

Abstract
Throughout the nervous system, ion gradients drive fundamental processes. Yet, the roles of interstitial ions in brain functioning is largely forgotten. Emerging literature is now revitalizing this area of neuroscience by showing that interstitial cations (K+, Ca2+ and Mg2+) are not static quantities but change dynamically across states such as sleep and locomotion. In turn, these state-dependent changes are capable of sculpting neuronal activity; for example, changing the local interstitial ion composition in the cortex is sufficient for modulating the prevalence of slow-frequency neuronal oscillations, or potentiating the gain of visually evoked responses. Disturbances in interstitial ionic homeostasis may also play a central role in the pathogenesis of central nervous system diseases. For example, impairments in K+ buffering occur in a number of neurodegenerative diseases, and abnormalities in neuronal activity in disease models disappear when interstitial K+ is normalized. Here we provide an overview of the roles of interstitial ions in physiology and pathology. We propose the brain uses interstitial ion signaling as a global mechanism to coordinate its complex activity patterns, and ion homeostasis failure contributes to central nervous system diseases affecting cognitive functions and behavior.
Keywords: Potassium, Calcium, Magnesium, Astrocytes, Neuromodulators, Huntington’s disease
1. Introduction
Ion gradients across plasma and organelle membranes are indispensable to cellular life and form the foundation for the physiology of excitable cells throughout the body. All cellular elements in the central nervous system (CNS) are exposed to the interstitial milieu, and the prevailing plasma membrane ion gradients drive vital functions including action potential firing, transmitter release, and synaptic transmission. Moreover, ion gradients provide an energy reservoir pivotal for cellular homeostasis and for driving uptake of energy metabolites and other essential solutes such as amino acids. In other words, brain function and ion gradients are inseparable.
A vast historical literature documents that the concentrations of certain cation species (potassium: K+, calcium: Ca2+ and magnesium: Mg2+) in the interstitial environment dynamically relate to neuronal excitability and activity (Aitken and Somjen, 1986; Baylor and Nicholls, 1969; Dingledine and Somjen, 1981; Heinemann et al., 1990, 1986, 1977; Heinemann and Lux, 1977; Karwoski et al., 1985; Kraig and Nicholson, 1978; Krnjević et al., 1982; Nicholson et al., 1978; Poolos et al., 1987; Poolos and Kocsis, 1990; Singer et al., 1976; Somjen, 1980; Utzschneider et al., 1992; Zanotto and Heinemann, 1983). Still, changes in the milieu of interstitial ions are not traditionally considered to be an integral part of neuronal signaling, circuit activity and global brain states, but rather a byproduct of neuronal activity. The persistence of this notion seems surprising, as even small changes in interstitial ion concentrations strongly affect the membrane potential (Fig. 1A), transmitter release, and excitability (Balestrino et al., 1986; Brocard et al., 2013; Egelman and Montague, 1999; Hablitz and Lundervold, 1981; Kamiya and Zucker, 1994; Matyushkin et al., 1995; Poolos and Kocsis, 1990; Rausche et al., 1990; Rusakov and Fine, 2003; Shih et al., 2013; Somjen and Müller, 2000; Tong et al., 2014; Utzschneider et al., 1992). In fact, the current convention is to consider the effects of channelopathies on neuronal membrane properties in isolation, disregarding their potential role on the interstitial ion environment (Bean, 2007).
Fig. 1. Overview of interstitial ions in neuronal activity.
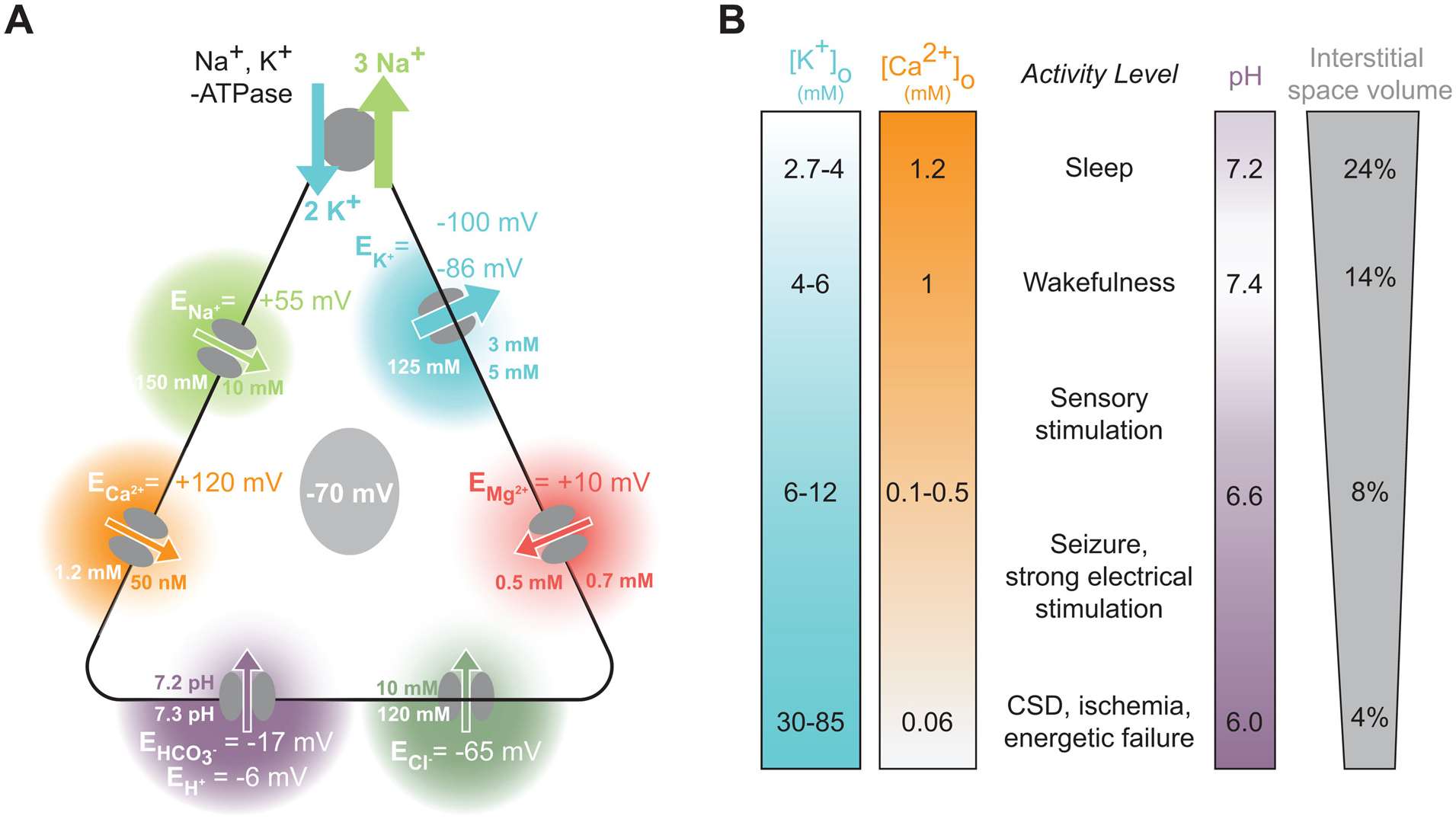
(A) A simplified model of free ion concentrations and their equilibrium potentials in neurons at rest. The resting membrane potential is principally established by the Na+, K+-ATPase, which creates a charge gradient by the ATP-dependent export of 3 Na+ ions for 2 K+ ions brought into the cell. The resting equilibrium potentials for Na+, Ca2+, H+, Cl−, Mg2+, and K+ are depicted, along with their typical intracellular and interstitial concentrations. Arrow directions and widths reflect the direction and strength of the driving forces at rest. (B) [K+]o, [Ca2+]o, pH, and interstitial space volume (ISV) are correlated both with brain state and local neuronal activity. [K+]o increases, [Ca2+]o, pH, and the interstitial cellular space volume decrease in the transition from sleep (top) to physiological activity and further to pathologic activity such as seizures and the complete energetic collapse occurring in cortical spreading depression (CSD) and ischemia (bottom). [Mg2+]o is lower in quiet wakefulness than in sleep, but it is not yet know whether neuronal activity further reduces its concentration (Ding et al., 2016). [Cl−]o exhibits a drop from ~120 mM to 50–60 mM in the setting of ischemia, but does not exhibit any major concentration shifts under more physiological conditions (Hansen, 1985).
