

Abstract
The ability to label active caspase-3 represents a useful pharmacodynamic strategy to determine the efficacy of anti-tumour drugs. Activity-based probes (ABPs) provide a method for the labelling of activated caspases and the recent development of hybrid combinatorial substrate libraries (HyCoSuL) has allowed for the generation of highly selective ABPs to discriminately label these proteases. Here using this approach, a novel caspase-3 selective ABP (CS1) has been developed and validated in apoptotic cells to selectively bind caspase-3 over the closely related caspase-7. However, a critical bottleneck for ABPs is their cell penetrance and therefore this cell-impermeable CS1 probe was subsequently formulated into PLGA-based nanoparticles (CS1-NPs). We demonstrate the ability of these particles to be taken up by the cells and facilitate intracellular delivery of the ABP to effectively label caspase 3 in response to apoptotic stimuli. This work forms the foundation of a novel approach for the labelling of caspase 3 and may have downstream utility to measure real time apoptosis in tumours and other organs.
Graphical Abstract
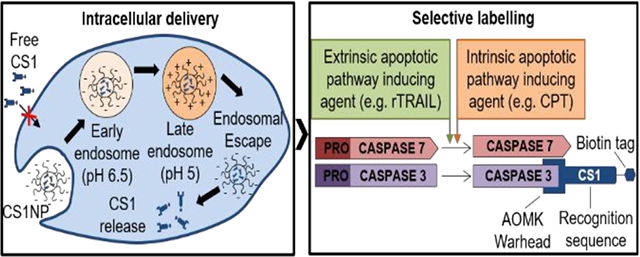
The formulation of the novel activity-based probe CS1 in PEG-PLGA nanoparticles allows intracellular selective labelling of caspase-3 over closely related caspase-7.
Introduction
Apoptosis is a form of programmed cell death which is fundamental in maintaining tissue homeostasis through providing a balance between cell proliferation and cell death. Apoptosis is a necessary process to eliminate unwanted, aged, damaged and mutated cells from the organism.(1) In cancer however, loss of apoptosis is an intrinsic feature of tumours, essential to ensure their development, progression, metastasis, and resistance to therapy.(2)
Given the fact that the majority of current chemotherapeutics ultimately lead to cell death, it is apparent that a system to visualise and quantify apoptosis is necessary to evaluate the efficacy of anti-cancer therapeutics. An interesting approach is the use of activity-based probes (ABPs) and quenched reporter substrates directed towards members of the caspase family.(3,4) Read-outs from ABPs or quenched reporter substrates are indicative of the activity of an enzyme, rather than its expression, and allow discrimination between the active and inactive population of a specific protease.(5) Most of the ABPs utilised to image apoptosis are developed to target caspase-3, as the core executioner caspase responsible for cell death.(6) Current commercially available quenched substrates utilised to image and quantify caspase-3 activity use the tetrapeptide Asp-Glu-Val-Asp (DEVD), a sequence that, even though it is used as the optimum substrate for caspase-3,(7) is also recognised by caspase-6, −7, and −8.(8–10) To generate more selective sequences for caspases, including caspase-3, we have previously employed a hybrid combinatorial substrate library (HyCoSuL) approach.(11) Similarly, the Wolan group has also used this strategy for the design of ABPs selective for caspase-3 over caspase-7.(12,13) Our caspase-directed ABPs covalently bind to the active population of the target caspase through the electrophilic acyloxymethyl ketone (AOMK) warhead, which has been reported as most suitable for caspase detection,(14) and give a measurable signal due to the presence of a tag group.(15) One major limitation of ABPs is their poor cell penetrance.(6,16) In addition, small molecule peptide inhibitors are usually taken up by cells through endocytosis pathways, thus labelling lysosomal rather than cytosolic proteases;(17,18) which for ABPs targeting caspases, lysosomal proteases represents off-targets to avoid. To translate the utility and efficacy of ABPs in cell-based and in vivo experiments, one possible strategy is to use nano-formulation delivery systems, because of their abilities to both deliver their cargo intracellularly and increase their half-life.(19,20) To date, nano-based systems that have been used to generate quenched substrates for real time imaging of caspase activity have been mostly developed using gold nanoparticles (AuNPs) for their optical imaging properties, highly suitable to exploit FRET, and the ability of the AuNP surface to easily bind biomolecules, allowing surface conjugation of DEVD-fluorophore substrates.(21–25)
Polymeric nanoparticles, based on materials such as PEGylated poly(lactic-co-glycolic acid) (PEG-PLGA) nanoparticles represent a clinically relevant drug delivery system (DDS), due to their ability to passively target tumours and proven biocompatibility.(26) Bioactive components or active pharmaceutical ingredients can be encapsulated in PLGA-based NPs. Subsequent release of the drug is the result of diffusion through the nanoparticle matrix combined with the spontaneous degradation of the PLGA in aqueous solutions, thus resulting in a controlled and sustained release.(27)
Bringing both the ABP and polymeric nanoparticle technologies together, herein a novel caspase-3 selective activity-based probe (CS1) is described. Its usefulness as a tool to monitor caspase-3 activity upon apoptotic stimuli is demonstrated. Furthermore, its encapsulation in PEG-PLGA nanoparticles (CS1-NP) is shown and its improved ability to label intracellular caspase-3 is revealed.
Results
We have developed a combinatorial approach for the synthesis of tetrapeptide substrates for members of the caspase family, with the aim of discovering new sequences recognizable by a single caspase species, thus overcoming the overlapping substrate specificity of currently available caspase substrates.(11) We recently reported a highly selective caspase 3 tetrapeptide substrate from screening a Hybrid Combinatorial Substrate Library (HyCoSuL).(11) This substrate, Ac-Asp-Glu-Thr(Bzl)-Asp-ACC incorporated the unnatural amino acid Thr(Bzl) in the P2 position. Using this new reference sequence as a starting point, a novel ABP, named CS1, was prepared (biotin-ahx-Asp-Glu(Me)-Thr(Bzl)-Asp-AOMK) (Figure 1). A key feature of this probe was the incorporation of a methylated Glu residue at the P3 position in order to make the compound more hydrophobic and aid subsequent entrapment in a hydrophobic PLGA-based nanoparticle. To validate CS1 in cell-based assays, a reference probe containing the DEVD tetrapeptide as recognition sequence was also synthesised (Figure 1) to use as a control (DEVD probe). Furthermore, the tag, spacer and warhead were constant between the two probes to allow comparison based on the recognition sequence only.
Figure 1.

Molecular structure of CS1 and DEVD ABPs. Elements constituting the probes are highlighted. The tag group is a biotin molecule; the spacer is the 6-aminohexanoic acid (6-ahx), the recognition sequence of CS1 is formed by a mixture of natural and unnatural amino acid: Asp-Glu(Me)-Thr(Bzl)-Asp; the recognition sequence of DEVD probe is the homonymous tetrapeptide: Asp-Glu-Val-Asp; the warhead responsible for the covalent binding with the catalytic thiol group of the active site is acyloxymethyl ketone (AOMK).
Detection of caspase-3 activity upon apoptotic stimuli in vitro
Following successful synthesis of CS1, the next step consisted in evaluating the ability of the probe to label active caspase-3 in cell lysates, upon apoptosis stimulation with clinically relevant agents. Apoptotic cell death in PC3 cells, a cell line derived from bone metastasis of grade IV prostate adenocarcinoma, was stimulated by pre-treatment with 2.5 μM entinostat for 20 hours followed by rTRAIL treatment for 4 hours.(28–30) Resultant activation of caspase-3/7 was monitored by assessing the fluorescence emitted from the peptidyl substrate Ac-DEVD-AMC when incubated with cell lysates (Figure 2A). Confirmation of apoptosis induction was established by Western blot analysis of caspase-3 activation and an endogenous natural substrate of caspase-3, poly (ADP-ribose) polymerase (PARP-1). Immuno-blot analysis of c-FLIP, an anti-apoptotic protein, has confirmed the ability of entinostat to sensitise PC3 cells to rTRAIL-induced apoptosis through downregulation of cFLIPL (Figure 2B). Hence, 2.5 μM entinostat for 20 hr followed by 4 hr of 40 ng mL−1 of rTRAIL was established as optimal treatment to obtain a marked increase in the levels of active caspase-3 in PC3 cells.
