

Abstract
Current efforts to find specific genodermatoses treatments and define precise pathogenesis mechanisms require appropriate surrogate models with human cells. Although transgenic and gene knockout mouse models for several of these disorders exist, they often fail to faithfully replicate the clinical and histopathological features of the human skin condition. We have established a highly efficient method for precise deletion of critical gene sequences in primary human keratinocytes, based on CRISPR-Cas9-mediated gene editing. Using this methodology, in the present study we generated a model of Netherton syndrome by disruption of SPINK5. Gene-edited cells showed absence of LEKTI expression and were able to recapitulate a hyperkeratotic phenotype with most of the molecular hallmarks of Netherton syndrome, after grafting to immunodeficient mice and in organotypic cultures. To validate the model as a platform for therapeutic intervention, we tested an ex vivo gene therapy approach using a lentiviral vector expressing SPINK5. Re-expression of SPINK5 in an immortalized clone of SPINK5-knockout keratinocytes was capable of reverting from Netherton syndrome to a normal skin phenotype in vivo and in vitro. Our results demonstrate the feasibility of modeling genodermatoses, such as Netherton syndrome, by efficiently disrupting the causative gene to better understand its pathogenesis and to develop novel therapeutic approaches.
Keywords: CRISPR/Cas9, human keratinocytes, genodermatosis, gene editing, disease modeling, skin equivalents., Netherton syndrome
Graphical Abstract
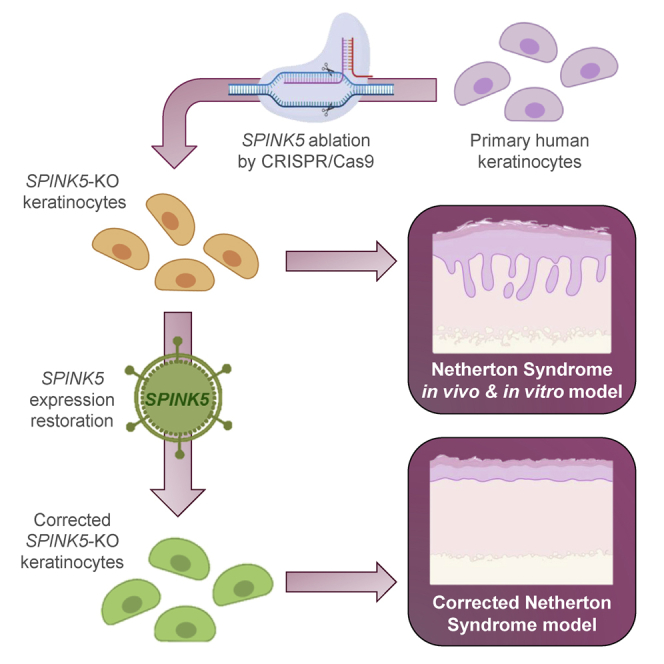
Exploiting the advantages of their highly efficient CRISPR-Cas9 gene editing strategy, in this study, Gálvez et al. abrogate SPINK5, the Netherton syndrome-causing gene, in primary human keratinocytes. SPINK5-knockout cells show disease hallmarks in vitro and in vivo, enabling the development of trustworthy models suitable for the evaluation of advanced therapies for genodermatoses.
Introduction
Skin disease studies in human subjects are restricted for ethical and practical reasons, and even more so in patients affected by rare and often devastating disorders of genetic origin (i.e., genodermatoses). Thus, the establishment of reliable models to address both pathogenic mechanisms and the efficacy of novel therapeutic approaches is of major interest and represents a great challenge. Although several genodermatoses murine models exist, it is not unusual that they pose important shortcomings ranging from early lethality to poor recapitulation of the actual human cutaneous phenotype. These problems may be overcome through the use of 3D organotypic cultures (OTCs) or transplantation of bioengineered skin produced with genodermatoses patient cells.1 However, important limitations persist, namely paucity of genodermatoses patients for cell isolation and the inaccuracy of working with non-isogenic, control-patient pairs. Both problems could be solved if proper gene loss of function (e.g., knockouts) is achieved on readily available cells from healthy individuals. Since RNAi-mediated gene silencing is rarely 100% effective at ablating the expression of a target gene product,2,3 gene disruption through genome editing is required to produce true null alleles.
We recently established the conditions to achieve gene correction by CRISPR-Cas9, facilitating non-homologous end joining (NHEJ) reframing of mutant COL7A1 with extraordinary efficacy in recessive dystrophic epidermolysis bullosa (RDEB) patient keratinocytes.4 In this study, we sought to determine whether such non-viral methodology could be adapted to disrupt a genodermatosis-causing gene, aiming at developing a trustworthy model of that disease.
Netherton syndrome (NS) is a debilitating condition characterized by a defective skin barrier with ichthyosiform erythroderma and allergic manifestations.5 NS is usually associated with a significant degree of mortality in the first year of life as a consequence of complications that include bronchopneumonia, sepsis, and hypernatremic dehydration due to the severe water loss through the faulty skin barrier.
NS is an autosomal recessive skin disorder, with an estimated prevalence of 1:200.000 at birth, due to mutations in the SPINK5 gene that encodes the lympho-epithelial Kazal-type inhibitor (LEKTI) protein, an inhibitor of the serine proteases kallikrein-related peptidase 5 (KLK5), KLK7, and KLK14, which play a fundamental role in the controlled desquamation of the stratum corneum.6, 7, 8, 9 Subsequently, uncontrolled kallikreins excessively degrade corneodesmosomes and cleave pro-elastase 2 (ELA2), which in turn leads to a defective skin barrier.8,10, 11, 12, 13 In patients with NS, mutations in SPINK5 cause the expression of truncated proteins with little or no LEKTI activity.14,15
Previously, we established a skin-humanized mouse model of NS, based on the engraftment in immunodeficient mice of bioengineered skins, generated with NS patients’ primary cells.5,16 The model fairly recapitulated the major NS features and served as a basis of a clinical protocol for gene therapy.17
Herein, we exploited the advantages of our very efficient CRISPR-Cas9 genome editing strategy to abrogate SPINK5 in human keratinocytes. SPINK5-knockout (ΔSPINK5) epidermal cell clones showed hallmarks of NS in vitro and in vivo, enabling the development of reliable models suitable for evaluation of relevant therapies.
