

Abstract
Scaffold proteins are thought to accelerate protein phosphorylation reactions by tethering kinases and substrates together, but there is little quantitative data on their functional effects. To assess the contribution of tethering to kinase reactivity, we compared intramolecular and intermolecular kinase reactions in a minimal model system. We find that tethering can enhance reaction rates in a flexible tethered kinase system, and the magnitude of the effect is sensitive to the structure of the tether. The largest effective molarity we obtained was ~0.08 μM, which is much lower than the effects observed in small molecule model systems and other tethered protein reactions. We further demonstrate that the tethered, intramolecular reaction only makes a significant contribution to observed rates when the scaffolded complex assembles at concentrations below the effective molarity. These findings provide a quantitative framework that can be applied to understand endogenous protein scaffolds and to engineer synthetic networks.
Introduction
Scaffold proteins assemble enzymes and their substrates into multiprotein complexes and are ubiquitous in signaling, metabolism, and protein homeostasis networks.1–3 These proteins are widely assumed to promote reactions by increasing local concentrations, but direct evidence for this model is limited. For example, the prototypical MAP kinase pathway scaffold protein Ste5 was originally identified through genetic screens as a protein essential for signaling in the yeast mating pathway. Although the protein sequence lacked any obvious catalytic domain, the finding that Ste5 binds multiple sequential kinases in the pathway led to the idea that Ste5 acted as a scaffold to promote kinase signaling reactions.4 Subsequent work revealed multiple functions beyond simple tethering, including complex allosteric effects on kinase activation.4–7 Similarly intricate behaviors in other scaffold systems, including 14-3-3 proteins and the mammalian MAP kinase pathway scaffolds KSR and JIP, indicate that many scaffold proteins have multiple functions,3,8–12 and the contribution to biological function from tethering effects is unclear.
Despite the complexities of natural scaffold protein functions, there is good evidence that tethering can enhance kinase activity and specificity. Both in vitro and in vivo data suggest that AKAP scaffold proteins, which coordinate PKA signaling, use flexible tethers to localize PKA activity and promote reactions with specific substrates.13 Biochemical studies on Src family tyrosine kinases suggest that SH2 and SH3 domains tether the kinase to its substrate to enhance multisite phosphorylation reactions, and the effects depend on tether length.14–16 Similarly, the adapter protein Cks1 binds phosphosites to tether the CDK near additional phosphorylation sites, and in vitro reaction rates depend sharply on the distance between the two sites.17 Furthermore, synthetic recruitment domains can be used to direct tyrosine kinase signaling to specific substrates in cells.18 This specificity presumably arises because tethering enhances reaction rates for the recruited substrate. Finally, a general coupling of binding energy to catalysis has been observed in kinase reactions catalyzed by Csk and Sky1p.19
MAP kinase docking motifs provide another line of evidence supporting the role of tethering in kinase activity and specificity. Docking motifs are modular binding elements that bind a remote site on the kinase surface, and these binding interactions can produce substantial increases in kinase activity, often by decreasing the KM for the reaction.20–24 However, docking peptides can also have allosteric effects on kinase activity.5,25 Some studies suggest that the energetic coupling between docking sites and the active site is small,26 but the possibility for unanticipated complexity in naturally-evolved systems makes it difficult to draw mechanistic conclusions about the contribution from tethering itself in these reactions.
Given the strong evidence from a variety of cellular and biochemical systems supporting a role for tethering effects on kinase activity, there is a compelling need for minimal, simplified biochemical models that can be used to systematically explore the functional properties of tethered kinase systems. Currently, we lack experimental systems to assess how large of a rate effect can be obtained from a scaffold protein, and how varying the structural properties of scaffold proteins affects reaction rates in protein assemblies. There is also substantial interest in engineering scaffold proteins for applications in synthetic biology and bioengineering.27–31 There have been notable successes in rewiring cell signaling networks with engineered scaffolds,28–30 but many attempts required screening large numbers of designs, and it is often unclear why plausible designs failed.28,29,32–34 Determining the criteria for effective protein scaffolds could enable more predictive rational design of scaffolded interactions.
To test the idea that proximity effects can increase reaction rates in kinase signaling reactions, and to develop a predictive framework for synthetic systems, we have constructed simplified model scaffolds using both covalent and non-covalent tethers that link a kinase to its substrate. We measured phosphorylation rates in both systems and compared them to the corresponding intermolecular, untethered kinase reaction (Figure 1). These comparisons allowed us to define an effective molarity for the intramolecular reaction (the ratio of the intramolecular to intermolecular rate constants), and to assess the effects of systematic variations of tether length on reactivity.
Figure 1. Models for scaffold-mediated kinase reactions.
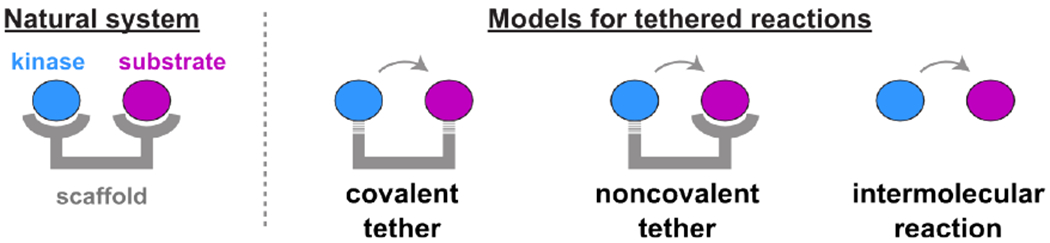
Natural scaffold proteins recruit kinases and their substrates (shown as blue and purple circles, respectively) to a scaffold (grey) using non-covalent protein interaction domains. To create a simplified scaffold model system, we recruited a kinase to a substrate using covalent and non-covalent tethers and compared them to an intermolecular reaction in the absence of scaffold.
Using this model system, we confirmed that a flexible tether can increase reaction rates, but the effects on kinase reactions are much smaller than expected compared to prior studies with small molecule and tethered protein reaction model systems.35–37 We also demonstrate that the relationship between the affinity of complex assembly and the effective molarity is a critical parameter that governs rates in scaffolded reactions. When the binding affinity is weaker than the effective molarity, the scaffold complex does not assemble at substrate concentrations where the intramolecular reaction can proceed faster than the intermolecular reaction. These results provide a possible explanation for why simple peptide tethering strategies often fail in synthetic signaling pathways. More broadly, our work provides the foundation for a quantitative framework to evaluate the molecular function of protein scaffolds in natural assemblies and synthetic systems.
Materials and Methods
Protein expression constructs
All PKA (mouse residues 15-351, Uniprot P05132)38 and SYNZIP-fused substrate constructs were cloned into E. coli expression vectors containing an N-terminal maltose binding protein (MBP) and a C-terminal His6 tag. SpyCatcher-fused substrate constructs were cloned into E. coli expression vectors containing an N-terminal TEV-cleavable His6 tag.39 DNA for the SYNZIPs was kindly provided by Amy Keating (MIT) and DNA for SpyCatcher was kindly provided by Lauren Carter (University of Washington). We used a peptide derived from cystic fibrosis transmembrane conductance regulator (residues 693-705, FGEKRKNSILNPI) as a substrate.40 All linkers were composed of Gly, Ser, and Thr residues. These linkers are uncharged and predicted to be disordered.37 A complete list of protein expression constructs is provided in Table 1 and protein sequence information is provided in the Supporting Information.
Table 1.
Protein expression plasmids
| Plasmid | Vector Backbonea | Expressed Protein |
|---|---|---|
| pES263 | pMBP-MG | MBP PKA-SpyTag H6 |
| pES367 | pBH4 | H6 Pep-2x-SpyCatcher |
| pES368 | pBH4 | H6 Pep-4x-SpyCatcher |
| pES264 | pBH4 | H6 Pep-8x-SpyCatcher |
| pES343 | pBH4 | H6 Pep-14x-SpyCatcher |
| pES340 | pBH4 | H6 Pep-28x-SpyCatcher |
| pES341 | pBH4 | H6 Pep-52x-SpyCatcher |
| pES339 | pBH4 | H6 Pep-80x-SpyCatcher |
| pES265 | pBH4 | H6 Pep-32x-SpyCatcher |
| pES266 | pBH4 | H6 PepΔS-32x-SpyCatcher |
| pES021 | pBH4 | H6 DHR10-PKA |
| pES028 | pMBP-MG | MBP Kemptide-SYNZIP2 H6 |
| pES010 | pMBP-MG | MBP Kemptide H6 |
| pES011 | pMBP-MG | MBP KemptideΔS H6 |
| pES044 | pMBP-MG | MBP Pep H6 |
| pES234 | pMBP-MG | MBP PKA-SYNZIP6 H6 |
| pES273 | pMBP-MG | MBP PKA H6 |
| pES311 | pMBP-MG | MBP Pep-SYNZIP5 H6 |
| pES346 | pMBP-MG | MBP Pep-SYNZIP5trunc1 H6 |
| pES349 | pMBP-MG | MBP Pep-SYNZIP5trunc2 H6 |
pMBP-MG is a modified version of pMAL-p2X (New England Biolabs) wit h an N-terminal TEV-cleavable MBP tag and a C-terminal His6 tag. pBH4 is a modified version of pET15b (Novagen) with an N-terminal TEV-cleavable His6 tag. pMBP-MG and pBH4 were described previously.6 Complete protein sequences are available in the Supporting Information.
