

Abstract
Poorly differentiated thyroid carcinomas (PDTC) in young individuals are rare and their clinical and histopathologic features, genetic mechanisms, and outcomes remain largely unknown. Here, we report a detailed characterization of a series of 6 PDTC in patients ≤ 21 years old defined by Turin diagnostic criteria studied for mutations and gene fusions characteristic of thyroid cancer using targeted next-generation sequencing (NGS) and whole exome sequencing (WES). All tumors had solid, insular or trabecular growth pattern and high mitotic rate, and 5 out of 6 tumors showed tumor necrosis. Targeted NGS assay identified somatic mutations in the DICER1 gene in 5 of 6 (83%) tumors, all of which were “hotspot” mutations encoding the metal-ion binding sites of the RNase IIIb domain of DICER1. WES was performed in 5 cases which confirmed all hotspot mutations and detected 2 tumors with additional inactivating DICER1 alterations. Of these two, one was a germline pathogenic DICER1 variant and the other had loss of heterozygosity for DICER1. No other mutations or gene fusions characteristic of adult well-differentiated thyroid cancer and PDTC (BRAF, RAS, TERT, RET/PTC and other) were detected. On follow-up, available for 5 patients, 3 patients died of disease 8-24 months after diagnosis, whereas 2 were alive with no disease. The results of our study demonstrate that childhood- and adolescent-onset PDTC are genetically distinct from adult-onset PDTC in that they are strongly associated with DICER1 mutations and may herald DICER1 syndrome in a minority. As such, all young persons with PDTC may benefit from genetic counselling. Furthermore, their clinically aggressive behavior contrasts sharply with the indolent nature of the great majority of thyroid tumors with DICER1 mutations reported to date.
Keywords: Poorly differentiated thyroid carcinoma, pediatric, children, adolescents, DICER1, whole exome sequencing, thyroid cancer
Poorly differentiated thyroid carcinomas (PDTC) are generally uncommon and represent approximately 1% of all thyroid malignancies1. They originate from follicular thyroid cells and have morphologic and biologic attributes that are intermediate between differentiated (papillary or follicular) and anaplastic (undifferentiated) thyroid carcinoma. The ten-year survival of these patients is 60-70%, significantly worse than well-differentiated carcinomas, but considerably better than anaplastic thyroid carcinomas1-3.
Historically, the histopathologic criteria used to diagnose PDTC have been a subject of controversy and have lacked a consensus. The Turin criteria for the diagnosis of PDTC were established at a consensus meeting held in 2006 and are the most widely accepted, having recently been adopted by the 2017 edition of the World Health Organization Classification of Tumors of the Endocrine Organs4,5. According to the Turin criteria, PDTC is defined as a tumor with solid, trabecular or insular growth that lacks papillary nuclear features and has at least one of the following: ≥ 3 mitoses per 10 high power fields (HPF), tumor necrosis or convoluted nuclei4. An alternative approach to the diagnosis of PDTC was proposed in a 2006 Memorial Sloan-Kettering Cancer Center (MSKCC) study in which PDTC was defined as any follicular cell-derived, non-anaplastic tumor with ≥ 5 mitoses per 10 HPF and/or necrosis6. The MSKCC criteria are less restrictive and, unlike the Turin criteria, include tumors that have papillary nuclear features and/or follicular architecture. Thus, while predictive of more aggressive biologic behavior, some tumors that meet MSKCC criteria are not exclusively poorly differentiated morphologically.
The overwhelming majority of PDTCs have occurred in adults with a mean patient age of 60 years4,7. PDTCs are exceedingly rare in children and adolescents with existing literature limited mainly to case reports8-28. The ability to extrapolate data from the literature is limited by the lack of uniform or clear diagnostic criteria, especially in reports that pre-date the publication of the Turin diagnostic criteria in 2007. Regarding the molecular mechanisms underlying PDTC, they have been studied almost exclusively in tumors from adults showing “early” genetic drivers characteristic of well-differentiated thyroid papillary and follicular carcinomas (e.g. BRAF, RAS mutations, RET, ALK fusions) and “late” driver mutations (TERT, TP53) characteristic of anaplastic carcinoma29-31. It remains unknown whether pediatric PDTC share genetic profiles with adult tumors or have distinct molecular mechanisms.
Here, we report detailed characterization of the clinical, pathologic and molecular features of PDTC in children and adolescents in a multi-institutional series of 6 patients. Our findings indicate that PDTC of childhood and adolescence appears to be a unique entity, distinct from morphologically similar tumors occurring in adults, and characterized by somatic DICER1 mutations found in most patients, with coexisting germline DICER1 mutations characteristic of DICER1 syndrome in a minority.
Materials and Methods
Case selection
This study was approved by the Human Studies Protection Office at Washington University, the Institutional Review Board of University of Pittsburgh and the McGill University Health Centre, Montreal. Pathology Department files of Washington University and the University of Pittsburgh, including consults, were searched for cases of PDTC occurring in patients ≤ 21 years of age. One additional case was identified at Vanderbilt University. Cases were included if the histologic features met Turin criteria for the diagnosis of PDTC4. The Turin criteria include the presence of solid/insular/trabecular growth pattern; absence of nuclear features of papillary thyroid carcinoma; and the presence of at least one of the following features: convoluted nuclei, mitotic activity of ≥ 3 mitoses per 10 high power field or tumor necrosis4. Six cases met Turin criteria and were included in the study. Clinical data was retrieved from the electronic medical record.
