

Abstract
Purpose:
To examine effect of oncolytic herpes simplex virus (oHSV) on NOTCH signaling in CNS tumors.
Experimental design:
Bioluminescence imaging, RPPA proteomics, fluorescence microscopy, reporter assays, and molecular biology approaches were used to evaluate NOTCH signaling. Orthotopic glioma- mouse models were utilized to evaluate effects in vivo.
Results:
We have identified that HSV-1 (oncolytic and wild type) infected glioma cells induce NOTCH signaling from inside of infected cells into adjacent tumor cells (inside out signaling). This was canonical NOTCH signaling which resulted in activation of RBPJ dependent transcriptional activity that could be rescued with dnMAML.
High-throughput screening of HSV-1-encoded cDNA and micro-RNA libraries further uncovered that HSV-1 miR-H16 induced NOTCH signaling. We further identified that factor inhibiting HIF-1 (FIH-1) is a direct target of miR-H16, and that FIH-1 downregulation by virus encoded miR-H16 induces NOTCH activity.
FIH-1 binding to Mib1 has been reported, but this is the first report that shows FIH-1 sequesters Mib1 to suppress NOTCH activation. We observed that FIH-1 degradation induced NOTCH ligand ubiquitination and NOTCH activity. Rembrandt and TCGA data analysis also uncovered a significant negative regulation between FIH-1 and NOTCH.
Furthermore, combination of oHSV with NOTCH blocking GSI had a therapeutic advantage in two different intracranial glioma models treated with oncolytic HSV, without affecting safety profile of the virus in vivo.
Conclusion:
To our knowledge this is the first report to identify impact of HSV-1 on NOTCH signaling and highlights the significance of combining oHSV and GSI for glioblastoma therapy.
Keywords: NOTCH signaling, oncolytic herpes simplex virus, glioma, gamma-secretase inhibitor, microRNA
Introduction
Glioblastoma (GBM) is the most aggressive primary brain tumor with a median survival of less than 2 years despite aggressive treatment [1, 2]. NOTCH signaling is a way of cell-cell communication wherein NOTCH ligand on signal sending cells binds to NOTCH receptor on adjacent cells leading to NOTCH receptor cleavage and subsequent activation of signal transduction in signal receiving cells [3]. The NOTCH signaling pathway is one of the core pathways frequently exploited by glioma cells for survival [4, 5], tumor progression [6, 7], stem cell maintenance [5], angiogenesis [8], invasion [9], and the development of resistance to chemotherapy and radiation [10, 11].
While Imlygic, an oncolytic herpes simplex virus-1 (oHSV) is approved for metastatic melanoma [12, 13], the clinical application of oHSVs for brain tumors is being investigated [14]. A clear understanding of the impact of these biotherapies on the glioma microenvironment is critical. While activation of NOTCH signaling following some virus infections has been noted [15], the impact of oHSV on NOTCH signaling is not known to our knowledge. Here we investigated the impact of oHSV treatment of glioma cells on NOTCH signaling. We observed that oHSV infection resulted in the activation of NOTCH signaling in tumor cells adjacent to infected cells. NOTCH activation was initiated by a virus encoded miRNA that targeted and reduced factor inhibiting HIF-1 (FIH-1) in infected cells. We further found that FIH-1 inhibited the activity of mind bomb1 (Mib1) to activate classical NOTCH signal transduction in adjacent cells. Blocking NOTCH signaling gamma secretase inhibitor (GSI) in conjunction with oHSV had a therapeutic advantage.
Methods
Ethics Statement
All experimental animals were housed and handled in accordance with the guideline of The University of Texas Health Science Center at Houston Center for Laboratory Animal Medicine and Care and were approved by institutional review board. Use of all patient derived de-identified GBM cells is approved by IRB.
Animal studies
All animal studies were reviewed and approved by University of Texas Health Science’s Center for laboratory animal Research (CLAMC). Briefly, U87ΔEGFR or GBM12 (2×105 cells) were stereotactically implanted in the right hemisphere of 6-8 week old mice as described [16]. Seven days (U87EGFR) or ten days (GBM12) later mice were randomized to be treated with 34.5ENVE (5 × 104 pfu), or PBS intratumorally. Two days prior to virus treatment mice were also treated with 10 mg/kg RO4929097 (Selleckchem, Houston, TX) or carrier by oral gavage every day.
For subcutaneous studies, U87ΔEGFR (1.5×106 cells) expressing RBP-Jk responsive luciferase reporter were implanted in the flanks of athymic nude mouse. Twenty days later, post tumor injection, mice were treated with PBS or 34.5ENVE virus (1×106 pfu/mouse) intra-tumorally, and six hours later were subject to IVIS in vivo imaging system (PerkinElmer, Waltham, MA).
Molecular biology reagents and assays
All cell lines and viruses used in this study are described in Supplementary Table 1. All primary and secondary antibodies used in the study are listed in Supplementary Table 2. Detailed protocols are in supplementary methods section. To generate control and NOTCH reporter cells, the indicated GBM cells were transfected with Cingal lenti negative control or RBP-Jk Reporter system (Qiagen, Germantown, MD) and selected with puromycin. For DN-MAML assays control or NOTCH reporter glioma cells were transduced with DN-MAML and overlaid on infected glioma cells. Luciferase activity was measured twelve hours post infection. For FIH-1 knockdown GBM cells were transfected with 100nM of siRNA (Negative control, Dharmacon, Lafayette, CO, D-001810-10-20) (HIF1AN, L-004073-03-0020) for 48 hours. For FIH-1 overexpression, GBM cells were transfected with plasmid expressing FIH-1 (Addgene, 21399 [17]) for 72 hours. For Mib1 knockdown experiments, cells were transfected with either negative control or Mib1 siRNA (Dharmacon, L-014033-00-0020) for 72 hours. Luciferase activity was evaluated using luciferase assay system or Dual-luciferase reporter system (Promega, Madison, WI), and normalized with BCA protein assay kit-reducing agent compatible (Thermo Fisher Scientific, Waltham, MA) or Renilla luciferase activity (Promega, E2241). All primer sequences are listed in Supplementary Table3.
RPPA analysis
U251T3 cells were transfected with negative control or HSV-1 miRNA-H16 using RNAiMAX. Seventy-two hours later, samples were harvested. RPPA data were generated by the RPPA core facility at MDACC (Houston, TX). The relative protein levels were normalized for protein loading and transformed to linear value. To stabilize the variance, the value was log2 transformed. Two-sided Student’s t test was performed to evaluate the statistical significance and p value was corrected by Benjamini-Hochberg method to adjust for the multiple testing. Adjusted p value less than 0.05 was considered as statistical significance.
3’UTR assay
3’UTR sequence of FIH-1 predicted to contain miR-H16 target sequence was PCR-amplified from genomic DNA and cloned into pGL3-Control Vector (Promega). Mutagenesis was performed using QuikChange Site-directed mutagenesis kit (Agilent, Santa Clara, CA) to produce the two different mutant FIH-1 3’UTR sequences. All primers used for 3’UTR assay are listed in Supplementary Table4. The indicated glioma cells were transfected with 250 ng of firefly plasmid and 20 ng of Renilla plasmid. Twenty-four hours later, 100nM of negative control or miR-H16 was transfected with RNAiMAX, and samples were collected after additional 48 hours. To evaluate the effect of oHSV on wild type or mutated FIH-1 3’UTR, 1×105 glioma cells were seeded and transfected with 250 ng of plasmids. Forty-eight hours later, cells were infected with oHSV for 24 hours. Luciferase signal was measured with Dual-Luciferase Reporter Assay System, and normalized with Renilla luciferase activity.
Ubiquitylation assay
Cells were transfected with 5µg each of plasmids encoding for DLL4(GeneCopoeia, Rockville, MD, EX-U1091-Lv241) and GFP tagged Ubiquitin {(GFP-Ub); Addgene, 11928}[18] using GeneJuice (Millipore, Burlington, MA). 24 hours later, siRNA or microRNA were transfected with RNAiMAX. 48 hours later, cells were immuno-precipitated with IgG or anti-GFP and Protein G-agarose (GenDEPOT). Immuno-precipitates were probed for DLL4 by western blot.
Cell cycle analysis
Cells were fixed with 70% ethanol. DNA was stained with 7-amino-actinomycin D (Miltenyi Biotec, Bergisch Gladbach, Germany) and RNase (100µg/ml, Qiagen). Cell cycle distribution of GFP-negative uninfected cells were analyzed by flow cytometry.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism (GraphPad, San Diego, CA). Student’s t-test were performed to test the difference in comparison between the two groups. Luciferase assays, qRT-PCR, immunofluorescent and immunohistological analyses, and flow cytometry were analyzed by the one-way analysis of variance test. Dunnett’s or Tukey’s post hoc test was applied for multiple comparisons. All statistical tests were two-sided. Kaplan-Meier curves were compared using the log-rank test. P-values <0.05 were considered statistically significant.
