

Abstract
Alterations of excitatory synaptic function are the strongest correlate to the pathologic disturbance of cognitive ability observed in the early stages of Alzheimer's disease (AD). This pathologic feature is driven by amyloid-β oligomers (Aβos) and propagates from neuron to neuron. Here, we investigated the mechanism by which Aβos affect the function of synapses and how these alterations propagate to surrounding healthy neurons. We used complementary techniques ranging from electrophysiological recordings and molecular biology to confocal microscopy in primary cortical cultures, and from acute hippocampal and cortical slices from male wild-type and amyloid precursor protein (APP) knock-out (KO) mice to assess the effects of Aβos on glutamatergic transmission, synaptic plasticity, and dendritic spine structure. We showed that extracellular application of Aβos reduced glutamatergic synaptic transmission and long-term potentiation. These alterations were not observed in APP KO neurons, suggesting that APP expression is required. We demonstrated that Aβos/APP interaction increases the amyloidogenic processing of APP leading to intracellular accumulation of newly produced Aβos. Intracellular Aβos participate in synaptic dysfunctions as shown by pharmacological inhibition of APP processing or by intraneuronal infusion of an antibody raised against Aβos. Furthermore, we provide evidence that following APP processing, extracellular release of Aβos mediates the propagation of the synaptic pathology characterized by a decreased spine density of neighboring healthy neurons in an APP-dependent manner. Together, our data unveil a complementary role for Aβos in AD, while intracellular Aβos alter synaptic function, extracellular Aβos promote a vicious cycle that propagates synaptic pathology from diseased to healthy neurons.
SIGNIFICANCE STATEMENT Here we provide the proof that a vicious cycle between extracellular and intracellular pools of Aβ oligomers (Aβos) is required for the spreading of Alzheimer's disease (AD) pathology. We showed that extracellular Aβos propagate excitatory synaptic alterations by promoting amyloid precursor protein (APP) processing. Our results also suggest that subsequent to APP cleavage two pools of Aβos are produced. One pool accumulates inside the cytosol, inducing the loss of synaptic plasticity potential. The other pool is released into the extracellular space and contributes to the propagation of the pathology from diseased to healthy neurons. Pharmacological strategies targeting the proteolytic cleavage of APP disrupt the relationship between extracellular and intracellular Aβ, providing a therapeutic approach for the disease.
Keywords: β- and γ-secretase inhibition, Alzheimer's disease, APP KO mice, APP processing, NMDA-dependent synaptic transmission, synaptic plasticity
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder characterized by a progressive cognitive impairment and by the presence of the following two characteristic lesions of the cerebral cortex: neurofibrillary tangles of tau and extracellular deposits of β-amyloid (Aβ) peptides as senile plaques (Cipriani et al., 2011). Extracellular senile plaques are formed from Aβ peptides of different lengths, as follows: the 40-residue peptide Aβ40 represents the most abundant physiological Aβ isoform in the brain, while the pathogenic 42-residue Aβ42 is elevated in AD brains (Näslund et al., 1994). Aβ results from the proteolytic processing of the amyloid precursor protein (APP), a transmembrane protein expressed in neuronal and non-neuronal cells in the CNS. In neurons, APPs are distributed in different subcellular compartments and processed by two routes designated as the nonamyloidogenic and amyloidogenic pathways, one precluding and the other promoting the generation of Aβ peptides (Haass et al., 2012). In the amyloidogenic pathway, APPs are internalized into endocytic compartments and subsequently cleaved by two proteases, β-secretase and γ-secretase, to generate Aβ (Kamenetz et al., 2003). APP primarily matures through the secretory pathway starting from the endoplasmic reticulum to the Golgi apparatus, where it is post-translationally modified before vesicular transport to the cell surface, though it can also be processed in the endoplasmic reticulum/intermediate compartment. Aβ produced in the endoplasmic reticulum is almost exclusively Aβ42 and is not destined for secretion (Greenfield et al., 1999), suggesting that several pools of Aβ are produced by neurons: one deposited extracellularly and the other accumulated intracellularly (LaFerla et al., 2007). Increased production of Aβ is thought to be the initial causative factor leading to the alteration of cognitive function, yet little is known about the contribution of these different pools of Aβ to the progression of the disease.
The ability of neurons to modulate excitatory synaptic strength is believed to be a cellular correlate of learning and memory. By studying the impact of Aβ oligomers (Aβos) on fast excitatory synaptic transmission mediated by the activation of postsynaptic ionotropic glutamate receptors AMPA and NMDA, we may improve our understanding of the causative effect of Aβos on cognitive processes. Extracellular application of diffusible Aβos on rat hippocampal slices blocks NMDA receptor-dependent long-term potentiation (LTP; Walsh et al., 2002). Other studies have confirmed the contribution of extracellular Aβos to alterations of learning and memory processes and synaptic failure (Shankar et al., 2007; Li et al., 2009; Wei et al., 2010). In contrast, several studies revealed that memory impairments in various transgenic models of AD are concomitant with intracellular accumulation of Aβos that precedes plaque formation (Oddo et al., 2003; Bayer and Wirths, 2010). This intracellular accumulation of Aβos is also observable in brains of AD patients with learning deficits (Gouras et al., 2000), suggesting that this specific feature is causal in the alteration of memory processes.
The present study aims at further understanding the impact of these two pools of Aβos on the excitatory neurotransmission. We reveal that extracellular Aβos promote APP processing and a subsequent accumulation of intracellular Aβos that is responsible for the synaptic dysfunctions.
Materials and Methods
Animals
All experiments were conducted in accordance with the European Community Council directives of November 24, 1986 (86/609/EEC) and with the French guidelines on the use of living animals in scientific investigations. Experiments were performed with male Swiss mice from Janvier, male wild-type (WT) C57BL/6 and male Amyloid Precursor Protein Knock-Out (KO) C57BL/6 mice (APPtm1Dbo; The Jackson Laboratory).
Preparation of human amyloid-β solution
The peptide used in these experiments was obtained from human recombinant Aβ1–42 peptide (Bachem) resuspended in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to 1 mm until complete resuspension (Stine et al., 2003). Aβos were prepared by diluting Aβ1-42 peptide to 1 mm in DMSO then to 100 μm in ice-cold HEPES and bicarbonate-buffered saline solution or in artificial CSF (ACSF; in mm: NaCl 119, KCl 2.5, NaH2PO4 1.25, MgSO4 1.3, CaCl2 2.5, NaHCO3 26, and glucose 11) with immediate vortexing and bath sonication and then incubated at 4°C for 24 h with mild agitation. Final solutions of Aβos were prepared by diluting the solution at 100 μm in ACSF, DMEM, or intracellular solution.
Purification of Aβ monomer
The Aβ monomer is purified on a C18 column (200 μl, 5 mg; SPE-Chromabond-HRX C18 ec, Macherey-Nagel). The column was equilibrated with 0.1% trifluoroacetic acid (TFA) in water. Immediately after dilution in DMSO, the Aβ sample was loaded and the column was washed three times with 0.1% TFA. Then, a gradient of acetonitrile from 30% to 60% was applied. Fractions (0.1 ml) were collected. The elution profile was determined by measuring the absorbance at 275 nm. The peak fraction was collected, and the concentration of peptide was determined by absorbance at 275 nm using ɛ275 nm = 1400 M−1 cm−1. The peptide is then stored at −80°C.
Production of histidine-tagged proteins
To make the plasmids for the fusion protein [Aβ-His (histidine)] of murine amyloid protein and the secreted soluble form of APP (sAPP-His), the cDNA containing the sequence for murine Aβ1-42 and human sAPP695α were obtained from synthetic oligonucleotides (containing a NdeI restriction site as forward primers and a PspXI restriction site as reverse primers; Sigma-Aldrich) using overlapping PCR. PCR products were then cloned into a pet28a-vector (Novagen, Merck-Millipore)and subsequently constructed as HIS-murine-Aβ-expressing plasmid (pet28a-murine–Aβ1-42) and HIS-sAPPα-expressing plasmid (pet28a-sAPPα). The resulting plasmids were verified by sequencing Escherichia coli BL21 (DE3) was transformed with the fusion protein plasmids (for either murine–Aβ1-42 or sAPPα) and a single colony chosen to grow a 250 ml starter culture in Luria broth (LB medium) overnight at 37°C. The next day, the 10 ml of culture was diluted in 1 L of LB culture medium. When the culture reached an OD600 of 0.8, isopropyl-β-d-thiogalactopyranoside was added to 1 mm concentration for induction. The culture was grown for an additional 4 h, and the cells harvested by centrifugation at 4000 × g for 20 min. The cell was resuspended in 10 ml of ice-cold PBS and lysed by sonication at ice-cold temperature. The cell extract was then centrifuged at 20,000 × g for 15 min at 4°C. For sAPPα purification, the supernatant was kept, whereas it was discarded for murine–Aβ1-42. In this case, the pellet was resuspended in 10 ml of 8 m urea in PBS and sonicated as previously described before centrifugation at 20,000 × g for 15 min at 4°C. The supernatant (5 ml) was diluted with 15 ml of binding buffer (PBS with 10 mm imidazole at pH 8.0). Before affinity purification using nickel-nitriloacetic acid (NTA) column purification, samples were filtered on 0.45 µm. The Ni-NTA column (3 ml of Protino Ni-NTA Agarose; Macherey-Nagel) was equilibrated with binding buffer before loading the sample on the column. Then the column was washed with the washing buffer (PBS with 30 mm imidazole at pH 8.0) with 5–10 column volumes. The protein was then eluted with the elution buffer (PBS with 500 mm imidazole at pH 7.4). The absorbance at 280 nm was used to monitor the elution, but the concentration of the fusion proteins was estimated by comparing the intensity of the band of the protein on SDS-PAGE with that of a known quantity of BSA. A final concentration of 100 μm was obtained, and aliquots were stored at −80°C. Aliquots from all subsequent purification steps were analyzed by SDS-PAGE, and the identities of sAPPα and murine Aβ1-42 were verified by Western blot using monoclonal antibodies against the N-terminal domain of APP (22C11) or Aβ sequence (4G8), respectively.
Cell lines
Mouse neuroblastoma N2a were cultured in DMEM (Sigma-Aldrich) supplemented with 10% fetal bovine serum (Millipore Sigma), as previously described (Gouras et al., 2010).
Primary culture of cortical neurons
Primary cortical neurons were prepared from Swiss embryonic mice [embryonic day 14 (E14) to E16), as previously described (Léveillé et al., 2008). Cerebral cortices were dissected, dissociated, and cultured in DMEM containing 5% fetal bovine serum, 5% horse serum, and 2 mm glutamine (all from Millipore Sigma) on 24-well plates (Falcon Becton Dickinson Labware Europe) for biochemical experiments. Neurons were seeded on 12 mm coverslips (Dominique Dutscher). Dishes and coverslips were coated with 0.1 mg/ml poly-d-lysine and 0.02 mg/ml laminin (Sigma-Aldrich). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2-95% air (Frandemiche et al., 2014) for 13–15 d in vitro (DIV) before use.
Brain slices preparation
Brain slices were prepared from 20- to 30-d-old mice for patch-clamp recordings and from 3-month-old mice for extracellular recordings. The brains of wild-type Swiss, wild-type C57BL/6 and APP KO mice were removed quickly, and 300-μm-thick sagittal slices containing both cortex and hippocampus were cut in the following ice-cold cutting solution (in mm): KCl 2.5, NaH2PO4 1.25, MgSO4 10, CaCl2 0.5, NaHCO3 26, sucrose 234, and glucose 11, saturated with 95% O2 and 5% CO2) with a Leica VT1200 blade microtome (Leica Microsystems). After the dissection, slices were kept in oxygenated ACSF at 37 ± 1°C for 30 min and then kept at room temperature for at least 1 h before recordings.
Electrophysiological recordings
For patch-clamp experiments, cortical neurons from cultures or somatosensory layer 5 pyramidal neurons were visualized in a chamber on an upright microscope with transmitted illumination and continuously perfused at 2 ml/min with an oxygenated Mg2+-free ACSF as follows (in mm): NaCl 119, KCl 2.5, NaH2PO4 1.25, CaCl2 2.5, NaHCO3 26, and glucose 11, at room temperature. Spontaneous EPSCs (sEPSC) were recorded at a membrane potential of −60 mV with borosilicate glass pipettes of 4–5 MΩ resistance filled with ∼30 μl of an intracellular solution, as follows (in mm): 117.5 CsMeSO4, 15.5 CsCl, 10 TEACl, 8 NaCl, 10 HEPES, 0.25 EGTA, 4 MgATP, and 0.3 NaGTP, at pH 7.3. Signals were acquired using a double EPC 10 Amplifier (HEKA Elektronik) filtered at 2 kHz, sampled at 10 kHz, and analyzed with Patchmaster software (HEKA Elektronik). Recordings were considered stable when the input and access resistances did not change >20% during the experiment. To isolate either NMDA or AMPA/kainite sEPSCs, we used Mg2+-free ACSF containing 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 10 μm), a potent inhibitor of non-NMDA glutamate receptor channels exhibiting IC50 values of 0.1–0.4 μm for AMPA and 1.7–8 μm for kainate receptors (Traynelis et al., 2010) that does not present any cross-reactivity for NMDA receptors (Goldstein and Litwin, 1993) or 2-amino-5-phosphonovalerate (d-APV; 100 μm) an NMDA receptor antagonist with an IC50 values of 0.28 μm (NR2A), 0.46 μm (NR2B), and 1.6 μm (NR2C), respectively. In control condition, we recorded sEPSCs in Mg2+-free ACSF 5 min after whole-cell configuration had been achieved (referred as T0) and 20 min after T0 (referred to as T20). In the extracellular Aβos (eAβos) condition, we recorded sEPSCs in Mg2+-free ACSF at T0 and T20, a perfusion with Mg2+-free ACSF containing Aβos (300 nm). Protocol was similar for d-APT (5 μm) and β-secretase inhibitor (1 μm). In the intracellular Aβos (iAβos) condition, the intracellular solution contained 300 nm Aβos. In this condition, we recorded sEPSCs in Mg2+-free ACSF at T0 and T20. The protocol was similar for 4G8 antibody diluted into the whole-cell recording intracellular solution (1:100 dilution). The final concentration of 4G8 antibody inside the patch pipette was 10 μg/ml. sEPSC and miniature EPSCs (mEPSC) analyses were performed on recordings of 180 s at −60 mV. The amplitude threshold was set at 8 pA, and all the detected events were accepted or rejected on the basis of visual examination. The average frequencies and amplitudes of these events were expressed in picoamperes for sEPSC amplitudes and in hertz for sEPSC frequencies and as percentage of the ratio between values measured at T20 over values at T0.
