

Abstract
Chronic inflammatory processes within the central nervous system (CNS) are in part responsible for the development of neurodegenerative and psychiatric diseases. These processes are associated with, among other things, the increased and disturbed activation of microglia and the elevated production of proinflammatory factors. Recent studies indicated that the disruption of the process of resolution of inflammation (RoI) may be the cause of CNS disorders. It is shown that the RoI is regulated by endogenous molecules called specialized pro-resolving mediators (SPMs), which interact with specific membrane receptors. Some SPMs activate formyl peptide receptors (FPRs), which belong to the family of seven-transmembrane G protein-coupled receptors. These receptors take part not only in the proinflammatory response but also in the resolution of the inflammation process. Therefore, the activation of FPRs might have complex consequences.
This review discusses the potential role of FPRs, and in particular the role of FPR2 subtype, in the brain under physiological and pathological conditions and their involvement in processes underlying neurodegenerative and psychiatric disorders as well as ischemia, the pathogenesis of which involves the dysfunction of inflammatory processes.
Keywords: Neuroinflammation, glial cells, resolution of inflammation, formyl peptide receptors, new pro-resolving agonists, Alzheimer’s disease, depression, ischemia
1. INTRODUCTION
Among the many hypotheses that attempt to explain the causes of CNS diseases, the hypothesis regarding the dysfunction of the immune system is of significance. It is believed that inflammatory processes in the brain, called neuroinflammation, can lead to the development of neurodegenerative diseases (e.g., Alzheimer’s and Parkinson’s diseases), mental disorders (e.g., depression and schizophrenia) and, to some extent, ischemic diseases. In general, acute inflammation is considered to be a beneficial process, while the dysfunction of the RoI leads to the development of the chronic inflammatory process. For this reason, in recent years, mechanisms and potential strategies that promote RoI have become a focus of interest. Indeed, based on the available literature, it can be postulated that both inflammatory activation and deficits in the RoI in the brain are significant causes of the development of CNS diseases.
The RoI is mediated by endogenous mediators called SPMs through interactions with specific membrane receptors. In the present review, special attention was paid to the role of G-protein-coupled receptors (GPCRs), which are expressed on immunocompetent cells in the brain, particularly formyl peptide receptors, in the RoI process.
Moreover, new strategies supporting their role in shifting from proinflammatory to anti-inflammatory activation were proposed as a potential tool for the pharmacotherapy of CNS diseases triggered by chronic inflammation.
2. OVERVIEW OF THE INFLAMMATORY RESPONSE
Inflammation is a pathophysiological response of the body to tissue dysfunction or homeostatic imbalance triggered by a variety of harmful stimuli, including toxins, infections, trauma, stress or ischemia, that elicit the activation of the immune system. This process is characterized by the classic cardinal signs described by the Roman scholar and encyclopedist Aurelius Celsus (25 – 50 B.C.), namely, pain (dolor), increased temperature (calor), redness (rubor), and swelling (tumor) [1]. The fifth symptom of inflammation, i.e., loss of function and organ damage (functio laesa) was described later by Galen (129 - 200 A.D) [2] (Fig. 1).
Fig. (1).

The classic cardinal signs of inflammation.
The molecular and cellular events of the inflammatory response are known and result in increased blood flow, capillary dilatation, leukocyte infiltration, and production of chemical mediators. Generally, several phases of inflammation, including initiation, propagation, and resolution, have been described [3]. Recently, there have been very interesting reports indicating that the phases of inflammation do not develop sequentially but rather overlap; thus, both inflammatory activation and resolution can co-occur. Of course, the intensity of these processes is different, and this determines whether inflammation progresses or is abated. This novel interpretation of the RoI attempts to explain the dual role of some factors in the course of neuroinflammation; for example, it is still an open question whether cyclooxygenase 2 (COX-2) is an inhibitory target or a source of resolution molecules [4].
Acute inflammation is a rapid and self-limiting process. It usually lasts a few days and disappears after the removal of the cause without major damage to the body. When inflammation is properly controlled, it results in a protection against the spread of infection or damage and is followed by a resolution phase in which the affected tissues are restored to their original structural and functional state [5, 6]. Inflammation is mainly characterized by the presence of neutrophils, which quickly migrate to the site of injury or infection, promote the recruitment of inflammatory monocytes, and produce proinflammatory factors, thereby allowing for appropriate neutralization of the harmful factor [7]. The trafficking and homing of neutrophils are mediated mainly by families of GPCRs, one of which recognizes chemokines and the other of which recognizes classic chemoattractants originating from pathogens or damaged host tissues. Although neutrophils are essential for the proper elimination of the harmful factor, an excess influx of leukocytes can be more dangerous than the infection or injury itself; therefore, neutrophils undergo apoptosis after performing their action at the site of inflammation [8] and are subsequently eliminated by macrophages.
Briefly, the major signs of acute inflammation resolution include: the limitation or cessation of blood-borne cell influx, the counter-regulation of chemokines and cytokines, the switching of the signaling pathways associated with leukocyte survival, the induction of leukocyte apoptosis, the subsequent removal of leukocytes through efferocytosis by macrophages, the reprogramming of macrophages from pro-inflammatory to anti-inflammatory activated cells, the return of nonapoptotic cells to the vasculature or lymphatic system, and finally the initiation of healing processes. Resolution is a multistage and complex process [9, 10], and the failure of one or more steps may be involved in prolonged inflammation and the pathogenesis of chronic inflammatory diseases mainly due to constant stimulation of the immune system, the overproduction of proinflammatory cytokines [e.g., interleukin (IL)-1β, IL-6, and (tumor necrosis factor (TNF)-α], oxidative stress, the destruction of tissues at the site of the inflammatory process, and impairments in returning to homeostasis [11-14].
3. NEUROINFLAMMATION: THE IMMUNE RESPONSE IN THE BRAIN
Generally, CNS inflammation differs from inflammation that occurs in peripheral tissues because it is the result of the collective effects of various brain cells (microglia, astrocytes, oligodendrocytes, and NG2 glia) and in some cases peripheral immune cells. Moreover, data demonstrated that glial cells play a dual role in this process depending on the course of the disease and inflammatory environment. Interestingly, the double-edged nature of neuroinflammation, especially when the process is acute (transient), suggests that controlling neuroinflammation may have a neuroprotective function in maintaining homeostasis. On the other hand, it is commonly accepted that, while the mechanisms that ultimately lead to brain disturbances are different in various diseases, chronic neuroinflammation is a prominent feature of the progressive nature of neurodegeneration [14,15]. It appears to result from the complexity of neuroinflammation, which, as mentioned above, requires a coordinated response of glial cells and peripheral immune cells through the release of proinflammatory cytokines, including IL-1 and IL-6, as well as caspase activation [16].
3.1. The Role of Glial Cells in the Course of Neuro-inflammation
In the brain, glial cells play a key role in the inflammatory response. They are the most abundant and widely distributed cells in the CNS, and they interact with neurons and immune cells as well as with blood vessels. They contribute to the monitoring of the physiological milieu and act as the first line of defense when the brain is exposed to different insults [17]. Glial cells participate in the activation, recognition, and modulation of immune reactions as well as the release of different factors (e.g., cytokines and chemokines), which support immune defense and are crucial for the coordination of various immune cells [18].
3.1.1. Microglia
Microglia, small phagocytic cells of myeloid origin that comprise 10 – 20% of all cells, are the main immunocompetent cells in the brain. A vast body of data suggested that microglia have a dual character: neuroprotective and neurotoxic. In fact, under normal conditions, the activation of microglia cells plays a protective role by regulating the response to pathogens and promoting tissue repair through the release of anti-inflammatory and neurotrophic factors. Moreover, microglia participate in ontogenetic brain development and in maintaining homeostasis, including being involved in the programmed death of neurons during development, the removal of cellular debris and dying cells, and the regulation of synaptic plasticity [19]. Microglia also play a crucial role in neuron-glia interactions, thus controlling the proper functioning of neurons. In the healthy adult brain, microglial cells are highly dynamic in the resting state. At the same time, they express low levels of markers, including major histocompatibility complex (MHC) class I and II molecules. However, microglial processes are highly active, and once activated, microglia and CNS-infiltrating monocyte-derived macrophages upregulate many surface molecules, including MHCII proteins and other costimulatory molecules, allowing them to act more efficiently than astrocytes as antigens for T cell presentation. Under pathological conditions, the chronic activation of immune responses can lead to the functional switch of microglia from a regulatory phenotype to a neurotoxic phenotype and the excessive release of proinflammatory cytokines, such as IL-1β, IL-18, TNF-α, and IL-6, chemokines and neurotoxic mediators, such as nitric oxide (NO), prostaglandin E2, superoxide anion, and excitatory amino acids [20]. Interestingly, the process of the acute and prolonged activation of microglia is phenotypically and functionally dynamic and may vary depending on the stage of disease or the status of the brain environment (Fig. 2).
Fig. (2).
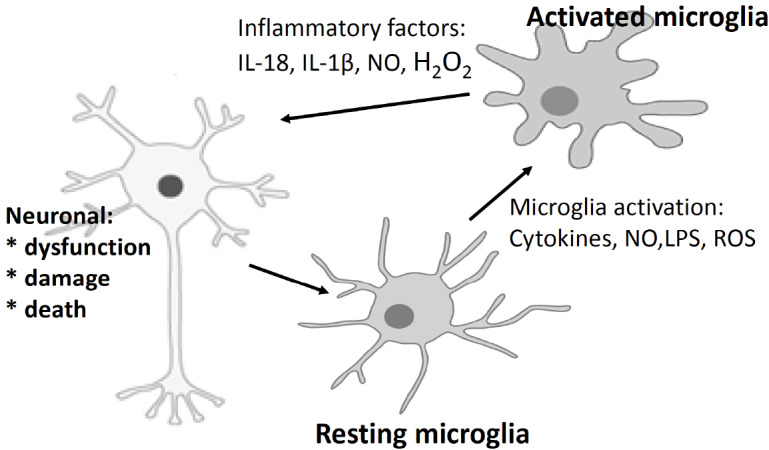
Microglia activated by stress, infection, and neurotoxins release many proinflammatory factors, such as IL-18, IL-1β, NO, and H2O2. These mediators, in turn, lead to neuronal dysfunction and, consequently, cell death.
3.1.2. Astrocytes
Astrocytes, the most abundant subtype of glial cells in the CNS, are essential for brain homeostasis, as they provide metabolites and growth factors for neurons and to support synapse formation and plasticity. Furthermore, astrocytes are also able to detect harmful signals, promote cytokine and chemokine secretion, and activate the immune defense. Generally, astrocytes are exposed simultaneously to a plethora of stimuli and activate various intracellular signaling pathways in the course of neuroinflammation. Thus, astrocyte responses are not a reaction to a single event but instead the net result of a complex and diverse network of intracellular activators [21]. Importantly, the activation of astrocytes often follows the primary activation of microglia, of which the IL-1 family of cytokines is a key mediator, in response to pathological conditions such as trauma, stroke, or neurodegenerative disorders [22]. In fact, the release of IL-1β, mainly from microglia, can occur rapidly and may increase the secretion of other cytokines, such as IL-6, from astrocytes to promote inflammation. Moreover, IL-1β might hinder the ability of astrocytes to reabsorb glutamic acid and promote the release of free radicals [23, 24]. Notably, IL-1Ra prevents astrocytes from causing pathological damage [25], showing that microglia might indirectly affect the function of astrocytes. In addition, microglial activation stimulates astrocytes to secrete IL-10 and TGF-β1 (transforming growth factor) [26], of which high levels initiate a feedback loop to reduce IL-1β release, thus inhibiting microglial activation and resulting in the RoI. Moreover, astrocytes may secrete growth substances, such as neural growth factor (NGF), cerebral growth factor (BGF), and basic fibroblast growth factor (bFGF), which have a significant role in the repair and growth of neurons as well as in brain development.
It should be mentioned that neuroinflammation is contingent upon the influx of immune cells from the peripheral blood to the brain. Experimental data have shown that a disturbance in the interaction between astrocytes and microglia plays a crucial role in blood-brain barrier (BBB) dysfunction [27]. The overexpression of inflammatory stimuli in the neurovascular unit may initiate a response that leads to the destruction of the BBB and to neuronal damage. In addition, reactive forms of microglia and astrocytes contribute to damage within the BBB, which becomes more permeable to peripheral leucocytes and activates glial cells to produce proinflammatory cytokines, chemokines, and reactive oxygen species (ROS) to become an additional source of inflammation-supporting molecules.
4. THE ROLE OF PRO-RESOLVING MOLECULES IN THE RESOLUTION OF INFLAMMATION
Accumulating evidence has suggested that chronic neuroinflammation plays a relevant role in the onset and progression of neurodegenerative diseases as well as psychiatric disorders. Furthermore, the combined dysregulation of glial activation and proinflammatory cytokine production may also be an urgent driver in the pathogenesis of ischemia and traumatic brain disorders, as they are associated with neuroimmunological abnormalities that contribute to neurodegeneration. To date, many potential neuroprotective agents, such as plant extracts, and strategies (e.g., autophagy activation), have been established [16, 28]. Generally, the prevention of neuropathologies crucially depends on the termination of inflammation, which is traditionally considered a passive process through which inflammation spontaneously subsides. However, as mentioned before, there is a growing appreciation that, like the initiation of inflammation, the RoI is a complex, active process aimed at restoring tissue integrity and function [5, 29]. The progression of this process requires proper endogenous activation that induces a switch from the release of proinflammatory molecules to the secretion of pro-resolving mediators, which comprise a wide variety of compounds, including gases, proteins, and lipids [30]. The latter group is most often studied and is termed SPMs (Fig. 3).
