j.ctrv.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner LM, Adams VR. Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. Onco Targets Ther. 2017;10:2097–2106. doi: 10.2147/OTT.S124008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campbell BB, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171(5):1042–1056.e10. doi: 10.1016/j.cell.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sweet-Cordero EA, Biegel JA. The genomic landscape of pediatric cancers: implications for diagnosis and treatment. Science. 2019;363(6432):1170–1175. doi: 10.1126/science.aaw3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grobner SN, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–327. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 64.Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. 2015;282(1821):20143085. doi: 10.1098/rspb.2014.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palmer DB. The effect of age on thymic function. Front Immunol. 2013;4:316. doi: 10.3389/fimmu.2013.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vakkila J, Jaffe R, Michelow M, Lotze MT. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: a major nosologic difference with adult tumors. Clin Cancer Res. 2006;12(7 pt 1):2049–2054. doi: 10.1158/1078-0432.CCR-05-1824. [DOI] [PubMed] [Google Scholar]
- 67.Aoki T, et al. Low frequency of programmed death ligand 1 expression in pediatric cancers. Pediatr Blood Cancer. 2016;63(8):1461–1464. doi: 10.1002/pbc.26018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pinto N, et al. Patterns of PD-1, PD-L1, and PD-L2 expression in pediatric solid tumors. Pediatr Blood Cancer. 2017;64(11):10.1002/pbc.26613. doi: 10.1002/pbc.26613. [DOI] [PubMed] [Google Scholar]
- 69.Curtin SC, Minino AM, Anderson RN. Declines in cancer death rates among children and adolescents in the United States, 1999-2014. NCHS Data Brief. 2016;(257):1–8. [PubMed] [Google Scholar]
- 70.Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012;14(suppl 5):v1–v49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sturm D, Pfister SM, Jones DTW. Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. J Clin Oncol. 2017;35(21):2370–2377. doi: 10.1200/JCO.2017.73.0242. [DOI] [PubMed] [Google Scholar]
- 72.Ostrom QT, et al. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooney T, et al. Contemporary survival endpoints: an International Diffuse Intrinsic Pontine Glioma Registry study. Neuro Oncol. 2017;19(9):1279–1280. doi: 10.1093/neuonc/nox107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jansen MH, van Vuurden DG, Vandertop WP, Kaspers GJ. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev. 2012;38(1):27–35. doi: 10.1016/j.ctrv.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 75.Filbin MG, Sturm D. Gliomas in children. Semin Neurol. 2018;38(1):121–130. doi: 10.1055/s-0038-1635106. [DOI] [PubMed] [Google Scholar]
- 76.Wang J, Garancher A, Ramaswamy V, Wechsler-Reya RJ. Medulloblastoma: from molecular subgroups to molecular targeted therapies. Annu Rev Neurosci. 2018;41:207–232. doi: 10.1146/annurev-neuro-070815-013838. [DOI] [PubMed] [Google Scholar]
- 77.Sabel M, et al. Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol. 2016;129(3):515–524. doi: 10.1007/s11060-016-2202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Plant AS, et al. Immunophenotyping of pediatric brain tumors: correlating immune infiltrate with histology, mutational load, and survival and assessing clonal T cell response. J Neurooncol. 2018;137(2):269–278. doi: 10.1007/s11060-017-2737-9. [DOI] [PubMed] [Google Scholar]
- 79.Griesinger AM, et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191(9):4880–4888. doi: 10.4049/jimmunol.1301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Margol AS, et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res. 2015;21(6):1457–1465. doi: 10.1158/1078-0432.CCR-14-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Teo WY, et al. Therapeutic implications of CD1d expression and tumor-infiltrating macrophages in pediatric medulloblastomas. J Neurooncol. 2014;120(2):293–301. doi: 10.1007/s11060-014-1572-5. [DOI] [PubMed] [Google Scholar]
- 82.Engler JR, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7(8):e43339. doi: 10.1371/journal.pone.0043339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mackay A, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell. 2018;33(5):829–842.e5. doi: 10.1016/j.ccell.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Witt DA, et al. Specific expression of PD-L1 in RELA-fusion supratentorial ependymoma: implications for PD-1-targeted therapy. Pediatr Blood Cancer. 2018;65(5):e26960. doi: 10.1002/pbc.26960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lieberman NAP, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol. 2019;21(1):83–94. doi: 10.1093/neuonc/noy145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vermeulen JF, et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology. 2018;7(3):e1398877. doi: 10.1080/2162402X.2017.1398877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murata D, et al. High programmed cell death 1 ligand-1 expression: association with CD8+ T-cell infiltration and poor prognosis in human medulloblastoma. J Neurosurg. 2018;128(3):710–716. doi: 10.3171/2016.11.JNS16991. [DOI] [PubMed] [Google Scholar]
