

Abstract
With more people reaching an advanced age in modern society, there is a growing need for strategies to slow down age-related neuropathology and loss of cognitive functions, which are a hallmark of Alzheimer's disease. Neuroprotective drugs and candidate drug compounds target one or more processes involved in the neurodegenerative cascade, such as excitotoxicity, oxidative stress, misfolded protein aggregation and/or ion dyshomeostasis. A growing body of research shows that a G-protein coupled zinc (Zn2+) receptor (GPR39) can modulate the abovementioned processes.
Zn2+ itself has a diverse activity profile at the synapse, and by binding to numerous receptors, it plays an important role in neurotransmission. However, Zn2+ is also necessary for the formation of toxic oligomeric forms of amyloid beta, which underlie the pathology of Alzheimer’s disease. Furthermore, the binding of Zn2+ by amyloid beta causes a disruption of zincergic signaling, and recent studies point to GPR39 and its intracellular targets being affected by amyloid pathology.
In this review, we present neurobiological findings related to Zn2+ and GPR39, focusing on its signaling pathways, neural plasticity, interactions with other neurotransmission systems, as well as on the effects of pathophysiological changes observed in Alzheimer's disease on GPR39 function.
Direct targeting of the GPR39 might be a promising strategy for the pharmacotherapy of zincergic dyshomeostasis observed in Alzheimer’s disease. The information presented in this article will hopefully fuel further research into the role of GPR39 in neurodegeneration and help in identifying novel therapeutic targets for dementia.
Keywords: Metal ions, brain, memory, cognitive, aging, glutamate, hippocampus
1. INTRODUCTION
The occurrence of amyloid beta (Aβ) plaques is considered the hallmark of Alzheimer’s disease (AD) and is the main factor for differential diagnosis of the disorder. However, it is still unknown why Aβ deposits start to form long before any neuropsychological symptoms become present. Moreover, even in patients with mild cognitive impairment (MCI) who are prone to late onset AD due to the apolipoprotein E ε4 genotype (which causes decreased clearance of Aβ), cognitive decline is only weakly associated with an Aβ plaque burden [1]. Since current advances in positron emission tomography (PET) scanning methods have enabled the detection of Aβ plaques in the brains of cognitively normal adults, there is a chance for preventive treatment to be developed [2]. Structural changes following Aβ deposition are also detectable by magnetic resonance imaging (MRI) in the preclinical asymptomatic phase of AD, which suggests that in some cases even initial brain volume loss could be halted before severe cognitive dysfunction occurs [3].
The only biomarker of AD that correlates well with cognitive decline is hyperphosphorylated tau protein [4] - the main cause of a spectrum of neurodegenerative disorders termed ‘tauopathies’. Over the last 20 years it has become apparent that oligomeric forms of Aβ (AβO) are more toxic than Aβ plaques, and not only promote tau pathology, but also cause oxidative stress, the loss of neurotrophic factors, synapse dysfunction and, eventually, nerve cell death [5]. This is why the field of AD research has shifted towards understanding the involvement of AβOs in functional, rather than, structural changes in the brain, placing synaptic communication at the center of the problem [6]. Biometals are among the factors responsible for AβO formation, and it is their binding by Aβ that additionally causes metal dyshomeostasis [7]. In this review, we focus on zinc ion (Zn2+), its interactions with Aβ and the ways in which the neurotransmitter properties of Zn2+ are disrupted by Aβ, eventually causing synapse loss, which is highly correlated with cognitive symptoms of AD. We also highlight the role of GPR39 – a G protein-coupled receptor for Zn2+ [8, 9]- and present current findings implicating a GPR39 role in AD-related neural functions. Understanding these interactions will hopefully help in designing novel dual-action compounds (i.e. drugs capable of addressing both Aβ toxicity and Zn2+ dyshomeostasis).
2. PHYSIOLOGICAL FUNCTIONS OF VESICULAR ZN2+ IN THE CNS
Zn2+ is a trace element that is involved in regulating the function of around 10% of human proteins [10]. In fact, it is crucial for all organisms, which may well be a legacy of its role in the origin of life [11]. Within the human body, Zn2+ is encountered in most abundance in the pancreas and the brain [12], where – among other functions - it is co-released with glutamate into synapses enabling neuronal communication [13]. Although this freely available pool of synaptic/vesicular Zn2+ constitutes only a fraction of the total Zn2+ in the brain, by binding to several different membrane receptors, the ion modulates synaptic connections [14-17]. The development of genetically modified mice lacking ZnT3 [18] – a membrane protein responsible for Zn2+ transport into the presynaptic terminals [19] enabled the influence of zincergic neurotransmission on psychological functions to be investigated [20]. Such studies highlighted the importance of vesicular Zn2+ in spatial [21] and fear memory [22], in exploratory behavior in a social context and in general anxiety [23], as well as in somatosensory discrimination abilities [24].
These observations correspond well with the localization of vesicular Zn2+ in the brain. The ion is predominantly found in the hippocampus, the cortex, the olfactory bulb and the amygdalae, where pools of Zn2+ reside within the axon terminals of glutamatergic neurons [13]. In the spinal cord and the cerebellum glycinergic and GABAergic neurons, respectively, also co-release Zn2+ into the synaptic cleft [25, 26]. However, the neurobiological mechanisms underlying the effects of zincergic neurotransmission on psychological functions can differ even within a single brain area.
This is probably best exemplified by the various ways in which Zn2+ affects neurotransmission at mossy fibers-CA3 (MF-CA3) and Schaffer collaterals-CA1 (SC-CA1) glutamatergic synapses of the hippocampus. During memory formation and retrieval, the pyramidal neurons of the CA3 area contribute to pattern completion - a process in which stimuli are experienced as being similar to previously encountered ones - while CA1 area pyramidal neurons are responsible for switching between the encoding and the retrieval of information [27]. The SC-CA1 and MF-CA3 pathways also use different modes of long-term potentiation (LTP; see Box 1 for details), with a classical postsynaptic N-methyl-D-aspartate receptor (NMDAR)-dependent LTP at SC-CA1 [28] and a presynaptic, NMDAR-independent form of LTP at MF-CA3 [29]. Remarkably, vesicular Zn2+ contributes to both forms of LTP in physiologically relevant concentrations at the synapse. By using a high-affinity Zn2+ chelator – ZX1 - Pan and colleagues demonstrated that vesicular Zn2+ is necessary for presynaptic LTP at MF-CA3 [14]. Lack of Zn2+ during high-frequency stimulation (HFS) blocks an increase in release probability of neurotransmitters from MF terminals and, subsequently, attenuates an increase in excitatory postsynaptic potentials (EPSPs) at the CA3 pyramidal neurons 60 min after HFS. At the same MF-CA3 synapse Zn2+ inhibits a form of calcium-mediated postsynaptic LTP, which is NMDAR-independent and becomes unmasked in ZnT3 knock-out (KO) mice. As well as increasing release probability, Zn2+ contributes to a progressive shift towards presynaptic MF-CA3 LTP by blocking the high-affinity GluN2A subunit of the NMDAR [30]. At the SC-CA1 synapses, where release probability is constitutively higher and less variable, Zn2+-dependent inhibition of GluN2A-containing NMDARs saturates quickly and negatively modulates postsynaptic LTP with different dynamics [30]. It is therefore intriguing that Zn2+ is also crucial for SC-CA1 LTP in vivo and that chelation of the ion from both extracellular and intracellular spaces causes CA1-dependent recognition memory deficits in rats [31]. A working hypothesis is that it is the intracellular Zn2+ that leads to SC-CA1 LTP. An increase in intracellular Zn2+ in CA1 pyramidal neurons could either be caused by the release of Zn2+ from intracellular stores or by the entry of Zn2+ through calcium ion (Ca2+)- and Zn2+ -permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) lacking the GluA2 subunit [32]. The former mechanism seems to be more plausible on the basis of studies involving the ZX1 chelator, which showed that even a single presynaptic action potential is sufficient for Zn2+ to inhibit AMPAR-mediated excitatory postsynaptic currents at the SC-CA1 level [17]. Thus, Zn2+ could prevent its own entry through the AMPAR during LTP formation at the SC-CA1 synapses. In the meantime, the same high-affinity Zn2+ chelator did not affect AMPAR-mediated postsynaptic currents at the MF-CA3 synapses [14], suggesting that Zn2+ does not inhibit AMPARs in the CA3 pyramidal neurons.