Fig. (3).
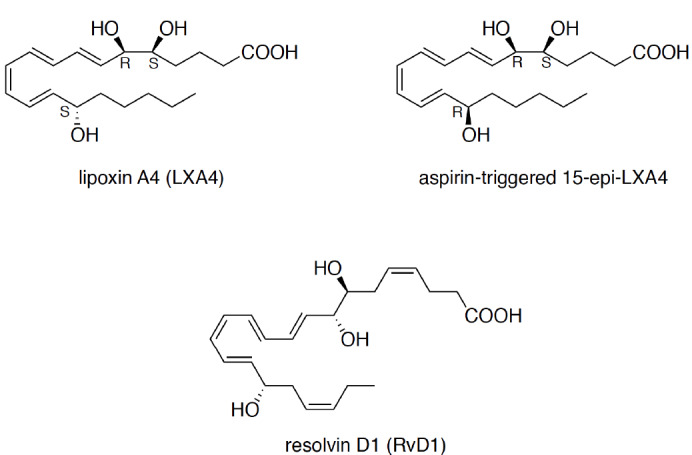
Structures of SPMs that bind at FPR2.
SPMs are produced through the oxidation of arachidonic acid (AA), 3-eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA). The first class of SPMs to be described was termed lipoxins and originates from AA. E-series resolvins are derived from EPA, while D-series resolvins comprise resolvins, maresins, and protectins generated from DHA. In humans, the production of lipid SPMs is catalyzed by epithelial cell-, eosinophil-, and monocyte-derived 15-lipoxygenase (15-LOX), leukocyte-derived 5-LOX, platelet-derived 12-LOX, and COX-2. These enzymes have been proven to be present in various cell types and to generate precursors of aspirin-triggered lipoxins, resolvins, and protectins in the acetylated form. SPMs exert their biological effects via G-protein-coupled receptors, including the following:
● Formyl peptide receptor 2 (also called lipoxin A4 receptor, FPR2/ALX) for lipoxin A4 (LXA4), annexin A1 (AnxA1), and resolvin D1 (RvD1).
● G-protein-coupled receptor 32 (GPR32) for lipoxin A4 and RvD1.
In addition, RvD1s act through chemokine-like receptor 1 (ChemR23), and RvE1s act through the leukotriene B4 receptor (LTB4R or BLT1) [29, 31, 32].
The main advantage of the biological activity of SPMs is that they do not block signal transduction within the inflammatory cascade; instead, they trigger processes that reduce the expression of proinflammatory molecules. Moreover, SPMs activate cascades that induce remodeling within sites damaged by inflammatory processes. Importantly, anti-inflammatory effects rely mostly on an inhibitory/blocking action, while pro-resolving effects are mediated by the activation and stimulation of specific inherent processes (e.g., apoptosis and efferocytosis); however, the RoI is the final result of both [33-35]. In addition, Peretti et al. recently noted that SPMs elicit “mild to moderate effects”, which, by establishing an equilibrium between proinflammatory and anti-inflammatory reactions, help to strike a balance [35].
According to the literature, ideal SPM’s, which combine both anti-inflammatory and pro-resolving activities, should fulfill the following fundamental criteria [3, 31, 36]: the cessation of inflammatory cell recruitment (e.g., tissue infiltration by neutrophils), the regulation of the secretion of cytokines and chemokines, the switching of macrophages from the classically activated phenotype to the alternatively activated phenotype, the induction of efferocytosis of apoptotic neutrophils (by macrophages), the removal of nonapoptotic cells, dendritic cells and macrophages from the site of inflammation, the modulation of the immune response, the instruction of suppressive immune cells and the adaptive immune response, the induction of tissue repair (the final step of resolution), and a return to homeostasis.
In the context of our article, lipoxins (LXs), which are metabolites of AA, are crucial. Briefly, LXA4 and lipoxin B4 (LXB4) are produced by the oxygenation of AA by 15-LOX and 5-LOX and subsequent enzymatic hydrolysis. On the other hand, new lipoxin analogs termed aspirin-triggered lipoxins (AT-LXA4) are derived from the aspirin-triggered pathway, in which aspirin alters COX-2 activity by increasing the acetylation of COX-2 [37]. The inactivation of lipoxin catalyzed by the enzyme 15-prostaglandin dehydrogenase can result in the synthesis of a series of other stable lipoxin analogs [38] that retain all of the biological functions of lipoxins. It is now widely recognized that the synthesis of lipoxins is enhanced during the inflammatory response, and their activation leads to the RoI through the promotion of phagocytosis of apoptotic neutrophils by macrophages, which potentiates the abatement of the inflammatory activation [39].
The concept that the biological action of lipoxins is mediated by GPCRs was developed in 1990 based on studies with pertussis toxin [40]. The first studies indicating a specific recognition site for LXA4 on polymorphonuclear leukocytes (PMNs) were conducted through the use of tritium-radiolabeled LXA4 in 1992 [41]. A research group led by Serhan identified cDNA for the human 7-transmembrane receptor (pINF114), which codes for functional high-affinity receptors for lipoxin A4 [42]. These receptors are termed FPR2/ALXR [43-48].
5. FORMYL PEPTIDE RECEPTOR FAMILY
Pattern recognition receptors (PRRs) located on immune cells are a type of receptor that plays a significant role in the mechanisms of innate immunity. These receptors recognize pathogen-associated molecular patterns (PAMPs) as important mechanisms for the elimination of various pathogens and their components by innate immunity [49]. N-formyl-methionyl-leucylphenylalanine (fMLP) is one of them, and it induces the chemotaxis of neutrophil granulocytes and monocytes [50, 51]; these effects are associated with interactions with formyl peptide receptors.
In humans, FPRs have been detected in the spinal cord, brain, anterior horn cells, hypoglossal nucleus neurons, choroid plexus, and epithelium [52]. FPRs have also been found in the cerebellar system, in the sensory system, on reticular activating system neurons, and in the ependyma. Hippocampal neurons, pyramidal cell neurons, end-plate pyramidal cells, astrocytes, Schwann cells of the peripheral nervous system, and cells in the parasympathetic system show a high level of FPR expression [53]. On the other hand, microglial cells isolated from normal adult humans express the gene for FPRs, but the level of the receptor protein is low, and no functional observation has been reported [54, 55]. In rodents, microglial cells lack a chemotactic response to fMLP [56], suggesting that these cells express a low level or no fMLP receptors on their surface. Cui and collaborators found that both murine primary microglial cells and the N9 cells constitutively express low levels of both the Fpr2 and Fpr1 genes in the resting state, and this expression is enhanced by treatment with TNF-α. However, resting murine microglial cells do not respond to fMLP or to other agonists of either FPR2 or FPR1. TNF-α treatment enables both primary and N9 cells to develop a potent chemotaxis response to agonists that act only on FPR2 in association with the surface expression of low-affinity fMLP binding sites [57].
5.1. Structural Characteristics of FPRs
FPRs belong to the largest and functionally diverse family of G-protein-coupled receptors. These receptors, irrespective of their function, have the same structural design. They are long single-chain proteins that span the plasma membrane seven times and are therefore often referred to as heptahelical (7-transmembrane) receptors (7-TMs) [58-60]. Their polypeptide chain is composed of the extracellular N-terminal domain, seven transmembrane helices (TM1-7) linked by intracellular and extracellular loops (IL1-3, EL1-3), and an intracellular C-terminus. In some receptors, an additional helix that is parallel to the inner surface of the plasma membrane has been detected [61, 62]. According to the GRAFS (glutamate, rhodopsin, adhesion, frizzled/taste2, and secretin) system recommended by the International Union of Basic and Clinical Pharmacology (IUPHAR), in which GPCRs are classified based on phylogenetic criteria, FPRs belong to the rhodopsin-like receptor family (class A) [63, 64]. Receptors of this family are characterized by relatively short N-terminal parts and transmembrane regions that participate in ligand recognition. To date, two very conserved and key motifs have been discovered in FPRs: the NPXXY motif in TM7, which is a part of a molecular switch that induces a conformational change that activates the receptor, and the E/DRY motif, which is located at the border between TM3 and TM2. The E/DRY motif acts as an ionic blockade and links TM3 and TM6 [65].
5.2. Human Formyl Peptide Receptors
To date, three members belonging to the FPR family have been identified, namely, FPR1, FPR2, and FPR3 [66- 70]. FPR1 and FPR2 show high homology and common functions.
The human formyl peptide receptor 1 (hFPR1), which is an fMLP target, was isolated from HL-60 cells, which are highly responsive to treatment with N-formylated peptides. hFPR2 is the result of gene amplification. However, evolutionarily, hFPR3 is the youngest member of this gene family and was created by gene duplication in the lineage leading to primates [71].
All members of the human FPR gene family are located on chromosome 19. Pairs of paralogous receptors exhibit a basic structure highly similar to that of human FPR. Human FPR2 has a 69% sequence homology with hFPR1. FPR3 sequence analysis has demonstrated that the receptor is most strongly related to FPR2, and this discovery suggests that FPR3 was created by FPR2 gene duplication [71, 72]. A comparison of human FPRs with their counterparts in primates has revealed a high level of homology that reaches 95-99% [73]. This finding highlights the functional significance of this receptor family. Interestingly, the highest variability levels of the FPR sequence are detectable in the extracellular loops of the FPR sequence, which suggests different ligand binding affinities/preferences of particular receptors and species [73,74]. FPR1 and FPR2 are expressed both on monocytes and neutrophils, whereas FPR3 expression is found only on monocytes. FPR1 is also expressed on astrocytes, microglia, and immature dendritic cells. FPR2 exhibits even greater expression both on cells in the CNS and in the periphery, including tumor cells [53, 52,76].
5.3. Mouse Formyl Peptide Receptors
Since the FPR1 gene was cloned in humans, this gene has also been identified in other mammals including rabbits, guinea pigs, horses, rats, and mice [44]. In spite of the general sequence homology, the genes coding for the FPR family in humans and other species significantly differ in terms of the number and sequence [71]. For instance, the mouse Fpr gene family comprises 8 known members (mFpr1, mFpr2, mFpr-rs1, mFpr-rs3, mFpr-rs4, mFpr-rs6, mFpr-rs7, and mFpr-rs8) located on mouse chromosome 17A3.2. Studies on mFprs have mostly focused on mFpr1 and mFpr2 gene products, which are expressed on mouse phagocytic leukocytes and are highly similar to their human counterparts. Targeted mFpr1 or mFpr2 deletion makes mice more susceptible to bacterial infections without affected viability or fertility [77, 78]. Under unstimulated conditions, mice with mFpr1 or mFpr2 gene knockout do not show behavioral disturbances. However, an array of models of diseases has shown that the development of disturbances in mFpr1-/- and mFpr2-/- mice is induced, suggesting a regulatory role for these receptors in inflammation. Similar to human FPR1, mFpr1 has been identified as a receptor with low affinity for classical fMLP [79]. In general, mFpr2 is not a high-affinity receptor for fMLP or other formyl peptides tested thus far [80]. However, it interacts with endogenous FPR2 agonists, including the amyloidogenic proteins SAA (serum amyloid A) [81] and β-amyloid plaques (Aβ) 42 [82]. In addition, mFpr2 has also been described as a receptor for F2L [83], which is a strong agonist of human FPR3 [84]. The gene products of five other mFprs, including mFpr-rs1, mFpr-rs3, mFpr-rs4, mFpr-rs6, and mFpr-rs7, have been described as mouse vomeronasal sensory receptors [79, 85]. The functions of mFpr-rs1 are still unclear. Until recently, it was believed that mFpr-rs1 has similar functions as those of human FPR2 because it was reported that an mFpr-rs1 variant encodes the mouse lipoxin A4 receptor [86]. However, functional and pharmacological tests on stably transfected cell lines showed that mFpr-rs1 barely responds to most of the agonists of human and mouse FPRs [87]. These observations indicate that mouse Fprs (especially mFpr1 and mFpr2) present many structural and pharmacological similarities to human FPRs.
5.4. Nomenclature of FPR Receptors
In the past, members of the FPR family were designated by different names (e.g., FPR1, FPR2, FPRL1, FPRH1, FPR-related receptor, HM63, FMLP-related receptor II for FPR2, FPRL2, FPRH2, and RMLP-related receptor I for FPR3). To standardize the terminology, the IUPHAR introduced a new nomenclature based on agonist-receptor interactions. Based on this system, members of the human FPR family are designated FPR1, FPR2, and FPR3 [44]. However, in the literature, human FPR2 is often termed LXA4R, ALX, or a combination of both of these terms (FPR2/ALX) due to its interaction with lipoxins A4 [42, 45, 47].