Expression and purification of SYNZIP constructs
Proteins were expressed in Rosetta (DE3) pLysS E. coli by inducing with 0.5 mM IPTG overnight at 18 °C. All proteins were purified by Ni-NTA chromatography. Briefly, ~5 mL cell culture pellet was resuspended in 25 mL lysis buffer (25 mM Tris, pH 8.0, 150 mM NaCl, 5 mM Imidazole, 2 mM MgCl2, 5 mM 2-mercaptoethanol, 5% glycerol, and containing one EDTA-free protease inhibitor tablet) and lysed by sonication. The subsequent lysate was cleared by centrifugation at 32,000xg for 50 minutes. Clarified lysate was incubated with Ni-NTA resin for 1 hour. The resin was washed four times with Buffer A (25 mM Tris, pH 8.0, 150 mM NaCl, 15 mM imidazole, 5% glycerol) and once with Buffer B (25 mM Tris, pH 8.0, 1 M NaCl, 5 mM imidazole, 5% glycerol). Protein was eluted using 25 mM Tris, pH 8.0, 150 mM NaCl, 250 mM imidazole, and 10% glycerol.
All proteins were dialyzed overnight into storage buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, and 2 mM DTT), aliquoted, and frozen at −80 °C. Protein concentrations were determined using a Bradford assay (Thermo Scientific).
Expression and Purification of SpyTag-SpyCatcher constructs
MBP-SpyTag-H6 and H6-SpyCatcher proteins were expressed separately in Rosetta (DE3) pLysS E. coli and purified on Ni-NTA resin as described above. Prior to conjugation, proteins were dialyzed overnight into storage buffer at 4 °C to ensure complete removal of ATP. Subsequently, MBP-SpyTag-H6 and H6-SpyCatcher were mixed at a molar ratio of 1:10 in 1xPBS for 16-24 hours and the efficiency of the conjugation reaction was assessed by SDS-PAGE.
To remove excess H6-SpyCather from the reaction mixture, we further purified the SpyTag-Catcher fusion on amylose resin; the conjugate was applied directly to amylose resin and allowed to bind for at least two hours. The resin was washed five times with amylose wash buffer (20 mM Tris, pH 8.0, 200 mM NaCl, 2 mM 2-mercaptoethanol) and bound protein was eluted using amylose wash buffer that contained 15 mM maltose. Proteins were dialyzed overnight into storage buffer, aliquoted, and frozen at −80 °C.
Kinetic assays
All kinetics measurements were performed in kinase assay buffer (40 mM Tris, pH 7.4, 200 mM NaCl, 10 mM MgCl2, and 0.05% IGEPAL) at room temperature (21 ± 1 °C). Phosphorylation was initiated by the addition of 100 μM ATP containing 0.02 μCi/μL γ-32P-ATP. This concentration of ATP is saturating in our system (Figure S1). Approximately 4.5 μL of the reaction was quenched at different time points by spotting onto nitrocellulose membrane and incubating in 0.5% phosphoric acid for at least 10 minutes. To remove excess ATP, the membranes were washed three to four times in 0.5% phosphoric acid, dried, and imaged on a GE Typhoon FLA 9000 imaging scanner. Kinetic parameters were determined using the initial rate method.
32P counts were normalized to concentration using an endpoint standard run in parallel to all experiments. Endpoint reactions contained 200 nM PKA, 1 μM MBP-Kemptide, 100 μM ATP containing 0.02 μCi/μL γ-32P-ATP in endpoint assay buffer (40 mM Tris pH 7.4, 100 μM EGTA, 10 mM MgCl2, 0.05% IGEPAL). This reaction rapidly proceeds to completion in <1 min (Figure S2, S3D) and time points were taken at 40 minutes.
Unimolecular rate constants (kintra) for SpyTag-mediated covalently tethered kinase-substrate complexes were measured by varying the covalently-tethered complex concentration over >30-fold range (from 0.005-0.16 μM for complexes containing Pep-SpyCatcher linkers <20 amino acids and 0.0125-0.2 μM for complexes with Pep-SpyCatcher linkers >20 amino acids). At low [substrate], the resulting data (Vobs vs. [Enzyme]) was linear and the slope of this data corresponds to the intramolecular rate constant kintra (Figure S4). The corresponding intermolecular reaction was measured using a fixed enzyme concentration (2.5 nM) and substrate concentrations that varied >200-fold range (from 0.02-5 μM). Initial rates were quantified as described above and the bimolecular rate constant (kcat/KM) was obtained from the slope of a linear fit to a plot of kobs vs. [substrate]. In separate experiments, we varied the enzyme concentration to confirm that the observed rates for the intermolecular reaction were also first order in enzyme concentration (Figure S5).
To make the tethered PKA-product complex, we allowed the tethered PKA-substrate complex to react to completion. Briefly, we incubated 2.5 μM tethered PKA-substrate complex with 250 μM ATP for two hours at room temperature. To measure reaction rates, the fully phosphorylated complex was diluted to 2.5 nM and mixed with 100 μM cold ATP containing 0.02 μCi/ μL γ-32P-ATP. The reaction was initiated by the addition of free substrate. Initial rates were quantified as described above and the bimolecular rate constant (kcat/KM) was obtained from the slope of a linear fit to a plot of kobs vs. [substrate].
For the non-covalent reactions, enzyme concentration was held constant at either 1 nM or 2.5 nM and substrate concentrations varied over >20-fold range (from 0.005-0.25 μM for tethered reactions and 0.01-0.25 μM for free reactions). In separate experiments, we varied the concentration of enzyme to verify that initial velocities scaled linearly with enzyme concentration (Figure S6).
Competition Assay
For the competitive reaction with covalently-tethered PKA-substrate complex and free substrate, 0.02 μM covalently-tethered complex was mixed with free substrate that varied over a >200-fold concentration range (from 0.02-5 μM). At a given time point, 9 μL of the reaction was quenched with 6 μL 5xSDS dye. Subsequently, 10 μL of the quenched sample was analyzed by SDS-PAGE. After electrophoresis, gels were washed with 0.5% phosphoric acid twice for five minutes and imaged on a GE Typhoon FLA 9000 gel scanner.
Fluorescence Polarization Binding Assays
Cys-containing proteins were labeled with Bodipy TMR using maleimide chemistry. 100 μM protein was reduced with 1 mM TCEP-HCl and incubated with at least a 10-fold molar excess of dye for 1 hour at room temperature. Free dye was removed by purification on a Ni-NTA column and/or a spin desalting column (Zeba). To determine labeling efficiency, proteins were separated by SDS-PAGE and subsequently imaged on a GE Typhoon FLA 9000 scanner at 532 nm.
1 nM labeled substrate was mixed with >700-fold range of unlabeled protein (from 1 to 700 nM) in binding buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 2 mM TCEP, 2 mM MgCl2, and 0.05% IGEPAL). Reactions were incubated at room temperature (21 ± 1 °C) for at least one hour in the dark. Polarization was measured in black 96-well plates using an EnVision 2105 multimode plate reader. The polarization of the peptide alone was subtracted from all measurements and the subsequent data was fit to a 1:1 binding model using equation 1:
| (eq. 1) |
where x represents the protein concentration, Y represents the fluorescence polarization, P1 represents the maximum fluorescence polarization, and P2 is the dissociation constant.
Competition binding assays were conducted as described above using 1 nM labeled substrate, 30 nM PKA-SYNZIP6, and an unlabeled competing substrate that varied >10,000 fold range (from 1 nM to 13 μM). Background fluorescence polarization from non-specific binding between substrate competitor and labeled substrate was subtracted from each measurement. To determine the IC50, the plot of fluorescence polarization vs. [competitor] was fit to equation 2.
| (eq. 2) |
Subsequently, the IC50 was related to KD using equation 3:
| (eq. 3) |
Results and Discussion
We used the catalytic subunit of protein kinase A (PKA) as a model kinase because it has been extensively characterized both structurally and biophysically.41–43 Furthermore, PKA can be purified in active form from E. coli and can phosphorylate short peptides in vitro.44 We chose a natural PKA phosphosite derived from cystic fibrosis transmembrane conductance regulator (CFTR) as a substrate, which has a reported KM value of 30 μM for PKA.40 We tested a smaller minimal peptide (residues 693-705) than was previously described; this peptide is phosphorylated by PKA on Ser700 but does not detectably saturate the kinase active site at concentrations up to 100 μM (Figure S3A–B & Table S1). These data indicate that the reaction between PKA and this peptide substrate is relatively weak and that the observed reaction will be bimolecular in the nM to low μM range, which provides a broad concentration range to assess tethering effects.