ThyroSeq v3 targeted NGS assay
Targeted next generation sequencing (NGS) analysis of thyroid cancer-related mutations and gene fusions was performed at the University of Pittsburgh Medical Center using a 112-gene ThyroSeq version 3 Genomic Classifier panel. It utilizes tumor DNA and RNA to test for 12,135 single nucleotide variations (SNVs) and insertions/deletions (indels) (COSMIC hotspots) and for more than 150 gene fusions (GF) implicated in thyroid cancer as previously described 32. In brief, manual microdissection of tumor-rich areas and paired normal tissue from unstained slides of formalin-fixed paraffin-embedded tissue was performed under a microscope with hematoxylin and eosin guidance. DNA and mRNA were isolated using MagNa Pure instrument (Roche). NGS libraries were generated using 10 ng DNA and 10 ng RNA using the Ion AmpliSeq Library kit 2.0. The libraries were sequenced on the Ion Proton or Ion GeneStudio S5 System (Thermo Fisher Scientific) according to the manufacturer’s protocol. The Torrent Suite Software v5.2.2, (Thermo Fisher Scientific) and an in-house developed software Variant Explorer v2 were used for data analysis and interpretation.
Whole exome sequencing
Normal and tumor tissue from 5 of the 6 PDTCs were subjected to whole exome sequencing (WES). FFPE-derived DNA (50 ng) of normal and tumor areas independently underwent exome capture using the SureSelect Human All Exon V6 kit from Agilent Technologies followed by 125-bp paired-end sequencing on an Illumina HiSeq 4000 sequencer. WES was performed at the McGill University and Génome Québec Innovation Centre (MUGQIC). Analysis methods are described in the Supplementary methods.
Sanger sequencing of the DICER1 gene
DICER1 mutations were further validated by Sanger sequencing using DICER1 transcript version ENST00000343455.7. Primers are available upon request. The quality of the amplified PCR product was evaluated by agarose gel electrophoresis. Then, bidirectional Sanger sequencing was performed using the BigDye Terminator Kit on ABI3730 (Thermo Fisher Scientific, Waltham, Maryland, USA).
mRNA analysis of the DICER1 germline variant
RNA was reverse transcribed into cDNA using SuperScriptIII first-strand cDNA synthesis (Thermo Fisher Scientific, Waltham, MA). DICER1 transcript NM_177438 was used to design cDNA-specific PCR primers for the exon 6-exon 7 boundaries as well as for the c.G5437A; p.E1813K mutation. Presence of a modified transcript and zygosity at the cDNA level in tumor–derived cDNA was tested using PCR followed by Sanger sequencing. Primers are available upon request.
Results
Clinical and pathologic features
The clinical and pathologic features of the 6 PDTC cases in our series are summarized in Table 1. The age of patients ranged from 14 to 19 years and the female to male ratio was 2:1. One patient (Case 1) had a history of prior radiation to the thyroid at the age of 4 months for a right neck cavernous hemangioma associated with Kasabach-Merritt syndrome (coagulopathy) that was refractory to medical management and embolization. None of the other patients had a history of prior radiation and no patient had a history of other cancers. No patient had a personal or family history of DICER1 syndrome associated tumors or other features of DICER1 syndrome. Case 1 had a non-immediate family history of “bone cancer” and Case 2 had a non-immediate family history of esophageal and breast cancer on the maternal side (paternal family history was not available in this case). Family history was not available for Cases 3, 5 and 6.
Table 1.
Clinical and pathologic features of pediatric poorly differentiated thyroid carcinoma
| Case # |
Patient age (yrs.) |
Sex | Prior radiation |
Differentiated component |
Tumor size (cm) |
Mitosis (per 10 HPF) |
Necrosis | TG/ TTF-1 IHC |
LVI | ETE | Margin | LN mets |
Distant mets |
Patient Survival (mo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 19 | F | + | No | 7.5 | 40 | + | +/+ | + | + | + | + | Lung/brain | DOD (24) |
| 2 | 17 | M | − | No | 9.0 | 37 | + | +/+ | + | + | + | − | Lung/finger/scalp | DOD (8) |
| 3 | 14 | F | − | Encapsulated FV PTC | 6.4 | 10 | − | NA | + | − | − | + | − | AWOD (53) |
| 4 | 17 | F | − | PTC | 3.1 | 22 | + | +/+ | + | − | − | − | − | AWOD (36) |
| 5 | 16 | M | − | Rare microfollicles | >7.5 | 9 | + | −/+ | + | + | + | + | Lung | DOD (10) |
| 6 | 14 | F | − | Encapsulated FV PTC | 5.0 | 21 | + | +/+ | + | − | − | − | − | NA |
FV PTC follicular variant papillary thyroid carcinoma; HPF high power field; TG thyroglobulin; TTF-1 thyroid transcription factor-1; NA not available; LVI lymphovascular invasion; ETE extrathyroidal extension; LN lymph node; mets metastases; mo months; DOD dead of disease; AWOD alive without disease
Of 5 patients with clinical follow-up available, 3 developed distant metastases and experienced a rapidly fatal clinical course (survival of 8 months to 2 years). Two other patients were alive with no disease 3-4 years after diagnosis.
All patients underwent total thyroidectomy. Pathology examination revealed large tumors ranging in size from 3.1 to 9.0 cm. Microscopically, architectural growth patterns ranged from solid and insular to trabecular (Fig. 1). Individual tumor cells had reduced amounts of cytoplasm, with relatively uniform nuclei and either dark or more vesicular chromatin. Mitotic activity was high in all tumors, ranging from 9 to 40 mitoses per 10 high power fields. Necrosis was identified in 5 of the 6 cases.
Fig 1.

Representative routine hematoxylin and eosin stained images of pediatric poorly differentiated thyroid carcinomas showing solid (A), insular (C) and trabecular (E) growth patterns. Mitotic activity (A) and necrosis (C) are present. Extrathyroidal extension with strap muscle invasion (B), vascular invasion (D) and positive margins (F) in Case 1. Only the 3 lethal cases showed extrathyroidal extension and positive margins, whereas lymphovascular invasion was present in all cases.