Yet, an emerging literature is challenging the view that interstitial ion changes are merely epiphenomenal; for example, ion changes have been causally implicated in the generation of rhythmic locomotor activity in the spinal cord (Brocard et al., 2013), in the sleep-wake cycle (Ding et al., 2016; Rasmussen et al., 2017), and in neuronal oscillations at various timescales (Bazhenov et al., 2004; Krishnan et al., 2018; Wang et al., 2012a, 2012b). Concurrently, multiple lines of new evidence point to dysregulation of interstitial ions playing a central role in the pathogenesis of a number of CNS diseases. For example, down-regulating of the astrocytic K+ channel, Kir4.1, occurs in a broad range of neurodegenerative diseases, including Huntington’s disease, amyotrophic lateral sclerosis, and multiple sclerosis (Benraiss et al., 2016; Kelley et al., 2018; Srivastava et al., 2012; Tong et al., 2014). This downregulation causes elevated interstitial K+ levels and abnormal neuronal activity (Tong et al., 2014). Recent work has also shown that disruption of the Kir4.1 channel in the lateral habenula, and the consequent abnormal K+ buffering, was linked to the development of depression-like symptoms in experimental animals (Cui et al., 2018).
Excellent review articles have discussed the consequences of acute pathological events in the CNS on interstitial ion concentrations (de Baaij et al., 2015; Fröhlich et al., 2008; Heinemann et al., 1986; Raimondo et al., 2015; Somjen, 2004, 2002). Furthermore, important work has previously summarized the effects of changes in interstitial ions and their role on neuronal activity (Nicholson, 1979; Syková, 1992), but there is no comprehensive review summarizing the most recent literature on the role of interstitial ions in normal brain physiology and brain state-dependent neuronal activity. Here we begin by considering how the prevailing interstitial concentrations of K+ ([K+]o), Ca2+ ([Ca2+]o) and Mg2+ ([Mg2+]o) change in the brain under different conditions and states, and how this in turn affects neurons and their firing mode. Next, we review current work implicating interstitial ions in controlling global brain states and state-dependent neuronal activity. These observations will serve as a basis for a discussion of emerging evidence implicating dysregulation of [K+]o in the development of several chronic CNS diseases. We conclude by outlining current experimental challenges to be overcome, and also propose key questions in the field that may be testable in the near future. Our overall synthesis suggests that concerted changes in interstitial ions should not be considered merely epiphenomena of neuronal activity, but instead constitute an integrated mechanism for regulating brain state-dependent neuronal activity. We contend that interstitial ion changes may be a simple, yet powerful, non-neuronal principle allowing the brain to globally control activity over longer timescales in diverse phenomena, notably across arousal states. We predict that disturbances in interstitial ion signaling will emerge as a common feature in diseases affecting mental states, including neurodegenerative and psychiatric diseases (Dietz et al., 2019).
2. Interstitial ions dynamically change and can regulate neuronal activity
2.1. Interstitial K+ strongly regulates neuronal activity
The brain tightly controls [K+]o within a range normally between 2.7 and 5 mM (Ding et al., 2016; Hansen, 1985; Monai et al., 2019; Nicholson, 1979; Rasmussen et al., 2019; Somjen, 2004, 2002; Syková, 1992). Changes in brain activity associate with alterations in [K+]o (Ding et al., 2016; Lux, 1974; Lux and Neher, 1973; Octeau et al., 2019; Rasmussen et al., 2019) (Fig. 1B and Table 1); a single action potential transiently induces a local increase in [K+]o of 0.01–0.02 mM (Dietzel et al., 1980; Syková et al., 1974). Local electrical and physiological sensory stimulation can increase [K+]o in the CNS by 0.05–2 mM, with complex paradigms such as painful tactile stimulation and rhythmic flexion of the knee joint driving increases of 1.7–2 mM (Table 1). Similarly, behavioral state transitions from sleep to awake, and from quiet to active wakefulness, associate with a 0.4–0.55 mM and 0.5–1 mM cortical [K+]o increase, respectively (Ding et al., 2016; Rasmussen et al., 2019). In pathological states, such as seizures, or during strong electrical stimulation, [K+]o readily increases by as much as 8–12 mM above baseline levels (Heinemann and Lux, 1977; Poolos et al., 1987; Poolos and Kocsis, 1990; Singer et al., 1976; Utzschneider et al., 1992). Finally, during the extreme conditions of mass neuronal depolarization seen only in pathologic energy failure of ischemia and cortical spreading depression, [K+]o can increase to as high as 55–85 mM, as all ionic gradients collapse (Heinemann and Lux, 1977; Somjen, 2002).
Table 1.
Stimulation and state-induced changes in interstitial ions alter neuronal activity. Representative changes in [K+]o and [Ca2+]o from a selection of studies demonstrating shifts in ions resulting from sensory stimulation, electrical stimulation (‘Stim.’), seizure induction, ischemia, spreading depression, brain state changes, and other manipulations. For studies that directly manipulated interstitial ion concentrations, the reported functional effect is noted.
| Model | Region(s) | Baseline (mM) | Stimulation | Δ (mM) | Functional Change | |
|---|---|---|---|---|---|---|
| [K+]o | ||||||
| Aitken and Somjen, 1986 | Rat | HC slice | 3.5 (ACSF) | Orthodromic stim. | ↑ 0.02–0.27 | |
| Branston et al., 1977 | Baboon | Ctx | 5.7 | Ischemia | ↑ ~25–75 | |
| Balestrino et al., 1986 | Rat | HC slice | 3.5 (ACSF) | Δ ACSF K+ | ↑ 1.5 ↓ 1.5 |
↑ 48% population response Depresses I/O response |
| Brocard et al., 2013 | Rat | Isolated SC | 4 (ACSF) | NMDA/serotonin | ↑ 1.5–2.1 | Fictive locomotion |
| Ding et al., 2016 * | Mouse | Ctx slice Ctx | ~2.8 3.7 (sleep) |
Neuromodulator cocktail Natural awakening Awake to isoflurane |
↑ 0.43 ↑ 0.40 ↓ 0.37 |