For LTP experiments, hippocampus was extracted from the slice and transferred in the microscope chamber. Oxygenated ASCF (in mm: NaCl 119, KCl 2.5, NaH2PO4 1.25, MgSO4 1.3, CaCl2 2.5, NaHCO3 26, and glucose 11) was continuously perfused into the chamber (2 ml/min) at 28°C. A borosilicate glass pipette filled with ACSF was attached to the measuring electrode, and a stimulation electrode was also mounted. To induce field EPSP (fEPSP) in the hippocampal CA1 region, the stimulating electrode was placed on the Schaffer collaterals, and the recording electrode was positioned in the striatum radiatum. Test stimuli were delivered once every 15 s, and the stimulus intensity was adjusted to produce 40–50% of the maximal response. A stable baseline was recorded for at least 15 min. LTP was induced by theta burst stimulation (involving five trains with 10 bursts of four pulses delivered at 100 Hz, an interburst interval of 200 ms, and a 20 s interval between each train). Signals were amplified with a double EPC 10 Amplifier (HEKA Elektronik). eAβos and β-secretase inhibitor were added to the perfusion ACSF 20 min before theta burst stimulation. Data in millivolts are normalized and expressed as a percentage of baseline.
Immunoblotting for neuronal lysates
At 13–15 DIV, cortical neurons in culture were treated with Aβos and β-secretase inhibitor intravenously (at 1 μm; Calbiochem) for 30 min. After treatment, neurons were rapidly transferred on ice and rinsed twice with ice-cold PBS then washed with PBS containing 0.1% saponin and rinsed again with ice-cold PBS. Neurons were then lysed with RIPA buffer (50 mm Tris-HCl, pH 7.4; 150 mm NaCl; 1% Triton X-100; 1% sodium deoxycholate; 0.1% SDS; 1 mm EDTA) containing a cocktail of protease and phosphatase inhibitors 1% (v/v). Lysates were dosed for proteins using the BCA method. Samples in loading buffer were boiled for 5 min, and equal amounts of protein (20 µg) were resolved on 4–20% gradient Bis-Tris polyacrylamide precast gels (Bio-Rad) in denaturing conditions. Proteins were transferred to a polyvinylidene difluoride 0.2 µm membranes (Millipore) for 2 h at 4°C. Membranes were blocked with Tris-buffered saline (10 mm Tris and 150 mm NaCl, pH 7.4) containing 0.01% Tween 20 and 5% nonfat dry milk for 1 h at room temperature. Membranes were then incubated overnight at 4°C with the following primary antibodies: APP full-length (1:2000 dilution; catalog #22C11, Millipore); APP C-terminal fragments (CTFs; 1:2000 dilution; catalog #Y188, Abcam); actin (1:2000 dilution; catalog #A2066, Sigma-Aldrich); and Aβ peptide (1:1000; catalog #4G8, Covance). Membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (1:40,000; Jackson ImmunoResearch and Immunotech) for 45 min at room temperature. Specific proteins were visualized with an enhanced chemiluminescence ECL Detection System (Bio-Rad). Chemiluminescence detection was performed with the Bio-Rad Chemidoc system and analyzed with the ImageJ software. In these experiments, Aβos were used at 100 nm.
Membrane/cytosol fractionation
To analyze subcellular distribution of APP fragments, we performed a membrane versus soluble/cytosolic fractionation protocol adapted from Florean et al. (2008). Briefly, cells were permeabilized for 3 min by 50 μm digitonin in an intracellular saline solution (in mm: 130 mm KCl, 10 mm NaCl, 20 mm HEPES, 1 mm MgSO4, and 5 mm succinate, at pH 7.2) containing 50 μm EGTA and protease/phosphatase cocktail inhibitors (Roche/Sigma). The supernatant was then collected and centrifuged for 30 min at 100,000 × g at 4°C. The resulting supernatant was kept as a soluble/cytosolic fraction. The neurons in the wells (membrane fractions) were washed twice with PBS before being collected in RIPA buffer.
Plasmids
pFR-Luciferase (pFR-Luc) reporter vector containing the Firefly Luciferase (FR-Luc) gene under the control of the yeast GAL4 activation sequence, phRL-thymidine kinase (TK) vector containing the renilla luciferase gene (phRL-TK) and pRC-CMV vector containing a cDNA encoding for human APP695 fused to the yeast transcription factor GAL4 (APP695-Gal4) were provided by Prof. R. Sadoul (Grenoble Institut des Neurosciences) and were previously described by Hoey et al. (2009).
The strategy used to obtain the double fluorescently tagged human APP695 Swedish (hAPPswe) mutant chimera was adapted from Sannerud et al. (2011). Using the mcherry-APP-HA-EYFP (enhanced yellow fluorescent protein) pcDNA3 vector (from the laboratory of Wim Annaert, Center for Brain and Disease Research, Leuven, Belgium), which contains the fluorescent mCherry within the ectodomain sequence of human APP695, we performed site-directed mutagenesis to introduce the following mutations (NL833/834 to KM) just before the Aβ sequence according to the manufacturer instructions (Phusion Site-Directed Mutagenesis Kit, Thermo Fisher Scientific). The resulting mcherry-hAPPswe-HA-EYFP pcDNA3 vector was verified by sequencing.
cDNAs of WT human APP695 (APPwt) and the Swedish mutant (APPswe) were cloned into pmcherry-N1 vector (SnapGene) using the BamHI and AgeI restriction sites. All constructions in pmCherry vector were verified by sequencing.
Neuronal transfection
Transfections were performed on cortical neuron cultures after 12 DIV with Mirus TransIT-2020 Transfection Reagent (Euromedex) according to the instructions of the manufacturer. Briefly, for each condition in a 24-well plate, 1 µg of plasmid containing mcherry-hAPPswe-HA-EYFP pcDNA3 vector was mixed with 1.5 µl of transit-2020 reagent in 50 µl of serum-free growth medium and incubated for 30 min at room temperature. Then the mixture was applied to cells, and cultures were returned to the incubator for 48 h.
Two-step transfection was used to study the impact of Aβ overproduction in the neighboring healthy cells. Three micrograms of WT human APP695 (APPwt-mCherry) or the Swedish mutant (APPswe-mCherry) plasmids [mixed with 1 m CaCl2 and HEPES-buffered saline (HBS) buffer] were first applied to cells for 30 min. Then, 3 μg of LifeActin-GFP plasmid (mixed with 1 m CaCl2 and HBS buffer) was added to the cells for 40 min. Transfection medium was replaced with conditioned growth medium, and cultures were returned to the incubator until use at DIV 14–15.
Dual-Glo luciferase reporter gene activity assay for quantification of γ-secretase activity
γ-Secretase activity was measured with the Dual-Glo luciferase reporter gene (Gralle et al., 2009). Twenty-four hours after seeding the clone of N2a into 3.5 cm dishes, cells were cotransfected with pFR-Luc (30 ng), APP695-Gal4 (300 ng), and phRL-TK (5 ng) using the cationic polymer transfection reagent Exgen (Euromedex) according to the protocol of the manufacturer. Dual-Glo luciferase activity assays were performed 48 h after transfection by the quantification of firefly and Renilla luciferase activities (Promega). In all experiments, Firefly Luciferase activity was normalized using the constitutive Renilla luciferase activity. In these experiments Aβos were used at 100 nm.
Immunostaining
After 48 h of transfection, cortical neurons were cultured for 2 h in serum-free growth medium and treated or not with Aβos 100 nm for 30 min. Cultured were then fixed with 4% paraformaldehyde/4% sucrose in 0.1 m PBS, at pH 7.3 for 10 min at 37°C. Cells were washed in PBS and processed for immunostaining. After saturation and permeabilization in 3% BSA/0.1% Triton X-100 in 0.1 m PBS for 30 min, cells were incubated with primary antibodies for 2 h, washed three times in 0.1% Triton X-100 in 0.1 m PBS, and incubated with Life Technologies Alexa Fluor 647-conjugated secondary antibody (1:1000; Thermo Fisher Scientific) for 1 h. After three washes in 0.1% Triton X-100 in 0.1 m PBS, cells were incubated for 5 min with Hoechst stain (catalog #33258, Sigma-Aldrich) for nuclei staining and mounted with DAKO fluorescent mounting medium (DAKO). To characterize subcellular compartments, the followingspecific antibodies were used: mouse monoclonal anti-EEA1 (1:500; G-4, Santa Cruz Biotechnology) for early endosomes, mouse monoclonal anti-LAMP2 antibody (1:500; Proteintech) for lysosomes and mouse monoclonal anti-58K Golgi protein (1:500; 58K-9, GeneTex) for Golgi apparatus.
Confocal imaging of transfected neurons
Transfected neurons were washed twice with HBSS solution and then fixed with 4% paraformaldehyde/5% sucrose in 0.1 m PBS, at pH 7.3 for 10 min at room temperature. Cells were washed in PBS and mounted in fluorescent mounting medium. Preparations were examined on a Zeiss LSM 710 confocal laser-scanning microscope, and images were acquired with a Zeiss Airyscan module with an oil-immersion Plan Apochromat 63× objective (numerical aperture 1.46) to improve lateral resolution (∼140 nm) and signal-to-noise ratios. For illustration, images were merged using ImageJ software (RRID:SCR_003070). Spine density was assessed by using NeuronStudio software, which performed an automatic tracing and reconstruction of neuron structures from confocal image stacks (Icahn School of Medicine at Mount Sinai, New York, NY).
Experimental design and statistical analysis
Statistical analyses were performed with GraphPad version 6.0 software. After determining whether the data follow normal distribution or not with a Shapiro–Wilk normality test, we performed, for normally distributed data, one-way or two-way ANOVA followed by Tukey's multiple-comparisons test. If the data did not display a normal distribution, we performed a Kruskal–Wallis followed by Dunn's multiple-comparisons test as the nonparametric test. Paired comparisons were assessed by Wilcoxon signed-rank tests and Mann–Whitney test for unpaired comparisons. Results are expressed as the mean ± SEM from independent experiments performed separately and corresponding to different cell cultures. Significance was set at 0.05. For electrophysiology experiments “n” corresponds to the number of cells recorded, and “N” to the number of mice used.
Drugs
NBQX, d-APV; (2R)-amino-5-phosphonopentanoate); N-[N-(3,5-difluorophenacetyl)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT), β-secretasepi inhibitor IV, and tetrodotoxin (TTX) werefrom Sigma-Aldrich. 4G8 antibody from BioLegend was diluted (1:100) into the recording solution and infused 20 min before the recording. The final concentration of 4G8 inside the pipette was 10 μg/ml.
Results
Extracellular Aβos perturb spontaneous synaptic activity in primary cultures of mouse cortical neurons
We studied the effect of eAβos at 300 nm on glutamatergic neurotransmission in murine primary cortical neurons. Aβ oligomers were prepared from commercially available peptides and resuspended in artificial CSF. When we performed a Western blot of this solution, we observed the presence of low-molecular weight oligomers of Aβ from dimers to tetramers (Fig. 1). As shown in Figure 2, A and B, eAβos did not modify AMPA/kainate sEPSC amplitudes (T0 = 23.35 ± 5.93 pA; T20 = 23.55 ± 4.62 pA; T20 vs T0: 117.68 ± 21.5%; p = 0,492; n = 10 neurons) but significantly decreased AMPA/kainate sEPSC frequencies (T0 = 0.59 ± 0.10 Hz; T20 = 0.37 ± 0.10 Hz; T20 vs T0: 60.71 ± 10.9%; p = 0.009; n = 10 neurons). Next, we evaluated the effect of eAβos on spontaneously occurring miniature excitatory postsynaptic current (mEPSC) recorded in the presence of TTX that blocks action potential formation and propagation. In contrast to sEPSCs, we did not detect any change on AMPA/kainate mEPSC frequencies (T0 = 0.35 ± 0.09 Hz; T20 = 0.37 ± 0.08 Hz; T20 vs T0: 92.32 ± 6.5%; p = 0.3750; n = 10 neurons; Fig. 2C,D). This result suggested that the reduced frequencies of sEPSCs recorded under eAβos is due to a reduction of neuronal excitability and not to a modification of presynaptic glutamate release probability. Interestingly, the application of eAβos markedly lowered both the amplitudes(T0 = 432.94 ± 63.98 pA; T20 = 252.38 ± 51.62 pA; T20 vs T0: 59.08 ± 7.4%; p = 0.0004; n = 17) and the frequencies (T0 = 0.15 ± 0.03 Hz; T20 = 0.07 ± 0.02 Hz; T20 vs T0: 66.65 ± 7.6%; p = 0.0009; n = 17; Fig. 2E,F) of NMDA sEPSCs. In contrast, a lower concentration of eAβos (100 nm) was still able to reduce NMDA sEPSC amplitudes (T20 vs T0: 63.74 ± 5.98%; p = 0.015; n = 7) but failed to decrease NMDA sEPSC frequencies (T20 vs T0: 96.03 ± 14.17%; p > 0.99; n = 7; Fig. 2F), suggesting a dose-dependent effect of eAβos on sEPSC frequencies. These data indicated that a 20 min exposure of eAβos altered neuronal excitability and selectively reduced NMDA sEPSC amplitudes in primary culture of cortical neurons. Perturbations of cellular excitability have already been reported in transgenic models of AD or after Aβos application (Brown et al., 2011; Marcantoni et al., 2014; Tamagnini et al., 2015). Our next objective was to investigate howeAβos selectively perturb NMDA sEPSC amplitudes.
Figure 1.