A definitive well-differentiated cancer component was observed in 3 tumors (Cases 3, 4 and 6) (Fig. 2). In Case 3, the PDTC was present as a focus within an encapsulated follicular variant of papillary thyroid carcinoma with capsular and lymphovascular invasion. Case 5 had only rare neoplastic microfollicles. Cases 1 and 2 were entirely poorly differentiated with no well-differentiated component found. Immunohistochemistry confirmed thyroid origin in these cases.
Fig 2.

Both Case 3 and Case 6 showed poorly differentiated thyroid carcinomas arising in encapsulated follicular variant of papillary thyroid carcinoma. In Case 3 (A), the poorly differentiated component was focal (top right), whereas in Case 6 (B), it was extensive. Transition zones between papillary thyroid carcinoma and the poorly differentiated component are shown for Case 3 (C) and Case 6 (D). Case 3 was completely encapsulated but showed lymphovascular invasion (E). Case 6 showed capsular invasion (F) in addition to lymphovascular invasion.
Lymph node metastases were present in 3 tumors (Case 1, 3 and 5). In Case 1 and 5, the nodal disease was extensive. Case 1 had 8 positive lymph nodes, in addition to a group of matted lymph nodes, with extensive extranodal extension in the lateral neck and upper mediastinum. The largest lymph node metastasis as well as the matted group of lymph nodes both measured 4 cm in greatest dimension. Case 5 had 45 lymph node metastases involving the central, right and left lateral neck compartments. Extranodal extension was present. The size of the largest metastasis was 11 cm. In contrast, only one of two perithyroidal lymph nodes contained a less than 1 mm metastasis (highlighted by a cytokeratin immunostain) in Case 3.
The 3 tumors associated with patient death (Cases 1, 2 and 5) shared common features. They were of a large size (7.5-9.0 cm) and all showed lymphovascular invasion, extrathyroidal extension, and positive resection margins (Fig. 1). None of these tumors revealed a definitive well-differentiated cancer component.
Targeted and whole exome sequencing analyses
Targeted next generation sequencing (Thyroseq v3) revealed DICER1 mutations in 5 of 6 tumors; all were “hotspot” mutations located at codons 1705, 1709, 1713, and 1813 (x2) of exons 24-25, coding for the metal binding sites of the RNase IIIb domain of the protein (Table 2). All mutations were confirmed by Sanger sequencing (Fig. 3). One tumor contained a second somatic mutation in the DICER1 gene. The mutations were confirmed to be somatic in origin by Sanger sequencing of paired non-tumor tissues available in 4 out of 5 cases.
Table 2.
DICER1 pathogenic variants in pediatric poorly differentiated thyroid carcinoma
| Case # |
Somatic mutations (VAF) |
Germline mutation (VAF) |
Other DICER1 alterations |
|---|---|---|---|
| 1 | p.1713Y(108/242) | - | - |
| 2 | p.E1813K (71/219) | c.735-8T>G (41/69) | - |
| 3 | #1 p.D1709N (78/210) #2 p.I1209fs (28/89) |
- | - |
| 4 | p.E1813Q (156/163) | - | LOH |
| 5 | - | - | - |
| 6 | p.E1705K* | NA | - |
VAF Variant allele frequency shown in number of reads of alternate allele over total number of reads; NA not available; LOH loss of heterozygosity
Sample 6 was not subjected to WES so there is no VAF to report
Fig 3.
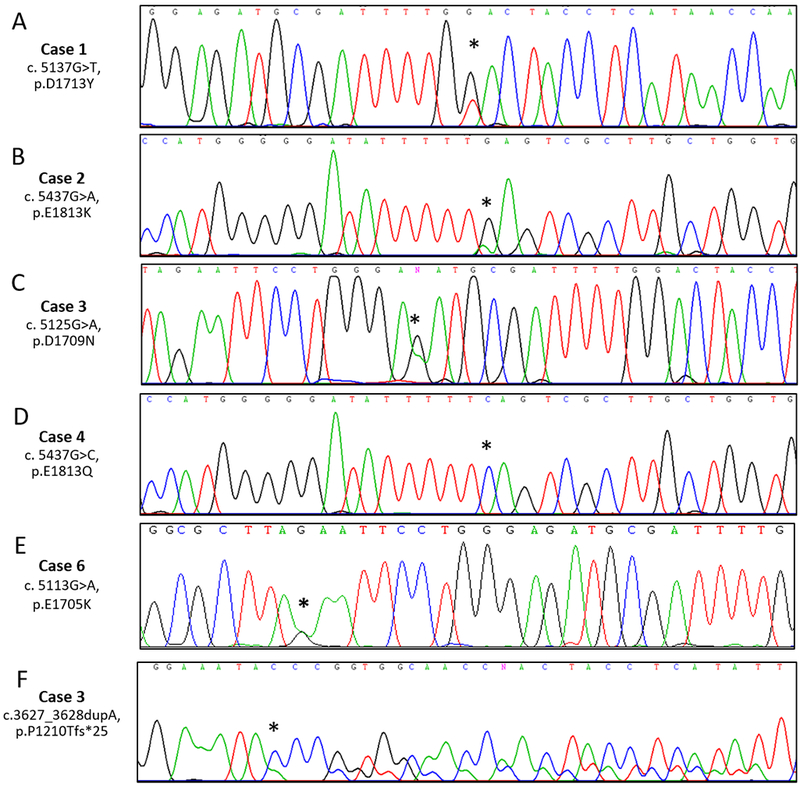
RNase IIIb and non-RNase IIIb domain DICER1 somatic mutations. RNase IIIb domain mutations in Cases 1-4 and 6 (A-E): A) Electropherogram of a G>T substitution at c.5137 resulting in a D1713Y missense mutation (Case 1) B) Electropherogram of a G>A substitution at c.5437 leading to an E1813K missense mutation (Case 2). C) Electropherogram of a G>A substitution at c.5125 causing a D1709N missense mutation (Case 3). D) Electropherogram of a G>C substitution at c.5437 causing an E1813Q missense mutation and loss of heterozygosity (Case 4). E): Electropherogram of a G>A substitution at c.5113 causing an E1705K missense mutation (Case 6). Non-RNase IIIb domain DICER1 mutation in Case 3: F) Electropherogram of an A duplication at c.3627 resulting in a frameshift mutation causing p.P1210Tfs*25.