Additional detailed description of methods can be found in the Supplementary Methods.
Results
oHSV treatment induces NOTCH signaling
Infection of glioma cells transduced with NOTCH reporter (RBP-Jk luciferase) revealed a significant induction of NOTCH activity in GBM cells following infection with oHSV (rHSVQ and 34.5ENVE) and wild type HSV-1. No significant induction of luciferase was observed in cells transduced with control CMV-luciferase (Figure 1 A–B, and supplementary Figure S1A). Infection of 10 different primary GBM and glioma cell lines with 34.5ENVE significantly induced the gene expression of NOTCH ligands and or NOTCH target genes relative to uninfected cells (Table 1). This NOTCH reporter and NOTCH target gene induction depended on live virus, as heat-inactivated oHSV did not induce NOTCH signaling (Supplementary Figure S1B–D). Dose response of viral infection on NOTCH activity 12 hours post infection (prior to virus burst) revealed a bell-shaped curve, indicating a dose-dependent increase in NOTCH activation until a threshold, after which there was a reduction (Figure 1C, and Supplementary Figure S1E). Since the cells were harvested prior to viral burst this was not accompanied by a significant amount of cell death, and so reduction in NOTCH signaling at high MOIs could not be attributed to cell death due to infection. NOTCH is a pathway of cell-cell communication and the bell-shaped dose response implied that infected cells induced NOTCH activity in adjacent uninfected cells. Hence reporter activity increased with increasing doses until a threshold after which most of the culture was infected and hence there were less uninfected cells to transduce signal. To test this we overlaid infected cells with uninfected NOTCH reporter cells in the presence of heparin to block reporter cell infection by any release virus (Figure 1D). When oHSV infected GBM cells were overlaid with uninfected NOTCH reporter cells, a robust increase in NOTCH signaling was observed in overlaid NOTCH reporter cells (Figure 1E, and Supplementary Figure 1F). This induction was not observed when control CMV-luciferase cells were overlaid on infected cells (Supplementary Figure S1G). Dose response curves of NOTCH activation in uninfected reporter cells overlaid on infected cells revealed a statistically significant correlation between virus dose and induction of NOTCH activity in overlaid uninfected glioma cells (Figure 1F, R2=0.7468). Together this shows that infected cells became signal transducing cells that led to NOTCH activity in adjacent uninfected tumor cells. Next, we evaluated if oHSV therapy induces NOTCH activity in vivo. Mice bearing NOTCH reporter transduced glioma cells (U87ΔEGFR) were treated with oHSV and monitored by IVIS imaging. A significant enhancement of NOTCH reporter activity in mice treated with an oHSV was observed in vivo (Figure 1G–H). This induction could not be attributed to tumor growth as virus treated tumors showed large areas of necrosis compared to control tumors (Supplementary Figure S2). Immunohistochemistry for NICD (NOTCH intracellular domain) also revealed significantly increased NICD-positive viable tumor cells in the oHSV-treated tumors relative to untreated tumors, indicating NOTCH activation post virotherapy in vivo (Figure 1I–J). Consistent with these finding, a retrospect analysis of published RNA-seq of newly transcribed RNA and ribosome profiling of HSV infected cells also showed induction of NOTCH ligand and target genes expression after HSV infection [19] (Supplementary Figure S3).
Fig.1. oHSV infection induces NOTCH signaling.
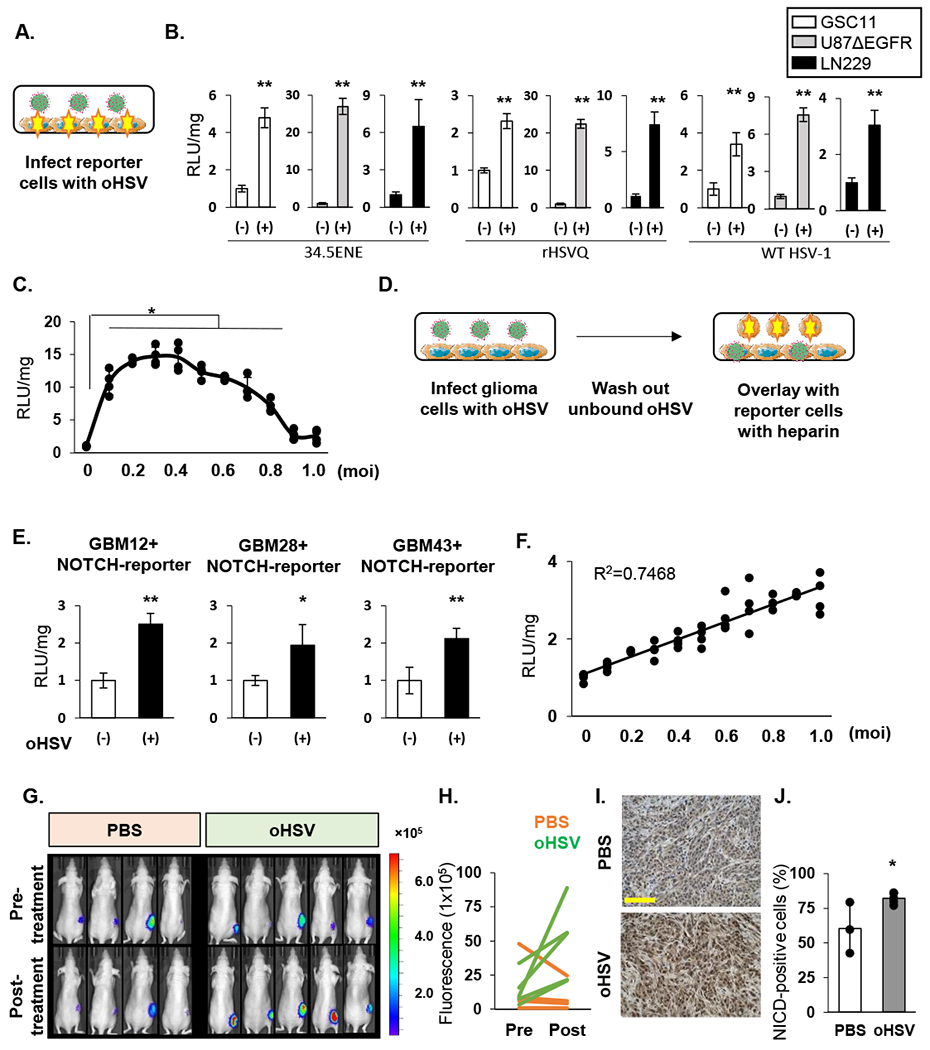
A: Schema of protocol in B. NOTCH reporter expressing cells (cells with stars), were infected with HSV (green) and relative luciferase activity (RLU) relative to uninfected cells was measured. B: Data shown are RLU in indicated cells post infection with 34.5ENVE virus (MOI = 0.1), rHSVQ virus (MOI = 0.1), or wild type F-strain HSV-1 (MOI = 0.3) (n ≥3). C: Dose response of viral infection to NOTCH reporter activity. U87ΔEGFR NOTCH reporter cells were infected with indicated dose of 34.5ENVE and changes in luciferase activity were measured (n=4/dose), 12 hours post infection, relative to uninfected cells. D: The schema of experimental set up in E to measure NOTCH activation in adjacent uninfected cells. The indicated primary GBM cells infected with 34.5ENVE (MOI=0.1), for an hour and unbound virus was washed off. NOTCH reporter cells were then overlaid and mean luciferase activity relative to uninfected cells ± SD (n ≥ 3 per group) is shown. F: Dose response of NOTCH reporter activity in reporter glioma cells overlaid on infected cells. Data points are changes in luciferase activity relative to uninfected cells (n = 4 per each point). Correlation was calculated by Pearson. G: Representative IVIS images of mice bearing subcutaneous tumors of U87ΔEGFR cells expressing NOTCH reporter (n=9) treated with either PBS or 34.5ENVE, pre and 6 hours post-treatment. H: Quantification of data shown in G. Data shown are change in luciferase activity pre and post treatment in mice injected with saline or 34.5ENVE. I-J: Immunohistochemistry of NICD staining in tissue from PBS and virus treated mice imaged above. I: Representative images of IHC showing increased intensity of NICD (brown) in viable tumor cells after virotherapy. *P<0.05, **P<0.01. Scale bar, 100µm
Table 1.
