

Abstract
Communication between myeloid cells and epithelium plays critical role in maintaining intestinal epithelial barrier integrity. Myeloid cells interact with intestinal epithelial cells (IECs) by producing various mediators; however, the molecules mediating their crosstalk remain incompletely understood. Here, we report that deficiency of angiogenin (Ang) in mouse myeloid cells caused impairment of epithelial barrier integrity, leading to high susceptibility to DSS‐induced colitis. Mechanistically, myeloid cell‐derived angiogenin promoted IEC survival and proliferation through plexin‐B2‐mediated production of tRNA‐derived stress‐induced small RNA (tiRNA) and transcription of ribosomal RNA (rRNA), respectively. Moreover, treatment with recombinant angiogenin significantly attenuated the severity of experimental colitis. In human samples, the expression of angiogenin was significantly down‐regulated in patients with inflammatory bowel disease (IBD). Collectively, we identified, for the first time to our knowledge, a novel mediator of myeloid cell‐IEC crosstalk in maintaining epithelial barrier integrity, suggesting that angiogenin may serve as a new preventive agent and therapeutic target for IBD.
Keywords: angiogenin, inflammatory bowel disease, intestinal epithelial cell, myeloid cell
Subject Categories: Molecular Biology of Disease, RNA Biology, Signal Transduction
Stromal cell‐secreted RNase angiogenin regulates tRNA and rRNA metabolism in the intestinal epithelium to protect against inflammation. Stromal cell‐secreted RNase angiogenin regulates tRNA and rRNA metabolism in the intestinal epithelium to protect against inflammation.
Introduction
Inflammatory bowel disease (IBD), typically including Crohn's disease (CD) and ulcerative colitis (UC), is characterized with epithelial barrier integrity impairment and intestinal inflammation (Torres et al, 2017; Ungaro et al, 2017). Epithelial barrier damage leads to an excessive exposure of mucosal tissue to microbial antigens, followed by leukocyte recruitment, soluble mediator release, and ultimately mucosal inflammation (Peterson & Artis, 2014; Martini et al, 2017). A key event in epithelial barrier maintenance is to balance survival and growth of IECs, which is orchestrated by both epithelium itself and recruited immune cells in mucosal tissue (Negroni et al, 2015; Luissint et al, 2016). Mucosal innate myeloid leukocytes, such as neutrophils, monocytes, and macrophages, have been found to reside near the epithelium and interact with IECs via mediators in normal condition and during inflammation (Bain & Mowat, 2014; Grainger et al, 2017). Therefore, understanding the crosstalk between myeloid cells and IECs will provide valuable insight into dysregulation of intestinal homeostasis and IBD pathogenesis.
Several pro‐inflammatory mediators, such as IFN‐γ and TNF‐α, are known to induce IEC apoptosis (Marini et al, 2003; Nava et al, 2010). By contrast, other mediators, such as IL‐6, promote IEC activation and survival (Grivennikov et al, 2009). However, therapeutic approaches designed to augment IEC functions by targeting these molecules have limited effectiveness, mainly due to the resultant immune imbalance in the intestine. For example, aside from enhancing IEC proliferation, IL‐27 also exerts a pro‐inflammatory effect by inducing T‐cell activation and TH1‐type cytokine production, limiting its direct application in IBD patients (Diegelmann et al, 2012). IL‐6 can stimulate proliferation and survival of IECs, but simultaneously activate other target cells, including antigen‐presenting cells and T cells (Atreya et al, 2000; Grivennikov et al, 2009). Therefore, it would be beneficial to identify novel myeloid cell‐derived factors that improve epithelial barrier integrity in IBD with more specific functions and less inflammatory side‐effects.
Angiogenin (ANG), a secretory vertebrate‐specific ribonuclease, enhances cell growth and survival by regulating intracellular RNA processing (Sheng & Xu, 2016). The secreted ANG is endocytosed in target cells via its receptor plexin‐B2 (PLXNB2) and undergoes context‐specific subcellular localization (Goncalves, Silberstein et al, 2016; Yu et al, 2017). Under growth conditions, ANG accumulates in the nucleus where it regulates rRNA transcription to promote cell proliferation (Moroianu & Riordan, 1994; Xu et al, 2002). Upon stress stimulation, ANG translocates to cytosol where it cleaves tRNAs to produce tRNA‐derived stress‐induced small RNAs (tiRNAs), resulting in reprogramming of protein translation and inhibition of apoptosome formation, thereby promoting cell survival (Fu et al, 2009; Ivanov et al, 2011; Goncalves et al, 2016; Li et al, 2018b). This protein may play a role in IBD pathogenesis. Two studies reported that serum ANG is significantly elevated in IBD patients (Koutroubakis et al, 2004; Oikonomou et al, 2011), while a third one found the opposite result (Magro et al, 2004). To explore the function of ANG in IBD, we first established their correlation in IBD patient samples and DSS‐induced experimental colitis and then uncovered that ANG mediates a crosstalk between myeloid cells and IECs to maintain epithelial barrier integrity.
Results
ANG is down‐regulated in IBD patient samples
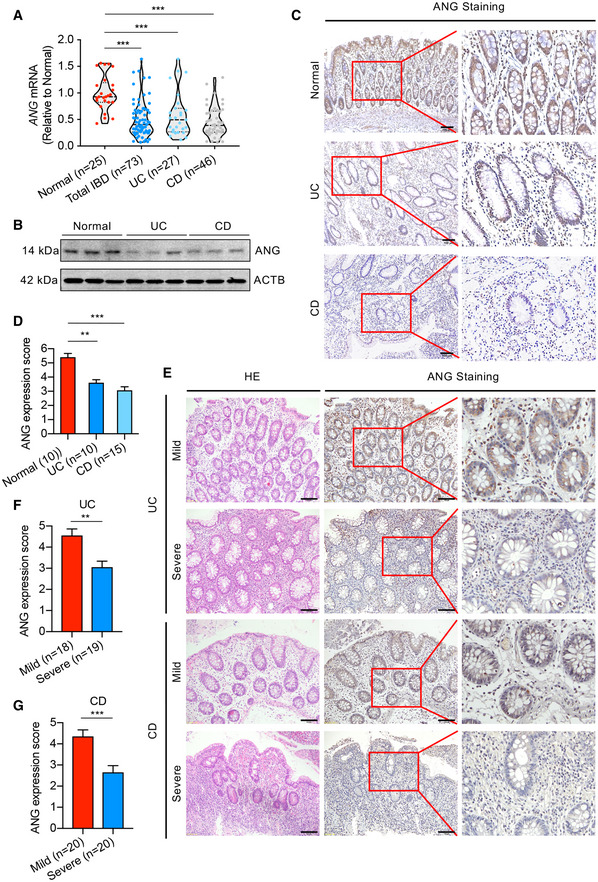
To establish the correlation of ANG with IBD progression, we first collected colonic tissue samples from IBD patients and measured ANG expression level. The results showed that ANG mRNA was significantly down‐regulated in IBD patients (including UC and CD) compared to normal controls (Fig 1A). Western blotting and immunohistochemical (IHC) staining confirmed that ANG protein was reduced in IBD samples (Fig 1B–D). We further evaluated ANG expression in mild and severe IBD patients with IHC staining (Fig 1E) and found that ANG levels were significantly decreased in severe colitis samples compared to those in the mild ones (Fig 1F and G). These data indicate that ANG expression is down‐regulated in IBD tissue and inversely correlated with IBD progression.
Figure 1. ANG is down‐regulated in IBD patient samples.
-
AmRNA expression of ANG in colonic tissues from normal controls (n = 25) and UC (n = 27) or CD (n = 46) patients.
-
BRepresentative Western blotting of ANG in colonic tissues from normal controls and UC or CD patients.
-
CRepresentative images showing ANG immunohistochemical (IHC) staining in normal control and UC or CD sample.
-
DCorresponding statistical analysis of ANG expression score in normal controls (n = 10) and UC (n = 10) or CD (n = 15) patients.
-
ERepresentative images showing hematoxylin–eosin (HE) staining and ANG IHC staining in mild and severe IBD samples.
-
F, GStatistical analyses of ANG expression scores in mild (n = 18) and severe (n = 19) UC (F) or mild (n = 20) and severe (n = 20) CD (G) patients.
Ang‐deficient mice are hyper‐susceptible to DSS‐induced experimental colitis
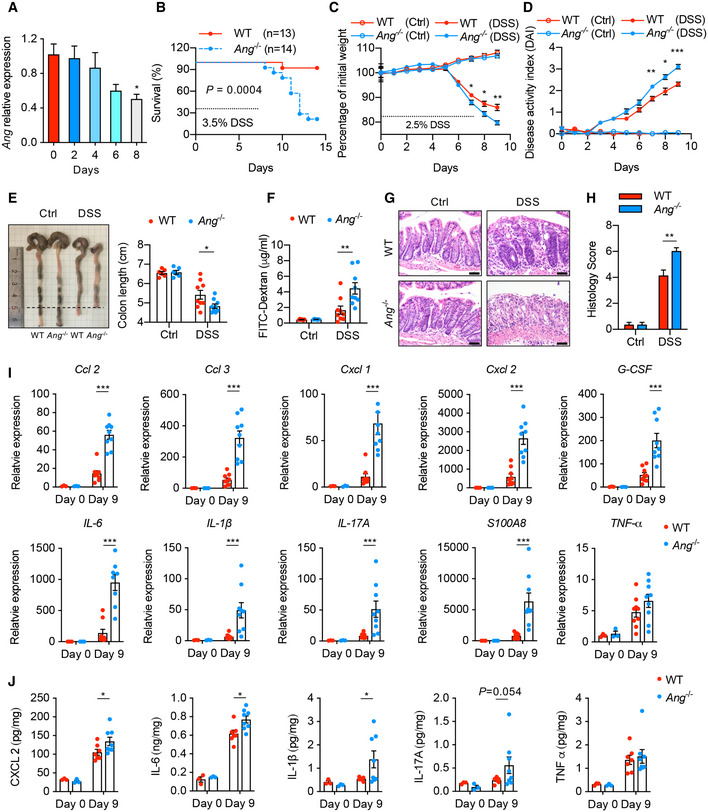
To validate the relationship between ANG and IBD, we established a colitis mouse model with DSS induction. Following 2.5% DSS treatment, Ang gradually declined in inflammatory colon tissue (Fig 2A), suggesting a potential functional role of ANG in colitis development. Although Ang −/− mice did not display any aberrant inflammation or overt colon phenotype at baseline, strikingly, their survival rate was significantly diminished compared to WT littermates after challenging with 3.5% DSS (Fig 2B). To carefully monitor disease progression, both mice were administered with 2.5% DSS for 7 days. Compared to their WT counterparts, Ang −/− mice exhibited obvious daily weight loss (Fig 2C), increased disease activity index (DAI) (Fig 2D), shorter colon length (Fig 2E), and enhanced intestinal permeability (Fig 2F) on day 9 when they were sacrificed. Consistent with these findings, hematoxylin and eosin (HE) staining showed a dramatically aggravated colonic histopathology, including inflammatory infiltrate and ulceration formation (Fig 2G and H). Meanwhile, Ang deficiency led to a significant increase in mRNAs and secretion of pro‐inflammatory cytokines in mucosal tissue (Fig 2I and J). These results reveal that ANG attenuates DSS‐induced colitis in mice. Combined with the down‐regulation of ANG in inflamed patient colon, our data suggest that ANG acts as a protective factor in the development of inflammation.
Figure 2. Ang deficiency enhances susceptibility to DSS‐induced colitis in mice.
-
AAng mRNA expression in colonic tissue from 2.5% DSS‐treated WT mice at indicated time point (n = 4 mice/time point).
-
BKaplan–Meier curve of 3.5% DSS‐treated Ang‐deficient mice (Ang −/−, n = 14) or littermate controls (WT, n = 13).
-
C, D(C) Body weight loss and (D) disease activity index (DAI) of WT and Ang −/− mice with (n = 9) or without (n = 6) 2.5% DSS treatment.
-
E–H(E) Colon length, (F) serum FITC‐dextran level, (G) representative HE staining image, and (H) histopathological score of colonic section from the WT and Ang −/− mice on day 9 with (n = 9) or without (n = 6) 2.5% DSS treatment.
-
IQuantitative mRNA expression of cytokine genes in colonic tissue from the WT and Ang −/− mice on day 0 (n = 3) or day 9 (n = 9) during 2.5% DSS treatment.
-
JSoluble cytokine level in supernatant of cultured colonic tissue isolated from WT and Ang −/− mice on day 0 (n = 3) or day 9 (n = 7) during 2.5% DSS treatment.