Results
Generation of ΔSPINK5 Human Keratinocytes
A non-viral NHEJ approach based on CRISPR-Cas9 was used to excise a genomic fragment encompassing SPINK5 exon 1 (E1), in order to generate a knockout model. The system requires the use of two CRISPR guide RNAs (gRNAs) recognizing sequences flanking the genome target (Figure 1A). Four different gRNA pairs were designed and tested for deletion efficacy of the intended SPINK5 sequences (Figure 1B). Human keratinocytes were nucleofected with ribonucleoprotein (RNP) complexes containing the different gRNA-Cas9 pairs. The corresponding sequence deletions were assessed by PCR amplification of a fragment spanning the RNP target sites (Figure 1C, left). All RNP pairs led to deletion, as shown by the presence of smaller bands with sizes congruent with the distance between Cas9 cutting sites (Figure 1C, right). Differences in deletion efficacy were found among the different RNP pairs (1+4 > 1+3 > 2+4 > 2+3). In keratinocytes treated with RNP 1+4, the most efficacious pair, the smallest band (464 bp corresponding to a 172-bp deletion) accounted for 81.3% of alleles as determined by densitometric quantitation of the PCR products (Figure 1C). Sanger sequencing analysis of the lower PCR bands demonstrated that the predicted repair event (i.e., precise rejoining of the Cas9 break ends) was mostly represented, although a slight contribution of other indel-bearing species could be detected (Figure S1). The presence of ΔSPINK5 alleles in a ratio higher than 50% indicated that homozygous deletion of SPINK5 E1 represented a frequent event.
Figure 1.

SPINK5 Disruption in Human Keratinocytes
(A) SPINK5-knockout strategy. Pairs of CRISPR gRNA guides (RNP1 and RNP2) were designed to allow excision of SPINK5 exon 1 (E1) by Cas9. (B) Sequences and alignment of the designed gRNA guides to the targeted SPINK5 sequences (1 and 2 targeting E1, and 3 and 4 targeting intron 1 [I1]). The protospacer adjacent motif (PAM) is indicated in a darker color, and the transcription start site is indicated in red. (C) PCR analysis of genomic DNA from ΔSPINK5 keratinocytes treated with the different RNP pairs. Distance between cutting sites, according to each RNP pair combination, is shown in the right panel, and the efficacy (densitometric) value of excision is indicated at the bottom of each lane. The RNP 1+4 and 1+3 pairs yield the highest proportion of excised E1 according to the intensity of the lower band (464–510 bp).
In order to determine whether SPINK5 gene disruption affected the expression of LEKTI, its expression was analyzed by immunofluorescence. Remarkably and consistently with the high degree of SPINK5 E1 deletion achieved with 1+4 and 1+3 guide pairs, LEKTI expression appeared barely detectable in bulk keratinocyte populations (Figure 2A). These results were confirmed by western blot analysis, which showed a decrease of both the 145-kDa full-length fragment and the 65-kDa bioactive cleavage LEKTI fragment (Figure 2B). Overall, these results demonstrate the feasibility and high efficacy of SPINK5 knockout in human keratinocytes using non-viral CRISPR-Cas9 RNP delivery.
Figure 2.

Effect of SPINK5 Disruption on LEKTI Expression
(A) LEKTI immunofluorescence (IF) of human keratinocytes treated with the different RNP pairs. Cells treated with RNP 1+4 and 1+3 show a significant decrease in LEKTI expression compared to their healthy isogenic counterparts (HK Ctrl), resembling NS keratinocytes (HKNS). (B) Western blot analysis confirms the reduction of full-length (145 kDa) and bioactive LEKTI (65 kDa) in ΔSPINK5 cell lysates, consistent with the exon 1 deletion rate and the IF data.
Isolation and Characterization of ΔSPINK5 Keratinocyte Clones
Despite the remarkable efficacy of SPINK5 knockout achieved in bulk human primary keratinocytes, a bona fide LEKTI null could only be attainable when both SPINK5 alleles were targeted. To obtain cells with homozygous gene deletion, several clones of the keratinocytes treated with CRISPR RNP 1+4 and 1+3 pairs were isolated and expanded. As predicted from the DNA and protein data of the polyclonal population, most of the clones presented a homozygous ΔSPINK5 deletion (Figure 3A). Selected homozygous clones were further analyzed for LEKTI expression by western blot (Figure 3B). As predicted, protein expression was completely abrogated in the gene-edited clones. Due to its better proliferation rate, clone 1+4.2 was chosen for NS modeling studies, and its homozygous ΔSPINK5 deletion was confirmed by Sanger sequencing (Figure S1A). To avoid senescence and loss of regenerative capacity of the selected clone, cells were immortalized using a retroviral vector carrying the human papillomavirus E6 and E7 coding genes (named as ΔSPINK5+E6E7), and the expression of E7 protein was assessed by western blot (Figure S2). The tumorigenic potential of the immortalized cells was assessed after subcutaneous injection in immunocompromised mice. Twelve weeks after injection, no tumor development was observed (data not shown).
Figure 3.

Genotype of ΔSPINK5 Clones and LEKTI Expression
(A) Ten out of 11 ΔSPINK5 isolated clones show a single PCR band of approximately 464/481 bp, consistent with homozygous exon 1 deletion induced by RNPs 1+4 or 1+3. (B) Absence of LEKTI expression is confirmed, by western blot, in the selected homozygous clones.
ΔSPINK5 Human Keratinocytes as a Model of NS
The ability to recapitulate the NS phenotype of the ΔSPINK5 keratinocytes was assessed in vivo after grafting bioengineered skin produced with ΔSPINK5 clone 1+4.2 and ΔSPINK5+E6E7 to immunodeficient mice. For a comparative analysis of the results, skin-humanized mouse models were also generated with cells from a clinically and histologically well-characterized NS patient (Figure S3), and healthy isogenic human keratinocytes were used to produce the ΔSPINK5 counterparts. Eight weeks after grafting, routine histological analysis of the different grafts (H&E staining) was performed (Figure 4A). Human-specific involucrin immunoperoxidase staining was used to demark the grafted areas (Figure 4B). Remarkably, the ΔSPINK5 grafts showed typical NS skin phenotype, i.e., acanthosis, stratum corneum detachment, and finger-like dermal papillae. Furthermore, the ΔSPINK5+E6E7 grafts showed an epidermal architecture consistent with that of NS grafts, similar to that achieved with the non-immortalized ΔSPINK5 cells despite a certain degree of atypia (Figure 4A).