The indicated NOTCH ligand and target genes’ changes after oHSV infection.
| DLL1 (ligand) | DLL4 (ligand) | Jag1 (ligand) | Hey1(target gene) | Hey2(target gene) | HeyL(target gene) | Hes1(target gene) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | |
| GBM30 | 11.831 | < 0.01 | 341.87 | < 0.01 | 1.980 | 0.032 | 1.576 | < 0.01 | 1.785 | 0.032 | 2.123 | < 0.01 | 14.296 | < 0.01 |
| GBM12 | 14.654 | 0.011 | 22.054 | 0.011 | 2.650 | 0.027 | 1.794 | 0.115 | 1.022 | 0.933 | 1.908 | < 0.01 | 7.268 | < 0.01 |
| GBM28 | 5.607 | < 0.01 | 29.772 | < 0.01 | 1.838 | 0.010 | 1.616 | < 0.01 | 1.788 | < 0.01 | 2.843 | < 0.01 | 2.194 | < 0.01 |
| GBM43 | 3.276 | 0.018 | 3.339 | < 0.01 | 1.588 | 0.048 | 1.138 | 0.418 | 1.796 | < 0.01 | 1.759 | 0.065 | 6.251 | < 0.01 |
| GSC8-11 | 3.444 | < 0.01 | 1.653 | 0.013 | 1.114 | 0.551 | 1.378 | 0.016 | 1.129 | 0.084 | 1.780 | 0.041 | 1.717 | 0.114 |
| GSC20 | 3.745 | < 0.01 | 94.352 | < 0.01 | 1.046 | 0.420 | 1.386 | < 0.01 | 1.758 | < 0.01 | 1.545 | 0.041 | 1.662 | < 0.01 |
| GSC23 | 12.732 | < 0.01 | 450.96 | < 0.01 | 1.610 | < 0.01 | 1.622 | < 0.01 | 1.593 | < 0.01 | 2.206 | < 0.01 | 2.400 | < 0.01 |
| LN229 | 29.645 | < 0.01 | 41.963 | < 0.01 | 2.0267 | < 0.01 | 1.254 | 0.020 | 0.740 | 0.111 | 2.615 | 0.062 | 3.089 | < 0.01 |
| U251T3 | 2.145 | 0.043 | 10.693 | < 0.01 | 5.174 | 0.021 | 2.453 | 0.032 | 6.464 | 0.019 | 4.131 | < 0.01 | 1.006 | 0.985 |
| U87ΔEGFR | 2.045 | 0.029 | 2.113 | 0.071 | 2.315 | 0.047 | 1.299 | 0.395 | 1.277 | 0.526 | 2.001 | < 0.01 | 3.316 | 0.045 |
The indicated primary glioma cells and established glioma cell lines were treated with 34.5ENVE or uninfected (MOI=0.2 for GBM30, GBM12, GBM28, GBM43, GSC8-11, GSC20 and GSC23, or 0.1 for LN229, U251T3 and U87ΔEGFR) for 12 hours. Gene expression were measured by quantitative real time PCR (n≥ 3). Fold exp: Fold expression change relative to uninfected cells.
HSV-1 encoded miRNA-H16 induces NOTCH activity in glioma
Since RBP-Jk is also known to activate transcription of genes independent of NICD [20], we tested if NOTCH reporter activity depended on canonical NOTCH activity. oHSV induced NOTCH activity was rescued when GBM cells were transfected with dominant negative mastermind-like (DN-MAML) known to inhibit NICD activity (Figure 2A) [21–23]. Western blot and staining of infected cells with an NICD specific antibody also indicated increased NOTCH receptor cleavage (Figure 2B). Quantification of immunofluorescent images of NICD positive cells also uncovered a significant increase in NICD expressing cells in infected cell cultures (mean[SD]=24.0%[7.1]) compared to control (mean[SD]=4.1[3.1]). Consistent with the notion that infected cells induced NOTCH activity in adjacent uninfected cells, a majority of NICD positive cells were localized in uninfected GFP-negative cells adjacent to infected cells (Figure 2 C inset, and quantified in 2D). The inset in Fig 2C shows a magnification of an infected (GFP positive cell) surrounded by adjacent NICD positive GFP negative tumor cells. These results are consistent with the notion that the infected tumor cells act as NOTCH “signal transducers” that educate surrounding glioma cells to become “signal acceptors” and activate NOTCH signaling.
Figure 2: HSV encoded miR-H16 induces NOTCH signaling.
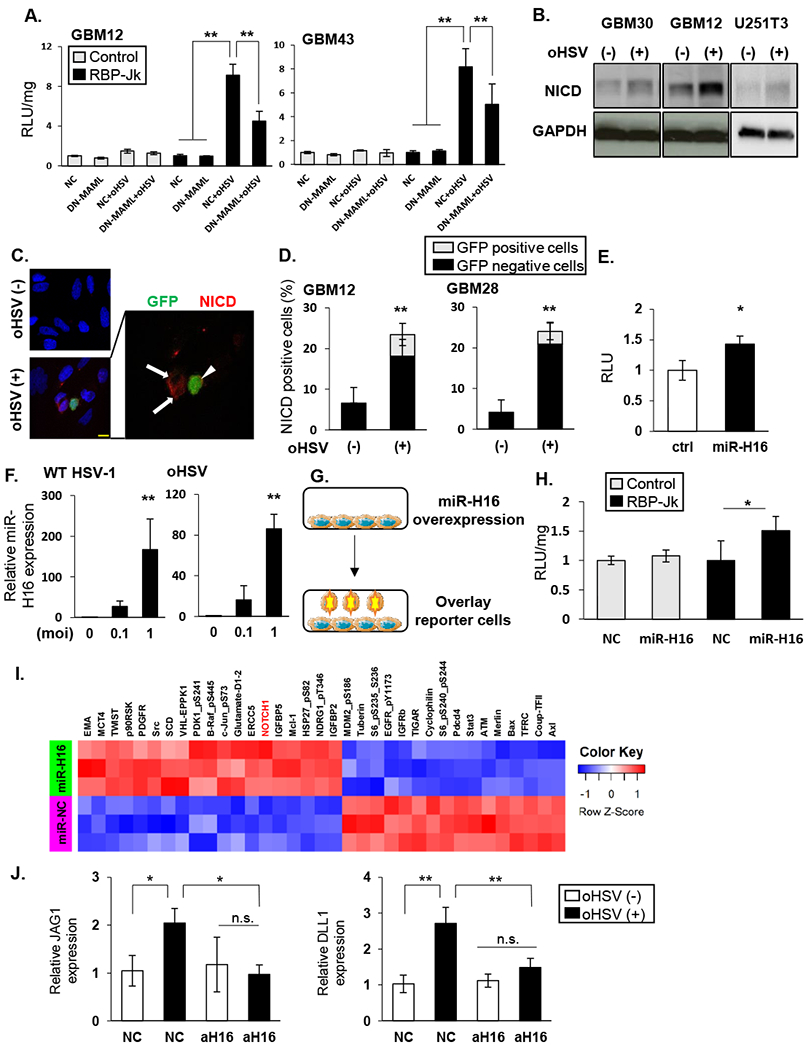
A: The indicated GBM cells transduced with DN-MAML or empty control plasmid (NC) were infected with 34.5ENVE (oHSV) for one hour. After unbound virus was washed out, they were overlaid with NOTCH reporter cells and change in luciferase activity was measured (RLU). B: Western blot of the indicated cell lysates probed for NOTCH intracellular domain (NICD) with 34.5ENVE (MOI = 0.1) (+) or without (−) infection. C: Immuno-fluorescent staining of NICD in infected glioma cells. Representative fluorescent microscopy images of GBM28 cells stained for NICD (red). DAPI (blue) shows nuclear staining, and GFP indicates virus-infected cells. Uninfected cells (arrow) around infected cells (arrowhead) showed NICD. D: Quantification of NICD staining with (+) or without (−) 34.5ENVE infection in indicated GBM cells. Mean NICD positive cells/view field ± SD. E: Relative NOTCH reporter activity in glioma cells transduced with control or HSV-1 encoded miR-H16 (n = 4/group). F: Expression of miR-H16 in glioma cells infected with 34.5ENVE or w.t. HSV-1 by qRT-PCR. G-H: miR-H16 transduced cells can activate NOTCH activity in adjacent cells. G: Schema of experimental set up in H. LN229 cells transfected with miR-H16 or control miRNA (NC) were overlaid with control (white bars) or NOTCH reporter cells (black bars). H: Mean change in relative luciferase activity measured 12 hours after reporter cell overlay (n = 4). I: Heatmap of significant changes in protein expression from Reverse Phase Protein Array (RPPA) analysis of control and miR-H16 transfected cells. Unsupervised hierarchical clustering of normalized RPPA data (n=3/group). J: GBM12 cells transduced with antagomir for miR-H16 (amiR-H16) or control (amiR-NC) were treated with 34.5ENVE (MOI = 0.1). Changes in NOTCH ligand expression was measured by qRT-PCR. Data shown are the mean ± SD relative to control cells (n≥ 3). *P<0.05, **P<0.01. Scale bar, 10µm
To identify the mechanism of virus induced NOTCH activation, we screened 65 viral genes cloned into an HSV expression library [24] for their ability to induce NOTCH signaling. None of the viral genes was found to induce NOTCH activity after transfection in glioma cells. Next we screened 21 HSV-1 encoded miRNAs for their ability to induce NOTCH signaling. A significant induction of NOTCH reporter activity was observed in cells transfected with miR-H16 (Figure 2E). Expression of miR-H16 in oHSV and wild type HSV-1 (F strain) infected glioma cells was confirmed by in multiple glioma cell lines infected with oHSV and wild type HSV-1 (Figure 2F, and Supplementary figure S4A–B). A significant induction in NOTCH activity was also observed in NOTCH reporter cells overlaid on miR-H16 transduced glioma cells (Figure 2G–H), indicating that similar to viral infection, miR-H16 transfection led to paracrine NOTCH induction in adjacent cells. Reverse Phase Protein Array (RPPA) proteomic analysis of miR-H16 transfected glioma cells compared to control miR-NC transfected glioma cells was performed to evaluate changes in signaling pathways by miR-H16 transfection. Unsupervised hierarchical clustering of the RPPA data uncovered NICD to be among the top candidate proteins to be significantly increased in the miR-H16 expressing cells compared to control (Figure 2I, and Supplementary table 5).