Deficiency of Ang in myeloid cells accounts for the aggravated colitis
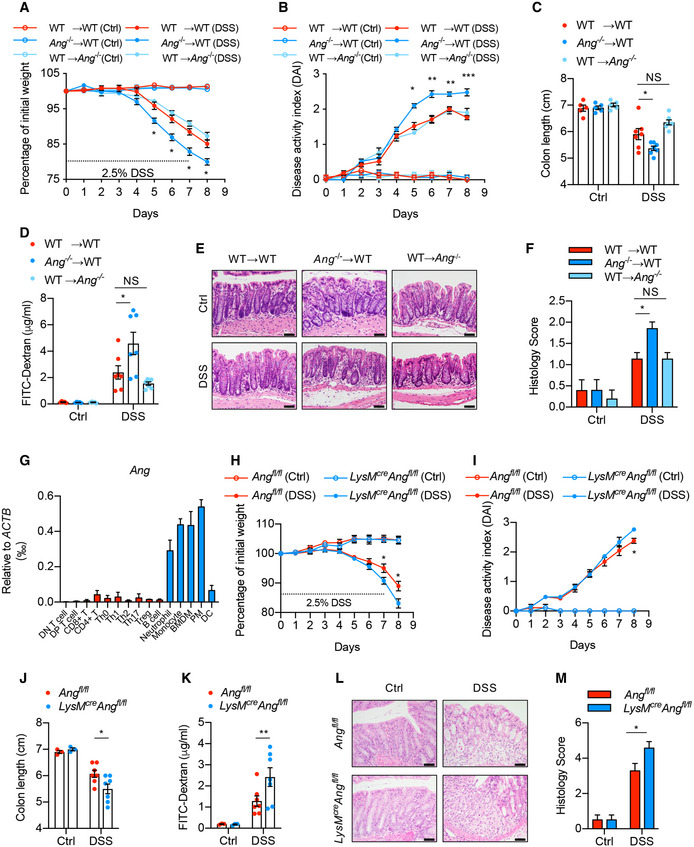
Hematopoietic stem and progenitor cells contribute to inflammation. Previous work has shown that ANG differentially regulates the fate of both cells (Goncalves et al, 2016), prompting us to ask whether the increased colitis observed in Ang −/− mice was due to the dysregulation of these two cell types. Therefore, we established reconstituted mouse models with either WT or Ang‐null hematopoietic cells by transferring WT (CD45.2) or Ang −/− (CD45.2) bone marrow‐derived cells to WT (CD45.1) recipients (WT→WT and Ang −/−→WT) and subjected them to DSS treatment. Similar to those observed in Ang −/− mice, the mice reconstituted with Ang‐deficient hematopoietic cells (Ang −/−→WT) exhibited exacerbated weight loss (Fig 3A), increased DAI score (Fig 3B), shorter colon length (Fig 3C), enhanced intestinal permeability (Fig 3D), more severe histological score (Fig 3E and F), and increased expression of cytokines in the colonic tissue (Fig EV1A), suggesting that hematopoietic cell‐derived ANG protects the mice against colitis progression. To further discriminate the relative contribution of primitive and differentiated hematopoietic cells, we assessed bone marrow reconstitution capacity. Consistent with a previous finding (Goncalves et al, 2016), Ang deficiency in hematopoietic cells did not change the differentiation capability of primitive cells, as reflected by similar percentage of myeloid cells, T cells, and B cells in WT recipients’ peripheral blood (Fig EV1B), suggesting that it is the ANG derived from differentiated immune cells responsible for the increased colitis. On the other hand, it is known that intestinal epithelial cells can also express and secrete ANG. To evaluate the contribution of epithelium‐derived ANG to colitis, we reconstituted Ang −/− mice with WT hematopoietic cells (WT→Ang −/−) or conditionally knocked out Ang in epithelial cells (i.e., Villin cre ;Ang fl/fl mice) and then subjected them to colitis induction. The data clearly showed that there were no differences between (WT→WT) and (WT→Ang −/−) (Fig 3A–F) or between WT and Villin cre ;Ang fl/fl mice (Fig EV2A–C), further supporting that hematopoietic rather than epithelial cell‐derived ANG protects mice from intestine inflammation.
Figure 3. Deficiency of Ang in myeloid cells accounts for the aggravated colitis.
-
A, B(A) Body weight loss and (B) DAI of WT→WT, Ang −/−→WT and WT→Ang −/− chimera mice with (n = 7) or without (n = 5) 2.5% DSS treatment.
-
C–F(C) Colon length, and (D) serum FITC‐dextran level, (E) representative HE staining image, and (F) histopathological score of colonic section from these chimera colitis mice on day 8 with (n = 7) or without (n = 5) 2.5% DSS treatment.
-
GAng mRNA expression in mouse lymphoid and myeloid cells; data represent two independent experiments (n = 2).
-
H, I(H) Body weight loss and (I) DAI of Ang fl/fl and LyzM cre ;Ang fl/fl mice with (n = 7) or without (n = 3) 2.5% DSS treatment.
-
J–M(J) Colon length, (K) serum FITC‐dextran level, (L) representative HE staining image, and (M) histopathological score of colonic section from the Ang fl/fl and LyzM cre ;Ang fl/fl mice on day 8 with (n = 7) or without (n = 3) 2.5% DSS treatment.
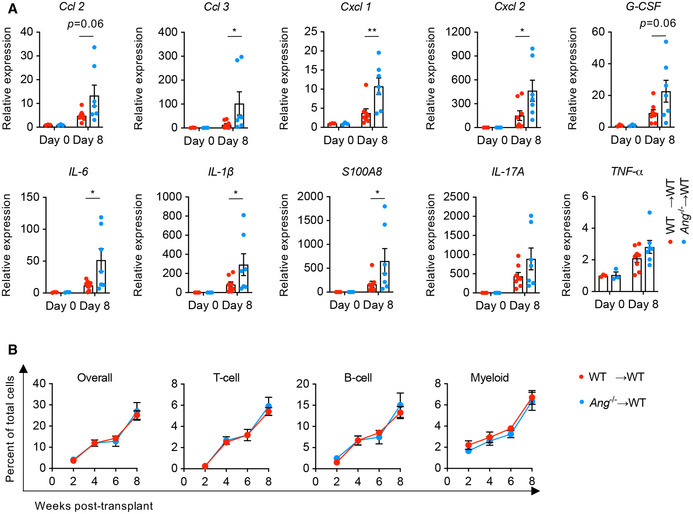
Figure EV1. Colonic cytokine production and multilineage reconstitution in chimera mice.
-
AQuantitative mRNA expression of cytokine genes in colonic tissue from WT→WT and Ang −/−→WT chimera mice on day 0 (n = 3) or day 8 (n = 7‐8) during DSS treatment.
-
BMultilineage reconstitution analysis in chimera mice (n = 5).
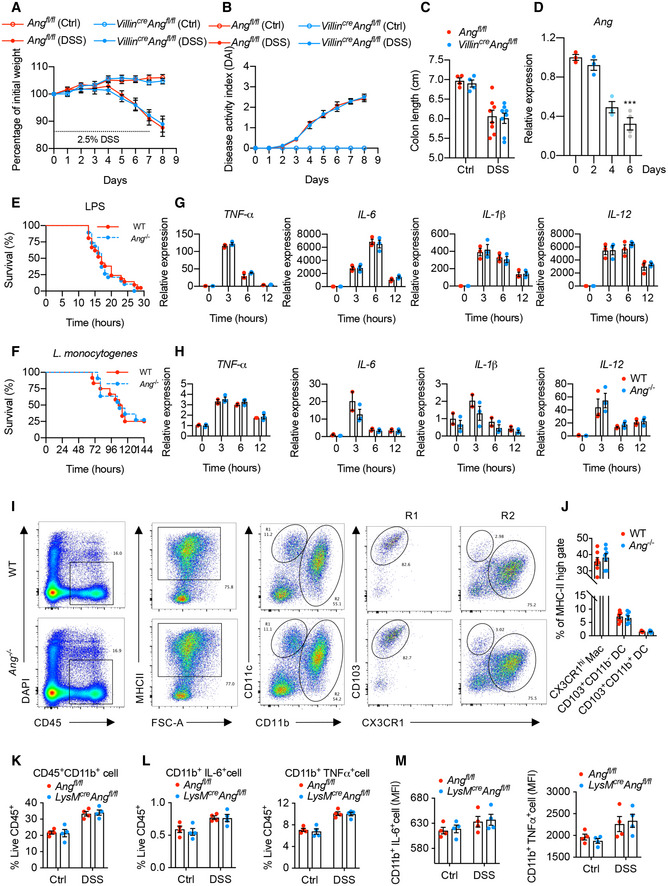
Figure EV2. Myeloid‐derived ANG plays a non‐cell‐autonomous role in attenuating DSS‐induced colitis.
-
A, B(A) Body weight loss and (B) DAI of Ang fl/fl and Villin cre ;Ang fl/fl mice with (n = 8) or without (n = 5) 2.5% DSS treatment.
-
CColon length of the Ang fl/fl and Villin cre ;Ang fl/fl mice on day 8 with (n = 8) or without (n = 5) 2.5% DSS treatment.
-
DAng mRNA expression in colonic myeloid cells from 2.5% DSS‐treated WT mice at indicated time point. (n = 3–4/time point).
-
EKaplan–Meier curve of WT (n = 21) or Ang −/− (n = 19) mice in LPS‐induced endotoxin shock model.
-
FKaplan–Meier curves of WT (n = 12) or Ang −/− (n = 12) mice in Listeria monocytogenes (LM)‐induced sepsis model.
-
G, HmRNA expression of cytokine genes in BMDMs from WT or Ang −/− mice administrated with (G) LPS or (H) synthetic RNA duplex poly(I:C) at indicated time point (n = 2–3/time point per group).
-
I, J(I) Representative plot of flow cytometry analysis and (J) the percentages of macrophage and dendritic cell subsets in cLP of WT or Ang −/− mice (n = 6) in steady state.
-
K, LThe percentages of CD45+CD11b+ (K), CD11b+IL6+, and CD11b+TNFα+ (L) subsets in LPS‐stimulated cLP of Ang fl/fl or LyzM cre ;Ang fl/fl mice (n = 4) with DSS treatment (day 0 and 4).
-
MMFI of CD11b+IL6+ and CD11b+TNFα+ in LPS‐stimulated cLP of Ang fl/fl or LyzM cre ;Ang fl/fl mice with DSS treatment (day 0 and 4) (n = 4).
To further identify the target cell types, we measured Ang expression levels in differentiated immune cells and found that it was highly expressed in myeloid cells, especially in monocytes and macrophages (Fig 3G), and was gradually reduced in colonic myeloid cells following DSS treatment (Fig EV2D). To directly determine the role of myeloid cell‐derived ANG in colitis mitigation, mice (LysM cre ;Ang fl/fl) with conditional Ang knockout in myeloid cells were generated and then administrated with DSS. These mice exhibited exacerbated colitis compared to controls (Fig 3H–M). Taken together, these data reveal that Ang expression in myeloid cells is critical for protecting mice against DSS‐induced colitis.
Myeloid‐derived ANG plays a non‐cell‐autonomous role in colitis attenuation
Myeloid cells play key role in innate immune response. To explore whether ANG participates in this process, we established lipopolysaccharide (LPS)‐triggered endotoxin shock and Listeria monocytogenes‐induced sepsis models and found that there were no significant differences in survival rate between WT and Ang −/− mice in both models (Fig EV2E and F). On the other hand, in vitro stimulation of bone marrow‐derived macrophages (BMDM) from both mice with LPS or synthetic RNA duplex poly(I:C) induced a similar extent of pro‐inflammatory cytokine production (Fig EV2G and H). These data indicate that Ang deficiency in myeloid cells has no significant influence on systemic innate immune response.
To determine whether ANG affects mononuclear phagocytes (MPs) composition in the colonic lamina propria (cLP), we did phenotypic analysis with flow cytometry. The data showed that there were no differences in the frequencies of CX3CR1hiCD11b+ macrophages, CD103+CD11b− dendritic cells (DCs), and CD103+CD11b+ DCs between WT and Ang −/− mice in the steady state (Fig EV2I and J), and 4‐day DSS treatment induced a comparable degree of myeloid cells infiltration into cLP in Ang fl/fl and LysM Cre ;Ang fl/fl mice (Fig EV2K). Furthermore, these mice exhibited a similar alteration in both the frequency and the mean fluorescence intensity (MFI, indicating the production of cytokines) of IL‐6+CD11b+ and TNF‐α+CD11b+ cells in cLP upon LPS stimulation (Fig EV2L and M), suggesting Ang has no influence on the activation of colonic myeloid cells under both steady and inflammatory states.