Figure 4.

Recapitulation of NS Phenotype by ΔSPINK5 Keratinocytes In Vivo
Skin grafts were produced with healthy donor keratinocytes (HK Ctrl), NS patient keratinocytes (HKNS), ΔSPINK5 clone 1+4.2 (HKΔSPINK5), and immortalized ΔSPINK5 clone (HKΔSPINK5+E6E7). (A) Histological analysis (H&E staining) of grafts produced with HKΔSPINK5 and HKΔSPINK5+E6E7 shows the recapitulation of the typical hyperkeratotic phenotype of NS. (B) Human-specific involucrin immunostaining (hINV) confirms the human origin of the grafts. Insets depict the staining at the mouse-human skin border. (C–F) Immunoperoxidase detection of (C) LEKTI, (D) keratin K10, and (E) psoriasin, as well as (F) in situ zymography, reveals that the skin equivalents regenerated from the ΔSPINK5 keratinocytes fairly resemble those of NS. Scale bars, (A–E) 100 μm, (F) 50 μm.
A comparative expression analysis of epidermal molecular markers was performed in the four groups of grafts. As predicted, neither NS, ΔSPINK5, nor ΔSPINK5+E6E7 grafts showed LEKTI expression (Figure 4C). Keratin K10 showed a patchy expression array in NS and ΔSPINK5 grafts that was also observed in ΔSPINK5+E6E7 grafts, although with a more heterogeneous pattern of staining reaching the basal layer (Figure 4D). Psoriasin, a marker associated with epidermal hyperproliferation, was barely detected in control grafts, but it was greatly increased in NS, ΔSPINK5, and ΔSPINK5+E6E7 grafts, presenting a similar expression pattern (Figure 4E). To assess whether the absence of LEKTI in NS and ΔSPINK5 keratinocytes resulted in exacerbated proteolytic activity in vivo due to uncontrolled kallikreins, in situ zymography was performed on frozen sections of the different grafts. Compared to the caseinolytic activity (green fluorescence) seen in grafts from healthy donor keratinocytes, increased proteolytic activities were detected in the upper layers of the hyperplastic epidermis in all LEKTI null graft sections (Figure 4F).
Histological examination of the dermal compartment of the different ΔSPINK5 grafts revealed also the existence of skin inflammation characterized by the presence of a vascular reaction and scattered neutrophils and eosinophils (Figure S4).
Overall, these results demonstrate that the skin regenerated from the ΔSPINK5 keratinocytes resembled to a great extent that of NS.
Use of the ΔSPINK5 Model for Proof-of-Concept Therapy Studies
We had previously conducted proof-of-concept and clinical gene therapy studies using lentiviral SPINK5 gene transfer to NS keratinocytes.5,17 In the present study, to validate the use of the model in the evaluation of therapeutic interventions, we tested the same ex vivo lentiviral approach with our ΔSPINK5 keratinocytes (ΔSPINK5+E6E7). Efficient gene transfer allowed a recovery of 61.3% of LEKTI expression (Figure S5). Skin equivalents prepared with gene-corrected ΔSPINK5+E6E7 keratinocytes were transplanted to immunodeficient mice, and grafts were analyzed 6 weeks post-grafting. Direct comparative examination of gene-corrected versus uncorrected ΔSPINK5+E6E7 grafts showed no obvious macroscopic signs of phenotypic reversion, albeit a tendency to present less scaly foci was found in the former (Figure S6). However, at the microscopic level, the gene-corrected skin regenerated in mice showed a striking normalization of the epidermal architecture, namely complete flattening of the epidermis, recovery of continuous K10 expression mostly at the suprabasal cell layers, and marked decrease of psoriasin expression and of kallikrein proteolytic activity (Figure 5).
Figure 5.

Validation of the ΔSPINK5 Model for Therapeutic Intervention In Vivo
Skin grafts were produced with healthy donor keratinocytes (HK Ctrl), immortalized ΔSPINK5 clone (HKΔSPINK5+E6E7), and SPINK5-transduced immortalized ΔSPINK5 clone (HKΔSPINK5+E6E7+SPINK5). (A) Histological analysis (H&E staining) of grafts produced with HKΔSPINK5+E6E7+SPINK5 shows the recovery of a normal epidermal architecture with marked reduction of rete ridges. (B) Human-specific involucrin immunostaining (hINV) confirms the human origin of the grafts. Insets depict the staining at the mouse-human skin border. (C) Restored expression of LEKTI closely follows the pattern of involucrin in the SPINK5 corrected grafts, verifying SPINK5 expression recovery. (D–F) Immunoperoxidase detection of (D) keratin K10 and (E) psoriasin, along with (F) in situ zymography, reveals that the skin equivalents regenerated from the SPINK5-transduced immortalized ΔSPINK5 keratinocytes fairly resemble those of healthy donor keratinocytes, with normal expression recovery of molecular markers and reduced proteolytic activity. Scale bars, (A–E) 100 μm, (F) 50 μm.
Given the successful recapitulation of the NS phenotype at the epidermal level in the in vivo skin-humanized model and its validity as a test system for therapeutic intervention, we explored whether valuable phenotypic data could also be obtained under a less complex experimental setting. To this end, in vitro OTCs produced either with gene-corrected (ΔSPINK5+E6E7+SPINK5) or uncorrected (ΔSPINK5+E6E7) keratinocytes were prepared and analyzed similarly to the in vivo grafts. The analysis of epidermal molecular markers, 2 weeks after exposure of the OTCs to the air-liquid interface, revealed the presence of clear hallmarks of NS in uncorrected OTCs (i.e., absence of LEKTI, irregular K10, and increased psoriasin expression), which were completely normalized in OTCs produced with gene-corrected cells (Figure 6). As seen in the in vivo skin model, gene-corrected OTCs also displayed markedly reduced caseinolytic activity, resembling that of OTCs prepared with healthy donor keratinocytes.
Figure 6.