Box 1. Long-term potentiation.
| Long-term potentiation (LTP) refers to a persistent strengthening of a synaptic connection in response to a specific firing pattern of presynaptic neurons. It was discovered in the rabbit hippocampus in 1966 by the Norwegian physiologist Terje Lømo and since then it has been widely studied as a neural substrate of memory and learning. Typically, at an excitatory synapse, a single action potential of a presynaptic neuron will cause an excitatory postsynaptic potential (EPSP) in the consecutive neuron. Both the amplitude and the duration of EPSPs can be altered by previous experience. In LTP, a high-frequency (100 Hz) train of stimuli, called a ‘tetanus’, causes a shift towards longer EPSPs with greater amplitudes, which are observed up to several days/weeks following tetanus. Different forms of LTP exist, with associative (NMDAR-dependent) and non-associative (NMDA-independent) LTP being one of the most prominent distinctions. In associative LTP, the presynaptic activity has to precede the firing of a postsynaptic neuron (“Hebb’s rule”). This enables the simultaneous occurrence of two events at the NMDARs - namely, the opening of an NMDAR channel and the expulsion of the Mg2+ ion, which blocks the channel at resting membrane potential. This causes a major depolarization of the neuronal membrane, as well as an influx of Ca2+ through the NMDARs and subsequent activation of downstream signaling pathways (ex. Ca2+/calmodulin-dependent protein kinase II - CAMKII, protein kinase C - PKC), which causes the recruitment of AMPARs from internal stores during early LTP and de novo AMPAR synthesis in late LTP. In non-associative LTP, the Ca2+ influx into the presynaptic terminal, together with Ca2+ release from its internal pools caused by the tetanus, leads to an increased glutamate release probability during future action potentials. Non-associative LTP is therefore independent from postsynaptic activity. |
This discrepancy is reflected in recent studies concerned with the role of Zn2+ in the degeneration of pyramidal neurons in the CA1 and CA3 areas [33] caused by excitotoxic insults following ischemia [34]. Both areas were affected by excessive intracellular accumulation of Zn2+, but the sources of Zn2+differed between the regions, with an AMPAR-mediated influx of Zn2+ in the CA3 area, and the mobilization of cytosolic Zn2+ from matallothionein III (a zinc-binding protein) in the CA1 area [33]. These results may reflect a quantitative difference in AMPAR availability between the areas, as zincergic synapses in the CA1 area contain around 40% fewer AMPARs than non-zincergic ones in the same region, while in the CA3 all of the AMPAR-containing synapses are zincergic [35]. However, it is also possible that distinct binding sites for Zn2+ in the AMPARs exist in the CA1 and CA3 regions, causing CA3 AMPARs not to be inhibited by Zn2+, but currently, there is no direct evidence to support this hypothesis. In relation to this, Blackmore and Trombley have provided a review of plausible evidence for a diverse expression of AMPAR subunits, as well as for the flip/flop splice variants of AMPAR genes being the reason for contradictory effects of Zn2+ on AMPAR transmission in different regions of the olfactory bulb [36].
NMDARs - the second major group of ionotropic receptors for glutamate and Zn2+ - are a vital component of functionally relevant synaptic connections and a molecular substrate of memory. NMDARs are inhibited by Zn2+ at multiple binding sites [37]. At low nano-molar concentrations, Zn2+ acts as an allosteric antagonist by binding to the N-terminal domain of the NMDAR containing the high-affinity GluN2A subunit [38]. At low micro-molar concentrations, Zn2+ is also capable of inhibiting low affinity GluN2B-containing NMDARs [39]. The action of Zn2+ at both sites is voltage-independent, but at higher concentrations (10-100 μM) the ion also inhibits NMDARs in a voltage-dependent manner irrespective of its subunit composition.
The modulatory potential of Zn2+ is further manifested in its influence on synaptic and extrasynaptic NMDARs. In the dorsal cochlear nucleus (DCN), which is an auditory pathway structure with a cerebellum-like circuitry, tonic extracellular levels of Zn2+ are between 1 and 10 nM and seem to be at the higher end of this range at extrasynaptic sites, where Zn2+ tonically inhibits GluN2A-comprised NMDARs [16]. No such effect was observed at synaptic GluN2A-containing NMDARs [16], where the release of vesicular Zn2+ either was necessary for inhibition [30] or was insufficient to inhibit synaptic GluN2A NMDARs due to high levels of glutamate, suggesting that only extrasynaptic GluN2A NMDARs are blocked by both tonic and vesicular Zn2+ [16]. Functionally, the location-dependent Zn2+ action on GluN2A NMDARs could be important in both brain development and aging since – contrary to the case with immature neurons - in mature neuronal connections the ratio of GluN2A to GluN2B is higher at the synaptic site, with an opposite proportion at extrasynaptic locations [40]. By inhibiting only extrasynaptic GluN2A NMDARs, Zn2+ would further emphasize this developmental switch. Therefore, an excessive accumulation of the ion during aging that leads to NMDA-dependent synaptic dysfunction [41] could possibly be attributed to excessive zincergic inhibition of synaptic GluN2A NMDARs in aged neurons.
As mentioned above, Zn2+ modulates LTP, which, like long-term depression (LTD: see Box 2 for details) – a process necessary for silencing irrelevant neuronal connections – is a neuronal substrate of some forms of memory. LTD is also an essential factor in brain development [42]; however, very little is known about how it is influenced by Zn2+. Since low-frequency stimulation-induced activation of extrasynaptic GluN2B-type NMDARs is necessary for LTD expression [43] and vesicular Zn2+ can reach 1-10 μM at extrasynaptic sites [44], this level of Zn2+should be sufficient to block LTD. Indeed, by exogenously applying 1-10 μM of Zn2+ in the CA1 region of hippocampal slices, Izumi et al. were able to inhibit LTD without affecting LTP [45]. Nevertheless, more studies are needed to elucidate this matter further.
Box 2. Long-term depression.
| Long-term depression (LTD) refers to a persistent decrease in the strength of a synaptic connection in response to prolonged (10-15 min) 1 Hz stimulation. LTD can occur at postsynaptic sites when EPSPs follow an action potential of the postsynaptic neuron. This implies that LTD contributes to the weakening of synapses which do not provide neurons with any functionally relevant input. Among the receptors that contribute to LTD, NMDARs and metabotropic glutamate receptors (mGluRs) are the most common. NMDAR-dependent LTD is triggered by Ca2+ influx that is less pronounced than in LTP, and which activates protein phosphatases such as calcineurin. The signaling cascade initiated by calcineurin leads to the dephosphorylation of AMPARs and their internalization, which renders the synapse less sensitive to glutamate-driven excitation. In mGluR-dependent LTD, different signaling pathways are involved - specifically, the mitogen-activated protein (MAP) kinases, such as extracellular signal regulated kinase (ERK) and p38 MAP kinase. Internalization of AMPARs remains the main outcome of mGluR-dependent LTD; however, it relies less on Ca2+signaling than NMDAR-dependent LTD. |
3. ZINCERGIC NEUROTRANSMISSION IN THE PATHOPHYSIOLOGY OF ALZHEIMER’S DISEASE
Measuring Zn2+ levels in AD patients’ bodies revealed a decrease of Zn2+ in either serum or hair samples [46]. In the brain however, a different pattern of Zn2+ dyshomeostasis is observed. A postmortem comparison of AD patients with age-matched controls revealed increased Zn2+ levels in the neocortex, which correlated with the severity of both the Aβ-related pathology and dementia [47]. Recent PET imaging studies of AD patients have confirmed that there are significant deficits in Zn2+ clearance from brain regions involved in cognitive symptoms of the disorder [48]. There is also clinical evidence for a specific form of AD with high peripheral Zn2+ deficiency, which affects younger individuals' brain areas that are typically spared at early stages of regular AD progression, and which, consequently, manifests itself differently during neuropsychological assessment [49]. While regular AD development is characterized by progressive memory loss, AD related to the peripheral Zn2+ deficiency includes such early symptoms as dyscalculia and aphasia.
On a cellular level, findings from preclinical pharmacological studies suggested that the binding of extracellular Zn2+ by Aβ might lead to a reduced influx of Zn2+ into postsynaptic neurons, and to the subsequent memory deficits observed in AD [50]. Studies of PBT2 – a Zn2+ ionophore which additionally blocks metal binding to Aβ - have shown that the compound improves the cognitive performance of transgenic AD model mice [50]. The Zn2+ deficiency hypothesis was further confirmed with the use of ZnT3 KO mice, which showed an age-dependent impairment of spatial memory [21]. This led to clinical PBT2 trials, which revealed some improvement in the executive functions of AD patients [51], but after additional Phase II testing, the trials were discontinued due to no statistically significant differences being found between the PBT2 and placebo groups.
It is now known that the mechanism through which PBT2 action occurs involves neuroprotection against NMDAR-induced neurotoxicity through preconditioning neurons to high levels of cytosolic Ca2+ during excitotoxicity [52]. By gating the entry of Zn2+ into the cell, PBT2 promotes Ca2+ release from intracellular stores, which prevents the cleavage of calcineurin and inhibits glycogen synthase kinase-3β (GSK3β) activity, both of which usually follow excessive Ca2+ entry through overstimulated NMDARs. It is important to note that the above-mentioned experiments were conducted in the absence of Aβ. Since Aβ facilitates NMDAR-dependent LTD via GSK3β [53], directly activates NMDARs [54], perforates neuronal membranes [55] and potentiates glutamate release [56], PBT2 preconditioning in the presence of Aβ could be insufficient, due to the additional Aβ–dependent Ca2+ influx. This could possibly explain the unsatisfactory clinical effects of the drug in AD patients. Furthermore, a recent study showed that physiological concentrations of extracellular Zn2+ (10nM) are essential for the in vivo uptake of Aβ and Zn2+ into hippocampal dentate granule cells and for the subsequent disruption of both LTP and object novelty recognition memory in rats [57]. Moreover, in vitro studies have shown that Aβ alone does not affect hippocampal LTP, pointing to Zn2+ as a necessary factor in Aβ’s impact on memory formation [57].