5.5. Conformational Changes Determine the Pro-resolving Activity of FPRs
A growing body of evidence has indicated that GPCRs are allosteric proteins that can adopt many conformations, some of which may promote continuous endocytosis in the absence of stimulation. More attention has been dedicated to processes that appear to be a crucial aspect of receptor regulation [88]. In many cases, FPR2 is constitutively internalized via the β-arrestin-dependent pathway and undergoes clathrin-dependent constitutive internalization [89]. Phosphorylation and internalization seem to be independent events, suggesting that constitutive endocytosis may not be a consequence of the basal activity of the receptor. Moreover, FPR2 is phosphorylated in an agonist-dependent manner, but the phosphorylation sites have not yet been identified [90]. Emerging literature has indicated that the formation of higher-order structures fulfills a vital role in the regulation of FPR family receptors. For instance, FPR1 forms homodimers and interacts with both FPR2 and FPR3 [88]. It is worth mentioning that the pro-resolving ligand AnxA1 and its N-terminal peptide Ac2-26 potentiate the formation of FPR1/FPR2 heterodimers and/or FPR2 homodimers, while proinflammatory ligands and antagonists do not exert such action. It is thought that the stabilization of the FPR2 oligomer may be significant for p38/MAPK/Hsp27 pathway activation [88, 91]. A conformational change in the ALX/FPR2 receptor (ligand-dependent) determines its pro-resolving action [88]. Moreover, the oligomerization of these receptors is not limited only to FPR family members but also involves surface receptors, such as the scavenger receptor MARCO (macrophage receptor with collagenous structure). Interactions between FPR1, FPR2, and MARCO have been demonstrated by bioluminescence and coimmunoprecipitation studies and are associated with agonist-induced changes in cyclic adenosine monophosphate (cAMP) levels and extracellular signal–regulated kinases (ERK1/2) phosphorylation. They can also play an important role in Aβ1-42-induced signal transduction in glial cells [92] (Fig. 4). Recently, in an excellent review, Raabe et al. discussed biased perspectives on FPRs. In the authors’ opinion, the unique character of FPR2 relies on the ability of its ligands to selectively activate subsets of downstream signaling pathways coupled to the receptor and inhibit others. This interpretation provides an elegant explanation as to why different FPR2 agonists do not cause the same effects; this receptor is exceptional due to its ability to shift from a proinflammatory to anti-inflammatory response while maintaining the former at a low but possibly life-saving level [93].
Fig. (4).

Ligand-biased signaling via formyl peptide receptors. A variety of ligands (e.g., lipoxin A4 (LXA4) and resolvin D1 (RvD1)) induce FPR2/ALX homodimerization and FPR2/FPR1 dimerization and lead to the activation of different downstream signaling cascades. Oligomerization is not limited to FPR family members and includes even non-GPCR surface receptors, such as the scavenger receptor MARCO. PMN: polymorphonuclear leukocyte.
5.6. New FPR Agonists as a Promising Tool for the Pharmacotherapy of CNS Diseases
The vital and fascinating characteristics of FPRs result from their ability to interact with structurally diverse ligands (lipids, proteins, and peptides), which can stimulate various cell type- and ligand-specific cellular responses downstream of receptor activation. This ability is caused by the biased agonism of FPRs rather than the existence of a variety of FPR subtypes. This feature reflects the ability of GPCRs to assume different conformational states, each linked to discrete cell effects. Therefore, the biased agonism provides an opportunity to potentially promote expected on-target signal transduction without on-target adverse outcomes. This paradigm has an impact on the current search for new drugs. In addition, the search of FPRs, finding new FPR ligands that are potentially more resistant to degradation then endogenous peptide ligands appears to be of special interest.
Several studies attempted to elucidate the molecular basis of this feature of FPRs. By using chimeric receptors, it has been demonstrated that distinct receptor domains might interact with distinct ligands, thus eliciting different downstream responses. In particular, lipoxin A4 has been shown to interact with the third extracellular loop of FPR2 to induce pro-resolving responses [94]. The interaction of AnxA1 with the N-terminal domain and the second extracellular loop is required for inducing Ca2+ mobilization and the modulation of gene expression. On the other hand, the proinflammatory ligand SAA might activate Ca2+ flux and ERK phosphorylation through its interaction with the first and second extracellular loop. Small-molecule FPR ligands, such as compound 43 (Fig. 5), which can penetrate into the transmembrane domains, have been proposed to interact with the first extracellular loop, the third helix, and the second intracellular loop [95]. In addition to naturally occurring peptides/proteins and endogenous lipids, various synthetic peptides, as well as nonpeptidic ligands belonging to different chemical classes, have been reported to date. The reader can refer to excellent reviews of FPR ligands reported in the literature [30, 96]. Here, we briefly discuss small-molecule nonpeptidic ligands that may be of interest for the development of future innovative therapies for chronic inflammatory conditions.
Fig. (5).

Structures of some “small-molecule” nonpeptidic FPR ligands.
Qin et al. [97] studied, for the first time, the effect of the biased agonism of two small-molecule FPR agonists, namely, compound 43 and compound 17b, on the cardioprotective profile in myocardial infarction. It was demonstrated that compound 17b, unlike compound 43, induces a lower Ca2+ flux relative to that induced by ERK1/2-Akt signal transduction. Since increased intracellular Ca2+ contributes to cardiomyocyte damage and is a key contributor to the influx of inflammatory neutrophils and macrophages, the biased agonism of compound 17b provides superior outcomes in in vivo models of myocardial infarction [97]. This study clearly suggests that exploiting biased signaling can offer new possibilities in balancing proinflammatory and anti-inflammatory pathways to retain the homeostatic environment in complex inflammatory diseases, and this approach can also be applied for drug development for CNS diseases.
We contributed to the field by identifying a series of ureidopropanamide derivatives as FPR2 agonists. The compounds formally originate from the gastrin-releasing peptide receptor antagonist PD-175266, which has been found to be a potent FPR1/FPR2 agonist [48, 98, 99]. Through appropriate structural modifications, we ultimately identified MR39 as an FPR2 agonist (Fig. 5). The in vitro pharmacokinetic properties of MR39 are favorable, as the compound is stable to oxidative metabolism in rat liver microsomes (t1/2 = 48 min) and displays good passive permeability through a monolayer of hCMEC/D3 cells, immortalized human brain microvascular endothelial cells that are considered an in vitro model of the BBB. In addition, MR39 reduced IL-1β and TNF-α levels in LPS-stimulated rat primary microglial cell cultures and thus showed protective and anti-inflammatory properties [100]. Considering that LXA4 is rapidly inactivated in vivo [101] and that there is no direct evidence that LXA4 can pass the BBB, MR39 represents a prospective tool for studying the therapeutic potential of FPR2 agonists for the pharmacotherapy of CNS diseases (Fig. 5).
6. FORMYL PEPTIDE RECEPTORS AS A NEW TARGET FOR THERAPY FOR SOME BRAIN PATHOLOGIES
Despite many years of multicenter studies, the efficacy of therapeutic interventions for some CNS diseases remains unsatisfactory. This seems to be due to the complex and multifactorial nature of their pathological basis. In this review, we focused our attention on Alzheimer’s disease, depression, and ischemic disease because they show different symptomatologies and different therapeutic requirements, although prolonged inflammation undoubtedly plays a key role in their etiopathogenesis. Moreover, the lack of appropriate pharmacological therapy for neuroinflammation is believed to hamper the efficacy of pharmacotherapy when it is available. Based on the available data, the multidimensional analysis was carried out to answer the question of whether supporting the endogenous RoI process through the regulation/modulation of FPR activity, including the use of pro-resolving and anti-inflammatory ligands, may be a promising therapeutic option for treating these diseases.
6.1. RoI Deficits in Alzheimer’s Disease
Dementia is a clinical syndrome that develops as a result of neurodegenerative processes in the brain. Alzheimer’s disease (AD), the most common form of dementia worldwide, has been reported for more than 100 years thanks to the studies of the German neuropathologist and psychiatrist Alois Alzheimer [102]. According to the World Health Organization (WHO) estimates, at present, 50 million people suffer from AD worldwide, and 10 million new cases are recorded every year. It has been projected that the number of AD patients will reach 82 million in 2030 and 152 million in 2050 [103].
The major histopathological hallmarks of AD include β-amyloid (Aβ) plaque formation, neurofibrillary tangles (NFTs), and progressive neuronal loss. However, a third core feature of AD was recently identified; postmortem studies of AD patient brains demonstrated an exaggerated inflammatory response [104-114]. It should be mentioned that the first studies in the 1980s revealed the presence of immune cells and their mediators in the vicinity of β-amyloid plaques [107] and demonstrated that medications used for diseases such as rheumatoid arthritis exhibit protective potential against AD. Moreover, the risk of developing AD is reduced to 50% in patients treated with nonsteroidal anti-inflammatory drugs (NSAIDs) in the long term [115-117]. Therefore, the results of these investigations support the hypothesis that chronic immune activation plays a key role, although indirectly, in the development of AD.
In fact, undisputed evidence of immune system activation in the brains of AD patients comes from postmortem studies of patients that suffered recent head trauma; the Aβ level and the IL-1β level are increased 1–3 weeks postinjury. These changes lead to the further elevation of the production of β-amyloid precursor [118, 119]. Moreover, it has been shown that increased IL-1β levels induce the synthesis of other cytokines, including IL-6, which stimulates the activation of cyclin-dependent kinase 5 (CDK5), a kinase associated with tau phosphorylation [120]. Since CDK5 is an autophagy-regulating kinase and its dysfunction is crucial in the development of neurodegenerative disorders, autophagy-targeted therapeutic approaches can be considered novel therapeutic strategies for AD treatment [28]. Advanced studies have indicated that neuroinflammation in AD patients is mostly related to microglial activation [121-123] and elevated levels of proinflammatory cytokines not only in the brain but also in the serum [124-126]. At the same time, postmortem studies documented a downregulation of anti-inflammatory factor expression in the brain tissue [127]. Interestingly, some data indicated that β -amyloid-induced neuronal death and synaptic impairment are in part mediated by astrocyte activation induced by numerous factors, including free saturated fatty acids, pathogens, and oxidative stress. As in the case of microglial activation, the activation of astrocytes leads to the release of proinflammatory cytokines as well as cyclooxygenase-2 and the receptor for advanced glycation end products/nuclear factor-kB (NF-κB) axis activation. These observations suggest a role for advanced glycation end products in age-related cognitive changes as well as a potential beneficial role of nutraceuticals in the prevention of chronic neuroinflammation and AD-related pathology [128].
Notable hallmarks of AD indicate that prolonged neuroinflammation is a consequence of mitochondrial dysfunction. In fact, data demonstrated that, in AD, the activity of various mitochondria-localized enzymes is decreased and that glucose utilization in the brain is reduced, which may indirectly reflect a consequence of mitochondrial impairment and bioenergetic failure [129]. Furthermore, in many ways, mitochondria represent the remnants of proteobacteria, and they contain immunogenic molecules, including mitochondrial DNA (mtDNA), adenosine triphosphate, cardiolipin, cytochrome c, and formyl peptides; these molecules act mainly through FPR1, which displays a high affinity for bacterial- and mitochondrial-derived peptides and may potentiate the immunological response [130]. Moreover, mitochondrial lysates increase the mRNA and protein expression of β-amyloid precursor protein (APP). Considering these observations and the fact that data concerning the impact of brain mitochondria dysfunction on the FPR-mediated pathways are scarce, such studies should provide a new understanding of this field and are strongly recommended [131].
Recently, an increasing body of data seemed to indicate that excess inflammatory processes in the brains of AD patients are linked to disturbances in the RoI and that these deficits are intensified during the aging processes. This hypothesis appears to be supported by preclinical data obtained in Balb mice, in which aging was shown to be associated with dysfunctional RoI and a greater increase in and slower clearance of recruited neutrophils following acute inflammatory challenge, resulting in higher levels of proinflammatory cytokines and deficits in pro-resolving factor production [132]. Wang et al. demonstrated in senescence-accelerated mice prone 8 (SAMP8), which is a murine model of accelerated aging that spontaneously exhibits β-amyloid overproduction, tau hyperphosphorylation, oxidative stress damage, and cognitive decline [133], that aging is indeed associated with a proinflammatory state [134]. Furthermore, experimental data indicate that changes in FPR2 activation play a role in these mechanisms. For instance, SAMP8 mice show increases in receptor levels and inflammatory process aggravation. Deficits in the levels of SPM’s for this receptor have also been observed. Moreover, in 9-month-old SAMP8 mice, there is a reduced level of leukocyte-type 12-lipoxygenase, which is positively correlated with the elevation of tau phosphorylation at Ser202/Thr205 (AT8), in the hippocampus. This finding suggests that the disturbance of pro-resolving processes and changes in FPR2 activation may influence the formation of Aβ and tau pathology.
The most recent data obtained in transgenic mice demonstrate that the application of FPR2 agonists may be a new approach for AD therapy through modulating the RoI process and FPR2 activation. In this study, lipoxin and its analogue aspirin-triggered lipoxin A4 were used. In Tg2576 mice bearing the Swedish double mutation in the human amyloid precursor protein, which shows the accelerated development of Aβ-related pathologies, ATL treatment reduces NF-κB activation and proinflammatory cytokine levels and increases anti-inflammatory cytokine IL-10 levels, thus enhancing the activation of the alternative microglia phenotype [135]. This change in the microglial phenotype is correlated with decreased synaptotoxicity and improvement of cognitive functions in Tg2576 mice.
Dunn et al. [136] reported that, in triple transgenic AD (3xTg-AD) mice, a transgenic mouse strain that expresses the Aβ-processing related mutations APPswe and PS1M146V as well as mutant aggregation tau (tauP301L), the formation of senile plaques and neurofibrillary tangles is hastened, while the LXA4 level in the brain is declined. Interestingly, 8 weeks of treatment with ATL leads to the restoration of cognitive functions and a reduction in Aβ levels and tau phosphorylation [136].