Figure 2.
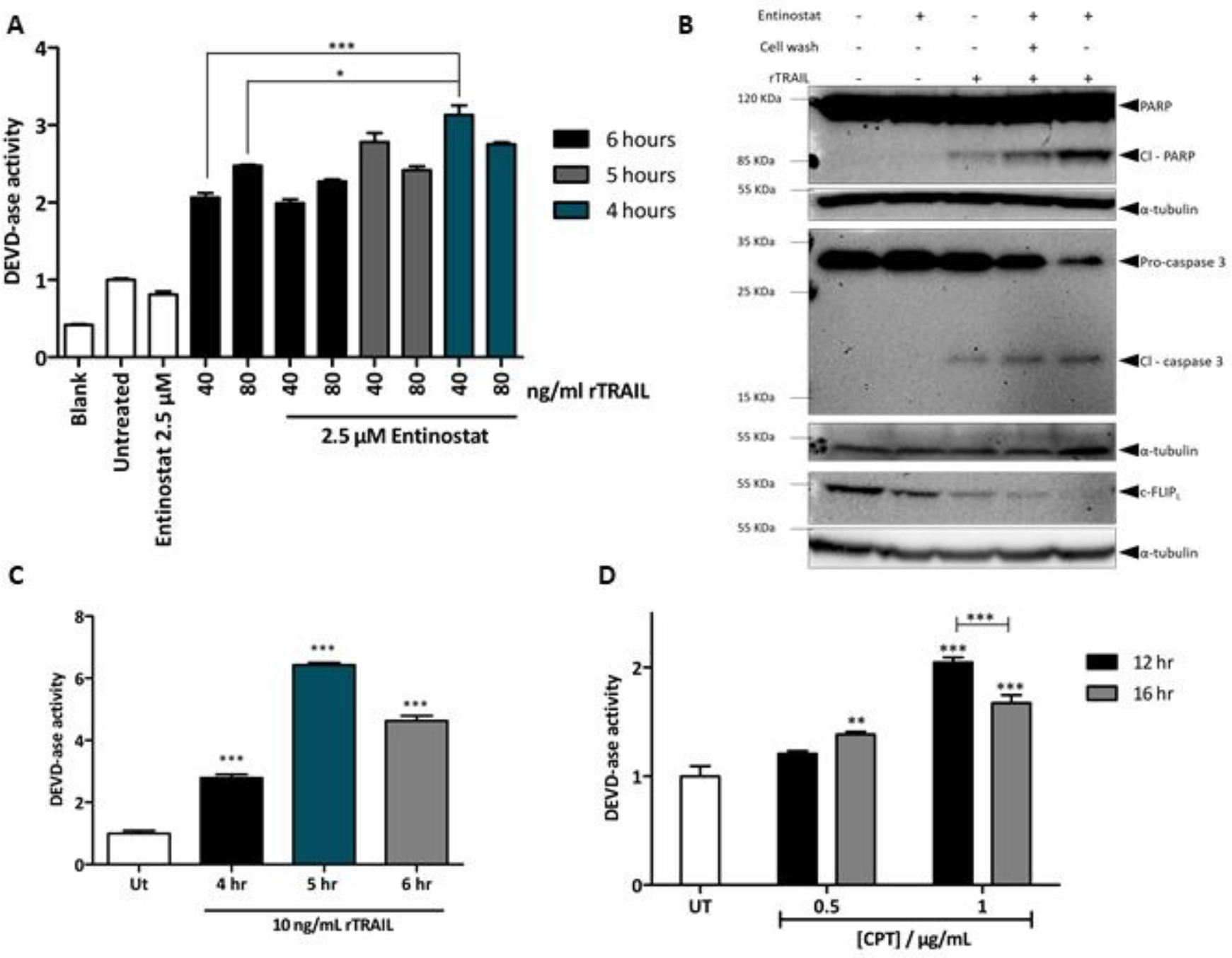
(A) Caspase-3/7 activity in PC3 cell lysates following rTRAIL treatment alone or in combination with 2.5 μM entinostat pre-treatment (24 hours), at different concentrations of rTRAIL (40 and 80 ng mL-1) and at different time points (4, 5, and 6 hours). Results are expressed as fold change to untreated control. Assay done in triplicate ± S.E.M. and representative of three different experiments. (B) Western blot analysis of PARP-1 cleavage (Cl PARP), caspase-3 activation and cFLIP expression in PC3 cells after treatment with 2.5 μM entinostat for 24 hours and 40 ng mL-1 of rTRAIL for 4 hours. b-tubulin was used as loading control (n = 2). (C) Caspase or DEVD-ase activity in HCT116 cells following 10 ng mL-1 of rTRAIL treatment for 4, 5, and 6 hours. Assay was done in triplicate and results expressed as fold change to untreated control ± S.E.M. (D) Caspase or DEVD-ase activity in MCF-7 cells following treatment with 0.5 and 1 μg mL-1 of CPT at 12 and 16 hours. Results expressed as fold change to untreated control. Assay done in triplicate ± S.E.M. and results are representative of three different experiments.
Another two cell lines where next examined; namely HCT116 and MCF-7 cells. The first is a human colon colorectal cancer cell line that has been shown to be sensitive to TRAIL induced apoptosis;(31) thus it was used as a positive control. The latter is a breast cancer cell line which has been shown to lack caspase-3.(32) Nevertheless, apoptosis stimulation in MCF-7 still results in cell death, with the activation of the executioner caspase-6 and −7.(33) For this reason, MCF-7 were chosen to assess the selectivity of CS1 for caspase-3 over caspase-7. Due to the relatively higher sensitivity of HCT116 cells to rTRAIL, 10 ng mL−1 of this apoptotic agent were enough to induce high levels of caspase-3/7 activity, with the optimal treatment of 5 hours showing more than 6-fold caspase activity compared to untreated control (Figure 2C).
TRAIL treatment in MCF-7 failed to generate an appropriate activation of caspase-7 (data not shown), thus it was decided to stimulate apoptosis using camptotechin (CPT), a topoisomerase II inhibitor that induces apoptosis through stimulation of the intrinsic apoptotic pathway.(34) CPT was shown to induce caspase-7 activity in the MCF-7 when treated with 1 μg mL−1 of CPT for 12 hours (Figure 2D).
CS1 labelling of caspase-3 in cell lysates
Following establishment of optimal treatment to induce apoptosis in PC3, HCT116 and MCF-7 cells, the ability of CS1 to label the executioner caspase-3 was next evaluated. This was done by incubating cell lysates from healthy and apoptotic cells with 1 and 10 μM of CS1 probe or DEVD probe. Western blot analysis using streptavidin-HRP was used to detect labelling of the enzymes by the probes, via binding to the biotin tag (Figure 3). In all three cell lines used, no labelling was detected when lysates from healthy cells were incubated with either probe at 10 μM concentration, due to absence of apoptotic stimuli and consequent lack of active caspase-3. Lysates from apoptotic PC3 cells incubated with the DEVD probe result in three distinct bands, corresponding to the two fragments of active caspase-3 (p17 and p19) and the active caspase-7. Indeed, this triple band pattern is characteristic of caspase-3 and −7 activation, in line with previous findings.(35) On the other hand, incubation of the same lysates with the CS1 probe results in two bands only, suggesting that CS1 only recognizes caspase-3 and not caspase-7 (Figure 3A).
Figure 3.