In contrast, Aβ alone has been shown to selectively promote LTD through GluN2B NMDARs by means of a metabotropic mechanism which does not require ionic flow through the channel [58]. Since in physiological conditions Zn2+ can suppress GluN2B NMDAR-dependent LTD [45], its binding by Aβ might additionally disturb Zn2+’s role in the regulation of synaptic plasticity. This provides another potential contribution of Aβ– this time a Ca2+-independent one - to the disturbance of zincergic neurotransmission and to synaptic dysfunction in AD.
As well as disrupting zincergic neurotransmission, the interaction between Zn2+ and Aß contributes to both senile plaque formation and the toxicity of soluble Aß oligomers. The first studies of Zn2+- induced Aß aggregation, which date back to 1994, showed that the different affinities of rodent and human Aß to Zn2+might explain why rats and mice do not develop age-dependent AD-like neurodegeneration [59]. More recently it has been shown that the binding of Zn2+ by Aß40 and Aß42 results in the formation of a species of soluble Aß oligomer that is more toxic than ADDL (Aß derived diffusible ligands), and that the Aβ oligomer inhibits hippocampal LTP more potently than ADDLs [60]. Zn2+-dependent oligomerization of Aß40-42, together with synapse targeting by the oligomer, requires the release of Zn2+ from presynaptic terminals during excitatory neurotransmission [61]. Therefore, vesicular Zn2+ may have been responsible for the findings of Bero et al., who have shown that susceptibility to the deposition of Aβ and to its toxicity is directly linked to the level of neuronal activity [62, 63].
The mechanism whereby Zn2+-induced Aβ accumulation and toxicity lead to neurodegeneration is unclear; however, it involves GluN2B NMDARs [61]. Opposite roles for synaptic GluN2A-and extrasynaptic GluN2B-type NMDARs in neuroprotection and neurodegeneration, respectively, have been proposed (for a review, see: [64]). According to this theory, stimulation of GluN2A-containing synaptic NMDARs promotes ‘prosurvival’ pathways through cAMP responsive element (CRE)-binding protein (CREB) and extracellular signal–regulated kinases 1/2 (ERK 1/2), while activation of extrasynaptic GluN2B NMDARs causes cytosolic Ca2+ overload and inactivation of CREB and ERK1/2. This distinction might not be clear-cut, as both subunits have been shown to promote ‘prosurvival’ pathways when stimulated separately, whereas only simultaneous coactivation of GluN2A-and GluN2B-type NMDARs leads to cell death [65]. Since Zn2+ negatively modulates both types of NMDARs, its chaperoning by Aβ should facilitate coactivation of the receptors and subsequent neurodegeneration. Moreover, with regard to Aβ production extrasynaptic NMDARs have been shown to make a sole contribution [66, 67]. Therefore, by enabling AβOs to colocalize with and activate GluN2B NMDARs [61], synaptic Zn2+ might lead to further up-regulation of Aβ production. This is consistent with the findings of other studies, which revealed a critical role of bursting activity in inducing conformational changes to presenilin 1 (a subunit of γ-secretase enzyme) and the subsequent up-regulation of Aß40 production [68].
4. THE POTENTIAL ROLE OF GPR39 IN AD
4.1. GPR39 Expression Pattern in the CNS
One target of vesicular Zn2+ stands out as a distinct Zn2+-sensing receptor. The human GPR39 gene is localized on chromosome 2 and comprises two exons [69]. The first exon encodes a short (non-functional) version of the receptor termed GPR39-1b, while both exons encode a full-length (functional) version – GPR39-1a [69]. Early mRNA expression studies of the rodent brain suggested that only GPR39-1b is widely expressed in the mammalian CNS, while GPR39-1a expression is slightly above the detection limit for the RT-PCR method [70].
In situ hybridization studies, which focused on specific brain sections, revealed the fully functional GPR39-1a form to be present in the mouse hippocampus and amygdala [71]. Popovics and Stewart suggested that regional specificity of GPR39-1a expression, as well as methodological differences, might explain these conflicting results [72]. Later functional studies of mouse hippocampal slices proved that GPR39 participates in zincergic neurotransmission in the hippocampus [8], leading the aforementioned authors to conclude that “the evidence for its involvement is nevertheless compelling” [72]. Our own GPR39 protein expression studies using rodent depression models corroborate the existence and the functional importance of the receptor in the murine hippocampus [73, 74]. Recent findings from another group showed alterations of GPR39 protein levels in the mouse nucleus accumbens to be linked to alcohol consumption [75]. Therefore, it is highly probable that the GPR39 receptor participates in neurotransmission in at least three subcortical structures responsible for mood, memory and decision-making.
Recent single-cell mRNA sequencing studies of human middle temporal gyrus revealed that GPR39 expression is restricted to certain types of GABAergic neurons and glial cells (astrocytes) in the human cortex [76]. The same study showed the expression of the GPR39 receptor to be very low in cortical glutamatergic cells, with no particular type of glutamatergic neuron corresponding to the receptor’s expression pattern [76]. A similar mRNA sequencing study showed an opposite tendency of GPR39 expression in the mouse neocortex [77]. While certain types of glutamatergic neurons readily expressed the GPR39 mRNA, GABAergic and glial cells showed very little or no expression [77]. These results fit well with the fact that only some neurons utilize Zn2+as a neurotransmitter, and suggest that caution should be used when translating results from animal GPR39 studies to human conditions, especially with respect to the neurobiology of cortical functions.
4.2. Signaling Pathways of GPR39
GPR39 is a G-protein coupled receptor (GPCR) which belongs to the ghrelin/neurotensin subfamily of rhodopsin-like (class A) metabotropic receptors [69]. When a ligand binds to an extracellular site, GPCRs undergo conformational changes which lead to the activation of intracellular G-proteins and to a subsequent signaling cascade that can affect a vast array of cell functions. In neurons, the GPR39 acts through the Gα/q and Gα/12/13 proteins, causing activation of the ERK1/2/MAPK or the PI3K/Akt pathway, and of the Rho/ROCK pathway, respectively [8, 78]. GPR39 is also known to act through the Gαs/cAMP/PKA pathway [79], although the involvement of this pathway in GPR39-mediated neurotransmission is yet to be confirmed. Both the Gαs pathway and Gαq pathway regulate gene transcription through CRE, while the Gα12/13 pathway acts through the serum response element (SRE). Additionally, basic cell research shows that the Gαq- and Gα12/13-dependent activity of the receptor is constitutive (ligand-independent) [80], while signal transduction through all three pathways is enhanced by ligand binding [72].
For some time after the discovery of GPR39 in 1997, it was thought that obestatin – a gastrointestinal hormone - was its endogenous ligand. However, in 2007 two groups established Zn2+ as an agonist of GPR39 [79, 81]. In cell cultures, Zn2+ can activate the GPR39 Gαs and Gαq pathways with effective concentration (EC50) values of 7 μM and 22 μM, respectively [79], which should be sufficient in the physiological conditions of a zincergic synapse. Indeed, Besser et al. have shown that an increase of Ca2+ release from endoplasmatic reticulum stores, and the subsequent ERK1/2/CAMKII phosphorylation in CA3 pyramidal neurons, is partially caused by vesicular Zn2+ acting through GPR39 [8]. This effect was mediated by the Gαq – phospholipase C (PLC) - IP3 pathway [8]. The constitutive activity of GPR39 through Gαq and/or Gα12/13 in overexpression systems is very high, but downstream effects are seen only with regard to PI3K/Akt and not ERK1/2 [80]. Moreover, an opposite effect has recently been shown in GPR39’s response to Zn2+ in neurons [82]. Thus, the ERK1/2 seems to be a ligand-dependent pathway for GPR39, while PI3K/Akt is likely a ligand-independent one. Mutual cross-inhibition of the two pathways has been described [83], and therefore could be responsible for such selectivity. As discussed below, GPR39-specific activity of the PKA inhibitor β (PKIB) could also promote pathway selectivity.
Although the physiological activity of the Gαs and Gα12/13 pathways is yet to be confirmed [84], basic cell research has provided interesting hypotheses about their roles in neuroprotection, about the desensitization of GPR39, and about ligand-mediated and constitutive activity of the receptor [78, 85, 86]. On the basis of their findings, Dittmer and colleagues proposed that constitutively active GPR39 protects hippocampal neurons from oxidative stress and glutamate toxicity through the Gα12/13-RhoA-SRE pathway, which leads to up-regulation of a neurotrophic glycoprotein – the pigment epithelium growth factor (PEDF) [78]. ERK1/2 activity is crucial for pretreatment with PEDF to be effective in immunizing neurons against oxidative stress in vitro [87], and - as previously mentioned - GPR39 does not seem to activate ERK1/2 in a ligand-independent manner. Again, this would suggest the existence of some regulatory mechanism to prevent overstimulation of ERK1/2 by both GPR39-Gα12/13-PEDF and –Gαq-PLC, as overactivity of ERK1/2 can cause neuronal death [88].
