

Abstract
Inter-individual transmission of cancer cells represents an intriguing and unexplored host-pathogen system, with significant ecological and evolutionary ramifications. The pathogen consists of clonal malignant cell lines that spread horizontally as allografts and/or xenografts. Although only nine transmissible cancer lineages in eight host species from both terrestrial and marine environments have been investigated, they exhibit evolutionary dynamics that may provide novel insights into tumor-host interactions particularly in the formation of metastases. Here we present an overview of known transmissible cancers, discuss the necessary and sufficient conditions for cancer transmission, and provide a comprehensive review on the evolutionary dynamics between transmissible cancers and their hosts.
Subject Areas: Biological Sciences, Evolutionary Biology, Evolutionary Ecology, Cancer
Graphical Abstract
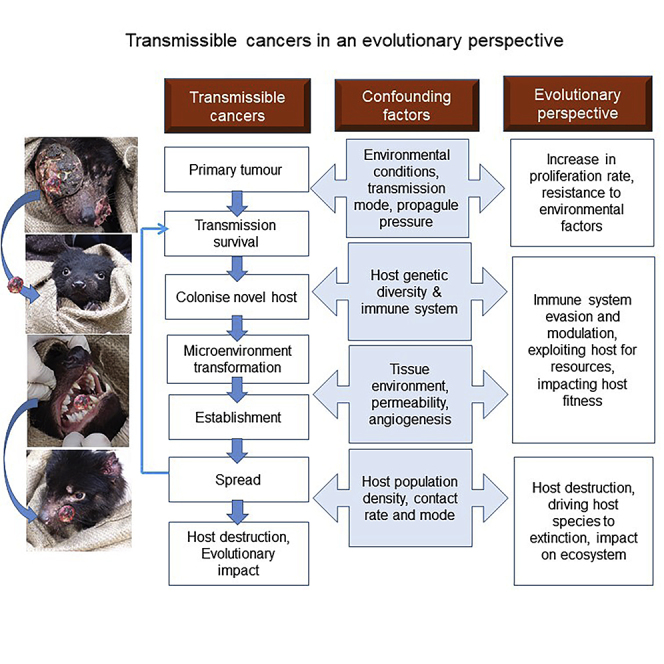
Biological Sciences; Evolutionary Biology; Evolutionary Ecology; Cancer
Introduction
Although rare, transmissible cancers are among the most intriguing and unexplored host-pathogen systems. The pathogens are clonal infectious malignant cell lines that originated in an individual within the host species or from a closely related species, and spread horizontally as allografts and/or xenografts. The vast majority of cancer cells that emerge in multicellular organisms die with their host, but evolutionary principles predict the existence and find cancer cell lineages that can escape the death by becoming contagious, acquire higher fitness, and consequently will be favored by selection (topic reviewed in Ujvari et al., 2017). Once the cancer cells have adapted to the normal barriers that prevent host-to-host transmission, they are subject to the evolutionary dynamics of infectious agents. They are, in effect, a new parasitic “species” (Dingli and Nowak, 2006). Currently, transmissible cancers have only been documented in three animal groups that occupy both terrestrial and marine environments and appear to result from the confluence of several environmental, host, and cell factors (the “perfect storm theory,” Ujvari et al., 2016a). Transmissible cancers have been proposed to have existed since the transition to multicellularity (Metzger and Goff, 2016). The evolution of multicellular organisms required that individual cells forgo their own reproductive interests, i.e., shifting the Darwinian unit of selection from individual cells to the entire multicellular community. However, the division of labor and specialization by differentiated cells in multicellular organisms also provided opportunities for these tissues to be colonized by fast proliferating cheater cells that take advantage of the benefits of multicellular tissue (e.g., blood flow) while not performing a differentiated function. To prevent colonization, multicellular organisms evolved defense mechanisms that may include sexual reproduction (Thomas et al., 2019) (Figure 1) and the development of immune systems (Metzger et al., 2015; Murchison, 2008; Murgia et al., 2006).
Figure 1.

Contagious Cancer Cells May Have Contributed to the Evolution of Sex
(A) Asexual reproduction results in low genetic diversity and high inter-individual similarity within a population that can lead to increased risk of vertical and horizontal transmission of contagious cells.
(B) Sexual reproduction results in greater genetic and inter-individual diversity in a population that limits the transmission probability of contagious cancer across individuals. Recognition of invading non-self cells is facilitated by higher inter-individual diversity. Selfish malignant cells regularly emerge in an organism, but unless the rare confluence of environmental and genetic factors occur (“perfect storm”), transmission of contagious cells will not occur (modified from Thomas et al., 2019).
The evolutionary importance of transmissible cancers as selective force is also demonstrated by their detectable impact on current vertebrate and invertebrate hosts. As described below, the devil facial tumor disease 1 (DFT1), previously referred to as DTFD, one of the three recognized mammalian contagious cancer lines, has caused >85% population decline in only 20 years as the Tasmanian devil species (Sarcophilus harrisii), once highly abundant, has been driven to the brink of extinction (Mccallum et al., 2009). Similarly, epizootic outbreaks of bivalve neoplasia have caused mass population declines in marine mollusk populations (Mateo et al., 2016). Such declines may have led to increased frequency of resistant individuals in the host population, which may have concomitantly contributed to the emergence of clonal cell lineages that are no longer able to infect the original host species, but are able to overcome immune defenses in a novel host (Metzger et al., 2016). It is also important to note that larger host population size may actually be the more likely scenario for host switching to occur. Perhaps success in one species could allow a larger population size of the cancer lineage, which might lead to a higher likelihood of a mutation that would allow a cross-species jump. An increased amount of cancer cells in the environment would provide more chances of cross-species interaction. In addition, from the literature in parasitology, other scenarios can be predicted. For example, a reduction in the host population may, at least temporarily, result in an increase in the parasite (here transmissible cancer) density (Combes et al., 2002). Then, a high parasite density on the principal host is likely to increase the number of infective propagules in the surrounding area and hence the probability of accidental encounter (and hence use) of alternative host species (Lootvoet et al., 2013; Poulin, 2007). Also, a high parasite (here transmissible cancer) density may increase overcrowding and within-host competition for limited resources, which may also favor alternative host use (Emelianov, 2007). Finally, the “principal host density” hypothesis suggests that when facing a decrease in the density of the principal host, parasites may colonize new host species to hedge their bets against the risk of co-extinction by reducing their dependence on a single resource (Bush and Kennedy, 1995; Koh et al., 2004).
Nevertheless, it is likely that this sequence has been recapitulated many times in the history of life on earth and may have contributed to extinction of the host species (Ní Leathlobhair et al., 2018). Thus, contagious cancers may represent an important, but so far understudied, selective force during organismal evolution, and therefore, an important component of many ecosystems (Russell et al., 2018).
Here we provide an overview of currently recognized transmissible cancers and discuss them from an evolutionary perspective.
Currently Known Transmissible Cancers
Nine independent transmissible cancers (one in dogs, two in Tasmanian devils, and six in six bivalve species) have so far been identified in the wild (Ujvari et al., 2017, Yonemitsu et al., 2019).
Canine Transmissible Venereal Tumor
Canine transmissible venereal tumor (CTVT) is caused by a sexually transmitted malignant cell line that affects dogs worldwide (Strakova and Murchison, 2014). Previous studies have proposed the cell line to be up to 11,000 years old (Strakova and Murchison, 2015), whereas phylogenetic analyses place the time of CTVT emergence between 4,000 and 8,500 years ago (Murgia et al., 2006; Murchison et al., 2014; Ostrander et al., 2016; Decker et al., 2015; Baez-Ortega et al., 2019) and the location to Asia (Baez-Ortega et al., 2019). CTVT most commonly affects sexually active dogs, as it spreads through sexual intercourse and licking and/or biting the affected areas. The cancer is mostly localized on the external genitalia (penis and foreskin in males and vulva in females), but in rare cases it can also manifest on the face (Das and Das, 2000; Mukaratirwa and Gruys, 2003; Papazoglou et al., 2001).