Western-blot analysis of PC3 (A) (entire blot shown in Figure S1), MCF-7 (B), and HCT116 (C) cell lysates incubated with DEVD probe or CS1 probe at 1 and 10 μM for 1 hour, at 37°C, shaking (500 rpm). Caspase-3 and caspase-7 activation was induced with previously validated treatments (Figure 2). Untreated cells were also incubated with the two probes as negative controls. SDS-PAGE was run in 15 % poly-acrylamide gels and, following protein transfer in immobilon membranes, the blots were probed with streptavidin-HRP to visualise caspase-3/−7 labelling. Equal loading was ensured by b-tubulin immuno-blot from the same gel.
The selectivity of CS1 for caspase-3 over caspase-7 was confirmed when lysates from apoptotic MCF-7 cells were incubated with the two probes; practically no labelling of caspase-7 was detected with CS1, while a much stronger labelling of caspase-7 was detected with DEVD probe (Figure 3B), highlighting the improved selectivity of the novel probe. Furthermore, it was possible to distinguish between the labelling efficiency of the two probes when these were incubated with lysates from apoptotic HCT116 (Figure 3C). As HCT116 are highly sensitive to TRAIL, it is to be expected that stimulation of apoptosis results in high levels of caspase-3 activation in the whole cell population equally, thus they represent an ideal cell line to estimate the labelling efficiency of a caspase directed ABP. It was noted that stronger intensity of the band resulted from the labelling of caspase-3 by CS1 compared to the band obtained from the labelling of caspase-3 by DEVD probe. This suggests that the CS1 probe is more efficient in caspase-3 labelling. The results infer that due to incorporation of unnatural amino acid in the recognition sequence, the CS1 probe is better recognised by caspase-3 compared to the DEVD probe, thus giving a stronger signal upon caspase-3 activation.
Finally, to exclude the possibility that the CS1 probe was capable of binding to lysosomal cysteine proteases, apoptotic HCT116 lysates pre-incubated with the broad spectrum cysteine cathepsin inhibitor E64-d, were probed with CS1 and no diminution of banding was observed, further confirming selectivity towards caspase 3 (Figure S2).
Encapsulation of CS1 in PEG-PLGA Nanoparticles
Next the encapsulation of the CS1 probe into PEG-PLGA nanoparticles through a single emulsion solvent evaporation (SESE) technique was undertaken. This methodology, previously optimised within our laboratory,(36) allows for the encapsulation of hydrophobic molecules into PLGA-based nanoparticle. The chosen nanoformulation was composed of a 75 % of PLGA and 25 % of mPEG-PLGA (polymer ratio 1:2, AK010). The mPEG surface coating allows for in vivo testing of this formulation, as PEGylation is the favourite strategy to minimise opsonisation and consequent uptake by cells of the MPS.(37,38) This formulation gives nanoparticles in which the amount of PEG on the surface is roughly 8.25 %, amount that we have previously shown be ideal to ensure stealth nanoparticles, quantified by nanoparticle uptake by a murine macrophage cell line (Raw 264.7).(36)
Following formation of CS1-NP and blank nanoparticles (BNP), characterisation studies were carried out through dynamic light scattering (DLS) analysis and results are summarised in table 1. The resulting particles are all in the size range of interest (~200 nm), with CS1-NP being approximately 10 nm bigger than BNP due to loading cargo. The polydispersity index (PDI) is also within the wanted limits (< 0.2), indicative of a monodisperse formulation, and an expected negative ζ-potential in ddH2O (−20 mV ca.). NPs in the size range of 100–200 nm will generally have more success in avoiding clearance and, at the same time, will exploit more efficiently the EPR effect.(39,40) The negative ζ-potential of CS1-NP is fundamental to undertake endosomal escape and release the CS1 ABP in the cytoplasmic compartment,(41) to access caspase 3.
Table 1.
Dynamic Light Scattering (DLS) analysis for BNP and CS1-NP made with SESE method. (n = 3)
| Formulation | d (nm) | PDI | Z-potential (mV) | Probe Loading (μg mg−1) | Encapsulation efficiency (%) |
|---|---|---|---|---|---|
| BNP | 188.07 ± 2.23 | 0.080 ± 0.020 | −23.87 ± 3.86 | - | - |
| CS1-NP | 198.59 ± 7.72 | 0.095 ± 0.050 | −21.49 ± 2.94 | 33.16 ± 7.20 | 16.57 ± 3.04 |
Nanoparticle formulations were re-suspended in ddH2O at 0.1 mg mL−1 prior analysis. Results expressed as mean of data collected from analysis of three independent formulations ± standard deviation (S.D.).
Scanning electron microscope (SEM) analysis of BNP and CS1NP confirmed the results of DLS analysis, showing nanoparticles with homogeneous size distribution in both formulations (Figure 4A). Furthermore, a stability study conducted over a period of six weeks has confirmed that both formulations (BNP and CS1-NP) were stable in terms of size, PDI, and surface charge, in three different storage conditions: room temperature (RT), 4°C and −20°C (Figure 4B).
Figure 4.
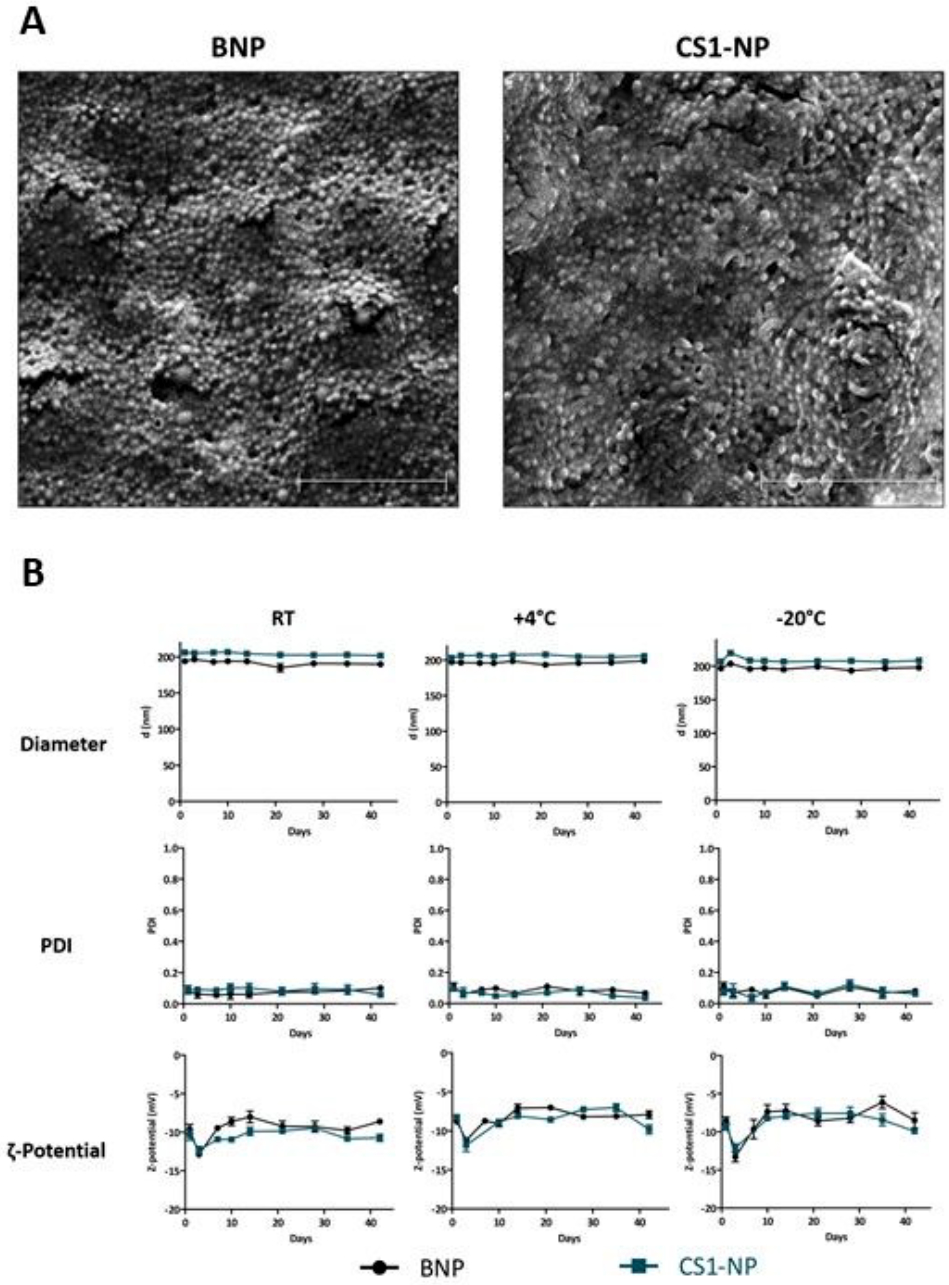
(A) Scanning Electron Microscope (SEM) analysis of BNP and CS1-NP resuspended in ddH2O at 5 mg mL−1, showing monodispersed size of the formulations. 2 μm scale bar. (B) Stability study of BNP and CS1-NP showing that both formulations are stable in suspension at room temperature (RT), at 4°C, or in dry pellet at −20°C. Both formulations maintain their size, PDI, and surface charge (Z-Pot) constant over a period of six weeks, in each of the storage conditions. Each measurement was done in triplicate and results are expressed as mean ± S.E.M.