Validation of the ΔSPINK5 Model for Therapeutic Intervention In Vitro
Fibrin-based organotypic cultures (OTCs) produced with healthy donor keratinocytes (HK Ctrl), NS patient keratinocytes (HKNS), immortalized ΔSPINK5 clone (HKΔSPINK5+E6E7), and SPINK5-transduced ΔSPINK5 clone (HKΔSPINK5+E6E7+SPINK5). (A) Histological analysis (H&E staining) shows that no major differences are observed between groups with regard to epidermal stratification and architecture. (B–E) Immunoperoxidase detection of (B) LEKTI, (C) keratin K10, and (D) psoriasin, as well as (E) in situ zymography, reveals that the OTCs produced with the ΔSPINK5 keratinocytes fairly resemble those of NS, and it shows normalized marker expression and reduced proteolytic activity in gene-corrected OTCs. Scale bar, 50 μm.
Globally, these results demonstrate that ΔSPINK5 keratinocyte derivatives (i.e., grafts or OTCs) represent useful platforms to test novel therapies for NS.
Discussion
Reliable models of genodermatoses based on the engraftment in immunodeficient mice of patient biopsies or bioengineered skin prepared with patient cells represent an advantage over murine models (e.g., transgenic or knockout), particularly when it comes to efficacy assessment of cell and gene therapies. Our group has provided trustworthy skin-humanized mouse models for epidermolysis bullosa, pachyonychia congenita, xeroderma pigmentosum, lamellar ichthyosis, and NS,5,18, 19, 20, 21, 22 contributing to the development of novel therapeutic strategies, some of which have already reached the clinical stage.17,23, 24, 25 However, one of the critical steps in the modeling based on skin bioengineering is the adequate provision of biopsies to obtain keratinocytes and fibroblasts. In addition to the paucity of available patients due to the inherent rarity of the disease, difficulties of biopsy procurement also involve ethical constrains, age of the patient, or availability of specialized physicians. Consequently, the study of genodermatoses, including their modeling, using patient cells has remained an effort mostly restricted to laboratories with roots in large dermatology centers. To avoid the dependence on patient biopsies, genetic manipulation of normal skin cells to alter the expression of diseases-causing genes can be considered. For disorders with recessive inheritance, abrogation of gene function is required.
Until recently, the predominant strategy to decrease the expression of a given gene relied on the use of RNAi technology. However, incomplete knockdown resulting from RNAi often leads to small amounts of protein, which is especially puzzling in cases where even low levels of protein are sufficient to provide a functional benefit.2,3 Currently, gene editing approaches using CRISPR-Cas9 technology have greatly facilitated the targeted disruption of genes allowing the generation of cell lines with fully abrogated gene function.26 However, to date, most knockout cell lines produced by this technology rely on constitutive Cas9 expression that is capable of inducing untoward genotoxic effects. In addition, to achieve biallelic gene disruption, cell cloning or isolation based on selectable markers is required, a possibility restricted mostly to cell lines. The previous low efficacy of gene editing achieved in primary human keratinocytes had precluded the possibility of isolation of large number of clones with the desired gene disruption. This drawback has now been solved thanks to an improved CRISPR-Cas9 strategy based on the use of double CRISPR RNA guides flanking the targeted DNA sequence delivered as RNPs by electroporation.4 This strategy, suited for hard-to-transfect cells27,28 and previously used by us to excise a mutation-bearing exon from COL7A1 (i.e., exon 80) causing recessive dystrophic epidermolysis bullosa, was shown to render a very high proportion of keratinocytes with biallelic gene disruption, enabling straightforward isolation of null clones. In this study, we used this gene editing strategy to generate a model of NS by deletion of a critical SPINK5 sequence (E1).
The extraordinary efficacy of the approach in keratinocytes resulted in a very high proportion (10 out of 11) of clones with homozygous SPINK5 disruption (Figure 3). The selected 1+4.2 clone clearly recapitulated the NS epidermal phenotype in grafted mice, a result that was our main goal of this study. Moreover, NS-like alterations were also found at the dermal level, suggesting that, despite the host (i.e., murine) origin of capillary and immune cells in this kind of model,29,30 the ΔSPINK5 epidermis was proficient to orchestrate an inflammatory response. This response, however, was likely not fully developed due to the lack of T cells in the immunodeficient mouse graft recipient.
In order to achieve a model enabling skin regeneration during a period of time sufficient to evaluate therapeutic interventions, we carried out immortalization of the gene-edited keratinocytes (ΔSPINK5+E6E7). In this regard, it could be argued that immortalization might invalidate the model by introducing additional genetic changes interfering with epidermal differentiation and/or proliferation. However, our results suggest that this was not the case in our ΔSPINK5 model, since the grafts from ΔSPINK5+E6E7 matched most of the histopathological features of NS grafts, including the response to a gene replacement therapy. In fact, restoration of LEKTI expression after lentiviral gene transfer to ΔSPINK5+E6E7 keratinocytes enabled full normalization of the epidermal phenotype of regenerated skin, despite a short-term (6- to 8-week) analysis.
Special concerns about the reduction and replacement of animal models in basic research and preclinical phases have encouraged the development of in vitro OTCs. Although they are simplified models, skin equivalents recapitulate, at least in part, a 3D architecture and represent valuable experimental platforms of lower cost and complexity than with animal models. In this regard, two different OTCs have been previously developed for NS, one with patient-derived keratinocytes5 and the other one with normal human keratinocytes transfected with SPINK5-interfering RNA.31 Both models partially recapitulated, with some degree of atypia, the NS phenotype and were used in preliminary screening of new therapies to treat NS, but they were not suitable for long-term studies. In fact, one of the main challenges of most OTCs is their limited lifespan. Therefore, current efforts have been made to optimize the formulation of dermal matrices and culture media to achieve greater durability.32,33
Despite the short half-life of our OTCs, the skin equivalent maintained its macroscopic and histological stability for up to 4 weeks and exhibited better epidermal architecture compared to the previous OTCs. Furthermore, it allowed the recapitulation of some distinctive hallmarks of NS and served to assess the protease activity reduction resulting from SPINK5 re-expression in ΔSPINK5+E6E7 keratinocytes (Figures 6B and 6E). Even so, partial recapitulation of the NS phenotype may suggest the need of other important cell lineages (such as immune cells) into the dermal matrix to more accurately mimic other pathological features of the disease.
There is a large number of monogenic genodermatoses, with only a small proportion being fully addressed in terms of models and therapies. In this study, we showed that efficient gene editing-mediated ablation of disease-causing genes and straightforward selection of gene-edited keratinocyte clones, enhanced by the use of Rho-associated protein kinase (ROCK) inhibitors in the cell culture,4,34, 35, 36 enables the production of genodermatoses-like primary epidermal cells. Our approach can contribute to increase the study and evaluation of novel therapeutic approaches for ultra-rare and previously neglected genodermatoses.
