

Abstract
Imaging the inventory of microbial small molecule interactions provides important insights into microbial chemical ecology and human medicine. Herein we demonstrate a new method for enhanced detection and analysis of metabolites present in interspecies interactions of microorganisms on surfaces. We demonstrate that desorption electrospray ionization-imaging mass spectrometry (DESI-IMS) using microporous membrane scaffolds (MMS) enables enhanced spatiochemical analyses of interacting microbes among tested sample preparation techniques. Membrane scaffolded DESI-IMS has inherent advantages compared to matrix-assisted laser desorption ionization (MALDI) and other IMS methods through direct IMS analyses of microbial chemistry in situ. This rapid imaging method yields sensitive MS analyses with unique m/z measurements when compared to liquid chromatography–electrospray ionization-mass spectrometry (LC–ESI-MS) via unmediated sampling by MMS DESI-IMS. Unsupervised segmentation imaging analysis of acquired DESI-IMS data reveals distinct chemical regions corresponding to intermicrobial phenomenon such as predation and communication. We validate the method by linking Myxovirescin A and DKxanthene-560 to their known biological roles of predation and phase variation, respectively. In addition to providing the first topographic locations of known natural products, we prioritize 54 unknown features using segmentation within the region of predation. Thus, DESI-IMS and unsupervised segmentation spatially annotates the known biology of myxobacteria and provides functional exploration of newly uncharacterized small molecules.
Graphical Abstract

A significant means by which microorganisms maintain interactions within ecological communities is through producing primary and secondary metabolites.1 In human–microbiome interactions, Escherichia coli was shown to secrete colibactin, which intercalates into host DNA. This demonstrated how human gut microbiota composition can be connected to carcinogenesis.2–4 In insects, volatile metabolites emitted from microbe–microbe interactions constitute a symbiotic relationship modulating insect behavior.5 Leaf cutting ants harbor symbiotic actinomycetes on their carapace that produce antifungal compounds, which allow the ants to prevent pathogenic strains from invading the fungal gardens they use as a food source.6 An anti-methicilin-resistant Staphylococcus aureus (MRSA) metabolite lugdunin is produced by Staphylococcus lugdunensis and mediates interspecies Staphylococcus interactions in human nares.7 The importance of microbial secondary metabolites in maintaining complex interkingdom, interspecies, and intergeneric chemical ecologies may be one reason that microbial natural products have become a major source of therapeutic antibiotics and anticancer agents in human medicine.
Historically, the discovery of secondary metabolites has been conducted via bioassay guided isolation from microbial liquid monocultures.8-10 However, recent innovations in natural product discovery have demonstrated novel secondary metabolite production from microbes in solid-phase growth.11 Indeed, microbial competition and other mixed culture phenomena induce broad changes in the measurable metabolome in comparison to individual monocultures.12-14 Typically, metabolomic changes are measured via reversed phase liquid chromatography–electrospray ionization-mass spectrometry (RPLC–ESI-MS).9,12,13 However, in these experiments, there is a loss of the spatial expression patterns of metabolites, which may reveal functional roles. Thus, developing tools that provide detailed spatial maps of secondary metabolites during coculture conditions on surfaces are an active area of research. Fully realized, these promise to both advance natural product discovery as well as detail the functional roles of primary and secondary metabolite dynamics in microbial chemical ecology.
Imaging mass spectrometry (IMS) is conducive to sampling microbes under planar solid-phase growth conditions. This technique can be used to assess chemical-spatial phenotypes by correlating the location of measured metabolites to observed microbial morphologies.11 These spatial correlations provide a significant advantage in assessing the functional roles of natural products. For instance, homospermidine lipids were correlated to myxobacteria fruiting body formation using matrix assisted laser desorption ionization (MALDI) imaging MS.11 Indeed, MALDI was the first technique used to map secondary metabolites from microorganisms.15 Subsequently, an array of imaging MS techniques have been developed including liquid extraction surface analysis (LESA), desorption electrospray ionization (DESI), and nanospray-DESI (nano-DESI) to analyze microbial metabolites. 16-22 DESI-IMS methods to sample microbial communities are of particular interest for screening purposes due to rapid data acquisition and minimal sample preparation associated with ambient sampling.22 Removing the vacuum requirements from ionization increases throughput and provides some flexibility in the state of the sample to be interrogated. Therefore, ambient sampling provides a means for the direct measurements of secondary metabolites within the microbial environment in situ. For these reasons, we describe DESI-IMS to spatially profile secondary metabolites of microbial communities in coculture conditions.