CS1-NP allows labelling of active caspase-3 in vitro.
The ability of the CS1-NP formulation to deliver the probe intracellularly was next evaluated. Following 24 hours of entinostat pre-treatment, the cells were treated with rTRAIL for 4 hours as established to induce activation of caspase 3, and 0.25 mg mL−1 or 0.5 mg mL−1 of nanoparticles were added to the cells at either the second, third, or fourth hour of a 4 hours apoptotic window as shown in Figure 5.
Figure 5.
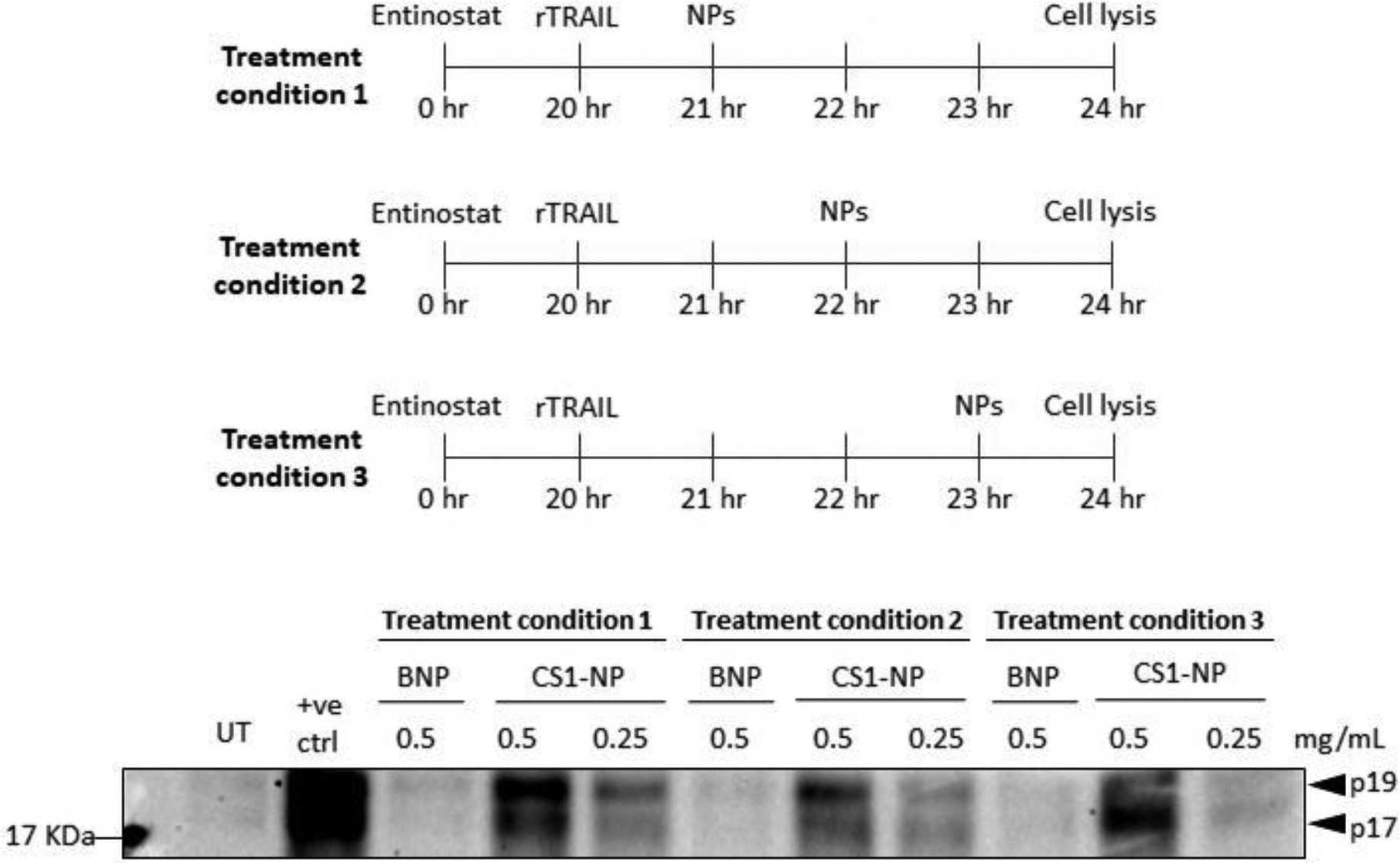
Schematic of experiment procedure and relative western blot analysis showing caspase-3 labelling by CS1-NP in apoptotic PC3 cells, highlighting the progression from p19 to p17 of active caspase-3. PC3 were treated with 2.5 μM entinostat for 20 hr, 40 ng mL−1 of rTRAIL for 4 hours and CS1-NP for 3, 2, and 1 hr prior cell lysis. Positive control made by incubating apoptotic cell lysates with 10 μM of CS1 for 1 hr, at 37°C, shaking.
To ensure detection of active caspase-3 labelling by the probe, a positive control was included and consisted of incubating lysates from apoptotic cells with 10 μM of CS1 probe (1hr, at 37°C, shaking). Western blot analysis with streptavidin-HRP allowed labelling of active caspase-3 in a dose-dependent manner (Figure 5). As expected, neither the BNP control nor untreated cells produced any labelling, and incubation of 0.25 mg mL−1 of CS1-NP resulted in a reduced labelling compared to 0.5 mg mL−1. Caspase-3 activation results in two form of the mature enzyme: the first form of caspase-3 is a complex of two p19 and two p12 fragments, following autocatalytic processing the fully mature form is a complex of two p17 and two p12 fragments.(42,43) The CS1-NP batch added for three out of four hours of apoptotic process distinctly labelled more of the 19 KDa fragment (p19) of caspase-3 compared to the smaller 17 KDa fragment (p17). This is consistent with the processing of caspase-3 where at early stages of apoptosis, the levels of p19 are higher than p17, and thus the ABP reveals preferentially this band. As the apoptotic process proceeds and the p17 becomes the predominant species, this becomes the main protein that was labelled (Figure 5, treatment condition 3). This was possible because the AOMK warhead of CS1 promote an irreversible and covalent binding with the catalytic thiol group of caspase-3, effectively behaving as an inhibitor, thus blocking any further processing of the enzyme.