Consistent with the induction of NOTCH reporter activity upon infection, miR-H16 transfection of glioma cells also resulted in a significant induction of NOTCH ligands and target genes (Table 2). To evaluate the requirement of miR-H16 expression by oHSV to induce NOTCH signaling we evaluated the effect on NOTCH activation in glioma cells transduced with an antago-miR-H16 (amiR-H16). The induction of NOTCH target genes after oHSV infection was reduced in cells transduced with amiR-H16, but not a negative control antagomiRNA (NC) (Figure 2J). These results indicated that miR-H16 induced NOTCH signaling after oHSV treatment and HSV-1 encoded miR-H16 as a novel inducer of the NOTCH signaling.
Table 2.
The indicated NOTCH ligand and target genes’ changes after miR-H16 transfection.
| JAG1(ligand) | DLL1(ligand) | DLL4(ligand) | HeyL(Target gene) | Hes1(Target gene) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | Fold exp. | p-value | |
| GBM30 | 1.849 | 0.019 | 1.019 | 0.962 | 0.837 | 0.450 | 2.217 | 0.016 | 1.703 | 0.022 |
| GBM12 | 2.499 | < 0.01 | 2.406 | 0.022 | 1.819 | 0.039 | 5.379 | 0.072 | 4.995 | 0.362 |
| GBM43 | 1.830 | 0.141 | 3.242 | 0.045 | 1.428 | 0.228 | 4.073 | 0.071 | 6.245 | < 0.01 |
| GSC2 | 2.648 | < 0.01 | 1.219 | 0.178 | 0.948 | 0.993 | 1.431 | 0.029 | 2.423 | 0.010 |
| GSC20 | 4.271 | < 0.01 | 1.652 | < 0.01 | 1.236 | 0.118 | 1.325 | 0.016 | 1.305 | < 0.01 |
| LN229 | 5.406 | 0.052 | 3.641 | 0.247 | 2.681 | 0.020 | 4.703 | 0.366 | 4.338 | 0.383 |
| U251T3 | 10.599 | 0.055 | 1.047 | 0.931 | 0.619 | 0.019 | 1.258 | 0.641 | 1.869 | 0.176 |
| U87ΔEGFR | 3.390 | 0.026 | 3.444 | 0.058 | 2.084 | 0.014 | 1.520 | 0.099 | 2.174 | 0.047 |
The indicated glioma cells were transfected with miR-NC or miR-H16 for 72 hours, and changes in gene expression were evaluated by quantitative real time PCR (n≥ 3). Fold exp: Fold expression change
miR-H16 targets FIH-1 to activate NOTCH signaling
To identify the downstream cellular target genes of miR-H16, we performed an in silico screen using two prediction tools: miRDB (Supplementary Table 6) and TargetScan (Supplementary Table 7). 92 overlapping genes were identified to contain potential miR-H16 targeting sites in their 3’UTR (Figure 3A). Of these 92 predicted miR-H16 target genes, ten have been previously implicated in NOTCH signaling and four of these ten genes (FIH-1, NLK, CBL, and NUMBL) have been predicted to regulate NOTCH activity [25–28]. Both NUMBL and CBL are ubiquitin ligases that regulate NICD stability and function in the signal-receiving cell. NLK is a kinase that can directly phosphorylate and inhibit NICD transcriptional activity in the signal receiving cell. FIH-1 is an asparaginyl hydroxylase that has been previously described to negatively regulate NOTCH activity in endothelial cells through an unknown mechanism [25, 29–31]. Analysis of REMBRANDT and TCGA datasets also uncovered a significant negative correlation between FIH-1 and NOTCH ligands and target genes in GBM patient samples (Figure 3B), implicating FIH-1 to be a negative regulator of NOTCH signaling. Thus we evaluated if miR-H16 and or HSV infection affected FIH-1 levels. Both miR-H16 transfection and oHSV-1 (34.5ENVE and HSVQ) infection reduced FIH-1 protein expression in transfected or infected glioma cells (Figure 3C–D). Further transfection of glioma cells with amiR-H16 rescued oHSV mediated reduction of FIH-1 protein (Figure 3E). This reduction of FIH-1 was directly mediated by miR-H16 as a luciferase-3’UTR containing FIH-1 predicted miR-H16 binding site, showed a significant reduction in luciferase expression after miR-H16 transfection (Figure 3F). A significant reduction in FIH-1 3’UTR reporter activity was also observed when cells were infected with oHSV (Figure 3G). This reduction was not observed when the binding site was mutated (Figure 3G and Supplementary Fig. S5A–B). Together these results indicate that FIH-1 is a direct target of miR-H16.
Figure 3: HSV-1 miR-H16 targets FIH-1 to activate NOTCH signaling.