Herein we have developed and evaluated a process for DESI-IMS based analysis of spatially resolved molecular patterns associated with interacting microbes grown on surfaces. The most promising workflow combines a microporous membrane scaffold (MMS) and an unsupervised image segmentation chemoinformatics platform for data analysis. Spatial segmentation has been applied to MALDI-IMS data of microbial interactions, setting a precedence for spatiochemical phenotyping of microorganisms in response to coculture conditions.23-25 Further, membrane scaffolds have been used in microbiology and DESI-IMS in particular to study biofilm formation and metabolite production.26,27 We developed a membrane scaffolded DESI-IMS workflow with unsupervised segmentation using the model predator–prey system of Myxococcus xanthus DK1622 and E. coli. Using the MMS DESI-IMS method, we observed the significant accumulation of the myxobacteria secondary metabolites DKxnathene-560 and Myxovirescin A within the region of interaction.28-31 To our knowledge DESI-IMS and unsupervised image segmentation provides the first topographic location of these metabolites during surface predation. We demonstrate the utility of the method in natural product discovery by measuring and spatially prioritizing unknown features within this designated region of predation. Therefore, DESI-IMS and unsupervised segmentation can be used to elucidate previously unreported molecules with implicated roles in secondary metabolism and predation. Comparing M. xanthus mutants and chemotypes, we validate previous reports of Myxovirescin A within myxobacteria predation of E. coli and further probe phase variation within this phenomenon via DKxanthene-560.28-31 In summary, we present a workflow that effectively evaluates microbe–microbe interactions and correlates spatial location of metabolites to biological function.
■ EXPERIMENTAL METHODS
Strains.
Escherichia coli B/R was obtained from a plate of the amoeba Dictyostelium discoideum AX-2. A D. discoideum AX-2 stock was obtained from the Dicty Stock Center at Northwestern University (Chicago, IL). The original plates containing both D. discoideum AX-2 and E. coli B/R were restreaked on CYE agar until pure E. coli B/R colonies were obtained. The Myxococcocus xanthus DK1622 and Δta1 strains were kindly donated to our laboratories by Dr. Daniel Wall (University of Wyoming, Laramie, WY). M. xanthus DK1622 undergoes phase variation, a process by which predominantly yellow or tan subpopulations appear due to changes in gene expression.28-30 Phenotypic studies show increased DKxanthene production resulting in the yellow pigment associated with yellow phase variants (PVs).28-30 We isolated two phase variants by streaking out M. xanthus Dk1622 and then picking individual colonies that were either yellow or tan in appearance.
Culture and Growth Conditions.
The prey E. coli was grown in casitone yeast extract (CYE) liquid medium (1% casitone, 0.5% yeast extract, 8 mM MgSO4, 10 mM MOPS, pH 7.6) for 16–24 h. After growth, E. coli was spun down and concentrated 10–20-fold. A volume of 50 μL of the prey was spotted onto 10% (carbon sources of yeast extract and casitone) CYE medium and allowed to dry for 1–2 h prior to placement of the predator myxobacterium. Predator seed cultures were inoculated into CYE liquid medium and shaken at 200 rpm for 48 h at 30 °C (New Brunswick Scientific, Innova 4900 Multitier Environmental Shaker). After growth, the cultures were spun down, concentrated 10–20-fold, and 50 μL was spotted adjacent to the dried E. coli. After the myxobacterium dried, each sample was placed into a 30 °C incubator and allowed to grow for 4 days. After the allotted time, they were stored at 4 °C until sampled.
Sample Preparation.
We evaluated three methods of sample preparation for the detection of microbially produced small molecules via DESI-IMS. Dried agar samples were prepared by first placing a glass slide in the Petri dish below the growth medium, such that predator and prey colonies were spotted on the agar directly above the glass slide. After growth, the samples were allowed to dry for 8 h. The glass slide was then removed from the Petri dish by detaching any medium not on top of it. The resulting dried medium and colonies were sampled.21 Imprinting samples were constructed by placing a glass slide on top of the grown microbial colonies on agar and applying uniform pressure for 10–20 s. The transferred colonies were allowed to dry for an hour before sampling.19 The microporous membrane scaffold (MMS) method entailed spotting predator and prey onto a sterile nylon membrane (Sigma-Aldrich, 0.45 μM pore size, 47 mm diameter) and placing the membrane on top of the medium. This system was kept intact within the incubator and during the growth period. Prior to sampling, the membrane was removed from the medium, dried for 10 min, and adhered to a glass slide using double-sided scotch tape. Membranes were incubated on agar without colonies to provide blanks for the MMS method.
Acquisitions.
A Waters Synapt G2S High Definition Mass Spectrometer (Waters Corporation, Milford MA) was used with a Waters x–y directional stage and DESI source described by Tillner et al.32 The system was mass calibrated with sodium formate salt clusters to a 95% confidence band and root-meansquare (RMS) residual mass ≤0.5 ppm. This resulted in experimental mass accuracies of generally 2–5 parts per million (ppm) ± 3 ppm and mass resolving powers ~11 000. Ion mobility calibrations were performed using polyalanine to achieve RMS CCS ≤ 0.5%. Optimized DESI source conditions were found to be −3 kV capillary voltage, 110 °C desolvation temperature, 0.5 mPa N2 gas flow, and a cone voltage of 40 V. The sprayer was set at a 70° angle, with the x, y, z, settings at −2, + 2, and +2.75, respectively. Ionization solvent was comprised of 90/10 acetonitrile/water (Optima grade, Fisher Scientific) solution with 0.1% NH4OH and 0.2 ng/μL leucine-enkephalin for lock mass and normalization purposes. Optical images were taken using a 12-megapixel camera. Imaging acquisitions were prepared using the HDImaging software. All images were acquired using a 50 μm × 200 μm pixel size with a raster rate of 100 μm/s equating to ~30 000 pixels per image at a 0.485 s scan rate. All analyses were performed in negative ion mode in the mass range of m/z 50–1200. Ion mobility (IM) experiments were performed using a nitrogen buffer gas. DESI-MS/MS experiments were individually performed to putatively identify natural products and metabolites. All acquisitions were performed in triplicate. Parameters for RPLC–ESI-IM-MS experiments can be found in the Supporting Information.