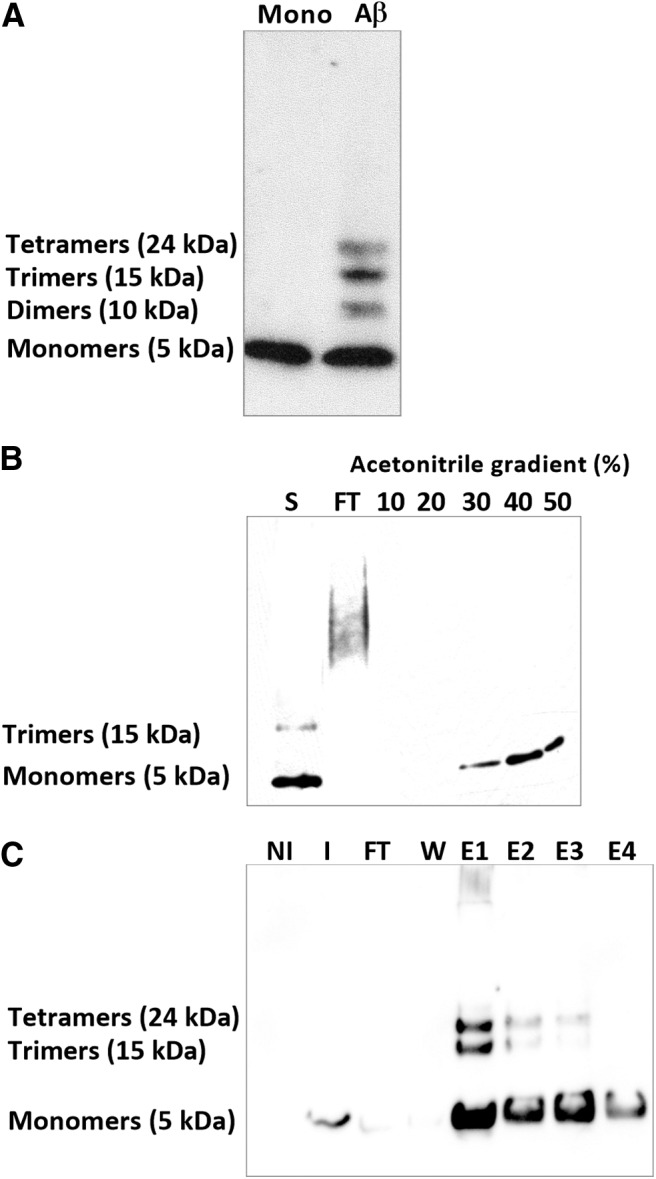
Analysis of experimental Aβ oligomers solution. A, Western blot analysis of experimental solutions of Aβ that display a composition of monomers and various oligomeric forms of Aβ (dimers to tetramers). B, SDS-PAGE analysis of Aβ monomers after purification on the C18 column. All fractions were electrophoresed on 15% tris-glycine gel. Aβ monomers are mainly eluted at 30% acetonitrile. S, Sample loaded; FT, flow through, peptides eluted at 30%, 40%, and 50% acetonitrile. C, SDS-PAGE analysis of murine Aβ after purification on affinity chromatography on a Nickel column. All fractions were electrophoresed on a 15% tris-glycine gel. Aβ monomers are eluted in PBS containing 250 mm imidazole. Solutions of murine Aβ display a composition of monomers, trimers, and tetramers. NI, Noninduced bacterial extract; I, induced bacterial extract FT, flow through; W, washes, peptides eluted in the four first fractions.
Figure 2.
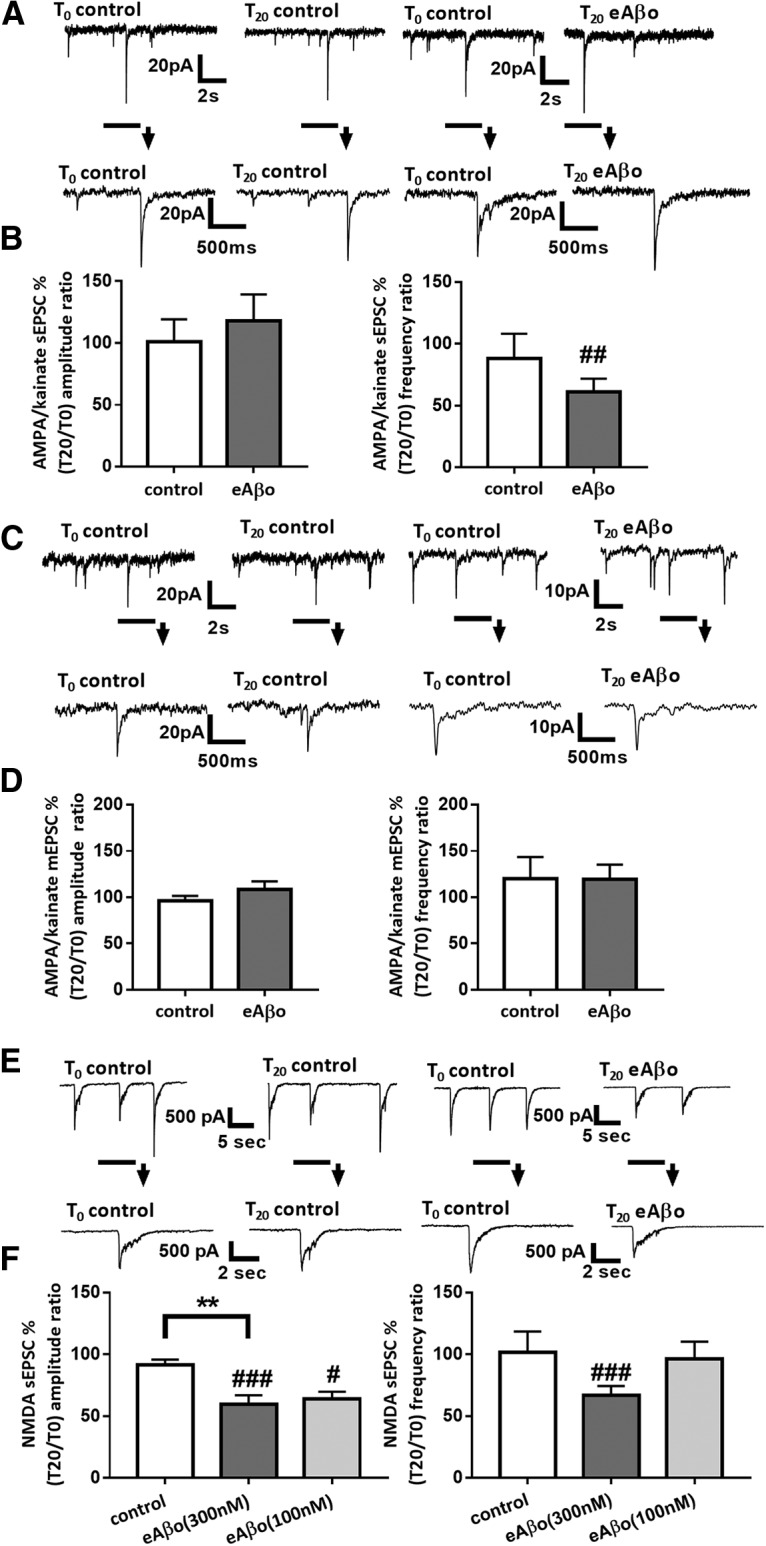
eAβos perturb spontaneous synaptic activity in cultures of mouse cortical neurons. A, Representative traces of AMPA/kainate sEPSCs at T0 and T20 in control condition or with eAβos (300 nm). B, Bar graphs (mean ± SEM) showing the T20/T0 ratio of AMPA/kainate sEPSC amplitudes and frequencies in control condition (white bars; n = 10 neurons, Wilcoxon W = −17(19, −36), p = 0.4316, for sEPSC amplitudes; and Wilcoxon W = −2(13, −15) p = 0.9375 for sEPSC frequencies); with eAβos (gray bars; n = 10 neurons, Wilcoxon W = −15(20, −35), p = 0.4922, for sEPSC amplitudes; and Wilcoxon W = −49(3, −52), p = 0.0098, for sEPSC frequencies). Control versus eAβos (Mann–Whitney U = 44(99, 111), p = 0.6842, for sEPSC amplitudes; and Mann–Whitney U = 38(117, 93), p = 0.3923, for sEPSC frequencies). C, Representative traces of AMPA/kainate mEPSCs at T0 and T20 in control condition or with eAβos (300 nm). D, Bar graphs (mean ± SEM) showing the T20/T0 ratio of AMPA/kainate mEPSCs amplitude and frequency in control condition (white bars; n = 7 neurons, Wilcoxon W = −14(7, −21), p = 0.2969, for mEPSC amplitudes; and Wilcoxon W = 4(5, −1), p = 0.5000, for mEPSC frequencies) or with eAβos (gray bars; n = 6 neurons, Wilcoxon W = 15(30, −15), p = 0.42, for mEPSC amplitudes; and Wilcoxon W = 10(19, −9), p = 0.46, for mEPSC frequencies). Control versus eAβos (Mann–Whitney U = 19 (47, 106), p = 0.13, for mEPSC amplitudes; and Mann–Whitney U = 34(64, 89), p = 0.96, for mEPSC frequencies). E, Representative traces of NMDA sEPSCs at T0 and T20 in control condition or with eAβos (300 nm). F, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitudes and frequencies in control condition (white bars; n = 13 neurons, Wilcoxon W = −55(18, −73), p= 0.0574, for sEPSC amplitudes; and Wilcoxon W = −21 (12, −33), p = 0.2383 for sEPSC frequencies); with eAβos (300 nm; gray bars; n = 17 neurons, Wilcoxon W = −137 (8, −145), p = 0.0004, for sEPSC amplitudes; and Wilcoxon W = −97(4, −101), p = 0.0009, for sEPSC frequencies) or with eAβos (100 nm; light gray bars; n = 7 neurons, Wilcoxon W = −28(0, −28), p = 0.0156, for sEPSC amplitudes; and Wilcoxon W = −1 (1, −2), p > 0.9999, for sEPSC frequencies). One-way ANOVA and Tukey's post hoc test for multiple comparisons (F(2,34) = 6.937; p = 0.0030; control vs eAβos (300 nm), p = 0.0028) for NMDA sEPSC amplitudes and Kruskal–Wallis followed by Dunn's multiple-comparisons test (6.473; p = 0.0393) for NMDA sEPSC frequencies. **p < 0.01; #p < 0.05, ##p < 0.01, ###p < 0.001 relative to the T0 recording normalized to 100%.
eAβos reduce NMDA sEPSC amplitudes in cortical slice neurons from WT but not APP KO mice
Previous studies highlighted the role of APP in extracellular Aβos-mediated toxicity and dysfunction (Lorenzo et al., 2000; Shaked et al., 2006, 2009; Fogel et al., 2014; Puzzo et al., 2017; Wang et al., 2017). In this regard, we investigated whether APP is required for eAβos altering NMDA-dependent synaptic transmission by using cortical slices from WT and APP KO mice. eAβos markedly reduced the amplitudes (T0 = 43.75 ± 7.83 pA; T20 = 29.48 ± 5.05 pA; T20 vs T0: 72.71 ± 6%; p = 0.0017; n = 16 neurons; N = 8 mice) and the frequencies (T0 = 0.82 ± 0.13 Hz; T20 = 0.53 ± 0.07 Hz; T20 vs T0: 71.88 ± 9.7%; p = 0.029; n = 16 neurons; N = 8 mice; Fig. 3A,B) of NMDA sEPSCs recorded in neurons from Swiss WT mice,confirming the data obtained in primary cultured neurons. Moreover, the lack of effect of extracellular application of Aβ monomers (300 nm) on both the amplitudes (T0 = 34.43 ± 4.87 pA; T20 = 37.71 ± 5.48 pA; T20 vs T0: 120.71 ± 19.92%; p = 0.74; n = 8 neurons; N = 4 mice) and the frequencies (T0 = 0.67 ± 0.17 Hz; T20 = 0.72 ± 0.21 Hz; T20 vs T0: 131.18 ± 31.39%; p = 0.64; n = 8 neurons; N = 4 mice; Fig. 3B) of NMDA sEPSCs suggested that the alterations are specially associated with eAβos. In contrast to C57BL/6J wild-type mice, where eAβos reduced both the amplitudes (T0 = 28.62 ± 5.19 pA; T20 = 19.67 ± 5.19 pA; T20 vs T0: 68.44 ± 9.19%; p = 0.031; n = 6 neurons; N = 3 mice) and the frequencies (T0 = 0.71 ± 0.26 Hz; T20 = 0.35 ± 0.17 Hz; T20 vs T0: 54.76 ± 9.95%; p = 0.031; n = 6 neurons; N = 3 mice; Fig. 3D) of NMDA sEPSCs, we did not detect any perturbations in the amplitudes of currents recorded in neurons from APP KO mice (T0 = 30.72 ± 6.88 pA; T20 = 34.22 pA ± 8.07 pA; T20 vs T0: 112.35 ± 9.07%; p = 0.25; n = 8 neurons, N = 5 mice), while we still observed a decrease in the frequencies (T0 = 0.57 ± 0.20 Hz; T20 = 0.19 ± 0.06 Hz; T20 vs T0: 49.34 ± 7.64%; p = 0.007; n = 8 neurons; N = 5 mice; Fig. 3C,D). Thus, the reduction of NMDA sEPSC amplitudes by eAβos required the presence of APP. The reduction of NMDA sEPSC frequencies appeared to be independent of APP expression.
Figure 3.
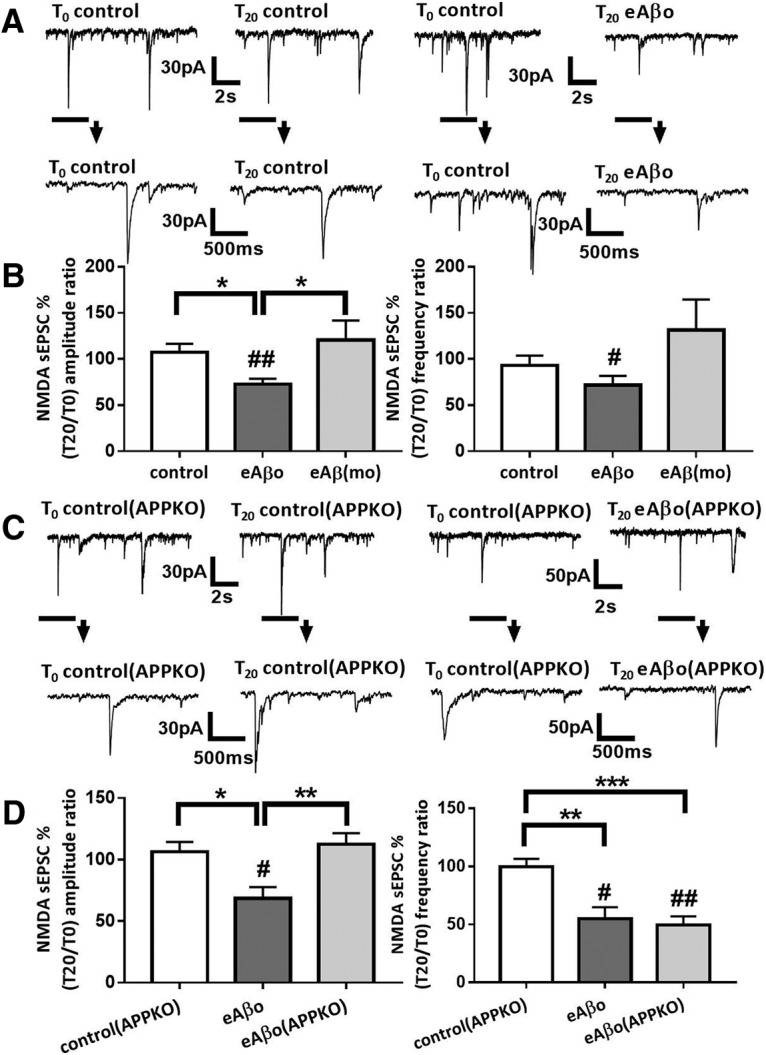
eAβos reduce NMDA sEPSC amplitude in cortical slice neurons from WT but not from APP KO mice. A, Representative traces of NMDA sEPSCs recorded in WT neurons from Swiss mice at T0 and T20 in control condition; with eAβos. B, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitude and frequency recorded in WT neurons from Swiss mice in control condition (white bars; n = 11 neurons, N = 9 mice, Wilcoxon W = 22(44, −22), p = 0.3652, for sEPSC amplitudes; and Wilcoxon W = −16(25, −41), p = 0.5195, for sEPSC frequencies); with eAβos (gray bars; n = 16 neurons, N = 8 mice, Wilcoxon W = −114(11, −125), p = 0.0017, for sEPSC amplitudes; and Wilcoxon W = −84(26, −110), p = 0.0290 for sEPSC frequencies); with eAβ monomers [eAβ(mo), 300 nm; light gray bars; n = 8 neurons, N = 4 mice, Wilcoxon W = 6(21, −15), p = 0.7422, for sEPSC amplitudes; and Wilcoxon W = 8(22, −14), p = 0.6406, for sEPSC frequencies). One-way ANOVA and Tukey's post hoc test for multiple comparisons (F(2,32) = 5.578, p = 0.0084; control vs eAβos; p = 0.0423, eAβos vs eAβ(mo), p = 0.0143] for NMDA sEPSC amplitudes and Kruskal–Wallis followed by Dunn's multiple-comparisons test (5.076, p = 0.0790, for NMDA sEPSC frequencies. C, Representative traces of NMDA sEPSCs recorded in APP KO neurons at T0 and T20 in control condition, with eAβos. D, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitude and frequency recorded in APP KO neurons in control condition (white bars; n = 6 neurons, N = 6 mice, Wilcoxon W = 1(11, −10), p > 0.9999, for sEPSC amplitudes; and Wilcoxon W = −3(9, −12), p = 0.8438, for sEPSC frequencies); in WT neurons from C57BL/6J mice with eAβos (gray bars; n = 6 neurons, N = 3 mice, Wilcoxon W = −21(0, −21), p = 0.0313, for sEPSC amplitudes; and Wilcoxon W = −21(0, −21), p = 0.0313, for sEPSC frequencies); in APP KO neurons with eAβos (light gray bars; n = 8 neurons, N = 5 mice, Wilcoxon W = 18(27, −9), p = 0.2500, for sEPSC amplitudes; and Wilcoxon W = −36(0, −36), p = 0.0078, for sEPSC frequencies). One-way ANOVA and Tukey's post hoc test for multiple comparisons [F(2,17) = 6.792; p = 0.0068; control(APP KO) vs eAβos p = 0.0298; eAβos vs eAβos (APP KO), p = 0.0072] for NMDA sEPSC amplitudes, and one-way ANOVA followed by Tukey's post hoc test for multiple comparisons [F(2,18) = 11.64; p = 0.0006; control(APP KO) vs eAβos, p = 0.0041; control(APP KO) vs eAβos (APP KO), p = 0.0008] for NMDA sEPSC frequencies. *p < 0.05, **p < 0.01, ***p < 0.001; #p < 0.05, ##p < 0.01 relative to the T0 recording normalized to 100%.