It has also been recently observed that human CHME3 microglia incubated with Aβ42 [(compared with those stimulated with lipopolysaccharide (LPS)] exhibit reduced phosphorylation of 5-lipoxygenase [137], a key enzyme involved in the regulation of leukotriene and lipoxin production, at Ser523. This diminished phosphorylation at Ser523 induces higher production of leukotrienes and reduces the formation of lipoxin, and these results are correlated with disturbances in the resolution of inflammation. Interestingly, neither Aβ42 nor LPS alter LXA4 or RvD1 levels in the cell culture medium. Moreover, unlike LPS, which induces an increase in FPR2, Aβ42 does not influence the FPR2 level. Thus, it appears that, compared with the effects of a proinflammatory factor (e.g., LPS), Aβ42 formation is associated with changes that lead to the distortion of the pro-resolving processes and to the development of chronic inflammation (Fig. 6).
Fig (6).

The most important targets of pro-resolution factors in Alzheimer's disease. (1) Neurotoxicity, e.g., dysregulated neurotransmission, glutamate and calcium signaling; (2) microglia and astrocyte activation, which leads to irreversible inflammation and neurodegeneration; (3) the phosphorylation of tau and the formation of neurofibrillary tangles; (4) the formation of Aβ plaques. Proresolving molecules increase the resolution of spontaneous reactions. (5) Aβ plaques can lead to the accumulation and activation of microglia, resulting in an increase in the synthesis of proinflammatory cytokines such as IL-1β and TNF-α. These cytokines can lead to the hyperphosphorylation of tau and a pathological cycle; increased Aβ formation and the persistent activation of microglia ultimately lead to chronic neuroinflammation and neurodegeneration. This schematic was adapted from Filep et al., 2018 [183].
Although age is one of the most relevant risk factors of late-onset AD, researchers have only recently begun to estimate the impact of aging on the resolution of inflammation in humans. Gangemi et al. measured the level of LXA4 in the urine and the levels of proinflammatory leukotrienes in 30 healthy volunteers divided into three age groups [138]. Compared with the youngest group, both older age groups showed much lower levels of LXA4 in the urine. Furthermore, the LXA4-to-leukotriene ratio was considerably decreased in the older groups. These studies suggest that the ability of our innate systems to shift proinflammatory leukotriene production towards the formation of pro-resolving lipoxins from arachidonic acid declines with age in humans.
In 2014, Wang and coworkers carried out one of the first trials in patients. They evaluated SPM levels and receptor expression in human CSF and brain tissue and found that neurodegenerative disturbances are related to dysfunctional RoI [139]. LXA4 and RvD1 levels were measured in CSF from patients with AD, mild cognitive impairment (MCI), and subjective cognitive impairment (SCI). Interestingly, LXA4 levels in the CSF and hippocampal tissue were significantly lower in the AD group than in the MCI and SCI groups. The researchers noted no changes in RvD1 levels in both the hippocampus and CSF. It is worth noting that they observed a positive correlation between LXA4 and RvD1 levels in the CSF and cognitive efficiency, which was measured with the mini-mental state examination [139]. In the same study, immunohistochemical staining of hippocampal tissue from AD patients (compared with that from control subjects) revealed higher levels of SPM receptors, FPR2, and ChemR23 (a RvE1 receptor). The levels of 15-LOX-2, a key enzyme engaged in LXA4 production, were elevated, while those of IL-10 were reduced [140, 141]. The most recent studies by Zhu and coworkers provided additional evidence of disturbed RoI in AD patients [142]. SPM levels in the entorhinal cortex of AD patients and control subjects were assessed 18-21 hours postmortem. Compared with those of age-matched controls, the levels of SPM maresin-1, protectin-1, and resolvin D5 were reduced, and these changes were correlated with the dysfunction of the RoI and chronic inflammation.
Recently, it has been postulated that chronic inflammation and RoI dysfunction in elderly AD patients may be potentiated by infectious (bacterial, viral, or fungal) agents, leading to the increased production and deposition of β-amyloid plaques as well as neurofibrillary tangles [143]. In fact, persistent and chronic infections play a role in inducing and amplifying chronic inflammation in AD. Annexin A1, one of the multiple ligands of FPR2, is incorporated into the budding virus membrane of the influenza A virus (IAV). Therefore, once the IAV infects a host cell, FPR2 signaling is activated, leading to an increase in viral replication and the dysregulation of the host immune response and inflammatory response. Interestingly, preclinical studies have proven that FPR2 modulators efficiently protect mice against influenza infections by inhibiting viral replication and deleterious inflammation, which is responsible for tissue injury at later stages of infection. In combination with antiviral drugs (e.g., oseltamivir), FPR2 antagonists might also have a much stronger effect in blocking IAV replication [144]. Therefore, antiviral (e.g., oseltamivir) monotherapy, as well as combined treatment with FPR2 modulators, may provide a new approach for the prevention of AD and/or AD-like pathologies as well as RoI normalization [134, 145].
Unfortunately, the crucial limitation of these promising studies is the low stability and bioavailability of SPMs; therefore, we recently focused on identifying new ureidopropanamide compounds with better pharmacokinetic profiles and evaluating their efficacy for the modulation of FPR2 in transgenic Fpr2-/- knock-out (KO) mice. Therefore, we decided to compare the potential of new ureidopropanamide-based FPR2 agonists for modulating neurodegenerative and inflammatory changes in an ex vivo model of inflammation. We tested their ability to modulate lactate dehydrogenase (LDH) release and metabolic activity in hippocampal organotypic cultures (OHCs). In fact, we evaluated the impact of MR39 (Fig. 5) on LDH release stimulated by LPS and/or oligomeric Aβ1-42 in both C57BL/6J (wild-type mice - WT) and Fpr2-/- OHCs (unpublished data). In these conditions, we found that LPS stimulated LDH release in WT and Fpr2-/- cultures, while oligomeric Aβ1-42 potentiated LDH release only in the OHCs obtained from WT animals. Furthermore, co-stimulation (LPS + oligomeric Aβ1-42) did not potentiate the impact of LPS on the LDH level. Importantly, we observed the pro-resolving properties of MR39 (1 µM dose) in WT OHCs only.
These studies are very promising and will be continued because finding new alternative modulators that reduce the levels of endogenous SPMs and an approach for promoting RoI mechanisms seem to be fully justified and are anticipated.
6.2. Does Depression Result from RoI Deficits?
In contrast to the wealth of studies supporting the presence of RoI dysfunction in AD, data on abnormalities in SPMs function and/or metabolism in other psychiatric disorders, such as depression and schizophrenia, which are also characterized by neurodegenerative processes, are quite scarce. However, for many years, it has been postulated that the pathogeneses of these diseases are complex, and that, in addition to changes in certain neurotransmitters (e.g., serotonin), the dysfunction of the endocrine system and inflammatory responses, including neutrophil infiltration into the brain and the increased expression of proinflammatory mediators, play a key role [146, 147].
In this context, much attention has been focused on depression. In fact, the first reports that revealed the role of inflammatory processes in the pathogenesis of depression were published by Maes et al. (1995, 2009), who demonstrated increased levels of inflammatory biomarkers, such as proinflammatory cytokines, IL-1β, IL-6, TNF-α, and IFN-γ in depressed patients [148, 149]. Following these initial reports, other articles confirmed the involvement of inflammation in the pathogenesis of depression. Among them, a study by Zorilla and collaborators [150], as well as one by Dowlati and collaborators [151], showed increased levels of IL-6, TNF-α, and C-reactive protein (CRP) in blood samples from depressed patients. Clinical studies confirmed that, in patients suffering from depression, plasma and cerebrospinal fluid (CSF) levels of IL-1β are elevated and that there is a positive correlation between the serum concentration of this cytokine and depression severity [152, 153]. On the other hand, decreases in the levels of IL-4, TGF-β [154], and IL-10 were observed [155], which suggests the dysfunction of the anti-inflammatory response.
Moreover, studies in animal models of depression revealed changes in immune system function. For instance, in rats exposed to repeated intermittent LPS injections (a neurobehavioral model of chronic depression), thymus weight and the proliferative activity of lymphocytes are significantly reduced, and these changes are accompanied by changes in interferon- γ (IFN-γ) and IL-10 synthesis in the periphery [156]. Experiments in a mouse model of restraint stress have revealed a higher expression of IL-1β and TNF-α in the hippocampus [157]. Moreover, in an animal model of depression based on chronic mild stress (CMS), in which adult animals are exposed to stress, the levels of IL-1β and IL-6 in the brain and IL-6 and TNF-α levels in the serum are increased [158]. In line with these observations, using a prenatal stress model of depression, we demonstrated enhanced microglial activity, increased expression of neurotoxic factors, including proinflammatory cytokines and chemokines (IL-1β, TNF-α, IFN-γ, and CCL2) and deficits in the neuron-glial interaction (e.g., CX3CL1-CX3CR1 and CXCL12-CCR4) in adult rats [159]. It should be noted that changes in immune activity indicative of neuroinflammation are prolonged and present both in young and adult animals and precede the manifestation of depressive behavioral deficits.
Importantly, the intracerebral administration of proteins that possess beneficial homeostatic properties (including IGF-1 and CX3CL1) normalize not only neuroinflammation but also behavioral changes [160]. Therefore, it can be suggested that the immune response of the brain preserves its ability to shift from a proinflammatory to an anti-inflammatory response. Moreover, these observations may indicate changes in the RoI and the potential role of deficits in FPRs, which, as promiscuous receptors, can be activated by structurally diverse agonists that elicit proinflammatory or pro-resolving effects depending on chemical structure.
To date, only a few studies have shown the involvement of FPRs in behavioral changes. In fact, some findings indicated that FPRs can modulate anxiety and fear-elicited responses. It has been shown that Fpr1-/- knockout mice exhibit increased exploratory activity, reduced anxiety-like behavior, and impaired fear memory [161]. Furthermore, the role of glucocorticoids in the modulation of behavioral deficits has been confirmed, which suggests the potential for FPRs to influence the hypothalamic-pituitary-adrenal (HPA) axis. Other studies have shown that mice lacking Fpr2/3 show increased exploratory behavior and reduced fear compared to those of WT mice, while these behavioral changes can be partially mimicked by the FPR2 antagonist Boc2 [162]. Interestingly, in both Fpr2/3-/- mice and Boc2-treated mice, reduced immunostaining for the phospho-p38/MAPK pathway, a key FPR downstream signaling pathway, has been found [162]. It has been demonstrated that, in mice, changes in anxiety-related behavior may result from the genetic deletion of Fpr2/3. Therefore, it appears that further studies aimed at discovering the mechanisms of ligand action on FPRs responses may be essential and provide better insight into the regulation of FPRs activation and RoI promotion.
However, only limited data are available so far. Various reports postulated that resolvin D1 exerts anti-inflammatory and pro-resolving actions in animal models of depression based on peripheral inflammation [34]. Resolvin D1 and D2 have been used as antidepressants in an animal stress model (the chronic unpredictable stress model), and their effectiveness has been proven [163]. Furthermore, in an LPS-induced depression model, RvD1 alleviates endotoxin-evoked depression-like behaviors via anti-inflammatory actions, including the inhibition of neutrophil chemotaxis and the expression of proinflammatory mediators [164].
Recently, it has also been shown that the pro-resolving lipids RvD1 and RvE1 downregulate LPS-induced pro-inflammatory cytokine (TNF-α, IL-6, and IL-1β) gene expression in microglia [165]. These data clearly indicate that these lipids are involved in the RoI. It is crucial to note that the mechanisms of action of the two types of resolvins are distinct, as RvE1 regulates the NF-κB signaling pathway, while RvD1 regulates miRNA expression [165].
Therefore, it may be postulated that SPMs represent promising novel therapeutic agents that are able to potentiate the RoI in the brain; however, their role in mood disorders remains to be investigated. The anti-inflammatory effects of docosahexaenoic acids and their derivatives in microglial cells were recently analyzed in a special review [166].
Based on these promising reports indicating the usefulness of SPMs, in our preliminary studies, we used ureidopropanamide FPR2 agonists to modulate the proinflammatory activation of glial cells observed in a prenatal stress model. The results obtained thus far indicate that, in primary microglia cultures, FPR2 agonists reduce LPS-induced cell mortality and lower LPS-induced proinflammatory cytokine levels, suggesting pro-resolving and anti-inflammatory properties. Moreover, the observed effects can be partly reversed by pretreatment with an FPR2 antagonist (unpublished data). We found that, in hippocampal organotypic cultures obtained from prenatally stressed animals, the expression of both FPR2 and proinflammatory cytokine genes is upregulated (unpublished data). However, these interesting findings undoubtedly require further confirmation in in vivo models, particularly in drug-resistant depression in which coexistent neuroinflammation has been described, for the development of a new supportive therapeutic strategy.
6.3. FPR as a Target for Brain Ischemia
Stroke remains the second leading cause of death and the main cause of long-lasting disability in adults despite enormous efforts in searching for efficient therapy [167, 168]. In general, two types of stroke, namely, hemorrhagic and ischemic stroke, have been identified; the latter accounts for 87% of all cases [169]. There are many causes of stroke, including cardiovascular disease, atherosclerosis of large vessels, atherosclerotic plaque rupture, and infarctions caused by the blockade of a deep artery [170].
Excitotoxicity, the distortion of the ionic balance, oxidative stress, and apoptosis are among the mechanisms that lead to cell death during brain ischemia. Their progression and intensity can slightly differ from case to case. On the other hand, it is known that these phenomena impair the function of neurons, glial cells, and blood vessels. Neurons, particularly those from the CA1 area of the hippocampus and from the cortex, cerebellar Purkinje cells, and oligodendrocytes appear to be more prone to injury and cell death than glial or endothelial cells [171]. Many studies demonstrated that inflammation plays an important role in the pathogenesis of ischemic stroke, while the extent of the affected area and ischemia duration are crucial factors related to outcomes. Since this inflammation is not induced by any specific immunogens, this inflammatory response is often termed sterile inflammation [172]. This process is characterized by the fast activation of microglial cells, the formation of pro-inflammatory mediators, including proinflammatory cytokines and chemokines, and the multistage infiltration of inflammatory cells, i.e., microglial cells, leukocytes, neutrophils, lymphocytes T, monocytes/macrophages, to the damaged nervous tissue. These events contribute to the aggravation of cell death [173, 174]. In tissue affected by ischemia, local inflammation involves astrocytes, activated microglia, and infiltrating monocytes and macrophages. Additional damage is generated by reactive oxygen species and the presence of other cells implicated in the inflammatory response (e.g., dendritic cells) [175].