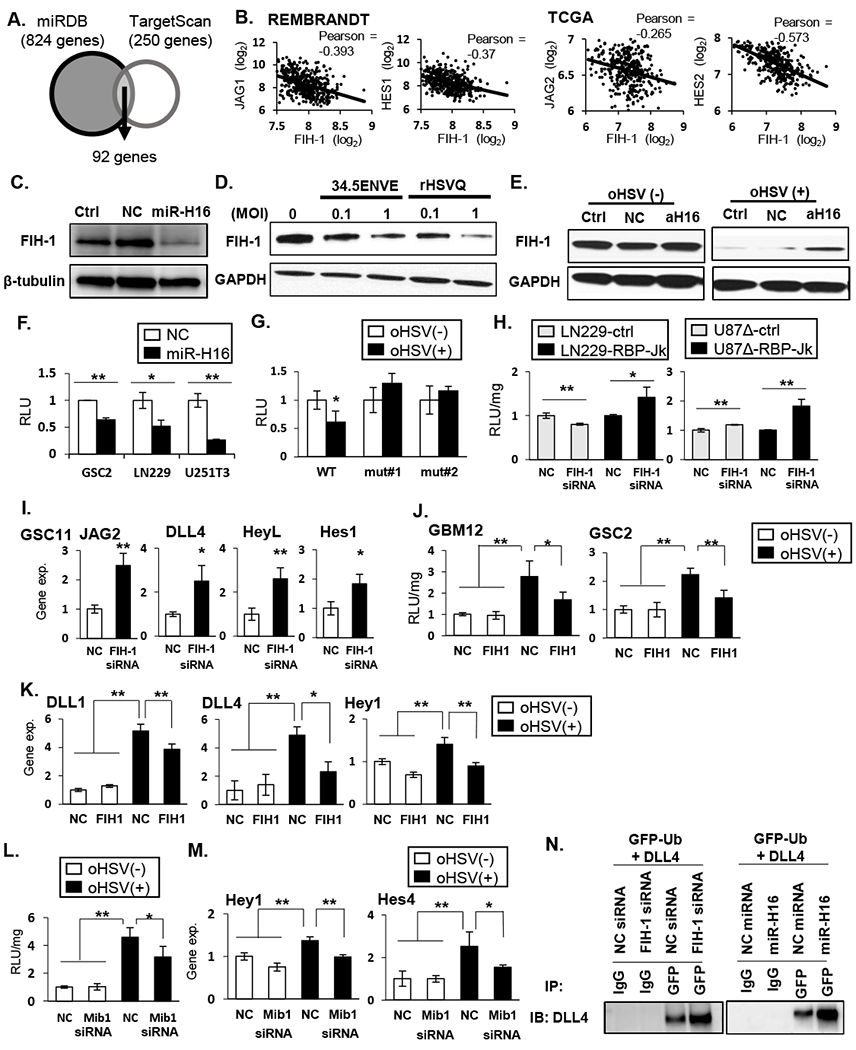
A: Schematic of overlap between genes predicted to be regulated by microRNA-H16 (miR-H16) in miRDB and TargetScan. B: Scatterplot of two-gene correlation between FIH-1 and NOTCH ligands and downstream target genes in REMBRANDT and TCGA. C: Western blot for FIH-1 expression in LN229 cells transfected with non-targeting control miRNA (NC) or miR-H16 (H16). D: Western blot analysis evaluating changes in FIH-1 after infection with 34.5 ENVE or rHSVQ. E: Antagomir for miR-H16 (amiR-H16) was transfected in LN229 cells 48 hours prior to infection with 34.5ENVE and probed for FIH-1 expression. F-G: FIH-1 3’UTR assay. Luciferase upstream of 3’ UTR sequence of FIH-1 containing the predicted wild type (WT) or mutant (mut#1 and mut#2) miR-H16 target site were transfected in indicated glioma cells. Relative changes in mean luciferase activity after miR-H16 transfection (F) or 34.5ENVE infection (G) is shown (n ≥ 3). H-I: knockdown of FIH-1 induced NOTCH reporter activity (H) and NOTCH target gene expression (I). J-K: Overexpression of FIH-1 reduced oHSV-induced NOTCH activity (J) and NOTCH target gene expression (K) (n≥ 3). L-M: Knockdown of mind bomb 1 (Mib1) by siRNA reduced virus induced NOTCH reporter activity (L) and target gene expression (M) in GBM12. Data are the mean ± SD of the relative change (n=4). N: Ubiquitylation of DLL4 in the presence of FIH-1 siRNA (left panel) or miR-H16 (right panel). Cells were transfected with GFP-tagged Ubiquitin. FIH-1 siRNA and miR-H16 increased DLL4 interaction with ubiquitin. *P<0.05, **P<0.01.
Next, we evaluated the effect of FIH-1 on activation of NOTCH signaling in glioma cells. FIH-1 siRNA transduction resulted in a significant induction of NOTCH reporter activity and NOTCH ligands, and target gene expression (Figure 3H–I, and Supplementary Figure 5C). Overexpression of FIH-1 in infected GSCs reduced oHSV induced NOTCH reporter activity and genes’ expression (Figure 3J–K). Together, this underscores the significant role of FIH-1 reduction in inducing NOTCH activity from the signal sending cell upon oHSV infection. While FIH-1 has been implicated as a negative regulator of NOTCH signaling its mechanism is not clear. FIH-1 has been described to interact with Mib1 [32, 33]. Mib1 mediated ubiquitination of receptor engaged NOTCH ligand ensues in the unveiling of gamma secretase cleavage sites of the bound NOTCH receptor [34, 35]. Thus, we hypothesized that FIH-1 binding negatively regulated Mib1 mediated NOTCH ligand ubiquitination.
Knockdown of Mib1 significantly reduced virus induced NOTCH reporter activity and NOTCH target gene expression (Figure 3L–M). Immunoprecipitation for GFP-tagged ubiquitin revealed increased ubiquitination of DLL4 cells upon FIH-1 siRNA transfection (left panel) and miR-H16 (right panel) transfection (Figure 3N). Collectively, these results show that miR-H16-mediated FIH-1 downregulation induced ubiquitylation of DLL4, thereby activating NOTCH signaling in surrounding cells.
Therapeutic benefit of combining oHSV with NOTCH blocking Gamma Secretase Inhibitors (GSI).
To evaluate the impact of combining oHSV-with NOTCH inhibition, we used two different gamma secretase inhibitors (GSIs): RO4929097 and DAPT. Treatment of infected glioma cells with either of these GSIs reduced oHSV-mediated NOTCH activation (Figure 4A–B). Immunofluorescent staining of infected primary GBM cells also showed a rescue of oHSV-induced NICD expression in the presence of GSI (Quantification of number of cells /view field staining for NICD in GFP-negative (black) or GFP-positive (grey) cells, Fig. 4C). To evaluate the impact of oHSV infection on GBM cell sensitivity to GSI, glioma cells were infected with or without GSI. Consistent with increased NOTCH activity upon infection, increased killing was observed in cells treated with both oHSV and GSI (Figure 4D and E, Supplementary Fig.S6). This effect could not be attributed to increased virus replication, as time-lapse quantification of virus spread (GFP-positive cell counts over time) revealed no difference between cultures treated with and without GSI (Supplementary FigS7). Together these results suggest that combining oHSV with GSI should have clinical benefit.
Fig.4. Therapeutic benefit of combining oHSV with γ-secretase inhibitors.
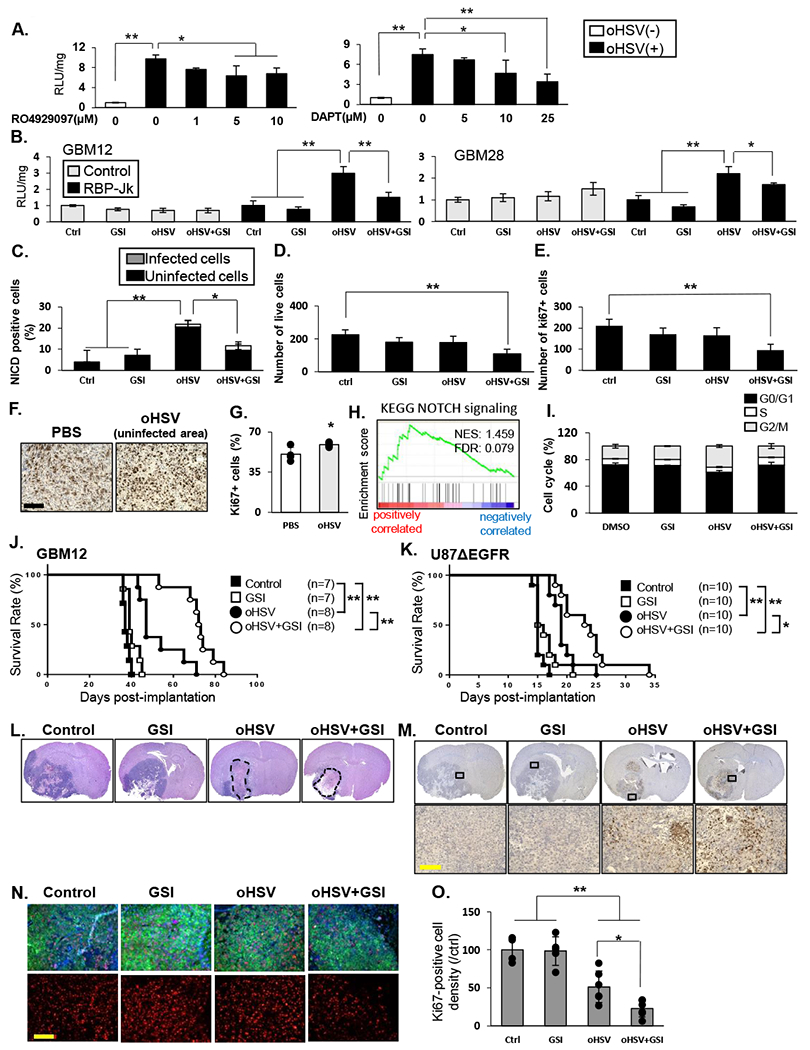
A: NOTCH reporter activity in U87EGFR NOTCH reporter cells after infection with 34.5 ENVE virus (MOI = 0.1) and treated with increasing dose of GSIs; RO4929097 or DAPT for 12 hours. Data shown are mean RLU ± SD (n≥ 3). B: Indicated glioma cells were treated with RO4929097 (10µM), and/or 34.5ENVE (MOI=0.1), and then overlaid with LN229 NOTCH reporter cells. Mean luciferase activity relative to no treatment ± SD is shown (n≥ 3). C: Quantification of GBM28 cells, 9 hours after infection with 34.5ENVE for number of cells staining positive for NICD with or without GSI. Data shown are the mean ± SD. D-E: GBM28 were treated with or without virus in the presence or absence of GSI and incubated for four days. Mean number of viable and proliferating (ki67-positive) cells ± SD (n=4) was measured using flow cytometry. F-G: Immunohistochemistry of ki67 in tissue from U87ΔEGFR subcutaneous mice model treated with PBS (n=4) or oHSV (n=5). Quantification of ki67 density in tumor adjacent to oncolytic virus destroyed plaques were compared to Ki67 density in random sections in untreated tumors. H: GSEA for KEGG_NOTCH signaling by ki67 expression in TCGA GBM database (n=416). I: GSI attenuated oHSV-mediated cell cycle progression. LN229 were treated as indicated for 72 hours. Cells in G0/G1, S and G2/M phases were determined by flow cytometry. Data shown are the mean ± SD (n=3). J-K: Kaplan-Meier analysis of mice implanted with orthotopic GBM12 (J) or U87ΔEGFR (K) cells treated with oHSV with or without GSI therapy. L: Representative hematoxylin and eosin staining of mice bearing GBM12 tumors treated as above, sacrificed twenty days after virus treatment. Both oHSV monotherapy and combination therapy group showed large necrotic areas (dashed line). M: Immunohistochemistry of GBM12 xenograft model showed HSV-1 replication in both of oHSV alone and combination treated group. N-O: Representative fluorescent microscopy images of GBM12 xenograft model immuno-stained for ki67 (red) and human HLA (green) and DAPI (blue) shows nuclear staining. Quantification of ki67 staining overall in entire tumor sections from mice treated as indicated. Data shown are average number of Ki67 +ve cells/ view field in randomly selected view fields/section. Immuno-fluorescent staining of GBM12 xenograft model showed a significant decrease in ki67-positive cell density in combination treated group. Data are shown as the mean ± SD (n = 4-6 in each group). *P<0.05, **P<0.01. Scale bar, 100µm