Data Processing and Analysis.
All raw imaging files were processed using HDImaging software. The 4 000 most abundant features of each experiment were investigated. Mass measurements for these features were lock mass adjusted in 2 min intervals throughout each experiment to the leucine-enkephalin internal standard. During this processing step for DESI-IM-IMS files, a temporary raw file was created that consists of the mobility drift time plotted against m/z. This file was directly imported into Progenesis QI. These 4 000 most abundant features result in an average of 1 670 features when peak picked across replicates. Further, ~300 features were observed in blank samples, leaving ~1 400 quality features to search and dereplicate against an in-house myxobacteria database consisting of 280 natural product entries as well as the online repositories KEGG, Chemspider, MassBank, E. coli Metabolome Database, Yeast Metabolome Database, Natural Product Updates, Natural Products Discovery Institute, and NIST. Tentative identifications were made on a threshold of 10 ppm mass accuracy and 80% isotopic similarity according to the level system proposed by Schrimpe-Rutlegdge et al.33 Level 2 identifications of natural products and other metabolites were made using DESI-MS/MS acquisitions (Figure S2).
Unsupervised segmentation used an imaging text file created by the HDImaging program. The generated text file was imported into R and Cardinal MSI. Feature intensities were normalized to the TIC and then subsequently normalized to the internal standard leucine-enkephalin intensity for each pixel across the image to account for substrate-dependent ionization or differential ionization in regards to the various structures and surfaces within an IMS experiment. Features were peak picked using a 10 ppm window. Prior to unsupervised segmentation via spatial shrunken centroids analysis, the initial number of families (k) and the shrinkage parameter (s) were determined empirically for each experiment as described by Bemis et al.33,34 The significant features within each segment of interest were dereplicated against online repositories and the myxobacteria database to prioritize unreported natural products.
■ RESULTS AND DISCUSSION
Sampling Methodology.
To effectively measure intermicrobial interactions using IMS, three regions representing the two microbial communities and their interface must first be resolved spatially (Figure 1A). Second, the measured metabolites from each of the separate regions must be unique. Thus, these regions that we refer to as predator, prey, and predation must have discrete locations and chemical profiles to successfully evaluate microbe–microbe interactions. While these regions are visually quite distinct, capturing these differences via IMS remains a challenge. This is mainly due to the relatively fragile nature of microbial colonies, which must be rigid for effective ionization. For this reason, sample preparation is the primary factor influencing the ability to analyze microbial communities. The extent of retained spatial and chemical information on the microbial environment ultimately determines the ability to distinguish between distinct phenotypic regions within intermicrobial interactions. Therefore, methods that avoid analyte delocalization and degradation during the sample preparation process will provide the most repeatable and comprehensive analysis. We evaluated the dried agar, imprinting, and MMS sample preparation methods depicted in Figure 1B using sensitivity, repeatability, and analyte delocalization as performance metrics. The optical images, which qualitatively represent the outcome of these preparative methods, demonstrate the varying levels of retained spatial and chemical information (Figure 1B).
Figure 1.

(A) Diagram of microbial predation experiments and annotation of optical image. (B) Visual representations of sample preparation methods and optical images of microbial colonies prior to acquisitions. (C) Ion images of DKxanthene-560 and lysoPE 16:1 of biological replicates across each method.
The delocalization of small molecules within predation experiments across the dried agar, imprinting, and MMS methods is shown by the ion images of DKxanthene-560 and lysophosphatidylethanolamine 16:1 (lysoPE 16:1) across biological replicates (Figure 1C). The ion images of the reported M. xanthus natural product DKxanthene-560 highlight the increased spatial preservation of the dried agar and MMS methods compared to imprinting. The MMS and dried agar methods avoid an indirect spatial readout, minimizing the delocalization of observed metabolites. The ion images of lysoPE 16:1 differentiated the dried agar and MMS methods. This lipid was investigated for its defined role as a SocA (a short-chain alcohol dehydrogenase) substrate within myxobacteria intercellular signaling and refined localization within predation experiments.36 Within this comparison, we observe more resolved and repeatable ion images from the MMS versus the dried agar method, which is attributed to the drying process of the latter. When agar gel is removed from humid culturing conditions and desiccated, it tends to shrink, resulting in some changes in metabolite abundance and localization. It is important to note that analyte recovery was not evaluated in this study. Varying localizations may be attributed to analyte extraction efficiency or ion suppression effects across each method. Substrate-dependent ionization and other sampling artifacts affecting signal intensity within each IMS image was addressed by normalizing feature intensities to the lock mass intensity for each pixel, which may account for the horizontal stripes within ion images (Figure 1C). Considering these factors, the MMS method showed the least amount of metabolite delocalization within ion images (Figure 1C). Therefore, this sampling technique avoids spatially perturbing the microbial environment, which allows for in situ IMS measurements of microbially produced small molecules in coculture conditions.