During apoptosis the cells undertake many morphological changes.(44) These changes could positively affect nanoparticle uptake by the cells. To evaluate the difference in nanoparticle uptake between healthy and apoptotic cells, confocal microscopy was carried out using nile-red loaded nanoparticles (Figure S3). When nile red NPs were incubated with PC3 cells for 3 hr, healthy cells have shown a reduced uptake compared to early apoptotic cells. An uptake assay using rhodamine nanoparticles (RNP) was conducted to confirm these findings, indicating an increased uptake of the NPs in apoptotic cells compared to healthy cells with both short (3 hr) and long (24 hr) RNP treatments (Figure S4).
Next, the ability of CS1-NP to accumulate and release CS1 intracellularly was evaluated. As apoptosis has shown to increase the NPs uptake (Figure S3 and S4), initial experiments were conducted washing the cells of uninternalized NPs before apoptosis stimulation (Figure S5). Although active caspase-3 labelling by CS1NPs can be detected up to 48 hr prior apoptosis stimulation, this procedure also results in reduced cell death induced by entinostat and rTRAIL, leading to weak labelling of active caspase-3 by CS1NPs. To exploit the synergy between the two pharmacological treatments, PC3 cells were incubated with CS1 and CS1-NPs 20 hours in concomitance with entinostat, followed by induction of apoptosis with rTRAIL for 4 hr (Figure 6A). Western blot analysis revealed a strong labelling of active caspase-3 by CS1-NP in a concentration-dependent fashion, whereas no labelling was obtained with free CS1. As a further control, incubation of CS1-NP with HCT116 cells in absence of apoptotic stimuli revealed no labelling of caspase 3, further confirming the reactivity of CS1 toward caspase-3 in only apoptotic cells (figure S6).
Figure 6.
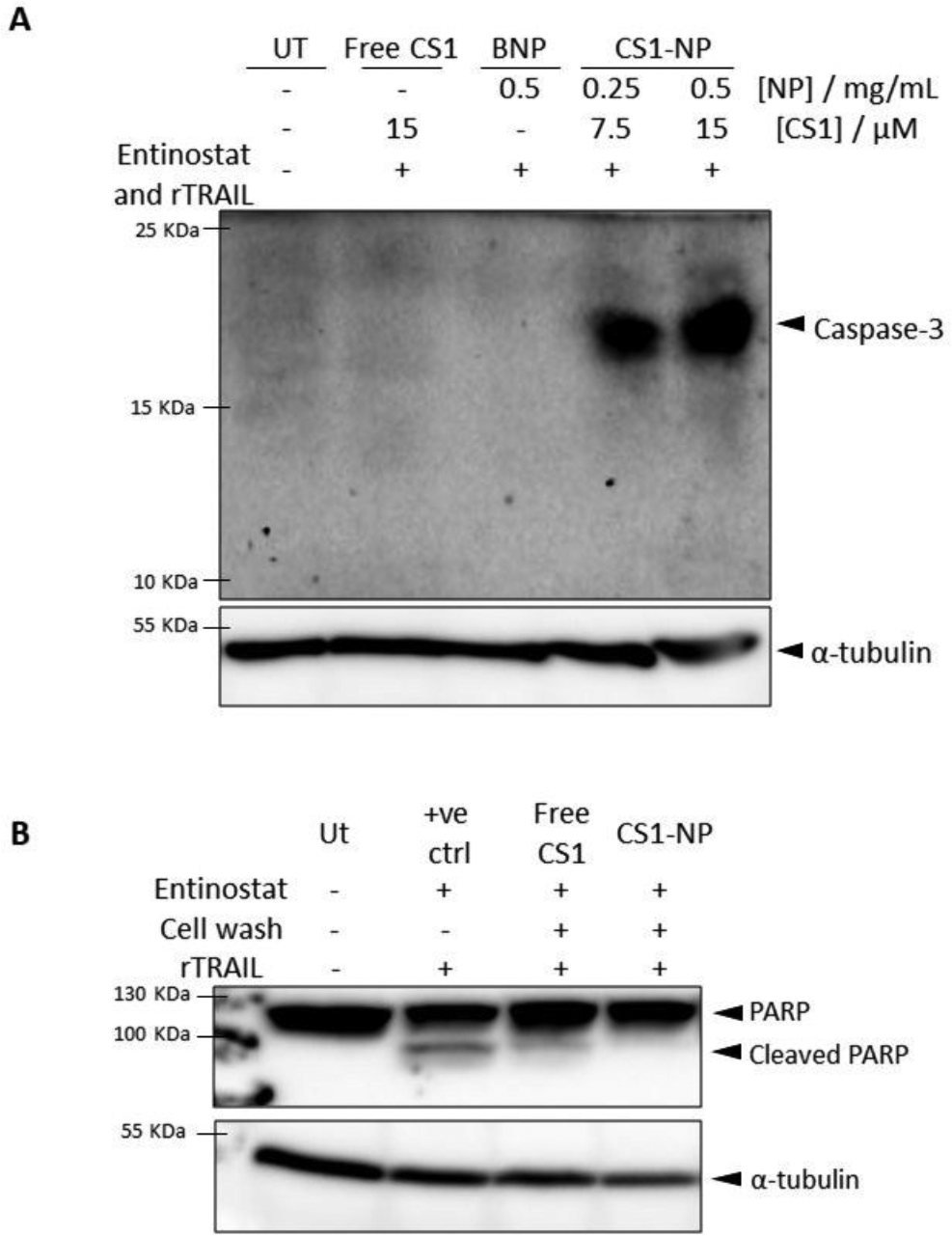
(A) Western blot analysis confirming inability of free CS1 to penetrate the cell membrane and label caspase-3, while its formulation into PLGA nanoparticles allow for the intracellular delivery of CS1 which, upon apoptosis stimulation, can label active caspase-3. PC3 cells were incubated with 2.5 μM entinostat for 24 hours and 40 ng mL−1 of rTRAIL to stimulate apoptosis. Free CS1, BNP, and CS1-NP were added to the cell for 24 hours with entinostat and were not removed before rTRAIL treatment to ensure high levels of active caspase-3. (B) Western blot analysis of PARP-1 cleavage, showing that CS1 irreversibly label active caspase-3. PC-3 cells were treated for 24 hours with entinostat and 4 hours with rTRAIL to induce PARP-1 cleavage. Cells were also incubated with 15 μM of CS1 or 0.5 mg mL−1 of CS1-NP for 24 hours in concurrence with entinostat, and uninternalized particles or probe were washed off with PBS before apoptosis stimulation with rTRAIL. Positive control (+ve ctrl) resulted from incubation of apoptotic cell lysates with CS1 (1 hour, 37°C, shaking). Tubulin was used as loading control.
Caspase-3 labelling by CS1 is an irreversible binding as the warhead covalently attaches to the thiol reactive group of the catalytic cysteine residue, thus resulting in the permanent inhibition of active caspase-3.(45) It is then expected that intracellular CS1 inhibit caspase-3 upon its activation, resulting in less cell death. Cells incubated with free CS1 and CS1NP for 24 hours were analysed for cleavage of PARP-1 (an endogenous substrate of caspase-3) (Figure 6B). Cells incubated with free CS1 still undergo PARP-1 cleavage, while cells treated with CS1-NPs do not show any PARP-1 cleavage, confirming the inability of free CS1 to effectively bind (and thus inhibit) caspase-3. Taken together, these results establish both the cell impermeability of CS1 and the rescue of this property by successful delivery of CS1 in the PLGA NPs.
Conclusions
In this study the in vitro validation of a selective caspase-3 ABPs (CS1), which discriminate between the closely related proteases caspase-3 and caspase-7 is presented. Furthermore, encapsulation of CS1 in PEG-PLGA NPs has allowed the intracellular delivery of CS1, releasing it in the cytoplasmic compartment where, upon apoptosis stimulation, successfully labelled the active form of caspase-3.