Experimental transplantation studies have revealed three distinct growth phases of CTVT (1) a progressive, (2) a static, and (3) a regressive stage (Das and Das, 2000; Mukaratirwa and Gruys, 2003). CTVT rarely metastasizes and responds well to treatment with vincristine (Nak et al., 2005).
Devil Facial Tumor Diseases
Similar to CTVT, direct contact is required for the transmission of the other two transmissible cancers in Tasmanian devils (S. harrisii), Tasmanian DFTDs. The first lineage of DFTD (DFT1 as some authors now refer to it) was discovered in 1996 in north eastern Tasmania (Hawkins et al., 2006), whereas the second and independently risen lineage (now reffered to as DFT2) was discovered in 2014 (Mccallum et al., 2009; Pye et al., 2015) in D'Entrecasteaux Peninsula (Figure 2). Both diseases are restricted to Tasmanian devils, which are endemic to this island, Tasmania, near the coast of Australia, and are transmitted via biting during social interactions (Pye et al., 2015). Both diseases present as large ulcerating tumors around the face and jaws, but DFT2 tumors often spread to other parts of the body (James et al., 2019). Despite the phenotypic similarity of the tumors they form, DFT1 and DFT2 cells are genetically, chromosomally, and histologically different (Pye et al., 2015). The presence of chromosome Y in DFT2 and remnants of an X chromosome in DFT1 indicate that the older cell line emerged in a female devil and the younger from a male (Murchison et al., 2012; Pye et al., 2015). DFT1 is a fatal cancer, frequently (70%) metastasizing to distant organs, with death typically occurring 6 to 9 months after appearance of the first lesions (Pyecroft et al., 2007). The typical disease course of DFT2 is not currently known. DFT1 has spread across Tasmania and reached epidemic proportion decimating the devil population, whereas DFT2 is currently confined to the D'Entrecasteaux Peninsula.
Figure 2.
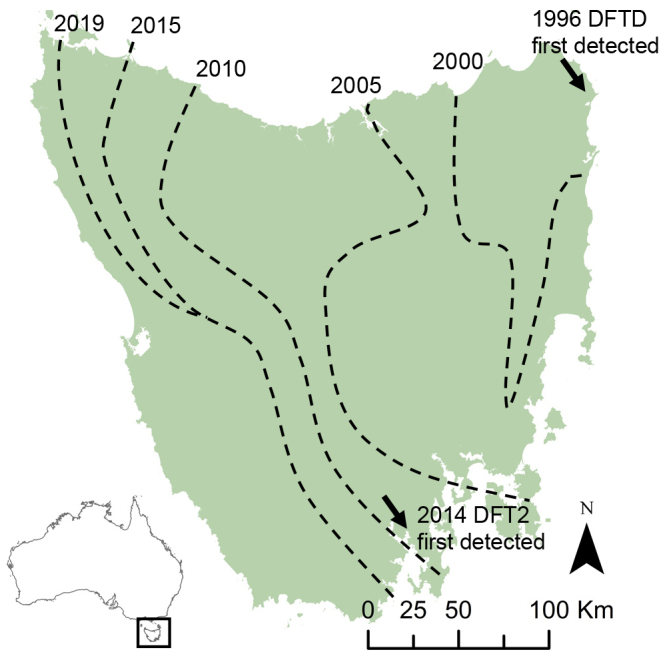
Distribution of Devil Facial Tumor Diseases (DFT1 and DFT2)
The first lineage of Tasmanian devil facial tumour diseases (previously referred to as DFTD, recently renamed as DFT1) was discovered in 1996 in the north eastern corner of Tasmania and spread across the island (Hawkins et al., 2006), whereas the second and independently risen lineage, DFT2, was discovered in 2014 and is currently confined to the D'Entrecasteaux Peninsula.
There are reports of the devils adapting to DFT1 as tumor regressions have been observed (see in details below) (Epstein et al., 2016; Hubert et al., 2018; Pye et al., 2016; Russell et al., 2018; Wright et al., 2017), but the Tasmanian devil remains classified as “Endangered” by the International Union for Conservation of Nature (https://www.iucnredlist.org/) and may face extinction in the coming decades (Mccallum et al., 2009).
Bivalve Transmissible Neoplasias
Abnormal proliferation of cells in bivalve hemolymph, i.e., disseminated neoplasia (DN, also referred to as hematopoietic or hemic neoplasia), has been widely described since the 1960s from various mollusks such as clams, mussels, oysters, and cockles across the world (Barber, 2004). Originally, a retrovirus or retrotransposon etiology was suspected (Oprandy and Chang, 1983), but recent studies demonstrated the horizontal transmission of clonal cells, and thus the presence of bivalve transmissible neoplasia (BTN) in soft-shell clams (Mya arenaria), in mussels (Mytilus trossulus), and in cockles (Cerastoderma edule) (Metzger et al., 2015, 2016), with currently two lineages being recognized in the last one. BTN cells have also been able to transmit across species, i.e., in golden carpet shell clams (Polititapes aureus) the cancer cells originated from the pullet shell clam (Venerupis corrugata) (Metzger et al., 2015, 2016) and a cancer line in the Pacific mussel Mytilus trossulus has spread to two other European and Chilean mussel species, Mytilus edulis and Mytilus chilensis (Yonemitsu et al., 2019). The distribution of transmissible BTN across continents most likely has been facilitated by a transmission mode that does not require direct contact between individuals; by human interference, e.g., the transplantation of seed stocks along the coast of the United States during the 1990s; as well as by marine transportation across the globe (Metzger et al., 2016, Yonemitsu et al., 2019).
DN and BTN are characterized in bivalves by the presence of large, invasive pleomorphic cells in the hemolymph, the circulatory fluid of bivalves. These neoplastic cells can cause displacement, compression, and necrosis within tissues due to disturbed apoptosis and phagocytic abilities (Aguilera, 2017). Neoplastic hemocytes in bivalves are also often aneuploid with an increased DNA content indicating a relative high chromosomal instability (Benadelmouna et al., 2018). In addition, diseased cells lose their phagocytic abilities, express a novel surface antigen, and display cytoplasmic sequestration of the TP53 tumor suppressor protein (Walker et al., 2011). Possible physiological effects of the disease include tissue emaciation, pale digestive gland, gonadal atrophy, and mantle recession (Cremonte et al., 2011), suggesting nutritional stress, reduced energy intake, or a possible metabolic burden imposed by the neoplastic cells on the host (Barber, 2004). As the disease is progressive, it often results in the death of the individual. Neoplastic diseases in bivalves appear at their highest frequency in polluted areas with high levels of heavy metals, polychlorinated biphenyls, and polycyclic aromatic hydrocarbons (Carballal et al., 2015).
Transmissible Cancers: the Inter-individual Metastasis
Many cancers are directly or indirectly caused by infectious agents (Aktipis et al., 2015; Vittecoq et al., 2015; Vittecoq et al., 2013; de Martel et al., 2012; Ewald and Swain Ewald, 2015), but for a cancer to be truly horizontally transmissible, the cancer cell itself must be able to migrate between hosts. The transplantation of live tumor cells (but not killed cells or filtrates) between individuals and the presence of characteristic marker chromosomes and cancer-specific genotypes in all tumor samples collected from different host populations across geographic regions provide evidence that the cancer cells are indeed transmitting as allografts.
The fitness of non-transmissible cancer cells is primarily determined by their capacity to proliferate, avoid immune recognition, undergo clonal expansion, and metastasize to novel organs to avoid competition at the primary tumor site. However, their survival is ultimately limited by the lifespan of the host. Cells that can persist in the host population by escaping the original host before its death will occupy an empty niche, and thus acquire higher fitness.