Preserving metabolite localizations in IMS sample preparation is necessary to spatially distinguish the regions in microbial predation experiments. However, sensitive and reproducible MS measurements are necessary to determine the underlying molecular changes within each region of the experiment. Imprinting and MMS methods exhibit increased sensitivity compared to dried agar with 340 and 355 features having signal-to-noise ratios (s/n) greater than or equal to 3, respectively (Table S1). The increased sensitivity of the imprinting and MMS method are attributed to avoiding the matrix effects associated with sampling from the agar. These methods are distinguished by the average mass spectrum and in particular the m/z range from 400 to 800, by which the MMS method yielded more comprehensive measurements (Figure S3). Lastly, across the normalized intensities of select features the dried agar, imprinting and MMS methods yielded 44.1%, 54.6%, and 20.2% covariances, respectively (Table S1). Thus, the MMS method not only provides the best spatial analyses but also the most sensitive and repeatable MS measurements. A summary of these results exhibits the comprehensive increase in sampling capabilities using the microporous membrane scaffolded DESI-IMS within microbial predation experiments (Table 1). The MMS method demonstrates sensitive MS measurements and retains analyte localization by directly sampling metabolites from the microbial environment with minimal matrix effects.
Table 1.
Sampling Metrics Across Methods
The MMS DESI-IMS method inherently provides a more native-like sampling of microbe–microbe interactions than comparable MALDI-IMS methods. MMS DESI-IMS does not require chemical modifications or matrix in order to sample microorganisms. Further, the developed method combines ambient sampling and membrane scaffolds for effective ionization without the drying steps and rigidity necessary for sampling under vacuum, which may disrupt the microbial environment. The MMS method even retains metabolites surrounding the microbial communities while avoiding spatial and chemical perturbations. It also avoids sampling agar, which is not rigid enough for DESI and further complicates the sample matrix. The direct analysis of in situ metabolites via the MMS method also provides orthogonal detection capabilities to RPLC–ESI-MS. M. xanthus–E. coli cocultures grown using the MMS method were extracted and analyzed using RPLC–ESI-MS to return 8,140 molecular features. When compared to 1,670 features from microporous membrane scaffolded DESI-IMS, 782 features are conserved across both techniques (Figure S1). These shared features represent the ability of the developed DESI-IMS method to analyze the primary features within a metabolomics experiment. However, the 888 features that are unique to the MMS method demonstrate the orthogonal sampling capabilities. These unique features are attributed to the direct spatial analysis of microbially produced metabolites, which avoids chemical artifacts, differences in analyte solubility, and time scales associated with extractions for RPLC–ESI-MS. The microporous membrane scaffolded DESI-IMS method exhibits localized, native-like sampling and sensitive, repeatable chemical measurements of small molecules in intermicrobial interactions.
Unsupervised Segmentation of Microbial Predation.
In situ spatial annotations and sensitive chemical measurements allowed for the development of an untargeted IMS data analytics workflow. To accomplish this, we used spatial shrunken centroids to perform unsupervised segmentation.34,35 Spatial shrunken centroids analysis provides unique segmentation, such that it entails statistical regularization to extract subsets of informative features (i.e., removing features that do not change across the image), which reduces the amount of features from ~1 700 to ~600 for spatial analyses.34,35 Spatial shrunken centroids also accounts for the spatial structure of the data during the segmentation process as opposed to post hoc correlations.34,35 This allows for the assignment of pixels into unique, homogeneous regions on the basis of mass spectral similarities across the most significant features within an IMS image.34,35 Thus, each of the unique segments resulting from this analysis have both unique locations and chemical profiles. The most significant features within each segment are determined using a “t-statistics” value. We used these “t-statistics” to determine the contributing features to each segment and rank them for untargeted spatial prioritization. Unsupervised segmentation of acquired IMS data from microbial predation experiments provides an unbiased evaluation of the homogeneous segments contributing to the heterogeneous phenotypes within these experiments. The assessment of chemical-spatial phenotypes using this technology facilitates the investigation of the underlying mechanisms by which microorganisms predate, communicate, and interact.
When unsupervised segmentation was applied to microbial predation experiments, we typically observed seven segments (including the membrane background) represented by the different colored regions within the segmentation results (Figure 2A). The ion images of the most significant features within each segment were overlaid using similar colors to confirm the authenticity and localization of output features (Figure 2A). The distinct localizations within the ion overlay image nearly match that of the segmentation results, which demonstrates accurate spatial analyses via segmentation (Figure 2A). We demonstrated the repeatability of unsupervised segmentation and annotated the primary features contributing to each segment across replicates (Figure S4). We also validated the ability of unsupervised segmentation to chemically distinguish these distinct regions via spatially directed principle component analysis (PCA) (Figure 2B). Six mass spectra taken across the area of each proposed segment group together within the PCA displaying the chemical homogeneity of each segment (Figure 2B). Further, each segment’s unique chemical profile is demonstrated by these groupings and their distinct separations (Figure 2B). Principle Component 1, which represents 46% of the data, differentiates the predator and prey communities (blue and green) from the region of interaction and those areas surrounding them (purple, cyan, orange, and yellow). This distinction asserts that the primary observed chemical difference lies between inactive and active secondary metabolism. These results validate the ability to use unsupervised segmentation as a chemoinformatics platform to spatially assess the chemical profile of discrete phenotypes within microbial predation experiments.