Changing overlying ACSF can drive sleep/wake rhythm changes |
| Harris et al., 1981 | Baboon | Ctx | 3.95 | Ischemia | ↑ ~25–65 | |
| Heinemann et al., 1977 | Cat | SCtx | 2.7–3.2 | Seizures Forepaw brushing |
↑ 4–5 ↑ 0.4–0.7 |
|
| Heinemann et al., 1990 | Cat | SC | 2.7–3.0 | Nerve stim. Leg brushing Rhythmic knee flexion Noxious/painful stimuli |
↑ 0.6 ↑ 0.4 ↑ 1.7 ↑ 2 |
|
| Lewis and Schuette, 1975 | Cat | HC | 4–5 | Stim. 40 Hz, 2 s | ↑ 5–6 | |
| Lux and Neher, 1973 | Cat | Ctx | 2.5–4 | Stim. (varied) | ↑ 0.1–15 | |
| Nicholson et al., 1978 | Cat | Cb | 3 | Stim. 5 Hz, 30 s | ↑ 1 | |
| Octeau et al., 2019 * | Mouse | Ctx and Str | 3 | Astrocytic channel-rhodopsin | ↑ 0.4 | Enhances MSN excitability |
| Poolos et al., 1987 | Rat | HC slice | 3 | Stim. 10–80 Hz, 1 s | ↑ 4 | ↑ excitability and conduction velocity below 6.0 mM [K+]o |
| Rasmussen et al., 2019 * | Mouse | SCtx, MCtx, VCtx | 3.5–3.7 | Quiet wakefulness to locomotion | ↑ 0.5–0.7 | Neuronal depolarization and enhanced I/O transformation |
| Singer et al., 1976 | Cat | VCtx | MRF stim. | ↑ 1.5–2.0 | ||
| Syková et al., 1974 | Rat | MRF | ~3 | Spontaneous bursts | ↑ 0.02–0.2 | |
| Tong et al., 2014 | WT Mouse R6/2 Mouse | Str | 1.5 2.9 | AAV2/5 Kir4.1 in R6/2 mice | ↓ 1.1 | ↑ [K+]o by 1.5 mM ↓ medium spiny neuron rheobase, ↑ input resistance, and ↑ firing |
| Utzschneider et al., 1992 | Rat | Excised dorsal root ganglion | 3 (ACSF) | Stim. 1 Hz, 10 s Stim. 5 Hz, 10 s Stim. 10 Hz, 10 s Stim. 50 Hz, 10 s |
no effect ↑ ~0.5 ↑ ~1.2 ↑ 2.20 |
90% of neurons were depolarized due to stimulation of axons of neighboring neurons |
| Wang et al., 2012a, 2012b | Mouse | HC/Cb slice Ctx | ~3.8 ~3.9 | Agonist-induced astrocytic Ca2+ waves | ↓ ~0.35–0.45 ↓ ~0.2–0.5 | ↓ [K+]o improves signal-to-noise ratio and reduces Purkinje cell bistable state oscillations |
| Huchzermeyer et al., 2008; Kann et al., 2011 | Rat | HC slice | 3 (ACSF) | 10 μM acetylcholine Stim. 20 Hz, 10 s |
↑ 0.4 ↑ 1.5 −2 |
|
| [Ca2+]o | ||||||
| Brocard et al., 2013 | Rat | Isolated SC | 1.2 | NMDA/serotonin | ↓ 0.26–0.36 | Fictive locomotor episode |
| Ding et al., 2016 * | Mouse | Ctx | 1.36 (sleep) | Natural awakening Awake to isoflurane |
↓ 0.13 ↑ 0.26 |
|
| Dingledine and Somjen, 1981 | Rat | HC slice | 1.2 | Δ [Ca2+]o | ↓ 0.1 | Shifts I/O curve slope by 15% |
| Heinemann et al., 1977 | Cat | SCtx | 1.2–1.5 | Forepaw brushing Penicillin-induced seizures |
↓ 0.1 ↓ 0.7 |
|
| Kraig and Nicholson, 1978 | Catfish | Cb | 2.2 | Spreading depression | ↓ ~1.4 | |
| Massimini and Amzica, 2001 | Cat | Ctx | 1.04 | Slow-wave oscillations | Peak: 1.18 Trough: 0.95 | |
| Nicholson et al., 1978 | Cat | Cb | 1.2 | Stim. 5 Hz, 30 s | ↓ 0.1 | |
| Xiong et al., 1997 | Culture | HC neurons | 1.5 (ACSF) | Δ ACSF | ↓ 1 | Increased excitability |
| Zanotto and Heinemann, 1983 | Rat | HC slice | 1.6 (ACSF) | Stim. 15 Hz, 10 s Glu/Asp iontophoresis |
↓ 0.5 ↓ > 1 |
Abbreviations: ACSF: artificial cerebrospinal fluid; Cb: Cerebellum; Ctx: Cortex; HC: Hippocampus; I/O: input-output; MCtx: Motor cortex; MRF: Mesencephalic reticular formation; SC: Spinal cord; SCtx: Somatosensory cortex; Str: Striatum; VCtx: Visual cortex.
indicates a study utilizing unanesthetized (awake or asleep) animals - all other in vivo studies were conducted under anesthesia. ‘(ACSF)’ indicates the reported concentration of an ion in the bath solution - not a true baseline value within the tissue.
Clearly, changes in neuronal activity are strongly correlated to [K+]o changes, but how may [K+]o in turn influence neuronal activity? Increases in [K+]o weakens the outward K+ driving force across the plasma membrane, resulting in a less negative K+ equilibrium potential and a consequent depolarization of the neuronal membrane potential (Fig. 2A). This [K+]o-driven depolarization reduces the threshold of synaptic barrages necessary for eliciting action potentials by reducing the distance to spike threshold, thus increasing excitability (Rasmussen et al., 2019; Tong et al., 2014) (Fig. 2A, B). Though neurons exhibit smaller changes in the membrane potential than would be predicted by the theoretical Nernst equation (Somjen, 2002), even subtle [K+]o increases of 0.5–1.5 mM are capable of depolarizing neurons by ~5 mV (Pan and Stringer, 1997; Rasmussen et al., 2019; Tong et al., 2014), which in turn can have profound effects on neuronal activity, especially in-vivo (Haider and Mccormick, 2009). Thus, such depolarizations seen with physiological increases in [K+]o are sufficient to lower the threshold for opening voltage-gated Ca2+ channels and enhance neurotransmitter release (Balestrino et al., 1986; Hablitz and Lundervold, 1981; Matyushkin et al., 1995; Rausche et al., 1990; Shih et al., 2013). Concurrently, due to decreased variability in the magnitude of presynaptic neurotransmitter release, increased neuronal firing synchrony may occur (Poolos et al., 1987) (Fig. 2C). Hence, physiological changes in [K+]o are sufficiently potent to regulate neuronal activity and synaptic transmission.
Fig. 2. Changes in [K+]o alters neuronal activity.
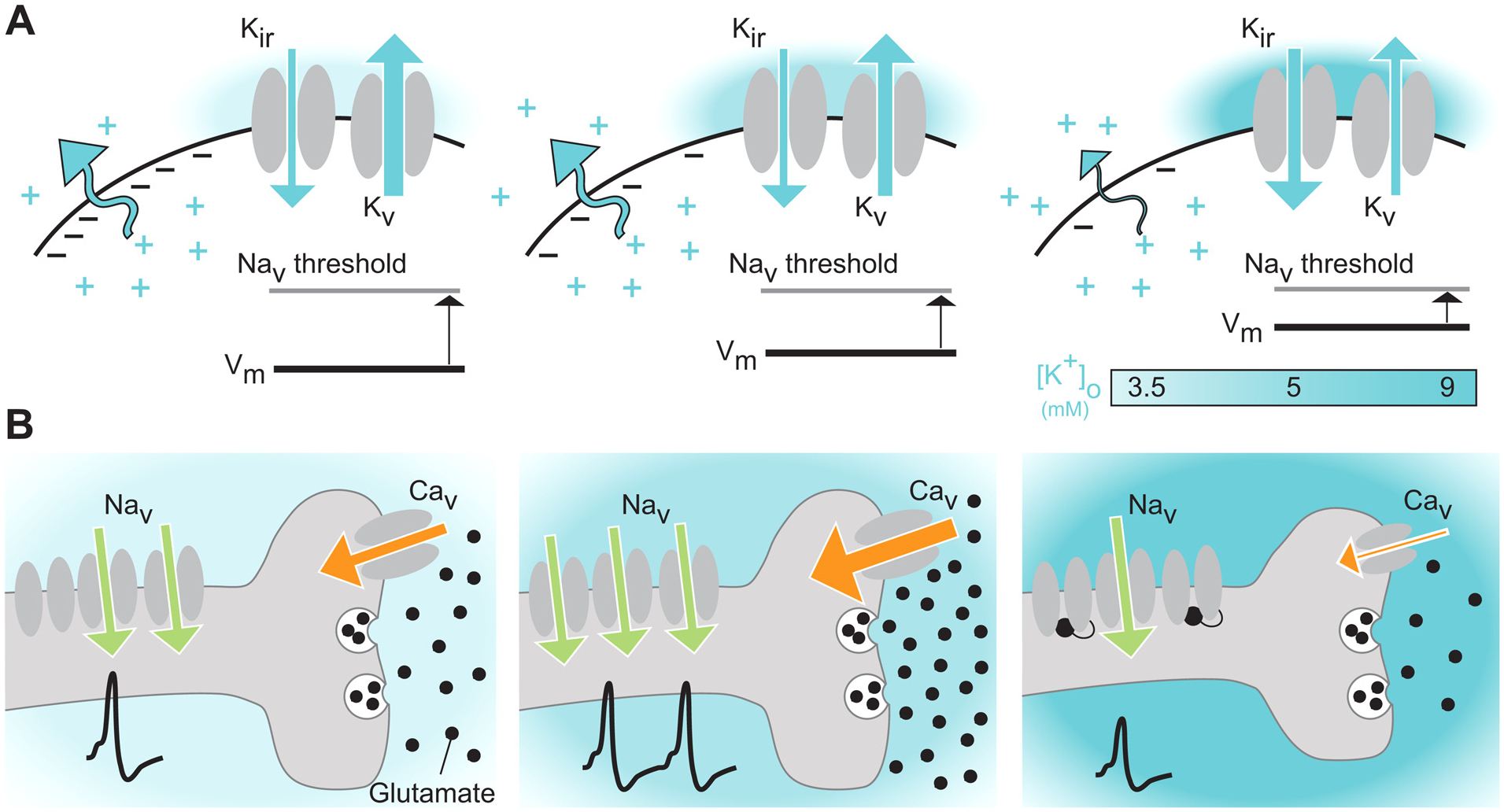
(A) Increases in [K+]o from resting levels (left), as seen in wakefulness and locomotion (middle) or pathological states (right), depolarize the neuronal membrane potential closer to the activation threshold of voltage-gated Na+ channels (Nav) in part through a decrease in outward-directed K+ currents and increases in regulatory inward-rectifying K+ currents (middle). (B) When [K+]o is increased from 3.5 to 5 mM (middle) the depolarized membrane potential may enable increased activation of voltage-gated Ca2+ channels (Cav), triggering higher levels of released glutamate. If [K+]o increases further, up to a level of roughly 9 mM, this depolarizes the membrane potential at a level that does not allow the Nav channels to reach their closed state, but rather locks them in their inactive state, leading to gradually fewer action potentials being fired, and as a result lower levels of glutamate being released.