It should be emphasized that, in contrast to the invariably deleterious effects of ischemia, cytokines and chemokines can play a dual role, and the final effect depends on which and how many specific mediators are produced, where they are released and at what time during ischemia or reperfusion they appear. In general, it is thought that TNF-α, IL-6, and IL-1β have emerged as central players in the orchestration of mediator responses. Next, during reperfusion, while efficient RoI processes are preserved, anti-inflammatory markers, including CXCL13, Ym1, TGFβ, and CD163, are expressed [174].
The most recent studies suggested a significant role for FPR ligands in the mechanisms that maintain the balance between proinflammatory and anti-inflammatory pathways to retain homeostasis following ischemic insult. Ligands such as AnxA1 and its mimetic peptides, e.g., N-terminal-derived Ac2-26, are of particular interest [176, 177]. The prevailing view is that AnxA1 regulates cell apoptosis, proliferation, and differentiation [178] but also diminishes leukocyte adhesion and migration, thus inhibiting proinflammatory cytokine release under ischemic conditions [175, 179]. Recent studies also demonstrated that the AnxA1/FPR axis plays a significant role in sealing the BBB in neonatal hypoxic-ischemic encephalopathy [180]. It is well known that the BBB is a critical gateway of communication between the periphery and the brain and that ischemia leads to BBB breakdown. Interestingly, neurodegenerative disorders, such as AD and amyotrophic lateral sclerosis (ALS), are associated with microvascular dysfunction and/or degeneration in the brain, neurovascular disintegration, defective BBB function, and vascular factors. BBB disruption has also been observed in postmortem studies of depressed patients. Therefore, the role of endogenous pro-resolving molecules, such as AnxA1, and their impact on FPRs may have a wider biological significance, which undoubtedly should be a subject of further research (Fig. 7).
Fig. (7).
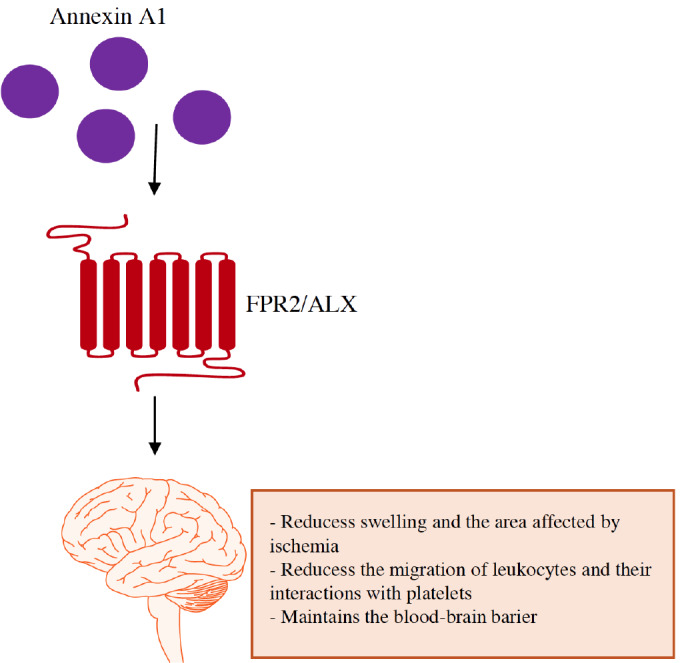
Annexin A1 and its peptide mimetics are possible therapeutic strategies for the treatment of ischemia based on their ability to regulate inflammation to lead to the resolution of inflammation, decreased tissue injury, and the promotion of resolution.
Transgenic mouse models are particularly useful for studying the role of FPRs in brain injury. In fact, such studies confirmed the involvement of mouse FPR2 and/or FPR3 in circulating neutrophils in mediating AnxA1-induced protection [179, 181]. A study by Vital et al. indicated that Fpr2/3-/- KO mice show an exaggerated inflammatory reaction to brain ischemia that involves the increased infiltration of leukocytes and platelets. An exacerbated cerebral inflammatory response as a consequence of the lack of FPR2 and FPR3 has also been demonstrated by other studies [179]. These studies suggested that FPR activity is crucial for the brain ischemia-induced inflammatory response [182]. On the other hand, the use of endogenous FPR2/3 agonists, such as AnxA1, after nervous tissue damage produces a neuroprotective effect by reducing detrimental effects of brain ischemia, including infarct volume, leukocyte adhesion, and inflammatory marker expression in the middle cerebral artery occlusion model. It appears that microglial cells contribute to these effects because, in addition to blood vessels, they are the main cells exhibiting high FPR2 expression in mice.
Bearing in mind that most of the available literature data derive from the studies with endogenous agonists of FPRs, it should be mentioned that there are several reports in which synthetic ligands, characterized by different stability and bioavailability, have been recently published. As an example, synthetic FPR2 ligands have been studied in models of cardioprotection, e.g., CGEN-855A [97]. They demonstrated that FPR1/FPR2 biased agonism can be crucial for the cardioprotective efficacy in the treatment of myocardial ischemia-reperfusion.
We performed preliminary experiments to assess the effects of FPR2 agonists in models of ischemia. In organotypic hippocampal cultures, oxygen-glucose deprivation (OGD), which is considered an in vitro model of ischemia, leads to the elevated expression of FPR2, while toxic OGD-induced effects can be modulated by FPR2 agonists (unpublished data). In light of the abovementioned data, it appears that there is still a need to support the RoI process to stimulate a shift between proinflammatory and anti-inflammatory pathways following an ischemic insult. Therefore, a better understanding of the basic mechanisms underlying FPR2 modulation may provide new potential therapeutic options for the treatment of ischemic diseases.
CONCLUSION
Neuroinflammation plays a dual role in the brain. On the one hand, transient inflammation terminated by a proper resolution process is beneficial and usually elicits neuroprotective effects. On the other hand, the persistence of this process is the cause of chronic inflammation, which leads to long-lasting disturbances in homeostasis and to the development of permanent neurotoxic or neurodegenerative changes that become an underlying cause of CNS diseases. In such circumstances, classic anti-inflammatory therapies do not induce beneficial effects. In addition, classic anti-inflammatory drugs, by strongly suppressing the pro-inflammatory response, fail to balance proinflammatory and anti-inflammatory components. Thus, the response to future insults that require immune activation is hampered, and the body is rendered defenseless, a condition that may be life-threatening. Therefore, a novel and innovative approach to modulating the inflammatory response is needed. This perspective article illustrates how a resolution-based strategy can offer new therapeutic opportunities, especially for CNS disorders characterized by prolonged neuroinflammation. Based on the data included in this review, FPRs, and FPR2, in particular, are potential novel targets for such therapeutic strategies. The characterization of the pharmacological properties of FPR2 agonists in both preclinical and clinical trials strongly suggests a prominent anti-inflammatory role in the context of the innate immune response and supports the possibility of developing a resolution-based approach. Lipid SPMs have been proven to possess particularly beneficial properties because they are able to exert both pro-resolving and anti-inflammatory actions. Hence, the search for ligands characterized by an adequate pharmacological profile and bioavailability, which may become widely used to promote endogenous the RoI through FPR activation, appears justified.
We hope that this narrative review has shed more light on the current knowledge of the role of FPRs in the RoI in brain disorders that are different in terms of clinical manifestation but are similar with respect to the involvement of neuroinflammation.
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- 7-TM
7-transmembrane receptors
- AA
Arachidonic acid
- AD
Alzheimer’s disease
- AnxA1
Annexin A1
- APP
β-amyloid precursor protein
- AT-LXA4
Aspirin-triggered lipoxins
- Aβ
β-amyloid plaques
- BBB
Blood-brain barrier
- bFGF
Basic fibroblast growth factor
- BGF
Brain growth factor
- cAMP
Cyclic adenosine monophosphate
- CDK5
Cyclin dependent kinase 5
- ChemR23
Chemokine-like receptor 1
- CNS
Central nervous system
- COX-2
Cyclooxygenase 2
- CSF
Cerebrospinal fluid
- DHA
Docosahexaenoic acid
- EPA
3-eicosapentaenoic acid
- ERK 1/2
Extracellular signal–regulated kinases
- fMLP
N-formyl-methionyl-leucylphenylalanine
- FPR2/ALX
Formyl peptide receptor/lipoxin A4 receptor
- GPCRs
G-protein-coupled receptors
- HPA
Hypothalamic-pituitary-adrenal
- IAV
Influenza A viruses
- IFN
Interferon
- IL
Interleukin
- KO
Knock-out mice
- LDH
Lactate dehydrogenase
- LOX
Lipooxygenase
- LPS
Lipopolysaccharide
- LTB4R or BLT1
Leukotriene B4 receptor
- LXA4
Lipoxin A4
- MARCO
Macrophage scavenger receptor
- MHC
Major histocompatibility complex
- NF-κB
Nuclear factor-kB
- NGF
Neural growth factor
- NO
Nitric oxide
- OGD
Oxygen-glucose deprivation
- PAMP
Pathogen-associated molecular patterns
- PRR
Pattern-recognition receptors
- RoI
Resolution of inflammation
- ROS
Reactive oxygen species
- RvD1
Resolvin D1
- SAA
Serum amyloid A
- SAMP8
Senescence-accelerated mice prone 8
- SPMs
Specialized pro-resolving mediators
- TGF
Transforming growth factor
- TNF
Tumor necrosis factor
- WT
Wild-type mice
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by a grant from the Alzheimer’s Association (AARG-NTF-18-565227) and by the Polish National Science Centre, grant no. 2017/26/M/NZ7/01048. Natalia Bryniarska is a holder of a scholarship from POWER no. POWR.03.02.00-00-I013/16.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Gallin J.I., Goldstein I.M., Snyderman R., editors. Inflammation: Basic principles and clinical correlates. New York: Raven Press; 1992. [Google Scholar]
- 3.Ortega-Gómez A., Perretti M., Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol. Med. 2013;5(5):661–674. doi: 10.1002/emmm.201202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serhan C.N. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol. Aspects Med. 2017;58:1–11. doi: 10.1016/j.mam.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan C., Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 6.Murakami M., Hirano T. The molecular mechanisms of chronic inflammation development. Front. Immunol. 2012;3:323. doi: 10.3389/fimmu.2012.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones H.R., Robb C.T., Perretti M., Rossi A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016;28(2):137–145. doi: 10.1016/j.smim.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Fox S., Leitch A.E., Duffin R., Haslett C., Rossi A.G. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2010;2(3):216–227. doi: 10.1159/000284367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feghali C.A., Wright T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997;2:d12–d26. doi: 10.2741/A171. [DOI] [PubMed] [Google Scholar]
- 10.Lintermans L.L., Stegeman C.A., Heeringa P., Abdulahad W.H. T cells in vascular inflammatory diseases. Front. Immunol. 2014;5:504. doi: 10.3389/fimmu.2014.00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 12.Ashley N.T., Weil Z.M., Nelson R. J. Inflammation: Mechanisms, Costs, and Natural Variation. Annu. Rev. Ecol. Evol. Syst. 2012;43:385–406. doi: 10.1146/annurev-ecolsys-040212-092530. [DOI] [Google Scholar]
- 13.Schett G., Neurath M.F. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat. Commun. 2018;9(1):3261. doi: 10.1038/s41467-018-05800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leszek J., Barreto G.E., Gąsiorowski K., Koutsouraki E., Ávila-Rodrigues M., Aliev G. Inflammatory mechanisms and oxidative stress as key factors responsible for progression of neurodegeneration: Role of brain innate immune system. CNS Neurol. Disord. Drug Targets. 2016;15(3):329–336. doi: 10.2174/1871527315666160202125914. [DOI] [PubMed] [Google Scholar]
- 15.Graeber M.B. Neuroinflammation: no rose by any other name. Brain Pathol. 2014;24(6):620–622. doi: 10.1111/bpa.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abushouk A.I., Negida A., Ahmed H., Abdel-Daim M.M. Neuroprotective mechanisms of plant extracts against MPTP induced neurotoxicity: Future applications in Parkinson’s disease. Biomed. Pharmacother. 2017;85:635–645. doi: 10.1016/j.biopha.2016.11.074. [DOI] [PubMed] [Google Scholar]
- 17.Yang Q.Q., Zhou J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia. 2019;67(6):1017–1035. doi: 10.1002/glia.23571. [DOI] [PubMed] [Google Scholar]
- 18.Mariani M.M., Kielian T. Microglia in infectious diseases of the central nervous system. J. Neuroimmune Pharmacol. 2009;4(4):448–461. doi: 10.1007/s11481-009-9170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michell-Robinson M.A., Touil H., Healy L.M., Owen D.R., Durafourt B.A., Bar-Or A., Antel J.P., Moore C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain. 2015;138(Pt 5):1138–1159. doi: 10.1093/brain/awv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kierdorf K., Prinz M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bylicky M.A., Mueller G.P., Day R.M. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid. Med. Cell. Longev. 2018;2018: 6501031. doi: 10.1155/2018/6501031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu W., Tang Y., Feng J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011;89(5-6):141–146. doi: 10.1016/j.lfs.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Hu S., Sheng W.S., Ehrlich L.C., Peterson P.K., Chao C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7(3):153–159. doi: 10.1159/000026433. [DOI] [PubMed] [Google Scholar]