Similar to the metastatic cascade of classical cancers, the spread of a transmissible cancer to a new host is a multi-step biological process in which the cancer cells must overcome distinct barriers and obstacles (Gatenby and Gillies, 2008; Ujvari et al., 2015) (Figure 3). Following the initial growth at the primary tumor sites (Nguyen et al., 2009), cancer cells acquire the capacity to disassemble the extracellular matrix and intravasate the lymphatic or blood microvessels, and to disseminate throughout the body of the host (i.e., metastasize). Although most of those circulating cells will die, some are able to survive transit by arresting in the capillary beds within distant organs and extravasate into the new host tissue (Nguyen et al., 2009; Hanahan and Weinberg, 2011). Following extravasation, the majority of invading cells perishes, and only a very small minority of metastatic cells ultimately colonize the new tissue site and form a clinically apparent tumor (Nguyen et al., 2009).
Figure 3.

Population Biology of Intra- and Inter-individual Metastasis
The generalized steps necessary for cancer cells to transmit to a new host are analogous to the steps of intra-individual metastasis (modified from Chen and Pienta, 2011). Similar to the classical metastatic cascade, following the initial growth at the primary tumor site, transmissible cancer cells have to survive transit, colonize, and establish in the new microenvironment of the novel host.
However, the total life cycle of a transmissible cancer is also affected by the evolutionary dynamics of infectious agents in which virulence and infectivity trade-offs are common. Most simply, the virulence of the disease cannot be so great that the host will not survive to transmit the disease. In the devil facial tumor, for example, the infected host can transmit the disease only if there are other devils in its territory, if the host is sufficiently healthy to socially interact with other devils, and if the inoculum of cancer cells during that interaction is sufficient to cause tumor formation in the new host. These are all highly dynamic variables. For example, as the tumor spreads throughout a population, the density of available, uninfected partners can decrease. If the tumor kills the host too quickly it may become too debilitated to perform the biting necessary to transmit the disease. Furthermore, if there is selection for resistance over time, the required inoculum size may increase substantially. Proliferative immortality (that is, for example, permitted by the continuous replenishment of chromosome ends via upregulated telomerase activity in DFT1 and CTVT, Chu et al., 2001; Ujvari et al., 2012) and the capacity for self-renewal allow the existence of these clonal cell lines.
Clearly, a key factor in these dynamics is the barriers for tumor transmission. Perhaps the most obvious of these include histocompatibility barriers (i.e., self/non-self recognition, not only within individuals of a species but also across species), as demonstrated by the mammalian tumors (CTVT and DFTDs) that have been proposed to have emerged due to the low genetic diversity of the host species (reviewed in Belov, 2011).
However, the transmitted cancer cells may also be limited by foreign environments that lack growth factors and nutrients and are subject to a host of immunological and non-immunologic host responses. Furthermore, contagious cancers must be transmitted by direct contact (DFTDs, CTVT) or environmental transport (BTN). This requires time outside of the host and tolerance to non-physiological conditions (Walther and Ewald, 2004). For example, because bivalves are filter feeders, BTN likely spreads through seawater. BTN cells therefore must be tolerant to a wide range of changes in the physical properties (e.g., temperature) and soluble factors within seawater. Indeed, bivalve neoplastic cells have been shown to survive at least 6 h under the broad range of environmental conditions encountered in marine habitats occupied by bivalve populations (i.e., variations of salinity, temperature, dissolved oxygen concentration, and pH on a diel temporal scale). Neoplastic cells of bivalves are able to survive typical estuarine conditions (salinity of 15, temperature of 24°C) for at least 48 h, and some cells are even able to remain viable in freezing conditions (Sunila and Farley, 1989).
How Transmissible Cancers Emerge: The Perfect Storm Theory
Although contagious cancer cells gain a clear fitness advantage over non-transmissible cell lines, the substantial barriers to horizontal transmission of cancer cells require a “perfect storm” with the confluence of multiple host (micro- and macro-environmental factors) and tumor cell traits (Ujvari et al., 2016a).
Propagule Pressure, Permissive Environments, and Phenotypic Plasticity Facilitate Cancer Cell Transmission
Conditions that are necessary for direct passage of cancer cells between hosts include tumor tissue properties that promote shedding of large numbers of malignant cells, some interactions between hosts that allow transmission, tumor cell (genetic and phenotypic) plasticity that permits survival during transmission and establishment in a new host, and a “permissible” host or host tissue.
Investigations of colonizing events by non-native species has found that outcomes depend on the “propagule pressure,” i.e., (1) the number of cells shed at introduction, (2) the number of introduction events, (3) and the temporal and spatial patterns of propagule arrival (Ujvari et al., 2016a).
Similar dynamics have been observed in cancer spread. For example, in the formation of metastatic disease from a primary site in the same host, the loss of cells at each step in the metastatic cascade is so substantial that no more than 1 cell in 10,000 will form a metastasis (Chambers et al., 2002). Similarly, in inter-individual metastatic cancers, the tumor inoculum needs to be very high (Barfield et al., 2015; Leggett et al., 2012). In DFT1, for example, under laboratory conditions, 105 to 106 DFT1 cells in immunocompromised mice are necessary for tumor formation (Kreiss et al., 2011). Experimental transmission of BTN showed that injecting 106 BTN cells induced cancer in 33% new hosts, whereas injecting 107 BTN cells resulted in 50% infection rate (House, 1997).
Establishment in, and adaptation to, a hostile host environment is governed by genetic similarity of the hosts, by the hosts' immune competence, and direct competition between cells from the host and cells from the cancer (Siddle and Kaufman, 2013). Contributing to host immune response, histocompatibility barriers generally prevent the transplantation of foreign grafts between individuals, or between species. The ability of DFT1 to infect >150,000 members of the same species has been attributed to the low genetic diversity of the Tasmanian devils (Belov, 2011, 2012). In addition, DFT1 cancer cells use epigenetic immune modulation to down-regulate genes involved in the antigen-processing pathways. This loss of cell surface expression of self/non-self recognizing major histocompatibility complex class I (MHC) molecules results in immune recognition evasion (Siddle and Kaufman, 2013; Siddle et al., 2013). Interestingly, the more recently emerged DFT2 cells have evolved a different immune escape strategy by expressing MHC alleles that are shared with hosts carrying tumors (Caldwell et al., 2018).
The mechanism(s) by which CTVT evades the host immune system are not completely known. However, experimental transplantation studies find that CTVT cancer cells initially secrete transforming growth factor β (TGF-β) that suppresses class I and II dog leukocyte antigen expression (DLA, equivalent of dog MHC) and natural killer cell activity. Tumor-infiltrating leukocytes produce interleukin-6 (IL-6) that counteracts, and eventually overcomes TGF-β, leading to the re-expression of DLAs on CTVT cells and finally to the elimination of tumor cells (Chiang et al., 2013; Hsiao et al., 2004). Importantly, during CTVT regression, most upregulated genes appeared to be of host origin and Frampton et al. (2018) proposed that the immune response to CTVT tumors was driven by a critical epigenetic remodeling of host tissue surrounding the tumor cells.
Epigenetic regulation, and thus phenotypic plasticity (change in phenotypic expression in response to environmental variations) not only facilitates immune system evasion but also can be essential facilitators for cancer cells to adapt to a range of environmental conditions both in transit and in the hosts (Ujvari et al., 2016a, 2016b). For example, phenotypic plasticity has been observed in the recently emerged DFT2 tumors, some expressing MHC class I molecules that are either shared with host devils or are non-classical MHC molecules, whereas in other DFT2 tumors the loss of MHC class I expression has been observed (Caldwell et al., 2018). Changes in epigenetic marks have been associated with the emergence and evolution of DFT1 cancer cells (Ingles and Deakin, 2015; Ujvari et al., 2013). Disintegration of a hypermethylated X chromosome followed by increasing loss of methylation of the DFT1 genome suggest that DFT1 is a dynamically changing entity with a high phenotypic plasticity and evolutionary potential (Ujvari et al., 2013, 2015). Although variation in mollusk methylome has already been demonstrated (e.g., during ontogenesis or in response to environmental stressors), it is still unknown whether epigenetic variation contributes to phenotypic plasticity in DN and BTN (Rondon et al., 2017).