- 88.Zhou Z, et al. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J Neurooncol. 2013;111(3):257–264. doi: 10.1007/s11060-012-1021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kjer-Nielsen L, et al. A structural basis for selection and cross-species reactivity of the semi-invariant NKT cell receptor in CD1d/glycolipid recognition. J Exp Med. 2006;203(3):661–673. doi: 10.1084/jem.20051777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steliarova-Foucher E, et al. International incidence of childhood cancer, 2001-10: a population-based registry study. Lancet Oncol. 2017;18(6):719–731. doi: 10.1016/S1470-2045(17)30186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matthay KK, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;2:16078. doi: 10.1038/nrdp.2016.78. [DOI] [PubMed] [Google Scholar]
- 92.Rudnick E, et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies—a report from the Children’s Cancer Group Study. Med Pediatr Oncol. 2001;36(6):612–622. doi: 10.1002/mpo.1138. [DOI] [PubMed] [Google Scholar]
- 93.Mina M, et al. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology. 2015;4(9):e1019981. doi: 10.1080/2162402X.2015.1019981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rahbar M, Mehrazma M, Karimian M. Tumor infiltrating cytotoxic CD8 T-cells predict clinical outcome of neuroblastoma in children. Indian J Med Paediatr Oncol. 2018;39(2):6 [Google Scholar]
- 95.Chowdhury F, Dunn S, Mitchell S, Mellows T, Ashton-Key M, Gray JC. PD-L1 and CD8+PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. OncoImmunology. 2015;4(10):e1029701. doi: 10.1080/2162402X.2015.1029701. [DOI] [Google Scholar]
- 96.Gowda M, et al. Distinct signatures of the immune responses in low risk versus high risk neuroblastoma. J Transl Med. 2011;9:170. doi: 10.1186/1479-5876-9-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wei JS, et al. Clinically relevant cytotoxic immune cell signatures and clonal expansion of T-cell receptors in high-risk MYCN-not-amplified human neuroblastoma. Clin Cancer Res. 2018;24(22):5673–5684. doi: 10.1158/1078-0432.CCR-18-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Majzner RG, et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer. 2017;123(19):3807–3815. doi: 10.1002/cncr.30724. [DOI] [PubMed] [Google Scholar]
- 99.Saletta F, et al. Programmed death-ligand 1 expression in a large cohort of pediatric patients with solid tumor and association with clinicopathologic features in neuroblastoma. JCO Precis Oncol. 2017;1(1):1–12. doi: 10.1200/PO.16.00049. [DOI] [PubMed] [Google Scholar]
- 100.van Groningen T, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. 2017;49(8):1261–1266. doi: 10.1038/ng.3899. [DOI] [PubMed] [Google Scholar]
- 101.von Stedingk K, Gisselsson D, Bexell D. Multidimensional intratumour heterogeneity in neuroblastoma. Oncotarget. 2019;10(1):3–5. doi: 10.18632/oncotarget.26524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karlsson J, et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat Genet. 2018;50(7):944–950. doi: 10.1038/s41588-018-0131-y. [DOI] [PubMed] [Google Scholar]
- 103.Anderson JL, Denny CT, Tap WD, Federman N. Pediatric sarcomas: translating molecular pathogenesis of disease to novel therapeutic possibilities. Pediatr Res. 2012;72(2):112–121. doi: 10.1038/pr.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Durfee RA, Mohammed M, Luu HH. Review of osteosarcoma and current management. Rheumatol Ther. 2016;3(2):221–243. doi: 10.1007/s40744-016-0046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chisholm JC, et al. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: a nomogram to better define patients who can be salvaged with further therapy. J Clin Oncol. 2011;29(10):1319–1325. doi: 10.1200/JCO.2010.32.1984. [DOI] [PubMed] [Google Scholar]
- 106.Amirian ES, Goodman JC, New P, Scheurer ME. Pediatric and adult malignant peripheral nerve sheath tumors: an analysis of data from the surveillance, epidemiology, and end results program. J Neurooncol. 2014;116(3):609–616. doi: 10.1007/s11060-013-1345-6. [DOI] [PubMed] [Google Scholar]