Taken together, although peculiarly expressed in the myeloid cells, Ang is dispensable for the inflammatory cytokine production in systemic innate immune response and the composition and activation of myeloid cells in local mucosal immune system, indicating that myeloid‐derived ANG plays a non‐cell‐autonomous role in the attenuation of DSS‐induced colitis.
ANG‐PLXNB2 axis is essential for the colitis attenuation
PLXNB2 was recently found to be a functional receptor to the secretory ANG protein (Yu et al, 2017), giving rise to the possibility that myeloid cell‐derived ANG may mitigate colitis in a paracrine manner via PLXNB2. To determine the potential ANG‐targeting cells in colonic tissue, we evaluated the expression of ANG and PLXNB2 in mouse colon and found that ANG was mainly expressed in cLP cells, whereas PLXNB2 was highly expressed in IECs (Fig 4A–C). Similar results were also observed in human colonic tissue (Fig EV3A and B). These data suggest a potential crosstalk between myeloid cells and IECs via the ANG‐PLXNB2 axis.
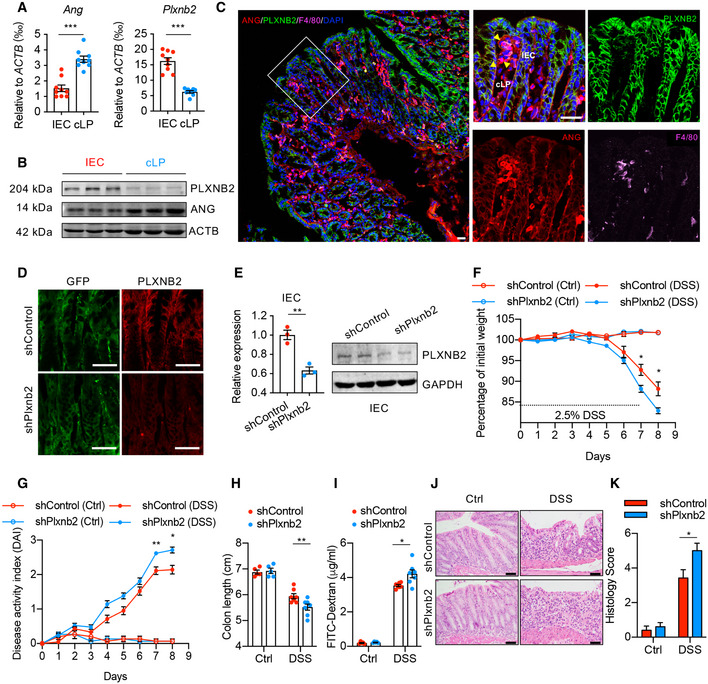
Figure 4. ANG‐PLXNB2 axis is essential for mitigating DSS‐induced colitis in mice.
-
A, B(A) Quantitative mRNA expression and (B) Western blotting of ANG and PLXNB2 in isolated IECs or colonic laminar propria (cLP) cells from WT mice (n = 9).
-
CRepresentative images showing immunofluorescence staining of ANG, PLXNB2, and macrophage surface marker F4/80 in frozen colonic section from WT mice.
-
DRepresentative images showing immunofluorescence staining of GFP and PLXNB2 in frozen colonic section from WT mice infected with adeno‐associated virus (AAV9).
-
EPLXNB2 expression in IECs from the AAV9‐infected WT mice (n = 3).
-
F, G(F) Body weight loss and (G) DAI of the AA9‐infected WT mice with (n = 7) or without (n = 5) 2.5% DSS treatment.
-
H–K(H) Colon length, (I) serum FITC‐dextran level, (J) representative HE staining image, and (K) histopathological score of colonic section from the AAV9‐infected WT mice on day 8 with (n = 7) or without (n = 5) 2.5% DSS treatment.
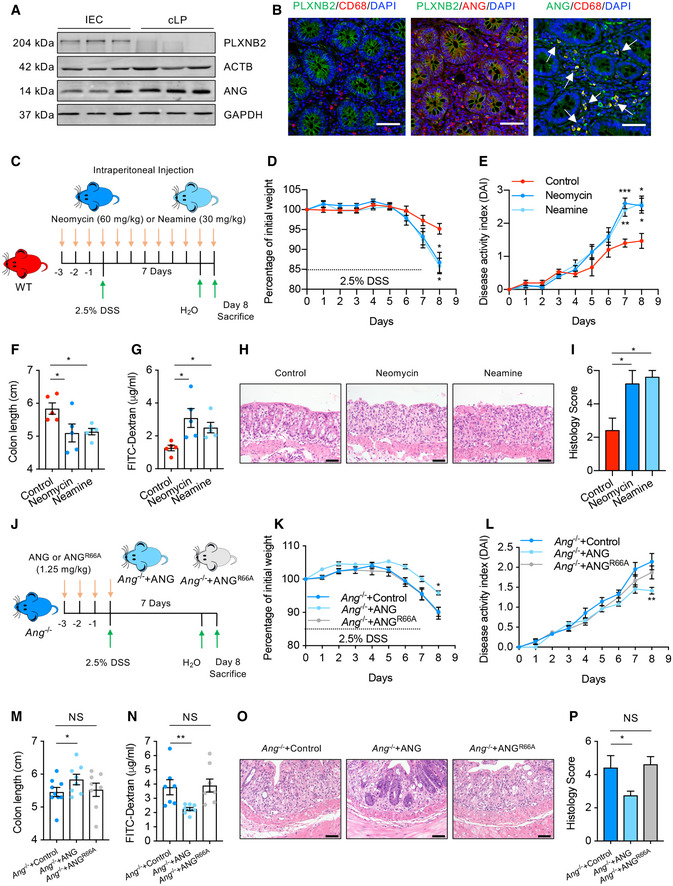
Figure EV3. Blockade of ANG‐PLXNB2 axis aggravates DSS‐induced colitis in mice.
-
AWestern blotting of ANG and PLXNB2 in isolated IECs or cLP cells from human colonic tissue.
-
BRepresentative images showing immunofluorescence staining of ANG, PLXNB2, and macrophage surface marker CD68 in frozen human colonic section derived from the same tissue sample.
-
CSchematic diagram of neomycin or neamine treatment in DSS‐induced colitis model (n = 5).
-
D, EThe effects of neomycin or neamine on body weight loss (D) and DAI (E) of WT colitis mice (n = 5).
-
F–I(F) Colon length, (G) serum FITC‐dextran level, (H) representative HE staining image, and (I) histopathological score of colonic section from neomycin‐ or neamine‐treated WT colitis mice on day 8 (n = 5).
-
JSchematic diagram of the ANG rescue experiment in Ang −/− colitis mice. The mice were pre‐treated with 1.25 mg/kg recombinant ANGWT or ANGR66A protein and then subjected to DSS induction (n = 8).
-
K, L(K) Body weight loss and (L) DAI of the ANGWT‐ or ANGR66A‐ pre‐treated Ang −/− colitis mice (n = 8).
-
M–P(M) Colon length, (N) serum FITC‐dextran level, (O) representative HE staining image, and (P) histopathological scores of colonic section from ANGWT‐ or ANGR66A‐pre‐treated Ang −/− colitis mice (n = 8) on day 8.
Therefore, we adopted three different approaches to manipulate the interaction between ANG and PLXNB2: (i) knocking‐down Plxnb2 with shRNA in WT mice, (ii) blocking their interaction using neomycin or neamine in WT mice, and (iii) ANG rescue experiment in Ang −/− mice with either ANG or its PLXNB2 binding deficient variant (ANGR66A). Interfering with the ANG‐PLXNB2 axis dramatically aggravated DSS‐induced colitis in WT mice (Figs 4D–K and EV3C–I), while only the recombinant ANG, but not the mutant ANGR66A, protected Ang −/− mice against colitis (Fig EV3J–P). These data clearly demonstrate the importance of ANG‐PLXNB2 axis in the crosstalk between myeloid cells and IECs.
ANG protects intestinal epithelial integrity by enhancing IEC survival and proliferation
The ANG‐PLXNB2 regulation axis mainly affects target cell behaviors. Therefore, we evaluated IEC apoptosis and proliferation in WT and Ang −/− mice during DSS treatment. Cleaved‐caspase 3 staining and TUNEL assay showed that Ang −/− mice exhibited dramatically increased apoptosis in differentiated IECs at days 2 and 4 of DSS treatment compared with WT littermates (Figs 5A and B, and EV4A and B), which was confirmed by flow cytometry analysis with annexin V staining (Fig 5C and D). Meanwhile, Ki67 and BrdU staining revealed a decreased IEC proliferation in colonic crypt at day 4 in Ang −/− mice (Figs 5E and F, and EV4C and D). Consistent with this finding, an increase in G0 phase and corresponding decrease in S/G2/M phases of cell cycle were observed in Ang −/− mice (Fig 5G and H). To further determine whether Ang deficiency in myeloid cells is responsible for the phenomena, we examined IEC behaviors in LysM cre ;Ang fl/fl or Plxnb2 knockdown mice and found that myeloid Ang deficiency or Plxnb2 knockdown resulted in elevated IEC apoptosis and decreased proliferation (Fig 5I–L). Together, these data demonstrate that myeloid cell‐derived ANG promotes IEC survival and proliferation via PLXNB2 in response to DSS treatment.
Figure 5. ANG protects intestinal epithelial integrity by enhancing IEC survival and proliferation.
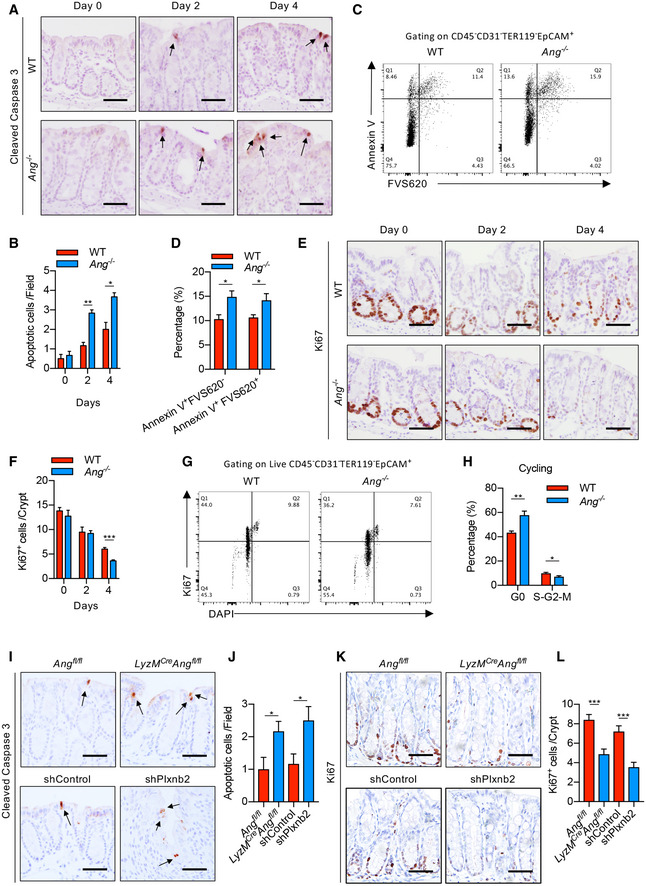
-
A, B(A) Representative images showing cleaved‐caspase 3 staining and (B) the number of apoptotic cells in colonic section from WT or Ang −/− colitis mice (n = 3) at indicated time point; arrows indicate cleaved‐caspase three positive cells; two fields per colon section are quantified.
-
C, DCell apoptotic status of IECs from WT or Ang −/− colitis mice (n = 5) on day 4.
-
E, F(E) Representative images showing Ki67 staining and (F) the number of proliferating cells per crypt in colonic section from WT or Ang −/− colitis mice (n = 3) at indicated time point; five representative crypts per colon section are quantified.
-
G, HCell cycle status of IECs from WT or Ang −/− colitis mice (n = 5) on day 4.
-
I, J(I) Representative images showing cleaved‐caspase 3 staining and (J) the number of apoptotic cells in colonic section from Ang fl/fl and LyzM Cre ;Ang fl/fl, or AAV‐shControl‐ and AAV‐shPlxnb2‐treated colitis mice on day 4 (n = 3); arrows indicate cleaved‐caspase three positive cells; two fields per colon section are quantified.
-
K, L(K) Ki67 staining and (L) the number of proliferating cells per crypt in representative colonic section from Ang fl/fl and LyzM Cre ;Ang fl/fl, or AAV‐shControl‐ and AAV‐shPlxnb2‐treated colitis mice on day 4 (n = 3); five representative crypts per colon section are quantified.
Figure EV4. ANG protects intestinal epithelial integrity to attenuate intestinal inflammation.