Reaction rates for covalently tethered kinase-substrate complexes
To covalently tether PKA to a peptide substrate, we expressed PKA and its substrate separately and covalently fused them after purification via SpyTag and SpyCatcher proteins, which spontaneously form an irreversible isopeptide bond.39 This approach allowed us to avoid premature phosphorylation of the substrate, which would likely occur with a genetically-fused kinase-substrate complex. We constructed C-terminal fusions of PKA to SpyTag (PKA-SpyTag) via an eight residue Gly/Ser (GS) linker and fused the peptide substrate to SpyCatcher (Pep-SpyCatcher) via a flexible linker (Figure S7). After expression and purification, PKA-SpyTag and Pep-SpyCatcher constructs were mixed to form a covalently tethered kinase-substrate complex (Figure 2).
Figure 2. Reactivity depends on the structural properties of the assembly.
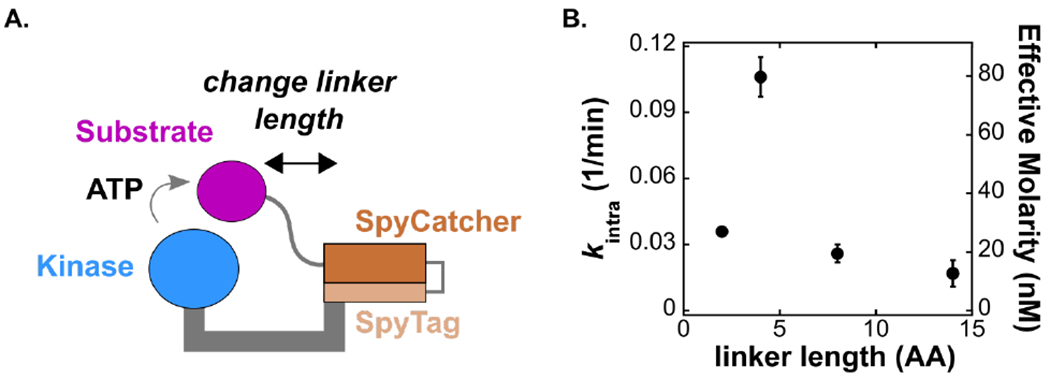
(A) Schematic of the covalently tethered complex. The SpyCatcher-SpyTag complex covalently links the kinase with its substrate. We varied the length of the linker that connects the substrate to SpyCatcher from 2 to 14 residues and measured the unimolecular rate constant (kintra). (B) Plot of kintra vs. number of residues. The secondary y-axis shows the effective molarity for each complex, determined using the bimolecular rate constant from the untethered reaction (Figure 3A). Error bars represent the standard error of kintra obtained from the fit of Vobs vs [tethered complex] as described in Figure S4.
With a covalently tethered complex in hand, we measured phosphorylation over time using γ-32P-ATP to detect phosphorylation and converted 32P counts to concentration using an endpoint standard that rapidly proceeds to completion (Figure S2). The plot of [phosphorylation] vs. time reaches an endpoint consistent with a single phosphorylation event per substrate (Figure S8). To ensure that phosphorylation is occurring at the desired CFTR peptide phosphosite and not a different Ser/Thr in a linker or folded protein domain, we performed control reactions with a Ser to Ala phosphosite point mutant (Figure S9A). This mutant is not significantly phosphorylated during the course of the reaction, indicating that the observed reaction represents a single phosphorylation event at the target CFTR peptide substrate (Figure S9B). Finally, to confirm that there were no unanticipated phosphosites in the longer Gly/Ser linkers used in some constructs, we performed additional untethered control reactions between PKA and peptide substrates fused to variable length of Gly/Ser linkers and observed no differences in observed rates (Figure S9C–D). Together, these data indicate that observed rates represent a single phosphorylation event at Ser700 of the CFTR peptide.
Tethered reaction rates are likely to depend on the length of the tether between the kinase and the substrate. Hence, we measured substrate phosphorylation rates for a series of covalently tethered complexes with variable Pep-SpyCatcher linker lengths ranging from 2 to 14 residues. For each complex, we measured initial rates of phosphorylation (Vobs) and calculated the unimolecular rate constant (kintra) from a plot of Vobs vs. [tethered complex]. These plots are linear at concentrations below 50 nM (Figure S4A), confirming that the observed reaction is intramolecular in this concentration range. However, at concentrations above 50 nM, the observed rates increased non-linearly, likely due to competition from a bimolecular phosphorylation reaction. To extract kintra, we fit the full dataset to a kinetic model that includes contributions from both the unimolecular and bimolecular reaction (Figure S4B). In parallel, we also fit the linear portion of the dataset at concentrations below 50 nM to a linear model. These fitting methods produce kintra values that are in close agreement (Figure S10).
As anticipated, we found that reaction rates depended on the structural properties of the tether. The observed values of kintra vary over a 6-fold range with different Pep-SpyCatcher linker lengths and display a maximum of 0.11 min−1 (1.8 × 10−3 s−1) with a 4-residue linker (Figure 2 and Table 2). Thus, the 4-residue linker appears to be sufficient to reach the kinase active site. The distance from the C-terminus of PKA to the C-terminus of a bound peptide is ~23 Å (Figure S7),38 and this distance can plausibly be spanned by the 4-residue linker because the construct also includes an 8-residue PKA-SpyTag linker. Pep-SpyCatcher linkers shorter and longer than 4 residues are suboptimal. The shorter 2-residue Pep-SpyCatcher linker may be unable to reach the active site of the kinase without distorting protein structure, while linkers longer than 4 residues likely result in a larger entropic barrier to finding the kinase active site.
Table 2.
Kinetic parameters for covalently tethered complexesa
| Linker length (AA) | kintra (s−1)a | Effective Molarity (μM)b |
|---|---|---|
| 2 | (6.1 ± 0.3) × 10−4 | 0.027 ± 0.002 |
| 4 | (1.8 ± 0.2) × 10−3 | 0.080 ± 0.007 |
| 8 | (4.4 ± 0.7) × 10−4 | 0.020 ± 0.003 |
| 14 | (2.9 ± 1.0) × 10−4 | 0.013 ± 0.005 |
| 28c | (6.6 ± 0.8) × 10−4 | 0.030 ± 0.004 |
| 52 | (2.2 ± 0.4) × 10−3 | 0.100 ± 0.018 |
| 80 | (1.9 ± 0.3) × 10−3 | 0.085 ± 0.013 |
Effective molarity is calculated by dividing kintra by (2.20 ± 0.03) × 104 M−1 s−1, the value of the corresponding bimolecular rate constant (kcat/KM) (Figure 3A). Errors in the effective molarity are propagated from the errors in kintra and kcat/KM.
To obtain the kintra value for this construct, we fit the Vobs data at low [tethered complex] to a line as described in Figure S4.
To determine how increasingly longer linkers affect reaction rates, we measured kintra values for a series of covalently-tethered complexes with Pep-SpyCatcher linkers ranging from 16 to 80 residues. We observed an unexpected bimodal behavior with increasing linker length; after the first peak in kintra at the 4-residue linker, there is a second peak of activity at 52 residues (Figure S11). The structural origins of this bimodal behavior are unknown, but we note that both activity peaks are similar in magnitude (kintra = ~2 × 10−3 s−1), and this value will serve as a key point of comparison between intra-and intermolecular reaction rates.
Effective molarity for the covalently-tethered reaction
To identify the conditions in which covalent tethering can increase reaction rates, we compared the intramolecular reaction to an intermolecular reaction containing PKA-SpyTag with a free, untethered substrate. For the free reaction, we measured initial rates over a >200-fold range of substrate concentration (0.02 – 5.1 μM) and a 10-fold range of enzyme concentration (Figure 3A and Figure S5, respectively). In these concentration ranges, observed rates scaled linearly with substrate and enzyme concentration, indicating that the observed reaction is bimolecular. For this reaction, there was no detectable saturation at high substrate concentration, as expected from the KM >100 μM for PKA and the peptide substrate (Figure S3B).40 We therefore fit kobs (i.e. Vobs/[E]) vs. [substrate] to a linear model, where kobs = (kcat/KM)[S] and obtained a bimolecular rate constant (kcat/KM) of 2.2 × 104 M−1 s−1.