Neurons overproducing secreted toxic Aβ affect neighboring neurons through APP
Several studies have shown that Aβ overproduction in one cell leads to Aβ secretion into the extracellular space affecting neighboring healthy cells and could explain the spreading of the pathology across the brain (Wei et al., 2010). Here, we examined the effects of a WT human APP695 (APPwt-mcherry) or the Swedish mutant (APPswe-mCherry), overexpressing neuron, which leads to increased secreted Aβ in the extracellular space (data not shown) on a healthy nearby neuron. The rationale behind this experiment is to see whether the amount of secreted Aβ correlates with the defects induced on the healthy neighboring neuron. For this purpose, we first transfected APPwt-mCherry or APPswe-mCherry plasmids in cultured corticalneurons then, 30 min later, we added LifeActin-GFP (LA-GFP), a peptide that specifically binds filamentous actin. This allowed us to have one neuron expressing both LA-GFP and one of the APP-mCherry (APP neuron) next to a neuron that only expresses LA-GFP (healthy neuron; Fig. 4A). We then examined the spine density of the dendrites of the healthy neuron depending on their distance from the APP neuron. Results showed that neurons overproducing secreted Aβ, namely APPwt-mCherry and APPswe-mCherry overexpressing neurons, decrease the spine density of the nearby healthy neuron. Indeed, the APPwt-mCherry neuron significantly decreased spine density of a healthy neuron from 0 (dendrites from both neurons are overlapping) to 20 µm (distance from APP neuron). APPswe-mCherry neuron had an even stronger impact on healthy neuron by decreasing its spine density from a range of 0–40 µm. These results suggest that, in order for the pathology to propagate from cell to cell, Aβ has to be secreted (Fig. 4B). Furthermore, the distance gradient of the synaptotoxic effect in neurons expressing APPswe suggests a relationship between the synaptotoxicity and the secretion level of Aβ.
Figure 4.

Overexpression of APPwt. APPswe in cortical cell cultures decreases the spine density of neighboring healthy wild-type neuron, in an APP-dependent manner. A, Representative confocal images of cultured cortical neurons where the neuron on the left is overexpressing either LA-GFP only (LA-GFP), APPwt-mCh (APPwt), APPswe-mCh (APPswe), or APP KO neurons that overexpress APPswe-mCh (APP KO+swe), and the neuron on the right is only overexpressing LA-GFP (healthy neuron). Scale bar, 10 µm. B, Bar graphs (mean ± SEM) show the spine density of healthy neurons depending on the distance from (LA-GFP, APPwt, APPswe, or APP KO+swe) neurons (n = at least 3 neurons/condition from three different cultures). Two-way ANOVA and Tukey's post hoc test for multiple comparisons. Spine density of healthy neurons according to the distance from the APP-overexpressing neuron (F(6,168) = 5.309; p < 0.0001; treatment: F(5,168) = 51.6, p < 0.0001, interaction: F(30,168) = 3.484, p < 0.0001). From 0 to 10 μm: LA-GFP versus APPwt, p < 0.0001; LA-GFP vs APPswe, p < 0.0001; APPswe vs APP KO+swe, p < 0.0001. From 20 to 30 μm: LA-GFP vs APPwt, p = 0.0487; LA-GFP vs APPswe, p < 0.0001. From 30 to 40 μm: LA-GFP vs APPswe, p < 0.0001. *p < 0.05, ***p < 0.001 when compared with control condition (both neurons only overexpress LA-GFP) at equivalent distance. ###p < 0.001 when healthy neurons in APP KO plus the swe condition are compared with healthy neurons in the APPswe condition.
Furthermore, we wanted to assess the role of APP in this spreading of synaptotoxic effects. We conducted the same set of experiments using APP knock-out primary cultured cortical neurons. Here, one APP KO neuron is overexpressing both APPswe-mCherry and LA-GFP, and the neighboring healthy APP KO neuron is only overexpressing LA-GFP. In these experiments, we did not observe any spreading of synaptotoxic effects induced by the APPswe-mCherry neuron on the surrounding neurons, suggesting the implication of APP expression in the spreading of the pathology (Fig. 4B).
eAβos induce APP processing through the amyloidogenic pathway
To gain more insight into the functional relationship between eAβos and APP, we blotted endogenous full-length APP and its cleavage products from primary cultures of cortical neurons exposed to eAβos for 30–360 min (Fig. 5A). A 30 min exposure to eAβos was sufficient to produce proteolytic CTFs whileAβ peptides were only detected after 6 h of eAβos exposure. According to these results, we then investigated the effects of eAβos on APP processing by analyzing the production of CTFs in presence of β-secretase inhibitors (Fig. 5B). The ratio of APP proteolytic CTFs over total full-length APP increased when primary cortical neurons were exposed to eAβos (1.71 ± 0.16 a.u.) compared with the control condition (0.78 ± 0.037 a.u.; p < 0.0001; n = 7 independent experiments). This increased proteolytic cleavage of APP due to Aβos was abolished by β-secretase inhibitor at 1 μm (0.63 ± 0.015 a.u.; p = 0.68; n = 4 independent experiments) compared with control (Fig. 5C). These data suggested that eAβos exposure promotes an increase of APP processing.
Figure 5.
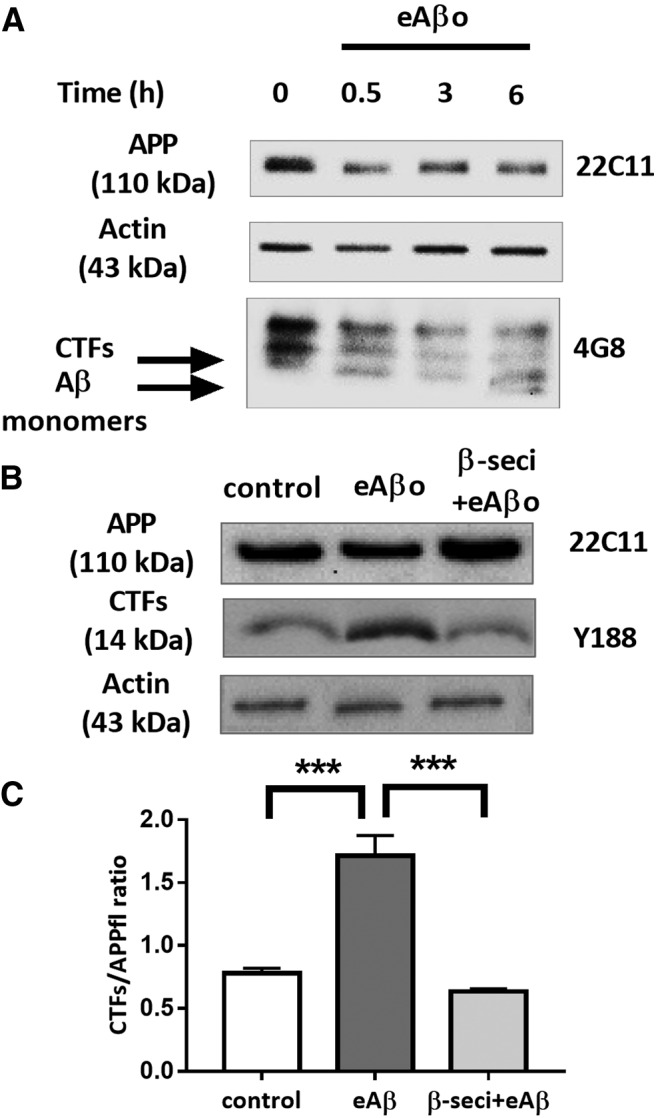
eAβos induce APP processing through amyloidogenic pathway. A, Representative Western blot of endogenous APP and its proteolytic fragments in a whole lysate extract of cortical neurons (14 DIVs) exposed to eAβos for 30 min. B, Top, Representative Western blot of endogenous APP and CTFs in a whole-lysate extract of cortical neurons (14 DIVs) exposed to eAβos with or without β-secretase inhibitor (β-seci; 1 μm) for 30 min. C, Quantification of APP full-length (APPfl) and APP proteolytic CTFs in control (n = 7 independent experiments), in the presence of eAβos (n = 7 independent experiments), in the presence of eAβos plus β-secretase inhibitor (n = 4 independent experiments). Results (mean ± SEM) are expressed as the ratio of APP CTFs over full-length APP. One-way ANOVA and Tukey's post hoc test for multiple comparisons (F(2,15) = 27.83, p < 0.0001; control vs eAβos, p < 0.0001; eAβos vs β-seci+eAβos p < 0.0001. ***p < 0.001.
To further investigate the functional relationship between eAβos and APP, we used a cell-based gene reporter assay to monitor γ-secretase-mediated cleavage of APP (Hoey et al., 2009). This technique uses a reporter APP695 fused at its C terminal to the transcription factor Gal4 (APP695-Gal4). The proteolytic cleavage of APP by α- or β- and γ-secretases produces an amyloid precursor protein intracellular domain-Gal4 fragment that can translocate into the nucleus in which Gal4 induces transcription of a transfected Gal4-dependent Firefly Luciferase reporter gene. γ-Secretase activity was quantified by normalizing FR-Luc luminescence using the constitutive Renilla-TK luciferase activity, which depends on the number of transfected cells. Preliminary experiments were performed to validate the γ-secretase test. These controls consisted in transfected cells with only FR-Luciferase and RL-TK Luciferase. In the presence of APP695-Gal4, Firefly Luciferase activity is ∼380 times stronger than the control, reflecting a clear increase in γ-secretase activity (p < 0.0001; n = 4 independent experiments; Fig. 6A). In the presence of DAPT (5 μm), γ-secretase activity was reduced by 70% when compared with APP695-Gal4 alone (p < 0.0001; n = 4 independent experiments; Fig. 6A). These results validate the γ-secretase test to measure the effect of eAβos on APPprocessing. When we applied eAβos to N2a cells expressing APP695-Gal4, we observed a time-dependent increase in luciferase activity (T60 vs control: 148 ± 2.47%; p < 0.0001; n = 4 independent experiments; Fig. 6B) but not with eAβ monomers (T60 vs control: 105.46 ± 1.04%; p = 0.26; n = 3 independent experiments; Fig. 6B). This result indicates that eAβos enhance APP processing by γ-secretase. Different binding sites for Aβ have been described on APP (Van Nostrand et al., 2002; Khalifa et al., 2010), and it has been shown that Aβ–APP interaction leads to APP dimerization (Fogel et al., 2014). This process promotes APPprocessing and Aβ production by the γ-secretase (Scheuermann et al., 2001; Munter et al., 2007). We tested whether sAPP interferes with the eAβos-induced processing of APP. We preincubated N2a cells expressing APP695-Gal4 with increasing concentration of sAPP (0.1, 1, and 5 nm) and then applied eAβos for 1 h (Fig. 6C). We observed a dose-dependent inhibition in γ-secretase-mediated APP processing in thepresence of sAPP (T60 vs sAPP 5 nm: 105.9 ± 1.16%; p < 0.0001; n = 3 independent experiments). These dataconfirmed that eAβos enhance the amyloidogenic processing of APP and suggest that sAPP may compete with Aβ for binding on APP, thus limiting Aβ-induced APP processing as suggested by a previous publication (Gralle et al., 2009). However, we cannot exclude that sAPP sequesters eAβos and subsequently reduced the effect of eAβos rather than exerting a direct competition between eAβos and sAPP for binding to APP.
Figure 6.