Figure 2.

(A) Optical image, unsupervised segmentation results, and ion overlays of prioritized features using segmentation. Features for ion overlays and replicates of segmentation results are shown in Figure S4. (B) Six mass spectra were extracted throughout the location of each segment with locations shown in PCA regions. The PCA of these mass spectra demonstrate the discrete chemical profiles of each segment.
The chemical-spatial phenotypes resulting from unsupervised segmentation were investigated using “t-statistics” and the average mass spectrum (Figure 3A). Statistically enriched features yielded positive “t-statistics”, while those systemically absent resulted in negative values. Within the well-studied M. xanthus DK1622 species, we were able to use these “t-statistic” values to annotate reported natural products and validate the prioritization of biologically appropriate molecules with associated segments. DKxanthene-560, which has been reported in phase variation and fruiting body formation, was used as a benchmark for secondary metabolism.28–30 The myxobacteria antibiotic Myxovirescin A produced in interactions with E. coli was used to determine the location of predation.31 Both these natural products were prioritized within the yellow interaction segment demonstrating the ability to discern the location of predation and activated secondary metabolism (Figure 3B). These positive controls define the yellow segment as the region where secondary metabolism was activated. Therefore, other measured features and unknowns including m/z 668.52 and m/z 596.47 may also be secondary metabolites associated with predation (Figure 3B). We observed 52 other unknown features within the predation segment when dereplicated against our in-house myxobacteria database and online repositories (Figure 3B).
Figure 3.
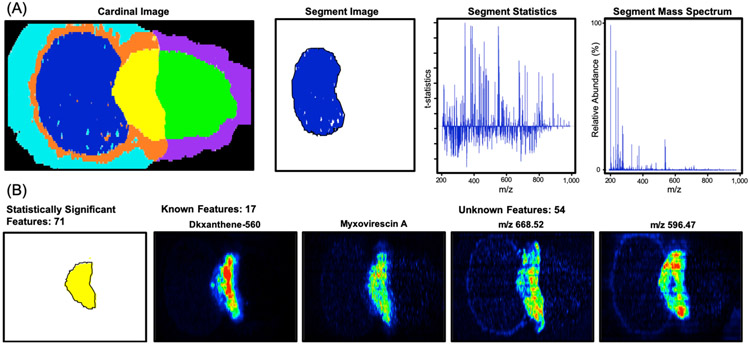
(A) Unsupervised segmentation results with outputs. (B) Predation segment with known and unknown features. Myxovirescin A and DKxanthene-560 represent known secondary metabolites, which validate predation and phase variation within the yellow segment. Unknowns highlight the application of this system to prioritize unreported natural products.
These previously unreported features suggest the potential for measurement and prioritization of novel natural products. Spatially prioritizing these unknowns has implications in drug discovery by removing features that are not localized to areas of activated secondary metabolism. Coupling spatial prioritization and dereplication simplifies the number of candidate masses from 4 000 total features to 54 unknowns associated with predation. This technology yields a natural product discovery workflow that incorporates spatial location and thus proposed function into the prioritization process (Figure S5). The segmentation results also validate the ability to successfully evaluate microbe–microbe interactions using microporous membrane scaffolded DESI-IMS and unsupervised segmentation. We were able to measure six chemically unique regions within these experiments where only three regions (predator, prey, and predation) were defined as necessary to effectively measure microbial relations (Figure 1A). These capabilities were validated across replicates with ion overlays, PCA, and the spatial prioritization of predation and secondary metabolism with biologically appropriate natural products.
Application to WT, M. xanthus Δta1 Mutant, and Phase Variants.
The developed method for characterizing microbe–microbe interactions was applied to wild type (WT), mutated M. xanthus Δta1 strains, and respective phase variants. M. xanthus Δta1 strains lack the megasynthetase responsible for Myxovirescin A (Antibiotic TA) production.37 Phase variation (PV) in myxobacteria results from environmental stimulation, which cues the expression of genes and secondary metabolites in a given population of cells, some of which are important for predation.28-30,38 For example, myxobacteria yellow PVs have increased DKxanthene production (resulting in a yellow pigment) and swarming abilities, whereas tan phase variants display little DKxanthene production and limited swarming abilities.28-30,38 Using the developed DESI-IMS sampling method and data analysis protocol, we distinguish between WT and M. xanthus Δta1 strains and associated phase variants using Myxovirescin A and DKxanthene-560, respectively.
Unsupervised segmentation was used to evaluate the distinct predations of the M. xanthus strains and PVs (Figure 4). In comparing WT and M. xanthus Δta1 strains, we measured the differential production of Myxovirescin A, which is demonstrated by the ion images in Figure 4. This distinction was observed across biological replicates of all sample types and validates the feature prioritization process as well as the capabilities to sample natural product antibiotics from microbial communities using the MMS DESI-IMS method (Figure S6). However, many features were conserved in the predation segment between M. xanthus Δta1 and WT. The unknown feature m/z 668.52 shares a similar localization to Myxovirescin A, which may indicate a role in predation (Figure 4). The conservation of this feature in both strains may be the product of an additional biosynthetic gene cluster activated during predation outside of the TA megasynthetase. It is of interest that M. xanthus Δta1 species were able to predate E. coli despite the mutation (Figure 4).37 While mutants predated slower than WT, this infers a variety of predatory mechanisms, which may be linked to measured unidentified features. More broadly, the measurement of unknown features associated with predation demonstrates the method’s capabilities toward natural product discovery.