The WES analysis was performed in 5 cases. It confirmed all DICER1 hotspot mutations detected by targeted NGS, confirmed a second somatic DICER1 mutation in Case 3 and identified a previously unrecognized loss of heterozygosity in Case 4 (further discussed below, Table 2). Furthermore, it also revealed that Case 2 possessed a germline DICER1 pathogenic variant (see below and Table 2) which, therefore, established the diagnosis of DICER1 syndrome in 1 of the 5 cases. No DICER1 mutations were found in Case 5 by WES, confirming the targeted NGS findings. WES analysis could not be performed in 1 case with a somatic DICER1 mutation (Case 6) due to lack of non-tumor DNA.
The one germline mutation, found in Case 2, was at a cryptic splice site in intron 6 (c.T735-8G) and predicted to affect the splicing of the transcript. In cDNA, the exon 6- exon 7 boundary showed no indication of an aberrant transcript however, a prominent allelic imbalance at the c.G5437A;p.E1813K hotspot mutation locus in this case indicated expression on only 1 allele, suggesting a non-sense mediated decay of an aberrant transcript (Fig. 4). Thus, the germline variant is provisionally classified as likely pathogenic, indicating that this patient had otherwise clinically inapparent DICER1 syndrome. In Case 4, in addition to the previous identified p.E1813Q hotspot mutation, ExomeAI detected allelic imbalance in chromosome 14 including the whole DICER1 locus, indicating loss of heterozygosity (LOH) in the tumor (Supplemental Fig. 1)33. No other common driver mutations or gene fusions characteristic of adult well-differentiated thyroid cancer or PDTC (BRAF, RAS, TERT, RET/PTC and other) were detected by Thyroseq v3 assay or WES.
Fig 4.
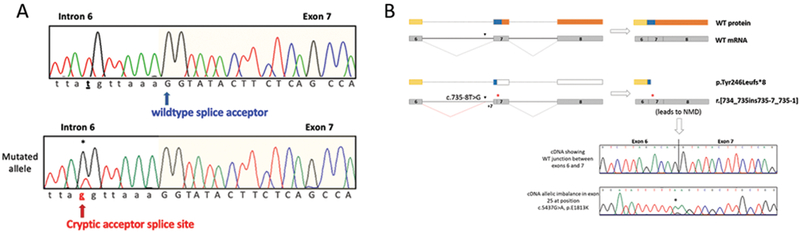
A) Chromatogram (upper) showing the location of the wild-type splice acceptor site before exon 7 in DNA from a control. Chromatogram (lower) showing the variant c.735-8T>G (asterisk) and cryptic new acceptor splice site in DNA from Case 2. Black bold with underlined lettering shows the WT base. Bold red lettering shows the altered base. Blue arrow points to the wildtype splice acceptor and red arrow indicates the location of the new cryptic acceptor site. B) Schematic representation of the expected protein with an intact exon 7 acceptor splice site compared to the mutated c.735-8T>G acceptor splice site. The red lines represent the aberrant splicing which retains 7bp from intron 6 and would be predicated to lead to a premature stop codon (red asterisk). If an aberrant transcript is present triggering its degradation by nonsense-mediated mRNA decay, no abnormal transcript would be detected in cDNA and the junction between Exon 6 and Exon 7 appeared to be intact. In the figure, upper chromatogram shows a wildtype junction between exons 6 and 7 in cDNA from case 4. Lower chromatogram shows allelic imbalance in exon 25 at position, c.5437G>A, p.E1813K in cDNA. In this scenario, chromatograms depicting the presence of the hotspot mutation and a wildtype splicing in cDNA suggests 1) that an aberrant transcript has been degraded and only the allele with the c.5437G>A, p.E1813K is retained and 2) the two variants are in trans. Yellow represents the DexD/H helicase domain. Blue represents the amino acids not belonging to a specific known domain.
The only other gene with alterations found in two or more cases was ATM. A somatic truncating mutation in ATM- c.583dupA;p.T195fs was found in Case 1. This case also harbored a germline heterozygous missense variant in ERCC2-c.G1606A;p.V536M that has been reported to have dominant negative activity in breast cancer34. In Case 4, LOH for the ATM gene was detected as well as a germline ATM variant (c.C146G;p.S49C) (Supplemental Table 1). However, this germline variant is not definitively pathogenic35. Additional germline and somatic alterations identified in single cases are shown in Table 3 and Supplemental Table 1; germline alterations found by WES analysis of each case and predicted to be pathogenic are shown in Supplemental Tables 2-6. Coverage metrics for the WES analysis in each case are shown in Supplemental Table 7.
Table 3.