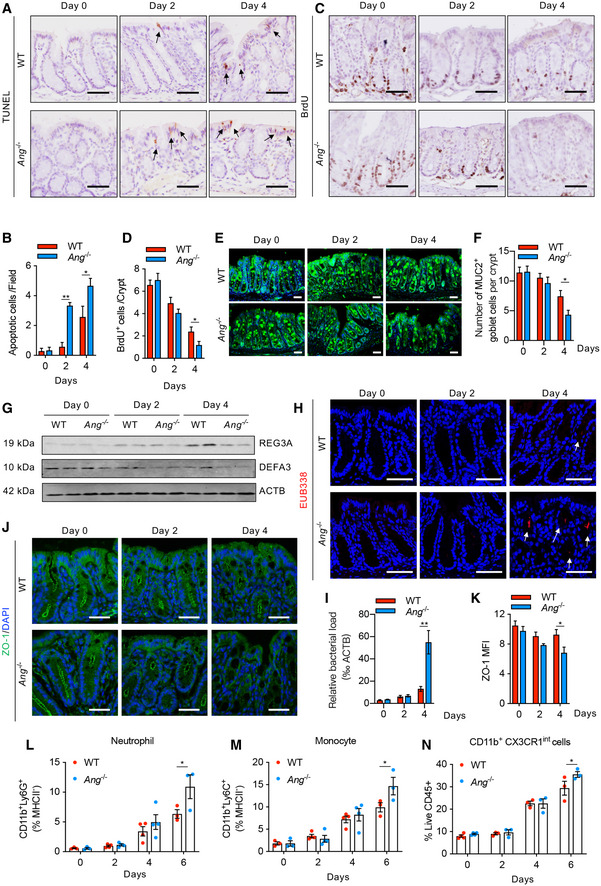
-
A, B(A) TUNEL staining and (B) quantification of apoptotic cells (arrows indicate positive cells; two fields per colon section are quantified; n = 3 mice/group).
-
C, D(C) BrdU incorporation in IECs and (D) quantification of proliferating cells per crypt (five representative crypts per colon section are quantified; n = 3 mice/group).
-
E, F(E) MUC2 staining and (F) quantification of MUC2‐positive cells per crypt (nuclei counterstained with DAPI dye; three representative crypts per colon section are quantified; n = 3 mice/group).
-
GWestern blotting of REG3A and DEFA3 in IECs (n = 2 mice/group).
-
HEUB338 staining by fluorescence in situ hybridization (FISH) in crypt (nuclei counterstained with Hoechst dye; arrows indicate positive EUB338 staining).
-
IRT–qPCR quantification of relative bacteria load in fresh colonic tissue (n = 4–6 mice/group).
-
J–M(J) ZO‐1 staining and (K) the corresponding mean fluorescence intensity (MFI) (nuclei counterstained with DAPI dye; three fields per colon section are quantified; n = 3 mice/group). Meanwhile, the percentages of neutrophil (L), monocyte (M) and CD11b+CX3CR1int (N) subsets in cLP of WT or Ang −/− colitis mice were calculated at indicated time point (n = 3–4/time point).
Meanwhile, the bacteria defense capability of epithelium was markedly reduced in Ang −/− mouse colon compared to WT counterpart following DSS treatment, as indicated by decreased production of MUC2 and antimicrobial molecules REG3A and DEFA3 (Fig EV4E–G), leading to more bacteria infiltration into the crypt of colonic tissue (Fig EV4H and I). Furthermore, the tight junction protein ZO‐1, an indicator of the colonic epithelial integrity (Marchiando et al, 2011), was also gradually decreased in Ang −/− mouse colon upon DSS treatment (Fig EV4J and K), indicating a severe damage to intestinal barrier integrity at the late stage of colitis induction, presumably allowing bacteria translocation into the colonic epithelia to trigger subsequent inflammatory cell infiltration. We did find significantly increased infiltration of myeloid inflammatory cells, including neutrophils, monocytes, and CD11b+CXCR1int cells in Ang −/− mouse colons (Fig EV4L–N). Taken together, these data indicate that myeloid cell‐derived ANG protects the epithelial barrier integrity against DSS‐induced epithelial damage via PLXNB2, thus attenuating the subsequent colitis progression.
ANG enhances IEC survival and proliferation via mediating different RNA production
Since different RNAs regulated by ANG control different cell behaviors (Sheng & Xu, 2016), we first assessed the ANG‐PLXNB2‐mediated tRNA metabolism. For this purpose, an in vitro crosstalk analysis system was established with colonic epithelial‐originated cell line HCT116 and macrophage‐like cell line THP‐1. As shown in Fig 6A and B, ANG and PLXNB2 were highly expressed in HCT116 and THP1 cells, respectively. To eliminate the influence of endogenous ANG, a HCT116KO (an ANG knockout cell line) was produced. Further analyses revealed that THP‐1‐derived ANG could interact with PLXNB2 in HCT116KO membrane and then translocate into cytosol (Fig 6C and D). We also created a TNF‐α‐induced apoptosis model and found that recombinant ANG (ANGWT), but not the receptor binding site deficiency variant ANGR66A or enzymatic inactivity variant ANGK40I, could significantly reduce HCT116KO apoptosis (Fig 6E). RNA analysis showed that only the recombinant ANGWT could produce more bulk tiRNAs as well as the 5′‐tiRNAAla (one type of tiRNA reported to participate in cell survival; Li et al, 2018a) in TNF‐α‐stimulated HCT116 cells (Fig 6F). Knockdown and overexpression experiments confirmed that 5′‐tiRNAAla was essential for ANG‐mediated protection against TNF‐α‐induced apoptosis (Fig 6G). RNA immunoprecipitation (RIP) assay showed a direct interaction between 5′‐tiRNAAla and the cytochrome c (Cyt c) in HCT116 (Fig 6H), leading to dramatically decreased Cyt c‐mediated caspase 9 and caspase 3 activation under TNF‐α stimulation (Fig 6I). These results suggest that ANG protects cells from apoptosis through PLXNB2‐mediated tRNA metabolism and caspase inhibition. We then evaluated rRNA transcription in HCT116 cells. Supplement with exogenous ANG indeed led to increased 47S pre‐rRNA transcription and enhanced cell proliferation, while blocking the ANG‐PLXNB2 interaction abrogated these phenomena (Fig 6J and K).
Figure 6. ANG enhances IEC survival and proliferation via mediating different RNA production.
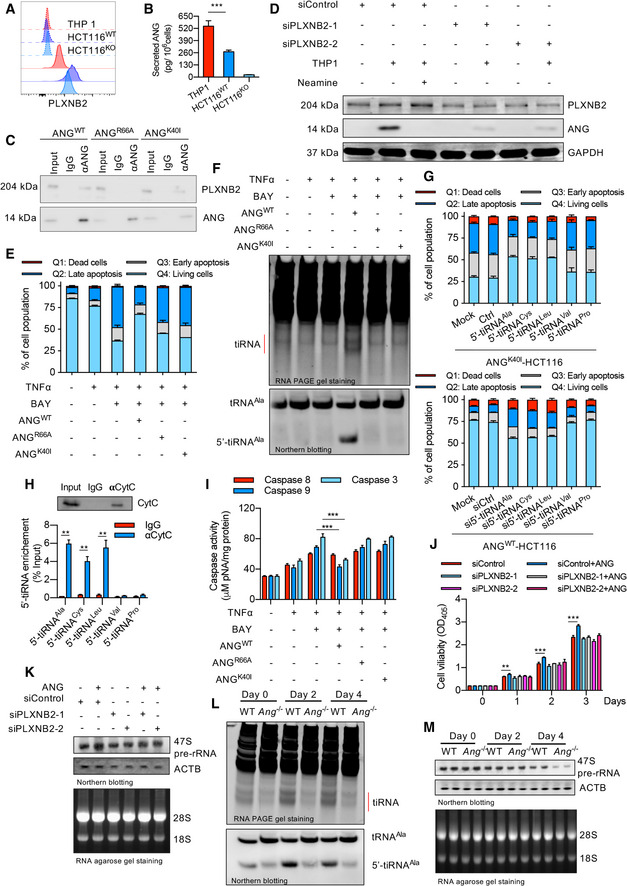
-
APLXNB2 expressions in HCT116 (including wild type HCT116 (HCT116WT) and ANG knockout HCT116 (HCT116KO)) and THP‐1 as detected by flow cytometry analysis. The upper three dash lines represent the intensity of unstained negative control of these cells, while the solid lines represent the intensity of PLXNB2 antibody staining.
-
BSecreted ANG level in conditioned medium from THP1, HCT116WT, or HCT116KO (n = 3).
-
CThe interaction between ANGWT, ANGR66A, or ANGK40I with PLXNB2 in HCT116KO detected by co‐immunoprecipitation assay.
-
DTHP‐1‐derived ANG translocating into HCT116KO through PLXNB2 in co‐cultured system as showed by Western blotting.
-
EFlow cytometry analysis of HCT116KO stained with annexin V and PI following different treatments as indicated.
-
FtiRNA and 5′‐tiRNAAla production in HCT116KO following different treatments as indicated.
-
GThe effect of 5′‐tiRNA knockdown or overexpression on TNF‐α‐induced apoptosis in ANGWT‐ or ANGK40I‐treated HCT116KO.
-
HThe interaction between 5′‐tiRNA and cytochrome c (Cyt c) in HCT116 cells as detected by RIP.
-
ICaspase activity of HCT116KO following different treatments as indicated.
-
JCell viability of HCT116KO following different treatments as indicated.
-
KDetection of 47S pre‐rRNA transcription level with Northern blotting in HCT116KO following different treatments as indicated.
-
LtiRNA and 5′‐tiRNAAla production in IECs isolated from WT and Ang −/− colitis mice at indicated time point.
-
M47S pre‐rRNA transcription level in IECs from WT and Ang −/− colitis mice at indicated time point.
To confirm the finding from this artificial crosstalk system, we further measured representative RNAs in isolated IECs from different mice used. First, to clarify the modulation of endogenous ribonuclease inhibitor protein (RNH1) on ANG activity, we analyzed the binding between RNH1 and ANG. Co‐IP assay revealed that ANG was sequestered by RNH1 under normal condition, but released in response to DSS treatment (Fig EV5A). Consistently, the total tiRNAs and 5′‐tiRNAAla levels were dramatically increased in IECs from DSS‐treated WT mice compared to its Ang −/− counterpart (Fig 6L). Second, we measured tiRNA productions in different in vivo conditions including LysM cre ;Ang fl/fl, bone marrow chimera (WT→WT, WT→Ang −/−, and Ang −/−→WT), and Plxnb2 knockdown mice. The results clearly indicated that myeloid‐derived ANG was responsible for tiRNA production via interacting with PLXNB2 under DSS treatment (Fig EV5B–D). Third, we also observed other differential expressed tiRNAs (such as 5′‐tiRNACys and 5′‐tiRNALeu) possessing similar function and mechanism of action to those of 5′‐tiRNAAla (Figs 6G and H, and EV5E). On the other hand, the 47S pre‐rRNA in IECs isolated from day 4 Ang −/− colon was lower than that from WT mice during inflammation (Fig 6M). Together, these data indicate that ANG promotes IEC survival and proliferation through differentially regulating RNA production under inflammatory condition.
Figure EV5. Myeloid‐derived ANG‐mediated tiRNA production in IECs via PLXNB2.
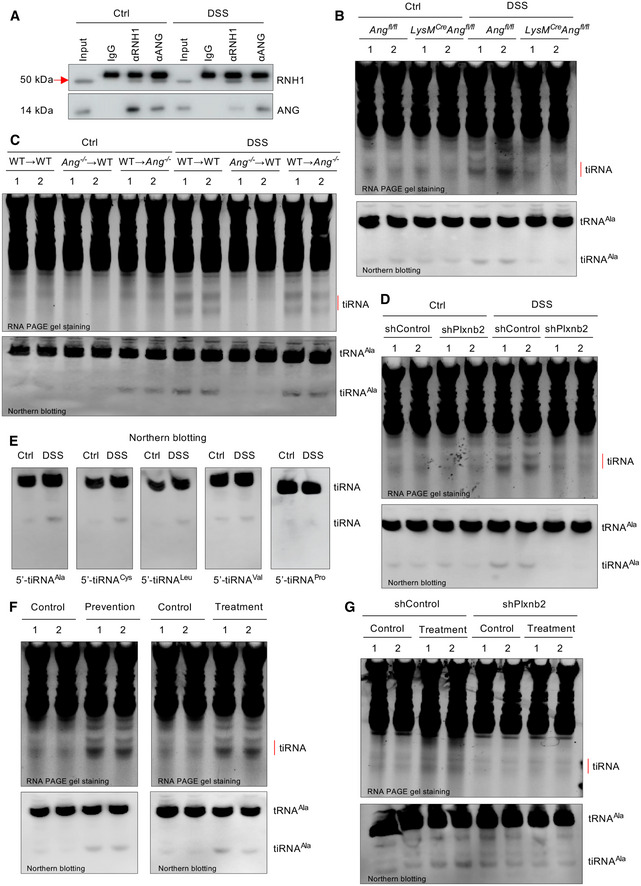
-
AThe interaction between ANG and its endogenous inhibitor RNH1 in IECs from DSS‐treated WT mice as detected with co‐immunoprecipitation assay.