Whilst much development has been afforded to the development of new cancer therapeutics, our ability to determine the effectiveness of many treatments is limited. The vast majority of current chemotherapeutic drugs act by inducing tumour cell death, and innovations that help understand if a particular treatment is working are of fundamental importance. This would allow clinicians to determine effectiveness and rapidly change to alternative therapies, thus increasing outcomes for each patient.
Activation of caspases represents potential pharmacodynamic biomarkers of cell death, and caspase-directed activity-based probes and quenched substrates have shown potential for the live monitoring and efficient evaluation of apoptotic cell death in cell lysates, living cells and animals.(3,46) However, most ABP and quenched substrates are not able to cross the cell membrane which requires a development of new strategies for the intracellular delivery of these imaging systems. To date, such strategies have involved the use of drug delivery system (DDS) like gold nanoparticles,(23,24) quantum dots,(47) dendrigraft poly-L-lysine,(48) and microspheres,(49) to which quenched substrates are conjugated on the surface of the DDS. Although these systems allow detection of caspase-3, the area available for conjugation of the imaging agent can represent a limitation. To address this problem, we sought to develop PLGA-based nanoparticles for the encapsulation of CS1 within the polymeric core, similarly to work previously conducted by our group for in vivo delivery of drugs in the cancer therapy.(36,50) In addition, the hydrolysis that PLGA undertakes in water results in the release of the encapsulated agent over time, allowing for its accumulation in the cytoplasmic compartment.(51) Furthermore, the entrapment of the ABP within the nanoparticle combined with the rapid endosomal escape that PEG-PLGA nanoparticles normally undertake,(41) results in extra protection of the ABP from labelling lysosomal proteases.
As the further goal is to utilise CS1-NPs for the real-time imaging of caspase-3 activation in response to a chemotherapeutic treatment, conversion of the biotinylated probe to FRET substrate will be fundamental to avoid inhibition of apoptosis. This will inevitably result in increased hydrophobicity of the probe, making its formulation into PLGA-based nanoparticles more efficient, and consequently allowing for in vivo imaging of caspase-3 activity in cancer models thanks to the physicochemical properties of the formulation here described. Nevertheless, the recognition sequence of CS1 has shown to be recognised by caspase-3 and not by caspase-7, making CS1 a tool to study the role of caspase-3 only, both in physiological and pathological processes.
In conclusion, this work displays the in vitro validation of a novel and caspase-3 selective activity-based probe. Its formulation in PEG-PLGA nanoparticles has allowed for the intracellular delivery of the poor cell permeable CS1 probe resulting in an efficient caspase-3 labelling upon induction of apoptosis.
Experimental Section
Reagents are listed in supporting information.
ABPs synthesis.
The synthesis of two ABPs considered in this study has been carried out using the same reagents and following the same steps previously described by Poreba et al.(16,52) Briefly, biotin-ahx-Asp(t-Bu)-Glu(t-Bu)-Val-OH (for the synthesis of the DEVD ABP) and biotin-ahx-Asp(t-Bu)-Glu(Me)-Thr(Bzl)-OH (for the synthesis of the CS1 ABP) peptides were synthesized using 2-chlorotrityl chloride resin and used for further synthesis without purification. In parallel, Boc-Asp(t-Bu)-AOMK warhead was synthesized. The conjugation of the warhead into the peptide fragment was carried out in solution and monitored with HPLC. Following removal of the protecting groups, probes were purified with HPLC to obtain biotin-ahx-DEVD-AOMK and biotin-ahx-DE(Me)T(Bzl)D-AOMK probes. Mass spectrometry analysis was applied after each synthetic step to confirm the molecular mass of obtained compounds (semi-products and final product) (Figure S7).
Nanoparticle formulation.
Blank nanoparticles (BNPs) and CS1 nanoparticles (CS1-NPs) were obtained with a single emulsion solvent evaporation procedure. Briefly, 75 % of PLGA and 25 % of mPEG were dissolved in DCM. CS1 probe (0.2 mg mg-1 of polymer) was dissolved in a mixture of DCM and MeOH (9:1, v/v) and added to the polymer. Dropwise addition of the organic phase in 1 % PVA in MES buffer pH 5, followed by pulse sonication (FB120 sonic dismembrator, Fisher Scientific) for 90 s, led to obtain the emulsion. This was left stirring for 5 hr and the nanoparticles were washed with three centrifugation-resuspension cycles (20,000 g, 20 min, 4°C). BNPs were obtained by dissolving in the organic phase the polymer only; rhodamine or nile red NPs were obtained by dissolving in the organic phase 10 mg of polymer (7.5 mg PLGA and 2.5 mg mPEG) with 0.1 mg of dye.
Nanoparticle characterisation.
BNPs and CS1NPs were resuspended in ddH2O at 0.1 mg mL−1 for Dynamic Light Scattering (DLS) analysis using NanoBrook Omni (Brookhaven Instruments Corporation). Entrapment efficiency was evaluated by HPLC (Varian Prostar, JVA Analytical). BNPs and CS1-NP were dissolved at 1 mg mL−1 in a mixture of ACN and DMSO (v/v). For scanning electron microscopy (SEM, QUANTA FEG 250, ThermoFisher Scientific), CS1NPs (5 mg mL−1) were added on copper tape in drops and left to dry overnight. CS1NPs were coated with gold before imaging using FEI Quanta FEG - Environmental Scanning Electron Microscope (E-SEM). The stability study was performed by DLS analysis as previously described. BNPs and CS1-NPs were resuspended in ddH2O at 1 mg mL−1 and left at room temperature or 4°C for a period of 6 weeks. NPs stored at −20°C were stored as dry pellets and resuspended in ddH2O the day of the analysis.
Cell culture.
Cell based work was performed in sterile conditions under a safety cabinet class II and cells were cultured in a humidified incubator at 37°C and 5 % CO2 (Sanyo). PC3 cells (ATCC) were cultured in RPMI medium, HCT116 (ATCC) were cultured in McCoy’s medium, MCF7 (ATCC) were cultured in high glucose DMEM, all media were supplemented with 10 % FCS and 1 % PenStrep. Cells were seeded in 6-well plates, let adhere overnight and treated the following day. Apoptosis was induced with following treatments: PC3 cells were treated with 2.5 μM entinostat for 20 hr, followed by 40–80 ng mL−1 of rTRAIL for 4 to 6 hr; HCT116 cells were treated with 10 ng mL−1 of rTRAIL for 4 to 6 hr; MCF7 cells were treated with 1 μg mL−1 of camptotechin (CPT) for 12 and 16 hr. Caspase-3 labelling with CS1-NPs was obtained by treating PC3 cells with 0.25 and 0.5 mg mL−1 of CS1-NPs for 1 to 48 hr. NPs uptake analysis was conducted by treating PC3 cells with 0.5 mg mL−1 of rhodamine loaded NPs or nile red loaded NPs, for 3 and 24 hr. At the endpoint, cells were detached mechanically and centrifuged (3 min, 300 g, RT). Cell pellets were resuspended in hypotonic lysis buffer (25 mM HEPES, 2 mM EDTA, 100 mM NaCl, 250 mM sucrose, 1 % IGEPAL CA-630, 1 mM PMSF, pH 7.4) and left on ice for 30 min. A final concentration of 10 mM DTT was added to the lysis buffer when lysing cells for western blot analysis of ABP labelling of caspase-3 in cell lysates. Following centrifugation of cell debris (10 min, 20000 g, 4°C), supernatants were transferred to new tubes and protein concentration quantified with BCA assay as per manufacturer instructions.
Caspase-3/7 fluorescent assay.
50–80 μg of protein were added in triplicate in a solid bottom 96-well black plate, volumes were equalised by addition of caspase buffer (50 mM HEPES, 1 mM EDTA, 1 % IGEPAL CA-630, 10 % sucrose, 10 mM DTT, pH 7.4) and the substrate Ac-DEVD-AMC was added to a final concentration of 50 μM. Fluorescence was read at 360/40Ex 450/40Em, over 1 hr using a Cytation5 plate reader (Biotek).