- 24.Thornton P., Pinteaux E., Gibson R.M., Allan S.M., Rothwell N.J. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J. Neurochem. 2006;98(1):258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- 25.Boutin H., LeFeuvre R.A., Horai R., Asano M., Iwakura Y., Rothwell N.J. Role of IL-1alpha and IL-1beta in ischemic brain damage. J. Neurosci. 2001;21(15):5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Cunha A., Jefferson J.A., Jackson R.W., Vitković L. Glial cell-specific mechanisms of TGF-beta 1 induction by IL-1 in cerebral cortex. J. Neuroimmunol. 1993;42(1):71–85. doi: 10.1016/0165-5728(93)90214-J. [DOI] [PubMed] [Google Scholar]
- 27.Abbott N.J., Rönnbäck L., Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 28.Uddin M.S., Stachowiak A., Mamun A.A., Tzvetkov N.T., Takeda S., Atanasov A.G., Bergantin L.B., Abdel-Daim M.M., Stankiewicz A.M. Autophagy and Alzheimer's Disease: From Molecular Mechanisms to Therapeutic Implications. Front Aging Neurosci. 2018;30:10–04. doi: 10.3389/fnagi.2018.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buckley C.D., Gilroy D.W., Serhan C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corminboeuf O., Leroy X. FPR2/ALXR agonists and the resolution of inflammation. J. Med. Chem. 2015;58(2):537–559. doi: 10.1021/jm501051x. [DOI] [PubMed] [Google Scholar]
- 31.Serhan C.N., Brain S.D., Buckley C.D., Gilroy D.W., Haslett C., O’Neill L.A., Perretti M., Rossi A.G., Wallace J.L. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21(2):325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stables M.J., Gilroy D.W. Old and new generation lipid mediators in acute inflammation and resolution. Prog. Lipid Res. 2011;50(1):35–51. doi: 10.1016/j.plipres.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Levy B.D., Clish C.B., Schmidt B., Gronert K., Serhan C.N. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 34.Serhan C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perretti M., Leroy X., Bland E.J., Montero-Melendez T. Resolution pharmacology: Opportunities for therapeutic innovation in inflammation. Trends Pharmacol. Sci. 2015;36(11):737–755. doi: 10.1016/j.tips.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Headland S.E., Norling L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015;27(3):149–160. doi: 10.1016/j.smim.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 37.Fiorucci S., Distrutti E., Mencarelli A., Rizzo G., Lorenzo A.R., Baldoni M., Del Soldato P., Morelli A., Wallace J.L. Cooperation between aspirin-triggered lipoxin and nitric oxide (NO) mediates antiadhesive properties of 2-(Acetyloxy)benzoic acid 3-(nitrooxymethyl)phenyl ester (NCX-4016) (NO-aspirin) on neutrophil-endothelial cell adherence. J. Pharmacol. Exp. Ther. 2004;309(3):1174–1182. doi: 10.1124/jpet.103.063651. [DOI] [PubMed] [Google Scholar]
- 38.Petasis N.A., Akritopoulou-Zanze I., Fokin V.V., Bernasconi G., Keledjian R., Yang R., Uddin J., Nagulapalli K.C., Serhan C.N. Design, synthesis and bioactions of novel stable mimetics of lipoxins and aspirin-triggered lipoxins. Prostaglandins Leukot. Essent. Fatty Acids. 2005;73(3-4):301–321. doi: 10.1016/j.plefa.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 39.Mitchell S., Thomas G., Harvey K., Cottell D., Reville K., Berlasconi G., Petasis N.A., Erwig L., Rees A.J., Savill J., Brady H.R., Godson C. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 2002;13(10):2497–2507. doi: 10.1097/01.ASN.0000032417.73640.72. [DOI] [PubMed] [Google Scholar]
- 40.Nigam S.K. Subcellular distribution of small GTP binding proteins in pancreas: identification of small GTP binding proteins in the rough endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 1990;87(4):1296–1299. doi: 10.1073/pnas.87.4.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiore S., Ryeom S.W., Weller P.F., Serhan C.N. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J. Biol. Chem. 1992;267(23):16168–16176. [PubMed] [Google Scholar]
- 42.Fiore S., Maddox J.F., Perez H.D., Serhan C.N. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 1994;180(1):253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bäck M., Powell W.S., Dahlén S.E., Drazen J.M., Evans J.F., Serhan C.N., Shimizu T., Yokomizo T., Rovati G.E. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br. J. Pharmacol. 2014;171(15):3551–3574. doi: 10.1111/bph.12665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye R.D., Boulay F., Wang J.M., Dahlgren C., Gerard C., Parmentier M., Serhan C.N., Murphy P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009;61(2):119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forsman H., Dahlgren C. Lipoxin A(4) metabolites/analogues from two commercial sources have no effects on TNF-alpha-mediated priming or activation through the neutrophil formyl peptide receptors. Scand. J. Immunol. 2009;70(4):396–402. doi: 10.1111/j.1365-3083.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- 46.Forsman H., Önnheim K., Andreasson E., Dahlgren C. What formyl peptide receptors, if any, are triggered by compound 43 and lipoxin A4? Scand. J. Immunol. 2011;74(3):227–234. doi: 10.1111/j.1365-3083.2011.02570.x. [DOI] [PubMed] [Google Scholar]
- 47.Hanson J., Ferreirós N., Pirotte B., Geisslinger G., Offermanns S. Heterologously expressed formyl peptide receptor 2 (FPR2/ALX) does not respond to lipoxin A4. Biochem. Pharmacol. 2013;85(12):1795–1802. doi: 10.1016/j.bcp.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 48.Planagumà A., Domenech T., Jover I., Ramos I., Sentellas S., Malhotra R., Miralpeix M. Lack of activity of 15-epi-lipoxin A4 on FPR2/ALX and CysLT1 receptors in interleukin-8-driven human neutrophil function. Clin. Exp. Immunol. 2013;173(2):298–309. doi: 10.1111/cei.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bianchi M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 50.Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 1982;155(1):264–275. doi: 10.1084/jem.155.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker E.L., Forouhar F.A., Grunnet M.L., Boulay F., Tardif M., Bormann B.J., Sodja D., Ye R.D., Woska J.R., Jr, Murphy P.M. Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 1998;292(1):129–135. doi: 10.1007/s004410051042. [DOI] [PubMed] [Google Scholar]
- 53.Cattaneo F., Guerra G., Ammendola R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010;35(12):2018–2026. doi: 10.1007/s11064-010-0301-5. [DOI] [PubMed] [Google Scholar]
- 54.Lacy M., Jones J., Whittemore S.R., Haviland D.L., Wetsel R.A., Barnum S.R. Expression of the receptors for the C5a anaphylatoxin, interleukin-8 and FMLP by human astrocytes and microglia. J. Neuroimmunol. 1995;61(1):71–78. doi: 10.1016/0165-5728(95)00075-D. [DOI] [PubMed] [Google Scholar]
- 55.Müller-Ladner U., Jones J.L., Wetsel R.A., Gay S., Raine C.S., Barnum S.R. Enhanced expression of chemotactic receptors in multiple sclerosis lesions. J. Neurol. Sci. 1996;144(1-2):135–141. doi: 10.1016/S0022-510X(96)00217-1. [DOI] [PubMed] [Google Scholar]
- 56.Yao J., Harvath L., Gilbert D.L., Colton C.A. Chemotaxis by a CNS macrophage, the microglia. J. Neurosci. Res. 1990;27(1):36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]
- 57.Cui Y.H., Le Y., Gong W., Proost P., Van Damme J., Murphy W.J., Wang J.M. Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J. Immunol. 2002;168(1):434–442. doi: 10.4049/jimmunol.168.1.434. [DOI] [PubMed] [Google Scholar]
- 58.Hilger D., Masureel M., Kobilka B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018;25(1):4–12. doi: 10.1038/s41594-017-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fredriksson R., Lagerström M.C., Lundin L.G., Schiöth H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63(6):1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 60.Perez D.M. The evolutionarily triumphant G-protein-coupled receptor. Mol. Pharmacol. 2003;63(6):1202–1205. doi: 10.1124/mol.63.6.1202. [DOI] [PubMed] [Google Scholar]
- 61.Venkatakrishnan A.J., Deupi X., Lebon G., Tate C.G., Schertler G.F., Babu M.M. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494(7436):185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 62.Sensoy O., Weinstein H. A mechanistic role of Helix 8 in GPCRs: Computational modeling of the dopamine D2 receptor interaction with the GIPC1-PDZ-domain. Biochim. Biophys. Acta. 2015;1848(4):976–983. doi: 10.1016/j.bbamem.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katritch V., Cherezov V., Stevens R.C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 2012;33(1):17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He R., Browning D.D., Ye R.D. Differential roles of the NPXXY motif in formyl peptide receptor signaling. J. Immunol. 2001;166(6):4099–4105. doi: 10.4049/jimmunol.166.6.4099. [DOI] [PubMed] [Google Scholar]
- 65.Bennett T.A., Maestas D.C., Prossnitz E.R. Arrestin binding to the G protein-coupled N-formyl peptide receptor is regulated by the conserved “DRY” sequence. J. Biol. Chem. 2000;275(32):24590–24594. doi: 10.1074/jbc.C000314200. [DOI] [PubMed] [Google Scholar]
- 66.Boulay F., Tardif M., Brouchon L., Vignais P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem. Biophys. Res. Commun. 1990;168(3):1103–1109. doi: 10.1016/0006-291X(90)91143-G. [DOI] [PubMed] [Google Scholar]
- 67.Boulay F., Tardif M., Brouchon L., Vignais P. The human N-formylpeptide receptor. Characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry. 1990;29(50):11123–11133. doi: 10.1021/bi00502a016. [DOI] [PubMed] [Google Scholar]
- 68.Ye R.D., Cavanagh S.L., Quehenberger O., Prossnitz E.R., Cochrane C.G. Isolation of a cDNA that encodes a novel granulocyte N-formyl peptide receptor. Biochem. Biophys. Res. Commun. 1992;184(2):582–589. doi: 10.1016/0006-291X(92)90629-Y. [DOI] [PubMed] [Google Scholar]
- 69.Murphy P.M., Ozçelik T., Kenney R.T., Tiffany H.L., McDermott D., Francke U. A structural homologue of the N-formyl peptide receptor. Characterization and chromosome mapping of a peptide chemoattractant receptor family. J. Biol. Chem. 1992;267(11):7637–7643. [PubMed] [Google Scholar]
- 70.Bao L., Gerard N.P., Eddy R.L., Jr, Shows T.B., Gerard C. Mapping of genes for the human C5a receptor (C5AR), human FMLP receptor (FPR), and two FMLP receptor homologue orphan receptors (FPRH1, FPRH2) to chromosome 19. Genomics. 1992;13(2):437–440. doi: 10.1016/0888-7543(92)90265-T. [DOI] [PubMed] [Google Scholar]
- 71.Muto Y., Guindon S., Umemura T., Kőhidai L., Ueda H. Adaptive evolution of formyl peptide receptors in mammals. J. Mol. Evol. 2015;80(2):130–141. doi: 10.1007/s00239-015-9666-z. [DOI] [PubMed] [Google Scholar]
- 72.Rabiet M.J., Macari L., Dahlgren C., Boulay F. N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J. Biol. Chem. 2011;286(30):26718–26731. doi: 10.1074/jbc.M111.244590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alvarez V., Coto E., Setién F., González-Roces S., López-Larrea C. Molecular evolution of the N-formyl peptide and C5a receptors in non-human primates. Immunogenetics. 1996;44(6):446–452. doi: 10.1007/BF02602806. [DOI] [PubMed] [Google Scholar]
- 74.Migeotte I., Communi D., Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17(6):501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 75.Becker E.L., Forouhar F.A., Grunnet M.L., Boulay F., Tardif M., Bormann B.J., Sodja D., Ye R.D., Woska J.R., Jr, Murphy P.M. Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 1998;292(1):129–135. doi: 10.1007/s004410051042. [DOI] [PubMed] [Google Scholar]
- 76.Le Y., Hu J., Gong W., Shen W., Li B., Dunlop N.M., Halverson D.O., Blair D.G., Wang J.M. Expression of functional formyl peptide receptors by human astrocytoma cell lines. J. Neuroimmunol. 2000;111(1-2):102–108. doi: 10.1016/S0165-5728(00)00373-8. [DOI] [PubMed] [Google Scholar]
- 77.Gao J.L., Lee E.J., Murphy P.M. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J. Exp. Med. 1999;189(4):657–662. doi: 10.1084/jem.189.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu M., Chen K., Yoshimura T., Liu Y., Gong W., Wang A., Gao J.L., Murphy P.M., Wang J.M. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2012;2:786. doi: 10.1038/srep00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hartt J.K., Barish G., Murphy P.M., Gao J.L. N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two N-formylpeptide receptor (FPR) subtypes. Molecular characterization of FPR2, a second mouse neutrophil FPR. J. Exp. Med. 1999;190(5):741–747. doi: 10.1084/jem.190.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He H.Q., Liao D., Wang Z.G., Wang Z.L., Zhou H.C., Wang M.W., Ye R.D. Functional characterization of three mouse formyl peptide receptors. Mol. Pharmacol. 2013;83(2):389–398. doi: 10.1124/mol.112.081315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang T.S., Wang J.M., Murphy P.M., Gao J.L. Serum amyloid A is a chemotactic agonist at FPR2, a low-affinity N-formylpeptide receptor on mouse neutrophils. Biochem. Biophys. Res. Commun. 2000;270(2):331–335. doi: 10.1006/bbrc.2000.2416. [DOI] [PubMed] [Google Scholar]