Environmental Factors and Instable Genomes May Contribute to the Emergence of Transmissible Cancers
Little is known about the origins and critical factors initiating transmissible cancers in nature. Although CTVT is the oldest known transmissible cancer cell lineage, its cellular origin remains uncertain. Various studies suggest a macrophage or a myeloid origin (reviewed in (Das and Das, 2000; Mukaratirwa and Gruys, 2003)), whereas the latest transcriptomic analysis proposed a skin cancer and cutaneous melanoma origin (Frampton et al., 2018). DFT1 and DFT2 appear to have both originated from cells of the peripheral nervous system (Stammnitz et al., 2018; Patchett et al., 2019). As melanoma and devil facial tumor cells are both proposed to have derived from neural crest cells, Frampton et al. (2018) have recently suggested that some transmissible cancers may share a common origin.
A combination of environmental factors and unstable genomes has been implicated in all three types of transmissible cancers. DFT1 and CTVT both carry the mutational signature of solar UV radiation (Decker et al., 2015; Murchison et al., 2012, 2014; Stammnitz et al., 2018). Most bivalves are intertidal species, and hence are exposed to UV radiation (Salvini-Plawen, 2019). Thus, it is feasible that UV exposure might have been a key initiator in all the known contagious cancers. Indeed, Baez-Ortega et al. (2019) have observed non-linear association between latitude and UV exposure and consequently location-specific variations in the mutational spectra of CTVT. In addition, bivalve neoplastic diseases appear at their highest frequency in heavily polluted areas (with heavy metals, polychlorinated biphenyls, and polycyclic aromatic hydrocarbons) (Carballal et al., 2015), which may have also contributed to genome instability and consequently genetic mutations facilitating abnormal cell proliferation.
Tug of War: Evolution of Transmissible Cancers and Their Hosts
Hosts Can Resist or Tolerate the Challenges by Contagious Cell Lines
Similar to classical pathogens, and in contrast to classical cancer cells, transmissible cancer cell lines have emerged from a common ancestor, maintaining their malignant genotypes and phenotypes across generations in the hosts' population. Therefore, their evolution follows fundamental Darwinian principles and resembles host-parasite interactions (described by the “Red-Queen” dynamics van Valen, 1973; Schmid-Hempel, 2011). As transmissible cancers negatively affect host fitness, by either impeding reproductive output (CTVT) or reducing survival (DFTD and BTN), they also impose strong selection pressure for resistance.
When infected by a pathogen, hosts can evolve defensive responses that can be broadly categorized as (1) resistance, defined as the ability of a host to limit or inhibit pathogen replication, thus reducing infection severity, and (2) tolerance, defined as the ability of an infected host to limit the impact of infection on host fitness (Raberg et al., 2007; Roy and Kirchner, 2000). When relying on resistance, a host can employ a multitude of, often costly, behavioral, morphological, physiological, and/or immunological defense mechanisms to limit and eliminate the negative effects of the pathogens (Råberg et al., 2009; Raberg et al., 2007; Roy and Kirchner, 2000). Unlike resistance, host tolerance does not limit infection, but reduces or compensates parasite-induced damages through reduced immunopathology, increased wound repair mechanisms, and a greater resilience to tissue injuries (Råberg et al., 2009; Raberg et al., 2007; Roy and Kirchner, 2000). Tolerance thus offsets and/or reduces the pathogen's impact at significantly lower fitness costs compared with responses involving resistance (Råberg et al., 2009; Raberg et al., 2007; Roy and Kirchner, 2000). Although resistance and tolerance may result in similar fitness benefit for the host, their evolutionary arcs proceed in different directions. That is, resistance produces a negative feedback loop by eventually eliminating the host alleles from the population that initially provided resistance against the infection (as the pathogen evolves faster and the host alleles that initially provided resistance become redundant over time), whereas tolerance generates a positive feedback loop that increases the chance for “tolerance genes” becoming fixed in the host population (Lough et al., 2015; Råberg et al., 2009; Raberg et al., 2007; Roy and Kirchner, 2000). As both tolerance and resistance are epidemiologically and ecologically costly (Restif and Koella, 2004), selection will therefore favor the most cost-effective strategy (resistance, tolerance, or the combination of the two) (Restif and Koella, 2004). Indeed, the combination of the two strategies has been observed in Tasmanian devil populations. Several studies have now shown rapid phenotypic and genetic adaptations to DFTD (Russell et al., 2018) including changes in reproductive strategy (transitioned from multiple reproductive cycles to a single breeding pattern, Jones et al., 2008), in gene expression variation (Ujvari et al., 2016c) and in allele frequency variation at immune and tumor suppressor genes that are involved in limiting tumor growth (Epstein et al., 2016; Wright et al., 2017) as well as genes underlying behavioral patterns (Hubert et al., 2018), which could potentially limit disease transmission. In some devil populations active immune responses and even tumor regression have been observed (Pye et al., 2016; Tovar et al., 2017), whereas in other populations tolerance has been detected in response to DFTD (Ruiz-Aravena et al., 2018). Thus the extreme selection pressure imposed by DFTD has allowed the Tasmanian devil population to evolve both resistance and tolerance responses in only four to six generations (Russell et al., 2018).
However, the transmissible cancer can also evolve adaptations to the host responses forming an “evolutionary arms race.” For example, the study by Decker et al. (2015) identified signatures of continuous adaptation by CTVT to its host, observing adaptive mutations in response to every increase in immune responses, including immune surveillance, self-antigen presentation, and apoptosis. Although spontaneous regression is uncommon in CTVT, dogs are able to tolerate the disease, and it appears that CTVT and the host have reached a stable evolutionary dynamic (Read et al., 2008). Nevertheless, CTVT responds to a single dose of vincristine, and Frampton et al. (2018) proposed that “CTVT is particularly susceptible to changes that break tolerance to this cancer.”
Intriguingly, although invertebrates, including mollusks, do not possess MHC molecules, they are equipped with potent self/non-self recognition mechanisms (Allam and Raftos, 2015). Mollusks are able to respond to transplants (Wei et al., 2017) and, in some cases, combat transformed malignant cells (Robert, 2010; Voskoboynik et al., 2013). For example, although DN can be fatal, up to 20% remission has been observed (Brousseau and Baglivo, 1991). Another study in the mussel (M. edulis) and clam (Mya arenaria) also showed that the hosts were capable of developing a response against the disease, resulting in a temporary remission (Elston et al., 1988; Weinberg et al., 1997). These studies indicate the presence of a thus far unknown immune response to DN cells, as well as potentially against BTN cells, and the ability of mussels to resist neoplasia.
The Evolution of Transmissible Cancer Cell Lines in Response to Challenges by the Host
Escaping the Red-Queen by Crossing the Species Barrier
Both CTVT and BTN can cross histocompatibility barriers between members of the same species and even between species. For example, jackals (Samso, 1965) and coyotes (Cockrill and Beasley, 1979) have been experimentally transfected with CTVT (reviewed in Mukaratirwa and Gruys, 2003), and cross-species transmission of BTN has been observed between two sympatric mollusks belonging to the same family (Veneridae) (Metzger et al., 2016), as well as between mollusk species across continents (Yonemitsu et al., 2019) (see earlier discussion). The ability of infectious cancers to transmit across species suggests two interesting evolutionary scenarios: (1) the original hosts have been able to recognize and evolve resistance against their own cells and (2) the cancer cells have potentially been able to overcome host resistance (thus “outrun the Red-Queen”) by spreading to a novel host species.
Intra- versus Inter-individual Metastasis: Where Should Evolution Take Transmissible Cancers
Transmissible cancers, in effect, are an inter-individual metastasis, and this poses fascinating evolutionary questions. In an infectious cancer, one might reasonably expect that higher proliferation rate and enhanced capacity to spread to different organs would favor a higher level of infectivity, and thus increased fitness of the cancer population. However, these same properties will also promote more rapid metastatic spread in the host leading to a more virulent cancer, which could lead to host disability or death before successful transmission. This would reduce the fitness of the cancer population. Consequently, a strategy that allows efficient inter-individual transmission and also reduced impact on host organs (thus prevention of intra-individual spread) may provide the highest fitness outcome to contagious cancer cell lines. As intra- and inter-individual metastasis might be driven by potentially similar mechanisms, deciphering the evolutionary trajectory of these intriguing parasitic cell lines remains a challenging but fascinating topic of evolutionary biology.