Studies of overexpression systems show that the only G-protein for which Zn2+ binding to GPR39 is a sine qua non condition is Gαs [72]. Gαs acts through cAMP/PKA second-messengers and regulates CRE-dependent transcription. The exact role of PKA in Zn2+-GPR39 mediated signaling is not certain since it has been shown that the PKIB dissociates from ligand-activated GPR39 and blocks PKA activity [86]. Furthermore, the same study showed that GPR39-bound PKIB enhances the receptor’s constitutive activity, which does not involve Gαs/cAMP/PKA [80]. There has been no investigation of the physiological functions of the feedback mechanism limiting the PKA activity in response to Zn2+ in neurons, but it could be part of a dynamic regulatory process responsible for pathway specificity in GPR39. In combination with the previously mentioned observations, we would like to propose a model of such a mechanism here (Fig. 1). In its constitutive conformation, the GPR39 interacts with PKIB [86], which facilitates the activation of PI3K/Akt pathway [89], leading to Akt phosphorylation [90] and subsequently causing inhibition of the protein kinase Raf - a crucial upstream component for ERK1/2 phosphorylation [91]. The ERK1/2 is further blocked by cytosolic PKA acting upon the Raf [92], until the GPR39 receptor binds to Zn2+, which is when PKIB dissociates from the GPR39, inhibits PKA and decreases the PI3K/Akt pathway activation [86]. This releases the Raf kinase, which can now be stimulated by the Gα/q-PLC-PKC pathway, promoting cross-inhibition of PI3K by ERK1/2 [83].
Fig. (1).
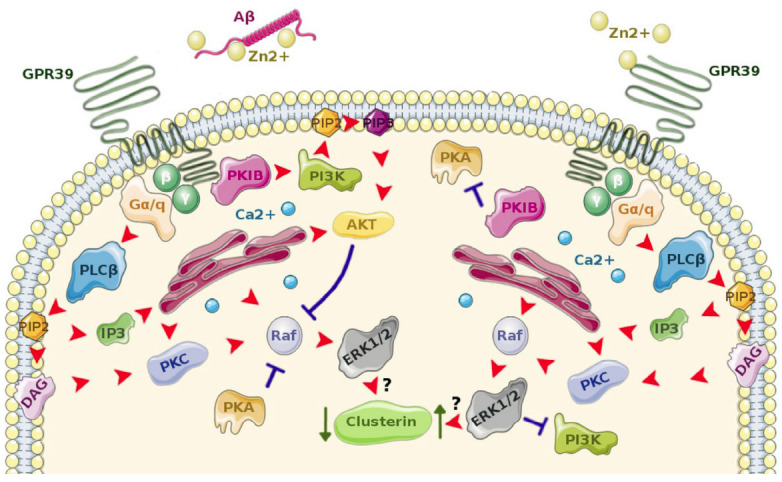
Schematic illustration of a hypothesized influence of Aβ on GPR39-mediated signaling pathways, which lead to down-regulation of expression of clusterin – a protein implicated in Alzheimer’s disease. In normal, physiological conditions (right) Zn2+ activates GPR39, which leads to up-regulation of clusterin expression via Gαq – PLCβ - ERK 1/2. Additionally, cross-inhibition of PI3K – Akt and cytosolic PKA by ERK 1/2 causes pathway specificity of GPR39 ligand-dependent signaling. In the presence of Aβ (left) ligand-independent activity of GPR39 is promoted. Here, GPR39-bound PKIβ cannot inhibit cytosolic PKA and activates PI3K – Akt, which leads to Raf – ERK 1/2 - clusterin inhibition. Red arrow – activation, blue ‘T’ – inhibition, AKT – protein kinase B, DAG – diacylglycerol, ERK 1/2 - extracellular signal–regulated kinase 1/2, IP3 - inositol 1,4,5-triphosphate, PI3K - phosphatidylinositol-3-kinase, PIP2 - phosphatidylinositol 4,5-bisphosphate, PIP3 - Phosphatidylinositol (3,4,5)-trisphosphate, PKA – protein kinase A, PKC – protein kinase C, PKIβ – protein kinase inhibitor β, PLCβ – phospholipase C β, Raf - proto-oncogene serine/threonine-protein kinase. (Figure composed using Servier Medical Art templates available at: http://smart.servier.com/).
Irrespective of the intracellular mechanism, the fact that GPR39 displays pathway specificity downstream of Gαq could be of use in drug development. For instance, if one was to promote the PI3K/Akt pathway in order to prevent GSK3β activation and subsequent tau phosphorylation [93], a neutral antagonist of the GPR39 could be utilized to this effect. In this respect, it is worth mentioning that at least two binding sites for Zn2+ exist on the GPR39 and that the ion has been shown to act as a positive allosteric modulator (PAM) of agonist compounds for the Gαq and β-arrestin pathways [94]. The same study showed that the reverse is also true and that agonist compounds can act as PAMs of zincergic GPR39 agonism through the Gαs pathway, although, as previously mentioned, this pathway is yet to be confirmed in neurons.
Another distinct feature of GPR39 is the way it is desensitized. Shimizu et al. have shown that overstimulation of GPR39 with Zn2+ (300 μM for 4 hours) leads to functional desensitization of the receptor and, by using biased agonists, they attributed this to down-regulation of GPR39 via the Gα12/13-RhoA-ROCK pathway, but not the Gαs, Gαq or β-arrestin pathways [85]. Although the constitutive activity of GPR39 through Gα12/13-RhoA is much more pronounced than in other receptors from the same family, it is insufficient to cause GPR39 internalization [80]. Since in the Shimizu et al. study the concentration of Zn2+ and the exposure time were toxic and much higher than in studies of hippocampal neurons (75 μM for 15 minutes = subtoxic), where functional desensitization of GPR39 was also observed [8], it is still unclear whether down-regulation of the receptor through the Gα12/13-RhoA-ROCK pathway is a mechanism that is responsible for the desensitization of GPR39 during synaptic transmission.
4.3. Physiological functions of GPR39 in the CNS
Synaptically released Zn2+ increases neurotransmitter release probability (Pr) at the MF-CA3 synapses in the hippocampus [14]. It is therefore intriguing that in the DCN synaptic Zn2+ has an opposite, GPR39-mediated effect [15]. One of the two types of primary output neurons of the DCN (i.e. the fusiform cells - FC) receive inputs from both granule cells (GC) and the auditory nerve; of these only the GC input is zincergic [17] and capable of synaptic plasticity [95]. During a single stimulus propagation through the GC-FC synapse, the release of Zn2+ blocks postsynaptic AMPARs in a manner similar to that observed in CA1 pyramidal neurons [17]. Additionally, in response to high frequency stimulation from GCs (which causes short-term facilitation - a form of transient synaptic plasticity), Zn2+ promotes endocannabinoid release (2-AG) from the postsynaptic site, which decreases Pr by binding to presynaptic CB1 receptors [15]. Crucially, this effect relies on the GPR39-Gαq-PLC pathway [15], and the amount of Zn2+ released from the GC terminals decreases in an experience-dependent manner in mice, thereby limiting its inhibitory modulation and helping to maintain the plasticity of the synapse without changing other neurotransmitter properties [17].
The GPR39-Gαq pathway is also involved in regulating the reversal potential for chloride ions (Cl-) in the hippocampal CA3 pyramidal neurons, and therefore in modulating the inhibitory action of the GABAA receptors [96]. Chorin et al. have shown that synaptic Zn2+ up-regulates the surface expression of the potassium ion (K+)/Cl- cotransporter 2 (KCC2), which is responsible for Cl- extrusion in neurons. Since this causes a more negative reversal potential for GABAA receptor-mediated currents, the Zn2+-mediated expression of KCC2 counteracts excessive excitability of neurons. Crucially, the up-regulation of KCC2 is downstream to ERK1/2 following GPR39-Gαq-PLC activation by Zn2+, and GPR39 KO mice are more susceptible to kainate-induced seizures [97]. Another transmembrane transporter protein regulated by GPR39-Gαq-PLC-ERK1/2 is the sodium ion/proton (Na+/H+) exchanger (NHE), which is responsible for pH homeostasis [98]. By up-regulating NHE activity, the GPR39 decreases intracellular acidification (that occurs after repeated firing of neurons) and increases extracellular acidification, but only to the point at which extracellular pH reaches ~ 6.5, which is when GPR39 is rendered insensitive to Zn2+ [98]. This negative-feedback mechanism highlights the role of GPR39 in maintaining neuronal homeostasis, but also provides a means of influencing acid-sensing ion channels (ASICs), which have recently been shown to participate in synaptic plasticity dysfunction in the CA1 region due to Aβ [99].