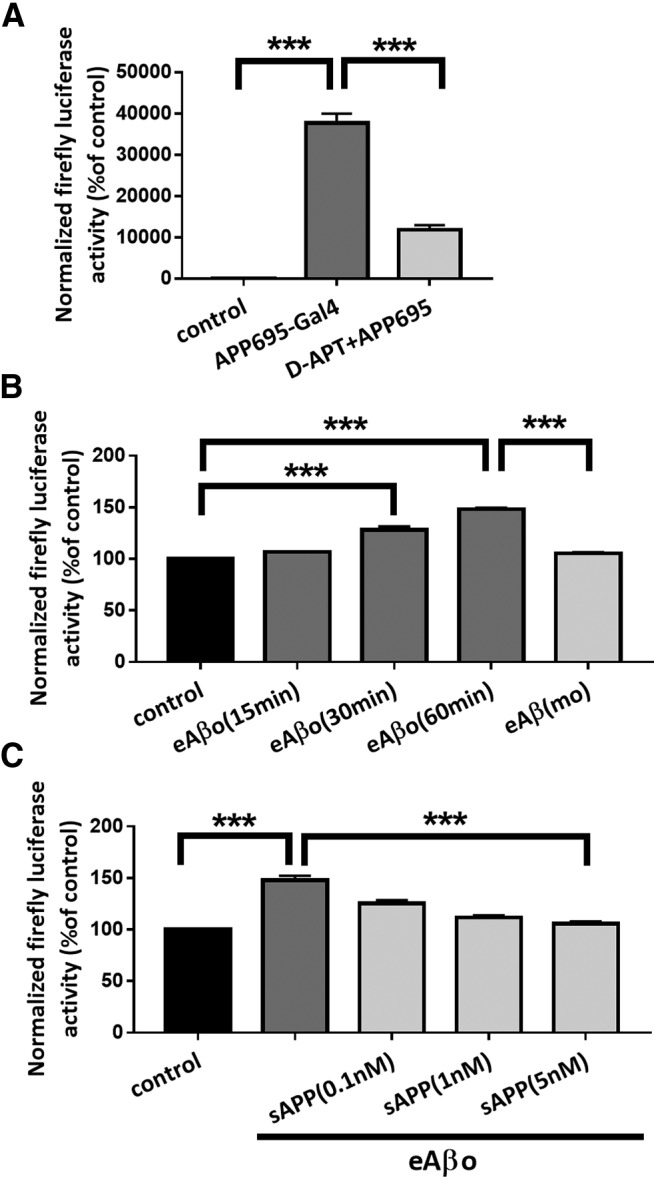
eAβos promote APP processing through γ-secretase activity. A, Quantification of Firefly Luciferase activity when N2a cells were cotransfected with pFR-Luc Firefly Luciferase reporter gene plasmid and phRL-TK Renilla Luciferase plasmid (control; n = 4 independent experiments); cotransfected with pFR-Luc Firefly Luciferase reporter gene plasmid, phRL-TK Renilla luciferase plasmid, and with a plasmid coding for APP695-Gal4 (n = 4independent experiments); cotransfected with pFR-Luc Firefly Luciferase reporter gene plasmid, phRL-TK Renilla Luciferase plasmid, and with a plasmid coding for APP695-Gal4 in the presence of DAPT (5 μm; n = 4 independent experiments). Results are expressed as a percentage of control (N2a not cotransfected with APP695-Gal4). One-way ANOVA and Tukey's post hoc test for multiple comparisons (F(2,9) = 174.3, p < 0.0001; control vs APP695-Gal4, p < 0.0001; APP695-Gal4 vs DAPT+APP695, p < 0.0001). B, Quantification of Firefly Luciferase activity when N2a cotransfected with pFR-Luc Firefly Luciferase reporter gene plasmid, phRL-TK Renilla Luciferase plasmid, and a plasmid coding for APP695-Gal4 were treated with eAβos for 15, 30, and 60 min (n = 4 independent experiments) and with eAβ(mo) for 60 min (n = 3 independent experiments). Results are expressed as a percentage of control (N2a not exposed to eAβos). One-way ANOVA and Tukey's post hoc test for multiple comparisons [F(4,14) = 139.9, p < 0.0001; control vs eAβos (30 min), p < 0.0001; control vs eAβos (60 min), p < 0.0001; eAβos (60 min) vs eAβ(mo), p < 0.0001]. C, Quantification of Firefly Luciferase activity when N2a cells cotransfected with pFR-Luc firefly luciferase reporter gene plasmid, phRL-TK Renilla luciferase plasmid, and a plasmid coding for APP695-Gal4 were pretreated with various concentration of secreted soluble APP fragment (sAPP) and then exposed with eAβos for 60 min (n = 3 independent experiments). Results are expressed as a percentage of control (N2a not exposed to eAβos). One-way ANOVA and Tukey's post hoc test for multiple comparisons [F(4,10) = 138.9, p < 0.0001; control vs eAβos, p < 0.0001; eAβos vs sAPP(5 nm), p < 0.0001]. ***p < 0.001.
Alteration of NMDA sEPSC amplitudes by eAβos depends on APP cleavage by γ-secretase and β-secretase
We then tested in cortical slice neurons whether perturbations of NMDA-dependent synaptic transmission by eAβos require APP processing. We first inhibited APP cleavage by inhibiting γ-secretase with DAPT added in the Mg2+-free ACSF. When we perfused this inhibitor alone at a concentration of 5 μm, we did not detect any modifications of either NMDA sEPSC amplitudes (T20 vs T0: 102.67 ± 3.51%; p = 0.81; n = 7 neurons; N = 4 mice) or frequencies (T20 vs T0: 91.29 ± 22.97%; p = 0.81; n = 7 neurons; N = 4 mice; Fig. 7A,B). In contrast, the reduction of NMDA sEPSC amplitudes induced by eAβos was abolished in the presenceof DAPT (T0 = 38.89 ± 7.3 pA; T20 = 44.53 ± 13.75 pA; T20 vs T0: 113.07± 16.12%; p > 0.9999; n = 10 neurons; N = 5 mice), while we still observed a reduction of NMDA sEPSC frequencies (T0 = 0.61 ± 0.14 Hz; T20 = 0.42 ± 0.10 Hz; T20 vs T0: 73.08 ± 9.61%; p = 0.027; n = 10 neurons; N = 5 mice; Fig. 7A,B).
Figure 7.
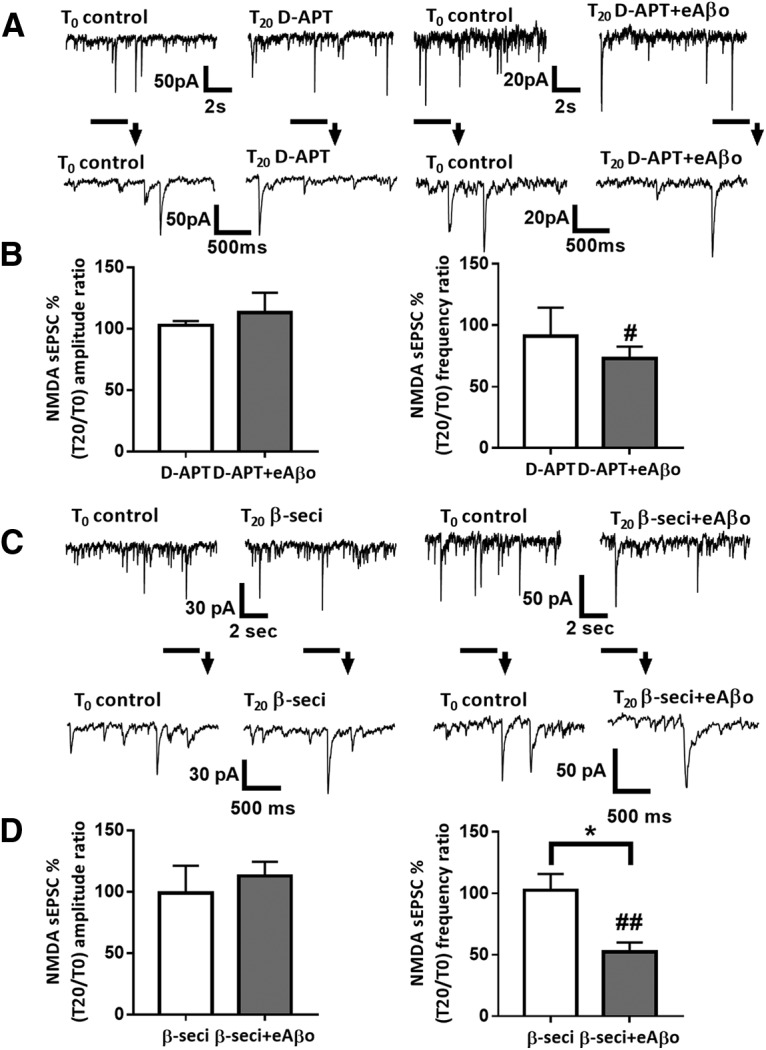
Alteration of NMDA sEPSCs amplitude in cortical slice neurons by eAβos depends on APP cleavage by γ-secretase and β-secretase. A, Representative traces of NMDA sEPSCs at T0 and T20 with DAPT (5 μm); with DAPT (5 μm) plus eAβos. B, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSCs amplitude and frequency with DAPT (white bars; n = 7 neurons, N = 4 mice, Wilcoxon W = 4(16, −12), p = 0.8125, for sEPSCs amplitude; and Wilcoxon W = −4(12, −16), p = 0.8125, for sEPSCs frequency); with DAPT plus eAβos (gray bars; n = 10 neurons, N = 5 mice, Wilcoxon W = 1(11, −10), p > 0.9999, for sEPSCs amplitude; and Wilcoxon W = −43(6, −49), p = 0.0273, for sEPSCs frequency). DAPT versus DAPT plus eAβos (Mann–Whitney U = 33(61, 92), p = 0.8868, for sEPSCs amplitude; and Mann–Whitney U = 30.5(67.5, 85.5), p = 0.6874, for sEPSCs frequency). C, Representative traces of NMDA sEPSCs at T0 and T20 with β-secretase inhibitor (1 μm); with β-secretase inhibitor (1 μm) plus eAβos. D, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitude and frequency with β-secretase inhibitor (white bars; n = 6 neurons, N = 4 mice, Wilcoxon W = 1(11, −10), p > 0.9999, for sEPSCs amplitude; and Wilcoxon W = 1(11, −10), p > 0.9999, for sEPSCs frequency); with β-secretase inhibitor plus eAβos (gray bars; n = 8 neurons, N = 3 mice, Wilcoxon W = 12 (24, −12), p = 0.4609, for sEPSCs amplitude; and Wilcoxon W = −36(0, −36), p = 0.0078, for sEPSCs frequency). β-Secretase inhibitor versus β-secretase inhibitor plus eAβos (Mann–Whitney U = 17(38, 67), p = 0.4136, for sEPSC amplitude; and Mann–Whitney U = 5(64, 41), p = 0.0127, for sEPSC frequency). *p < 0.05; #p < 0.05, ##p < 0.01 relative to the T0 recording normalized to 100%.
Because γ-secretase is involved in both amyloidogenic and nonamyloidogenic pathways, we investigated the pivotal role of APP processing in the effect of eAβos by performing experiments with the β-secretase inhibitor added in the Mg2+-free ACSF. At a concentration of 1 μm, β-secretase inhibitor affected neither NMDA sEPSC amplitudes (T20 vs T0: 98.85 ± 20.68%; p > 0.9999; n = 6 neurons; N = 3 mice) nor frequencies (T20 vs T0: 102.58 ± 12.17%; p > 0.9999; n = 6 neurons; N = 4 mice; Fig. 7C,D). The reduction of NMDA sEPSC amplitudes induced by eAβos was abolished when we inhibited β-secretase cleavage of APP (T0 = 32.91 ± 6.15 pA; T20 = 35.11 ± 5.71 pA; T20 vs T0: 112.67 ± 11.62%; p = 0.46; n = 8 neurons; N = 3 mice), while we still observed a reduction of NMDA sEPSC frequencies (T0 = 0.74 ± 0.09 Hz; T20 = 0.39 ± 0.09 Hz; T20 vs T0: 52.14 ± 8.39%; p = 0.007; n = 8 neurons; N = 3 mice; Fig. 7C,D). Thus, the alteration of NMDA sEPSC amplitudes by eAβos involved APP processing toward the amyloidogenic pathway.
eAβos inhibition of long-term potentiation requires β-secretase cleavage of APP
Activity-dependent facilitation of excitatory synaptic transmission is a prevalent mechanism of synaptic plasticity underlying learning and memory processes in the mammalian brain. This is particularly well documented in the CA1 region of the hippocampus (Malenka and Nicoll, 1999). For instance, theta burst stimulation protocol produces an NMDA-dependent LTP at CA3–CA1 synapses (Grover et al., 2009). Since several studies reported that hippocampal LTP is highly sensitive to Aβos, we investigated whether this synaptic plasticity perturbation induced by eAβos requires APP processing toward the amyloidogenic pathway. We applied a theta burst stimulation protocol to induce LTP at CA3–CA1 synapses on acute hippocampal slices from WT mice. eAβos applied 20 min before theta burst stimulation significantly reduced the potentiation (for the last 5 min of recording: 130.09 ± 5.78 in eAβos condition; n = 12 slices; N = 8 mice; gray circle) compared with the control condition (200.83 ± 15.14%; p = 0.0003; n = 11 slices; N = 6 mice; Fig. 8A, white circle). When we applied the β-secretase inhibitor IV (1 μm), we prevented the alteration of LTP induced by eAβos (183.63 ± 11.35; p = 0.021; n = 8 slices; N = 4 mice; gray triangle), while this inhibitor had no effect by itself when perfused 20 min before theta burst stimulation (194.38 ± 13.48; p = 0.96; n = 9 slices; N = 5 mice; Fig. 8B, white triangle). These results demonstrated that LTP perturbation induced by eAβos in the CA1 region of the hippocampus involved β-secretase cleavage of APP.
Figure 8.