Transmissible Cancer Cell Lines Survive over Evolutionary Time by Overcoming Genomic Decay
As transmissible cancer cell lines have emerged and evolved from one ancestral cell, they propagate by asexual reproduction resulting in accumulation of deleterious mutations (termed “Muller's ratchet") leading to a decline in fitness. However, CTVT has been shown to avoid an “information catastrophe” (Eigen and Schuster, 1977) by capturing and incorporating host mitochondrial DNA (mtDNA) (Rebbeck et al., 2009), as well as via negative selection in captured mtDNA and occasional mtDNA recombination and re-assortment (Strakova et al., 2016). Although more work is needed to confirm mitochondrial replacement in bivalves, the sequence data from some of the Chilean BTN samples indicated the replacement of part of the cancer mtDNA genome with sequence from the host mtDNA (Yonemitsu et al., 2019). However, host mtDNA capture has not been observed in the DFTDs perhaps because the diseases are, evolutionarily speaking, relatively young and may not yet be subject to Muller's ratchet. More likely, DFTDs employ other mechanisms, such as increasing chromosome numbers or duplication associated with ectopic gene conversion to overcome genomic decay (Marais et al., 2010). The increased ploidy can stabilize the genomic configuration of clonal cells and hence promote the selection of polyploid cancer cells, which are also able to sustain higher mutation rates. Polyploidy can therefore mask recessive deleterious mutations and ameliorate genomic decay (Orr, 1995). As elevated chromosome numbers have been observed in all transmissible cancers, polyploidy may provide an adaptive advantage (Metzger et al., 2015; Mukaratirwa and Gruys, 2003; Ujvari et al., 2014). Positive selection has been shown to outweigh negative selection during the evolution of classical, non-transmissible cancer development (Martincorena et al., 2017). The lack of negative selection on coding (potentially fatal) point mutations in clonal cancer cells indicates that either the majority of genes are dispensable in these cell lineages or that the deleterious effect is being buffered by diploidy/polyploidy and/or by the inherent resilience and redundancy of cellular pathways (Martincorena et al., 2017). This could also explain the capacity of long-lived transmissible cancer cell lines, such as CTVT, to accumulate and tolerate extreme levels of hypermutation (Baez-Ortega et al., 2019; Murchison et al., 2012, 2014). In a recent study, Baez-Ortega et al. (2019) found that in contrast to short-lived cancers where positive selection is the primary driving force, neutral genetic drift appears be the dominant evolutionary force operating in CTVT (but see Decker et al. (2015)). As discussed earlier in the article, the lack of negative selection may result in the accumulation of deleterious mutations; however, the increased chromosome numbers in cancer cells most likely buffer the effect of negative selection and facilitate the maintenance of cancer cells' fitness.
Conclusions
“Just like people often don't realize the benefits of vaccines when the disease becomes rare, evolutionary biologists may not recognize the critical role of transmissible cancers in shaping animal evolution as these cancers are very rare [Prof James DeGregori]”.
So far there are very few infectious cancer clones described. This could be due to several factors: (1) we just have not looked hard enough, (2) they did exist but their hosts were driven to extinction, and (3) they are exceedingly rare because “perfect storms” are rare, i.e., the barriers are very stringent. Although transmissible cancers appear to be rare in nature, they represent a fascinating and important eco-evolutionary phenomenon that may provide novel insights into human cancers. It is likely that during the history of life on earth, several contagious cancers have evolved on numerous occasions and may have contributed to some species extinctions. Similar to classical host-parasite interactions (described by the “Red-Queen” dynamics (van Valen, 1973; Schmid-Hempel, 2011)), both hosts and contagious cancer cells will evolve continuously to adapt to their opponent's innovations generating an evolutionary arms race. These strategies and counterstrategies may be recapitulated over time as the tumor-host interactions progress from in situ growth to progression at the primary site to elements of the metastatic cascade.
Limitations of the Study
Additional articles may have been published on the topic since submission of the manuscript.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Beata Ujvari, Beata.ujvari@deakin.edu.au.
Materials Availability
Not applicable as this is a review article.
Data and Code Availability
Not applicable as this is a review article.
Acknowledgments
This work was supported by an ARC Linkage (LP170101105), Australia; Deakin SEBE_RGS_2019, Australia; an ANR TRANSCAN (ANR-18-CE35-0009), France; a Holsworth Wildlife Research Endowment, Australia; and a CNRS “International Associated Laboratory Grant”, France.
Author Contributions
Conceptual development and literature review of bivalve disseminated neoplasia sections were conducted by A.M.D., G.B., and A.S.; of Tasmanian devil facial tumor disease sections, by R.H., A.-L.G., and E.D.; and of canine venereal tumor disease sections, by N.R., N.M., and B.U. Evolutionary perspectives and parallels with metastatic cancers were developed by M.G., F.T., and R.A.G. Graphical abstract was prepared by A.D. and B.U. All authors contributed to writing and final editing of the manuscript. Funding was obtained by R.H., F.T., N.R., A.S., and B.U.
Declaration of Interests
The authors declare no competing interests.
References
- Aguilera F. Neoplasia in mollusks: what does it tell us about cancer in humans? - a review. J. Genet. Disord. 2017;1:1–10. [Google Scholar]
- Aktipis C.A., Boddy A.M., Jansen G., Hibner U., Hochberg M.E., Maley C.C., Wilkinson G.S. Cancer across the tree of life: cooperation and cheating in multicellularity. Phil. Trans. R. Soc. B. 2015;370:20140219. doi: 10.1098/rstb.2014.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam B., Raftos D. Immune responses to infectious diseases in bivalves. J. Invertebr. Pathol. 2015;131:121–136. doi: 10.1016/j.jip.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Baez-Ortega A., Gori K., Strakova A., Allen J.L., Allum K.M., Bansse-Issa L., Bhutia T.N., Bisson J.L., Briceño C., Castillo Domracheva A. Somatic evolution and global expansion of an ancient transmissible cancer lineage. Science. 2019;365:eaau9923. doi: 10.1126/science.aau9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber B.J. Neoplastic diseases of commercially important marine bivalves. Aquat. Living Resour. 2004;17:449–466. [Google Scholar]
- Barfield M., Orive M.E., Holt R.D. The role of pathogen shedding in linking within- and between-host pathogen dynamics. Math. Biosci. 2015;270:249–262. doi: 10.1016/j.mbs.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov K. The role of the major histocompatibility complex in the spread of contagious cancers. Mamm. Genome. 2011;22:83–90. doi: 10.1007/s00335-010-9294-2. [DOI] [PubMed] [Google Scholar]
- Belov K. Contagious cancer: lessons from the devil and the dog. BioEssays. 2012;34:285–292. doi: 10.1002/bies.201100161. [DOI] [PubMed] [Google Scholar]
- Benadelmouna A., Saunier A., Ledu C., Travers M.A., Morga B. Genomic abnormalities affecting mussels (Mytilus edulis-galloprovincialis) in France are related to ongoing neoplastic processes, evidenced by dual flow cytometry and cell monolayer analyses. J. Invertebr Pathol. 2018;157:45–52. doi: 10.1016/j.jip.2018.08.003. [DOI] [PubMed] [Google Scholar]