Materials and Methods
Skin Biopsies, Keratinocyte Culture, Isolation of Clones, and Immortalization
Skin biopsies were obtained, after informed consent, from healthy donors or NS patients in accordance with the Declaration of Helsinki and the approval of the Ethics Committees of the collaborating hospitals.
Primary keratinocytes were isolated from skin biopsies and plated onto lethally irradiated 3T3-J2 cells (feeder layer) and cultured in keratinocyte growth complete medium (KCa), a 2:1 mix of DMEM and Ham’S F12 medium (Gibco-BRL, Barcelona, Spain) containing 10% fetal bovine serum (FBS) (HyClone, Thermo Fisher Scientific, Waltham, MA, USA), 1% penicillin/streptomycin, and supplemented with 8 ng/mL cholera toxin, 5 μg/mL insulin, 2.4 ng/mL adenine, 0.4 μg/mL hydrocortisone, 1.3 ng/mL triiodothyronine, and 10 ng/mL epidermal growth factor (EGF) (Sigma-Aldrich, St. Louis, MO, USA).
To obtain isolated clones, cells were plated at low density in 100-mm plates (103 cells/plate) with 2 × 106 lethally irradiated 3T3 feeder cells per plate. Cell clones were then collected using polystyrene cloning cylinders (Sigma-Aldrich) and expanded in growth medium supplemented with Y-27632 ROCK inhibitor (10 μM, Sigma-Aldrich).
Immortalization of gene-edited human keratinocytes was performed as previously described.37 Briefly, a 20%–30% confluence keratinocyte cell culture was incubated overnight with the supernatant produced by HEK293T cells transfected with the retroviral vector plasmid pLXSN16E6E7 (Addgene, Watertown, MA, USA) together with plasmids containing necessary sequences for viral encapsidation (pNG-VL3-MLVgag-pol, pNGVL3-4070; Addgene). Cells were subsequently subjected to serial passage until the 3T3 feeder layer was no longer required.
CRISPR-Cas9 Delivery
gRNA CRISPR guides were designed targeting SPINK5 E1 (gRNA1, 5′-GTGCAGTATGACTGAACTCG-3′; and gRNA2, 5′-TGCACCAGCTGAGCAATGCA-3′) and SPINK5 intron 1 (gRNA3, 5′-GCTTTGAGATGTGTGGCAAG-3′; and gRNA4, 5′-GCCACACATCTCAAAGCCCT-3′) sequences. Synthetic RNAs and recombinant Cas9 were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA), and ribonucleoprotein (RNP) complexes were delivered to the primary keratinocytes by nucleofection using the Neon transfection system 10 μL kit (Thermo Fisher Scientific).
Primary cells were resuspended at 1.5 × 105 cells/10 μL of resuspension buffer R for each reaction. RNP complexes were added to each sample (72.7 pmol of CRISPR RNA [crRNA; gRNA]/trans-activating crRNA [tracrRNA], 10.9 pmol of Cas9, 6.6:1 molar ratio). The electroporation conditions were as follows: 1,700 V/20 ms/1 pulse. After electroporation, cells were seeded into six-well plates containing a feeder layer.
Genotyping of Gene-Edited Keratinocytes
Genomic DNA was isolated by isopropanol precipitation of keratinocyte lysates (lysis buffer was 20 mM Tris-HCl [pH 8], 5 mM EDTA, 1% SDS, 400 mM NaCl, and 20 μg/μL proteinase K [Roche, Basel, Switzerland]) and resuspended in Tris/EDTA (TE) buffer. Approximately 50 ng of genomic DNA was used for PCR amplification. PCR fragments spanning the Cas9 target sites within SPINK5 were generated with primers 5′-AGGGTCTGTGGACTCCATCA-3′ (forward) and 5′-AGCAAGAGGAAGATGAAGAAGAATCT-3′ (reverse).
The PCR program was as follows: 94°C for 5 min; 5 cycles of 94°C for 30 s, 65°C for 30 s, and 72°C for 45 s, decreasing annealing temperature 1°C every cycle; followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 45 s, then 72°C for 7 min. PCR products were analyzed in 1.5% agarose gel. The molecular weight marker was IX (Sigma-Aldrich). For sequencing, PCR products were treated with Illustra ExoProStar (GE Healthcare, UK), sequenced using BigDye Terminator version (v)1.1 cycle sequencing kit (Thermo Fisher Scientific), and examined on a 3730 DNA analyzer (Life Technologies, Carlsbad, CA, USA). Chromatograms were analyzed using Sequencher software (Gene Codes, Ann Harbor, MI, USA). Bio-Rad’s Image Lab software 6.0 was used for PCR band densitometry.
Western Blot Analysis
Keratinocytes were lysed in protein extraction buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, and 5 mM EDTA) containing proteinase inhibitors cocktail (cOmplete Mini protease inhibitor cocktail, Roche) and phosphatase inhibitors cocktail (PhosSTOP, Roche). For each sample, 20 μg of total protein was resolved on NuPAGE 4%–12% Bis-Tris gel (Invitrogen, Carlsbad, CA, USA) and electrotransferred onto nitrocellulose membranes (Invitrogen).
For LEKTI analysis, blots were probed with a polyclonal anti-LEKTI antibody38 at 1:1,000 dilution, and cell immortalization was assessed with an anti-E7 monoclonal antibody (Santa Cruz Biotechnology. Dallas, TX, USA) at 1:1,000 dilution. An antibody against β-actin (Abcam, Cambridge, UK) was used as loading control. Visualization was performed by incubating the membrane with horseradish peroxidase (HRP)-conjugated anti-immunoglobulin G (IgG) rabbit antibody (Amersham, Burlington, MA, USA) or anti-IgG mouse antibody (Jackson ImmunoResearch Laboratories. Cambridge, UK) at 1:5,000 dilution and Clarity western ECL (enhanced chemiluminescence) blotting substrate (Bio-Rad).