Figure 3. Intermolecular reactions readily outcompete the covalently tethered reaction.
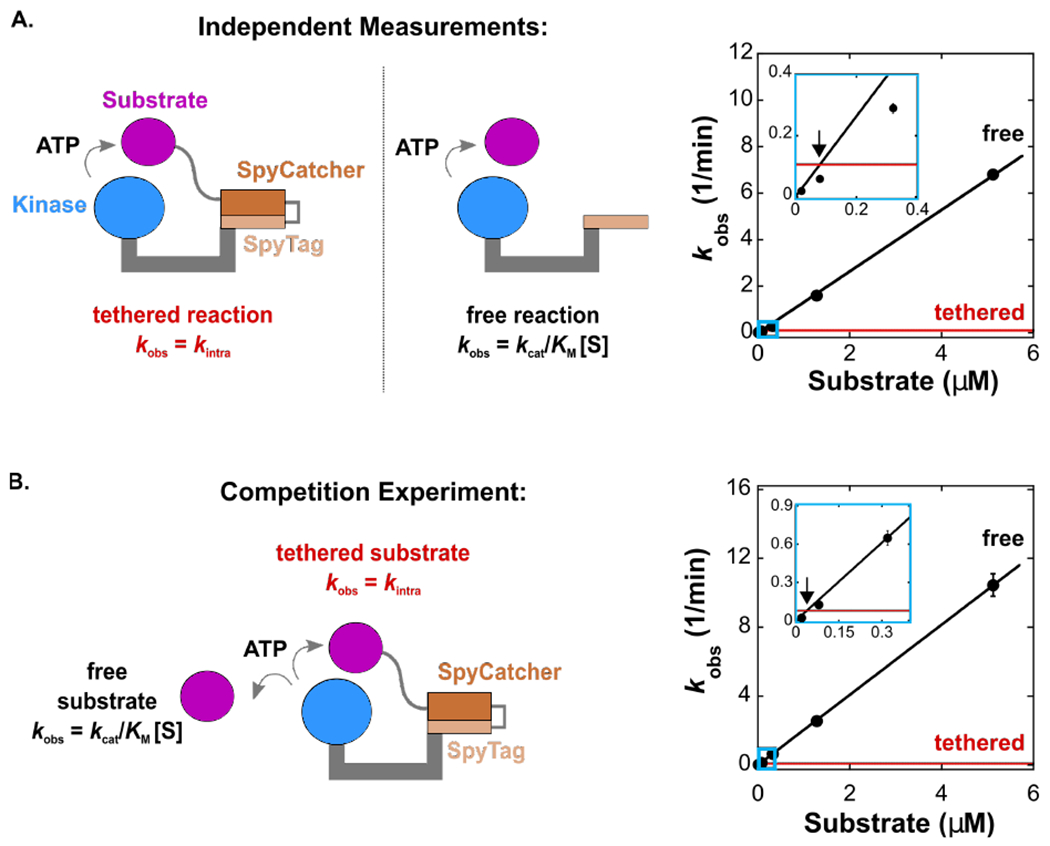
(A)Schematic of the tethered (4 aa linker) and free reaction and corresponding plot of kobs vs. [substrate] for the tethered (red) and free (black) reactions. Inset is marked with light blue rectangle. The observed effective molarity is 0.08 μM.
(B) Schematic of a competitive reaction with both tethered (4 aa linker) and free substrates present, and corresponding plot of kobs vs. [substrate] for the tethered (red) and free (black) reactions. Inset is marked with light blue rectangle. The observed effective molarity is 0.04 μM.
Each [product] vs time trace was measured from separate reactions in duplicate. For (A) and (B), error bars represent the standard error for kobs obtained from a linear fit to [product] vs time for both datasets. The error for kintra is smaller than the thickness of the red line (see Table 2).
The effective molarity is the substrate concentration at which the intermolecular and intramolecular rates are equal and can be obtained by finding the point at which the rate constant kobs is equal for the two reactions. For the free reaction, kobs = (kcat/KM)[S] and increases linearly with increasing [substrate]. For the tethered reaction, kobs = kintra and is constant. Plotting these values together gives a pair of lines that intersect at the effective molarity. For the optimal 4-residue tethered complex, the observed effective molarity is 0.08 μM (Figure 3A). At substrate concentrations <0.08 μM, the intermolecular reaction is slower than the covalently-tethered reaction, while at substrate concentrations >0.08 μM the intermolecular reaction is faster. As with the observed value of kintra, the effective molarity displays a maximum with a 4-residue Pep-SpyCatcher linker (Figure 2B). Similar effective molarity trends with a maximum at an intermediate linker length have been observed in tethered protein-ligand model systems.15
To evaluate the significance of an effective molarity of 0.08 μM, we can make comparisons to other systems where these values have been measured. Small molecule reactions have effective molarities that can range above 108 M,35,45 while tethered protein-ligand interactions have measured and predicted values in the mM range,36,37,46 and protein kinase docking interactions have estimated effective molarities in the μM to mM range (Table S2).20,22,26,47 Further, we can obtain a rough estimate of an effective concentration for a tethered substrate by using the covalent tether length to define a spherical volume where the substrate is confined, which gives a value of >4 mM for the 4-residue linker and predicts that effective concentrations should vary >600-fold for the range of linkers lengths used here (Figure S12). Based on these comparisons, the effective molarity of 0.08 μM for our optimal tethered kinase-substrate system is unexpectedly small. We note that this approach to calculate effective concentration is vastly oversimplified, most notably because this model does not account for conformations that are sterically occluded by PKA or the SpyTag domains. These effects could increase rates by confining the substrate to a smaller volume or decrease rates by positioning the substrate in an unfavorable location. More sophisticated polymer models may be necessary to accurately describe the ensembles of conformations available to the system.37 Also, effective molarity is a complex parameter that also depends on orientation effects from rotational and vibrational degrees of freedom.45 There is limited data available for effective molarities in tethered enzymatic protein-protein reactions, and it remains to be seen whether there is a general difference in behavior between tethered enzymatic reactions and binding interactions. Larger effective molarities in kinase-substrate reactions could potentially be obtained with a scaffold that precisely constrains the orientation of the substrate relative to the active site.
The covalently-tethered system does not form a stable product-bound complex
One potential tradeoff associated with scaffold-mediated rate enhancements is that, by promoting interactions between a kinase and a substrate, the scaffold may stabilize the product-bound state and inhibit multiple turnovers of the enzyme. To test this possibility, we measured reaction rates in a competitive reaction between free substrate and substrate covalently tethered to enzyme. If product inhibition by the tethered product limits turnover, phosphorylation of the free peptide by the tethered PKA-substrate complex should be slower than the reaction of untethered substrate with free PKA.
To measure rates for a tethered and free substrate in the same reaction, we incubated the covalently tethered PKA-substrate complex with varying concentrations of free substrate and monitored phosphorylation of both substrates simultaneously using SDS-PAGE and autoradiography (Figure S13). We used 20 nM tethered complex, a concentration at which the bimolecular reaction between two tethered complexes is insignificant (Figure S4). From the observed rate of phosphorylation of the free substrate, we obtained a plot of kobs vs. [substrate] and a bimolecular rate constant (kcat/KM) of 3.3 × 104 M−1 s−1 (Figure 3B). In the same reaction, we measured the phosphorylation rate of the tethered substrate and used kintra = Vobs/[ES]tethered to obtain a unimolecular rate constant of 1.3 × 10−3 s−1. The values of kcat/KM and kintra from this competition experiment are similar to our previous measurements of these values in separate reactions (2.2 × 104 M−1 s−1 and 1.8 × 10−3 s−1, respectively) (compare Figure 3A and 3B). In particular, the identical kintra value indicates the tethered complex is still functional for the intramolecular reaction in these conditions, and the absence of any decrease in kcat/KM indicates that the tethered substrate does not stably occupy the active site and inhibit enzyme turnover.
To independently confirm that tethered product does not inhibit enzyme turnover, we also prepared a fully phosphorylated tethered complex by allowing the covalently tethered PKA-substrate complex to react to completion. When the tethered PKA-product complex is mixed with varying concentrations of free substrate, the observed bimolecular rate constant (kcat/KM) is 2.3 × 104 M−1 s−1, indistinguishable from the kcat/KM of 2.2 × 104 M−1 s−1 for the free reaction (Figure S14).
Importantly, these results directly demonstrate the conditions in which an untethered reaction outcompetes the tethered reaction. The plot of kobs vs. [substrate] gives a pair of lines that intersect at an effective molarity of 0.04 μM (Figure 3B), similar to the value of 0.08 μM obtained when the rates are measured in separate reactions (Figure 3A). At concentrations above the effective molarity, even when a substrate is covalently tethered to PKA, the free reaction outcompetes the tethered reaction.