-
B–DtiRNA and 5′‐tiRNAAla production in IECs isolated from (B) Ang fl/fl or LyzM cre ;Ang fl/fl, (C) bone marrow chimera or (D) Plxnb2 knockdown mice (n = 2) in response to DSS treatment (day 0, 4).
-
E5′‐tiRNA detection in IECs isolated from DSS‐treated mice (day 6) by Northern blotting.
-
FtiRNA and 5′‐tiRNAAla production in IECs isolated from DSS‐treated mice on day 6 in the ANG prevention or treatment model. The same‐day mice treated with DSS but without ANG supplement serve as a control (n = 2 mice/group).
-
GtiRNA and 5′‐tiRNAAla production in IECs isolated from DSS‐treated WT or Plxnb2 knockdown mice (n = 2) on day 6 with or without ANG injection.
Source data are available online for this figure.
ANG is a preventive and therapeutic factor for colitis
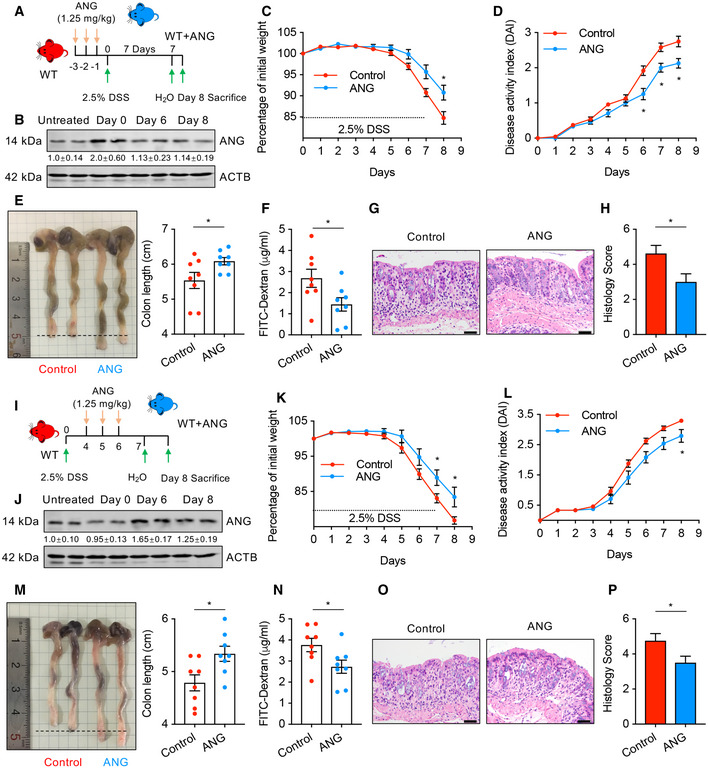
Our finding that ANG regulates IEC behaviors to maintain intestinal epithelial barrier integrity during mucosal inflammation suggests that ANG may have clinical application in IBD. To assess this possibility, we first test the potential of ANG as a preventive measure of colitis. After pre‐treating WT mice with recombinant ANG protein for 3 successive days, followed by administration with 2.5% DSS for the next 7 days (ANG prevention model) (Fig 7A), we found that the ANG‐pre‐treated mice exhibited improved clinical signs of DSS‐induced colitis (Fig 7C–H), supporting the preventive potential of ANG for colitis. Furthermore, we evaluated whether ANG has any therapeutic effect on established colitis. To this end, WT mice were initially treated with DSS, and at day 4 when mucosal inflammation was evidenced, recombinant ANG was consecutively given for 3 days (ANG treatment model) (Fig 7I). The results showed that treatment dramatically attenuated the progression of established colitis (Fig 7K‐P), testifying its potency as a therapeutic approach. We further confirmed that exogenous ANG can successfully reach the colonic tissue following intraperitoneal injection as shown by elevated local ANG levels in the day 0 mice of the prevention model (Fig 7B) and the day 6 mice of the treatment model (Fig 7J), where this protein executed its biological functions such as producing tiRNAs through PLXNB2 (Fig EV5F and G). Taken together, our data indicate that ANG could serve as both a preventive agent and a therapeutic reagent for DSS‐induced colitis.
Figure 7. Recombinant ANG protects mice from DSS‐induced colitis.
-
ASchematic diagram of the ANG prevention model. Mice (n = 8) were pre‐treated with 1.25 mg/kg recombinant ANG protein and then subjected to DSS induction.
-
BWestern blotting of ANG in colonic tissue from WT mice (n = 2) at indicated time point in the ANG prevention model. The grayscale of ANG band in WT mice with no recombinant ANG and DSS treatment (untreated) is arbitrarily set as 1.
-
C, D(C) Body weight loss and (D) DAI of the pre‐treated WT colitis mice (n = 8).
-
E–H(E) Colon length, (F) serum FITC‐dextran level, (G) representative HE staining image, and (H) histopathological score of colon section from the pre‐treated WT colitis mice on day 8 (n = 8).
-
ISchematic diagram of the ANG treatment model. Mice (n = 8) were treated with DSS for 7 days and simultaneously administrated with 1.25 mg/kg recombinant ANG during days 4–6.
-
JWestern blotting of ANG in colonic tissue from WT (n = 2) mice at indicated time point in the ANG treatment model. The grayscale of ANG band in WT mice with no recombinant ANG and DSS treatment (untreated) is arbitrarily set as 1.
-
K, L(K) Body weight loss and (L) DAI of the treated WT colitis mice (n = 8).
-
M–P(M) Colon length, (N) serum FITC‐dextran level, (O) representative HE staining image, and (P) histopathological score of colon sections from the treated WT colitis mice on day 8 (n = 8).
Discussion
Epithelial barrier integrity is critical for intestinal homeostasis and acts as an indicator of IBD therapy, which predicts lower hospitalization rate, sustained clinical remission, and resection‐free survival (Neurath & Travis, 2012). However, the molecular mechanisms underlying integrity maintenance are not fully understood. Here, we reported a novel regulatory pathway driven by myeloid cell‐secreted ANG that is critical for maintaining epithelial barrier integrity. Our results indicate that modulation of this ANG‐driven pathway could be a novel preventive and/or therapeutic approach for IBD.
Although anti‐inflammation‐based therapies show great potential in IBD treatment, their effectiveness is limited due to the undesirable side‐effects. For example, anti‐inflammatory reagents (such as antibodies against TNF‐α, IL‐12, or IL‐23) and cytokine signaling blockers (such as Tofacitinib) are reported to be effective in a selective subset of patients (Neurath, 2014; Sales‐Campos et al, 2015). This may reflect the fact that the mucosal inflammatory networks are dynamically changing during IBD pathogenesis, allowing compensatory pathways to be rewired in response to the blockade of a single target (Neurath, 2014). In contrast, successful regeneration of the intestinal mucosa results in long‐term remission and low risk of surgical treatment in clinical patients, indicating an unexpected contribution of IECs to IBD therapy (Okamoto & Watanabe, 2016). Here, we found that ANG promotes IEC survival and growth to maintain epithelial barrier integrity and that administration of exogenous ANG could significantly attenuate the severity of colitis. More intriguingly, ANG does not trigger inflammatory cytokine production in systemic innate immune response nor change the composition of MPs in the local mucosal immune system, indicating that ANG treatment does not cause inflammatory side‐effects. Therefore, ANG may be a potential candidate for clinical use to prevent and even repair epithelial barrier damage.
Our data show that ANG is down‐regulated in IBD patient samples, and the extent of decline is negatively correlated with IBD progression, suggesting that ANG expression is dynamically regulated along with the alteration of the inflammatory milieu. With the onset of intestinal inflammation, ANG expression is down‐regulated by a yet‐to‐be determined mechanism, and this insufficient ANG level under pathologic condition, in turn, may lead to more epithelial barrier impairment, creating a vicious cycle which promotes IBD progression. Thus, further elucidating the precise regulatory mechanism of ANG expression during inflammation will be beneficial for the development of IBD prevention or treatment strategies.
A success in epithelial barrier restitution depends upon rapid protection of IECs from cell death and an increase in IEC proliferation (Gunther et al, 2013). Myeloid cells have been reported to secrete protective factors that function through either preserving IEC survival, i.e., IL‐22 and Prostaglandin E2 (Nishihara et al, 2003; Grivennikov et al, 2009; Pickert et al, 2009), or inducing IEC proliferation, i.e., IL‐10, insulin‐like growth factor 1, and hepatocyte growth factor (D'Angelo et al, 2013; Quiros et al, 2017; Youssif et al, 2018). Unlike these mediators, our data clearly indicate that ANG is able to simultaneously activate both, implying a dual function of ANG in maintaining epithelial barrier integrity. Cytosolic ANG cleaves tRNAs to produce tiRNAs to increase the survival of IECs, while nucleolar ANG enhances rRNA transcription to promote IEC proliferation. Although cytosolic ANG is sequestered by its endogenous inhibitor RNH1 to prevent RNA cleavage, it could dissociate from RNH1 binding and enrich in stress granules to produce tiRNAs (Pizzo et al, 2013). In this study, we demonstrated that ANG was blocked by RNH1 in IECs and thus has no influence on basal intestinal homeostasis; however, the interaction between ANG and RNH1 was greatly impaired upon DSS treatment, leading to the activation of ANG and subsequent production of tiRNAs to promote IEC survival. This finding suggests that, in addition to ANG expression level, activation of its ribonuclease activity is also essential for IECs to deal with stress conditions. Overall, the dual role of ANG could lead to improved efficiency to prevent and/or repair epithelial barrier damage.
The IECs are actually a mixture of multiple specialized cell types, including differentiated epithelial cells, transit amplifying cells (TACs) which are the most actively dividing cells, and can migrate out of the stem cell niche to differentiate into various cell lineages of mature IECs (Clevers, 2013), Paneth cells, and intestinal stem cells (ISCs) which locate at the base of crypts in the intestine and undergo continuous and rapid renewal (Barker, 2014). Our data clearly showed that the apoptotic cells are located to the differentiated IEC region at days 2 and 4 of DSS treatment (Fig 5A), while the Ki67‐positive cells are in the colonic crypt (Fig 5E), suggesting that the first wave of apoptosis occurs in differentiated IECs and the proliferating cells are TACs or stem cells. Further study is warranted to determine the specific cell types regulated by ANG.
Taken together, our findings demonstrate ANG as a novel myeloid‐IEC crosstalk mediator maintaining epithelial barrier integrity to ameliorate intestinal inflammation. Through a direct interaction with PLXNB2, myeloid cell‐secreted ANG promotes IEC survival and proliferation by regulating tiRNA production and rRNA transcription, respectively. Our work not only elucidates a novel mechanism by which myeloid cells regulate IEC behaviors, but also provide clues to develop new preventive and therapeutic measures for IBD.
Materials and Methods
Human samples
Non‐IBD control, CD, and UC patient samples were collected at the Inflammatory Bowel Disease Center, Sir Run Run Shaw Hospital affiliated to Zhejiang University School of Medicine. The diagnosis of CD or UC was performed according to a standard combination of clinical, endoscopic, histological, and radiologic criteria. The partial Mayo score (PMS) for UC and the Harvey‐Bradshaw index (HBI) for CD were used to assess disease status. Active disease was defined as a PMS > 2 in UC or HBI > 5 in CD. UC samples with mild (PMS range of 2–4) and severe (PMS range of 7–9) disease stages and CD samples with mild (HBI range of 5–7) and severe (HBI > 16) scores were taken for analysis. The determination of colitis severity was also assessed by pathological evidence of mucosal inflammation. Basic information about patients is summarized in Tables EV1, EV2 and EV3. Ethics approval for use of human samples was obtained from the Ethics Committee of Zhejiang University School of Medicine.
ANG IHC staining and score
Non‐IBD control, CD, and UC patient samples were immunohistochemically stained with mouse anti‐angiogenin monoclonal antibody 26‐2F (Yu et al, 2017) and scored using Constantine's protocol. Briefly, under high magnification, integrated staining intensity and the percentage of positive cells were semi‐quantitatively scored. Staining intensity was scored as follows: 0 = no color; 1 = yellow; 2 = brown‐yellow; and 3 = brown. The proportion of positive cells was graded as follows: 0 = positive cells < 10%; 1 = positive cells between 10% and 40%; 2 = positive cells between 40% and 70%; and 3 = positive cells ≥ 70%. The staining intensity score and proportion of positive cells score were added up.