Interestingly, increases in [K+]o appears to differentially affect excitatory glutamatergic and inhibitory GABAergic synaptic transmission. Previous work has shown that local increases in [K+]o within the synaptic cleft, e.g. following NMDA receptor activation, boost the presynaptic influx of Ca2+ causing enhanced glutamate release and potentiate the amplitude of excitatory postsynaptic potentials; suggesting that activity-dependent changes in [K+]o can signal locally within synapses in a retrograde manner (Shih et al., 2013). This finding is in accord with experiments showing that a 0.8 mM decrease in [K+]o suppresses excitatory synaptic transmission by reducing the frequency of spontaneous excitatory postsynaptic potentials (Wang et al., 2012a). In contrast to this effect on excitatory transmission, inhibitory postsynaptic currents were largely unaffected by lowering [K+]o by 0.8 mM (Wang et al., 2012a). This finding appears congruent with recordings obtained from cultured neurons, showing that increasing [K+]o from 4 to 10 mM selectively dampened evoked glutamatergic transmission while leaving inhibitory transmission intact (He et al., 2002). Sub-sequent work from the same group, performed in acute hippocampal brain slices, documented that increases in [K+]o of 8–10 mM from 2.5 mM selectively depressed excitatory postsynaptic currents relative to inhibitory currents (Meeks and Mennerick, 2004). Although the [K+]o increases employed in these experiments fall outside of the normal, physiological range (Fig. 1B and Table 1), it underscores the differential effect of [K+]o changes on glutamatergic versus GABAergic neurons. This difference likely reflects that inhibitory postsynaptic potentials rely on conductance of chloride ions, which is insensitive to minor changes in interstitial [K+]o. In contrast, excitatory postsynaptic potentials involve transmembrane fluxes of multiple cations, including K+ (Browne et al., 2001). Furthermore, voltage-gated Na+ channels in the axons of glutamatergic (compared to GABAergic) axons appears to encompass different sensitivities to [K+]o-induced depolarization (He et al., 2002; Meeks and Mennerick, 2004) and different Na+ channel axonal distribution profiles (Hu and Jonas, 2014). To complicate matters even more, [K+]o modulates the activity patterns of neurons through direct modulation of a multitude of ion channels (Amzica and Steriade, 2000; Bazhenov et al., 2004; Frace et al., 1992; Korn et al., 1987; Shin and Carlen, 2008; Somjen and Müller, 2000; Sturm et al., 2005; Wang et al., 2012b). Since the complement of ion channels expressed by a neuron determines its firing properties (Goaillard et al., 2009; Marder and Goaillard, 2006), and considering the complexity of neuronal expression profiles in, for example, the cerebral cortex (Tasic et al., 2016), this makes predicting the outcome of a given [K+]o change on neuronal circuit dynamics in-vivo a non-trivial task. When reviewing the literature, it is also important to remember that much of our current knowledge on how [K+]o affects individual neurons and small ensembles stems from experiments performed in-vitro. In the future, it will be important to determine how changes in [K+]o affect specific cell types in awake behaving mice, and how this is mediated by neural circuit activity.
2.2. Interstitial Ca2+ shapes neuronal activity and synaptic transmission
Ca2+ levels are tightly regulated in the brain, with [Ca2+]o typically ranging from 0.9–1.6 mM (Hansen, 1985; Somjen, 2004). During normal brain activity, changes in [Ca2+]o tend to be small; sensory stimulation, slow-wave sleep oscillations, sleep-wake transitions, and spinal locomotor-like activity can decrease [Ca2+]o in a range of 0.02–0.3 mM (Brocard et al., 2013; Ding et al., 2016; Dingledine and Somjen, 1981; Massimini and Amzica, 2001; Nicholson et al., 1978; Somjen, 1980) (Fig. 1B and Table 1). However, during strong electrical stimulation or in pathological states, such as epileptic seizures, cortical spreading depression or ischemia, [Ca2+]o can decrease by 0.4–1.0 mM to as low as 0.06 mM (Amzica et al., 2002; Heinemann et al., 1986, 1977; Kraig and Nicholson, 1978; Krnjević et al., 1982; Nicholson et al., 1978, 1977; Somjen, 1980; Zanotto and Heinemann, 1983) (Fig. 1B).
In turn, changes in [Ca2+]o can alter neuronal activity through bivalent excitatory and inhibitory mechanisms: Increases in [Ca2+]o generally dampen neuronal excitability while decreases usually excite neurons. Several explanations for this seemingly paradoxical effect have been suggested. First, many ion channels, including voltage-gated Na+, K+, and Ca2+ channels, contain exposed, negatively charged residues within their extracellular domains. These charges locally offset the membrane potential, resulting in a more depolarized effective potential across the channel (Fig. 3). Interactions of interstitial cations with these anionic residues neutralize the charge through surface charge screening (Frankenhaeuser and Hodgkin, 1957; Hille, 1978; Hille et al., 1975; Somjen, 2004). In this process, interstitial Ca2+ ions cluster alongside negatively charged residues on the extracellular domains of voltage-gated ion channels, thus increasing the effective polarization across that channel. This charge screening hyperpolarizes the effective membrane potential, and modulates the kinetics and activity of the ion channels (Madeja, 2000). As a result, decreasing [Ca2+]o can result in net depolarization of the membrane potential (Fig. 3). In addition, reductions in [Ca2+]o can activate none-selective cation channels located on the soma or axon terminals, causing depolarization (Smith et al., 2004; Xiong et al., 1997). Complementing these inhibitory effects, increases in [Ca2+]o can be excitatory. Intraaxonal Ca2+ influx through voltage-gated Ca2+ channels is required for docking and fusion of neurotransmitter-filled vesicles (Eggermann et al., 2012; Südhof, 2013). It is thus well known that [Ca2+]o modulates the probability of neurotransmitter release (Borst, 2010; Ohana and Sakmann, 1998). Within the physiological range of 0.8–1.2 mM, decreasing [Ca2+]o reduces the driving force for Ca2+ entry into the terminals, resulting in increased rates of synaptic failure, reduced neurotransmitter release, and weakened postsynaptic potentials (Balestrino et al., 1986; Dingledine and Somjen, 1981; Egelman and Montague, 1999; Kamiya and Zucker, 1994; Rusakov and Fine, 2003; Somjen and Müller, 2000) (Fig. 3).
Fig. 3. Lowering [Ca2+]o alters neuronal activity.

(A) Schematic diagram of a neural circuit characterized by an activity-dependent local decrease in [Ca2+]o. (B) Lowering [Ca2+]o can reduce the strength of synaptic signaling. As [Ca2+]o falls, the reduced driving force for Ca2+ influx through voltage-gated Ca2+ channels (Cav) leads to weakened presynaptic glutamate release, and, in extreme cases, action potential failure. Green arrow directions and widths reflect the direction and strength of the driving force for primarily Na+ influx; excitatory postsynaptic potential (EPSP) amplitudes are shown by schematic membrane potential traces. (C) The effective membrane potential across voltage-gated channels is partly governed by the high concentration of negatively charged residues on these channels’ interstitial domains. Under conditions of physiological [Ca2+]o (1.2 mM, left) the negatively-charged residues are largely neutralized by divalent cations (both Ca2+ and Mg2+). As a result, the local effective membrane potential is similar to the soma. Conversely, lowering the [Ca2+]o to, for example 0.6 mM as occurs during seizures, exposes the negative on the channels’ interstitial domains resulting in a local depolarizing the membrane and increasing neuronal activity through voltage-gated channels.