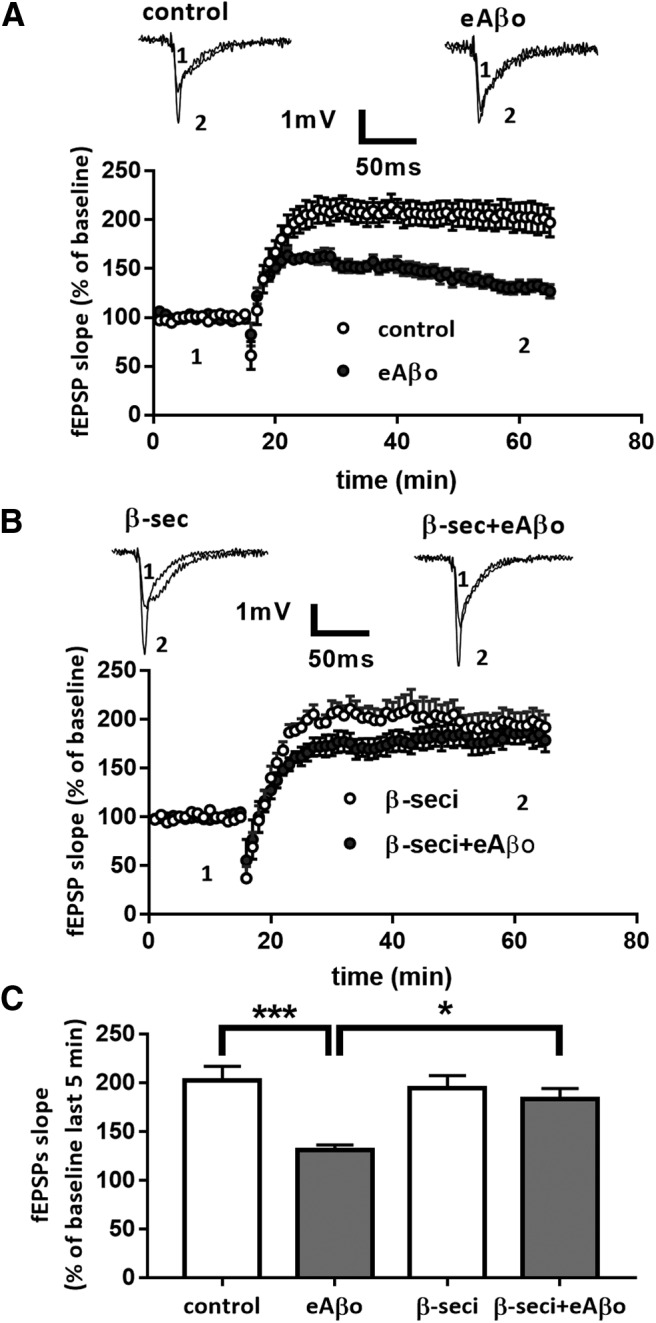
β-Secretase inhibition prevents the long-term plasticity inhibition induced by eAβos. A, Data are the mean (±SEM), and they are expressed as percentages of fEPSP slope baseline in the control condition (white circle; n = 11 slices, N = 6 mice); with eAβos (gray circle; n = 12 slices, N = 8 mice). Representative traces from one experiment are shown. They were extracted at the times indicated (1, 2) on the graph. B, Data are the mean (±SEM), and they are expressed as percentages of fEPSP slope baseline with β-secretase inhibitor (1 μm; white triangle; n = 9 slices, N = 5 mice); with β-secretase inhibitor plus eAβos (1 μm; gray triangle; n = 8 slices, N = 4 mice). Representative traces from one experiment are shown. They were extracted at the times indicated (1, 2) on the graph. C, Summary bar graph depicting the effect of various experimental conditions on LTP. On the graph, data are the mean (±SEM), and they are expressed as percentages of fEPSP slope baseline measured during the 5 last min of recordings in control condition (white bar); with eAβos (gray bar); with β-secretase inhibitor (white bar); and with β-secretase inhibitor plus eAβos (gray bar). One-way ANOVA and Tukey's post hoc test for multiple comparisons: F(3,36) = 8.288, p = 0.0003; control vs eAβos, p = 0.0003; eAβos vs β-seci+eAβos, p = 0.0218. *p < 0.05, ***p < 0.001.
eAβos-induced processing of APP leads to accumulation of cytosolic Aβos
Several studies reported that extracellular Aβos application promotes a production followed by an accumulation of Aβ inside the neurons (Yang et al., 1999; Tampellini et al., 2009), and that intraneuronal accumulation of Aβ is toxic and precedes its extracellular deposition in patients and mouse models of AD (Gouras et al., 2000; Wirths et al., 2001; Takahashi et al., 2002; Oddo et al., 2006). Accordingly, we tested whether eAβos could modify the processing of APP and lead to cytosolic accumulation of APP fragments (Fig. 9). To visualize the processing of APP, we transfected primary cortical neurons with fluorescently tagged APP construct (mcherry-APPswe-EYFP). In this construction, mCherry fluorescent protein was inserted into the ectodomain and YFP fused to the C-terminal domain of APP695 expressing the Swedish mutation (APPswe). Thus, unprocessed APP or partially processed APP (β-secretase cleavage only) appeared in yellow while the presence of red or green puncta reveals processed APP. The location of APP processing to produce Aβ peptides has been ascribed to many organelles, including the endoplasmic recticulum, the trans-Golgi network, endosomes, and lysosomes (Small and Gandy, 2006; Thinakaran and Koo, 2008). These organelles serve as sorting stations for proteins that traffic through the endocytic and secretory pathways. To characterize the site of APP processing, we performed confocal analysis of the fluorescence obtained with specific markers for early endosomes (EEA1), lysosomes (Lamp2), and the Golgi apparatus (58K Golgi protein; Fig. 9B). In presence of eAβos, the number of yellow puncta per neuron significantly increased compared with control (99.7 ± 8.70 vs 41.6 ± 3.14; p < 0.001; n = 10–14), while there was no significant difference regarding the red/green puncta (Fig. 9C). In control cells, yellow puncta localized predominantly to the early endosome as visualized by the EEA1 labeling (39.80 ± 11.11%) with only a subset of yellow puncta colocalized with the lysosome marker Lamp2 (4.15 ± 1.48%) or the Golgi marker 58K (7.065 ± 2.37%; Fig. 9A,C). In eAβos-treated neurons, yellow puncta were redistributed from early endosomes to Golgiapparatus (for early endosomes: 39.80 ± 11.11% in control cells vs 13.72 ± 6.58% in eAβos-treated cells, p = 0.0481; for Golgi: 7.065 ± 2.37% in control cells vs 34.50 ± 6.35% in eAβos-treated cells, p = 0.0495; n = 5–10). Concerning processed APP (red/green puncta), we observed that only 2% are colocalized with early endosomes. The processing of APP occurred preferentially in lysosomes in control conditions (20.57 ± 8.08% vs 6.93 ± 2.49% in eAβos-treated cells) and in Golgi apparatus in the presence of eAβos (62.92 ± 20.24% vs 0% in control cells; p = 0.0017; n = 5-9). These data indicated that eAβos lead to APP accumulation and processing in the Golgi apparatus.
Figure 9.

eAβos-induced processing of APP leads to the accumulation of cytosolic Aβ oligomers. A, Airyscan images of cortical neurons transfected with mcherry-APPsw-EYFP, treated or not with eAβos for 30 min and immunostained with EEA1, LAMP2, and 58K Golgi protein antibodies (blue).White rectangle areas indicate higher magnification used for fluorescent puncta quantification. Red arrows and red stars indicate respectively colocalized yellow puncta or red puncta with EEA1 or LAMP2 vesicles, yellow arrows, and yellow stars indicate, respectively, noncolocalized yellow puncta or red puncta with EEA1 or LAMP2 vesicles. Wide-field scale bar, 5 µm. Magnification scale bar, 1 µm. B, Number of yellow puncta or red/green puncta in cortical neurons treated (gray bars) or not (black bars) with eAβos. For yellow puncta, one-way ANOVA and Tukey's post hoc test for multiple comparisons (F(3,44) = 83.58, p < 0.0001; control vs eAβos, p < 0.001. C, Proportion of yellow or red/green puncta in the different subcellular compartments in cortical neurons treated (gray bars) or not (black bars) with eAβos. For yellow puncta, one-way ANOVA and Tukey's post hoc test for multiple comparisons (F(5,35) = 7.35, p < 0.0001; for early endosomes: control vs eAβos, p = 0.0481; for Golgi apparatus: control vs eAβos, p = 0.0495). For red/green puncta, one-way ANOVA and Tukey's post hoc test for multiple comparisons (F(5,29) = 7.089, p = 0.0002; for Golgi apparatus: control vs eAβos, p = 0.0017. *p < 0.05, **p < 0.01, ***p < 0.001. Data are presented as the mean ± SEM. D, Western blot analysis of APP full-length and APP fragments in membrane or cytosol fractions obtained from cortical neurons exposed or not with eAβos for 30 min.
Next, we investigated the consequences of APP processing in the production ofsoluble/cytosolic Aβ peptides. Control or treated cells were permeabilized with digitonin. The two resulting protein fractions (membrane-bound and soluble/cytosolic) were collected separately and analyzed by Western blot, using 22C11 (directed against the N terminal of APP) and 4G8 (directed against Aβ) antibodies (Fig. 9D).While APP was found in membrane-bound fractions, sAPP appeared in the soluble/cytosolic fractions of eAβos-treated neurons as revealed by the 22C11 antibodies. Furthermore, we also observed an accumulation of Aβ oligomers (trimers and tetramers forms) and a band at 50 kDa that may represent large SDS-stable oligomers in the soluble/cytosol fractions of eAβos-treated neurons compared with control. Together, these data provide evidence that eAβos led to APP processing through the secretory pathway and to a cytosolic accumulation of Aβ oligomers.
Application of intracellular Aβ oligomers perturbs spontaneous synaptic activity in cultures of mouse cortical neurons
From these observations, we decided to evaluate the effects of cytosolic accumulation of Aβos on both AMPA/kainate and NMDA-dependent synaptic transmission by infusing Aβos directly into the cytosol of neurons via the patch pipette. We infused Aβos at 300 nm into cultured neurons through the recording patch pipette (iAβos). In this condition, we recorded sEPSCs in Mg2+-free ACSF at T0 and T20. After 20 min of iAβos, we did not observe any modifications of AMPA/kainate sEPSC amplitudes and frequencies (T0 = 21.65 ± 1.89 pA; T20 = 23.50 ± 4.65 pA; T20 vs T0: 103.93 ± 13.1%; p = 0.96; n = 11 neurons; and T0 = 0.69 ± 0.11 Hz; T20 = 0.65 ± 0.14 Hz; T20 vs T0: 108.93 ± 27.6%; p = 0.46; n = 11 neurons, respectively; Fig. 10A,B). In contrast, iAβos reduced NMDA sEPSC amplitudes (T0 = 681.06 ± 72.23 pA; T20 = 375.06 ± 54.32 pA; T20 vs T0: 54.50 ± 5.1%; p = 0.0039; n = 9 neurons) without affecting NMDA sEPSC frequencies (T0 = 0.10 ± 0.02 Hz; T20 = 0.10 ± 0.02 Hz; T20 vs T0: 97.36 ± 13.5%; p = 0.71; n = 9 neurons; Fig. 10C,D). Similar to eAβos, 20 min of iAβos induced a strong reduction of NMDA but not of AMPA/kainate sEPSC amplitudes in primary cultures of cortical neurons.
Figure 10.
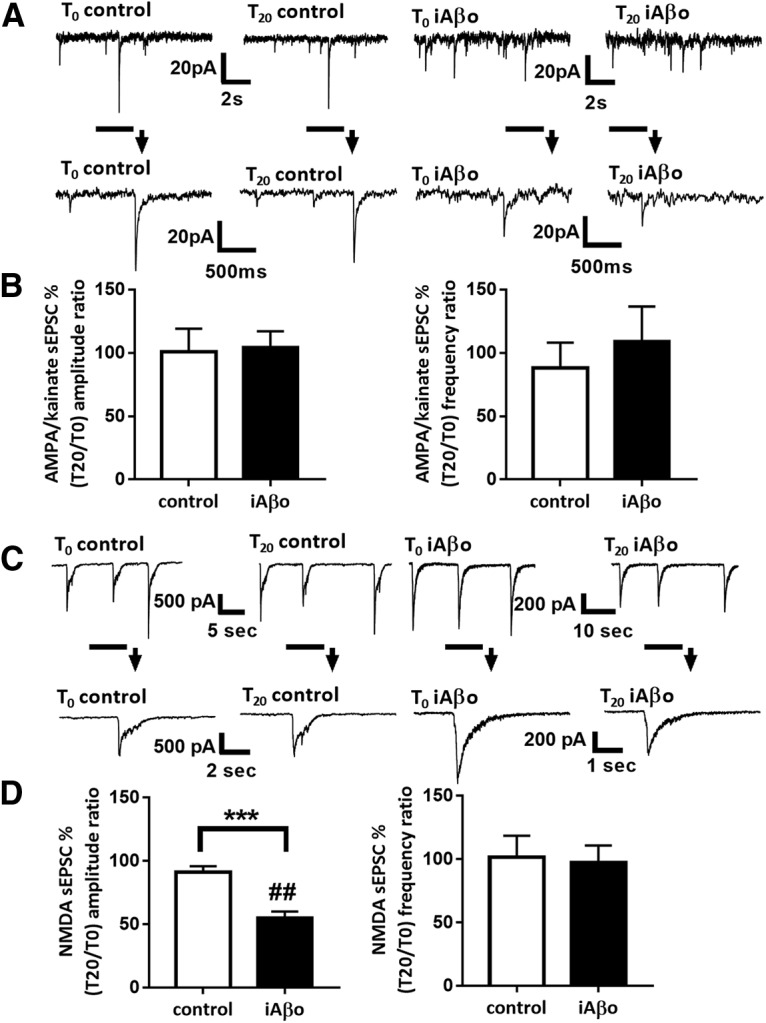
iAβos perturb spontaneous synaptic activity in cultures of mouse cortical neurons. A, Representative traces of AMPA/NMDA sEPSCs at T0 and T20 in control condition or with iAβos (300 nm). B, Bar graphs (mean ± SEM) showing the T20/T0 ratio of AMPA/kainate sEPSC amplitude and frequency in control condition (white bars; n = 10 neurons, Wilcoxon W = −17(19, −36), p = 0.43, for sEPSC amplitudes; and Wilcoxon W = −2(13, −15), p = 0.93, for sEPSC frequencies) or with iAβos (black bars; n = 11 neurons, Wilcoxon W = 2(34, −32), p = 0.9658, for sEPSC amplitudes; and Wilcoxon W = −18(24, −42), p = 0.46, for sEPSC frequencies). Control versus iAβos for AMPA sEPSCs (Mann–Whitney U = 47(102, 129), p = 0.6047, for sEPSC amplitudes; and Mann–Whitney U = 50 (105, 126), p = 0.7561, for sEPSC frequencies). C, Representative traces of NMDA sEPSCs at T0 and T20 in control condition; with iAβos (300 nm). D, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitudes and frequencies in control condition (white bars; n = 13 neurons, Wilcoxon W = −55(18, −73), p = 0.0574, for sEPSC amplitudes; and Wilcoxon W = −21(12, −33), p = 0.2383, for sEPSC frequencies); with iAβos (black bars, n = 9 neurons, WilcoxonW = −45(0, −45), p = 0.0039, for sEPSC amplitudes; and Wilcoxon W = −5(11.5, −16.5), p = 0.7188, for sEPSC frequencies). Control versus iAβos for NMDA sEPSCs (Mann–Whitney U = 6(202, 51), p = 0.0001, for sEPSC amplitudes; and Mann–Whitney U = 54.5(145.5, 107.5), p = 0.8041, for sEPSC frequencies). ***p < 0.001; ##p < 0.01, relative to the T0 recording normalized to 100%.