A non-covalent tether can accelerate the kinase reaction at low substrate concentrations
Natural scaffold proteins use non-covalent protein-protein interactions to recruit multiple signaling proteins to the macromolecular complex. To understand how protein-protein interactions affect tethering, we constructed a model system where the substrate can bind non-covalently to a remote binding site on the kinase (Figure 1). For simplicity, the kinase remains covalently fused to the scaffold in our system. This approach allows us to make a direct comparison between the non-covalently tethered reaction and the corresponding free/untethered reaction. It also avoids the complications that arise in a non-covalent three component kinase/substrate/scaffold system where there are additional bound and unbound states to consider (i.e. kinase and substrate segregated on different scaffold proteins).48 We expect that the observed effects from this model system should be generalizable to more complex multivalent assemblies.
Based on a minimal kinetic model, we predict that to observe a significant contribution from a tethered reaction, the substrate-scaffold interaction needs to be strong enough to appreciably bind at concentrations below the effective molarity (Scheme 1). In the minimal model, the substrate binds to the scaffold via a remote binding interaction and can then be phosphorylated in an intramolecular reaction. The free substrate can also bind directly to the active site of the kinase and react via an intermolecular reaction without engaging the remote binding site. The kinetic model indicates that the intramolecular, scaffold-mediated reaction (Vintra) is faster than the intermolecular reaction (Vinter) when the effective molarity is greater than KD + [S], where KD is the affinity of the substrate for the scaffold (Figure 4). If the affinity of the substrate for the scaffold is weak compared to the effective molarity (i.e. KD > EM), there is no substrate concentration at which the intramolecular reaction is faster than the intermolecular reaction because the scaffolded complex does not significantly accumulate at concentrations below the effective molarity (Figure 4B). In contrast, if the affinity is strong relative to the effective molarity (i.e. KD < EM, Figure 4C), then the intramolecular reaction will outcompete the intermolecular reaction at low substrate concentrations (i.e. when [S] < EM - KD). Even in this scenario, the intermolecular reaction will still outcompete the intramolecular reaction at sufficiently high substrate concentrations. The substrate concentration at which this switch occurs is determined by the effective molarity (Figure 4). Practically, our kinetic model suggests that we will observe the largest effects from tethering with a scaffold that recruits a substrate with a KD < ~0.08 μM, the effective molarity that we observed in the covalently-tethered kinase-substrate system (Figure 2). A similar conceptual analysis was recently described for tethered protein-ligand interactions.49
Scheme 1.
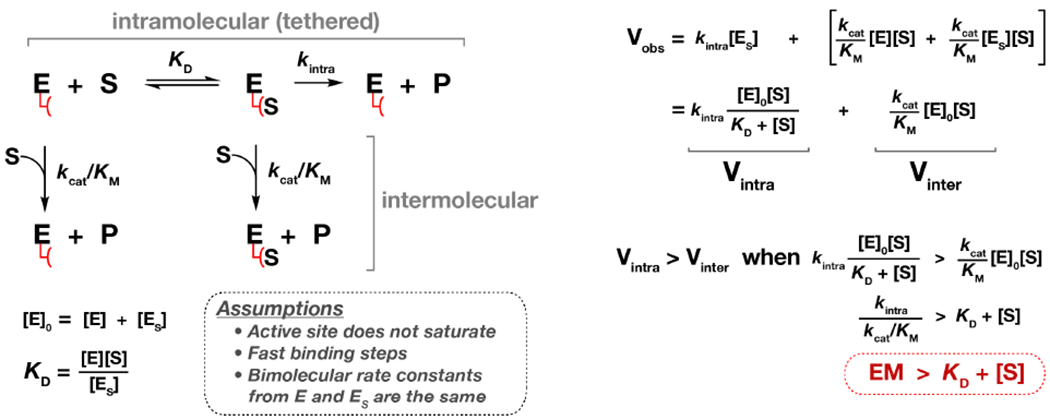
Minimal kinetic scheme for a noncovalent tethered reaction.
The enzyme has a remote tether that can recruit a substrate, which then reacts with the active site through an intramolecular mechanism. Alternatively, the substrate can react with the enzyme through an intermolecular reaction by binding directly to the enzyme active site without engaging the tether. For simplicity, the intermolecular pathway does not include an E-S complex with substrate occupying the active site, because we did not detect saturation in any untethered reaction between PKA and the substrate peptide (Figure 2). We also assumed that the binding step to the tether is fast compared to kintra. Finally, we assumed that the bimolecular rate constants (kcat/KM) for E and ES are the same. With these simplifying assumptions, the condition for when the intramolecular reaction (Vintra) is faster than the intermolecular reaction (Vinter) is: EM > KD + [S]. See Supporting Information for a complete derivation.
Figure 4. Predicted rates versus substrate concentration for various tethering strategies.
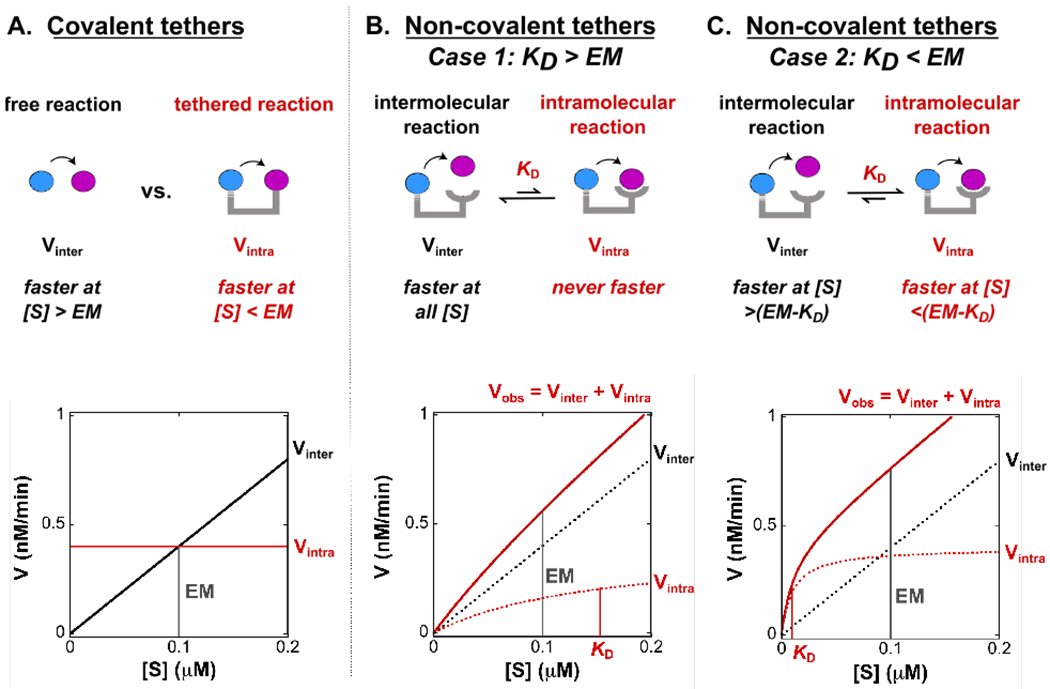
(A) Observed rates vs. substrate concentration for a bimolecular reaction between kinase and free substrate (solid black line), and a covalently tethered kinase-substrate reaction (solid red line). The effective molarity (EM) is the substrate concentration at which Vinter and Vintra are equal (indicated by grey line).
(B) Observed rates vs. substrate concentration for a model scaffold system where the remote tether binds to the substrate with a KD > EM. There is no substrate concentration at which the rate of the intramolecular reaction (Vintra, red dotted line) is faster than the intermolecular reaction (Vinter, black dotted line). The observed rate (Vobs, solid red line) will include contributions from both the intramolecular and intermolecular reactions (i.e. Vobs = Vintra+Vinter).
(C) Observed rates vs. substrate concentration for a model scaffold system where the substrate binds to the remote tether with a KD < EM. At low substrate concentrations (i.e. when [S] < EM - KD), the intramolecular reaction (Vintra, red dotted line) is faster than the intermolecular reaction (Vinter, black dotted line) because the complex assembles at concentrations below the EM. The total observed rate (Vobs, solid red line) will include contributions from both the intramolecular and intermolecular reactions (i.e. Vobs = Vintra+Vinter).
For all plots the effective molarity is indicated by a vertical grey line and is 0.1 μM. KD values in B and C are indicated by a vertical red line and are 0.15 μM and 0.01 μM, respectively.