- 82.Tiffany H.L., Lavigne M.C., Cui Y.H., Wang J.M., Leto T.L., Gao J.L., Murphy P.M. Amyloid-beta induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J. Biol. Chem. 2001;276(26):23645–23652. doi: 10.1074/jbc.M101031200. [DOI] [PubMed] [Google Scholar]
- 83.Gao J.L., Guillabert A., Hu J., Le Y., Urizar E., Seligman E., Fang K.J., Yuan X., Imbault V., Communi D., Wang J.M., Parmentier M., Murphy P.M., Migeotte I. F2L, a peptide derived from heme-binding protein, chemoattracts mouse neutrophils by specifically activating Fpr2, the low-affinity N-formylpeptide receptor. J. Immunol. 2007;178(3):1450–1456. doi: 10.4049/jimmunol.178.3.1450. [DOI] [PubMed] [Google Scholar]
- 84.Migeotte I., Riboldi E., Franssen J.D., Grégoire F., Loison C., Wittamer V., Detheux M., Robberecht P., Costagliola S., Vassart G., Sozzani S., Parmentier M., Communi D. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J. Exp. Med. 2005;201(1):83–93. doi: 10.1084/jem.20041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tiffany H.L., Gao J.L., Roffe E., Sechler J.M., Murphy P.M. Characterization of Fpr-rs8, an atypical member of the mouse formyl peptide receptor gene family. J. Innate Immun. 2011;3(5):519–529. doi: 10.1159/000327718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takano T., Fiore S., Maddox J.F., Brady H.R., Petasis N.A., Serhan C.N. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J. Exp. Med. 1997;185(9):1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Z.G., Ye R.D. Characterization of two new members of the formyl peptide receptor gene family from 129S6 mice. Gene. 2002;299(1-2):57–63. doi: 10.1016/S0378-1119(02)01012-0. [DOI] [PubMed] [Google Scholar]
- 88.Cooray S.N., Gobbetti T., Montero-Melendez T., McArthur S., Thompson D., Clark A.J., Flower R.J., Perretti M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA. 2013;110(45):18232–18237. doi: 10.1073/pnas.1308253110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu Z.Q., Zhang X., Scott L. Regulation of G protein-coupled receptor trafficking. Acta Physiol. (Oxf.) 2007;190(1):39–45. doi: 10.1111/j.1365-201X.2007.01695.x. [DOI] [PubMed] [Google Scholar]
- 90.Maaty W.S., Lord C.I., Gripentrog J.M., Riesselman M., Keren-Aviram G., Liu T., Dratz E.A., Bothner B., Jesaitis A.J. Identification of C-terminal phosphorylation sites of N-formyl peptide receptor-1 (FPR1) in human blood neutrophils. J. Biol. Chem. 2013;288(38):27042–27058. doi: 10.1074/jbc.M113.484113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Filep J.G. Biasing the lipoxin A4/formyl peptide receptor 2 pushes inflammatory resolution. Proc. Natl. Acad. Sci. USA. 2013;110(45):18033–18034. doi: 10.1073/pnas.1317798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brandenburg L.O., Konrad M., Wruck C.J., Koch T., Lucius R., Pufe T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1-42-induced signal transduction in glial cells. J. Neurochem. 2010;113(3):749–760. doi: 10.1111/j.1471-4159.2010.06637.x. [DOI] [PubMed] [Google Scholar]
- 93.Raabe C.A., Gröper J., Rescher U. Biased perspectives on formyl peptide receptors. Biochim. Biophys. Acta Mol. Cell Res. 2019;1866(2):305–316. doi: 10.1016/j.bbamcr.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 94.Chiang N., Fierro I.M., Gronert K., Serhan C.N. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 2000;191(7):1197–1208. doi: 10.1084/jem.191.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bena S., Brancaleone V., Wang J.M., Perretti M., Flower R.J. Annexin A1 interaction with the FPR2/ALX receptor: identification of distinct domains and downstream associated signaling. J. Biol. Chem. 2012;287(29):24690–24697. doi: 10.1074/jbc.M112.377101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schepetkin I.A., Khlebnikov A.I., Giovannoni M.P., Kirpotina L.N., Cilibrizzi A., Quinn M.T. Development of small molecule non-peptide formyl peptide receptor (FPR) ligands and molecular modeling of their recognition. Curr. Med. Chem. 2014;21(13):1478–1504. doi: 10.2174/0929867321666131218095521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qin C.X., May L.T., Li R., Cao N., Rosli S., Deo M., Alexander A.E., Horlock D., Bourke J.E., Yang Y.H., Stewart A.G., Kaye D.M., Du X.J., Sexton P.M., Christopoulos A., Gao X.M., Ritchie R.H. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 2017;8:14232. doi: 10.1038/ncomms14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schepetkin I.A., Kirpotina L.N., Khlebnikov A.I., Leopoldo M., Lucente E., Lacivita E., De Giorgio P., Quinn M.T. 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: molecular modeling of chiral recognition by FPR2. Biochem. Pharmacol. 2013;85(3):404–416. doi: 10.1016/j.bcp.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lacivita E., Schepetkin I.A., Stama M.L., Kirpotina L.N., Colabufo N.A., Perrone R., Khlebnikov A.I., Quinn M.T., Leopoldo M. Novel 3-(1H-indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]propanamides as selective agonists of human formyl-peptide receptor 2. Bioorg. Med. Chem. 2015;23(14):3913–3924. doi: 10.1016/j.bmc.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stama M.L., Ślusarczyk J., Lacivita E., Kirpotina L.N., Schepetkin I.A., Chamera K., Riganti C., Perrone R., Quinn M.T., Basta-Kaim A., Leopoldo M. Novel ureidopropanamide based N-formyl peptide receptor 2 (FPR2) agonists with potential application for central nervous system disorders characterized by neuroinflammation. Eur. J. Med. Chem. 2017;141:703–720. doi: 10.1016/j.ejmech.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clish C.B., Levy B.D., Chiang N., Tai H.H., Serhan C.N. Oxidoreductases in lipoxin A4 metabolic inactivation: a novel role for 15-onoprostaglandin 13-reductase/leukotriene B4 12-hydroxydehydrogenase in inflammation. J. Biol. Chem. 2000;275(33):25372–25380. doi: 10.1074/jbc.M002863200. [DOI] [PubMed] [Google Scholar]
- 102.Alzheimer A., Stelzmann R.A., Schnitzlein H.N., Murtagh F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde. Clin. Anat. 1995;8(6):429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 103.Doody R.S., Farlow M., Aisen P.S. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N. Engl. J. Med. 2014;370(15):1460. doi: 10.1056/NEJMc1402193. [DOI] [PubMed] [Google Scholar]
- 104.Akama K.T., Van Eldik L.J. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J. Biol. Chem. 2000;275(11):7918–7924. doi: 10.1074/jbc.275.11.7918. [DOI] [PubMed] [Google Scholar]
- 105.Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G.M., Cooper N.R., Eikelenboom P., Emmerling M., Fiebich B.L., Finch C.E., Frautschy S., Griffin W.S., Hampel H., Hull M., Landreth G., Lue L., Mrak R., Mackenzie I.R., McGeer P.L., O’Banion M.K., Pachter J., Pasinetti G., Plata-Salaman C., Rogers J., Rydel R., Shen Y., Streit W., Strohmeyer R., Tooyoma I., Van Muiswinkel F.L., Veerhuis R., Walker D., Webster S., Wegrzyniak B., Wenk G., Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol. Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Combs C.K., Johnson D.E., Karlo J.C., Cannady S.B., Landreth G.E. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000;20(2):558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Griffin W.S., Stanley L.C., Ling C., White L., MacLeod V., Perrot L.J., White C.L., III, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1989;86(19):7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Griffin W.S.T., Sheng J.G., Roberts G.W., Mrak R.E. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J. Neuropathol. Exp. Neurol. 1995;54(2):276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 109.McGeer P.L., Akiyama H., Itagaki S., McGeer E.G. Immune system response in Alzheimer’s disease. Can. J. Neurol. Sci. 1989;16(4) Suppl.:516–527. doi: 10.1017/S0317167100029863. [DOI] [PubMed] [Google Scholar]
- 110.McGeer P.L., McGeer E.G. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Brain Res. Rev. 1995;21(2):195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 111.Mrak R.E., Griffin W.S.T. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2007;66(8):683–686. doi: 10.1097/nen.0b013e31812503e1. [DOI] [PubMed] [Google Scholar]
- 112.Mrak R.E., Sheng J.G., Griffin W.S.T. Glial cytokines in Alzheimer’s disease: review and pathogenic implications. Hum. Pathol. 1995;26(8):816–823. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tuppo E.E., Arias H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005;37(2):289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 114.Walters A., Phillips E., Zheng R., Biju M., Kuruvilla T. Evidence for neuroinflammation in Alzheimer’s disease. Prog. Neurol. Psychiatry. 2016;20:25–31. doi: 10.1002/pnp.444. [DOI] [Google Scholar]
- 115.Beard C.M., Waring S.C., O’Brien P.C., Kurland L.T., Kokmen E. Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease: a case-control study in Rochester, Minnesota, 1980 through 1984. Mayo Clin. Proc. 1998;73(10):951–955. doi: 10.4065/73.10.951. [DOI] [PubMed] [Google Scholar]
- 116.Breitner J.C., Gau B.A., Welsh K.A., Plassman B.L., McDonald W.M., Helms M.J., Anthony J.C. Inverse association of anti-inflammatory treatments and Alzheimer’s disease: initial results of a co-twin control study. Neurology. 1994;44(2):227–232. doi: 10.1212/WNL.44.2.227. [DOI] [PubMed] [Google Scholar]
- 117.Rich J.B., Rasmusson D.X., Folstein M.F., Carson K.A., Kawas C., Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology. 1995;45(1):51–55. doi: 10.1212/WNL.45.1.51. [DOI] [PubMed] [Google Scholar]
- 118.Goldgaber D., Harris H.W., Hla T., Maciag T., Donnelly R.J., Jacobsen J.S., Vitek M.P., Gajdusek D.C. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. USA. 1989;86(19):7606–7610. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Plassman B.L., Havlik R.J., Steffens D.C., Helms M.J., Newman T.N., Drosdick D., Phillips C., Gau B.A., Welsh-Bohmer K.A., Burke J.R., Guralnik J.M., Breitner J.C. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55(8):1158–1166. doi: 10.1212/WNL.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 120.Quintanilla R.A., Orellana D.I., González-Billault C., Maccioni R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004;295(1):245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 121.Cagnin A., Brooks D.J., Kennedy A.M., Gunn R.N., Myers R., Turkheimer F.E., Jones T., Banati R.B. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 122.Fan Z., Brooks D.J., Okello A., Edison P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain. 2017;140(3):792–803. doi: 10.1093/brain/aww349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Parbo P., Ismail R., Hansen K.V., Amidi A., Mårup F.H., Gottrup H., Brændgaard H., Eriksson B.O., Eskildsen S.F., Lund T.E., Tietze A., Edison P., Pavese N., Stokholm M.G., Borghammer P., Hinz R., Aanerud J., Brooks D.J. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain. 2017;140(7):2002–2011. doi: 10.1093/brain/awx120. [DOI] [PubMed] [Google Scholar]
- 124.Swardfager W., Lanctôt K., Rothenburg L., Wong A., Cappell J., Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry. 2010;68(10):930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 125.Bishnoi R.J., Palmer R.F., Royall D.R. Serum interleukin (IL)-15 as a biomarker of Alzheimer’s disease. PLoS One. 2015;10(2):e0117282. doi: 10.1371/journal.pone.0117282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lai K.S.P., Liu C.S., Rau A., Lanctôt K.L., Köhler C.A., Pakosh M., Carvalho A.F., Herrmann N. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry. 2017;88(10):876–882. doi: 10.1136/jnnp-2017-316201. [DOI] [PubMed] [Google Scholar]
- 127.Walker D.G., Dalsing-Hernandez J.E., Campbell N.A., Lue L.F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp. Neurol. 2009;215(1):5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sawikr Y., Yarla N.S., Peluso I., Kamal M.A., Aliev G., Bishayee A. Neuroinflammation in alzheimer’s disease: The preventive and therapeutic potential of polyphenolic nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017;108:33–57. doi: 10.1016/bs.apcsb.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 129.Wilkins H.M., Swerdlow R.H. Relationships between mitochondria and neuroinflammation: Implications for alzheimer’s disease. Curr. Top. Med. Chem. 2016;16(8):849–857. doi: 10.2174/1568026615666150827095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dahlgren C., Gabl M., Holdfeldt A., Winther M., Forsman H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem. Pharmacol. 2016;114:22–39. doi: 10.1016/j.bcp.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 131.Raoof M., Zhang Q., Itagaki K., Hauser C.J. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J. Trauma. 2010;68(6):1328–1332. doi: 10.1097/TA.0b013e3181dcd28d. [DOI] [PubMed] [Google Scholar]