Somatic mutations in other genes in the MSKCC-Impact panel (468 genes)
| Gene | Case | Mutation | Oncogenic Classification (CGI) |
COSMIC ID |
VAF | SIFT | Poly phen2 |
Mutation Taster |
Revel | MCAP | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATM | 1 | c.583dupA; p.T195fs | Predicted Driver Tier 1 | NA | 24/31 | NA | NA | NA | NA | NA | NA |
| CDC73 | 1 | c.1013delA; p.Q338fs | Predicted Driver Tier 1 | NA | 57/138 | NA | NA | NA | NA | NA | NA |
| MAP2K2 | 2 | c.C383G; p.P128R | Predicted Driver Tier 1 | NA | 86/322 | 0,1.00,D | 1.0,D | 1.0,D | 0.916 | 0.693 | 26.8 |
| RBM10 | 2 | c.C2065T; p.R689C | Predicted Driver Tier 1 | COSM1315532 | 35/154 | 0,1.00,D | 1.0,D | 1.0,D | 0.466 | 0.864 | 34 |
| ARID1A | 2 | c.5624_5625insCCCTGCCC; p.P1875fs | Predicted Driver Tier 1 | NA | 73/284 | NA | NA | NA | NA | NA | NA |
| FLT3 | 2 | c.G854T; p.W285L | Predicted Driver Tier 1 | NA | 30/115 | 0.22,0.78,T | 0.737,P | 0.967,D | 0.47 | 0.0465 | 26.9 |
| EGFR | 4 | c.C2506G; p.R836G | Predicted Driver Tier 1 | NA | 32/162 | 0,1.00,D | 1.0,D | 1.0,D | 0.856 | 0.289 | 34 |
| TP53 | 4 | c.G631T; p.E211X | Predicted Driver Tier 1 | COSM11078, COSM308326 | 129/136 | NA | NA | NA | NA | NA | 35 |
VAF variant allele frequency shown in number of reads of alternate allele over total number of reads; D damaging; B benign, P probably damaging, T tolerated, NA not applicable
DNA was amplified with primers flanking from exon 2 to exon 27 (Supplemental Table 1)
Discussion
In this study, we uncover the genetic mechanisms of pediatric PDTC and demonstrate that these tumors typically carry DICER1 mutations and lack molecular alterations characteristic of PDTC in adults. Furthermore, we show that pediatric PDTC may represent a manifestation of DICER1 syndrome.
DICER1 plays a central role in RNA interference including in the processing of microRNAs, and thus has a broad impact on gene expression. Germline loss-of-function non-“hotspot” mutations, when accompanied by a second, somatic “hit” to DICER1, cause DICER1 syndrome, an inherited cancer syndrome with variable expressivity for which a “two-hit” model has been proposed36. “Hotspot” DICER1 mutations affecting the metal binding sites of the RNase IIIb domain are nearly all somatic in origin, likely due to deleterious effects during development36,37. Rarely, they may occur as germline mutations in non-tumorous tissues in a mosaic state, but in DICER1 syndrome, they occur as second “hits” in associated tumors36,37. Patients with DICER1 syndrome may develop pleuropulmonary blastoma, cystic nephroma, nasal chondromesenchymal hamartoma, embryonal rhabdomyosarcoma and ovarian sex-cord stromal tumors, among other tumors types, often at a young age38,39. Multinodular thyroid goiter is also a frequent finding in DICER1 syndrome; 75% of affected women and 17% of men develop multinodular disease by age 4040. The patients are also at elevated risk of developing differentiated thyroid neoplasms which uniformly have been reported to have an indolent clinical course40,41. These differentiated thyroid cancers contain second-hit “hotspot” mutations in DICER140,42,43.
Two PDTCs with DICER1 mutations in patients with DICER1 syndrome were recently reported in the literature; however, upon review of the published scanned whole slides of the tumors, we found that they did not meet Turin diagnostic criteria for PDTC41. Furthermore, the 2 reported tumors had no high-risk pathologic features and behaved in a clinically indolent manner, which is more in keeping with differentiated thyroid carcinoma.
Outside of DICER1 syndrome, DICER1 mutations are rare in thyroid neoplasms and, to date, have been found nearly exclusively in well-differentiated tumors (papillary thyroid carcinoma, follicular carcinoma and follicular adenoma)41,44-48. Of note, somatic “hotspot” DICER1 mutations may be more common in pediatric compared to adult differentiated thyroid tumors, seen in 10% of pediatric papillary thyroid carcinomas in one series47. All were “low-risk” tumors according to the American Thyroid Association (ATA) guidelines47,49. Although usually mutually exclusive with other driver mutations, a rare example of papillary thyroid carcinoma with a new WNK1-B4GALNT3 fusion that also harbored a DICER1 “hotspot” mutation has been reported45.
Genetic analysis of pediatric PDTC, as defined by the Turin criteria, has not been previously reported. Here, we show that DICER1 mutations define a rare group of thyroid carcinomas that are poorly differentiated, occur mainly in adolescents and may have more clinically aggressive behavior than the existing literature suggests. Although none of our patients had other clinical manifestations or family history of DICER1 syndrome, one patient was found to have a germline intronic likely pathogenic variant consistent with clinically inapparent DICER1 syndrome. This indicates a need for genetic counselling of children and adolescents with PDTC, as the tumor type may be the first manifestation of DICER1 syndrome in a subset of these patients. Two other PDTCs had biallelic inactivation of DICER1, in line with the “two-hit” model of DICER1 inactivation in oncogenesis. Only 1 of the 6 pediatric PDTC in our series had no DICER1 mutations.