To construct a non-covalently tethered kinase substrate system, we used heterodimeric coiled-coil interactions to recruit the substrate to the kinase via a flexible tether (Figure 5A). We used the heterodimeric coiled-coils SYNZIP 5 and SYNZIP 6 because this pair is well-characterized structurally and biophysically and has been used for many applications in synthetic biology and protein design.50–52 We fused PKA to SYNZIP 6 (PKA-SYNZIP6) and the peptide substrate to SYNZIP5 (Pep-SYNZIP5). We measured a KD of 0.01 μM for the Pep-SYNZIP5-PKA-SYNZIP6 interaction using fluorescence anisotropy (Figure S15); this binding affinity is consistent with previous measurements for SYNZIP5/6 affinity.51 Importantly, this affinity is below the predicted effective molarity of 0.08 μM, so we expect to observe a significant contribution from the tethered reaction at substrate concentrations <0.08 μM, but almost no effect on rates at substrate concentrations substantially above the effective molarity.
Figure 5. Non-covalent tethering enhances the rate of phosphorylation and depends on the relationship between effective molarity and KD.
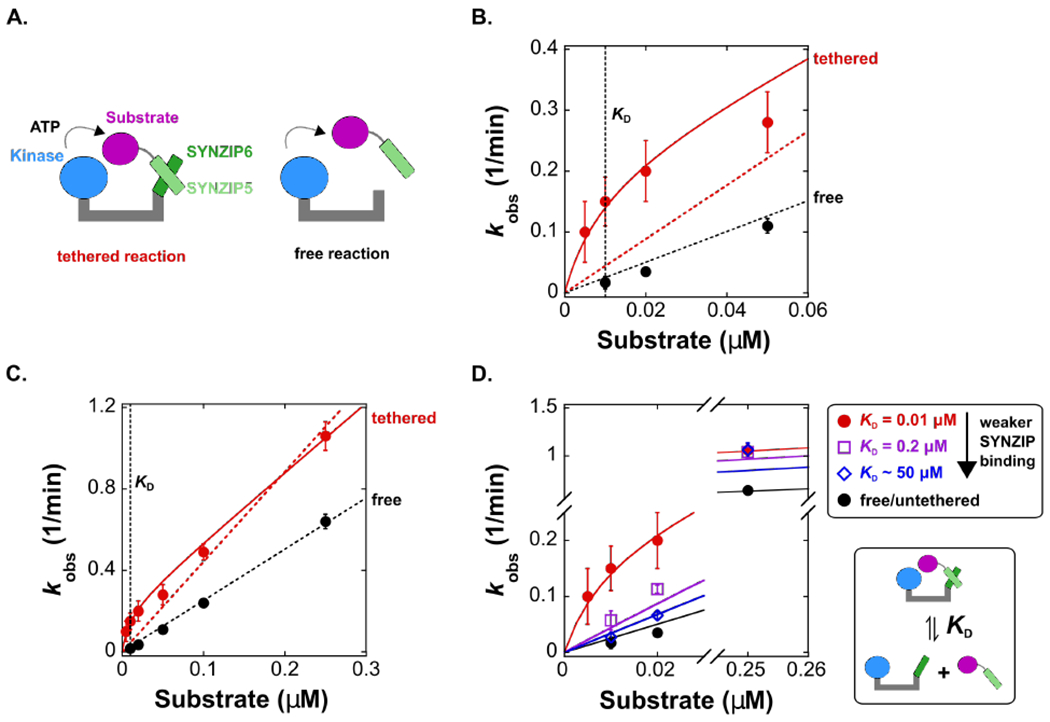
(A) Schematic of a model scaffold with a non-covalent tether that recruits a substrate to a kinase. The kinase is fused to SYNZIP6 (PKA-SYNZIP6) and the substrate is fused to SYNZIP5, which interact to form the tethered complex. In the corresponding free reaction, we used a kinase without SYNZIP6.
(B) Plot of kobs vs. [substrate] for the tethered and free reactions for [substrate] < 0.06 μM. At concentrations below the effective molarity, the values of kobs for the tethered reaction are significantly larger than the untethered reaction. The solid red line is a fit to a kinetic model (Scheme 1 and Supporting Information) that includes a contribution from kintra, which accurately fits the tethered reaction data. The dotted lines are fits to a model where kobs = (kcat/KM)[S] (this linear fit forces the line through zero). Both fits include data at higher substrate concentrations (full dataset is shown in panel C), and for the tethered reaction this linear model fails to account for the observed rate increase at low substrate concentrations. For the untethered reaction, the data scale linearly with [substrate]. The reaction of PKA-SYNZIP6 is also first order in enzyme concentration (Figure S6).
(C) Plot of kobs vs. [substrate] over an expanded range of substrate concentrations up to 0.25 μM. The dotted lines are fits to a simple linear model (kobs = kcat/KM [S]), and the solid red line is a fit to the complete model that includes kintra (Scheme 1 and Supporting Information). For the untethered reaction, the slope corresponds to a bimolecular rate constant (kcat/KM) of 4.2 × 104 M−1 s−1. For the tethered reaction, the value of kcat/KM obtained from the fit to the complete model (solid red line) is 5.6 × 104 M−1 s−1. This value differs from the untethered reaction by a factor of 1.3, likely due to prep-to-prep variations in the total activity of PKA-SYNZIP6 versus free PKA.
(D) Plot of kobs vs. [substrate] for the tethered reaction with the SYNZIP 5/6 pair and with two SYNZIP5 truncations that weaken the binding affinity (increasing KD) (Figure S19). At [substrate] < 0.03 μM, values of kobs decrease as KD increases. At [substrate] = 0.25 μM, well above the effective molarity, values of kobs are indistinguishable for substrates with different SYNZIP interaction affinities. The solid lines represent kinetic models using the values of kintra and kcat/KM from the untruncated pair in Figure 5C, and the corresponding KD value from each truncated pair. For SYNZIP5trunc2, we used a KD of 50 μM (using larger KD values does not change the prediction, see Figure S18B & C). Each [product] vs time trace was measured from separate reactions in duplicate. Error bars in B-D represent the standard error for kobs obtained from a linear fit to [product] vs time for both datasets.
To test this prediction, we compared phosphorylation rates (kobs, where kobs = Vobs/[E]) for the tethered and free reactions over a range of substrate concentrations above and below the anticipated effective molarity (Figure 5). As expected, we observed a significant increase in kobs for the tethered reaction, particularly at substrate concentrations <0.05 μM (Figure 5B). The observed values of kobs for the tethered reaction are biphasic with [substrate] and correspond closely to the predictions of the kinetic model that includes contributions from an intramolecular reaction (Scheme 1). At low substrate concentrations, kobs is dominated by contributions from the intramolecular reaction, while at high substrate concentrations kobs is dominated by contributions from an intermolecular reaction of the tethered complex with free substrate (Figure 5B & C). For the untethered reaction, kobs scaled linearly with substrate and enzyme concentrations, consistent with a bimolecular reaction (Figure 5B, 5C, & Figure S6). Thus, tethering can enhance kinase reaction rates at substrate concentrations below the effective molarity.
We performed several controls to confirm that the observed reactions reflect tethered phosphorylation of Ser700 of the CFTR target peptide. First, we did not detect any phosphorylation of off-target sites in the SYNZIP interaction domains or linkers that could potentially confound the assay (Figures S3B, S5, & Table S3). To confirm that product release from the SYNZIP tethering interaction is not limiting the observed rates, we performed control reactions under single turnover conditions and obtained the same value of kobs as observed in multi-turnover conditions (Figure S16 & S17).
Fitting the data to kinetic models allows us to calculate the effective molarity and define the conditions under which tethering produces significant rate enhancements. For the tethered reaction, the fit provides an intramolecular rate constant (kintra) of 3.5 × 10−3 s−1 for a substrate non-covalently tethered to enzyme (Figure 5B & C), similar to the kintra value of 1.8 × 10−3 s−1 for covalently tethered substrate (Table 2). Using this kintra value and the kcat/KM value of 4.2 × 104 M−1 s−1 derived from the free reaction (Figure 5B & C), we calculated an effective molarity of 0.08 μM for the non-covalently tethered system, identical to the effective molarity obtained for the covalent system (Table 2). At substrate concentrations <0.07 μM (i.e. when [S] < EM - KD; EM = 0.08 μM and KD = 0.01 μM), the substrate binds to the scaffold and primarily reacts with the kinase through the intramolecular, tethered reaction. Conversely, at substrate concentrations >0.07 μM (i.e. when [S] > EM - KD), the substrate predominantly reacts through a bimolecular pathway, even when PKA is fully occupied by a tethered substrate. Hence, we only observe significant effects from tethering with substrate concentrations less than ~70 nM.