Increased compensatory proliferation in uninfected tumor areas adjacent to infected plaques in vivo after oHSV treatment has been described before [36]. Immunohistochemistry of an in vivo subcutaneous model showed a significant increase in cell proliferation in uninfected areas of oHSV-treated glioma compared with PBS-treated glioma (Figure 4F–G,). NOTCH signaling is known to regulate the cell cycle of glioma cells, and GSI induced G1 arrest [37]. Thus, we hypothesized that oHSV-mediated NOTCH signaling would increase cell cycle progression in surrounding cells. Gene set enrichment analysis of TCGA data (n=416) revealed significant correlation between ki67 expression and NOTCH signaling (Figure 4H, NES=1.459, FDR=0.079). Consistent with this, in vitro cell cycle analysis revealed that uninfected cells in oHSV-treated sample showed high S and G2/M phase (G0/G1; mean[SD]=61.0%[2.5], S; mean[SD]=7.6%[0.5], G2/M; mean[SD]=31.3%[2.0]). Furthermore, combination of GSI with oHSV reduced cell cycle progression (G0/G1; mean[SD]=71.7%[4.3], S; mean[SD]=11.5%[0.6], G2/M; mean[SD]=16.8%[3.7]) (Figure 4I).
We next assessed the effect of combining oHSV therapy with GSI in vivo. Mice bearing intracranial gliomas (GBM12 or U87EGFR) were treated with either PBS or GSI in the presence or absence of oHSV. While GSI as a monotherapy had no effect on survival compared to mice treated with PBS, it significantly enhanced survival of mice treated with oHSV in both tumor models (GBM12 with oHSV; median survival time (MST) = 47 days, with combination therapy; MST = 72.5 days) (U87 ΔEGFR with oHSV; MST = 19 days, with combination therapy; MST = 23 days) (Figure 4 J–K). Histological analysis revealed large necrotic areas in both the oHSV monotherapy and oHSV + GSI combination groups, with no difference in HSV-1 staining (Fig. 4L and M, Supplementary Fig. 8A). Consistent with the in vitro results, the density of Ki67+ cells was significantly reduced in tumors treated with both GSI and oHSV compared to tumors treated with either therapy alone (Figure 4 N–O). Immunofluorescent staining for cleaved caspase-3 also indicated increasing of tumor cell death in combination therapy group (Supplementary Fig. 8B and C). While a detailed toxicological analysis was not performed on these mice, no obvious toxicity attributable to the combination of GSI with oHSV was observed in the mice treated with RO4929097 (with/without virus). Thus oHSV treatment of tumors induced NOTCH activation in residual tumor that rendered previously unresponsive tumors sensitive to GSI. This study underscores the importance of testing the combination of GSI and oHSV therapy in patients with brain tumors.
Discussion
Collectively our results show that intracranial tumors that did not respond to GSI treatment as a single agent become sensitive to GSI therapy after oncolytic virus therapy. The activation of NOTCH in cancer stem cells has been reported to promote tumor growth, invasion, angiogenesis, and therapy resistance [38], [10, 39], and its blockade reduces tumor growth and augments therapy [40, 41]. NOTCH activation is also known to be induced by several viruses [42]. For example, KSHV (human herpesvirus 8) activates NOTCH signaling [43] to enhance activation of transcription of its own genome [44]. During pulmonary respiratory syncytial virus (RSV) infection, activation of NOTCH by the virus induced NOTCH ligands has been implicated in maintenance of Treg cells [45]. While oHSV is now an FDA approved therapy for metastatic melanoma, its impact on NOTCH signaling has not been investigated. To our knowledge this is the first report to show that oHSV infection of glioma cells results in the activation of NOTCH signaling.
HSV-1 lytic life cycle includes virion entry into the cell, after which the capsid is transported to the nucleus for viral gene transcription, followed by genome replication, virion assembly and egress. Along with viral gene expression, 21 viral miRNA are also expressed during the viral life cycle [46]. The role of these miRNAs in viral life cycle is understudied. Here we discovered that HSV-1 induced NOTCH activity via HSV-1 encoded miR-H16, which has been shown to be abundantly expressed during productive infection [47, 48]. Proteomic RPPA analysis of miR-H16 transduced cells also revealed the activation of NOTCH pathway among the top signaling pathways affected by miR-H16 transfection. miR-H16 was found to directly target 3’UTR of FIH-1. FIH-1 was originally described as a cellular hydroxylase that hydroxylates the N803 residue of the HIF1α C-terminal activation domain to inhibit its activity. More recently it has been shown to have other substrates such as Iκβ and NICD [49] [25]. FIH-1 is thought to regulate NOTCH activity by directly affecting NICD in signal sending cells. The observation that FIH-1 regulates NOTCH signaling from inside signal sending cells has not been previously reported to our knowledge. While FIH-1 has been shown to directly interact with Mib1, here we report for the first time the impact of this interaction on regulating NOTCH signaling. Mib1 is a ubiquitin ligase that ubiquitinates NOTCH ligands which increases their trafficking into endosomes. This trafficking induce NOTCH receptor cleavage and activation in adjacent cells (Figure 5). Here we show that FIH-1 sequesters Mib1, and reduction of FIH-1 liberates Mib1 to ubiquitinate, NOTCH ligands and thus activate NOTCH signaling. Our finding that down-regulation of FIH-1 by the miR-H16 results in ubiquitylation of NOTCH ligands and increased NOTCH signaling is in agreement with these findings, implicating FIH-1 to negatively regulate NOTCH activity from signal sending cells to signal receiving cells [29].
Fig. 5. Graphical abstract of the study showing mechanism of HSV-1 induced NOTCH activation.