Figure 4.
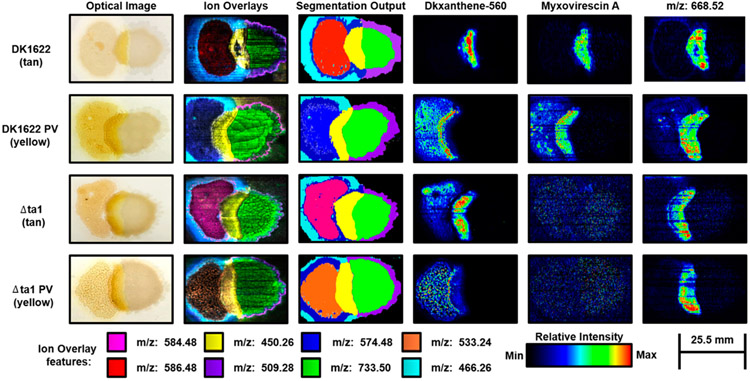
Optical images, ion overlays, and unsupervised segmentation results of WT and M. xanthus Δtal strains and respective phase variants (PV). Colored boxes represent features in ion overlays.
Further analysis of unsupervised segmentation results by comparing PVs provides insight into the role of PV within predation (Figure 4). In tan PVs, we observed DKxanthene-560 and m/z 668.52 as well as Myxovirescin A in WT within the predation segment. Since DKxanthene-560 and Myxovirescin A are colocalized in tan PVs, this suggests that predation and phase variation occur at similar localizations and time scales within the predation process. In yellow PVs, however, DKxanthene-560 was not colocalized to Myxovirescin A and m/z 668.52, which infers that DKxanthene-560 is not directly involved in predation. This comparison suggests that predation may cause the switch from tan PV to yellow PV. Yellow PVs have been shown to be more motile, resistant to heat, and active than tan variants, which is a predatory advantage.28-30,38 Also, PV has been reported in other processes requiring secondary metabolism such as sporulation, which suggests a higher amount of secondary metabolism activated within yellow PVs.28-30,38 Our data support a model whereby tan PVs undergo phase variation prior to predation (Figure 4).
Despite annotating the differences associated with the WT and M. xanthus Δta1 strains along with respective PVs, the majority of metabolic spatial expressions represented by segments were conserved throughout sample types, which is shown in the similar localizations in ion overlays and unsupervised segmentation outputs (Figure 4). These similarities across all sample types elucidate some of the dynamic biosynthetic processes associated within M. xanthus predation and the chemical-spatial phenotypes that facilitate them. For example, the cyan segment may be associated with myxobacteria continuing to explore the surrounding environment despite predation. This segment and others may provide insight into myxobacteria predation as M. xanthus species have been reported to undergo a series of processes during predation including predataxis or cellular reversals to increase contact with prey organisms, fruiting body formation and “wolfpack” motility.10,39 Thus, the combination of unsupervised segmentation and microporous membrane scaffolded DESI-IMS sampling presents the opportunity to unbiasedly investigate the molecular mediators by which microorganisms interact with others and the surrounding environment.
■ CONCLUSIONS
We demonstrate that DESI-IMS using MMS is an effective sampling method for evaluating interspecies microbial interactions. The robust and minimalistic preparation via this method provided sensitive analyses of secondary metabolites while preserving the in situ localization of microbially produced small molecules. The low covariance of metabolite intensities and repeatable ion images across replicates afforded consistent access to the chemical and spatial information between interspecies metabolite exchange. Capitalizing on this advantage, we developed a chemoinformatics platform capable of discerning chemically unique phenotypic regions using unsupervised segmentation. This imaging analysis presents a powerful tool in exploring the chemical ecology of microorganisms in an unbiased manner. Segmentation results were validated using ion overlays and PCA as well as the spatial prioritization of biologically appropriate natural products. These results highlight the ability to unbiasedly elucidate the molecular mediators in microbial relations by correlating spatially resolved and chemically unique regions to phenotypes within predation. The combination of microporous membrane scaffolded DESI-IMS and unsupervised segmentation present unprecedented capabilities to investigate the roles of small molecules within microbial chemical ecology.
The application of this innovation to WT and M. xanthus Δta1 strains along with yellow and tan phase variants validated the sampling and feature prioritization processes using the natural products DKxanthene-560 and Myxovirescin A. We used Myxovirescin A to differentiate between the WT and mutant strains and validate the predation segment within our analyses. DKxanthene-560 was used as a marker for phase variation and activated secondary metabolism. By comparing these secondary metabolites, we were able to probe the role of phase variation in myxobacteria predation, such that tan PVs turn to yellow PVs to predate more effectively. These assertions reflect the ability of this method to unbiasedly assess underlying ecological roles of natural products. We also add to the known myxobacteria chemistry by sampling unreported natural products with suggested roles in predation and secondary metabolism via our assignment of the predation segment. Lastly, other segments and features were measured outside of the predation region, which combine to represent the multiplicity of phenotypes within microbial predation. These segments may or may not be contributing to the overarching predatory process, but represent the ability of this technology to probe the chemical mechanisms of microbial behavior.