Recombinant ANG production
Mouse and human recombinant ANG proteins were generated in E. coli expression system and purified by HPLC as described previously (Shapiro et al, 1988). Angiogenic and ribonucleolytic activities of each batch of ANG preparation were confirmed (Li et al, 2013). ANG variants (K40I and R66A) were generated through sit‐directed mutagenesis with the Fast Mutagenesis System kit (TransGen Biotech), followed by expression in E. coli expression system and HPLC purification.
In vitro and in vivo treatment with ANG, ANG variants, neomycin, and neamine
Unless otherwise indicated, 300 ng/ml ANG or ANG variants and 100 μM neomycin or neamine were used for in vitro treatment. For in vivo treatment, 1.25 mg/kg ANG or ANG variants and 60 mg/kg neomycin or 30 mg/kg neamine were injected intraperitoneally at the indicated time point.
Mice
Generation and characterization of Ang −/− and Ang conditional knockout mice were previously described (Goncalves et al, 2016; Silberstein et al, 2016). B6.129P2−Lyz2 tm1(cre)Ifo/J (004781, The Jackson Laboratory) and B6.Cg‐Tg (Vil1‐cre) 997Gum/J (004586, The Jackson Laboratory) were used to delete the conditional floxed Ang allele specifically in myeloid cells and epithelial cells, respectively. CD45.1 (B6.SJL) was a gift from Dr. Lingrong Lu (Department of Immunology, Zhejiang University School of Medicine). Mice were maintained and bred in specific pathogen‐free condition at Laboratory Animal Center of Zhejiang University and were allocated to experimental groups on the basis of their genotype and randomized into different treatment groups. For in vivo experiments, the investigator was blinded toward the genotype, but not the grouping. All animal studies were performed in compliance with the guide for the care and use of laboratory animals, and the protocol was approved by the Medical Experimental Animal Care Commission of Zhejiang University.
Experimental colitis induction
Mice were administrated with dextran sulfate sodium (DSS) in drinking water to induce acute colitis, as previously described (Chassaing et al, 2014). In brief, sex‐matched 8‐week‐old mice were fed with DSS (2.5% w/vol; molecular weight: 36–50 kDa; MP Biomedicals) in drinking water for 1 week, followed by regular drinking water until the end of the study, and colonic tissue was collected on the indicated day. For survival analysis, mice were given 3.5% DSS for 1 week, followed by regular drinking water until the end of the study.
Bone marrow chimera
Bone marrow transfer was used to create Ang −/− chimera mice wherein the genetic deficiency of Ang was confined to either circulating cells (Ang −/−→WT) or non‐hematopoietic tissue (WT→Ang −/−) as described previously (Song et al, 2014). Briefly, 6‐week‐old recipient WT (CD45.1 or CD45.2) or Ang −/− (CD45.2) mice were lethally irradiated with 10 Gy and then injected in tail vein with 100 μl fresh mixed bone marrow cells (1 × 108/ml) from WT or Ang −/− donor mice. After 8 weeks of bone marrow transplantation, mouse blood was collected for reconstitution capacity analysis using flow cytometry and then administrated with 2.5% DSS to induce colitis.
Disease activity index score
The clinical sign of mouse colitis was evaluated based on the Disease Activity Index (DAI) score that includes body weight loss, occult blood, and stool consistency as previously described (Zaki et al, 2010). Mice were scored blindly during the colitis experiment. In brief, weight loss score was determined as follows: 0, no weight loss; 1, loss of 1–5% original weight; 2, loss of 6–10% original weight; 3, loss of 11–20% original weight; and 4, loss of > 20% original weight. Stool score was determined as follows: 0, well‐formed pellets; 1, semi‐formed stools that did not adhere to the anus; 2, pasty semi‐formed stool that adhered to the anus; and 3, liquid stools that adhered to the anus. Bleeding score was determined as follows: 0, no blood by using Hemoccult (Beckman Coulter) analysis; 1, positive Hemoccult; 2, visible blood traces in stool; and 3, gross rectal bleeding.
Histological score
Colon tissue was embedded in paraffin, cut into 4‐μm sections, and then stained with hematoxylin and eosin (HE) for histological analysis. Histological assessment of colitis was performed blindly by board‐certified pathologist based on the previously described criteria that consist of the extent and severity of inflammation and ulceration of the mucosa (Zaki et al, 2011). Briefly, severity score for inflammation was as follows: 0, normal (within normal limits); 1, mild (small, focal, or widely separated, limited to lamina propria); 2, moderate (multifocal or locally extensive, extending to submucosa); and 3, severe (transmural inflammation with ulcers covering > 20 crypts). Score for ulceration was as follows: 0, normal (no ulcers); 1, mild (1–2 ulcers involving up to a total of 20 crypts); 2, moderate (1–4 ulcers involving a total of 20–40 crypts); and 3, severe (more than 4 ulcers or over 40 crypts).
FITC‐dextran permeability assay
Intestinal permeability was evaluated by oral administration of fluorescein isothiocyanate (FITC)‐labeled dextran as previously described (Huang et al, 2014). In brief, mice were fasted for 4 h and then treated with FITC‐dextran tracer (4,000 Da, 0.6 mg/g body weight, Sigma‐Aldrich) intragastrically in 100 μl phosphate‐buffered solution (PBS). After 4 h, mouse blood was collected to obtain the hemolysis‐free serum for fluorescence intensity detection using Thermo Scientific™ Varioskan Flash fluorescence spectrophotometer (excitation of 488 nm and emission of 520 nm). A standard curve for FITC‐dextran by serially diluting a known amount of FITC‐dextran in PBS was prepared for the calculation of concentration of FITC‐dextran in mouse serum.
Adeno‐associated virus (AAV) infection and Plxnb2 knockdown
For in vivo AAV administration, mice were fasted overnight and pre‐treated with 20 mM N‐acetyl‐L‐cysteine (NAC: a mucolytic agent for the intestinal mucosal surface, Sigma‐Aldrich) as previously described (Polyak et al, 2012). Briefly, mouse colon was washed with intrarectal injection of 300 μl of 20 mM NAC using stainless steel straight round‐tip microsyringe and allowed to drain without sedation for 30 min. Then, mice were anesthetized with inhaled isoflurane and 5 × 1010 physical particles of AAV in 100 μl of PBS were given through enema. After 3‐week AAV infection, these mice were subjected to DSS administration. For ANG treatment in these mice, recombinant ANG was injected on days 4–6 during DSS treatment. The PLXNB2 knockdown efficiency was detected with RT–qPCR, Western blotting, and immunofluorescence staining. The transgene plasmids carrying control shRNA or Plxnb2 shRNA and their corresponding physical AAVs were constructed by Shanghai SunBio Medical Biotechnology CO. The shRNA sequences are listed in Table EV5.
Ex vivo colon organ culture
Colon was harvested and cut with scissor longitudinally. After washed with ice‐cold PBS three times, 5 mm distal colon was cultured in 500 μl of RPMI 1640 complete medium for 24 h at 37°C. Cytokines in culture supernatants were measured by mouse IL‐6, IL‐17A, and CXCL 2 Platinum ELISA kits (eBioscience) and Duoset mouse IL‐1β and TNF‐α ELISA kits (R&D Systems) according to the manufacturers’ instruction.
Endotoxin shock and bacterial infection mouse models
The endotoxin shock mouse model was established by intraperitoneal injection of lipopolysaccharides from Escherichia coli O111:B4 (LPS, 15 mg/kg body weight, Sigma‐Aldrich), and then, mouse survival proportion was recorded every 2 h until the end of the study. For bacterial infection, mice were injected intravenously with 5 × 104 L. monocytogenes, and the survival proportion was recorded as described above.
Cell isolation from Intestinal epithelia and lamina propria
Isolation of intestinal epithelial cells or colonic lamina propria cells was performed with established method reported previously (Song et al, 2011; Bain et al, 2013). Briefly, after removing extra‐intestinal fat tissue and blood vessel, colons were flushed of their luminal contents with cold PBS, opened longitudinally, cut into 2‐cm pieces, and then incubated with Hank's balanced salt solution (HBSS, Corning) without Ca2+ and Mg2+ (Thermo Fisher Scientific) containing 5% FBS, 3 mM EDTA (Sigma‐Aldrich), and 1 mM DTT (Sigma‐Aldrich) for 30 min with shaking at 250 rpm to obtain the epithelial cells for RNA isolation and flow cytometry analysis. The remaining colon tissue was further cut into smaller 1‐mm2 pieces and then digested by 1 mg/ml Collagenase IV (Sigma‐Aldrich) and 0.1 mg/ml DNase I (Roche) at 37°C with shaking for 40 min at 37°C. The suspension was washed with cold PBS and passed through a 100‐μm cell strainer followed by a 40‐μm cell strainer. After centrifugation at 600 g for 5 min, the fraction was resuspended in PBS containing 5% FBS for the following RNA isolation and the FACS analysis.
Flow cytometry analysis
For analysis of cLP cells in the steady state, cells were incubated with Fc block/anti‐CD16/32 (2.4G2, BioLegend) 10 min and then stained for 30 min on ice with antibodies against CD45 AF700 (30‐F11, BD), MHCII APC (M5/114.15.2, BioLegend), CD11c PE‐Cy7 (N418, BioLegend), CD11b FITC (M1/70, BioLegend), CD103 BV650 (M290, BD), and CX3CR1 PE (FAB5825P, R&D).
For analysis of cLP cells during DSS‐induced colitis, cells were incubated with Fc block/anti‐CD16/32 (2.4G2, BioLegend) 10 min and then stained for 30 min on ice with antibodies against CD45 AF700 (30‐F11, BD), MHCII BV650 (M5/114.15.2, BD), CD11c BV421 (N418, BD), CD11b FITC (M1/70, BioLegend), F4/80 AF647 (T45‐2342, BD), Ly‐6C PE‐Cy7 (HK1.4, BioLegend), and Ly‐6G PE (1A8, eBioscience).
For analysis of intracellular IL‐6 and TNF‐α in myeloid cells, cells were incubated with either LPS (1 μg/ml) plus ionomycin (500 ng/ml) for 5 h in the presence of Golgi‐stop Brefeldin A (1,000×, BioLegend) and then stained for 30 min on ice with cell surface marker antibodies against CD45 AF700 (30‐F11, BD), CD11b FITC (M1/70, BioLegend), and Fixable Viability Dye eFluor‐506 (FVD506, eBioscience). Cells were then fixed and permeabilized using Cytofix/Cytoperm Fixation/Permeabilization Kit (BD) per manufacturer's instruction and stained with IL‐6 PE (MQ2‐13A5, eBioscience) and TNF‐α APC (Mab11, eBioscience).
For lineage analysis in chimerism, peripheral blood was obtained by retro‐orbital bleeding and depleted of red blood cells. Sample were stained for 30 min on ice with antibodies against CD45.1 APC (A20, BD), CD45.2 APC‐Cy7 (104, BD), CD11b FITC (M1/70, BioLegend), Gr‐1 PE (RB6‐8C5, BioLegend), CD45R/B220 PerCP‐Cy5.5 (RA3‐6B2, BD), and CD3ε BV510 (145‐2C11, BD).
For analysis of cell surface PLXNB2 in THP‐1 and HCT116, cells were maintained in RPMI 1649 containing 10% FBS, washed with PBS twice, and detached with Trypsin/EDTA solution (Thermo Fisher Scientific). After centrifuged at 600 g, cell pellets were collected, washed twice with cold PBS, resuspended in PBS containing 5% FBS at a density of 1 × 106/ml, and then kept on ice. For each staining, cells were stained with mouse anti‐PLXNB2 (ab193355, Abcam) and IgG isotype control antibody for 30 min. After washed with cold PBS containing 5% FBS, cells were incubated with PE‐conjugated secondary antibody at 1:100 dilution (550767, BD) on ice for 30 min in dark. Cells were the washed twice with PBS containing 5% FBS and resuspended in 400 μl PBS containing 5% FBS for flow cytometry analysis.
For cell cycle analysis, IECs were incubated with Fc block/anti‐CD16/32 (2.4G2, BioLegend) 10 min and then stained for 30 min on ice with cell surface marker antibodies against CD45 PE (30‐F11, BioLegend), CD31 PE (390, BioLegend), TER119 PE (TER119, BioLegend), EpCAM APC (G8.8, eBioscience), and Fixable Viability Stain 620 (FVS620, BD). Cells were then fixed and permeabilized using Cytofix/Cytoperm Fixation/Permeabilization Kit (BD) per manufacturer's instruction and stained with Ki67 PE‐CY7 (B56, BD; 1:10 in BD Perm/Wash buffer) and DAPI (2 μg/ml for 10 min prior to analysis).