4.4. Potential Role of GPR39 in AD
To date, there is only one study directly linking the impact of Aβ on neuronal function with the GPR39 receptor. Abramovitch-Dahan et al. showed not only that pretreatment with exogenous Aβ impairs ligand-dependent GPR39-Ca2+ signaling in both human neuroblastoma cells and mouse cortical neurons, but also that it decreases phosphorylation of ERK1/2 [82]. Moreover, Aβ alone did not affect the levels of phosphorylated ERK1/2 and the potentially detrimental effects of Aβ on ligand-dependent GPR39 signaling were overcome when Aβ was applied with excessive Zn2+, clearly suggesting a disruption of zincergic metabotropic neurotransmission by Aβ. What further highlights the link between Aβ and ligand-dependent GPR39 signaling is the fact that activation of PI3K/Akt cascade by Zn2+ was independent of GPR39 and was also not disrupted by Aβ. Additionally, Zn2+-dependent GPR39 activity caused the expression of cytoplasmic clusterin - a chaperone glycoprotein, mutations of which convey a higher risk of progression from MCI to AD than Apoe4 [100]. At least 3 different forms of clusterin, with separate and often contradictory functions are found in neurons. In general, both secretory and cytoplasmic clusterin are thought to be protective, while nuclear clusterin is associated with cell death through apoptosis [101]. The exact mechanism of clusterin’s interaction with Aβ is yet to be elucidated. However, a growing body of research shows, that while clusterin participates in Aβ clearance from the brain, at the same time even the protective forms of the protein can induce the formation of extracellular toxic AβOs and activate intracellular apoptotic pathways when exposed to Aβ [102].
Abramovitch-Dahan et al. found that neurons can up-regulate cytoplasmic clusterin production in response to Aβ irrespective of GPR39, but this effect, as well as the GPR39-mediated clusterin up-regulation, was blocked when Zn2+ was administered with Aβ. This confirms a “two-edged-sword” quality of ion dyshomeostasis in AD (i.e. the ability of Zn2+ to exacerbate pathological Aβ activity and, at the same time, cause other detrimental effects as a result of its depletion). Given the opposite roles of clusterin in cell fate, as described above, it may seem hard to determine whether the inhibition of clusterin expression by Zn2+ -Aβ would be protective or detrimental for neurons. However, Abramovitch-Dahan et al. provide evidence for the elevated expression of cytoplasmic clusterin in response to an oxidative insult caused by hydrogen peroxide (H2O2), swaying the conclusion towards a protective role. Furthermore, as previously mentioned, GPR39 immunizes hippocampal cell lines against H2O2 stress by constitutively up-regulating PEDF [78]. It is therefore probable that by disrupting GPR39-mediated zincergic neurotransmission, Aβ deprives neurons of a protective mechanism specifically related to maintaining homeostasis in the dynamic environment of a synapse (Fig. 1).
The impact of Aβ on GPR39 would also affect two transporter proteins regulated by the receptor. By blocking GPR39 activity, Aβ should down-regulate KCC2 and subsequently cause a depolarizing shift in the GABAA receptor-related Cl- reversal potential, possibly facilitating excitotoxicity, which is one of the postulated pathomechanisms in AD [103]. Indeed, AD11 transgenic mice, which display high levels of mouse Aβ plaques, as well as age-dependent neurodegeneration and cognitive deficits, also exhibit down-regulated KCC2 mRNA and a depolarizing switch of GABAA currents in CA1 pyramidal neurons [104]. The authors of this study interpret these results in terms of a regulatory mechanism, a claim supported by the fact that memantine - a drug used in pharmacotherapy of AD - also down-regulates KCC2 through brain-derived neurotrophic factor (BDNF) [105], which decreases KCC2 expression in mature neurons by activating the tyrosine kinase receptor B (TrkB) [106]. However, this mechanism might not be beneficial, since the high doses of memantine used in the Molinaro et al. [105] study have also been shown to be epileptogenic [107]. Therefore another possibility is expressed in the view that Aβ-related changes in KCC2 expression might explain the higher prevalence of epilepsy observed in AD patients, possibly through disruption of GPR39 signaling [84]. More information on this possible link can be found in [108]. Even less is known about the potential role of the second transporter protein controlled by GPR39 (i.e. NHE) in AD. As a major player in regulating extracellular acidification at the synapse, NHE should eventually fall under scientific scrutiny in AD-related research, especially in light of recent evidence of the involvement of ASICs in the synaptic dysfunction observed in AD [109].
The development of GPR39 KO mice has enabled the receptor’s role in AD to be investigated not only on a cellular level, but also on a behavioural one. To date, very little is known about the phenotype displayed by such animals. Studies performed in our laboratory point to a depressive-like behaviour of GPR39 KO mice in standard preclinical drug-screening tests (i.e. the forced swimming test / the tail suspension test) [73, 74, 110]. Moreover, in response to the strong acute stress caused by forced swimming, the GPR39 KO mice show a significant reduction of both BDNF and CREB proteins in their hippocampi, but not in their frontal cortices, suggesting a neuroprotective role of GPR39 in a structure that is highly vulnerable to AD pathology. Taken together with observations of altered levels of BDNF in the serum of AD patients, these results point to another mechanism behind the possible involvement of GPR39 in AD [108]. Clearly, more work is needed in this field of inquiry.
CONCLUSION
Studies concerned with the role of metal ions in AD have provided substantial empirical support for the concept of a double-edged sword mode of interaction between metal ions and Aβ in AD neuropathology [111]. In this review we focused on functional changes in zincergic neurotransmission caused by Aβ, emphasizing the potential role of a Zn2+-sensing receptor, GPR39, in AD in order to provide a comprehensive look at possible targets for pharmacological intervention. One of the unresolved issues in this respect is the question of other endogenous ligands of GPR39, since fish express a functional form of GPR39 without a Zn2+-binding site, which may have been preserved in mammals [72]. Moreover, GPR39 is capable of interacting with and affecting the function of other GPCRs - for instance, the 5-HT1A and GalR1 receptors [112], which might be important in 5-HT1A-targeted AD pharmacotherapy. Even the short isoform of the receptor (GPR39-1b), which is considered inactive and does not bind Zn2+, is now known to attenuate the activity of a neurotensin receptor (NTSR1) [113] that is significantly down-regulated in the temporal lobes of AD patients [114]. In conclusion, we believe that the location of GPR39 in brain areas affected by AD pathology (e.g. the hippocampus), together with its sensitivity to Aβ-induced Zn2+ dyshomeostasis, suggests that GPR39 may become a promising target for preclinical research aimed at pharmacological interventions in AD.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This study was supported by a grant from the National Science Centre (2015/19/B/NZ7/00255), and by the statutory funds of the Faculty of Pharmacy, Jagiellonian University Medical College, Poland.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Villemagne V.L., Pike K.E., Chételat G., Ellis K.A., Mulligan R.S., Bourgeat P., Ackermann U., Jones G., Szoeke C., Salvado O., Martins R., O’Keefe G., Mathis C.A., Klunk W.E., Ames D., Masters C.L., Rowe C.C. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann. Neurol. 2011;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reiman E.M., Langbaum J.B., Tariot P.N., Lopera F., Bateman R.J., Morris J.C., Sperling R.A., Aisen P.S., Roses A.D., Welsh-Bohmer K.A., Carrillo M.C., Weninger S. CAP--advancing the evaluation of preclinical Alzheimer disease treatments. Nat. Rev. Neurol. 2016;12(1):56–61. doi: 10.1038/nrneurol.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raskin J., Cummings J., Hardy J., Schuh K., Dean R.A. Neurobiology of alzheimer’s disease: Integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr. Alzheimer Res. 2015;12(8):712–722. doi: 10.2174/1567205012666150701103107. https://doi.org/1875-5828/1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingelsson M., Fukumoto H., Newell K.L., Growdon J.H., Hedley-Whyte E.T., Frosch M.P., Albert M.S., Hyman B.T., Irizarry M.C. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62(6):925–931. doi: 10.1212/01.WNL.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 5.Cline E.N., Bicca M.A., Viola K.L., Klein W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimers Dis. 2018;64(s1):S567–S610. doi: 10.3233/JAD-179941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spires-Jones T.L., Hyman B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82(4):756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts B.R., Ryan T.M., Bush A.I., Masters C.L., Duce J.A. The role of metallobiology and amyloid-β peptides in Alzheimer’s disease. J. Neurochem. 2012;120(Suppl. 1):149–166. doi: 10.1111/j.1471-4159.2011.07500.x. [DOI] [PubMed] [Google Scholar]