Western blot analysis.
For ABP labelling of caspase-3 in cell lysates, 75 μg of lysates were incubated with the ABP (f.c. 1 and 10 μM, 37°C, 500 rpm, 1 hr). Positive controls for CS1-NP western blot analysis was made by incubating apoptotic cell lysates with 10 μM CS1 for 1 hr, at 37°C, with shaking at 500 rpm. Samples were denatured with 5x Laemmli buffer (0.3 M TRIS, 20 % glycerol, 25 % P-mercaptoethanol, 10 % SDS, 0.1% bromophenol blue) and incubated at 95°C for 10 min. Samples were resolved in 12 %, 15 %, or 17.5 % acrylamide gels, proteins were transferred to a polyvinylidene fluoride membrane (PVDF, Immobilon-P, 0.45 μm pore size, Merck Millipore) by semi-dry transfer (PowerPac 1000, Biorad) and blocked with 5 % bovine serum albumin in TBS-T (50 mM TRIS, 150 mM NaCl, 0.1 % tween-20, pH 7.4–7.6) for 2 hr, at RT, shaking. For western blot analysis of ABP labelling of caspase-3, membranes were incubated with streptavidin-HRP (1:2,000; 30–45 min; RT; shaking). For immunoblot analysis, membranes were incubated in primary antibody (1:1,000) overnight and in secondary antibody (1:10,000; 1 hr; RT; shaking) the following day. Proteins were detected by incubating the membranes in the dark with ECL plus for 5 minutes and imaged with ChemDoc XRS+ (Biorad).
Confocal analysis.
PC3 cells were seeded in 8-well confocal chamber slides at 25,000 cells/well and let adhere overnight. Following treatments, cells were washed with cold PBS (3x) and fixed with cold 4 % PFA in PBS for 20 min. Cells were washed with cold PBS (3x) and cell membranes were permeabilised with 0.5 % Triton X in PBS (5 min), followed by three further PBS washes. Nuclei were stained with DAPI and coverslips applied. Slides were viewed on a SP8 confocal microscope (Leica Microsystems) equipped with LAS AF software and images were captured with a x40 lens.
Uptake assay.
Following treatment with rhodamine NPs, PC3 cells were detached with trypsin and centrifuged at 300 g for 3 min at RT. Cells were washed with PBS in three cycles of resuspension-centrifugation. Cell pellets were resuspended in 200 μL of lysis buffer (0.2 M NaOH, 0.5 % triton X-100 in PBS) and left on ice. Protein concentration was assessed with BCA assay and 30 μg of protein added to a solid bottom 96-well black plate in triplicate, volumes were equalized with PBS. Fluorescence was recorded at 516/20Ex 557/20Em using a Cytation5 plate reader (Biotek).
Statistical analysis.
Statistical analysis were performed using GraphPad Prism5 software using one-way analysis of variance (ANOVA) when only one variable was considered, and two-way ANOVA when two variables were considered. Statistical significance is indicated as follow: * p < 0.05, ** p < 0.01, *** p < 0.001.
Supplementary Material
Acknowledgements
FC was supported by the Belfast-Manchester Movember Centre of Excellence (CE013_2-004), funded in partnership with Prostate Cancer UK. The Scott laboratory is supported by the Health and Social Care in Northern Ireland (HSCNI) (STL/5010/14, Medical Research Council grant MC_PC_15013). The Drag laboratory is supported by the Foundation for Polish Science and National Science Centre in Poland.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
References
- 1.Elmore S Apoptosis: a review of programmed cell death. Toxicol Pathol [Internet]. 2007;35(4):495–516. Available from: http://tpx.sagepub.com/content/35/4/495.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldar S, Khaniani MS, Derakhshan SM, Baradaran B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pacific J Cancer Prev. 2015;16(6):2129–44. [DOI] [PubMed] [Google Scholar]
- 3.Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, et al. Noninvasive optical imaging of apoptosis by caspase-targeted activity-based probes. Nat Med [Internet]. Nature Publishing Group; 2009;15(8):967–73. Available from: 10.1038/nm.1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poreba M, Szalek A, Rut W, Kasperkiewicz P, Rutkowska-Wlodarczyk I, Snipas SJ, et al. Highly sensitive and adaptable fluorescence-quenched pair discloses the substrate specificity profiles in diverse protease families. Sci Rep [Internet]. Nature Publishing Group; 2017;7(January):1–13. Available from: 10.1038/srep43135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.FonoviV M, Bogyo M. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev Proteomics [Internet]. 2008;5(5):721–30. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2997944&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poreba M, Szalek A, Kasperkiewicz P, Rut W, Salvesen GS, Drag M. Small Molecule Active Site Directed Tools for Studying Human Caspases. Chem Rev. 2015;115(22):12546–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thornberry NA, Rano TA, Peterson P, Rasper DM, Timkey T, Garcia-calvo M, et al. A Combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B. J Biol Chem. 1997;272(29):17907–11. [DOI] [PubMed] [Google Scholar]
- 8.McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15(2):322–31. [DOI] [PubMed] [Google Scholar]
- 9.Pereira NA, Song Z. Some commonly used caspase substrates and inhibitors lack the specificity required to monitor individual caspase activity. Biochem Biophys Res Commun [Internet]. Elsevier Inc.; 2008;377(3):873–7. Available from: 10.1016/j.bbrc.2008.10.101 [DOI] [PubMed] [Google Scholar]
- 10.Benkova B, Lozanov V, Ivanov IP, Mitev V. Evaluation of recombinant caspase specificity by competitive substrates. Anal Biochem [Internet]. Elsevier Inc.; 2009;394(1):68–74. Available from: 10.1016/j.ab.2009.07.012 [DOI] [PubMed] [Google Scholar]
- 11.Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS, Drag M. Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ [Internet]. Nature Publishing Group; 2014;21(9):1482–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24832467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vickers CJ, Gonza GE, Wolan DW. Selective Detection of Caspase - 3 versus Caspase - 7 Using Activity-Based Probes with Key Unnatural Amino Acids. ACS Chem Biol. 2013;8:1558–66. [DOI] [PubMed] [Google Scholar]
- 13.Vickers CJ, Gonza GE, Wolan DW. Discovery of a Highly Selective Caspase - 3 Substrate for Imaging Live Cells. ACS Chem Biol. 2014;9:2199–203. [DOI] [PubMed] [Google Scholar]
- 14.Powers JC, Asgian JL, James KE. Irreversible Inhibitors of Serine , Cysteine , and Threonine Proteases. Chem Rev. 2002;102:4639–750. [DOI] [PubMed] [Google Scholar]
- 15.Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr Opin Chem Biol. 2007;11(1):20–8. [DOI] [PubMed] [Google Scholar]
- 16.Poreba M, Groborz K, Navarro M, Snipas SJ, Drag M, Salvesen GS. Caspase selective reagents for diagnosing apoptotic mechanisms. Cell Death Differ [Internet]. Springer US; 2018; Available from: 10.1038/s41418-018-0110-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poreba M, Solberg R, Rut W, Turk B, Salvesen GS, Drag M, et al. Counter Selection Substrate Library Strategy for Developing Specific Protease Substrates and Probes Counter Selection Substrate Library Strategy for Developing Specific Protease Substrates and Probes. Cell Chem Biol [Internet]. Elsevier Ltd; 2016;23(8):1023–35. Available from: 10.1016/j.chembiol.2016.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Syed S, Bachmann MH, Blum G, Bogyo M. Functional Imaging of Legumain in Cancer Using a New Quenched Activity-Based Probe. J Am Chem Soc. 2013;135:174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: Focus on cancer. Int J Nanomedicine. 2014;9(1):467–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J Control Release [Internet]. Elsevier B.V.; 2012;161(2):175–87. Available from: 10.1016/j.jconrel.2011.09.063 [DOI] [PubMed] [Google Scholar]