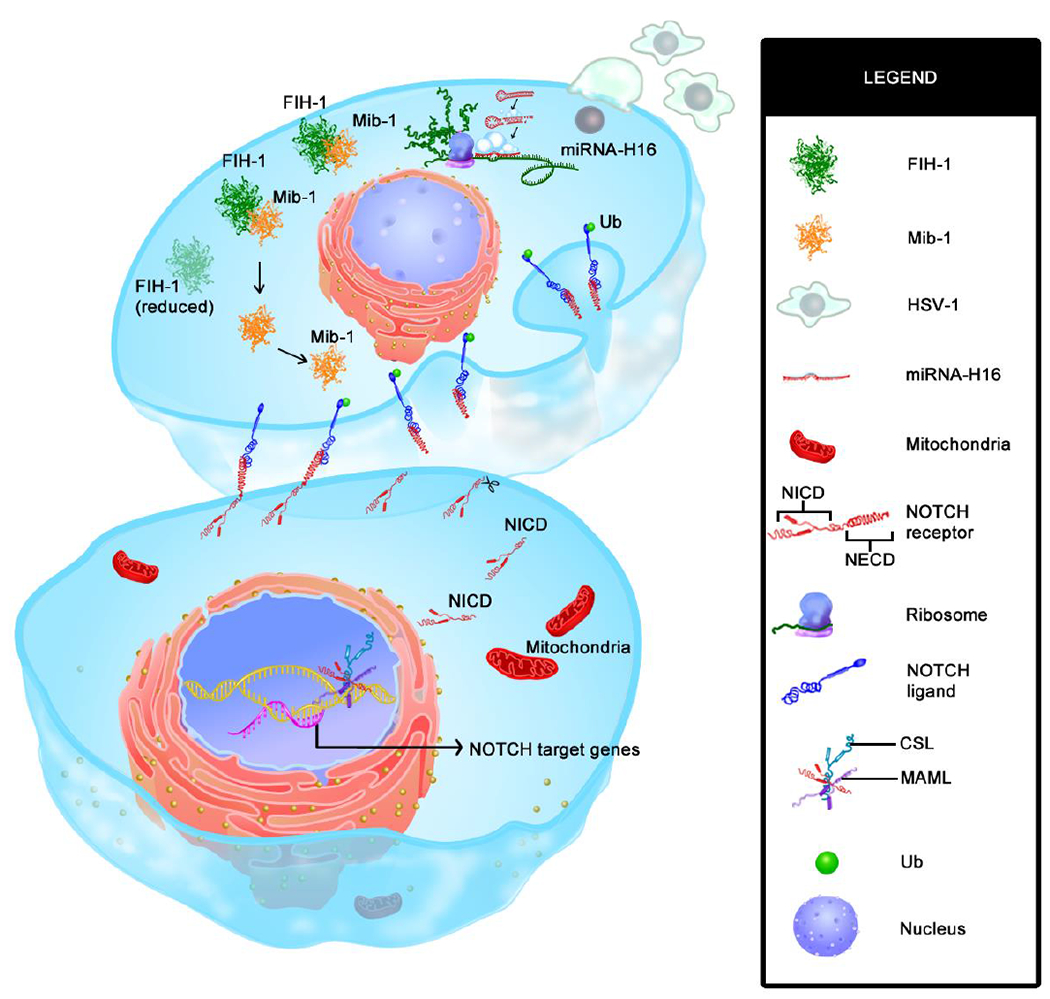
Infection of glioma cells with oHSV converts it into a signal sending cell that engages with adjacent signal receiving cell to promote gamma secretase mediated cleavage of NOTCH receptor to initiate NOTCH activation in bottom uninfected signal receiving cell. Image shows two adjacent tumor cells (top is infected and bottom cell is uninfected). Upon HSV-1 infection (top cell) virus encoded microRNA-H16 (miR-H16) targets FIH-1 3’UTR (green) which results in reduced amount of FIH-1 protein (faded green in top cell). FIH-1, normally binds and sequester mind bomb 1 (Mib1, shown in orange) which is known to ubiquitinate (green dot) Notch ligand (blue NOTCH ligand at the bottom of the top cell) bound to NOTCH receptor on adjacent cell. Reduction of FIH-1 release Mib1 to ubiquitinate NOTCH ligands. Ubiquitinated NOTCH ligand then traffics to endosomes, causing a physical pull that then exposes NOTCH receptor proteolytic cleavage sites, and cleavage by a disintegrin and metalloprotease (ADAM) Protease and γ secretase (scissors in bottom cell) resulting in eventual release of NOTCH intracellular domain (NICD). Upon release, NICD traffics to the nucleus to initiate NOTCH target gene transcription.
Inhibition of gamma secretase with RO4929097, a GSI to block NOTCH cleavage and activity resulted in sensitization of glioma cells to GSI after oHSV therapy and increased survival for glioma-bearing mice. Several GSI have been tested in clinical trials for various tumor types including glioma (Clinicaltrials.gov #NCT00572182, NCT01122901, NCT01119599 [50], NCT01189240 [51]), and numerous oHSV viruses are being tested in patients for safety and efficacy. Therefore, combining oHSV therapy with GSI can be rapidly translated into the clinic. Gamma secretase complex was first implicated to proteolytically process amyloid-β protein precursor, but since then 90 different substrates including NOTCH receptors have been identified. Thus gamma secretase inhibition is likely to have a wider effect than just blockade of NOTCH signaling. While here we have used molecular approaches (such as dnMAML, RBP-J driver reporters and detection of NICD etc) to clearly show NOTCH pathway activation with oHSV, future studies such as genetic deletion studies for NOTCH ligands/receptors etc will unveil the impact of NOTCH inhibition on the therapeutic benefit of GSI inhibitors with oHSV therapy [52].
In summary, we have discovered a novel role for the HSV-1 encoded miR-H16 as an activator of the NOTCH signaling pathway in GBM, and show that this natural HSV-1 mechanism can be exploited to sensitize neighboring uninfected glioma cells to GSI therapy, ultimately improving oHSV therapy for GBM.
Supplementary Material
Key points:
Oncolytic HSV treatment induce NOTCH signaling via HSV-1 encoded microRNA-H16
NOTCH activation in uninfected cells involve in cell proliferation
Inhibition of NOTCH by gamma-secretase inhibitor improve oncolytic HSV efficacy
Translational Relevance.
HSV-1 derived oncolytic viruses are increasingly being used in patients with melanoma (FDA approved therapy) and CNS malignancies as investigational agents. This is the first study to observe that HSV-1 therapy modifies brain tumor microenvironment to activate NOTCH signaling in adjacent tumor cells in vitro and in vivo. Mechanistically we observed that virus encoded miR-H16 targets FIH-1 mRNA and is important for NOTCH activation. miR-H16 mediated reduction of FIH-1 increased Mib1 mediated mono-ubiquitination of NOTCH ligands, which activated NOTCH signaling in adjacent NOTCH receptor bearing cells. Translationally treatment of mice bearing intracranial glioma with oHSV converted brain tumor cells that were resistant to gamma secretase inhibition, sensitive. Combination of NOTCH blocking gamma secretase inhibitor with oHSV improved therapeutic outcome in vivo. This study uncovers changes in brain tumor biology upon oHSV treatment and highlights the significance of combining oHSV with strategies to block NOTCH signaling.
Acknowledgements
We thank Li Xin (University of Washington) and Xiang Zhang (Baylor College of Medicine) for providing DN-MAML plasmid. pcDNA-FIH1 was a gift from Eric Metzen (Addgene plasmid # 21399; http://n2t.net/addgene:21399; RRID: Addgene_21399). p3HA-hMIB1 was a gift from Vanessa Redecke (Addgene plasmid #33317; http://n2t.net/addgene:33317; RRID: Addgene_33317). GFP-Ub was a gift from Nico Dantuma (Addgene plasmid #11928; http://n2t.net/addgene:11928; RRID: Addgene_11928). The images in Fig 5 were drawn by Beth Watson and Cynthia Reyna in UTHealth’s Multimedia Scriptorium, an on campus support group for faculty and staff.
Funding:
This work was supported by National Institute of Health (NIH) grants R01 NS064607, P01CA163205, R01 CA150153 to BK, and by the American Cancer Society Research Scholar Grant to JYY.
Abbreviations:
- ADAM
a disintegrin and metalloprotease
- FIH-1
factor inhibiting HIF-1
- Mib1
mind bomb 1
- miR-H16
microRNA-H16
- NICD
NOTCH intracellular domain
Footnotes
Conflict of interest disclosure statement:
The authors have declared that no conflict of interest exists.
Reference.