Only one tumor with a DICER1 mutation has previously been reported among over 80 adult PDTC sequenced using assays that include the DICER1 gene29,50,51. This was a case of PDTC defined by Turin criteria in a 22-year-old woman with a p.E1813K DICER1 mutation, reported by Landa et al (2016)29. This case, together with the cases in the current series, provide strong evidence that PDTC in young patients represent a genetically homogeneous group of tumors developing via molecular pathways different from adult PDTC. The latter commonly harbor initiating BRAF or RAS mutations, which are not found in pediatric PDTC 29,50. Despite distinct origins, adult and pediatric PDTC may, however, share late genetic events. A TP53 mutation was present in one pediatric PDTC, which, along with TERT promoter mutations, is a common late genetic event in adult PDTC29,50. Two of the DICER1-mutated PDTCs in our series additionally harbored somatic alterations in ATM, a cell-cycle checkpoint and DNA repair response gene, as well as germline ATM variants, one of which is not definitely pathogenic. ATM mutations have been reported in 5-10% of adult PDTCs and anaplastic thyroid carcinomas but only in 1-2% of differentiated thyroid carcinomas, suggesting that ATM mutations may represent another late molecular alteration in PDTC seen in both pediatric and adult tumors29,46.
An important feature of the pediatric PDTCs in our series was the observed aggressive clinical behavior with a rapidly fatal clinical course (≤ 2 years) in 3 of our patients. Our review of the literature yielded 27 patients with PDTC that were ≤ 21 years of age (Supplemental Table 8)8-28. Scanned slides from 2 tumors were available for review and indicated that these 2 cases did not meet Turin criteria for the diagnosis of PDTC41. These were excluded, leaving 25 cases. Most patients were adolescents, with only a few PDTCs occurring in children < 10 years of age. Of 20 patients with clinical follow-up available, only 3 died of disease and 1 was alive with disease at last follow-up9,14,23,26. Four patients were alive but the disease status was not known, and the remaining 12 patients were alive and free of disease (overall survival of 85%)8-15,17,19-21. The majority of cases reported in the literature predated the publication of the Turin diagnostic criteria in 20074. An attempt was made to apply Turin criteria to all cases in the literature, but definitive confirmation of Turin criteria was possible in only 4 tumors, 3 of which had clinical follow-up information available14,21,22. Combining these 3 cases with 5 patients with clinical follow-up in the current series, 4 of 8 (50%) pediatric patients with PDTC defined by Turin criteria died of disease14,21,22,28. Although the number of patients remain relatively small, these findings suggest that (i) the outcomes for pediatric PDTC may be worse than the existing literature suggests, and (ii) the overall survival of children with PDTC may be more in line with that of adults.
Prognostic features of pediatric PDTC are difficult to ascertain from our small number of cases. However, it is of interest to note that the 2 patients in our series with clinical follow-up who are alive and free of disease had more localized disease with no extrathyroidal extension and negative margins of resection. Furthermore, these tumors had a component of well-differentiated papillary thyroid carcinoma. One of the 2 cases showed tumor encapsulation, a feature that has been associated with a better prognosis in adults who have high-grade thyroid malignancies52. Evaluation of these characteristics in a larger number of pediatric PDTC may yield more definitive prognostic information.
Thyroid hyperplasia and neoplasia represent the most common phenotype in individuals with germline DICER1 mutations; 75% of women and 17% of men have multinodular goiter (nodular hyperplasia) and/or removal of the thyroid by age 4040. Germline mutation carriers have a 16-18 fold increased risk for thyroid cancer compared with the general population40. Overall, previously reported DICER1-mutated thyroid carcinomas (both those with germline and somatic-only mutations) have nearly universally behaved in an indolent manner. The exception was a recent case report of a carcinosarcoma (anaplastic thyroid carcinoma with heterologous elements) that harbored an E1705K DICER1 hotspot mutation in a tumor from a 45-year-old woman who died of disease after 4 months53. Our findings and this case suggest that DICER1-mutated differentiated thyroid tumors may be at risk for high-grade transformation, although this is likely an uncommon event. Further evidence of the potential for high-grade transformation from a differentiated thyroid carcinoma is the occurrence of PDTC in association with papillary thyroid carcinoma in 3 of our cases. This report demonstrates that the thyroid gland can express the full spectrum of neoplastic pathology from benign, often cystic, hyperplasias to poorly differentiated aggressive cancers seen in the other more fully described DICER1-associated neoplasms in the lung, ovary and kidney. As with these other organs, it appears that additional genetic events, beyond the biallelic combination of one DICER1 loss of function and a “hotspot” mutation, may be necessary for high-grade transformation to take place. For lung and kidney, the most common additional genetic event associated with high-grade transformation is mutations and/or loss of TP53, seen in one of the five DICER1-associated PDTCs tested in this study54-56.
In summary, PDTC of childhood and adolescence is very rare and a historical lack of uniform diagnostic criteria and molecular studies has hindered our understanding of its biology and behavior. Our findings indicate that they may represent a distinct tumor type genetically different from adult-onset PDTC that is strongly associated with DICER1 mutations and may herald DICER1 syndrome in a subset. The clinically aggressive behavior of these tumors contrasts sharply with the indolent nature of most thyroid tumors with DICER1 mutations reported to date.
Supplementary Material
Acknowledgements
This work was funded by the CIHR Foundation Grant FDN-148390 to W.D.F. and the National Institutes of Health grant CA181150 to Y.E.N. B.R. is a Young Investigator from the ALSF and the Mina Neri Foundation for Childhood Cancer.
Footnotes
Disclosure/Conflict of Interest
Y.E.N. owns intellectual property and receives royalties related to ThyroSeq. T.J.G. is a Consultant for Interspace Diagnostics. All other authors have nothing to disclose.