- 107.Wong HH, et al. Desmoplastic small round cell tumour: characteristics and prognostic factors of 41 patients and review of the literature. Clin Sarcoma Res. 2013;3(1):14. doi: 10.1186/2045-3329-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer. 2009;115(15):3537–3547. doi: 10.1002/cncr.24424. [DOI] [PubMed] [Google Scholar]
- 109.van Erp AEM, et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8(+) lymphocytes in primary sarcomas is subtype dependent. Oncotarget. 2017;8(41):71371–71384. doi: 10.18632/oncotarget.19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McEachron TA, Triche TJ, Sorenson L, Parham DM, Carpten JD. Profiling targetable immune checkpoints in osteosarcoma. Oncoimmunology. 2018;7(12):e1475873. doi: 10.1080/2162402X.2018.1475873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Koirala P, et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci Rep. 2016;6:30093. doi: 10.1038/srep30093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kather JN, et al. CD163+ immune cell infiltrates and presence of CD54+ microvessels are prognostic markers for patients with embryonal rhabdomyosarcoma. Sci Rep. 2019;9(1):9211. doi: 10.1038/s41598-019-45551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berghuis D, et al. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol. 2011;223(3):347–357. doi: 10.1002/path.2819. [DOI] [PubMed] [Google Scholar]
- 114.Haworth KB, et al. Immune profiling of NF1-associated tumors reveals histologic subtype distinctions and heterogeneity: implications for immunotherapy. Oncotarget. 2017;8(47):82037–82048. doi: 10.18632/oncotarget.18301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kabir TF, Chauhan A, Anthony L, Hildebrandt GC. Immune checkpoint inhibitors in pediatric solid tumors: status in 2018. Ochsner J. 2018;18(4):370–376. doi: 10.31486/toj.18.0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dyson KA, et al. Emerging trends in immunotherapy for pediatric sarcomas. J Hematol Oncol. 2019;12(1):78. doi: 10.1186/s13045-019-0756-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Geoerger B, et al. Phase II study of ipilimumab in adolescents with unresectable stage III or IV malignant melanoma. Eur J Cancer. 2017;86:358–363. doi: 10.1016/j.ejca.2017.09.032. [DOI] [PubMed] [Google Scholar]
- 118.Grill J, et al. Phase II, open-label, randomized, multicenter trial (HERBY) of bevacizumab in pediatric patients with newly diagnosed high-grade glioma. J Clin Oncol. 2018;36(10):951–958. doi: 10.1200/JCO.2017.76.0611. [DOI] [PubMed] [Google Scholar]
- 119.Mathew M, Enzler T, Shu CA, Rizvi NA. Combining chemotherapy with PD-1 blockade in NSCLC. Pharmacol Ther. 2018;186:130–137. doi: 10.1016/j.pharmthera.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 120.Barker CA, Postow MA. Combinations of radiation therapy and immunotherapy for melanoma: a review of clinical outcomes. Int J Radiat Oncol Biol Phys. 2014;88(5):986–997. doi: 10.1016/j.ijrobp.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Moriya S, et al. A retrospective study of bevacizumab for treatment of brainstem glioma with malignant features. J Clin Neurosci. 2018;47:228–233. doi: 10.1016/j.jocn.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 122.Chen Y, et al. RANKL blockade prevents and treats aggressive osteosarcomas. Sci Transl Med. 2015;7(317):317ra197. doi: 10.1126/scitranslmed.aad0295. [DOI] [PubMed] [Google Scholar]
- 123.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol. 2012;30:95–114. doi: 10.1146/annurev-immunol-020711-075035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Khan IS, et al. Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J Exp Med. 2014;211(5):761–768. doi: 10.1084/jem.20131889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mackall CL, Merchant MS, Fry TJ. Immune-based therapies for childhood cancer. Nat Rev Clin Oncol. 2014;11(12):693–703. doi: 10.1038/nrclinonc.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cesano A, Warren S. Bringing the next generation of immuno-oncology biomarkers to the clinic. Biomedicines. 2018;6(1):14. doi: 10.3390/biomedicines6010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yuan J, et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J Immunother Cancer. 2016;4:3. doi: 10.1186/s40425-016-0107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lin GL, Nagaraja S, Filbin MG, Suva ML, Vogel H, Monje M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2018;6(1):51. doi: 10.1186/s40478-018-0553-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gowda M, Payne KK, Godder K, Manjili MH. HLA-DR expression on myeloid cells is a potential prognostic factor in patients with high-risk neuroblastoma. Oncoimmunology. 2013;2(10):e26616. doi: 10.4161/onci.26616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.