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med, 2005. 352(10): p. 987–96. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Taillibert S, Kanner A, Read W, Steinberg DM, Lhermitte B, et al. , Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Jama, 2017. 318(23): p. 2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guruharsha KG, Kankel MW, and Artavanis-Tsakonas S, The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet, 2012. 13(9): p. 654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai EC, Notch signaling: control of cell communication and cell fate. Development, 2004. 131(5): p. 965–73. [DOI] [PubMed] [Google Scholar]
- 5.Park NI, Guilhamon P, Desai K, McAdam RF, Langille E, O’Connor M, et al. , ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell, 2017. 21(2): p. 209–224.e7. [DOI] [PubMed] [Google Scholar]
- 6.Xu P, Yu S, Jiang R, Kang C, Wang G, Jiang H, et al. , Differential expression of Notch family members in astrocytomas and medulloblastomas. Pathol Oncol Res, 2009. 15(4): p. 703–10. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Cui Y, Gao G, Zhao Z, Zhang H, and Wang X, Notch1 is an independent prognostic factor for patients with glioma. J Surg Oncol, 2011. 103(8): p. 813–7. [DOI] [PubMed] [Google Scholar]
- 8.Qiu XX, Chen L, Wang CH, Lin ZX, Chen BJ, You N, et al. , The Vascular Notch Ligands Delta-Like Ligand 4 (DLL4) and Jagged1 (JAG1) Have Opposing Correlations with Microvascularization but a Uniform Prognostic Effect in Primary Glioblastoma: A Preliminary Study. World Neurosurg, 2016. 88: p. 447–58. [DOI] [PubMed] [Google Scholar]
- 9.Xu P, Zhang A, Jiang R, Qiu M, Kang C, Jia Z, et al. , The different role of Notch1 and Notch2 in astrocytic gliomas. PLoS One, 2013. 8(1): p. e53654. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, et al. , Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell, 2017. 20(2): p. 233–246.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yahyanejad S, Theys J, and Vooijs M, Targeting Notch to overcome radiation resistance. Oncotarget, 2016. 7(7): p. 7610–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehman H, Silk AW, Kane MP, and Kaufman HL, Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer, 2016. 4: p. 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohlhapp FJ and Kaufman HL, Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin Cancer Res, 2016. 22(5): p. 1048–54. [DOI] [PubMed] [Google Scholar]
- 14.Bernstock JD, Wright Z, Bag AK, Gessler F, Gillespie GY, Markert JM, et al. , Stereotactic Placement of Intratumoral Catheters for Continuous Infusion Delivery of Herpes Simplex Virus −1 G207 in Pediatric Malignant Supratentorial Brain Tumors. World Neurosurg, 2019. 122: p. e1592–e1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu R, Li X, Tulpule A, Zhou Y, Scehnet JS, Zhang S, et al. , KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood, 2010. 115(4): p. 887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoo JY, Haseley A, Bratasz A, Chiocca EA, Zhang J, Powell K, et al. , Antitumor efficacy of 34.5ENVE: a transcriptionally retargeted and “Vstat120”-expressing oncolytic virus. Mol Ther, 2012. 20(2): p. 287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, et al. , Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci, 2003. 116(Pt 7): p. 1319–26. [DOI] [PubMed] [Google Scholar]
- 18.Dantuma NP, Groothuis TA, Salomons FA, and Neefjes J, A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol, 2006. 173(1): p. 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutkowski AJ, Erhard F, L’Hernault A, Bonfert T, Schilhabel M, Crump C, et al. , Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun, 2015. 6: p. 7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Q, Wu Q, Kim L, Miller TE, Liau BB, Mack SC, et al. , RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J Clin Invest, 2016. 126(7): p. 2757–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitagawa M, Notch signalling in the nucleus: roles of Mastermind-like (MAML) transcriptional coactivators. J Biochem, 2016. 159(3): p. 287–94. [DOI] [PubMed] [Google Scholar]
- 22.Valdez JM, Zhang L, Su Q, Dakhova O, Zhang Y, Shahi P, et al. , Notch and TGFbeta form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitor cell activity. Cell Stem Cell, 2012. 11(5): p. 676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. , Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol, 2016. 18(6): p. 632–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai HS, Griffin N, Bolyard C, Mao HC, Zhang J, Cripe TP, et al. , The Fc Domain of Immunoglobulin Is Sufficient to Bridge NK Cells with Virally Infected Cells. Immunity, 2017. 47(1): p. 159–170 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, et al. , Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A, 2008. 105(9): p. 3368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jehn BM, Dittert I, Beyer S, von der Mark K, and Bielke W, c-Cbl binding and ubiquitin-dependent lysosomal degradation of membrane-associated Notch1. J Biol Chem, 2002. 277(10): p. 8033–40. [DOI] [PubMed] [Google Scholar]
- 27.Ishitani T, Hirao T, Suzuki M, Isoda M, Ishitani S, Harigaya K, et al. , Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat Cell Biol, 2010. 12(3): p. 278–85. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Heredia JM, Verdugo Sivianes EM, Lucena-Cacace A, Molina-Pinelo S, and Carnero A, Numb-like (NumbL) downregulation increases tumorigenicity, cancer stem cell-like properties and resistance to chemotherapy. Oncotarget, 2016. 7(39): p. 63611–63628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiriakidis S, Henze AT, Kruszynska-Ziaja I, Skobridis K, Theodorou V, Paleolog EM, et al. , Factor-inhibiting HIF-1 (FIH-1) is required for human vascular endothelial cell survival. Faseb j, 2015. 29(7): p. 2814–27. [DOI] [PubMed] [Google Scholar]
- 30.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. , Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell, 2005. 9(5): p. 617–28. [DOI] [PubMed] [Google Scholar]
- 31.Wilkins SE, Hyvarinen J, Chicher J, Gorman JJ, Peet DJ, Bilton RL, et al. , Differences in hydroxylation and binding of Notch and HIF-1alpha demonstrate substrate selectivity for factor inhibiting HIF-1 (FIH-1). Int J Biochem Cell Biol, 2009. 41(7): p. 1563–71. [DOI] [PubMed] [Google Scholar]
- 32.So JH, Kim JD, Yoo KW, Kim HT, Jung SH, Choi JH, et al. , FIH-1, a novel interactor of mindbomb, functions as an essential anti-angiogenic factor during zebrafish vascular development. PLoS One, 2014. 9(10): p. e109517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tseng LC, Zhang C, Cheng CM, Xu H, Hsu CH, and Jiang YJ, New classes of mind bomb-interacting proteins identified from yeast two-hybrid screens. PLoS One, 2014. 9(4): p. e93394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh M, Kim CH, Palardy G, Oda T, Jiang YJ, Maust D, et al. , Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev Cell, 2003. 4(1): p. 67–82. [DOI] [PubMed] [Google Scholar]
- 35.Koo BK, Lim HS, Song R, Yoon MJ, Yoon KJ, Moon JS, et al. , Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development, 2005. 132(15): p. 3459–70. [DOI] [PubMed] [Google Scholar]
- 36.Huszthy PC, Immervoll H, Wang J, Goplen D, Miletic H, Eide GE, et al. , Cellular effects of oncolytic viral therapy on the glioblastoma microenvironment. Gene Ther, 2010. 17(2): p. 202–16. [DOI] [PubMed] [Google Scholar]
- 37.Hai L, Zhang C, Li T, Zhou X, Liu B, Li S, et al. , Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-kappaB(p65) pathway. Cell Death Dis, 2018. 9(2): p. 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Xu SL, Duan JJ, Yi L, Guo YF, Shi Y, et al. , Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat Neurosci, 2019. 22(1): p. 91–105. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. , Notch promotes radioresistance of glioma stem cells. Stem Cells, 2010. 28(1): p. 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vermezovic J, Adamowicz M, Santarpia L, Rustighi A, Forcato M, Lucano C, et al. , Notch is a direct negative regulator of the DNA-damage response. Nat Struct Mol Biol, 2015. 22(5): p. 417–24. [DOI] [PubMed] [Google Scholar]
- 41.Hiddingh L, Tannous BA, Teng J, Tops B, Jeuken J, Hulleman E, et al. , EFEMP1 induces gamma-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget, 2014. 5(2): p. 363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vazquez-Ulloa E, Lizano M, Sjoqvist M, Olmedo-Nieva L, and Contreras-Paredes A, Deregulation of the Notch pathway as a common road in viral carcinogenesis. Rev Med Virol, 2018. 28(5): p. e1988. [DOI] [PubMed] [Google Scholar]
- 43.DeCotiis JL and Lukac DM, KSHV and the Role of Notch Receptor Dysregulation in Disease Progression. Pathogens, 2017. 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Persson LM and Wilson AC, Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J Virol, 2010. 84(3): p. 1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ting HA, de Almeida Nagata D, Rasky AJ, Malinczak CA, Maillard IP, Schaller MA, et al. , Notch ligand Delta-like 4 induces epigenetic regulation of Treg cell differentiation and function in viral infection. Mucosal Immunol, 2018. 11(5): p. 1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun L and Li Q, The miRNAs of herpes simplex virus (HSV). Virol Sin, 2012. 27(6): p. 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du T, Han Z, Zhou G, and Roizman B, Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc Natl Acad Sci U S A, 2015. 112(1): p. E49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, et al. , Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol, 2010. 84(9): p. 4659–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, et al. , Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci U S A, 2006. 103(40): p. 14767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu R, Shimizu F, Hovinga K, Beal K, Karimi S, Droms L, et al. , Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin Cancer Res, 2016. 22(19): p. 4786–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan E, Supko JG, Kaley TJ, Butowski NA, Cloughesy T, Jung J, et al. , Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J Neurooncol, 2016. 130(3): p. 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haapasalo A and Kovacs DM, The many substrates of presenilin/gamma-secretase. J Alzheimers Dis, 2011. 25(1): p. 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.