The developed technology presents an innovation in annotating microbially produced small molecules by combining the sensitive MS measurements and imaging capabilities of DESI-IMS. Using unsupervised segmentation, location can be used as a means for prioritization, which significantly reduces the number of prioritized features and provides insight into biological function. MMS DESI-IMS method is not limited to coculture experiments, but can be applied to measuring the chemical crosstalk within other systems such as host–pathogen relations, tumor and tissue heterogeneity, and microbiome studies. The presented microporous membrane scaffolded DESI-IMS method and unsupervised segmentation is a novel method for molecular discovery and the unbiased assessment of small molecules within chemical ecology.
Supplementary Material
Figure S1, Venn diagram showing the number of conserved and unique features between LC–MS and DESI-MS
Figure S2, fragmentation spectra of identified metabolites
Figure S3, average mass spectra of dried agar, imprinting, and microporous membrane scaffold sampling methods
Figure S4, biological replicates of unsupervised segmentation from MMS DESI-IMS
Figure S5, natural product discovery workflow combining unsupervised segmentation and dereplication
Figure S6, production of reported natural products Myxovirescin A and DKxanthene-560 in biological replicates of WT and M. xanthus Δta1 phase variants
Figure S7, drift time vs relative intensity of WT and M. xanthus Δta1 phase variants (PDF)
Table S1, calculated values annotating sampling efficiency across methods (n = 3)
Table S2, Observed features with regions resulting from unsupervised segmentation. Columns correspond to: “t-statistic” value denoting their significance in the region, standard deviation of “t-statistic” values across replicates (n = 3), identification, molecular formula, adduct, mass accuracy in parts per million (ppm), and normalized intensity. (XLSX)
ACKNOWLEDGMENTS
Financial support for aspects of this research was provided by the National Institutes of Health (Grants NIH NIGMS R01GM092218, NCI R03CA222452, and NIH NIGMS 1F32GM128344), the U.S. Environmental Protection Agency under Assistance Agreement 83573601, and the U.S. Army Research Office and the Defense Advanced Research Projects Agency (DARPA) under Cooperative Agreement W911 NF-14-2-0022. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the funding agencies and organizations
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b02992.
REFERENCES
- (1).Mcfall-Ngai M; Hadfield MG; Bosch TC; Carey HV; Domazet-Loso T; Douglas AE; Dubilier N; Eberl G; Fukami T; Gilbert SF; et al. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (9), 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wilson MR; Jiang Y; Villalta PW; Stornetta A; Boudreau PD; Carrá A; Brennan CA; Chun E; Ngo L; Samson LD; et al. Science 2019, 363 (6428), No. eaar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vizcaino MI; Crawford JM Nat. Chem 2015, 7 (5), 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Xue M; Kim CS; Healy AR; Wernke KM; Wang Z; Frischling MC; Shine EE; Wang W; Herzon SB; Crawford JM Science 2019, 365 (6457), No. eaax2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fischer CN; Trautman EP; Crawford JM; Stabb EV; Handelsman J; Broderick NA eLife 2017, 6, e18855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Currie CR; Scott JA; Summerbell RC; Malloch D Nature 1999, 398 (6729), 701–704. [Google Scholar]
- (7).Zipperer A; Konnerth MC; Laux C; Berscheid A; Janek D; Weidenmaier C; Burian M; Schilling NA; Slavetinsky C; Marschal M; et al. Nature 2016, 535 (7613), 511–516. [DOI] [PubMed] [Google Scholar]
- (8).Newman DJ; Cragg GM J. Nat. Prod 2016, 79 (3), 629–661. [DOI] [PubMed] [Google Scholar]
- (9).Covington BC; Mclean JA; Bachmann BO Nat. Prod. Rep 2017, 34 (1), 6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Findlay BL ACS Chem. Biol 2016, 11 (6), 1502–1510. [DOI] [PubMed] [Google Scholar]
- (11).Hoffmann M; Auerbach D; Panter F; Hoffmann T; Dorrestein PC; Muller R ACS Chem. Biol 2018, 13 (1), 273–280. [DOI] [PubMed] [Google Scholar]
- (12).Covington BC; Spraggins JM; Ynigez-Gutierrez AE; Hylton ZB; Bachmann BO Appl. Environ. Microbiol 2018, 84 (19), No. e01125–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Derewacz DK; Covington BC; Mclean JA; Bachmann BO ACS Chem. Biol 2015, 10 (9), 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Derewacz DK; Goodwin CR; McNees CR; McLean JA; Bachmann BO Proc. Natl. Acad. Sci. U. S. A 2013, 110 (6), 2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yang YL; Xu Y; Straight P; Dorrestein PC Nat. Chem. Biol 2009, 5 (12), 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Watrous J; Roach P; Alexandrov T; Heath BS; Yang JY; Kersten RD; van der Voort M; Pogliano K; Gross H; Raaijmakers JM; et al. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (26) , E1743–E1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Watrous J; Roach P; Heath B; Alexandrov T; Laskin J; Dorrestein PC Anal. Chem 2013, 85 (21), 10385–10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Araujo FDS; Vieira RL; Molano EPL; Maximo HJ; Dalio RJD; Vendramini PH; Araujo WL; Eberlin MN RSC Adv. 2017, 7 (48), 29953–29958. [Google Scholar]