[Ca2+]o has profound effects on neuronal properties, including the membrane potential and synaptic transmission. However, it is worth pointing out here that much of our current knowledge on [Ca2+]o in the brain stems from studies with Ca2+-sensitive microelectrodes. While early studies proposed that only large-amplitude, synchronous neuronal activity can elicit changes in [Ca2+]o, more recent work has suggested that under normal conditions there may be transient (< 1 s) [Ca2+]o decreases which are spatially confined to the synaptic cleft (Borst and Sakmann, 1999; Cohen and Fields, 2004; Egelman and Montague, 1999; Vassilev et al., 1997). Ca2+-sensitive microelectrodes cannot track changes at this temporal or spatial resolution due to properties of the available Ca2+ ionophores. It should here be noted that Ca2+-sensitive microelectrodes have been produced with faster detection kinetics (Pumain et al., 1983) but, due to technical difficulties in production, this method was never widely used. Thus, as more sensitive and easy-to-use techniques for measuring [Ca2+]o emerge, we expect that a multitude of processes shaping local and global [Ca2+]o shall prove to be involved in regulating neuronal activity at all timescales.
2.3. Interstitial Mg2+ gates synaptic plasticity and sculpts neuronal activity
In the brain, [Mg2+]o is maintained at levels of 0.7–1.3 mM (Ames et al., 1964; Bradbury et al., 1968; Ding et al., 2016; Hansen, 1985; Sun et al., 2009). Despite the known importance of Mg2+ for normal brain function (Kirkland et al., 2018), it has yet to be sufficiently established whether changes in neural activity are directly related to changes in [Mg2+]o. It is, however, known that [Mg2+]o tracks circadian rhythms and changes in hibernation in a range of phylogenetically diverse organisms (Bijak, 1989; Feeney et al., 2016; Suomalainen, 1938). Elevation of brain Mg2+ leads to improved learning abilities, working memory, and short- and long-term memory in rats (Slutsky et al., 2010), all possibly mediated by the positive effect of Mg2+ on sleep quality (Nielsen and Johnson, 2010). Notably, it was recently shown that [Mg2+]o changes as a function of the sleep-wake cycle, with increases of 0.1–0.2 mM upon entering natural sleep or anesthesia (Ding et al., 2016). It is possible that, rather than having a simpler relation to local neuronal activity, [Mg2+]o changes on a much longer timescale, potentially regulated by blood-brain-barrier permeability changes across circadian rhythms and the sleep-wake cycle (Cuddapah et al., 2019; Ding et al., 2016; Feeney et al., 2016; Zhang et al., 2018).
[Mg2+]o has a broadly inhibitory impact on neuronal activity (Furukawa et al., 2009; Kelly et al., 1969; Smith et al., 1989). For example, when [Mg2+]o is lowered from 1.2 to 0.8 mM in-vitro, synaptic responses increase in the isolated spinal cord (Czeh and Somjen, 1989), while conversely the firing rate of cultured hippocampal neurons is drastically reduced by increasing [Mg2+]o from 0.8 to 3.8 mM (Penn et al., 2016). The most studied role of [Mg2+]o in the CNS concerns its voltage-dependent block of the NMDA receptor, restricting its opening to more depolarized potentials (Hsiao et al., 2002; Mayer et al., 1984; Nowak et al., 1984). Thus, a large body of work has shown that [Mg2+]o powerfully modulates the induction of long-term potentiation (LTP); increasing [Mg2+]o in artificial cerebrospinal fluid hampers the induction of LTP in hippocampal slices, and conversely, lower [Mg2+]o facilitates LTP (Calabresi et al., 1992; Dunwiddie and Lynch, 1979; Jung et al., 1990; Malenka et al., 1992; Malenka and Nicoll, 1993). The NMDA receptor is involved not only in LTP, but also in sensory feature selectivity through local dendritic events. Synaptic inputs sufficiently clustered in space and time can trigger NMDA receptor-dependent spikes within the dendrites (Branco and Häusser, 2011; Gambino et al., 2014; Losonczy and Magee, 2006; Schiller et al., 2000): This is thought to enhance feature selectivity and responsiveness (Lavzin et al., 2012; Palmer et al., 2014; Smith et al., 2013). Although the role of [Mg2+]o in generating dendritic spikes in-vivo has yet to be explored, we speculate that altered [Mg2+]o could regulate the activation of NMDA receptors (but see also Slutsky et al., 2010). Furthermore, [Mg2+]o inhibits the opening of voltage-gated ion channels through surface-charge screening (Frankenhaeuser and Hodgkin, 1957; Hille, 1978; Hille et al., 1975; Madeja, 2000; Somjen, 2004), as explained above for [Ca2+]o (Fig. 3C). However, while [Mg2+]o tends to generally dampen neuronal activity, its ability to inhibit hyperpolarizing K+ currents gives [Mg2+]o a more complex role (Terlau et al., 1996). While it is clear that [Mg2+]o is a powerful regulator of neuronal activity and synaptic transmission, whether dynamic changes in [Mg2+]o contribute to altered neural circuit function and brain state-dependent activity is largely unexplored. One reason for this is the lack of effective tools for measuring [Mg2+]o levels in-vivo. Mg2+-sensitive microelectrodes are far from selective (Ding et al., 2016) and neither chemical nor genetic Mg2+-sensitive fluorescent probes exist. If this technical limitation were overcome, many as yet unsolved questions about local and global dynamics of [Mg2+]o would undoubtedly be answered.
3. Interstitial ions are capable of regulating the firing regime of neurons
So far, we have summarized how interstitial ion concentrations change as a function of brain activity, and how individual ion species in turn modulate and shape neuronal activity in various ways. One of the purposes of this review is to explain how interstitial ions are involved in regulating brain state-dependent neuronal activity. Each brain state has characteristic patterns of neuronal network oscillations in many brain areas, which can be measured electrophysiologically by means of electroencephalography (EEG) (Berger, 1931) or the local field potential (LFP) (Buzsáki et al., 2012; Gervasoni et al., 2004). The frequency of these oscillations covers more than three orders of magnitude, from slow and intermediate oscillations in the delta (0.5–4 Hz), theta (3–8 Hz) and beta (13–30 Hz) ranges to fast oscillations in the gamma (30–90 Hz) and ultrafast (90–200 Hz) ranges (Bartos et al., 2007; Buzsaki and Draguhn, 2004; Uhlhaas and Singer, 2010). By obtaining EEG or LFP measurements, while humans or animals transition between different brain states, we now have a very well-defined characterization of how distinct neuronal oscillations change as a function of state. For example, the shift from sleep to awake is associated with a marked suppression of delta oscillations (Berger, 1931; Harris and Thiele, 2011; Kryger et al., 2017; Lee and Dan, 2012; Steriade et al., 2001), while gamma oscillations are prominently enhanced when animals transition from quiet to active wakefulness (Crochet and Petersen, 2006; Harris and Thiele, 2011; McGinley et al., 2015b; Poulet and Crochet, 2019; Poulet and Petersen, 2008), or during attentional processing (Gregoriou et al., 2015, 2014, 2009). Neuronal oscillations emerge from the concerted activity of ensembles of neurons, and numerous studies have established a strong link between such oscillations and the activity of individual neurons. For example, during periods of slow-wave sleep, regular burst firing is prevalent among cortical neurons, whereas during waking, the activity is generally dominated by irregular trains of action potentials (Domich et al., 1986; Evarts, 1964; Hirsch et al., 1983; Livingstone and Hubel, 1981; Steriade et al., 2001, 1993b, 1986). Hence, to understand how interstitial ions might be involved in regulating brain state-dependent neuronal activity and network oscillations, we need first to consider how ions regulate the firing pattern of individual neurons.
3.1. Neurons fire action potentials in two characteristic modes: Tonic and burst
Neurons in the thalamus (Hirsch et al., 1983; Jahnsen and Llinás, 1984; Livingstone and Hubel, 1981; Steriade et al., 1986) and cortex (Evarts, 1964; McCormick et al., 1985; Steriade et al., 2001, 1993a) exhibit tonic and burst firing. In tonic firing, the membrane potential returns to baseline between each of a series of action potentials without silent periods, whilst in burst firing, neurons fire multiple action potentials in rapid succession during a period of membrane potential depolarization, followed by a silent period (Murray Sherman, 2001). A neuron must possess depolarizing mechanisms capable of raising and holding the membrane potential at a lasting upstate in order to burst, i.e. it must be able to support multiple action potentials in rapid sequence. This depolarization must in turn enhance opposing hyperpolarizing mechanisms to pull the membrane potential back into the downstate. Lastly, there must be depolarizing mechanisms which are activated by this hyperpolarization, to drive the membrane potential out of the downstate and into the next upstate. The transition between tonic and burst firing is determined by the precise activation pattern of several types of ion channels (Kadala et al., 2015; Murray Sherman, 2001). Hence, shifting ion gradients due to changes in interstitial ions can powerfully regulate the firing mode of individual neurons.