iAβos reduce NMDA current amplitudes in cortical slice neurons from both WT and APP KO mice
Next, we checked whether iAβos-induced alterations of excitatory neurotransmission in cortical slice neurons from WT and APP KO mice. iAβos reduced NMDA sEPSC amplitudes (T0 = 36.30 ± 6.27 pA; T20 = 22.29 ± 5.43 pA; T20 vs T0: 63.80 ± 8.2%; p = 0.007; n = 8 neurons; N = 6 mice) without affecting NMDA sEPSC frequencies (T0 = 0.68 ± 0.14 Hz; T20 = 0.61 ± 0.22 Hz; T20 vs T0: 81.67 ± 16.4%; p = 0.38; n = 8 neurons; N = 6 mice; Fig. 11A,B) recorded in cortical slice neurons from Swiss WT mice. In a dose–response experiment, we observed that the reduction induced by iAβos was still observed at 50 nm but not at 10 nm (data not shown). This reduction of NMDA sEPSC amplitudes seemed to require Aβos since we failed to observe any modifications of NMDA currents when neurons were infused with a solution containing Aβ monomers (300 nm; T0 = 35.05 ± 7.00 pA; T20 = 24.32 ± 3.58 pA; T20 vs T0: 83.75 ± 9.6%; p = 0.13; n = 10 neurons; N = 4 mice; Fig. 11B). These findings strengthened our previous observation that eAβos effects are due to iAβos accumulation originating from the processing ofmurine APP. Subsequently, because the murine Aβ peptide exhibits a different sequence than human Aβ, we tested the infusion of murine Aβ oligomers (300 nm) on NMDA-dependent sEPSCs. Similar to human Aβos, we observed a reduction of the amplitudes (T0 = 32.33 ± 3.21 pA;T20 = 22.14 ± 1.74 pA; T20 vs T0: 72.86 ± 8.02%; p = 0.019; n = 9 neurons; N = 4 mice) but not of the frequencies (T0 = 0.51 ± 0.06 Hz; T20 = 0.43 ± 0.06 Hz; T20 vs T0: 85.60 ± 6.6%; 0.p = 0.078; n = 9 neurons; N = 4 mice; Fig. 11B) of NMDA-dependent sEPSCs recorded in neurons infused with murine Aβos (iAβ(mu)). In an additional set of experiments, we tested the influence of intracellular accumulation of Aβos in APP KO mice. Interestingly, iAβos also affected the amplitudes (T0 = 35.49 ± 5.07 pA; T20 = 24.10 ± 4.03 pA; T20 vs T0: 77,10 ± 6.39%; p = 0.023; n = 8 neurons; N = 3 mice), but not the frequencies (T0 = 0.63 ± 0.12 Hz; T20 = 0.64 ± 0.10 Hz; T20 vs T0: 105.96 ± 8.03%; p = 0.54; n = 8 neurons; N = 3 mice; Fig. 11C,D) of sEPSCs recorded in cortical slice neurons from APP KO mice. These data demonstrated that a cytosolic accumulation of Aβos significantly affected NMDA-dependent synaptic transmission in both neuronal cultures and cortex slices.
Figure 11.
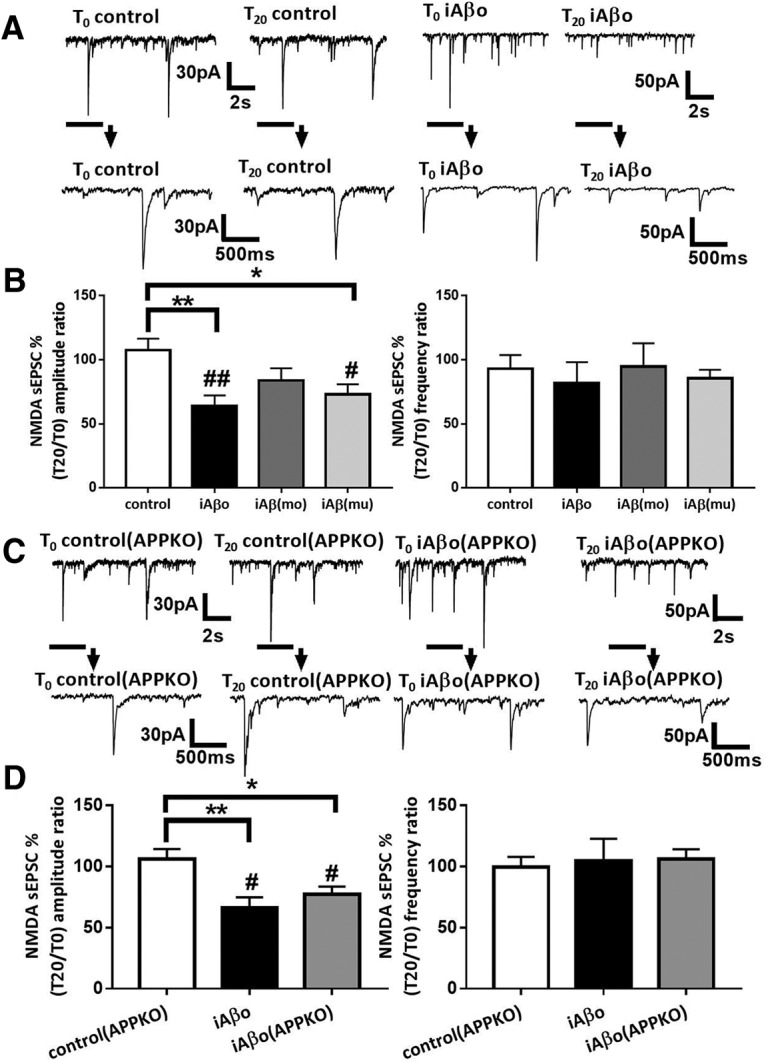
iAβos reduces NMDA currents amplitude in cortical slice neurons from both WT and APP KO mice. A, Representative traces of NMDA sEPSCs recorded in WT neurons from Swiss mice at T0 and T20 in control condition; with iAβos. B, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitude and frequency recorded in WT neurons from Swiss mice in control condition (white bars; n = 11 neurons, N = 9 mice, Wilcoxon W = 22(44, −22), p = 0.3652, for sEPSC amplitudes; and Wilcoxon W = −16(25, −41), p = 0.5195, for sEPSC frequencies); with iAβos (black bars; n = 8, N = 6 mice, Wilcoxon W = −36(0, −36), p = 0.0078, for sEPSC amplitudes; and Wilcoxon W = −14(11, −25), p = 0.3828, for sEPSC frequencies); with iAβmo (gray bars; n = 8, N = 4 mice, Wilcoxon W = −31(12, −43), p = 0.1309, for sEPSC amplitudes; and Wilcoxon W = −9(23, −32), p = 0.6953, for sEPSC frequencies); and with murine iAβ (iAβ(mu)), 300 nm; gray bar; n = 9 neurons, N = 4 mice, Wilcoxon W = −39(42, −3), p = 0.0195, for sEPSC amplitudes; and Wilcoxon W = 26(31, −5), p = 0.0781, for sEPSC frequencies). One-way ANOVA and Tukey's post hoc test for multiple comparisons (F(3,34) = 4.555, p = 0.0087; control vs iAβos, p = 0.0090; control vs (iAβ(mu)), p = 0.0412, for NMDA sEPSCs amplitudes) and one-way ANOVA followed by Tukey's post hoc test for multiple comparisons (F(3,34) = 0.1971, p = 0.8977 for NMDA sEPSC frequencies). C, Representative traces of NMDA sEPSCs recorded in APP KO neurons at T0 and T20 in control condition, with iAβos. D, Bar graphs (mean ± SEM) showing the T20/T0 ratio of NMDA sEPSC amplitudes and frequencies recorded in APP KO neurons in control condition (white bars; n = 6 neurons, N = 6 mice, Wilcoxon W = 1(11, −10), p > 0.9999, for sEPSC amplitudes; and Wilcoxon W = −3(9, −12), p = 0.8438, for sEPSC frequencies); in WT neurons from C57BL/6J mice with iAβos (black bars; n = 7 neurons, N = 3 mice Wilcoxon W = −28(0, −28), p = 0.0156, for sEPSC amplitudes; and Wilcoxon W = 3(12, −9), p = 0.8438, for sEPSC frequencies), and APP KO neurons with iAβos (gray bars; n = 8 neurons, N = 3 mice, Wilcoxon W = −32(2, −34), p = 0.0234, for sEPSC amplitudes; and Wilcoxon W = 10(23, −13), p = 0.5469, for sEPSCs frequencies). One-way ANOVA and Tukey's post hoc test for multiple comparisons [F(2,18) = 6.754, p = 0.0065; control(APP KO) vs iAβos, p = 0.0058; control(APP KO) vs iAβos (APP KO), p = 0,0382, for NMDA sEPSC amplitudes; and one-way ANOVA followed by Tukey's post hoc test for multiple comparisons (F(2,18) = 0.07,348, p = 0.9294, for NMDA sEPSC frequencies). *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 relative to the T0 recording normalized to 100%.
Intracellular infusion of cortical neurons with an antibody directed againstAβ prevents the inhibition ofNMDA-dependent synaptic transmission induced by eAβos
Since murine iAβos promoted a decrease of NMDA sEPSC amplitudes, we investigated whether eAβos-induced reduction of NMDA synaptic transmission was linked to an accumulation of newly produced endogenous Aβos. For this purpose, the 4G8 antibody directed against the sequence 17–24 of Aβ peptide was infused in the neurons through the recording patch pipette. The 4G8 antibody has been shown previously to completely prevent the block of LTP by Aβos through a rapid and direct neutralization of the peptide (Klyubin et al., 2005). Remarkably, 4G8 antibody (1:100, 10 μg/ml) infusion into cortical slice neurons affected neither NMDA sEPSC amplitudes (T20 vs T0: 91.22 ± 4.5%; p = 0.68; n = 6 neurons; N = 3 mice) nor frequencies (T20 vs T0: 91.31 ± 3%; p = 0.43; n = 6 neurons; N = 3 mice), whereas it prevented the inhibition of NMDA sEPSC amplitudes induced by eAβos (T0 = 30.36 ± 6.74 pA; T20 = 29.09 ± 5.96 pA; T20 vs T0: 97.13 ± 2.52%; p = 0.195; n = 8 neurons; N = 6 mice), but not the frequencies reduction (T0 = 0.90 ± 0.24 Hz; T20 = 0.50 ± 0.16 Hz; T20 vs T0: 58.49 ± 3.71%; p = 0.023; n = 8 neurons; N = 6 mice; Fig. 12A,B). These data suggested that NMDA current alterations induced by eAβos depend on newly produced Aβ that accumulates intracellularly after APP processing.
Figure 12.
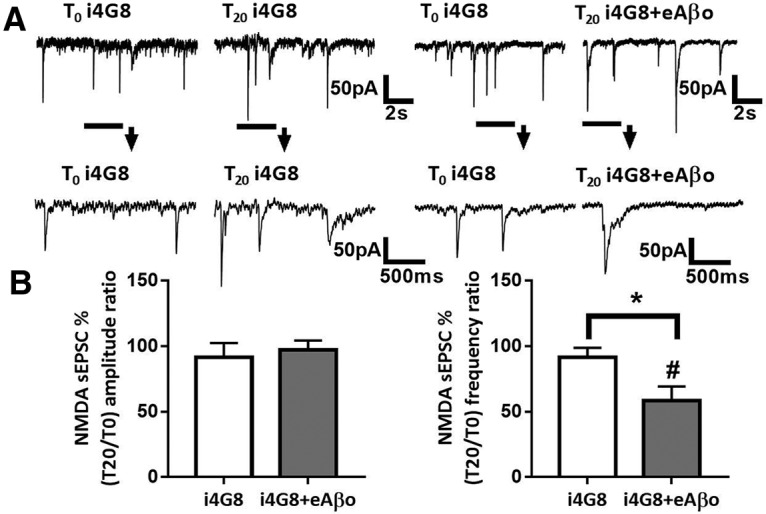
Inhibition of NMDA-dependent synaptic transmission by eAβos is prevented by an antibody directed against Aβ infused into neurons. A, Representative traces of NMDA sEPSCs at T0 and T20 with i4G8 antibody (1:100, 10 μg/ml); with i4G8 antibody (1:100, 10 μg/ml) plus eAβos. B, Bar graphs (mean ± SEM) showing the T20/T0 ratio of sEPSC amplitude and frequency with i4G8 antibody (white bars; n = 6 neurons, N = 3 mice, Wilcoxon W = −5(8, −13), p = 0.68, for sEPSC amplitudes; and Wilcoxon W = −7(4, −11), p = 0.43, for sEPSC frequencies); i4G8 antibody plus eAβos (black bars; n = 8 neurons, N = 6 mice, Wilcoxon W = −20(8, −28), p = 0.19, for sEPSC amplitudes; and Wilcoxon W = −32(2, −34), p = 0.02, for sEPSC frequencies). i4G8 versus i4G8 plus eAβos (Mann–Whitney U = 21(42, 63), p = 0.75, for sEPSC amplitudes; and Mann–Whitney U = 8(61,44), p = 0.04, for sEPSC frequencies). *p < 0.05; #p < 0.05 relative to the T0 recording normalized to 100%.
Discussion
The impact of extracellular Aβos on excitatory synaptic transmission has been extensively studied to further understand the mechanism of action of Aβos in the disruption of learning and memory processes associated with AD. If a consensus has emerged that validates the idea that Aβos application negatively affects excitatory neurotransmission, questions remain about the molecular mechanism involved.
In our study, we demonstrated that 20 min of eAβos significantly affect glutamatergic synaptic transmission by inducing a selective decrease of NMDA sEPSC amplitudes and frequencies. The reduction of NMDA sEPSC amplitudes but not the frequencies requires the presence of APP and the activity of β- and γ-secretases, two enzymes involved in the amyloidogenic pathway. The processing of APP driven by eAβos promotes a subsequent accumulation of Aβos into the cytosol, leading to the reduction of synaptic NMDA currents.