- (19).Tata A; Perez C; Campos ML; Bayfield MA; Eberlin MN ; Ifa DR Anal. Chem 2015, 87 (24), 12298–12305. [DOI] [PubMed] [Google Scholar]
- (20).Menezes RC; Kai M; Krause K; Matthäus C; Svatoš A; Popp J; Kothe E Anal. Bioanal. Chem 2015, 407 (8), 2273–2282. [DOI] [PubMed] [Google Scholar]
- (21).Angolini CFF; Vendramini PH; Araujo FDS; Araujo WL; Augusti R; Eberlin MN; de Oliveira LG Anal. Chem 2015, 87 (13), 6925–6930. [DOI] [PubMed] [Google Scholar]
- (22).Yan C; Parmeggiani F; Jones EA; Claude E; Hussain SA; Turner NJ; Flitsch SL; Barran PE J. Am. Chem. Soc 2017, 139 (4), 1408–1411. [DOI] [PubMed] [Google Scholar]
- (23).Song C; Mazzola M; Cheng X; Oetjen J; Alexandrov T; Dorrestein PC; Watrous J; van der Voort M; Raaijmakers JM Sci. Rep 2015, 5, 12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Alexandrov T BMC Bioinf. 2012, 13, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rath CM; Alexandrov T; Higginbottom SK; Song J; Milla M; Fischbach MA; Sonnenburg JL; Dorrestein PC Anal. Chem 2012, 84 (21), 9259–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ravindran SJ; Kumar R; Srimany A; Philip L; Pradeep T Anal. Chem 2018, 90 (1), 988–997. [DOI] [PubMed] [Google Scholar]
- (27).Powell LC; Pritchard MF; Ferguson EL; Powell KA; Patel SU; Rye PD; Sakellakou SM; Buurma NJ; Brilliant CD; Copping JM; Menzies GE; Lewis PD; Hill KE; Thomas DW NPJ Biofilms Microbiomes. 2018, 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Meiser P; Weissman KJ; Bode HB; Krug D; Dickschat JS; Sandmann A; Muller R Chem. Biol 2008, 15 (8), 771–781. [DOI] [PubMed] [Google Scholar]
- (29).Dziewanowska K; Settles M; Hunter S; Linquist I; Schilkey F; Hartzell PL PLoS One 2014, 9 (4), No. e95189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Meiser P; Bode HB; Müller R Proc. Natl. Acad. Sci. U. S. A 2006, 103 (50), 19128–19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Xiao Y; Wei X; Ebright R; Wall DJ Bacteriol. 2011, 193 (18), 4626–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tillner J; Wu V; Jones EA; Pringle SD; Karancsi T; Dannhorn A; Veselkov K; McKenzie JS; Takats Z J. Am. Soc. Mass Spectrom 2017, 28 (10), 2090–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Schrimpe-Rutledge AC; Codreanu SG; Sherrod SD; McLean JA J. Am. Soc. Mass Spectrom 2016, 27 (12), 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bemis KD; Harry A; Eberlin LS; Ferreira CR; van de Ven SM; Mallick P; Stolowitz M; Vitek O Mol. Cell. Proteomics 2016, 15 (5), 1761–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bemis KD; Harry A; Eberlin LS; Ferreira C; van de Ven SM; Mallick P; Stolowitz M; Vitek O Bioinformatics 2015, 31 (14), 2418–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Avadhani M; Geyer R; White DC; Shimkets LJ J. Bacteriol 2006, 188 (24), 8543–8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Korp J; Vela Gurovic MS; Nett M Beilstein J. Org. Chem 2016, 12, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dahl JL; Ulrich CH; Kroft TL J. Bacteriol 2011, 193 (19), 5081–5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Muñoz-Dorado J; Marcos-Torres FJ; García-Bravo E; Moraleda-Muñoz A; Pérez J Front. Microbiol 2016, 7, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, Venn diagram showing the number of conserved and unique features between LC–MS and DESI-MS
Figure S2, fragmentation spectra of identified metabolites
Figure S3, average mass spectra of dried agar, imprinting, and microporous membrane scaffold sampling methods
Figure S4, biological replicates of unsupervised segmentation from MMS DESI-IMS
Figure S5, natural product discovery workflow combining unsupervised segmentation and dereplication
Figure S6, production of reported natural products Myxovirescin A and DKxanthene-560 in biological replicates of WT and M. xanthus Δta1 phase variants
Figure S7, drift time vs relative intensity of WT and M. xanthus Δta1 phase variants (PDF)
Table S1, calculated values annotating sampling efficiency across methods (n = 3)
Table S2, Observed features with regions resulting from unsupervised segmentation. Columns correspond to: “t-statistic” value denoting their significance in the region, standard deviation of “t-statistic” values across replicates (n = 3), identification, molecular formula, adduct, mass accuracy in parts per million (ppm), and normalized intensity. (XLSX)