References
- 1.Asioli S, Erickson LA, Righi A, Jin L, Volante M, Jenkins S, et al. Poorly differentiated carcinoma of the thyroid: validation of the Turin proposal and analysis of IMP3 expression. Mod Pathol. 2010;23:1269–78. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, et al. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. 2003;21:3226–35. [DOI] [PubMed] [Google Scholar]
- 3.Ibrahimpasic T, Ghossein R, Carlson DL, Nixon I, Palmer FL, Shaha AR, et al. Outcomes in patients with poorly differentiated thyroid carcinoma. J Clin Endocrinol Metab. 2014;99:1245–52. [DOI] [PubMed] [Google Scholar]
- 4.Volante M, Collini P, Nikiforov YE, Sakamoto A, Kakudo K, Katoh R, et al. Poorly differentiated thyroid carcinoma: the Turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am J Surg Pathol. 2007;31:1256–64. [DOI] [PubMed] [Google Scholar]
- 5.Tallini G, Asioli S, Aubert S, Carcangiu ML, Chernock RD, Fellegara G, et al. Poorly differentiated thyroid carcinoma In: WHO Classification of Tumours of Endocrine Organs, 4th ed. Lyon: International Agency for Research on Cancer, 2017. [Google Scholar]
- 6.Hiltzik D, Carlson DL, Tuttle RM, Chuai S, Ishill N, Shaha A, et al. Poorly differentiated thyroid carcinomas defined on the basis of mitosis and necrosis: a clinicopathologic study of 58 patients. Cancer. 2006;106:1286–95. [DOI] [PubMed] [Google Scholar]
- 7.Gnemmi V, Renaud F, Do Cao C, Salleron J, Lion G, Wemeau JL, et al. Poorly differentiated thyroid carcinomas: application of the Turin proposal provides prognostic results similar to those from the assessment of high-grade features. Histopathology. 2014;64:263–73. [DOI] [PubMed] [Google Scholar]
- 8.Kotiloglu E, Kale G, Senocak ME. Follicular thyroid carcinoma with a predominant insular component in a child: a case report. Tumori. 1995;81:296–8. [DOI] [PubMed] [Google Scholar]
- 9.Hassoun AA, Hay ID, Goellner JR, Zimmerman D. Insular thyroid carcinoma in adolescents: a potentially lethal endocrine malignancy. Cancer. 1997;79:1044–8. [DOI] [PubMed] [Google Scholar]
- 10.Pilotti S, Collini P, Mariani L, Placucci M, Bongarzone I, Vigneri P, et al. Insular carcinoma: a distinct de novo entity among follicular carcinomas of the thyroid gland. Am J Surg Pathol. 1997;21:1466–73. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez JM, Parrilla P, Moreno A, Sola J, Pinero A, Ortiz S, et al. Insular carcinoma: an infrequent subtype of thyroid cancer. J Am Coll Surg. 1998;187:503–8. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi Y, Daa T, Kashima K, Yokoyama S, Nakayama I, Noguchi S. Mutations of p53 in thyroid carcinoma with an insular component. Thyroid. 1999;9:377–81. [DOI] [PubMed] [Google Scholar]
- 13.Lam KY, Lo CY, Chan KW, Wan KY. Insular and anaplastic carcinoma of the thyroid: a 45-year comparative study at a single institution and a review of the significance of p53 and p21. Ann Surg. 2000;231:329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo CY, Lam KY, Wan KY. Insular thyroid carcinoma in adolescents. Eur J Surg Acta Chir. 2000;166:585–8. [DOI] [PubMed] [Google Scholar]
- 15.Zettinig G, Kaserer K, Passler C, Flores JA, Niederle B, Dudczak R. Advanced insular thyroid carcinoma in a fourteen-year-old girl: twenty-four years of follow-up. Thyroid. 2000;10:435–7. [DOI] [PubMed] [Google Scholar]
- 16.Rijhwani A, Satish GN. Insular carcinoma of the thyroid in a 10-year-old child. J Pediatr Surg. 2003;38:1083–5. [DOI] [PubMed] [Google Scholar]
- 17.Yusuf K, Reyes-Mugica M, Carpenter TO. Insular carcinoma of the thyroid in an adolescent: a case report and review of the literature. Curr Opin Pediatr. 2003;15:512–5. [DOI] [PubMed] [Google Scholar]
- 18.Kumagai A, Namba H, Mitsutake N, Saenko VA, Ohtsuru A, Ito M, et al. Childhood thyroid carcinoma with BRAFT1799A mutation shows unique pathological features of poor differentiation. Oncol Rep. 2006;16:123–6. [PubMed] [Google Scholar]
- 19.Prommegger R, Häussler B, Gabriel M, Ensinger C, Sauper T, Hager J. Insular-type component follicular thyroid carcinoma in a 10-year-old girl--case report. J Pediatr Surg. 2006;41:e5–7. [DOI] [PubMed] [Google Scholar]
- 20.Donnellan KA, Carron JD, Bigler SA, Wein RO. Metastatic insular thyroid carcinoma in the pediatric patient. Am J Otolaryngol. 2009;30:61–4. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y-L, Ting W-H, Wey S-L, Chen CK, Huang CY, Cheng SP, et al. Poorly differentiated thyroid carcinoma in a 9-year-old boy: case report. J Pediatr Endocrinol Metab. 2011;24:783–6. [PubMed] [Google Scholar]
- 22.Hod R, Bachar G, Sternov Y, Shvero J. Insular thyroid carcinoma: a retrospective clinicopathologic study. Am J Otolaryngol. 2013;34:292–5. [DOI] [PubMed] [Google Scholar]
- 23.Liu L, Li D, Wang H,Yang X, Yu Y, Gao M. Multifocality predicts poor outcome of patients with insular thyroid cancer: a clinicopathological study. Int J Clin Exp Pathol. 2015;8:11212–7. [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsutake N, Fukushima T, Matsuse M, Rogounovitch T, Saenko V, Uchino S, et al. BRAF(V600E) mutation is highly prevalent in thyroid carcinomas in the young population in Fukushima: a different oncogenic profile from Chernobyl. Sci Rep. 2015;5:16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norlén O, Charlton A, Sarkis LM, Henwood T, Shun A, Gill AJ, et al. Risk of malignancy for each Bethesda class in pediatric thyroid nodules. J Pediatr Surg. 2015;50:1147–9. [DOI] [PubMed] [Google Scholar]
- 26.Sironi M, Collini P, Cantaboni A. Fine needle aspiration cytology of insular thyroid carcinoma. A report of four cases. Acta Cytol. 1992;36:435–9. [PubMed] [Google Scholar]