- 132.Arnardottir H.H., Dalli J., Colas R.A., Shinohara M., Serhan C.N. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J. Immunol. 2014;193(8):4235–4244. doi: 10.4049/jimmunol.1401313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang X., Puerta E., Cedazo-Minguez A., Hjorth E., Schultzberg M. Insufficient resolution response in the hippocampus of a senescence-accelerated mouse model--SAMP8. J. Mol. Neurosci. 2015;55(2):396–405. doi: 10.1007/s12031-014-0346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pallas M., Camins A., Smith M.A., Perry G., Lee H.G., Casadesus G. From aging to Alzheimer’s disease: unveiling “the switch” with the senescence-accelerated mouse model (SAMP8). J. Alzheimers Dis. 2008;15(4):615–624. doi: 10.3233/JAD-2008-15408. [DOI] [PubMed] [Google Scholar]
- 135.Medeiros R., Kitazawa M., Passos G.F., Baglietto-Vargas D., Cheng D., Cribbs D.H., LaFerla F.M. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am. J. Pathol. 2013;182(5):1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dunn H.C., Ager R.R., Baglietto-Vargas D., Cheng D., Kitazawa M., Cribbs D.H., Medeiros R. Restoration of lipoxin A4 signaling reduces Alzheimer’s disease-like pathology in the 3xTg-AD mouse model. J. Alzheimers Dis. 2015;43(3):893–903. doi: 10.3233/JAD-141335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhu M., Wang X., Schultzberg M., Hjorth E. Differential regulation of resolution in inflammation induced by amyloid-β42 and lipopolysaccharides in human microglia. J. Alzheimers Dis. 2015;43(4):1237–1250. doi: 10.3233/JAD-141233. [DOI] [PubMed] [Google Scholar]
- 138.Gangemi S., Pescara L., D’Urbano E., Basile G., Nicita-Mauro V., Davì G., Romano M. Aging is characterized by a profound reduction in anti-inflammatory lipoxin A4 levels. Exp. Gerontol. 2005;40(7):612–614. doi: 10.1016/j.exger.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 139.Wang X., Zhu M., Hjorth E., Cortés-Toro V., Eyjolfsdottir H., Graff C., Nennesmo I., Palmblad J., Eriksdotter M., Sambamurti K., Fitzgerald J.M., Serhan C.N., Granholm A.C., Schultzberg M. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2015;11:40–50. doi: 10.1016/j.jalz.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.de Waal Malefyt R., Abrams J., Bennett B., Figdor C.G., de Vries J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991;174(5):1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Moore K.W., de Waal Malefyt R., Coffman R.L., O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 142.Zhu M., Wang X., Hjorth E., Colas R.A., Schroeder L., Granholm A.C., Serhan C.N., Schultzberg M. Pro-Resolving Lipid Mediators Improve Neuronal Survival and Increase Aβ42 Phagocytosis. Mol. Neurobiol. 2016;53(4):2733–2749. doi: 10.1007/s12035-015-9544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ashraf G.M., Tarasov V.V., Makhmutova A., Chubarev V.N., Avila-Rodriguez M., Bachurin S.O., Aliev G. The Possibility of an Infectious Etiology of Alzheimer Disease. Mol. Neurobiol. 2019;56(6):4479–4491. doi: 10.1007/s12035-018-1388-y. [DOI] [PubMed] [Google Scholar]
- 144.Schloer S., Hübel N., Masemann D., Pajonczyk D., Brunotte L., Ehrhardt C., Brandenburg L.O., Ludwig S., Gerke V., Rescher U. The annexin A1/FPR2 signaling axis expands alveolar macrophages, limits viral replication, and attenuates pathogenesis in the murine influenza A virus infection model. FASEB J. 2019: fj201901265R. doi: 10.1096/fj.201901265R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tarasov V.V., Kudryashov N.V., Chubarev V.N., Kalinina T.S., Barreto G.E., Ashraf G.M., Aliev G. Pharmacological Aspects of Neuro-Immune Interactions. Curr. Pharm. Des. 2018;24(1):15–21. doi: 10.2174/1381612823666170829135115. [DOI] [PubMed] [Google Scholar]
- 146.Aguilar-Valles A., Kim J., Jung S., Woodside B., Luheshi G.N. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol. Psychiatry. 2014;19(5):599–606. doi: 10.1038/mp.2013.137. [DOI] [PubMed] [Google Scholar]
- 147.Wohleb E.S., Franklin T., Iwata M., Duman R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016;17(8):497–511. doi: 10.1038/nrn.2016.69. [DOI] [PubMed] [Google Scholar]
- 148.Maes M. Evidence for an immune response in major depression: a review and hypothesis. Prog. Neuropsychopharmacol. Biol. Psychiatry. 1995;19(1):11–38. doi: 10.1016/0278-5846(94)00101-M. [DOI] [PubMed] [Google Scholar]
- 149.Maes M., Yirmyia R., Noraberg J., Brene S., Hibbeln J., Perini G., Kubera M., Bob P., Lerer B., Maj M. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab. Brain Dis. 2009;24(1):27–53. doi: 10.1007/s11011-008-9118-1. [DOI] [PubMed] [Google Scholar]
- 150.Zorrilla E.P., Luborsky L., McKay J.R., Rosenthal R., Houldin A., Tax A., McCorkle R., Seligman D.A., Schmidt K. The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav. Immun. 2001;15(3):199–226. doi: 10.1006/brbi.2000.0597. [DOI] [PubMed] [Google Scholar]
- 151.Dowlati Y., Herrmann N., Swardfager W., Liu H., Sham L., Reim E.K., Lanctôt K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry. 2010;67(5):446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 152.Maes M. Major depression and activation of the inflammatory response system. Adv. Exp. Med. Biol. 1999;461:25–46. doi: 10.1007/978-0-585-37970-8_2. [DOI] [PubMed] [Google Scholar]
- 153.Raison C.L., Capuron L., Miller A.H. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sutcigil L., Oktenli C., Musabak U., Bozkurt A., Cansever A., Uzun O., Sanisoglu S.Y., Yesilova Z., Ozmenler N., Ozsahin A., Sengul A. Pro- and anti-inflammatory cytokine balance in major depression: effect of sertraline therapy. Clin. Dev. Immunol. 2007;2007:76396. doi: 10.1155/2007/76396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hernández M.E., Mendieta D., Martínez-Fong D., Loría F., Moreno J., Estrada I., Bojalil R., Pavón L. Variations in circulating cytokine levels during 52 week course of treatment with SSRI for major depressive disorder. Eur. Neuropsychopharmacol. 2008;18(12):917–924. doi: 10.1016/j.euroneuro.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 156.Kubera M., Curzytek K., Duda W., Leskiewicz M., Basta-Kaim A., Budziszewska B., Roman A., Zajicova A., Holan V., Szczesny E., Lason W., Maes M. A new animal model of (chronic) depression induced by repeated and intermittent lipopolysaccharide administration for 4 months. Brain Behav. Immun. 2013;31:96–104. doi: 10.1016/j.bbi.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 157.Diz-Chaves Y., Astiz M., Bellini M.J., Garcia-Segura L.M. Prenatal stress increases the expression of proinflammatory cytokines and exacerbates the inflammatory response to LPS in the hippocampal formation of adult male mice. Brain Behav. Immun. 2013;28:196–206. doi: 10.1016/j.bbi.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 158.Xiu L.J., Lin H.M., Wei P.K. The effect of chronic mild stress on tumor-bearing rats’ behavior and its mechanism. Neurosci. Lett. 2010;473(1):1–4. doi: 10.1016/j.neulet.2009.06.031. [DOI] [PubMed] [Google Scholar]
- 159.Ślusarczyk J., Trojan E., Głombik K., Budziszewska B., Kubera M., Lasoń W., Popiołek-Barczyk K., Mika J., Wędzony K., Basta-Kaim A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front. Cell. Neurosci. 2015;9:82. doi: 10.3389/fncel.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ślusarczyk J., Trojan E., Wydra K., Głombik K., Chamera K., Kucharczyk M., Budziszewska B., Kubera M., Lasoń W., Filip M., Basta-Kaim A. Beneficial impact of intracerebroventricular fractalkine administration on behavioral and biochemical changes induced by prenatal stress in adult rats: Possible role of NLRP3 inflammasome pathway. Biochem. Pharmacol. 2016;113:45–56. doi: 10.1016/j.bcp.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 161.Gao J.L., Schneider E.H., Dimitrov E.L., Haun F., Pham T.M., Mohammed A.H., Usdin T.B., Murphy P.M. Reduced fear memory and anxiety-like behavior in mice lacking formylpeptide receptor 1. Behav. Genet. 2011;41(5):724–733. doi: 10.1007/s10519-011-9467-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gallo I., Rattazzi L., Piras G., Gobbetti T., Panza E., Perretti M., Dalley J.W., D’Acquisto F. Formyl peptide receptor as a novel therapeutic target for anxiety-related disorders. PLoS One. 2014;9(12): e114626. doi: 10.1371/journal.pone.0114626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ishikawa Y., Deyama S., Shimoda K., Yoshikawa K., Ide S., Satoh M., Minami M. Rapid and sustained antidepressant effects of resolvin D1 and D2 in a chronic unpredictable stress model. Behav. Brain Res. 2017;332:233–236. doi: 10.1016/j.bbr.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 164.Deyama S., Ishikawa Y., Yoshikawa K., Shimoda K., Ide S., Satoh M., Minami M. Resolvin D1 and D2 Reverse Lipopolysaccharide-Induced Depression-Like Behaviors Through the mTORC1 Signaling Pathway. Int. J. Neuropsychopharmacol. 2017;20(7):575–584. doi: 10.1093/ijnp/pyx023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rey C., Nadjar A., Buaud B., Vaysse C., Aubert A., Pallet V., Layé S., Joffre C. Resolvin D1 and E1 promote resolution of inflammation in microglial cells in vitro. Brain Behav. Immun. 2016;55:249–259. doi: 10.1016/j.bbi.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 166.Layé S., Nadjar A., Joffre C., Bazinet R.P. Anti-inflammatory effects of omega-3 fatty acids in the brain: Physiological mechanisms and relevance to pharmacology. Pharmacol. Rev. 2018;70(1):12–38. doi: 10.1124/pr.117.014092. [DOI] [PubMed] [Google Scholar]
- 167.Durukan A., Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007;87(1):179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 168.Markus H. Stroke: causes and clinical features. Medicine (Baltimore) 2008;36:586–591. doi: 10.1016/j.mpmed.2008.08.009. [DOI] [Google Scholar]
- 169.Doyle K.P., Simon R.P., Stenzel-Poore M.P. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55(3):310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sommer C.J. Ischemic stroke: experimental models and reality. Acta Neuropathol. 2017;133(2):245–261. doi: 10.1007/s00401-017-1667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lo E.H., Dalkara T., Moskowitz M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 172.Kalogeris T., Baines C.P., Krenz M., Korthuis R.J. Ischemia/Reperfusion. Compr. Physiol. 2016;7(1):113–170. doi: 10.1002/cphy.c160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jin R., Yang G., Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol. 2010;87(5):779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fang M., Zhong L., Jin X., Cui R., Yang W., Gao S., Lv J. ; Li B., Liu T. Effect of inflammation on the process of stroke rehabilitation and poststroke depression. Front. Psychiatry. 2019;10:184. doi: 10.3389/fpsyt.2019.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ansari J., Kaur G., Gavins F.N.E. Therapeutic potential of annexin A1 in ischemia reperfusion injury. Int. J. Mol. Sci. 2018;19(4): E1211. doi: 10.3390/ijms19041211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Perretti M. The annexin 1 receptor(s): is the plot unravelling? Trends Pharmacol. Sci. 2003;24(11):574–579. doi: 10.1016/j.tips.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 177.Gavins F.N. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol. Sci. 2010;31(6):266–276. doi: 10.1016/j.tips.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Solito E., McArthur S., Christian H., Gavins F., Buckingham J.C., Gillies G.E. Annexin A1 in the brain--undiscovered roles? Trends Pharmacol. Sci. 2008;29(3):135–142. doi: 10.1016/j.tips.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 179.Vital S.A., Becker F., Holloway P.M., Russell J., Perretti M., Granger D.N., Gavins F.N. Formyl-peptide receptor 2/3/lipoxin A4 receptor regulates neutrophil-platelet aggregation and attenuates cerebral inflammation: Impact for therapy in cardiovascular disease. Circulation. 2016;133(22):2169–2179. doi: 10.1161/CIRCULATIONAHA.115.020633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gussenhoven R., Klein L., Ophelders D.R.M.G., Habets D.H.J., Giebel B., Kramer B.W., Schurgers L.J., Reutelingsperger C.P.M., Wolfs T.G.A.M. Annexin A1 as neuroprotective determinant for blood-brain barrier integrity in neonatal hypoxic-ischemic encephalopathy. J. Clin. Med. 2019;8(2): E137. doi: 10.3390/jcm8020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gavins F.N., Sawmynaden P., Chatterjee B.E., Perretti M. A twist in anti-inflammation: annexin 1 acts via the lipoxin A4 receptor. Prostaglandins Leukot. Essent. Fatty Acids. 2005;73(3-4):211–219. doi: 10.1016/j.plefa.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 182.Smith H.K., Gil C.D., Oliani S.M., Gavins F.N.E. Targeting formyl peptide receptor 2 reduces leukocyte-endothelial interactions in a murine model of stroke. FASEB J. 2015;29(5):2161–2171. doi: 10.1096/fj.14-263160. [DOI] [PubMed] [Google Scholar]
- 183.Filep J.G., Sekheri M., El Kebir D. Targeting formyl peptide receptors to facilitate the resolution of inflammation. Eur. J. Pharmacol. 2018;833:339–348. doi: 10.1016/j.ejphar.2018.06.025. [DOI] [PubMed] [Google Scholar]