For apoptosis analysis, IECs were incubated with Fc block/anti‐CD16/32 (2.4G2, BioLegend) 10 min and then stained for 30 min on ice with cell surface marker antibodies against CD45 PE (30‐F11, BioLegend), CD31 PE (390, BioLegend), TER119 PE (TER119, BioLegend), EpCAM APC (G8.8, eBioscience), annexin V‐FITC (Lianke), and Fixable Viability Stain 620 (FVS620, BD).
For sorting colonic myeloid cells, cLP cells were incubated with Fc block/anti‐CD16/32 (2.4G2, BioLegend) 10 min and then stained for 30 min on ice with antibodies against CD45 AF700 (30‐F11, BD) and CD11b FITC (M1/70, BioLegend).
For all analyses, 4′,6‐diamidino‐2‐phenylindole (DAPI, BD), 7‐aminoactinomycin d (7‐AAD, BD), or Fixable Viability Stain 620 (FVS620, BD) were used as viability dyes according to manufacturer's instruction. Cells were analyzed using LSR Fortessa flow cytometer (BD) and sorted with FACS ARIA II SORP (BD).
Immunohistochemical staining and TUNEL assay
The Ki67 or 5‐bromo‐2′‐deoxyuridine (BrdU) staining was used for cell proliferation analysis. For Ki67 staining, paraffin sections were stained with anti‐Ki67 antibody (ab16667, Abcam). For BrdU staining, mice were i.p. with 100 mg/kg BrdU (B9285, Sigma‐Aldrich) in PBS for 4 h and then sacrificed. Paraffin sections were stained for BrdU with BrdU In‐situ Detection Kit (BD). For each analysis, Ki67‐positive or BrdU‐positive cells were scored in 15 representative full crypts from three animals of each group. For cell apoptosis analysis, cleaved‐caspase 3 staining or terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was adopted. For cleaved‐caspase 3 staining, paraffin sections were stained using anti‐cleaved‐caspase 3 (#9664, CST). The TUNEL assay was performed with the In Situ Cell Death Kit (Roche). For each analysis, cleaved‐caspase 3‐positive or TUNEL‐positive cells in six representative high power fields (HPF, 40×) from three animals of each group were counted. Immunohistochemical and TUNEL assay were performed in the Histomorphology Platform, Zhejiang University according to the manufacturer's instruction, and the slides were examined and analyzed with Olympus microscope blindly by a board‐certified pathologist.
Fluorescence in situ hybridization and immunofluorescence
Colon paraffin sections were stained at 46°C with hybridization buffer containing Cy3‐conjugated EUB338 probe (the sequence is listed in Table EV5) and then analyzed with a Nikon confocal microscope system A1 (Nikon Instruments). Tissue immunofluorescence was performed with murine colon frozen sections which were fixed in 4% paraformaldehyde (PFA) for 24 h, dehydrated in 30% sucrose for 12 h, and then embedded with O.C.T. (Sakura). Seven μm cryo‐sections were prepared to be balanced at RT for 1 h and then blocked with blocking solution (0.3% Triton in PBS supplemented 0.5% BSA) for 30 min. The slides were incubated with primary antibodies including rat anti‐PLXNB2 (1:200, AF6836, R&D systems), rabbit anti‐PLXNB2 (1:200, 10602‐1‐AP, Proteintech), mouse anti‐ANG (1:200, home‐made), rabbit anti‐ANG (1:200, home‐made), mouse anti‐CD68 (1:200, ab955, Abcam), rabbit anti‐MUC2 (1:200, 27675‐1‐AP, Proteintech), rabbit anti‐ZO‐1 (1:200, 21773‐1‐AP, Proteintech), and chicken anti‐GFP (1:200, ab13970, Abcam) overnight. After washed with PBT (0.3% Triton in PBS) for 10 min twice, the slides were stained with secondary antibodies including Donkey anti‐Mouse Alexa Fluor488 (1:250, A21202, Life Technology), Donkey anti‐Mouse Alexa Fluor555 (1:250, A31570, Life Technology), Goat anti‐Rat Alexa Fluor555 (1:250, ab150158, Abcam), Donkey anti‐Rabbit Alexa Fluor488 (1:250, ab150073, Abcam), Goat anti‐Rabbit Alexa Fluor555 (1:250, ab150078, Abcam), Donkey anti‐Chicken Alexa Fluor 488 (1:500, 1703‐545‐1550, Jackson ImmunoResearch), or anti‐Mouse F4/80 AF647 (1:250, T45‐2342, BD) for 3 h at RT. ZO‐1 fluorescence intensity analysis and MUC2‐positive goblet cell number count were performed using ImageJ software.
Quantification of bacterial load in colonic tissue
The colonic tissue was homogenized in liquid nitrogen with mortar and pestle. The bacterial and cellular genomic DNAs were extracted from colonic tissue by QIAamp DNA Mini Kit (51604, Qiagen) according to the manufacturer's instruction. The relative bacterial load was determined by qPCR using the universal bacterial primer and Actb as the internal reference. The primers information is listed in Table EV4.
Cell culture, stimulation and transfection
Bone marrow cells were isolated from femurs and tibias of 8‐week‐old WT and Ang −/− mice and then cultured in Dulbecco's modified Eagle's medium (DMEM) complemented with 10% fetal bovine serum (FBS, Gibco Invitrogen) and 20 ng/ml recombinant murine macrophage colony‐stimulating factor (M‐CSF, PeproTech) for 5 days to prepare bone marrow‐derived macrophages (BMDMs). After stimulation with LPS (100 ng/ml, Sigma‐Aldrich) or poly(I:C) (10 μg/ml, Sigma‐Aldrich), BMDMs were harvested at different time points to isolate RNAs for evaluating the cytokine expressions with RT–qPCR. THP‐1 and HCT116 were maintained in RPMI 1640 (Hyclone) supplemented with 10% FBS. For co‐culture experiment, HCT116 was seeded in a six‐well plate (Corning) at amount of 50 × 104 cells for 6 h and then washed twice with complete RPMI before adding 1 ml 30 × 104 THP‐1 into each well. After co‐culture for 24 h, medium containing THP‐1 was removed, and HCT116 was harvested after washed twice with complete RPMI for immunofluorescence assay and protein isolation. All cells were incubated at 37°C in a humidified 5% CO2 atmosphere. Transfection of siRNA was performed with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol, and non‐targeting siRNA (siCtrl) was used as a negative control. The siRNA targeting specific tiRNA was designed using siExplorer (Gress et al, 2016) and synthesized by Shanghai Genepharma. The knockdown efficiency was verified by Northern blotting at 48 h after transfection (Appendix Fig S1B). The siRNA sequences are listed in Table EV5.
Co‐immunoprecipitation analysis
Cell lysate was prepared with lysis buffer (100 mM Tris–HCl, pH 7.5, 100 mM NaCl, 20 mM MgCl2, 1% Triton X‐100, 1 mM PMSF, and protease inhibitor cocktail) and cleared by centrifugation at 16,000 g for 30 min at 4°C. Protein concentration was determined using a modified Bradford protocol. An aliquot of cell lysate containing 400 μg of total protein was incubated with 5 μg affinity‐purified Goat anti‐ANG polyclonal antibody (AF265, R&D Systems) or normal IgG as control at 4°C overnight, followed by incubation with Protein G‐conjugated agarose beads for 1 h. After washing the beads several times, the mixture was centrifuged at 12,000 g for 1 min. The pellet was resuspended in 100 μl laemmli buffer. The immune precipitates were then analyzed by Western blotting.
RNA immunoprecipitation analysis
Experiment was performed according to the protocol provided by the kit (17‐700, Millipore). Briefly, cells after treatment were harvested for lysate preparation. The lysate was then incubated with anti‐Cyt C antibody (MAB898, R&D Systems) and immunoprecipitated with protein A/G magnetic beads. The precipitated tiRNAs were detected by TaqMan‐based qPCR, and the proteins in the precipitated product were detected with Western blotting. The primers and probes for tiRNA detection were obtained from GenePharm and listed in Table EV6.
Enzyme‐Linked Immunosorbent Assay
The ANG level in the culture supernatant of cells was quantitated using a double antibody enzyme‐linked immunosorbent assay (ELISA) method. Briefly, supernatant was collected from the cells and the volume was normalized to the cell numbers. ELISA plate was first coated with 1 μg of mouse anti‐angiogenin monoclonal antibody 26‐2F per well and blocked with 5 mg/ml BSA in PBS, then added 100 μl sample, and further incubated at 4°C overnight. After washing with PBS five times, the plate was incubated with 100 μl of rabbit anti‐angiogenin polyclonal antibody (1 μg/ml, home‐made) per well at RT for 2 h, then washed four times with PBS, and incubated with an alkaline phosphatase‐labeled goat anti‐rabbit antibody (1.25 μg/ml, #31340, Thermo scientific) at RT for 1 h. Following four times washing with PBS, each well was added with 100 μl of 5 mg/ml p‐nitrophenyl phosphate in 0.1 M diethanolamine containing 10 mM MgCl2 (pH 9.8) and then measured the absorbance at 410 nm. A standard curve of recombinant human ANG at concentrations ranging from 50 to 1000 pg/well was performed each time on every plate.
RNA isolation, RT–qPCR, and Northern blotting
Total RNAs were extracted using TRIzol reagent (Invitrogen). Total RNAs from colonic tissue of DSS‐treated mice were purified via precipitation with lithium chloride reported previously (Kerr et al, 2012). The mRNA level was evaluated by reverse transcription (RT) reaction with Moloney Murine Leukemia Virus (M‐MLV) reverse transcriptase (Invitrogen), followed by quantitative PCR (qPCR) with SYBR Premix Ex Taq (TaKaRa). The qPCR was performed on a Roche 480 real‐time PCR system (Roche). For 47S pre‐rRNA or β‐actin mRNA Northern blotting, 10 μg total RNAs was separated by electrophoresis on 1% agarose/2% formaldehyde gel and transferred to a positively charged nylon membrane by capillary transfer. For tiRNA Northern blotting, 5 μg of total RNAs was separated by electrophoresis on 15% urea acrylamide‐polyacrylamide (PAGE) gel (Invitrogen) and transferred to a positively charged nylon membrane by semi‐dry transfer. After UV‐crosslink, the hybridization was performed with Digoxin (DIG)‐labeled DNA probes specific for 47S pre‐rRNA, β‐actin mRNA, and tiRNAs. Blots were detected using DIG Northern Starter Kit from Roche Applied Science following the manufacturer's instruction. The probe sequence is listed in Table EV5.
Western blotting
Cells or fresh tissues were lysed with RIPA lysis buffer consisting of 150 mM NaCl, 10 mM Tris, pH 7.5, 1% NP40, 1% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1× protease inhibitor cocktail (Roche). After separated by electrophoresis on 10‐15% SDS‐polyacrylamide gel, the proteins were transferred to nitrocellulose membranes (Whatman), blocked with 3% bovine serum albumin (BSA) in Tris‐buffered saline and Tween 20, and then blotted with the primary antibody followed by the IRDye 800CW‐labeled goat anti‐rabbit or mouse secondary antibody (Li‐COR Biosciences). The blot was visualized by an Odyssey infrared imaging system (Li‐COR) and then quantified using densitometry. The antibodies used for Western blotting in this study including Rabbit anti‐ANG (1 μg/ml, home‐made), Rabbit anti‐PLXNB2 (10602‐1‐AP, Proteintech), Rabbit anti‐Cyto c (10993‐1‐AP, Proteintech), Rabbit anti‐REG3A (DF6825, Affinit), Rabbit anti‐DEFA3 (DF8528, Affinit), Rabbit anti‐RNH1(10345‐1‐AP, Proteintech), Rabbit anti‐GAPDH (#5174, CST), and Rabbit anti‐ACTB (20536‐1‐AP, Proteintech).
Cell viability assay
The siRNA‐transfected cells were seeded at 96‐well plates in triplicate and treated with or without recombinant ANG for 1, 2, and 3 days, respectively. The cell viability was analyzed by CCK‐8 reagent kit (Dojindo Molecular Technologies) following the manufacturer's protocol.
Cell apoptosis detection
Cells were harvested and subjected to annexin V and PI double staining. After incubated with 5 μl of annexin V‐FITC and PI solution (Lianke) according to manufacturer's instruction for 10 min, cells were immediately run on the flow cytometer (Beckman Coulter).