- 8.Besser L., Chorin E., Sekler I., Silverman W.F., Atkin S., Russell J.T., Hershfinkel M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009;29(9):2890–2901. doi: 10.1523/JNEUROSCI.5093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hershfinkel M., Moran A., Grossman N., Sekler I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc. Natl. Acad. Sci. USA. 2001;98(20):11749–11754. doi: 10.1073/pnas.201193398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Gladyshev V.N. Comparative genomics of trace element dependence in biology. J. Biol. Chem. 2011;286(27):23623–23629. doi: 10.1074/jbc.R110.172833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulkidjanian A.Y. On the origin of life in the zinc world: 1. Photosynthesizing, porous edifices built of hydrothermally precipitated zinc sulfide as cradles of life on Earth. Biol. Direct. 2009;4:26. doi: 10.1186/1745-6150-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frederickson C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989;31:145–238. doi: 10.1016/S0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- 13.Frederickson C.J., Moncrieff D.W. Zinc-containing neurons. Biol. Signals. 1994;3(3):127–139. doi: 10.1159/000109536. [DOI] [PubMed] [Google Scholar]
- 14.Pan E., Zhang X.A., Huang Z., Krezel A., Zhao M., Tinberg C.E., Lippard S.J., McNamara J.O. Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron. 2011;71(6):1116–1126. doi: 10.1016/j.neuron.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Rosello T., Anderson C.T., Schopfer F.J., Zhao Y., Gilad D., Salvatore S.R., Freeman B.A., Hershfinkel M., Aizenman E., Tzounopoulos T. Synaptic Zn2+ inhibits neurotransmitter release by promoting endocannabinoid synthesis. J. Neurosci. 2013;33(22):9259–9272. doi: 10.1523/JNEUROSCI.0237-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson C.T., Radford R.J., Zastrow M.L., Zhang D.Y., Apfel U-P., Lippard S.J., Tzounopoulos T. Modulation of extrasynaptic NMDA receptors by synaptic and tonic zinc. Proc. Natl. Acad. Sci. USA. 2015;112(20):E2705–E2714. doi: 10.1073/pnas.1503348112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalappa B.I., Anderson C.T., Goldberg J.M., Lippard S.J., Tzounopoulos T. AMPA receptor inhibition by synaptically released zinc. Proc. Natl. Acad. Sci. USA. 2015;112(51):15749–15754. doi: 10.1073/pnas.1512296112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole T.B., Wenzel H.J., Kafer K.E., Schwartzkroin P.A., Palmiter R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA. 1999;96(4):1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmiter R.D., Cole T.B., Quaife C.J., Findley S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA. 1996;93(25):14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAllister B.B., Dyck R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017;80:329–350. doi: 10.1016/j.neubiorev.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Adlard P.A., Parncutt J.M., Finkelstein D.I., Bush A.I. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010;30(5):1631–1636. doi: 10.1523/JNEUROSCI.5255-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martel G., Hevi C., Friebely O., Baybutt T., Shumyatsky G.P. Zinc transporter 3 is involved in learned fear and extinction, but not in innate fear. Learn. Mem. 2010;17(11):582–590. doi: 10.1101/lm.1962010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoo M.H., Kim T.Y., Yoon Y.H., Koh J.Y. Autism phenotypes in ZnT3 null mice: Involvement of zinc dyshomeostasis, MMP-9 activation and BDNF upregulation. Sci. Rep. 2016;6:28548. doi: 10.1038/srep28548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patrick Wu H.P., Dyck R.H. Signaling by synaptic zinc is required for whisker-mediated, fine texture discrimination. Neuroscience. 2018;369:242–247. doi: 10.1016/j.neuroscience.2017.11.020. [DOI] [PubMed] [Google Scholar]
- 25.Birinyi A., Parker D., Antal M., Shupliakov O. Zinc co-localizes with GABA and glycine in synapses in the lamprey spinal cord. J. Comp. Neurol. 2001;433(2):208–221. doi: 10.1002/cne.1136. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z., Danscher G., Kim Y.K., Dahlstrom A., Mook Jo S. Inhibitory zinc-enriched terminals in the mouse cerebellum: double-immunohistochemistry for zinc transporter 3 and glutamate decarboxylase. Neurosci. Lett. 2002;321(1-2):37–40. doi: 10.1016/S0304-3940(01)02560-5. [DOI] [PubMed] [Google Scholar]
- 27.Leal S.L., Yassa M.A. Integrating new findings and examining clinical applications of pattern separation. Nat. Neurosci. 2018;21(2):163–173. doi: 10.1038/s41593-017-0065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C., Malenka R.C. NMDA receptor-dependent long-term potentiation and long-term depression. LTP/LTD; 2012. pp. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicoll R.A., Schmitz D. Synaptic Plasticity at Hippocampal Mossy Fibre Synapses. Nat. Rev. Neurosci. Nat. Pub. 2005;6(11):863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- 30.Vergnano A.M., Rebola N., Savtchenko L.P., Pinheiro P.S., Casado M., Kieffer B.L., Rusakov D.A., Mulle C., Paoletti P. Zinc dynamics and action at excitatory synapses. Neuron. 2014;82(5):1101–1114. doi: 10.1016/j.neuron.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 31.Takeda A., Suzuki M., Tempaku M., Ohashi K., Tamano H. Influx of extracellular Zn(2+) into the hippocampal CA1 neurons is required for cognitive performance via long-term potentiation. Neuroscience. 2015;304:209–216. doi: 10.1016/j.neuroscience.2015.07.042. [DOI] [PubMed] [Google Scholar]
- 32.Takeda A., Tamano H. The impact of synaptic Zn2+ dynamics on cognition and its decline. Int. J. Mol. Sci. 2017;18(11):2411. doi: 10.3390/ijms18112411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medvedeva Y.V., Ji S.G., Yin H.Z., Weiss J.H. Differential vulnerability of CA1 versus CA3 pyramidal neurons after ischemia: Possible relationship to sources of Zn2+ accumulation and its entry into and prolonged effects on mitochondria. J. Neurosci. 2017;37(3):726–737. doi: 10.1523/JNEUROSCI.3270-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah N.H., Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 2014;5(1):38–58. doi: 10.1007/s12975-013-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sindreu C.B., Varoqui H., Erickson J.D., Pérez-Clausell J. Boutons containing vesicular zinc define a subpopulation of synapses with low AMPAR content in rat hippocampus. Cereb. Cortex. 2003;13(8):823–829. doi: 10.1093/cercor/13.8.823. [DOI] [PubMed] [Google Scholar]
- 36.Blakemore L.J., Trombley P.Q. Zinc as a neuromodulator in the central nervous system with a focus on the olfactory bulb. Front. Cell. Neurosci. 2017;11:297. doi: 10.3389/fncel.2017.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paoletti P., Vergnano A.M., Barbour B., Casado M. Glutamatergic Synapses. Neuroscience. Pergamon January. 2009;12:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 38.Romero-Hernandez A., Simorowski N., Karakas E., Furukawa H. Molecular basis for subtype specificity and high-affinity zinc inhibition in the GluN1-GluN2A NMDA receptor amino-terminal domain. Neuron. 2016;92(6):1324–1336. doi: 10.1016/j.neuron.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogt K., Mellor J., Tong G., Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26(1):187–196. doi: 10.1016/S0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 40.Gladding C.M., Raymond L.A. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol. Cell. Neurosci. 2011;48(4):308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 41.Shetty M.S., Sharma M., Sajikumar S. Chelation of hippocampal zinc enhances long-term potentiation and synaptic tagging/capture in CA1 pyramidal neurons of aged rats: implications to aging and memory. Aging Cell. 2017;16(1):136–148. doi: 10.1111/acel.12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piochon C., Kano M., Hansel C. LTD-like molecular pathways in developmental synaptic pruning. Nat. Neurosci. 2016;19(10):1299–1310. doi: 10.1038/nn.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papouin T., Ladépêche L., Ruel J., Sacchi S., Labasque M., Hanini M., Groc L., Pollegioni L., Mothet J.P., Oliet S.H.R. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150(3):633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 44.Frederickson C.J., Giblin L.J., Krezel A., McAdoo D.J., Mueller R.N., Zeng Y., Balaji R.V., Masalha R., Thompson R.B., Fierke C.A., Sarvey J.M., de Valdenebro M., Prough D.S., Zornow M.H. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 2006;198(2):285–293. doi: 10.1016/j.expneurol.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 45.Izumi Y., Auberson Y.P., Zorumski C.F. Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J. Neurosci. 2006;26(27):7181–7188. doi: 10.1523/JNEUROSCI.1258-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ventriglia M., Brewer G.J., Simonelli I., Mariani S., Siotto M., Bucossi S., Squitti R. Zinc in alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 2015;46(1):75–87. doi: 10.3233/JAD-141296. [DOI] [PubMed] [Google Scholar]