- 27.Mao XC, Yu WQ, Shang JB, Wang KJ. Clinical characteristics and treatment of thyroid cancer in children and adolescents: a retrospective analysis of 83 patients. J Zhejiang Univ Sci B. 2017;18:430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Win TT, Othman NH, Mohamad I. Poorly differentiated thyroid carcinoma: A hospital-based clinicopathological study and review of literature. Indian J Pathol Microbiol. 2017;60:167–71. [DOI] [PubMed] [Google Scholar]
- 29.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest. 2016;126:1052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly LM, Barila G, Liu P, Evdokimova VH, Trivedi S, Panebianco F, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci. 2014;111:4233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin Cancer Res. 2018;24:3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikiforova MN, Mercurio S, Wald AI, Barbi de Moura M, Callenberg K, Santana-Santos L, et al. Analytical performance of the ThyroSeq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer. 2018;124:1682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nadaf J, Majewski J, Fahiminiya S. ExomeAI: detection of recurrent allelic imbalance in tumors using whole-exome sequencing data. Bioinforma. 2015;31:429–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rump A, Benet-Pages A, Schubert S, Kuhlmann JD, Janavicius R, Machackova E, et al. Identification and Functional Testing of ERCC2 Mutations in a Multi-national Cohort of Patients with Familial Breast- and Ovarian Cancer. PLoS Genet. 2016;12:e1006248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stredrick DL, Garcia-Closas M, Pineda MA, Bhatti P, Alexander BH, Doody MM, et al. The ATM missense mutation p.Ser49Cys (c.146C>G) and the risk of breast cancer. Hum Mutat. 2006;27:538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Research. 2015;4:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein S, Lee H, Ghahremani S, Kempert P, Ischander M, Teitell MA, et al. Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet. 2014;51:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72. [DOI] [PubMed] [Google Scholar]
- 39.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan NE, Bauer AJ, Schultz KAP, Doros L, Decastro RM, Ling A, et al. Quantification of Thyroid Cancer and Multinodular Goiter Risk in the DICER1 Syndrome: A Family-Based Cohort Study. J Clin Endocrinol Metab. 2017;102:1614–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Tuin K, de Kock L, Kamping EJ, Hannema SE, Pouwels MJM, Niedziela M, et al. Clinical and Molecular Characteristics May Alter Treatment Strategies of Thyroid Malignancies in DICER1 Syndrome. J Clin Endocrinol Metab. 2019;104:277–84. [DOI] [PubMed] [Google Scholar]
- 42.de Kock L, Wang YC, Revil T, Badescu D, Rivera B, Sabbaghian N, et al. High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J Med Genet. 2016;53:43–52. [DOI] [PubMed] [Google Scholar]
- 43.Rutter MM, Jha P, Schultz KAP, Sheil A, Harris AK, Bauer AJ, et al. DICER1 Mutations and Differentiated Thyroid Carcinoma: Evidence of a Direct Association. J Clin Endocrinol Metab. 2016;101:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H, et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS Genet. 2016;12:e1006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa V, Esposito R, Ziviello C, Sepe R, Bim LV, Cacciola NA, et al. New somatic mutations and WNK1-B4GALNT3 gene fusion in papillary thyroid carcinoma. Oncotarget. 2015;6:11242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wasserman JD, Sabbaghian N, Fahiminiya S, Chami R, Mete O, Acker M, et al. DICER1 mutations are frequent in adolescent-onset papillary thyroid carcinoma. J Clin Endocrinol Metab. 2018;103:2009–15. [DOI] [PubMed] [Google Scholar]
- 48.Gullo I, Batista R, Rodrigues-Pereira P, Soares P, Barroca H, do Bom-Sucesso M, et al. Multinodular Goiter Progression Toward Malignancy in a Case of DICER1 Syndrome: Histologic and Molecular Alterations. Am J Clin Pathol. 2018;149:379–86. [DOI] [PubMed] [Google Scholar]
- 49.Francis GL, Waguespack SG, Bauer AJ, Angelos P, Benvenga S, Cerutti JM, et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2015;25:716–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sykorova V, Dvorakova S, Vcelak J, Vaclavikova E, Halkova T, Kodetova D, et al. Search for new genetic biomarkers in poorly differentiated and anaplastic thyroid carcinomas using next generation sequencing. Anticancer Res. 2015;35:2029–36. [PubMed] [Google Scholar]
- 51.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivera M, Ricarte-Filho J, Patel S, Tuttle M, Shaha A, Shah JP, et al. Encapsulated thyroid tumors of follicular cell origin with high grade features (high mitotic rate/tumor necrosis): a clinicopathologic and molecular study. Hum Pathol. 2010;41:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J, Sarita-Reyes C, Kindelberger D, Zhao Q. A rare malignant thyroid carcinosarcoma with aggressive behavior and DICER1 gene mutation: a case report with literature review. Thyroid Res. 2018;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pugh TJ, Yu W, Yang J, Field AL, Ambrogio L, Carter SL, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene. 2014;33:5295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seki M, Yoshida K, Shiraishi Y, Shimamura T, Sato Y, Nishimura R, et al. Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma. Cancer Res. 2014;74:2742–9. [DOI] [PubMed] [Google Scholar]
- 56.Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol. 2014;27:1267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.