3.2. Interstitial K+ regulates burst and tonic firing modes
A large body of evidence has documented the ability of [K+]o to regulate the firing of neurons. Increasing [K+]o above 7 mM in a wide range of preparations and experimental models is sufficient to drive the transition from tonic to burst firing (Bazhenov et al., 2004; Fröhlich et al., 2008; Jensen et al., 1994; Korn et al., 1987; Pan and Stringer, 1997; Rybak et al., 2003). However, many of these studies were performed in brain slices and focused on the role of [K+]o in epilepsy and seizure generation, which is associated with elevated [K+]o levels. A [K+]o of 7 mM certainly exceeds what is usually measured in-vivo under physiological conditions (Ding et al., 2016; Hansen, 1985; Monai et al., 2019; Rasmussen et al., 2019; Somjen, 2004) (Fig. 1B and Table 1). However, a number of experimental and theoretical studies have explored the effects of changing [K+]o within the normal physiological range. For example, when increasing [K+]o from 4 to 6 mM, a higher proportion of cells in a locomotor-related network of the isolated spinal cord of neonatal mice generated burst firing (Brocard et al., 2013). Corroborating this, theoretical modeling has shown that simulated neurons are in a tonic firing mode when [K+]o is 4.85 mM, occupy a bistable state with coexistent tonic and burst firing when [K+]o is between 5.45 and 6.35 mM, and stably burst fire when [K+]o exceeds 6.35 mM (Frohlich et al., 2006). Another modeling study explored the role of [K+]o at lower levels, similar to the concentrations measured invivo across the sleep-wake cycle (Ding et al., 2016). Here the authors found that when [K+]o was 3.9 mM, the simulated neuron was in a stable burst firing mode with highly rhythmic and repeatedly occurring up- and downstates, with a strong prevalence of delta oscillation in the membrane potential (Rasmussen et al., 2017). Increasing [K+]o to 4.4 mM slightly depolarized the membrane potential and elicited a chaotic activity state with burst firing periods occurring at irregular intervals and for variable durations, and this correlated with a marked suppression of delta oscillations. Further increasing [K+]o to 4.9 mM elicited a tonic membrane potential depolarization and transition to a stable tonic firing mode, associated with a further suppression of delta oscillations and a notable increase in gamma oscillations (Rasmussen et al., 2017) (Fig. 4). Finally, a recent study implicated lowered [K+]o levels in observations of enhanced burst firing in the lateral habenula (Cui et al., 2018): decreasing [K+]o caused hyperpolarization of the membrane potential, which in turn drove the transition from tonic to burst firing. Altogether, the impact of changes in [K+]o is evidently somewhat complex, and whether a given change in [K+]o associates with tonic or burst firing seems to depend on the absolute [K+]o. However, within the physiologically-relevant range of 2.7–5 mM, it appears that an increase in [K+]o is often associated with the emergence of tonic firing, whilst a decrease in [K+]o may be more likely to facilitate burst firing. This bivalent effect could be mediated by a direct membrane potential depolarizing effect and a reduction in outward K+ currents with increasing [K+]o levels (Fig. 2A). However, it should be emphasized that this conclusion is without doubt too simplistic, and future in-vivo studies are needed in order to determine the exact effect of [K+]o changes on the firing patterns in different neuronal cell types, in different layers, in different brain regions, and under different behavioral scenarios.
Fig. 4. Changing interstitial ion concentrations regulates the neuronal firing pattern.
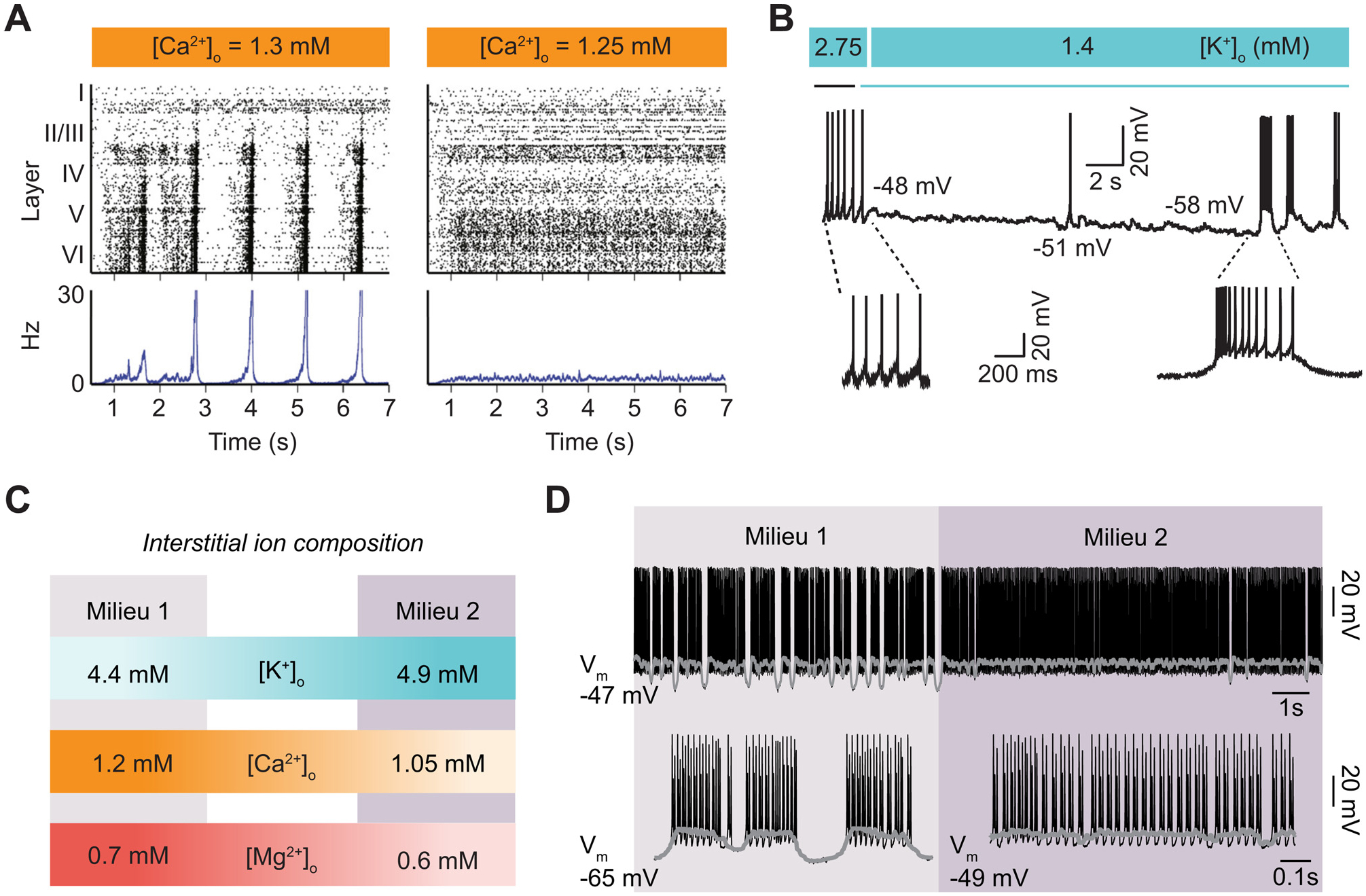
(A) Panels show raster plots (upper) and peristimulus time histogram (lower) of spontaneous activity for an in-silico neural network when [Ca2+]o is changed from 1.3 to 1.25 mM: When the [Ca2+]o is higher the neural activity is in synchrony and exhibits burst-like patterned activity, whereas when the [Ca2+]o is lower activity is asynchronous and neurons fire in a tonic-like pattern. The panel is reproduced based on Markram et al., 2015. (B) Recording showing how a lateral habenula neuron transform from tonic-firing to burst-firing mode when [K+]o is reduced from 2.75 mM to 1.4 mM. Panel is reproduced based on Cui et al., 2018. (C) Diagram depicting the effect on neuronal firing of two physiological interstitial ion milieus. Milieu 1: [K+]o =4.4 mM, [Ca2+]o =1.2 mM, and [Mg2+]o =0.7 mM. Milieu 2: [K+]o =4.9 mM, [Ca2+]o =1.05 mM, and [Mg2+]o =0.6 mM. (D) Representative membrane potential dynamics when a modeled neuron is exposed to the two interstitial ion milieus (top). Expansion of the membrane potential dynamics showing a clear shift from burst to tonic firing. Figure is reproduced based on Rasmussen et al., 2017.