A scaffold-substrate interaction that is weak relative to the EM does not accelerate the reaction
Our model predicts that observed rates from a tethered reaction should depend on the binding affinity between the substrate and the scaffold. If the affinity of the substrate for the scaffold is weak relative to the effective molarity (KD ≫ EM), then the intermolecular reaction is faster than the intramolecular reaction at all substrate concentrations (Figure 4B). At intermediate binding affinities (KD ~ EM), we expect to observe detectable rate effects from tethering. These effects will be largest at low substrate concentrations below the effective molarity; at concentrations above the effective molarity, the intermolecular reaction will be faster than the intramolecular reaction regardless of the value of KD (Figure 4 & S18). To test these predictions, we measured kobs for PKA-SYNZIP6 and weaker binding variants at substrate concentrations of 0.01 and 0.02 μM, concentrations well below the effective molarity (~0.1 μM), and at 0.25 μM, a concentration well above the effective molarity.
To weaken the binding affinity of the tethering interaction, we made truncations of SYNZIP5. We identified two mutants, SYNZIP5trunc1 and SYNZIP5trunc2, with weaker binding affinities of 0.2 μM and ~50 μM (Figure S19), respectively, compared to 0.01 μM for the cognate pair (Figure S15). As expected, for substrate concentrations below the effective molarity (i.e. 0.01 or 0.02 μM), phosphorylation rates (kobs) decrease as KD increases (Figure 5D). At a substrate concentration above the effective molarity (i.e. 0.25 μM), kobs values are relatively unaffected by binding affinity. These kobs values are captured by our model using the values of kintra and kcat/KM derived from the cognate SYNZIP5/6 pair in Figure 5C, and the KD value for each truncated pair. Moreover, this data confirms a key prediction from our model: as KD increases, there will be less tethered complex assembled and the intramolecular, tethered reaction will therefore make smaller contributions to observed rates. Thus, the relationship between the effective molarity and the KD is a critical parameter that determines the magnitude of the contribution, if any, from a tethered reaction.
Conclusions
Our results show that tethering a kinase and a substrate together can increase phosphorylation reaction rates. We show experimentally that the affinity for complex assembly plays a central role in the observed rates; if the affinity is weak compared to the effective molarity, a scaffolded complex will not assemble at concentrations where the intramolecular reaction is faster than the intermolecular reaction (Figure 5). This observation follows from a simple, intuitive kinetic model (Figure 4). Somewhat unexpectedly, however, the value of the effective molarity that we observe with a flexible scaffold model, ~0.1 μM, is relatively small compared to effective molarities for small molecule and other tethered protein reactions.13,14,20,22,23,26,53 Natural protein-protein affinities span a broad range from low nM to high μM,29,30 and many scaffold-mediated interactions in signaling networks could be well above a 0.1 μM effective molarity threshold.29,30 Endogenous scaffold proteins could potentially overcome this problem with multivalent interactions, which would result in tighter affinities and ensure that complexes assemble in a concentration range where scaffold-mediated reactions can make a significant contribution to reaction rates. Alternatively, natural scaffold proteins can have many functions beyond tethering,1,3,54,55 and it may be that tethering-mediated rate enhancements are simply not the primary function of many scaffold proteins. Additional quantitative biochemical studies with natural scaffold systems will be necessary to explore these possibilities.
Any scaffold-mediated tethering effects that do occur should also depend on cellular concentrations; at substrate concentrations are well above the effective molarity, tethered reaction rates should be small compared to competing untethered reactions (Figure 5). In principle, we could assess whether cellular concentrations of PKA, substrates, and scaffold proteins exist in a range where we would predict significant tethering effects. In practice, however, estimating cellular concentrations for particular cell lines or tissues is challenging. Mass spectrometry data can provide cellular protein abundances, but converting these values to concentration requires knowing how much total protein is free in solution, folded, and active. Defining an appropriate volume is equally challenging, as cells are not well-mixed solutions. Although determining what functional effects are relevant in a given cell is not trivial, we emphasize that the value of in vitro biochemical studies lies more towards allowing us to describe the possible space of functional behaviors that are accessible to biological systems.
While the effective molarities obtained in our minimal model system are small, it is plausible that natural systems could achieve faster tethered reaction rates with different tether designs, or with ordered structures that precisely orient kinase and substrate. Our understanding of scaffold protein structure remains limited, however, and many scaffold proteins are thought to contain intrinsically disordered regions.13,56,57 Future experiments could test whether larger effective molarities can be obtained in engineered systems with different flexible or well-ordered tether designs. Indeed, a recent paper describes an alternative PKA tethering strategy with different orientations, recruitment domains, and substrates that results in significantly faster tethered reaction rates.58 Further studies are needed to determine why conceptually similar design strategies produced distinct behaviors, and to identify the critical structural features responsible for the differing effects. We note that even in a system with different observed rate effects, the relationship between effective molarity and binding affinity that we have described and experimentally characterized here should apply as a useful predictive framework.
While many questions remain open, our work suggests several important conclusions with broad implications. First, many scaffold proteins have been defined by qualitative assays that indicate binding to a kinase and substrate. In the absence of quantitative data for reaction rates and binding constants, however, any inference that the function of these scaffold proteins is to accelerate reaction rates should be viewed with caution. Second, when attempts are made to engineer scaffolds in synthetic networks, careful consideration should be given to both linker structure and interaction affinities. Further studies on both natural and model scaffold proteins will help to expand our understanding of complex signaling networks and improve our ability to engineer effective synthetic systems.
Supplementary Material
Acknowledgements
We thank Dustin Maly, Mike Gelb, Geeta Narlikar, Wendell Lim, Dan Herschlag, Maire Gavagan, Magnus Kjaergaard, and members of the Zalatan group for comments and discussion. This work was supported by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund (#1010814 to J.G.Z.) and NIH R35 GM124773 (J.G.Z.). E.B.S. was funded by a Washington Research Foundation Innovation Postdoctoral Fellowship at the Institute for Protein Design.
Footnotes
Accession Codes
PKA Uniprot P05132
Supporting Information
Supporting information is available free of charge online.
Rate constants for untethered PKA reactions and comparisons to literature values, rate constants and calculated effective molarities for kinase docking interactions from literature values, kinetic assay endpoint standards, KM for ATP, structural schematics, single-turnover kinetics for covalently tethered reactions, off-target phosphorylation controls, measurement of kintra for covalently tethered complexes, data with linkers up to 80 aa, simple effective concentration model calculation, covalently tethered competition reaction data, controls for product inhibition, fluorescence polarization binding assays, observed rates versus enzyme concentration, progress curves for non-covalently tethered reactions, model predictions for non-covalently tethered reactions with varying binding affinity, protein sequences, and a complete derivation of the equation shown in Scheme 1.