Bioengineered Skin Generation and Grafting to Immunodeficient Mice
Animal studies were approved by our institutional animal care and use committee according to national and European legal regulations (protocol PROEX 187/15). Keratinocytes were seeded on fibrin dermal equivalents containing normal human fibroblasts, prepared as previously described.39 After keratinocytes reached confluence, bioengineered skin equivalents (n = 6) were grafted onto the back of 7-week-old female immunodeficient mice (nu/nu, NMRI background), purchased from Elevage Janvier (France), as previously described.39 Mice were sacrificed at different time points after grafting, and grafts were harvested and embedded in Tissue-Tek OCT compound or paraffin for skin histology and immunohistochemistry analyses.
Organotypic Skin Cultures
To establish the in vitro OTCs, human dermal fibroblasts (1 × 105) were resuspended in a fibrin matrix derived from pig plasma cryoprecipitate diluted in growth medium, supplemented with 800 μg/mL Amchafibrin (Fides Ecopharma, Barcelona, Spain) and 2.5 U of human thrombin diluted in 0.025 mM CaCl2 (Sigma-Aldrich). The mixture was placed in a six-well transwell plate with 1-μm pore polyester membrane (Falcon cell culture insert, Corning Life Sciences, NY, USA). After gelation (2 h at 37°C), human keratinocytes (1 × 106 cells/well) were seeded onto the gel. When keratinocytes reached confluence, the insert was transferred to deep-well plates (Corning Life Sciences) and cultured at the air-liquid interface for 2 weeks to allow proper epithelium differentiation. To this end, the growth medium was removed from the surface of the culture and differentiation medium (2:1 of DMEM/Ham’S F-12 mixture supplemented with 0.5% FBS, 5 μg/mL insulin, 8 ng/mL cholera toxin, 0.4 μg/mL hydrocortisone, 2.4 ng/mL adenine, 1.3 ng/mL triiodothyronine, 1% penicillin/streptomycin, 1.5 ng/mL EGF, and 50 μg/mL ascorbic acid) was added under the insert. The differentiation medium was renewed twice a week for 2 weeks until OTC harvest. In order to confirm tissue stability, some OTCs remained in culture up to 4 weeks.
Immunofluorescence and Immunohistochemical Staining
For immunofluorescence detection of LEKTI in keratinocytes, cells grown on glass coverslips were fixed in 4% paraformaldehyde for 10 min at room temperature. Cells were incubated in Tris-buffered saline (TBS) with 1% BSA (Sigma-Aldrich) for 1 h at 37°C, and in polyclonal anti-LEKTI antibody38 at 1:1,000 dilution. Secondary antibody (Alexa Fluor 594, Invitrogen) was used at 1:2,000 dilution for 30 min at 37°C. Preparations were mounted using Mowiol mounting medium (Hoechst, Somerville, NJ, USA) containing 0.25 ng/mL DAPI (Sigma-Aldrich).
Formalin-fixed paraffin sections (4–6 μm) were stained with hematoxylin and eosin (Gill 2 hematoxylin and eosin Y, alcoholic; Thermo Fisher Scientific, Altrincham, UK) following a standard procedure to determine tissue architecture. Epidermal molecular markers were studied by immunoperoxidase staining. Primary antibodies used were as follows: anti-involucrin (Sigma-Aldrich) at 1:100 dilution, anti-LEKTI38 at 1:1,000 dilution, anti-K10 (Santa Cruz Biotechnology) at 1:100 dilution, and anti-psoriasin (Imgenex, San Diego, CA, USA) at 1:200 dilution. Secondary antibody anti-rabbit or anti-mouse IgG (H+L) (Jackson ImmunoResearch Laboratories) was used at 1:500 dilution. The ABC peroxidase kit (Vector Laboratories, Burlingame, CA, USA) was used for immunohistochemical detection.
In Situ Zymography
To detect skin proteolytic activity, frozen sections were incubated overnight at 37°C with 10 μg/mL casein conjugated with BODIPY-FL using the EnzChek Ultra protease assay kit (Invitrogen) in a buffer containing 10 mM Tris-HCl (pH 7.8). The fluorescence was detected under a fluorescence microscope (λ490 nm).
SPINK5 Lentiviral Transduction
Lentiviral vector preparations were produced by transfecting HEK293T cells by calcium phosphate precipitation,19 using packaging plasmids encoding the viral capsid (pMDLgpRRE, pRSV Rev, pMD2.VSVG [Addgene]) and the plasmid vector SPINK5/EGFP or the control version devoid of SPINK5.38 Infectious lentiviruses were harvested at 24 and 48 h post-transfection and filtered through 0.45-μm pore cellulose acetate filters (Merck, Darmstadt, Germany). Immortalized keratinocytes were infected with SPINK5/EGFP or control EGFP lentiviral supernatants in two infection cycles of 7 h at 37°C.
Author Contributions
Conceptualization: V.G., M.C., and F.L.; Investigation: V.G., E.C.-S., J.B., A.M., and R.M.; Project Administration and Funding Acquisition: M.D.R. and F.L.; Resources: S.L., W.-L.D., and A.V.; Writing – Original Draft: V.G., M.C., and F.L.; Writing – Review & Editing: V.G., E.C.-S., J.B., M.C., and F.L.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The study was funded by Spanish grants PI14/00931 and PI17/01747 from the Instituto de Salud Carlos III (to F.L.); grant SAF2017-86810-R from the Ministry of Economy and Competitiveness (to M.D.R.), all co-funded with European Regional Development Funds (ERDF); and grant Avancell-CM (S2017/BMD-3692). Authors are indebted to Blanca Duarte, Almudena Holguín, and Nuria Illera for grafting experiments, and to Jesús Martínez and Edilia De Almeida for animal maintenance and care. W.-L.D. is a Great Ormond Street Hospital Children’s Charity Senior Lecturer.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.031.
Contributor Information
Marta Carretero, Email: marta.carretero@ciemat.es.
Fernando Larcher, Email: fernando.larcher@ciemat.es.