Among the potential membrane receptors of Aβos (Jarosz-Griffiths et al., 2016), several reports described that the peptide interacts with the extracellular domain of APP (Shaked et al., 2006, 2009; Fogel et al., 2014; Puzzo et al., 2017). Such an interaction may occur in our model since eAβos-induced γ-secretase activity is abolished by the extracellular application of sAPPα that can interact with Aβos and compete with its binding on APP. These results are strengthened by the observation that the effects of eAβos on NMDA sEPSCs are prevented in cortical neurons from APP KO mice. In a recent article, Wang et al. (2017) demonstrate that Aβos-containing AD brain extracts induced blockade of hippocampal LTP and depends on APP expression. Several studies have described a selective reduction of NMDA receptor function by Aβos via a decreased membrane expression of memory-related NMDA receptors (Snyder et al., 2005), even if a direct reduction of NMDA receptor function (Chen and Roche, 2007) without alterations of GluN2A and GluN2B expression at the synaptic level cannot be excluded (Frandemiche et al., 2014). Our results also showed that mutant APP overexpression, which leads to increased production of Aβ in the extracellular space induces synaptotoxicity in the nearby “healthy” neurons. This effect seemed to be proportional to the amount of Aβ produced. Indeed, APPs we-overexpressing neurons, which display an increased production of Aβ when compared with APPwt-overexpressing neurons, induced a more pronounced synaptotoxic effect on the healthy neuron. These results suggest that a relationship exists between the neuronal production of Aβ and the severity of the synaptotoxic effect observed in healthy neurons. When we conducted the same experiments using APP KO cortical neurons, the healthy APP KO neuron was not affected by the nearby Aβ-secreting neuron. This suggests that the effects observed on the healthy wild-type neuron require APP expression. Together, these data reveal that genetic ablation of APP prevents Aβos-mediated synaptic dysfunction. Subsequently, we showed that the effect of eAβos on NMDA currents requires the presence of not only APP but also its cleavage through the amyloidogenic pathway. Indeed, the impacts of eAβos on NMDA sEPSC amplitudes and LTP in the hippocampus are reversed by treatment with γ- and/or β-secretase inhibitors, highlighting a causal role of the amyloidogenic pathway in these processes and strengthening the therapeutic interest of the pharmacological blockade of the APP processing pathway. Our observations are strengthened by a biochemical assay in which we observed that eAβos promote APP processing toward the amyloidogenic pathway, revealing a vicious circle triggered by eAβos that may contribute to the transmission of the disease from sick to healthy neurons. In a recent article, Puzzo et al. (2017), also confirm the requirement of APP expression in Aβos-mediated synaptic dysfunction even if APP processing does not seem to be involved. In the amyloidogenic pathway, APPs are internalized into endocytic compartments and subsequently cleaved to generate Aβ. In pathologic conditions, the accumulation of Aβos inside the endocytic system perturbs the function and induces a loss of membrane permeability of endosomal/lysosomal compartments (Yang et al., 1998; Willén et al., 2017). The loss of membrane permeability is correlated with a release of Aβos into the cytosol (Yang et al., 1998). Our data highlight another mechanism in which eAβos also promote APP processing during the secretory pathway resulting in the accumulation of cytosolic Aβ oligomers (Fig. 7). From these observations, we evaluated the effects of cytosolic Aβos accumulation by infusing Aβos (300 nm) directly into the cytosol of neurons via the patch pipette. We observed that, similar to eAβos, 20 min of iAβos induce a reduction of NMDA sEPSC amplitudes in neurons from both cultures and acute cortical slices. This effect was still detected at 50 nm iAβos but lost at 10 nm (data not shown). These results demonstrate that a cytosolic infusion of Aβos may disrupt synaptic glutamatergic transmission and subsequently the ability of neurons to induce synaptic plasticity in AD pathology. The implication of intracellular Aβos in the physiopathological pathways that sustain AD-related cellular alteration has been identified in many animal models of AD. Consistent with our data, previous reports related that Aβos application on neuronal culture promotes production and intracellular accumulation of Aβ (Yang et al., 1999; Tampellini et al., 2009). Similarly, the Osaka (E613Del) mutation of APP identified in a Japanese pedigree showing Alzheimer's-type dementia is associated with massive intracellular Aβos accumulation that impairs organelle transport and induces dramatic dendritic spine loss in neurons likely contributing to synaptic pathology in AD (Umeda et al., 2015). How intracellular accumulation of Aβos perturb these neuronal functions remains to be fully demonstrated. Several studies described the presence of intracellular Aβ and recently, high-resolution imaging techniques have localized Aβ oligomers inside postsynaptic densities of excitatory synapses in AD mouse models (Gouras et al., 2010; Capetillo-Zarate et al., 2011; Pickett et al., 2016) that may uncouple NMDA receptors function and/or synaptic expression.
The findings showing that reduction of current frequencies was not normalized by treatment with inhibitors of Aβ-producing secretases suggest that eAβos may exert differential effects on the presynaptic and postsynaptic compartments of the synapse. Indeed, our data reveal that eAβos reduced the frequency of spontaneous but not miniature AMPA/kainate EPSCs recorded in the presence of TTX, suggesting an alteration of neuronal excitability. Perturbations of cellular excitability have already been reported in transgenic models of AD or after Aβos application. Indeed, it has been shown that Aβ-overproducing transgenic mice present a reduction in somatic Na+ current (Brown et al., 2011) or an increase in K+ maximal conductance (Tamagnini et al., 2015). Similarly, in another transgenic mouse model of AD, neurons involved in the major input to the entorhinal cortex exhibited a decreased firing frequency during depolarization, as well as an increased spike frequency adaptation (Marcantoni et al., 2014). Thus, a perturbation of action potential properties might lead to the Aβos-driven reduction of sEPSC frequencies described in our study, but additional experiments will be required to confirm this hypothesis. More particularly, it was reported that Aβ oligomers modulate voltage-gated calcium and potassium channels directly or indirectly, by changing the properties of the membrane (Lioudyno et al., 2012). Thus, a direct effect of eAβos on sodium or potassium channels could reduce sEPSC frequencies through a perturbation of action potential properties.
In summary, our study suggests that APP processing promoted by eAβos is followed by cytosolic accumulation of Aβ oligomers responsible for the alteration of glutamatergic neurotransmission. These results strengthen the point of view that synaptic dysfunction in AD is closely linked to the accumulation of intracellular Aβos. The result showing that the intracellular infusion of 4G8 antibody inhibits the negative impact of eAβos reinforces this scheme. Moreover, APP processing also produces a releasable pool of Aβos into the extracellular space that may contribute to the transfer of pathology from pathologic to healthy neurons and to a larger scale to the spreading of AD in different brain structures. Finally, it will be necessary to further determine how the iAβ-dependent reduction of NMDAR sEPSC amplitude is responsible for Aβ-induced cognitive dysfunction in Alzheimer's disease. Pharmacological strategies targeting this functional relationship between extracellular and intracellular Aβos may represent a novel therapeutic approach to the early stage of the disease.
Footnotes
This work was supported by Grenoble Alpes University; Institut National de la Santé et de la Recherche Médicale; Région Auvergne-Rhône-Alpes; Grant ANR-15-IDEX-02 NeuroCoG from Agence Nationale de la Recherche (F.L.), in the framework of the “Investissements d'avenir” program and Grant ANR MALAAD program; GREnoble Excellence in Neurodegeneration (GREEN); R.P. was a fellow of Fondation pour la Recherche Médicale; and Fondation Vaincre Alzheimer. We would like to acknowledge Prof. Remy Sadoul for providing plasmids to quantify gamma secretase activity. We also thank Dr. Mireille Albrieux and Prof. Michel Vignes for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Bayer TA, Wirths O (2010) Intracellular accumulation of amyloid-beta—a predictor for synaptic dysfunction and neuron loss in Alzheimer's disease. Front Aging Neurosci 2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Chin J, Leiser SC, Pangalos MN, Randall AD (2011) Altered intrinsic neuronal excitability and reduced Na+ currents in a mouse model of Alzheimer's disease. Neurobiol Aging 32:2109.e1–14. 10.1016/j.neurobiolaging.2011.05.025 [DOI] [PubMed] [Google Scholar]
- Capetillo-Zarate E, Gracia L, Yu F, Banfelder JR, Lin MT, Tampellini D, Gouras GK (2011) High-resolution 3D reconstruction reveals intra-synaptic amyloid fibrils. Am J Pathol 179:2551–2558. 10.1016/j.ajpath.2011.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Roche KW (2007) Regulation of NMDA Receptors by Phosphorylation. Neuropharmacology 53:362–368. 10.1016/j.neuropharm.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani G, Dolciotti C, Picchi L, Bonuccelli U (2011) Alzheimer and his disease: a brief history. Neurol Sci 32:275–279. 10.1007/s10072-010-0454-7 [DOI] [PubMed] [Google Scholar]
- Florean C, Zampese E, Zanese M, Brunello L, Ichas F, De Giorgi F, Pizzo P (2008) High content analysis of γ-secretase activity reveals variable dominance of presenilin mutations linked to familial Alzheimer's disease. Biochim Biophys Acta 1783:1551–1560. 10.1016/j.bbamcr.2008.03.012 [DOI] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O'Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, Isacoff EY, Hirsch JA, Slutsky I (2014) APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep 7:1560–1576. 10.1016/j.celrep.2014.04.024 [DOI] [PubMed] [Google Scholar]
- Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lanté F, Buisson A (2014) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci 34:6084–6097. 10.1523/JNEUROSCI.4261-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JM, Litwin LC (1993) NBQX is a selective non-NMDA receptor antagonist in rat hippocampal slice. Mol Chem Neuropathol 18:145–152. 10.1007/BF03160028 [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Aβ42 accumulation in human brain. Am J Pathol 156:15–20. 10.1016/S0002-9440(10)64700-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E (2010) Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol 119:523–541. 10.1007/s00401-010-0679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralle M, Botelho MG, Wouters FS (2009) Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem 284:15016–15025. 10.1074/jbc.M808755200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H (1999) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A 96:742–747. 10.1073/pnas.96.2.742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Kim E, Cooke JD, Holmes WR (2009) LTP in hippocampal area CA1 is induced by burst stimulation over a broad frequency range centered around delta. Learn Mem 16:69–81. 10.1101/lm.1179109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2:a006270. 10.1101/cshperspect.a006270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey SE, Williams RJ, Perkinton MS (2009) Synaptic NMDA receptor activation stimulates α-secretase amyloid precursor protein processing and inhibits amyloid-β production. J Neurosci 29:4442–4460. 10.1523/JNEUROSCI.6017-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM (2016) Amyloid-β receptors: the good, the bad, and the prion protein. J Biol Chem 291:3174–3183. 10.1074/jbc.R115.702704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R, Point W (2003) APP processing and synaptic function. Neuron 37:925–937. 10.1016/S0896-6273(03)00124-7 [DOI] [PubMed] [Google Scholar]
- Khalifa NB, Van Hees J, Tasiaux B, Huysseune S, Smith SO, Constantinescu SN, Octave JN, Kienlen- Campard P (2010) What is the role of amyloid precursor protein dimerization? Cell Adh Migr 4:268–272. 10.4161/cam.4.2.11476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Walsh D, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ (2005) Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat Med 11:556–561. 10.1038/nm1234 [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci 8:499–509. 10.1038/nrn2168 [DOI] [PubMed] [Google Scholar]
- Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A (2008) Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J 22:4258–4271. 10.1096/fj.08-107268 [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801. 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioudyno MI, Broccio M, Sokolov Y, Rasool S, Wu J, Alkire MT, Liu V, Kozak JA, Dennison PR, Glabe CG, Lösche M, Hall JE (2012) Effect of synthetic Aβ peptide oligomers and fluorinated solvents on Kv1.3 channel properties and membrane conductance. PLoS One 7:e35090 10.1371/journal.pone.0035090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA (2000) Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci 3:460–464. 10.1038/74833 [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999) Long-term potentiation a decade of progress? Science 285:1870–1874. 10.1126/science.285.5435.1870 [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Raymond EF, Carbone E, Marie H (2014) Firing properties of entorhinal cortex neurons and early alterations in an Alzheimer's disease transgenic model. Pflugers Arch 466:1437–1450. 10.1007/s00424-013-1368-z [DOI] [PubMed] [Google Scholar]
- Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G (2007) GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J 26:1702–1712. 10.1038/sj.emboj.7601616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD, Tjernberg LO, Silberring J, Gandy SE, Winblad B, Greengard P (1994) Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci U S A 91:8378–8382. 10.1073/pnas.91.18.8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421. 10.1016/s0896-6273(03)00434-3 [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM (2006) A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol 168:184–194. 10.2353/ajpath.2006.050593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett EK, Koffie RM, Wegmann S, Henstridge CM, Herrmann AG, Colom-Cadena M, Lleo A, Kay KR, Vaught M, Soberman R, Walsh DM, Hyman BT, Spires-Jones TL (2016) Non-fibrillar oligomeric amyloid-β within synapses. J Alzheimers Dis 53:787–800. 10.3233/JAD-160007 [DOI] [PubMed] [Google Scholar]
- Puzzo D, Piacentini R, Fá M, Gulisano W, Li Puma DD, Staniszewski A, Zhang H, Tropea MR, Cocco S, Palmeri A, Fraser P, D'Adamio L, Grassi C, Arancio O (2017) LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. Elife 6:e26991 10.7554/eLife.26991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, Veerle B, Coen K, Munck S, De Strooper B, Schiavo G Annaert W (2011) ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A 108:E559–E568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G (2001) Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J Biol Chem 276:33923–33929. 10.1074/jbc.M105410200 [DOI] [PubMed] [Google Scholar]
- Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH (2006) Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597-624). FASEB J 20:1254–1256. 10.1096/fj.05-5032fje [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked GM, Chauv S, Ubhi K, Hansen LA, Masliah E (2009) Interactions between the amyloid precursor protein C-terminal domain and G proteins mediate calcium dysregulation and amyloid beta toxicity in Alzheimer's disease. FEBS J 276:2736–2751. 10.1111/j.1742-4658.2009.06997.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini B (2007) Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866–2875. 10.1523/JNEUROSCI.4970-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Gandy S (2006) Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52:15–31. 10.1016/j.neuron.2006.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051–1058. 10.1038/nn1503 [DOI] [PubMed] [Google Scholar]
- Stine WB Jr, Dahlgren KN, Krafft GA, LaDu MJ (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 278:11612–11622. 10.1074/jbc.M210207200 [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161:1869–1879. 10.1016/s0002-9440(10)64463-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnini F, Novelia J, Kerrigan TL, Brown JT, Tsaneva-Atanasova K, Randall AD (2015) Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front Cell Neurosci 9:372. 10.3389/fncel.2015.00372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK (2009) Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Aβ-related synaptic alterations. J Neurosci 29:9704–9713. 10.1523/JNEUROSCI.2292-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283:29615–29619. 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Hongjie Y, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda T, Ramser EM, Yamashita M, Nakajima K, Mori H, Silverman MA, Tomiyama T (2015) Intracellular amyloid β oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol Commun 3:51. 10.1186/s40478-015-0230-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand WE, Melchor JP, Keane DM, Saporito-Irwin SM, Romanov G, Davis J, Xu F (2002) Localization of a fibrillar amyloid beta-protein binding domain on its precursor. J Biol Chem 277:36392–36398. 10.1074/jbc.M204676200 [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539. 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Wang Z, Jackson RJ, Hong W, Taylor WM, Corbett GT, Moreno A, Liu W, Li S, Frosch MP, Slutsky I, Young-Pearse TL, Spires-Jones TL, Walsh DM (2017) Human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J Neurosci 37:11947–11966. 10.1523/JNEUROSCI.2009-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R (2010) Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 13:190–196. 10.1038/nn.2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willén K, Edgar JR, Hasegawa T, Tanaka N, Futter CE, Gouras GK (2017) Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener 12:61. 10.1186/s13024-017-0203-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306:116–120. 10.1016/s0304-3940(01)01876-6 [DOI] [PubMed] [Google Scholar]
- Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG (1998) Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Aβ1-42 pathogenesis. J Neurosci Res 52:691–698. [DOI] [PubMed] [Google Scholar]
- Yang AJ, Chandswangbhuvana D, Shu T, Henschen A, Glabe CG (1999) Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42. J Biol Chem 274:20650–20656. 10.1074/jbc.274.29.20650 [DOI] [PubMed] [Google Scholar]