References
- (1).Langeberg LK, and Scott JD (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol 16, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bhattacharyya RP, Remenyi A, Yeh BJ, and Lim WA (2006) Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem 75, 655–680. [DOI] [PubMed] [Google Scholar]
- (3).Good MC, Zalatan JG, and Lim WA (2011) Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Elion EA (2001) The Ste5p scaffold. J. CellSci 114, 3967–3978. [DOI] [PubMed] [Google Scholar]
- (5).Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, and Lim WA (2006) The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311, 822–826. [DOI] [PubMed] [Google Scholar]
- (6).Good M, Tang G, Singleton J, Remenyi A, and Lim WA (2009) The Ste5 Scaffold Directs Mating Signaling by Catalytically Unlocking the Fus3 MAP Kinase for Activation. Cell 136, 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zalatan JG, Coyle SM, Rajan S, Sidhu SS, and Lim WA (2012) Conformational control of the Ste5 scaffold protein insulates against MAP kinase misactivation. Science 337, 1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Morrison DK, and Davis RJ (2003) Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol 19, 91–118. [DOI] [PubMed] [Google Scholar]
- (9).McKay MM, Freeman AK, and Morrison DK (2014) Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases 2, 276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dard N, and Peter M (2006) Scaffold proteins in MAP kinase signaling: more than simple passive activating platforms. BioEssays 28, 146–156. [DOI] [PubMed] [Google Scholar]
- (11).Park E, Rawson S, Li K, Kim B-W, Ficarro SB, Pino GG-D, Sharif H, Marto JA, Jeon H, and Eck MJ (2019) Architecture of autoinhibited and active BRAF-MEK1–14-3– 3 complexes. Nature 575, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kondo Y, Ognjenovic J, Banerjee S, Karandur D, Merk A, Kulhanek K, Wong K, Roose JP, Subramaniam S, and Kuriyan J (2019) Cryo-EM structure of a dimeric B-Raf:14– 3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD, and Gonen T (2013) Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. eLife 2, e01319–e01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pellicena P, Stowell KR, and Miller WT (1998) Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J. Biol. Chem 273, 15325–15328. [DOI] [PubMed] [Google Scholar]
- (15).Pellicena P, and Miller WT (2001) Processive Phosphorylation of p130Cas by Src Depends on SH3-Polyproline Interactions. J. Biol. Chem 276, 28190–28196. [DOI] [PubMed] [Google Scholar]
- (16).Miller MPSAAWT (1910) A Peptide Model System for Processive Phosphorylation by Src Family Kinases. Biochemistry 39, 1–7. [DOI] [PubMed] [Google Scholar]
- (17).Kõivomägi M, Örd M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ERM, Rubin SM, and Loog M (2013) Multisite phosphorylation networks as signal processors for Cdk1. Nature 20, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sharma A, Antoku S, Fujiwara K, and Mayer BJ (2003) Functional interaction trap: a strategy for validating the functional consequences of tyrosine phosphorylation of specific substrates in vivo. Mol. Cell. Proteomics 2, 1217–1224. [DOI] [PubMed] [Google Scholar]
- (19).Lieser SA, Aubol BE, Wong L, Jennings PA, and Adams JA (2005) Coupling phosphoryl transfer and substrate interactions in protein kinases. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1754, 191–199. [DOI] [PubMed] [Google Scholar]
- (20).Fernandes N, and Allbritton NL (2009) Effect of the DEF motif on phosphorylation of peptide substrates by ERK. Biochem. Biophys. Res. Commun 387, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sheridan DL, Kong Y, Parker SA, Dalby KN, and Turk BE (2008) Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem 283, 19511–19520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fernandes N, Bailey DE, VanVranken DL, and Allbritton NL (2007) Use of Docking Peptides to Design Modular Substrates with High Efficiency for Mitogen-Activated Protein Kinase Extracellular Signal-Regulated Kinase. ACS Chem. Biol 2, 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Remenyi A, Good MC, Bhattacharyya RP, and Lim WA (2005) The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Molecular Cell 20, 951–962. [DOI] [PubMed] [Google Scholar]
- (24).Rainey MA, Callaway K, Barnes R, Wilson B, and Dalby KN (2005) Proximity-induced catalysis by the protein kinase ERK2. J. Am. Chem. Soc 127, 10494–10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tokunaga Y, Takeuchi K, Takahashi H, and Shimada I (2014) Allosteric enhancement of MAP kinase p38a’s activity and substrate selectivity by docking interactions. Nature 21, 704–711. [DOI] [PubMed] [Google Scholar]
- (26).Lee S, Warthaka M, Yan C, Kaoud TS, Ren P, and Dalby KN (2011) Examining docking interactions on ERK2 with modular peptide substrates. Biochemistry 50, 9500–9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bashor CJ, Helman NC, Yan S, and Lim WA (2008) Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 319, 1539–1543. [DOI] [PubMed] [Google Scholar]
- (28).Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ, and Keasling JD (2009) Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol 27, 753–759. [DOI] [PubMed] [Google Scholar]
- (29).Whitaker WR, Davis SA, Arkin AP, and Dueber JE (2012) Engineering robust control of two-component system phosphotransfer using modular scaffolds. Proc. Natl. Acad. Sci. U.S.A 109, 18090–18095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Park SH (2003) Rewiring MAP Kinase Pathways Using Alternative Scaffold Assembly Mechanisms. Science 299, 1061–1064. [DOI] [PubMed] [Google Scholar]
- (31).Moon J, and Park S-H (2014) Reassembly of JIP1 Scaffold Complex in JNK MAP Kinase Pathway Using Heterologous Protein Interactions. PLoS One 9, e96797–e96797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hobert EM, and Schepartz A (2012) Rewiring Kinase Specificity with a Synthetic Adaptor Protein. J. Am. Chem. Soc 134, 3976–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dueber JE, Yeh BJ, Chak K, and Lim WA (2003) Reprogramming control of an allosteric signaling switch through modular recombination. Science 301, 1904–1908. [DOI] [PubMed] [Google Scholar]
- (34).Ryu J, and Park S-H (2015) Simple synthetic protein scaffolds can create adjustable artificial MAPK circuits in yeast and mammalian cells. Sci. Signal 8, ra66–ra66. [DOI] [PubMed] [Google Scholar]
- (35).Kirby AJ (1980) Effective molarities for intramolecular reactions. Adv. Phys. Org. Chem 17, 183–278. [Google Scholar]
- (36).Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, and Whitesides GM (2007) Dependence of Effective Molarity on Linker Length for an Intramolecular Protein-Ligand System. J. Am. Chem. Soc 129, 1312–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sørensen CS, and Kjaergaard M (2019) Effective concentrations enforced by intrinsically disordered linkers are governed by polymer physics. Proc. Natl. Acad. Sci. U.S.A 116, 23124–23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Das A, Gerlits O, Parks JM, Langan P, Kovalevsky A, and Heller WT (2015) Protein Kinase A Catalytic Subunit Primed for Action: Time-Lapse Crystallography of Michaelis Complex Formation. Structure 23, 2331–2340. [DOI] [PubMed] [Google Scholar]
- (39).Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, and Howarth M (2012) Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. U.S.A 109, E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Picciotto MR, Cohn JA, Bertuzzi G, Greengard P, and Nairn AC (1992) Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem 267, 12742–12752. [PubMed] [Google Scholar]
- (41).Madhusudan Trafny, Xuong EA, Adams NH, Eyck JA, Ten LF, Taylor SS, and Sowadski JM (1994) cAMP-dependent protein kinase: crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 3, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Whitehouse S, Feramisco JR, Casnellie JE, Krebs EG, and Walsh DA (1983) Studies on the kinetic mechanism of the catalytic subunit of the cAMP-dependent protein kinase. J. Biol. Chem 258, 3693–3701. [PubMed] [Google Scholar]
- (43).Taylor SS, Knighton DR, Zheng J, Sowadski JM, Gibbs CS, and Zoller MJ (1993) A template for the protein kinase family. Trends Biochem. Sci 18, 84–89. [DOI] [PubMed] [Google Scholar]
- (44).Adams JA, and Taylor SS (1993) Phosphorylation of peptide substrates for the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem 268, 7747–7752. [PubMed] [Google Scholar]
- (45).Page MI, and Jencks WP (1971) Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. U.S.A 68, 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Van Valen D, Haataja M, and Phillips R (2009) Biochemistry on a Leash: The Roles of Tether Length and Geometry in Signal Integration Proteins. Biophys. J 96, 1275–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Robinson MJ, Harkins PC, Zhang J, Baer R, Haycock JW, Cobb MH, and Goldsmith EJ (1996) Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5’-triphosphate. Biochemistry 35, 5641–5646. [DOI] [PubMed] [Google Scholar]
- (48).Levchenko A, Bruck J, and Sternberg PW (2000) Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. U.S.A 97, 5818–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sørensen CS, Jendroszek A, and Kjaergaard M (2019) Linker Dependence of Avidity in Multivalent Interactions Between Disordered Proteins. J. Mol. Biol 431, 1–12. [DOI] [PubMed] [Google Scholar]
- (50).Reinke AW, Grant RA, and Keating AE (2010) A Synthetic Coiled-Coil Interactome Provides Heterospecific Modules for Molecular Engineering. J. Am. Chem. Soc 132, 6025–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Thompson KE, Bashor CJ, Lim WA, and Keating AE (2012) SYNZIP Protein Interaction Toolbox: in Vitro and in Vivo Specifications of Heterospecific Coiled-Coil Interaction Domains. ACS Synth. Biol 1, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Park WM, Bedewy M, Berggren KK, and Keating AE (2017) Modular assembly of a protein nanotriangle using orthogonally interacting coiled coils. Sci. Rep 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kondo Y, Ognjenovic J, Banerjee S, Karandur D, Merk A, Kulhanek K, Wong K, Roose JP, Subramaniam S, and Kuriyan J (2019) Cryo-EM structure of a dimeric B-Raf:14– 3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nussinov R, Ma B, and Tsai C-J (2013) A broad view of scaffolding suggests that scaffolding proteins can actively control regulation and signaling of multienzyme complexes through allostery. Biochim. Biophys. Acta 1834, 820–829. [DOI] [PubMed] [Google Scholar]
- (55).Mayer BJ, Blinov ML, and Loew LM (2009) Molecular machines or pleiomorphic ensembles: signaling complexes revisited. J. Biol 8, 81.1–81.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Noutsou M, Duarte AMS, Anvarian Z, Didenko T, Minde DP, Kuper I, de Ridder I, Oikonomou C, Friedler A, Boelens R, Rudiger SGD, and Maurice MM (2011) Critical Scaffolding Regions of the Tumor Suppressor Axin1 Are Natively Unfolded. J. Mol. Biol 405, 773–786. [DOI] [PubMed] [Google Scholar]
- (57).Cortese MS, Uversky VN, and Keith Dunker A (2008) Intrinsic disorder in scaffold proteins: Getting more from less. Prog. Biophys. Mol. Biol 98, 85–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Dyla M, and Kjaergaard M (2020) Intrinsically disordered linkers control tethered kinases via effective concentration. bioRxiv doi: 10.1101-2020.04.04.023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.