Supplemental Information
References
- 1.Chacón-Solano E., Guerrero-Aspizua S., Martínez-Santamaría L., Del Río M., Larcher F. Organotypic and humanized animal models of genodermatoses. In: Marques A.P., Pirraco R.P., Cerqueira M.T., Reis R.L., editors. Skin Tissue Models. Elsevier; 2018. pp. 77–102. [Google Scholar]
- 2.Evers B., Jastrzebski K., Heijmans J.P.M., Grernrum W., Beijersbergen R.L., Bernards R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016;34:631–633. doi: 10.1038/nbt.3536. [DOI] [PubMed] [Google Scholar]
- 3.Morgens D.W., Deans R.M., Li A., Bassik M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016;34:634–636. doi: 10.1038/nbt.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonafont J., Mencía Á., García M., Torres R., Rodríguez S., Carretero M., Chacón-Solano E., Modamio-Høybjør S., Marinas L., León C. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-mediated gene editing. Mol. Ther. 2019;27:986–998. doi: 10.1016/j.ymthe.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di W.L., Larcher F., Semenova E., Talbot G.E., Harper J.I., Del Rio M., Thrasher A.J., Qasim W. Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of Netherton syndrome-derived skin grafts. Mol. Ther. 2011;19:408–416. doi: 10.1038/mt.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chavanas S., Bodemer C., Rochat A., Hamel-Teillac D., Ali M., Irvine A.D., Bonafé J.L., Wilkinson J., Taïeb A., Barrandon Y. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 7.Bitoun E., Micheloni A., Lamant L., Bonnart C., Tartaglia-Polcini A., Cobbold C., Al Saati T., Mariotti F., Mazereeuw-Hautier J., Boralevi F. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum. Mol. Genet. 2003;12:2417–2430. doi: 10.1093/hmg/ddg247. [DOI] [PubMed] [Google Scholar]
- 8.Egelrud T., Brattsand M., Kreutzmann P., Walden M., Vitzithum K., Marx U.C., Forssmann W.G., Mägert H.J. hK5 and hK7, two serine proteinases abundant in human skin, are inhibited by LEKTI domain 6. Br. J. Dermatol. 2005;153:1200–1203. doi: 10.1111/j.1365-2133.2005.06834.x. [DOI] [PubMed] [Google Scholar]
- 9.Mägert H.J., Kreutzmann P., Ständker L., Walden M., Drögemüller K., Forssmann W.G. LEKTI: a multidomain serine proteinase inhibitor with pathophysiological relevance. Int. J. Biochem. Cell Biol. 2002;34:573–576. doi: 10.1016/s1357-2725(01)00179-0. [DOI] [PubMed] [Google Scholar]
- 10.Bonnart C., Deraison C., Lacroix M., Uchida Y., Besson C., Robin A., Briot A., Gonthier M., Lamant L., Dubus P. Elastase 2 is expressed in human and mouse epidermis and impairs skin barrier function in Netherton syndrome through filaggrin and lipid misprocessing. J. Clin. Invest. 2010;120:871–882. doi: 10.1172/JCI41440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borgoño C.A., Michael I.P., Komatsu N., Jayakumar A., Kapadia R., Clayman G.L., Sotiropoulou G., Diamandis E.P. A potential role for multiple tissue kallikrein serine proteases in epidermal desquamation. J. Biol. Chem. 2007;282:3640–3652. doi: 10.1074/jbc.M607567200. [DOI] [PubMed] [Google Scholar]
- 12.Deraison C., Bonnart C., Lopez F., Besson C., Robinson R., Jayakumar A., Wagberg F., Brattsand M., Hachem J.P., Leonardsson G., Hovnanian A. LEKTI fragments specifically inhibit KLK5, KLK7, and KLK14 and control desquamation through a pH-dependent interaction. Mol. Biol. Cell. 2007;18:3607–3619. doi: 10.1091/mbc.E07-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Descargues P., Deraison C., Prost C., Fraitag S., Mazereeuw-Hautier J., D’Alessio M., Ishida-Yamamoto A., Bodemer C., Zambruno G., Hovnanian A. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J. Invest. Dermatol. 2006;126:1622–1632. doi: 10.1038/sj.jid.5700284. [DOI] [PubMed] [Google Scholar]
- 14.Bitoun E., Chavanas S., Irvine A.D., Lonie L., Bodemer C., Paradisi M., Hamel-Teillac D., Ansai S., Mitsuhashi Y., Taïeb A. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J. Invest. Dermatol. 2002;118:352–361. doi: 10.1046/j.1523-1747.2002.01603.x. [DOI] [PubMed] [Google Scholar]
- 15.Ong C., O’Toole E.A., Ghali L., Malone M., Smith V.V., Callard R., Harper J.I. LEKTI demonstrable by immunohistochemistry of the skin: a potential diagnostic skin test for Netherton syndrome. Br. J. Dermatol. 2004;151:1253–1257. doi: 10.1111/j.1365-2133.2004.06180.x. [DOI] [PubMed] [Google Scholar]
- 16.Di W.-L., Mellerio J.E., Bernadis C., Harper J., Abdul-Wahab A., Ghani S., Chan L., Martinez-Queipo M., Hara H., McNicol A.M. Phase I study protocol for ex vivo lentiviral gene therapy for the inherited skin disease, Netherton syndrome. Hum. Gene Ther. Clin. Dev. 2013;24:182–190. doi: 10.1089/humc.2013.195. [DOI] [PubMed] [Google Scholar]
- 17.Di W.-L., Lwin S.M., Petrova A., Bernadis C., Syed F., Farzaneh F., Moulding D., Martinez A.E., Sebire N.J., Rampling D. Generation and clinical application of gene-modified autologous epidermal sheets in Netherton syndrome: lessons learned from a phase 1 trial. Hum. Gene Ther. 2019;30:1067–1078. doi: 10.1089/hum.2019.049. [DOI] [PubMed] [Google Scholar]
- 18.Gache Y., Baldeschi C., Del Rio M., Gagnoux-Palacios L., Larcher F., Lacour J.P., Meneguzzi G. Construction of skin equivalents for gene therapy of recessive dystrophic epidermolysis bullosa. Hum. Gene Ther. 2004;15:921–933. doi: 10.1089/hum.2004.15.921. [DOI] [PubMed] [Google Scholar]
- 19.Di Nunzio F., Maruggi G., Ferrari S., Di Iorio E., Poletti V., Garcia M., Del Rio M., De Luca M., Larcher F., Pellegrini G., Mavilio F. Correction of laminin-5 deficiency in human epidermal stem cells by transcriptionally targeted lentiviral vectors. Mol. Ther. 2008;16:1977–1985. doi: 10.1038/mt.2008.204. [DOI] [PubMed] [Google Scholar]