- Brousseau D.J., Baglivo J.A. Field and laboratory comparisons of mortality in normal and neoplastic Mya arenaria. J. Invertebr. Pathol. 1991;57:59–65. doi: 10.1016/0022-2011(91)90041-n. [DOI] [PubMed] [Google Scholar]
- Bush A., Kennedy C.R. Host fragmentation and helminth parasites: hedging your bets against extinction. Int. J. Parasitol. 1995;24:1333–1343. doi: 10.1016/0020-7519(94)90199-6. [DOI] [PubMed] [Google Scholar]
- Caldwell A., Coleby R., Tovar C., Stammnitz M.R., Kwon Y.M., Owen R.S., Tringides M., Murchison E.P., Skjødt K., Thomas G.J. The newly-arisen Devil facial tumour disease 2 (DFT2) reveals a mechanism for the emergence of a contagious cancer. Elife. 2018;7:e35314. doi: 10.7554/eLife.35314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballal M.J., Barber B.J., Iglesias D., Villalba A. Neoplastic diseases of marine bivalves. J. Invertebr. Pathol. 2015;131:83–106. doi: 10.1016/j.jip.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Chambers A.F., Groom A.C., Macdonald I.C. Metastasis: dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Chen K.W., Pienta K.J. Modeling invasion of metastasizing cancer cells to bone marrow utilizing ecological principles. Theor. Biol. Med. Model. 2011;8:36. doi: 10.1186/1742-4682-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang H.-C., Liao A.T.-C., Jan T.-R., Wang Y.-S., Lei H.-J., Tsai M.-H., Chen M.-F., Lee C.-Y., Lin Y.-C., Chu R.-M. Gene-expression profiling to identify genes related to spontaneous tumor regression in a canine cancer model. Vet. Immunol. immunopathol. 2013;151:207–216. doi: 10.1016/j.vetimm.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Chu R.M., Lin C.Y., Liu C.C., Yang S.Y., Hsiao Y.W., Hung S.W., Pao H.N., Liao K.W. Proliferation characteristics of canine transmissible venereal tumor. Anticancer Res. 2001;21:4017–4024. [PubMed] [Google Scholar]
- Cockrill J.M., Beasley J.N. Transmission of transmissible venereal tumor of the dog to the coyote. Am. J. Vet. Res. 1979;40:409–410. [PubMed] [Google Scholar]
- Combes C., de Buron I., Connors V. The University of Chicago Press; 2002. Parasitism: The Ecology and Evolution of Intimate Interactions. Bibliovault OAI Repository. [Google Scholar]
- Cremonte F., Vazquez N., Silva M. Gonad atrophy caused by disseminated neoplasia in Mytilus chilensis cultured in the beagle channel, tierra del fuego province, Argentina. J. Shellfish Res. 2011;30:845–849. [Google Scholar]
- Das U., Das A. Review of canine transmissible venereal sarcoma. Vet. Res. Commun. 2000;24:545–556. doi: 10.1023/a:1006491918910. [DOI] [PubMed] [Google Scholar]
- de Martel C., Ferlay J., Franceschi S., Vignat J., Bray F., Forman D., Plummer M. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–615. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- Decker B., Davis B.W., Rimbault M., Long A.H., Karlins E., Jagannathan V., Reiman R., Parker H.G., Drögemüller C., Corneveaux J.J. Comparison against 186 canid whole-genome sequences reveals survival strategies of an ancient clonally transmissible canine tumor. Genome Res. 2015;25:1646–1655. doi: 10.1101/gr.190314.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingli D., Nowak M.A. Cancer biology: infectious tumour cells. Nature. 2006;443:35–36. doi: 10.1038/443035a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M., Schuster P. The hypercycle. A principle of natural self-organization. Part A: emergence of the hypercycle. Naturwissenschaften. 1977;64:541–565. doi: 10.1007/BF00450633. [DOI] [PubMed] [Google Scholar]
- Elston R.A., Kent M., Drum A.S. Progression, lethality and remission of hemic neoplasia in the bay mussel Mylilus edulis. Dis. Aquat. Organisms. 1988;4:135–142. [Google Scholar]
- Emelianov I. How adaptive is parasite species diversity? Int. J. Parasitol. 2007;37:851–860. doi: 10.1016/j.ijpara.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Epstein B., Jones M., Hamede R., Hendricks S., Mccallum H., Murchison E.P., Schonfeld B., Wiench C., Hohenlohe P., Storfer A. Rapid evolutionary response to a transmissible cancer in Tasmanian devils. Nat. Commun. 2016;7:12684. doi: 10.1038/ncomms12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald P.W., Swain Ewald H.A. Infection and cancer in multicellular organisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20140224. doi: 10.1098/rstb.2014.0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton D., Schwenzer H., Marino G., Butcher L.M., Pollara G., Kriston-Vizi J., Venturini C., Austin R., de Castro K.F., Ketteler R. Molecular signatures of regression of the canine transmissible venereal tumor. Cancer Cell. 2018;33:620–633.e6. doi: 10.1016/j.ccell.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby R.A., Gillies R.J. A microenvironmental model of carcinogenesis. Nat. Rev. Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hawkins C.E., Baars C., Hesterman H., Hocking G.J., Jones M.E., Lazenby B., Mann D., Mooney N., Pemberton D., Pyecroft S. Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol. Conserv. 2006;131:307–324. [Google Scholar]
- House M. Doctor of Philosophy Oregon State University; 1997. Transmission of Disseminated Neoplasia in the Soft Shell Clam Mya Arenaria. [Google Scholar]
- Hsiao Y.-W., Liao K.-W., Hung S.-W., Chu R.-M. Tumor-infiltrating lymphocyte secretion of IL-6 antagonizes tumor-derived TGF-β1 and restores the lymphokine-activated killing activity. J. Immunol. 2004;172:1508–1514. doi: 10.4049/jimmunol.172.3.1508. [DOI] [PubMed] [Google Scholar]
- Hubert J.N., Zerjal T., Hospital F. Cancer- and behavior-related genes are targeted by selection in the Tasmanian devil (Sarcophilus harrisii) PLoS One. 2018;13:e0201838. doi: 10.1371/journal.pone.0201838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingles E.D., Deakin J.E. Global DNA Methylation patterns on marsupial and devil facial tumour chromosomes. Mol. Cytogenet. 2015;8:74. doi: 10.1186/s13039-015-0176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S., Jennings G., Kwon Y.M., Stammnitz M., Fraik A., Storfer A., Comte S., Pemberton D., Fox S., Brown B. Tracing the rise of malignant cell lines: distribution, epidemiology and evolutionary interactions of two transmissible cancers in Tasmanian devils. Evol. Appl. 2019;12:1772–1780. doi: 10.1111/eva.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M.E., Cockburn A., Hamede R., Hawkins C., Hesterman H., Lachish S., Mann D., Mccallum H., Pemberton D. Life-history change in disease-ravaged Tasmanian devil populations. Proc. Natl. Acad. Sci. U S A. 2008;105:10023–10027. doi: 10.1073/pnas.0711236105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh L.P., Dunn R.R., Sodhi N.S., Colwell R.K., Proctor H.C., Smith V.S. Species coextinctions and the biodiversity crisis. Science. 2004;305:1632. doi: 10.1126/science.1101101. [DOI] [PubMed] [Google Scholar]
- Kreiss A., Tovar C., Obendorf D.L., Dun K., Woods G.M. A murine xenograft model for a transmissible cancer in tasmanian devils. Vet. Pathol. 2011;48:475–481. doi: 10.1177/0300985810380398. [DOI] [PubMed] [Google Scholar]
- Leggett H.C., Cornwallis C.K., West S.A. Mechanisms of pathogenesis, infective dose and virulence in human parasites. PLoS Pathog. 2012;8:e1002512. doi: 10.1371/journal.ppat.1002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lootvoet A., Blanchet S., Gevrey M., Buisson L., Tudesque L., Loot G. Patterns and processes of alternative host use in a generalist parasite: insights from a natural host-parasite interaction. Funct. Ecol. 2013;27:1403–1414. [Google Scholar]