- 47.Religa D., Strozyk D., Cherny R.A., Volitakis I., Haroutunian V., Winblad B., Naslund J., Bush A.I. Elevated cortical zinc in Alzheimer disease. Neurology. 2006;67(1):69–75. doi: 10.1212/01.wnl.0000223644.08653.b5. [DOI] [PubMed] [Google Scholar]
- 48.DeGrado T.R., Kemp B.J., Pandey M.K., Jiang H., Gunderson T.M., Linscheid L.R., Woodwick A.R., McConnell D.M., Fletcher J.G., Johnson G.B., Petersen R.C., Knopman D.S., Lowe V.J. First PET imaging studies with 63Zn-Zinc citrate in healthy human participants and patients with alzheimer disease. Mol. Imaging. 2016: 15153601211667379. doi: 10.1177/1536012116673793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bredesen D.E. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging (Albany NY) 2015;7(8):595–600. doi: 10.18632/aging.100801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adlard P.A., Cherny R.A., Finkelstein D.I., Gautier E., Robb E., Cortes M., Volitakis I., Liu X., Smith J.P., Perez K., Laughton K., Li Q.X., Charman S.A., Nicolazzo J.A., Wilkins S., Deleva K., Lynch T., Kok G., Ritchie C.W., Tanzi R.E., Cappai R., Masters C.L., Barnham K.J., Bush A.I. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59(1):43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 51.Faux N.G., Ritchie C.W., Gunn A., Rembach A., Tsatsanis A., Bedo J., Harrison J., Lannfelt L., Blennow K., Zetterberg H., Ingelsson M., Masters C.L., Tanzi R.E., Cummings J.L., Herd C.M., Bush A.I. PBT2 rapidly improves cognition in Alzheimer’s Disease: additional phase II analyses. J. Alzheimers Dis. 2010;20(2):509–516. doi: 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- 52.Johanssen T., Suphantarida N., Donnelly P.S., Liu X.M., Petrou S., Hill A.F., Barnham K.J. PBT2 inhibits glutamate-induced excitotoxicity in neurons through metal-mediated preconditioning. Neurobiol. Dis. 2015;81:176–185. doi: 10.1016/j.nbd.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 53.Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M., Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Texidó L., Martín-Satué M., Alberdi E., Solsona C., Matute C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49(3):184–190. doi: 10.1016/j.ceca.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Sepulveda F.J., Parodi J., Peoples R.W., Opazo C., Aguayo L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS One. 2010;5(7):e11820. doi: 10.1371/journal.pone.0011820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabogo D., Rauw G., Amritraj A., Baker G., Kar S. ß-amyloid-related peptides potentiate k+-evoked glutamate release from adult rat hippocampal slices. Neurobiol. Aging. 2010;31(7):1164–1172. doi: 10.1016/j.neurobiolaging.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 57.Takeda A., Tamano H., Tempaku M., Sasaki M., Uematsu C., Sato S., Kanazawa H., Datki Z.L., Adlard P.A., Bush A.I. Extracellular Zn2+ is essential for amyloid β1-42-induced cognitive decline in the normal brain and its rescue. J. Neurosci. 2017;37(30):7253–7262. doi: 10.1523/JNEUROSCI.0954-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kessels H.W., Nabavi S., Malinow R. Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA. 2013;110(10):4033–4038. doi: 10.1073/pnas.1219605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bush A.I., Pettingell W.H., Multhaup G. d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science. 1994;265(5177):1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 60.Lee M.C., Yu W.C., Shih Y.H., Chen C.Y., Guo Z.H., Huang S.J., Chan J.C.C., Chen Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018;8(1):4772. doi: 10.1038/s41598-018-23122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deshpande A., Kawai H., Metherate R., Glabe C.G., Busciglio J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 2009;29(13):4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bero A.W., Yan P., Roh J.H., Cirrito J.R., Stewart F.R., Raichle M.E., Lee J.M., Holtzman D.M. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 2011;14(6):750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bero A.W., Bauer A.Q., Stewart F.R., White B.R., Cirrito J.R., Raichle M.E., Culver J.P., Holtzman D.M. Bidirectional relationship between functional connectivity and amyloid-β deposition in mouse brain. J. Neurosci. 2012;32(13):4334–4340. doi: 10.1523/JNEUROSCI.5845-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11(10):682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X., Hollern D., Liao J., Andrechek E., Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013;4(3):e560–e11. doi: 10.1038/cddis.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bordji K., Becerril-Ortega J., Nicole O., Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ß production. J. Neurosci. 2010;30(47):15927–15942. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rush T., Buisson A. Reciprocal disruption of neuronal signaling and Aβ production mediated by extrasynaptic NMDA receptors: a downward spiral. Cell Tissue Res. 2014;356(2):279–286. doi: 10.1007/s00441-013-1789-1. [DOI] [PubMed] [Google Scholar]
- 68.Dolev I., Fogel H., Milshtein H., Berdichevsky Y., Lipstein N., Brose N., Gazit N., Slutsky I. Spike bursts increase amyloid-β 40/42 ratio by inducing a presenilin-1 conformational change. Nat. Neurosci. 2013;16(5):587–595. doi: 10.1038/nn.3376. [DOI] [PubMed] [Google Scholar]
- 69.McKee K.K., Tan C.P., Palyha O.C., Liu J., Feighner S.D., Hreniuk D.L., Smith R.G., Howard A.D., Van der Ploeg L.H. Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics. 1997;46(3):426–434. doi: 10.1006/geno.1997.5069. [DOI] [PubMed] [Google Scholar]
- 70.Egerod K.L., Holst B., Petersen P.S., Hansen J.B., Mulder J., Hökfelt T., Schwartz T.W. GPR39 splice variants versus antisense gene LYPD1: expression and regulation in gastrointestinal tract, endocrine pancreas, liver, and white adipose tissue. Mol. Endocrinol. 2007;21(7):1685–1698. doi: 10.1210/me.2007-0055. [DOI] [PubMed] [Google Scholar]
- 71.Jackson V.R., Nothacker H-P., Civelli O. GPR39 receptor expression in the mouse brain. Neuroreport. 2006;17(8):813–816. doi: 10.1097/01.wnr.0000215779.76602.93. [DOI] [PubMed] [Google Scholar]
- 72.Popovics P., Stewart A.J. GPR39: a Zn(2+)-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell. Mol. Life Sci. 2011;68(1):85–95. doi: 10.1007/s00018-010-0517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Młyniec K., Doboszewska U., Szewczyk B., Sowa-Kućma M., Misztak P., Piekoszewski W., Trela F., Ostachowicz B., Nowak G. The involvement of the GPR39-Zn(2+)-sensing receptor in the pathophysiology of depression. Studies in rodent models and suicide victims. Neuropharmacology. 2014;79:290–297. doi: 10.1016/j.neuropharm.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 74.Młyniec K., Starowicz G., Gaweł M., Frąckiewicz E., Nowak G. Potential antidepressant-like properties of the TC G-1008, a GPR39 (zinc receptor) agonist. J. Affect. Disord. 2016;201:179–184. doi: 10.1016/j.jad.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 75.Cuzon Carlson V.C., Ford M.M., Carlson T.L., Lomniczi A., Grant K.A., Ferguson B., Cervera-Juanes R.P. Modulation of Gpr39, a G-protein coupled receptor associated with alcohol use in non-human primates, curbs ethanol intake in mice. Neuropsychopharmacology. 2019;44(6):1103–1113. doi: 10.1038/s41386-018-0308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hodge R.D., Trygve E.B., Jeremy A.M., Kimberly A.S., Eliza R.B., Lucas T.G., Jennie L.C., Brian L., Osnat P., Zizhen Y., Jeroen E., Thomas H., Boaz P.L., Soraya I.S., Brian A., Allison B.E.S.L. Conserved cell types with divergent features between human and mouse cortex. bioRxiv. 2018:1–112. doi: 10.1101/3848266. [DOI] [Google Scholar]
- 77.Tasic B., Yao Z., Graybuck L.T., Smith K.A., Nguyen T.N., Bertagnolli D., Goldy J., Garren E., Economo M.N., Viswanathan S., Penn O., Bakken T., Menon V., Miller J., Fong O., Hirokawa K.E., Lathia K., Rimorin C., Tieu M., Larsen R., Casper T., Barkan E., Kroll M., Parry S., Shapovalova N.V., Hirschstein D., Pendergraft J., Sullivan H.A., Kim T.K., Szafer A., Dee N., Groblewski P., Wickersham I., Cetin A., Harris J.A., Levi B.P., Sunkin S.M., Madisen L., Daigle T.L., Looger L., Bernard A., Phillips J., Lein E., Hawrylycz M., Svoboda K., Jones A.R., Koch C., Zeng H. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563(7729):72–78. doi: 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dittmer S., Sahin M., Pantlen A., Saxena A., Toutzaris D., Pina A.L., Geerts A., Golz S., Methner A. The constitutively active orphan G-protein-coupled receptor GPR39 protects from cell death by increasing secretion of pigment epithelium-derived growth factor. J. Biol. Chem. 2008;283(11):7074–7081. doi: 10.1074/jbc.M704323200. [DOI] [PubMed] [Google Scholar]
- 79.Holst B., Egerod K.L., Schild E., Vickers S.P., Cheetham S., Gerlach L.O., Storjohann L., Stidsen C.E., Jones R., Beck-Sickinger A.G., Schwartz T.W. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology. 2007;148(1):13–20. doi: 10.1210/en.2006-0933. [DOI] [PubMed] [Google Scholar]