3.3. Interstitial Ca2+ regulates burst and tonic firing
That [Ca2+]o regulates neuronal firing is well known to all electrophysiologists, as [Ca2+]o is routinely modified in experimental settings to suppress activity during brain slice preparation, or to enhance synaptic transmission and plasticity. Corroborating this concept, recent studies have explored the effects of changing [Ca2+]o on the firing of individual neurons and neuronal assemblies. Earlier work demonstrated that increasing [Ca2+]o from 1 to 5 mM caused thalamic neurons to switch from tonic to burst firing in-vitro (Formenti et al., 2001). More recently, decreasing [Ca2+]o from 1 to 0.1 mM in simulations elicited a pronounced change from regular burst firing, with rhythmic periods of up- and downstates, to chaotic firing with long periods of tonic firing interleaved with occasional downstates (Rasmussen et al., 2017). In the same work, decreasing [Ca2+]o from 1.2 to 1.05 mM, while also decreasing [Mg2+]o and increasing [K+]o, was sufficient to invoke the transition from burst to tonic firing (Fig. 4). Similarly, simulations using a reconstructed cortical microcircuit showed that lowering [Ca2+]o from 2 to 1.3 mM triggered the switch from burst-like to tonic-like firing, with a shift in the activity across neurons from synchronous to asynchronous (Markram et al., 2015); even slight reductions from 1.4 to 1.25 mM, which are well within the physiological range (Hansen, 1985; Somjen, 2004) (Fig. 1B and Table 1), triggered this transition (Markram et al., 2015). Comparable findings were recently obtained, showing that decreasing [Ca2+]o from 1.35 to 1.2 mM caused neuronal activity to transition from a state of regular, burst-like firing to a state of irregular, tonic-like firing, and this transition was further characterized by a shift from a supercritical to a subcritical state (Nolte et al., 2019). Collectively, these data seem to suggest that within the physiological range, higher [Ca2+]o associate with burst firing and periods of synchrony, while lower [Ca2+]o associate with tonic firing and desynchronous activity (Markram et al., 2015; Newton et al., 2019). However, it should be noted that there is experimental evidence for [Ca2+]o having opposing effects on tonic and burst firing: When [Ca2+]o was lowered from 1.2 to 0.9 mM, neurons in a locomotor-related network within the isolated spinal cord of neonatal mice showed increased proportions in the bursting mode (Brocard et al., 2013). It is still unknown whether this discrepancy reflects fundamental differences between how interstitial cations regulate neuronal firing in the telencephalon versus the spinal cord or reflects developmental differences or other factors. However, it appears reasonable to conclude that changes in [Ca2+]o over the range encountered in-vivo can have profound and causal effects on the firing pattern of neurons. These effects are likely brought about when decreases in [Ca2+]o cause membrane potential depolarization (Fig. 3C) along with reduced activity of Ca2+-activated outward K+ currents.
3.4. Interstitial Mg2+ may regulate burst and tonic firing modes
Although less studied than [K+]o and [Ca2+]o, there is evidence that [Mg2+]o can modulate the firing of neurons. Lowering [Mg2+]o in slice preparations is commonly used as a model to elicit and study epileptiform activity. Numerous studies have thus employed artificial cerebrospinal fluid containing 0 mM Mg2+ for triggering burst-like epileptiform activity (Golomb et al., 2006; Mody et al., 1987; Petersen et al., 2017; Robinson et al., 1993). However, in the brain, [Mg2+]o is usually within the range of 0.7–1.3 mM (Ames et al., 1964; Bradbury et al., 1968; Ding et al., 2016; Hansen, 1985; Sun et al., 2009). Unfortunately, few studies have explored the effects of more subtle changes in [Mg2+]o alone on the firing pattern of neurons. One modeling study testing the effect of changing [Mg2+]o from 0.7 to 0.6 mM reported no notable change in the firing mode of individual neurons (Rasmussen et al., 2017), although when this decrease was combined with increased [K+]o and decreased [Ca2+]o, it elicited a transition from burst to tonic firing (Fig. 4). However, this effect seemed to be more driven by the changes in [K+]o and [Ca2+]o rather than [Mg2+]o per se (Rasmussen et al., 2017). Another modeling study testing more extreme [Mg2+]o changes in the range of 0.5–3.5 mM showed that burst firing in dopaminergic neurons was stronger at high [Mg2+]o (Oster et al., 2015). In accord, work in cortical slices showed that increasing [Mg2+]o from 1.3 to 10 mM maintained spontaneously bursting neurons in the bursting state (Wenger Combremont et al., 2016), suggesting that [Mg2+]o in this range may indeed support the emergence of burst firing. Together, it seems clear that altering [Mg2+]o can influence the firing of neurons. While a large body of evidence suggests that pathological epileptiform activity can be elicited by removing or dramatically reducing [Mg2+]o from the interstitial milieu, it is not yet sufficiently established if and how more subtle and physiologically relevant changes in interstitial [Mg2+]o may contribute to the firing mode of neurons. Although the currently available, albeit sparse, data generally seem to suggest that increasing [Mg2+]o supports burst firing, further studies are needed to clarify this issue. This could be driven by the membrane potential hyperpolarizing effect of increasing [Mg2+]o due to factors such as surface-charge screening and reduced NMDA receptor activation, similar to [Ca2+]o (Fig. 3). Alternately, [Mg2+]o within the normal physiological range might be more involved in stabilizing overall neuronal activity levels and gating long-term synaptic plasticity, rather than regulating the temporal firing pattern of individual neurons.
3.5. Interstitial pH changes as a function of neuronal activity and state
Outside of the principle interstitial ions, other important factors to consider is interstitial pH (pHo) and interstitial space volume. In the brain, pHo is regulated by a combination of transmembrane solute fluxes and chemical buffering in the interstitium (for an in-depth review of brain pH see Chessler 2003). Homeostatic mechanisms tightly regulate pHo, maintaining a proton (H+) concentration equivalent to a mean pH of 7.3 in brain interstitial fluids (Cragg et al., 1977; Javaheri et al., 1983; Kraig et al., 1983), thus roughly 0.1–0.2 pH units below arterial levels. This environment is principally regulated by the blood-brain-barrier being impermeable to metabolic acids and bases, while remaining permeable to lipophilic CO2 gas, which is in equilibrium with carbonic acid. As such, pHo is tightly correlated with respiration, meaning that increased breathing rates (i.e. hyperventilation) reduce blood CO2 levels and consequently tend to alkalinize neural tissue (Meyer and Gotoh, 1960). This works so well that hyperventilation is routinely used during EEG recordings to screen for seizure disorders (Seneviratne et al., 2017): Hyperventilation evoked absence seizures in 67% of patients in a pediatric cohort of children suffering absence seizures (Dalby, 1969), although triggering interictal and ictal events by hyperventilation is less successful in adult cohorts and in other seizure disorders (Holmes et al., 2004). Certain diseases, such as metabolic acidosis, may lead to compensatory increases in respiration and paradoxical alkalinization in the brain (Posner and Plum, 1967). Similarly, metabolic demand alters pHo, with levels decreasing by 0.1–0.2 during sensory stimulation, and as much as 0.3 during periods of high (8–12 mM) [K+]o (Kraig et al., 1983) and seizure-like activity (Blennow et al., 1985) (Fig. 1B). Under the substantial metabolic stress of cortical spreading depression and ischemia, pHo is reported to fall to as low as 6.6 (Mutch and Hansen, 1984), while hyperglycemic loading causes decreases down to 5.8–6.1 through the production of lactic acid (Siemkowicz and Hansen, 1981). Following pharmacological, electrical, or physiological sensory stimulation, rapid interstitial alkalosis is followed by long-lasting acidosis. These shifts are driven by changes in transmembrane acid/base currents, with alkalinizing fluxes being linked to enhanced activity of the plasma membrane Ca2+/H+-ATPase, HCO3− exchanger, channel-mediate H+ flux, and glutamate uptake (Chessler 2003). These early alkalinizing currents are ‘muffled’ by acid transport through glial Na+/HCO3− exchangers (Grichtchenko and Chesler, 1994a, 1994b) and co-release from synaptic vesicles (DeVries, 2001; Zhang et al., 2010), while long-term acidic shifts may occur through changes in classic mechanisms including the Na+/H+ co-transporter or metabolic shifts including accumulation of CO2 (Voipio and Kaila, 1993) and release of lactate (Schurr et al., 1999). How these mechanisms operate across microdomain to regional volumes remains an area of active research.