- Lough G., Kyriazakis I., Bergmann S., Lengeling A., Doeschl-Wilson A.B. Health trajectories reveal the dynamic contributions of host genetic resistance and tolerance to infection outcome. Proc. Biol. Sci. 2015;282:20152151. doi: 10.1098/rspb.2015.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G.A.B., Campos P.R.A., Gordo I. Can intra-Y gene conversion oppose the degeneration of the human Y chromosome? A simulation study. Genome Biol. Evol. 2010;2:347–357. doi: 10.1093/gbe/evq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I., Raine K.M., Gerstung M., Dawson K.J., Haase K., van Loo P., Davies H., Stratton M.R., Campbell P.J. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171:1029–1041.e21. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo D.R., Maccallum G.S., Davidson J. Field and laboratory transmission studies of haemic neoplasia in the soft-shell clam, Mya arenaria, from Atlantic Canada. J. Fish Dis. 2016;39:913–927. doi: 10.1111/jfd.12426. [DOI] [PubMed] [Google Scholar]
- Mccallum H., Jones M., Hawkins C., Hamede R., Lachish S., Sinn D.L., Beeton N., Lazenby B. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology. 2009;90:3379–3392. doi: 10.1890/08-1763.1. [DOI] [PubMed] [Google Scholar]
- Metzger M.J., Goff S.P. A sixth modality of infectious disease: contagious cancer from devils to clams and beyond. PLOS Pathog. 2016;12:e1005904. doi: 10.1371/journal.ppat.1005904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger M.J., Reinisch C., Sherry J., Goff S.P. Horizontal transmission of clonal cancer cells causes leukemia in soft-shell clams. Cell. 2015;161:255–263. doi: 10.1016/j.cell.2015.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger M.J., Villalba A., Carballal M.J., Iglesias D., Sherry J., Reinisch C., Muttray A.F., Baldwin S.A., Goff S.P. Widespread transmission of independent cancer lineages within multiple bivalve species. Nature. 2016;534:705–709. doi: 10.1038/nature18599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaratirwa S., Gruys E. Canine transmissible venereal tumour: cytogenetic origin, immunophenotype, and immunobiology. A review. Vet. Q. 2003;25:101–111. doi: 10.1080/01652176.2003.9695151. [DOI] [PubMed] [Google Scholar]
- Murchison E.P. Clonally transmissible cancers in dogs and Tasmanian devils. Oncogene. 2008;27:S19–S30. doi: 10.1038/onc.2009.350. [DOI] [PubMed] [Google Scholar]
- Murchison E.P., Schulz-Trieglaff O.B., Ning Z., Alexandrov L.B., Bauer M.J., Fu B., Hims M., Ding Z., Ivakhno S., Stewart C. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell. 2012;148:780–791. doi: 10.1016/j.cell.2011.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison E.P., Wedge D.C., Alexandrov L.B., Fu B., Martincorena I., Ning Z., Tubio J.M.C., Werner E.I., Allen J., de Nardi A.B. Transmissible dog cancer genome reveals the origin and history of an ancient cell lineage. Science. 2014;343:437–440. doi: 10.1126/science.1247167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia C., Pritchard J.K., Kim S.Y., Fassati A., Weiss R.A. Clonal origin and evolution of a transmissible cancer. Cell. 2006;126:477–487. doi: 10.1016/j.cell.2006.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nak D., Nak Y., Cangul T., Tuna B. A clinico-pathological study on the effect of vincristine on transmissible venereal tumour in dogs. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2005;52:366–370. doi: 10.1111/j.1439-0442.2005.00743.x. [DOI] [PubMed] [Google Scholar]
- Nguyen D.X., Bos P.D., Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Ní Leathlobhair M., Perri A.R., Irving-Pease E.K., Witt K.E., Linderholm A., Haile J., Lebrasseur O., Ameen C., Blick J., Boyko A.R. The evolutionary history of dogs in the Americas. Science. 2018;361:81. doi: 10.1126/science.aao4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprandy J.J., Chang P.W. 5-Bromodeoxyuridine induction of hematopoietic neoplasia and retrovirus activation in the soft-shell clam, Mya arenaria. J. Invertebr. Pathol. 1983;42:196–206. doi: 10.1016/0022-2011(83)90062-9. [DOI] [PubMed] [Google Scholar]
- Orr H.A. Somatic mutation favors the evolution of diploidy. Genetics. 1995;139:1441–1447. doi: 10.1093/genetics/139.3.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander E.A., Davis B.W., Ostrander G.K. Transmissible tumors: breaking the cancer paradigm. Trends Genet. 2016;32:1–15. doi: 10.1016/j.tig.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazoglou L., Koutinas A., Plevraki A.G., Tontis D. Primary intranasal transmissible venereal tumour in the dog: a retrospective study of six spontaneous cases. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2001;48:391–400. doi: 10.1046/j.1439-0442.2001.00361.x. [DOI] [PubMed] [Google Scholar]
- Patchett A., Coorens T., Darby J., Wilson R., Mckay M., Kamath K., Rubin A., Wakefield M., Mcintosh L., Mangiola S. Two of a kind: transmissible Schwann cell cancers in the endangered Tasmanian devil (Sarcophilus harrisii) Cell Mol. Life Sci. 2019;77:1847–1858. doi: 10.1007/s00018-019-03259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin R. Princeton University Press; 2007. Evolutionary Ecology of Parasites: from Individuals to Communities. [Google Scholar]
- Pye R., Hamede R., Siddle H.V., Caldwell A., Knowles G.W., Swift K., Kreiss A., Jones M.E., Lyons A.B., Woods G.M. Demonstration of immune responses against devil facial tumour disease in wild Tasmanian devils. Biol. Lett. 2016;12:20160553. doi: 10.1098/rsbl.2016.0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye R.J., Pemberton D., Tovar C., Tubioc J.M.C., Dund K.A., Fox S., Darbya J., Hayes D., Knowles G.W., Kreiss A. A second transmissible cancer in Tasmanian devils. Proc. Nat. Acad. Sci. U S A. 2015 doi: 10.1073/pnas.1519691113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyecroft S.B., Pearse A.M., Loh R., Swift K., Belov K., Fox N., Noonan E., Hayes D., Hyatt A., Wang L. Towards a case definition for devil facial tumour disease: what is it? EcoHealth. 2007;4:346–351. [Google Scholar]
- Råberg L., Graham A.L., Read A.F. Decomposing health: tolerance and resistance to parasites in animals. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2009;364:37–49. doi: 10.1098/rstb.2008.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raberg L., Sim D., Read A.F. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science. 2007;318:812–814. doi: 10.1126/science.1148526. [DOI] [PubMed] [Google Scholar]
- Read A.F., Graham A.L., Råberg L. Animal defenses against infectious agents: is damage control more important than pathogen control. PLoS Biol. 2008;6:e1000004. doi: 10.1371/journal.pbio.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck C.A., Thomas R., Breen M., Leroi A.M., Burt A. Origins and evolution of a transmissible cancer. Evolution. 2009;63:2340–2349. doi: 10.1111/j.1558-5646.2009.00724.x. [DOI] [PubMed] [Google Scholar]
- Restif O., Koella J.C. Concurrent evolution of resistance and tolerance to pathogens. Am. Nat. 2004;164:E90–E102. doi: 10.1086/423713. [DOI] [PubMed] [Google Scholar]
- Robert J. Comparative study of tumorigenesis and tumor imminity in invertebrates and nonmammlian vertebrates. Dev. Comp. Immunol. 2010;34:915–925. doi: 10.1016/j.dci.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon R., Grunau C., Fallet M., Charlemagne N., Sussarellu R., Chaparro C., Montagnani C., Mitta G., Bachère E., Akcha F., Cosseau C. Effects of a parental exposure to diuron on pacific oyster spat methylome. Environ. Epigenetics. 2017;3:dvx004. doi: 10.1093/eep/dvx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B.A., Kirchner J.W. Evolutionary dynamics of pathogen resistance and tolerance. Evolution. 2000;54:51–63. doi: 10.1111/j.0014-3820.2000.tb00007.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Aravena M., Jones M.E., Carver S., Estay S., Espejo C., Storfer A., Hamede R.K. Sex bias in ability to cope with cancer: tasmanian devils and facial tumour disease. Proc. R. Soc. B: Biol. Sci. 2018;285:20182239. doi: 10.1098/rspb.2018.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell T., Madsen T., Thomas F., Raven N., Hamede R., Ujvari B. Oncogenesis as a selective force: adaptive evolution in the face of a transmissible cancer. Bioessays. 2018;40 doi: 10.1002/bies.201700146. [DOI] [PubMed] [Google Scholar]