Caspase activity detection
The caspase activity was detected using a caspase activity assay kit (Beyotime) according to manufacturer's instruction. Briefly, cells were seeded in dishes and incubated for 24 h. The culture medium was replaced with fresh culture medium supplemented with different recombinant ANG, ANG variants, TNF‐α, or NF‐κB inhibitor BAY. After 24 h, cells were harvested and resuspended in pyrolysis liquid. The lysate was centrifugated for 15 min at 4 °C. Finally, 40 μl of the supernatant was removed to a 96‐well plate, followed by mixturing with 10 μl Ac‐DEVD‐pNA (acetyl‐Asp‐Glu‐Val‐Asp p‐nitroanilide, for caspase3), Ac‐IETD‐pNA (acetyl‐Ile‐Glu‐Thr‐Asp p‐nitroanilide, for caspase8), or Ac‐LEHD‐pNA (acetyl‐Leu‐Glu‐His‐Asp p‐nitroanilide, for caspase9). After 1 h incubation at 37°C, the absorbance at 405 nm was detected by Microplate Reader (Thermo Fisher scientific).
Statistical analysis
All analyses were performed with GraphPad Prism 8 (GraphPad Software). Data were presented as mean with standard error of the mean (SEM) or violin plot. The difference between two groups was assessed using unpaired two‐tailed Student's t‐test or Mann–Whitney test for non‐normally distributed data. The difference between variables was assessed by one‐way ANOVA with Dunnett's multiple comparisons test or Kruskal–Wallis with Dunn's multiple comparisons test for nan‐normally distributed data. Equality of survivor function was determined by log‐rank test. A P‐value < 0.05 was considered as statistically significant; P‐value results were denoted by asterisks in the figures (*P < 0.05, **P < 0.01 and ***P < 0.001). The detailed statistical analysis and replicate numbers for each experiment were described in the figure legends, and the exact P‐value can be found in the corresponding source data file.
Author contributions
RB, JS, and ZX conceived of and designed the project. RB, DS, WZ, MC, XS, ZY, and YL performed experiments. RB, DS, JS, and ZX analyzed, discussed, and interpreted data. WZ, XGe, and LL were in charge of recruiting patients/controls and collected tissue samples. WZ, XGa, and GH provided essential study materials, reagents and patient samples. RB, JS, and ZX wrote the manuscript. All authors revised and approved the final version of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Prof. Hong Deng (Zhejiang University) for helpful discussion on pathologic analysis; Dr. Chaofeng Han (Second Military Medical University) and Dr. Wenlong Lin (Zhejiang University) for critical suggestion to the experimental design; and Yanwei Li, Shuangshuang Liu, and Yingying Huang from the Core Facilities of Zhejiang University School of Medicine for technical assistance in flow cytometry and microscopy analyses. This study was supported by the following grants: National Natural Science Foundation of China (No. 31770867, No. 31570786, No. 81790631, No. 81602557, and No. 31741026); Zhejiang Provincial Natural Science Foundation of China (No. LY18H030006 and LY17H160021); and Fundamental Research Funds for the Central Universities (No. 2017FZA7006).
EMBO Journal (2020) 39: e103325
Contributor Information
Jinghao Sheng, Email: jhsheng@zju.edu.cn.
Zhengping Xu, Email: zpxu@zju.edu.cn.
References
- Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C et al (2000) Blockade of interleukin 6 trans signaling suppresses T‐cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo . Nat Med 6: 583–588 [DOI] [PubMed] [Google Scholar]
- Bain CC, Scott CL, Uronen‐Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, Mowat AM (2013) Resident and pro‐inflammatory macrophages in the colon represent alternative context‐dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol 6: 498–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Mowat AM (2014) Macrophages in intestinal homeostasis and inflammation. Immunol Rev 260: 102–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N (2014) Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 15: 19–33 [DOI] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, Vijay‐Kumar M (2014) Dextran sulfate sodium (DSS)‐induced colitis in mice. Curr Protoc Immunol 104: Unit 15 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2013) The intestinal crypt, a prototype stem cell compartment. Cell 154: 274–284 [DOI] [PubMed] [Google Scholar]
- D'Angelo F, Bernasconi E, Schafer M, Moyat M, Michetti P, Maillard MH, Velin D (2013) Macrophages promote epithelial repair through hepatocyte growth factor secretion. Clin Exp Immunol 174: 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diegelmann J, Olszak T, Goke B, Blumberg RS, Brand S (2012) A novel role for interleukin‐27 (IL‐27) as mediator of intestinal epithelial barrier protection mediated via differential signal transducer and activator of transcription (STAT) protein signaling and induction of antibacterial and anti‐inflammatory proteins. J Biol Chem 287: 286–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2009) Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett 583: 437–442 [DOI] [PubMed] [Google Scholar]
- Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, Yang H, Scadden DT, Hu GF (2016) Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell 166: 894–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger JR, Konkel JE, Zangerle‐Murray T, Shaw TN (2017) Macrophages in gastrointestinal homeostasis and inflammation. Pflugers Arch 469: 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gress A, Ramensky V, Buch J, Keller A, Kalinina OV (2016) StructMAn: annotation of single‐nucleotide polymorphisms in the structural context. Nucleic Acids Res 44: W463–W468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose‐John S, Cheroutre H, Eckmann L et al (2009) IL‐6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis‐associated cancer. Cancer Cell 15: 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Neumann H, Neurath MF, Becker C (2013) Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut 62: 1062–1071 [DOI] [PubMed] [Google Scholar]
- Huang LY, He Q, Liang SJ, Su YX, Xiong LX, Wu QQ, Wu QY, Tao J, Wang JP, Tang YB et al (2014) ClC‐3 chloride channel/antiporter defect contributes to inflammatory bowel disease in humans and mice. Gut 63: 1587–1595 [DOI] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P (2011) Angiogenin‐induced tRNA fragments inhibit translation initiation. Mol Cell 43: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr TA, Ciorba MA, Matsumoto H, Davis VR, Luo J, Kennedy S, Xie Y, Shaker A, Dieckgraefe BK, Davidson NO (2012) Dextran sodium sulfate inhibition of real‐time polymerase chain reaction amplification: a poly‐A purification solution. Inflamm Bowel Dis 18: 344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutroubakis IE, Xidakis C, Karmiris K, Sfiridaki A, Kandidaki E, Kouroumalis EA (2004) Serum angiogenin in inflammatory bowel disease. Dig Dis Sci 49: 1758–1762 [DOI] [PubMed] [Google Scholar]
- Li S, Sheng J, Hu JK, Yu W, Kishikawa H, Hu MG, Shima K, Wu D, Xu Z, Xin W et al (2013) Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis 16: 387–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chen Y, Sun D, Bai R, Gao X, Yang Y, Sheng J, Xu Z (2018a) Angiogenin prevents progranulin A9D mutation‐induced neuronal‐like cell apoptosis through cleaving tRNAs into tiRNAs. Mol Neurobiol 55: 1338–1351 [DOI] [PubMed] [Google Scholar]
- Li S, Xu Z, Sheng J (2018b) tRNA‐derived small RNA: a novel regulatory small non‐coding RNA. Genes (Basel) 9: e246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luissint AC, Parkos CA, Nusrat A (2016) Inflammation and the intestinal barrier: leukocyte‐epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology 151: 616–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magro F, Araujo F, Pereira P, Meireles E, Diniz‐Ribeiro M, Velosom FT (2004) Soluble selectins, sICAM, sVCAM, and angiogenic proteins in different activity groups of patients with inflammatory bowel disease. Dig Dis Sci 49: 1265–1274 [DOI] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, Montrose MH, Turner JR, Watson AJ (2011) The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 140: 1208–1218.e1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini M, Bamias G, Rivera‐Nieves J, Moskaluk CA, Hoang SB, Ross WG, Pizarro TT, Cominelli F (2003) TNF‐alpha neutralization ameliorates the severity of murine Crohn's‐like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci USA 100: 8366–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini E, Krug SM, Siegmund B, Neurath MF, Becker C (2017) Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 4: 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu J, Riordan JF (1994) Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc Natl Acad Sci USA 91: 1677–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner‐Smidt K, Neumaier I et al (2010) Interferon‐gamma regulates intestinal epithelial homeostasis through converging beta‐catenin signaling pathways. Immunity 32: 392–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negroni A, Cucchiara S, Stronati L (2015) Apoptosis, necrosis, and necroptosis in the gut and intestinal homeostasis. Mediators Inflamm 2015: 250762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Travis SP (2012) Mucosal healing in inflammatory bowel diseases: a systematic review. Gut 61: 1619–1635 [DOI] [PubMed] [Google Scholar]
- Neurath MF (2014) Cytokines in inflammatory bowel disease. Nat Rev Immunol 14: 329–342 [DOI] [PubMed] [Google Scholar]
- Nishihara H, Kizaka‐Kondoh S, Insel PA, Eckmann L (2003) Inhibition of apoptosis in normal and transformed intestinal epithelial cells by cAMP through induction of inhibitor of apoptosis protein (IAP)‐2. Proc Natl Acad Sci USA 100: 8921–8926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikonomou KA, Kapsoritakis AN, Kapsoritaki AI, Manolakis AC, Tiaka EK, Tsiopoulos FD, Tsiompanidis IA, Potamianos SP (2011) Angiogenin, angiopoietin‐1, angiopoietin‐2, and endostatin serum levels in inflammatory bowel disease. Inflamm Bowel Dis 17: 963–970 [DOI] [PubMed] [Google Scholar]
- Okamoto R, Watanabe M (2016) Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J Gastroenterol 51: 11–21 [DOI] [PubMed] [Google Scholar]
- Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153 [DOI] [PubMed] [Google Scholar]
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S et al (2009) STAT3 links IL‐22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206: 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo E, Sarcinelli C, Sheng J, Fusco S, Formiggini F, Netti P, Yu W, D'Alessio G, Hu GF (2013) Ribonuclease/angiogenin inhibitor 1 regulates stress‐induced subcellular localization of angiogenin to control growth and survival. J Cell Sci 126: 4308–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak S, Mach A, Porvasnik S, Dixon L, Conlon T, Erger KE, Acosta A, Wright AJ, Campbell‐Thompson M, Zolotukhin I et al (2012) Identification of adeno‐associated viral vectors suitable for intestinal gene delivery and modulation of experimental colitis. Am J Physiol Gastrointest Liver Physiol 302: G296–G308 [DOI] [PubMed] [Google Scholar]
- Quiros M, Nishio H, Neumann PA, Siuda D, Brazil JC, Azcutia V, Hilgarth R, O'Leary MN, Garcia‐Hernandez V, Leoni G et al (2017) Macrophage‐derived IL‐10 mediates mucosal repair by epithelial WISP‐1 signaling. J Clin Invest 127: 3510–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales‐Campos H, Basso PJ, Alves VB, Fonseca MT, Bonfa G, Nardini V, Cardoso CR (2015) Classical and recent advances in the treatment of inflammatory bowel diseases. Braz J Med Biol Res 48: 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro R, Harper JW, Fox EA, Jansen HW, Hein F, Uhlmann E (1988) Expression of Met‐(‐1) angiogenin in Escherichia coli: conversion to the authentic less than Glu‐1 protein. Anal Biochem 175: 450–461 [DOI] [PubMed] [Google Scholar]
- Sheng J, Xu Z (2016) Three decades of research on angiogenin: a review and perspective. Acta Biochim Biophys Sin 48: 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, Mercier F, Baryawno N, Severe N, Bachand J, Spencer JA et al (2016) Proximity‐based differential single‐cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell 19: 530–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y (2011) IL‐17RE is the functional receptor for IL‐17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol 12: 1151–1158 [DOI] [PubMed] [Google Scholar]
- Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N et al (2014) Alterations in the microbiota drive interleukin‐17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40: 140–152 [DOI] [PubMed] [Google Scholar]
- Torres J, Mehandru S, Colombel JF, Peyrin‐Biroulet L (2017) Crohn's disease. Lancet 389: 1741–1755 [DOI] [PubMed] [Google Scholar]
- Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel JF (2017) Ulcerative colitis. Lancet 389: 1756–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZP, Tsuji T, Riordan JF, Hu GF (2002) The nuclear function of angiogenin in endothelial cells is related to rRNA production. Biochem Biophys Res Commun 294: 287–292 [DOI] [PubMed] [Google Scholar]
- Youssif C, Cubillos‐Rojas M, Comalada M, Llonch E, Perna C, Djouder N, Nebreda AR (2018) Myeloid p38alpha signaling promotes intestinal IGF‐1 production and inflammation‐associated tumorigenesis. EMBO Mol Med 10: e8403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Goncalves KA, Li S, Kishikawa H, Sun G, Yang H, Vanli N, Wu Y, Jiang Y, Hu MG et al (2017) Plexin‐B2 mediates physiologic and pathologic functions of angiogenin. Cell 171: 849–864.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32: 379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Malireddi RK, Body‐Malatrapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, Kanneganti TD (2011) The NOD‐like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20: 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7