- 80.Holst B., Holliday N.D., Bach A., Elling C.E., Cox H.M., Schwartz T.W. Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 2004;279(51):53806–53817. doi: 10.1074/jbc.M407676200. [DOI] [PubMed] [Google Scholar]
- 81.Yasuda S., Miyazaki T., Munechika K., Yamashita M., Ikeda Y., Kamizono A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J. Recept. Signal Transduct. Res. 2007;27(4):235–246. doi: 10.1080/10799890701506147. [DOI] [PubMed] [Google Scholar]
- 82.Abramovitch-Dahan C., Asraf H., Bogdanovic M., Sekler I., Bush A.I., Hershfinkel M. Amyloid β attenuates metabotropic zinc sensing receptor, mZnR/GPR39, dependent Ca2+, ERK1/2 and Clusterin signaling in neurons. J. Neurochem. 2016;139(2):221–233. doi: 10.1111/jnc.13760. [DOI] [PubMed] [Google Scholar]
- 83.Mendoza M.C., Er E.E., Blenis J. The Ras-ERK and PI3K-MTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011;36(6):320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hershfinkel M. The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease. Int. J. Mol. Sci. 2018;19(2):E439. doi: 10.3390/ijms19020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shimizu Y., Koyama R., Kawamoto T. Rho kinase-dependent desensitization of GPR39; a unique mechanism of GPCR downregulation. Biochem. Pharmacol. 2017;140:105–114. doi: 10.1016/j.bcp.2017.06.115. [DOI] [PubMed] [Google Scholar]
- 86.Kovacs Z., Schacht T., Herrmann A-K., Albrecht P., Lefkimmiatis K., Methner A. Protein kinase inhibitor β enhances the constitutive activity of G-protein-coupled zinc receptor GPR39. Biochem. J. 2014;462(1):125–132. doi: 10.1042/BJ20131198. [DOI] [PubMed] [Google Scholar]
- 87.Sanchez A., Tripathy D., Yin X., Luo J., Martinez J., Grammas P. Pigment epithelium-derived factor (PEDF) protects cortical neurons in vitro from oxidant injury by activation of extracellular signal-regulated kinase (ERK) 1/2 and induction of Bcl-2. Neurosci. Res. 2012;72(1):1–8. doi: 10.1016/j.neures.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Subramaniam S., Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277(1):22–29. doi: 10.1111/j.1742-4658.2009.07367.x. [DOI] [PubMed] [Google Scholar]
- 89.Dou P., Zhang D., Cheng Z., Zhou G., Zhang L. PKIB promotes cell proliferation and the invasion-metastasis cascade through the PI3K/Akt pathway in NSCLC cells. Exp. Biol. Med. (Maywood) 2016;241(17):1911–1918. doi: 10.1177/1535370216655908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dabanaka K., Chung S., Nakagawa H., Nakamura Y., Okabayashi T., Sugimoto T., Hanazaki K., Furihata M. PKIB expression strongly correlated with phosphorylated Akt expression in breast cancers and also with triple-negative breast cancer subtype. Med. Mol. Morphol. 2012;45(4):229–233. doi: 10.1007/s00795-011-0565-0. [DOI] [PubMed] [Google Scholar]
- 91.Zimmermann S., Moelling K. Phosphorylation and Regulation of Raf by Akt (Protein Kinase B). Science (80-.) 1999;286(5445):1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 92.Dumaz N., Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J. Biol. Chem. 2003;278(32):29819–29823. doi: 10.1074/jbc.C300182200. [DOI] [PubMed] [Google Scholar]
- 93.Kremer A., Louis J.V., Jaworski T., Van Leuven F. GSK3 and alzheimer’s disease: Facts and fiction. Front. Mol. Neurosci. 2011;4:17. doi: 10.3389/fnmol.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sato S., Huang X-P., Kroeze W.K., Roth B.L. Discovery and characterization of novel GPR39 agonists allosterically modulated by zinc. Mol. Pharmacol. 2016;90(6):726–737. doi: 10.1124/mol.116.106112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oertel D., Young E.D. What’s a cerebellar circuit doing in the auditory system? Trends Neurosci. 2004;27(2):104–110. doi: 10.1016/j.tins.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 96.Chorin E., Vinograd O., Fleidervish I., Gilad D., Herrmann S., Sekler I., Aizenman E., Hershfinkel M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 2011;31(36):12916–12926. doi: 10.1523/JNEUROSCI.2205-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gilad D., Shorer S., Ketzef M., Friedman A., Sekler I., Aizenman E., Hershfinkel M. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol. Dis. 2015;81:4–13. doi: 10.1016/j.nbd.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ganay T., Asraf H., Aizenman E., Bogdanovic M., Sekler I., Hershfinkel M. Regulation of neuronal pH by the metabotropic Zn(2+)-sensing Gq-coupled receptor, mZnR/GPR39. J. Neurochem. 2015;135(5):897–907. doi: 10.1111/jnc.13367. [DOI] [PubMed] [Google Scholar]
- 99.Mango D., Nisticò R. Role of ASIC1a in Aβ-induced synaptic alterations in the hippocampus. Pharmacol. Res. 2018;131:61–65. doi: 10.1016/j.phrs.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 100.Lacour A., Espinosa A., Louwersheimer E., Heilmann S., Hernández I., Wolfsgruber S., Fernández V., Wagner H., Rosende-Roca M., Mauleón A., Moreno-Grau S., Vargas L., Pijnenburg Y.A., Koene T., Rodríguez-Gómez O., Ortega G., Ruiz S., Holstege H., Sotolongo-Grau O., Kornhuber J., Peters O., Frölich L., Hüll M., Rüther E., Wiltfang J., Scherer M., Riedel-Heller S., Alegret M., Nöthen M.M., Scheltens P., Wagner M., Tárraga L., Jessen F., Boada M., Maier W., van der Flier W.M., Becker T., Ramirez A., Ruiz A. Genome-wide significant risk factors for Alzheimer’s disease: Role in progression to dementia due to Alzheimer’s disease among subjects with mild cognitive impairment. Mol. Psychiatry. 2017;22(1):153–160. doi: 10.1038/mp.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Charnay Y., Imhof A., Vallet P.G., Kovari E., Bouras C., Giannakopoulos P. Clusterin in neurological disorders: Molecular perspectives and clinical relevance. Brain Res. Bull. 2012;88(5):434–443. doi: 10.1016/j.brainresbull.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 102.Li X., Ma Y., Wei X., Li Y., Wu H., Zhuang J., Zhao Z. Clusterin in Alzheimer’s disease: a player in the biological behavior of amyloid-beta. Neurosci. Bull. 2014;30(1):162–168. doi: 10.1007/s12264-013-1391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zádori D., Veres G., Szalárdy L., Klivényi P., Toldi J., Vécsei L. Glutamatergic dysfunctioning in Alzheimer’s disease and related therapeutic targets. J. Alzheimers Dis. 2014;42(Suppl. 3):S177–S187. doi: 10.3233/JAD-132621. [DOI] [PubMed] [Google Scholar]
- 104.Lagostena L., Rosato-Siri M., D’Onofrio M., Brandi R., Arisi I., Capsoni S., Franzot J., Cattaneo A., Cherubini E. In the adult hippocampus, chronic nerve growth factor deprivation shifts GABAergic signaling from the hyperpolarizing to the depolarizing direction. J. Neurosci. 2010;30(3):885–893. doi: 10.1523/JNEUROSCI.3326-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Molinaro G., Battaglia G., Riozzi B., Di Menna L., Rampello L., Bruno V., Nicoletti F. Memantine treatment reduces the expression of the K+/Cl- cotransporter KCC2 in the hippocampus and cerebral cortex, and attenuates behavioural responses mediated by GABA(A) receptor activation in mice. Brain Res. 2009;1265:75–79. doi: 10.1016/j.brainres.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 106.Schulte J.T., Wierenga C.J., Bruining H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018;90(March):260–271. doi: 10.1016/j.neubiorev.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 107.Löscher W., Hönack D. High doses of memantine (1-amino-3,5-dimethyladamantane) induce seizures in kindled but not in non-kindled rats. Naunyn Schmiedebergs Arch. Pharmacol. 1990;341(5):476–481. doi: 10.1007/BF00176343. [DOI] [PubMed] [Google Scholar]
- 108.Khan M.Z. A possible significant role of zinc and GPR39 zinc sensing receptor in Alzheimer disease and epilepsy. Biomed. Pharmacother. 2016;79(24):263–272. doi: 10.1016/j.biopha.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 109.Gonzales E.B., Sumien N. Acidity and acid-sensing ion channels in the normal and alzheimer’s disease brain. J. Alzheimers Dis. 2017;57(4):1137–1144. doi: 10.3233/JAD-161131. [DOI] [PubMed] [Google Scholar]
- 110.Mlyniec K. Zinc in the glutamatergic theory of depression. Curr. Neuropharmacol. 2015;13(4):505–513. doi: 10.2174/1570159X13666150115220617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kepp K.P. Alzheimer’s disease: How metal ions define β-amyloid function. Coord. Chem. Rev. 2017;351:127–159. doi: 10.1016/j.ccr.2017.05.007. [DOI] [Google Scholar]
- 112.Tena-Campos M., Ramon E., Borroto-Escuela D.O., Fuxe K., Garriga P. The zinc binding receptor GPR39 interacts with 5-HT1A and GalR1 to form dynamic heteroreceptor complexes with signaling diversity. Biochim. Biophys. Acta. 2015;1852(12):2585–2592. doi: 10.1016/j.bbadis.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 113.Yasuda S., Ishida J. GPR39-1b, the 5-transmembrane isoform of GPR39 interacts with neurotensin receptor NTSR1 and modifies its function. J. Recept. Signal Transduct. Res. 2014;34(4):307–312. doi: 10.3109/10799893.2014.885050. [DOI] [PubMed] [Google Scholar]
- 114.Gahete M.D., Rubio A., Córdoba-Chacón J., Gracia-Navarro F., Kineman R.D., Avila J., Luque R.M., Castaño J.P. Expression of the ghrelin and neurotensin systems is altered in the temporal lobe of Alzheimer’s disease patients. J. Alzheimers Dis. 2010;22(3):819–828. doi: 10.3233/JAD-2010-100873. [DOI] [PubMed] [Google Scholar]