- 21.Lin SY, Chen NT, Sun SP, Chang JC, Wang YC, Yang CS, et al. The protease-mediated nucleus shuttles of subnanometer gold quantum dots for real-time monitoring of apoptotic cell death. J Am Chem Soc. 2010;132(24):8309–15. [DOI] [PubMed] [Google Scholar]
- 22.Chen WH, Xu XD, Jia HZ, Lei Q, Luo GF, Cheng SX, et al. Therapeutic nanomedicine based on dual-intelligent functionalized gold nanoparticles for cancer imaging and therapy invivo. Biomaterials [Internet]. Elsevier Ltd; 2013;34(34):8798–807. Available from: 10.1016/j.biomaterials.2013.07.084 [DOI] [PubMed] [Google Scholar]
- 23.Sun IC, Lee S, Koo H, Kwon IC, Choi K, Ahn CH, et al. Caspase sensitive gold nanoparticle for apoptosis imaging in live cells. Bioconjug Chem. 2010;21(11):1939–42. [DOI] [PubMed] [Google Scholar]
- 24.Jun Y, Sheikholeslami S, Hostetter DR, Tajon C, Craik CS, Alivisatos AP. Continuous imaging of plasmon rulers in live cells reveals early-stage caspase-3 activation at the single-molecule level. Proc Natl Acad Sci U S A. 2009;106(42):17735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao W, Ji L, Li L, Cui G, Xu K, Li P, et al. Bifunctional combined Au-Fe 2O 3 nanoparticles for induction of cancer cell-specific apoptosis and real-time imaging. Biomaterials. 2012;33(14):3710–8. [DOI] [PubMed] [Google Scholar]
- 26.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel). 2011;3(3):1377–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surfaces B Biointerfaces. 2010;75(1):1–18. [DOI] [PubMed] [Google Scholar]
- 28.Rokhlin OW, Guseva N, Tagiyev A, Knudson CM, Cohen MB. Bcl-2 oncoprotein protects the human prostatic carcinoma cell line PC3 from TRAIL-mediated apoptosis. Oncogene. 2001;20:2836–43. [DOI] [PubMed] [Google Scholar]
- 29.Perego P, Zuco V, Gatti L, Zunino F. Sensitization of tumor cells by targeting histone deacetylases. Biochem Pharmacol [Internet]. Elsevier Inc.; 2012;83(8):987–94. Available from: 10.1016/j.bcp.2011.11.010 [DOI] [PubMed] [Google Scholar]
- 30.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galligan L, Longley DB, Miranda M, R WT, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther [Internet]. 2005;4(12):2026–36. Available from: http://mct.aacrjournals.org/cgi/doi/10.1158/15357163.MCT-05-0262 [DOI] [PubMed] [Google Scholar]
- 32.Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene. 2002;21(57):8843–51. [DOI] [PubMed] [Google Scholar]
- 33.Liang Y, Yan C, Schor NF. Apoptosis in the absence of caspase 3. Oncogene. 2001;20(45):6570–8. [DOI] [PubMed] [Google Scholar]
- 34.Chu C, Xu J, Cheng D, Li X, Tong S, Yan J, et al. Anti-Proliferative and Apoptosis-Inducing Effects of Camptothecin-20(s)-O-(2-pyrazolyl-1)acetic Ester in Human Breast Tumor MCF-7 Cells. Molecules [Internet]. 2014;19(4):4941–55. Available from: http://www.mdpi.com/1420-3049/19/4/4941/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berger AB, Witte MD, Denault J-B, Sadaghiani AM, Sexton KMB, Salvesen GS, et al. Identification of Early Intermediates of Caspase Activation Using Selective Inhibitors and Activity-Based Probes. Mol Cell [Internet]. 2006;23(4):509–21. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1097276506004357 [DOI] [PubMed] [Google Scholar]
- 36.Schmid D, Jarvis GE, Fay F, Small DM, Greene MK, Majkut J, et al. Nanoencapsulation of ABT-737 and camptothecin enhances their clinical potential through synergistic antitumor effects and reduction of systemic toxicity. Cell Death Dis [Internet]. Nature Publishing Group; 2014;5(10):e1454 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25299779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari R, Lupi M, Colombo C, Morbidelli M, D’Incalci M, Moscatelli D. Investigation of size, surface charge, PEGylation degree and concentration on the cellular uptake of polymer nanoparticles. Colloids Surfaces B Biointerfaces [Internet]. Elsevier B.V; 2014;123:639–47. Available from: 10.1016/j.colsurfb.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 38.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev [Internet]. Elsevier B.V.; 2015;99:28–51. Available from: 10.1016/j.addr.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J Control Release. 2000;65(1–2):271–84. [DOI] [PubMed] [Google Scholar]
- 40.Maeda H The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41(00):189–207. [DOI] [PubMed] [Google Scholar]
- 41.Panyam J, Zhou W-Z, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-coglycolide) nanoparticles: implications for drug and gene delivery. FASEB J [Internet]. 2002;16(10):1217–26. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12153989 [DOI] [PubMed] [Google Scholar]
- 42.Kavanagh E, Rodhe J, Burguillos MA, Venero JL, Joseph B. Regulation of caspase-3 processing by cIAP2 controls the switch between pro-inflammatory activation and cell death in microglia. Cell Death Dis [Internet]. Nature Publishing Group; 2014;5(12):e1565–9. Available from: 10.1038/cddis.2014.514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferreira KS, Kreutz C, MacNelly S, Neubert K, Haber A, Bogyo M, et al. Caspase-3 feeds back on caspase-8 , Bid and XIAP in type I Fas signaling in primary mouse hepatocytes. Apoptosis. 2012;17:503–15. [DOI] [PubMed] [Google Scholar]
- 44.Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ [Internet]. 2003;10(1):76–100. Available from: http://www.nature.com/doifinder/10.1038/sj.cdd.4401160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brady KD, Giegel DA, Grinnell C, Lunney E, Talanian RV., Wong W, et al. A catalytic mechanism for caspase-1 and for bimodal inhibition of caspase-1 by activated aspartic ketones. Bioorganic Med Chem. 1999;7(4):621–31. [DOI] [PubMed] [Google Scholar]
- 46.Edgington LE, Van Raam BJ, Verdoes M, Wierschem C, Salvesen GS, Bogyo M. An optimized activity-based probe for the study of caspase-6 activation. Chem Biol [Internet]. Elsevier Ltd; 2012;19(3):340–52. Available from: 10.1016/j.chembiol.2011.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren D, Wang J, You Z. Long-term monitoring of capase-3 activity in living cells based on the FRET probe composed of quantum dot, nanogold and EGF. RSC Adv [Internet]. Royal Society of Chemistry; 2014;4(97):54907–18. Available from: http://xlink.rsc.org/?DOI=C4RA07913B [Google Scholar]
- 48.Liu Y, Hu Y, Guo Y, Ma H, Li J, Jiang C. Targeted imaging of activated caspase-3 in the central nervous system by a dual functional nano-device. J Control Release [Internet]. Elsevier B.V.; 2012;163(2):203–10. Available from: 10.1016/j.jconrel.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 49.Cárdenas-Maestre JM, Pérez-Lõpez AM, Bradley M, Sánchez-Martín RM. Microsphere-based intracellular sensing of caspase-3/7 in apoptotic living cells. Macromol Biosci. 2014;14(7):923–8. [DOI] [PubMed] [Google Scholar]
- 50.Schmid D, Fay F, Small DM, Jaworski J, Riley JS, Tegazzini D, et al. Efficient drug delivery and induction of apoptosis in colorectal tumors using a death receptor 5-targeted nanomedicine. Mol Ther [Internet]. 2014;22(12):2083–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25200008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev. 2007;59:718–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poreba M, Salvesen GS, Drag M. Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat Protoc [Internet]. Nature Publishing Group; 2017;12(10):2189–214. Available from: 10.1038/nprot.2017.091 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.