- Salvini-Plawen L. Encyclopedia Britannica; 2019. Mollusk. [Google Scholar]
- Samso A. Docteur ès Sciences Naturalles thesis; 1965. Recherches Experimentales sur le Sarcome de Sticker. [Google Scholar]
- Schmid-Hempel P. Oxford University Press; 2011. Evolutionary Parasitology: The Integrated Study of Infections, Immunology, Ecology, and Genetics. [Google Scholar]
- Siddle H.V., Kaufman J. How the devil facial tumor disease escapes host immune responses. OncoImmunology. 2013;2:e25235. doi: 10.4161/onci.25235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddle H.V., Kreiss A., Tovar C., Yuen C.K., Cheng Y., Belov K., Swift K., Pearse A.-M., Hamede R., Jones M.E. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl. Acad. Sci. 2013;110:5103–5108. doi: 10.1073/pnas.1219920110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stammnitz M.R., Coorens T.H.H., Gori K.C., Hayes D., Fu B., Wang J., Martin-Herranz D.E., Alexandrov L.B., Baez-Ortega A., Barthorpe S. The origins and vulnerabilities of two transmissible cancers in tasmanian devils. Cancer Cell. 2018;33:607–619.e15. doi: 10.1016/j.ccell.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakova A., Murchison E.P. The changing global distribution and prevalence of canine transmissible venereal tumour. BMC Vet. Res. 2014;10:168. doi: 10.1186/s12917-014-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakova A., Murchison E.P. The cancer which survived: insights from the genome of an 11,000 year-old cancer. Curr. Opin. Genet. Dev. 2015;30:49–55. doi: 10.1016/j.gde.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Strakova A., Ní Leathlobhair M., Wang G.-D., Yin T.-T., Airikkala-Otter I., Allen J.L., Allum K.M., Bansse-Issa L., Bisson J.L., Castillo Domracheva A. Mitochondrial genetic diversity, selection and recombination in a canine transmissible cancer. Elife. 2016:e14552. doi: 10.7554/eLife.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunila I., Farley C.A. Environmental limits for survival of sarcoma cells from the soft-shell clam Mya arenaria. Dis. Aquat. Organ. 1989;7:111–115. [Google Scholar]
- Thomas F., Madsen T., Giraudeau M., Misse D., Hamede R., Vincze O., Renaud F., Roche B., Ujvari B. Transmissible cancer and the evolution of sex. PLoS Biol. 2019;17:e3000275. doi: 10.1371/journal.pbio.3000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar C., Pye R.J., Kreiss A., Cheng Y., Brown G.K., Darby J., Malley R.C., Siddle H.V.T., Skjødt K., Kaufman J. Regression of devil facial tumour disease following immunotherapy in immunised Tasmanian devils. Sci. Rep. 2017;7:43827. doi: 10.1038/srep43827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari B., Gatenby R.A., Thomas F. The evolutionary ecology of transmissible cancers. Infect. Genet. Evol. 2016;39:293–303. doi: 10.1016/j.meegid.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Ujvari B., Gatenby R.A., Thomas F. Transmissible cancers, are they more common than thought? Evol. Appl. 2016;9:633–634. doi: 10.1111/eva.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari B., Gatenby R.A., Thomas F. Chapter 12 - transmissible cancer: the evolution of interindividual metastasis. In: Ujvari B., Roche B., Thomas F., editors. Ecology and Evolution of Cancer. Academic Press; 2017. pp. 167–175. [Google Scholar]
- Ujvari B., Hamede R., Peck S., Pemberton D., Jones M., Belov K., Madsen T. Immunoglubolin dynamics and cancer prevalence in Tasmanian devils (Sarcophilus harrisii) Sci. Rep. 2016;6:25093. doi: 10.1038/srep25093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari B., Papenfuss A.T., Belov K. Transmissible cancers in an evolutionary context. Inside Cell. 2015;1:1–10. doi: 10.1002/bies.201670904. [DOI] [PubMed] [Google Scholar]
- Ujvari B., Pearse A.-M., Peck S., Harmsen C., Taylor R., Pyecroft S., Madsen T., Papenfuss A.T., Belov K. Evolution of a contagious cancer: epigenetic variation in devil facial tumour disease. Proc. R. Soc. B: Biol. Sci. 2013;280:20121720. doi: 10.1098/rspb.2012.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari B., Pearse A.-M., Swift K., Hodson P., Hua B., Pyecroft S., Taylor R., Hamede R., Jones M., Belov K., Madsen T. Anthropogenic selection enhances cancer evolution in Tasmanian devil tumours. Evol. Appl. 2014;7:260–265. doi: 10.1111/eva.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari B., Pearse A.-M., Taylor R., Pyecroft S., Flanagan C., Gombert S., Papenfuss A.T., Madsen T., Belov K. Telomere dynamics and homeostasis in a transmissible cancer. PLoS One. 2012;7:e44085. doi: 10.1371/journal.pone.0044085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Valen L. A new evolutionary law. Evol. Theor. 1973;1:1–30. [Google Scholar]
- Vittecoq M., Ducasse H., Arnal A., Møller A.P., Ujvari B., Jacqueline C.B., Tissot T., Missé D., Bernex F., Pirot N. Animal behaviour and cancer. Anim. Behav. 2015;101:19–26. [Google Scholar]
- Vittecoq M., Roche B., Daoust S.P., Ducasse H., Missé D., Abadie J., Labrut S., Renaud F., Gauthier-Clerc M., Thomas F. Cancer: a missing link in ecosystem functioning? Trends Ecol. Evol. 2013;28:628–635. doi: 10.1016/j.tree.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Voskoboynik A., Newman A.M., Corey D.M., Sahoo D., Pushkarev D., Neff N.F., Passarelli B., Koh W., Ishizuka K.J., Palmeri K.J. Identification of a colonial chordate histocompatibility gene. Science. 2013;341:384–387. doi: 10.1126/science.1238036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C.W., van Beneden R.J., Muttray A.F., Böttger S.A., Kelley M.L., Tucker A.E., Thomas W.K. Chapter one - p53 superfamily proteins in marine bivalve cancer and stress biology. In: Michael L., editor. Advances in Marine Biology. Academic Press; 2011. pp. 3–29. [DOI] [PubMed] [Google Scholar]
- Walther B.A., Ewald P.W. Pathogen survival in the external environment and the evolution of virulence. Biol. Rev. Camb Philos. Soc. 2004;79:849–869. doi: 10.1017/S1464793104006475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Fan S., Liu B., Zhang B., Su J., Yu D. Transcriptome analysis of the immune reaction of the pearl oyster Pinctada fucata to xenograft from Pinctada maxima. Fish Shellfish Immunol. 2017;67:331–345. doi: 10.1016/j.fsi.2017.06.030. [DOI] [PubMed] [Google Scholar]
- Weinberg J.R., Leavitt D.F., Lancaster B.A., Capuzzo J.M. Experimental field studies with Mya arenaria(Bivalvia) on the induction and effect of hematopoietic neoplasia. J. Invertebr. Pathol. 1997;69:183–194. doi: 10.1006/jipa.1996.4641. [DOI] [PubMed] [Google Scholar]
- Wright B., Willet C.E., Hamede R., Jones M., Belov K., Wade C.M. Variants in the host genome may inhibit tumour growth in devil facial tumours: evidence from genome-wide association. Sci. Rep. 2017;7:423. doi: 10.1038/s41598-017-00439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemitsu M.A., Giersch R.M., Polo-Prieto M., Hammel M., Simon A., Cremonte F., Avilés F.T., Merino-Véliz N., Burioli E.A.V., Muttray A.F. A single clonal lineage of transmissible cancer identified in two marine mussel species in South America and Europe. Elife. 2019;8:e47788. doi: 10.7554/eLife.47788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable as this is a review article.