- 20.García M., Llames S., García E., Meana A., Cuadrado N., Recasens M., Puig S., Nagore E., Illera N., Jorcano J.L. In vivo assessment of acute UVB responses in normal and xeroderma pigmentosum (XP-C) skin-humanized mouse models. Am. J. Pathol. 2010;177:865–872. doi: 10.2353/ajpath.2010.091096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aufenvenne K., Rice R.H., Hausser I., Oji V., Hennies H.C., Rio M.D., Traupe H., Larcher F. Long-term faithful recapitulation of transglutaminase 1-deficient lamellar ichthyosis in a skin-humanized mouse model, and insights from proteomic studies. J. Invest. Dermatol. 2012;132:1918–1921. doi: 10.1038/jid.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.García M., Larcher F., Hickerson R.P., Baselga E., Leachman S.A., Kaspar R.L., Del Rio M. Development of skin-humanized mouse models of pachyonychia congenita. J. Invest. Dermatol. 2011;131:1053–1060. doi: 10.1038/jid.2010.353. [DOI] [PubMed] [Google Scholar]
- 23.Mavilio F., Pellegrini G., Ferrari S., Di Nunzio F., Di Iorio E., Recchia A., Maruggi G., Ferrari G., Provasi E., Bonini C. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006;12:1397–1402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 24.Hirsch T., Rothoeft T., Teig N., Bauer J.W., Pellegrini G., De Rosa L., Scaglione D., Reichelt J., Klausegger A., Kneisz D. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551:327–332. doi: 10.1038/nature24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eichstadt S., Barriga M., Ponakala A., Teng C., Nguyen N.T., Siprashvili Z., Nazaroff J., Gorell E.S., Chiou A.S., Taylor L. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4 doi: 10.1172/jci.insight.130554. e130554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang F., Guo T., Jiang H., Li R., Wang T., Zeng N., Dong G., Zeng X., Li D., Xiao Y. A comparison of CRISPR/Cas9 and siRNA-mediated ALDH2 gene silencing in human cell lines. Mol. Genet. Genomics. 2018;293:769–783. doi: 10.1007/s00438-018-1420-y. [DOI] [PubMed] [Google Scholar]
- 27.Kim S., Kim D., Cho S.W., Kim J., Kim J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M., Carte J., Chen W., Roark N., Ranganathan S. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 29.Luchetti M.M., Moroncini G., Jose Escamez M., Svegliati Baroni S., Spadoni T., Grieco A., Paolini C., Funaro A., Avvedimento E.V., Larcher F. Induction of scleroderma fibrosis in skin-humanized mice by administration of anti-platelet-derived growth factor receptor agonistic autoantibodies. Arthritis Rheumatol. 2016;68:2263–2273. doi: 10.1002/art.39728. [DOI] [PubMed] [Google Scholar]
- 30.Guerrero-Aspizua S., García M., Murillas R., Retamosa L., Illera N., Duarte B., Holguín A., Puig S., Hernández M.I., Meana A. Development of a bioengineered skin-humanized mouse model for psoriasis: dissecting epidermal-lymphocyte interacting pathways. Am. J. Pathol. 2010;177:3112–3124. doi: 10.2353/ajpath.2010.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S., Olt S., Schoefmann N., Stuetz A., Winiski A., Wolff-Winiski B. SPINK5 knockdown in organotypic human skin culture as a model system for Netherton syndrome: effect of genetic inhibition of serine proteases kallikrein 5 and kallikrein 7. Exp. Dermatol. 2014;23:524–526. doi: 10.1111/exd.12451. [DOI] [PubMed] [Google Scholar]
- 32.Stark H.J., Szabowski A., Fusenig N.E., Maas-Szabowski N. Organotypic cocultures as skin equivalents: a complex and sophisticated in vitro system. Biol. Proced. Online. 2004;6:55–60. doi: 10.1251/bpo72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boehnke K., Mirancea N., Pavesio A., Fusenig N.E., Boukamp P., Stark H.J. Effects of fibroblasts and microenvironment on epidermal regeneration and tissue function in long-term skin equivalents. Eur. J. Cell Biol. 2007;86:731–746. doi: 10.1016/j.ejcb.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 34.McMullan R., Lax S., Robertson V.H., Radford D.J., Broad S., Watt F.M., Rowles A., Croft D.R., Olson M.F., Hotchin N.A. Keratinocyte differentiation is regulated by the Rho and ROCK signaling pathway. Curr. Biol. 2003;13:2185–2189. doi: 10.1016/j.cub.2003.11.050. [DOI] [PubMed] [Google Scholar]
- 35.Strudwick X.L., Lang D.L., Smith L.E., Cowin A.J. Combination of low calcium with Y-27632 rock inhibitor increases the proliferative capacity, expansion potential and lifespan of primary human keratinocytes while retaining their capacity to differentiate into stratified epidermis in a 3D skin model. PLoS ONE. 2015;10:e0123651. doi: 10.1371/journal.pone.0123651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duarte B., Miselli F., Murillas R., Espinosa-Hevia L., Cigudosa J.C., Recchia A., Del Río M., Larcher F. Long-term skin regeneration from a gene-targeted human epidermal stem cell clone. Mol. Ther. 2014;22:1878–1880. doi: 10.1038/mt.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chamorro C., Almarza D., Duarte B., Llames S.G., Murillas R., García M., Cigudosa J.C., Espinosa-Hevia L., Escámez M.J., Mencía A. Keratinocyte cell lines derived from severe generalized recessive epidermolysis bullosa patients carrying a highly recurrent COL7A1 homozygous mutation: models to assess cell and gene therapies in vitro and in vivo. Exp. Dermatol. 2013;22:601–603. doi: 10.1111/exd.12203. [DOI] [PubMed] [Google Scholar]
- 38.Di W.-L., Semenova E., Larcher F., Del Rio M., Harper J.I., Thrasher A.J., Qasim W. Human involucrin promoter mediates repression-resistant and compartment-specific LEKTI expression. Hum. Gene Ther. 2012;23:83–90. doi: 10.1089/hum.2011.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larcher F., Dellambra E., Rico L., Bondanza S., Murillas R., Cattoglio C., Mavilio F., Jorcano J.L., Zambruno G., Del Rio M. Long-term engraftment of single genetically modified human epidermal holoclones enables safety pre-assessment of cutaneous gene therapy. Mol. Ther. 2007;15:1670–1676. doi: 10.1038/sj.mt.6300238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.