Generally, interstitial acidification reduces neuronal activity in the brain while alkalization tends to increase it (Chesler, 2003; Somjen and Tombaugh, 1998) because H+ inhibits currents through voltage- and ligand-gated ion channels (Taira et al., 1993; Traynelis and Cull-Candy, 1990). While voltage-gated K+ and Na+ channels are also inhibited by decreasing pH, the predominant effect of pHo on presynaptic terminals occurs through voltage-gated Ca2+ channels: Assuming a baseline voltage-clamp measured conductance of 100% at a pHo of 7.4, decreasing the pHo to 6.5 decreases conductance to 40%, while alkalinizing pHo to 8 increases the conductance to 120% (Tombaugh and Somjen, 1996). Similarly, ligand-gated ion channels are sensitive to changes in pHo, with decreases driving reduced NMDA receptor currents (Traynelis and Cull-Candy, 1990) while simultaneously increasing GABAergic currents (Pasternack et al., 1996; Robello et al., 1994); decreases in pHo are thus capable of decreasing the excitation:inhibition ratio. The net result is that decreasing pHo within the range of sensory stimulation can reduce postsynaptic population spikes by as much as 30–50% (Balestrino and Somjen, 1988). Intracellular pH in neurons tracks that of the interstitial space during activity, while astrocytes exhibit strikingly different behavior. Starting from a resting pH of 0.1–0.2 below that of neurons (Rose and Ransom, 1997), astrocytes alkalinize during activity (Chesler and Kraig, 1987), potentially through the electrogenic Na+/HCO3− exchanger NBCe1 (Bevensee et al., 1997; Raimondo et al., 2016). By promoting glycolysis (Ruminot et al., 2011), maintaining intracellular Na+ levels, and providing feedback acidification of the interstitial space, astrocytes dampen neuronal activity. The finding that pHo decreases from 7.4 in anesthesia to 7.25 during quiet wakefulness (Ding et al., 2016), suggests that pHo may be regionally regulated in a brain state-dependent manner, and raises the possibility that astrocytes play a key role in regulating the interrelations between K+ buffering, pH, and brain metabolism to sculpt local neuronal activity.
3.6. The interstitial space volume
Astrocytes are key to buffering activity-dependent rises in [K+]o. A less appreciated aspect of interstitial ion concentration changes is the effect on astrocytic volume (Hertz, 1965; Kuffler et al., 1966; Orkand et al., 1966). Astrocytes swell as they buffer and redistribute K+ through the glial syncytium. This swelling reduces the interstitial space volume by up to 5% during slow, 10 Hz electrical stimulation (Syková, 2003), 20% during stimulation of the optic nerve (Ransom et al., 1985), 30–50% during epileptiform activity and early hypoxia (Dietzel et al., 1980; Syková et al., 1994), and by 80% in ischemia (Syková et al., 1994) (Fig. 1B). This activity-dependent decrease in interstitial space directly parallels that of [K+]o in a manner reflecting local efflux, and swelling of synaptic terminals and glial cells (Dietzel et al., 1980). In hypoosmotic states and ischemia these changes in interstitial space volume are accompanied by significant shifts in the tortuosity of the interstitial space (Sykova et al., 1994 and Sykova and Nicholson 2008). However, interstitial space volume may change without concurrent changes in tortuosity, as seen in a pilocarpine seizure model in rat somatosensory cortex (Slais et al., 2008), as well as in work demonstrating an ~60% increase in interstitial space volume, from 13% to 21% of total brain volume, upon transitioning from awake into sleep (Xie et al., 2013). This suggests that substantial changes in interstitial space volume, but not tortuosity, may be actively regulated in healthy physiology in a brain state-dependent fashion.
What effect do changes in interstitial space volume have on neuronal activity? At the fundamental level, decreased interstitial space will favor increased uptake of glutamate, enhanced ionic regulation, and more rapid clearance of [K+]o from the synaptic cleft, simply due to reduced ion flux (Nagelhus and Ottersen, 2013; Syková and Nicholson, 2008). Beyond this simple geometric effect, the volume of the interstitial space can play a substantial role in regulating diffusion of small molecules, compartmentalizing interstitial changes, and increasing local signaling (Nicholson and Phillips, 1981). Shrinkage of the interstitial space can impact the spillover of ions and neurotransmitters from nearby synapses, properly known as volume transmission. This may play a significant role in the decorrelation of neuronal activity seen in active wakefulness, and in the enhanced synchrony observed in slow-wave sleep, though this has yet to be experimentally confirmed. Intriguingly, the volume of interstitial space may be able to regulate neuronal activity in the absence of neurotransmitters in a process known as ephaptic transmission. Ephaptic transmission is the term for excitability transmitted between adjacent neurons which is mediated by local electric fields. Early work found that ephaptic transmission could drive synchronous neuronal activity even in the absence of synaptic activity (Taylor and Dudek, 1982). Within the last decade, this non-classical mode of neurotransmission has been found to regulate phase-locking in cortical neurons (Anastassiou et al., 2011; Anastassiou and Koch, 2015), drive slow periodic rhythms of the hippocampus (Chiang et al., 2019), and enable synchronization of adjacent Purkinje cells in the cerebellum (Han et al., 2018). As norepinephrine is capable of altering the interstitial space volume between sleep and the awake state (Xie et al., 2013), active regulation of the interstitial space may play a substantial role in governing neuronal activity and transmission during different states.
4. Coordinated interstitial ion changes and global brain states
As discussed in Section 3, the patterned, electrical behavior of individual neurons is reflected in the neuronal oscillations measured across neuronal assemblies, ultimately determining the prevailing brain state. However, the internal brain state is constantly fluctuating along a continuum, even in the absence of overt behavioral changes (Harris and Thiele, 2011; Lee and Dan, 2012; McGinley et al., 2015b; Poulet and Crochet, 2019): From a synchronized state characterized by strong, low-frequency delta oscillations to a desynchronized state where these are suppressed and high-frequency gamma oscillations become more pronounced. In turn, the brain state profoundly influences the operating mode of the brain by virtue of changes in, for example, spontaneous neuronal activity, the ability to execute motor commands, and responses to external sensory stimuli (Cao and Händel, 2019; Destexhe et al., 1999; Livingstone and Hubel, 1981; Massimini et al., 2005; Polack et al., 2013).
The best-characterized brain state transition is undoubtedly the transition between sleep and wakefulness, and these states can easily be distinguished by measuring the EEG or LFP. During sleep, neuronal activity is synchronized and dominated by slow 0.5–1 Hz delta oscillations, while during wakefulness activity generally becomes desynchronized and delta oscillations are suppressed (Berger, 1931; Harris and Thiele, 2011; Kryger et al., 2017; Lee and Dan, 2012). The function of the highly synchronized neuronal oscillations during sleep is a matter of active research. One idea that has gained traction in recent years is that sleep is critical for learning and memory (Diekelmann and Born, 2010; Maquet, 2001; Stickgold, 2005). In one study, using transcranial stimulation to increase delta oscillations during sleep in human subjects enhanced the retention of declarative memory: This indicates that these slow oscillations contribute to memory consolidation (Marshall et al., 2006). Another idea is that during sleep and periods of synchronized, slow neuronal oscillations, the brain is cleared from toxic waste products via a macroscopic clearance system - the glymphatic system - formed by astrocytes (Iliff et al., 2012; Xie et al., 2013). Brain state not only varies between sleep and awake, but recent experiments have indicated a more complex picture, in which cortical state also varies within wakefulness (Harris and Thiele, 2011; McGinley et al., 2015b; Petersen, 2019; Poulet and Crochet, 2019). As such, it is increasingly acknowledged that the awake state is comprised of at least two sub-states, namely quiet awake and active awake, related to the level of arousal and motor behavior (McGinley et al., 2015b; Poulet and Crochet, 2019). Thus, when animals transition from quiet to active wakefulness, this correlates with suppression of delta oscillations and a concomitant enhancement of gamma oscillations (Niell and Stryker, 2010; Rasmussen et al., 2019; Reimer et al., 2014; Vinck et al., 2015). The state-dependent enhancement of gamma oscillations has received particular attention, because their relationship to higher brain function is an area of intense, active research (for in-depth reviews on gamma oscillations see Bartos et al., 2007; Buzsaki and Draguhn, 2004; Buzsáki and Wang, 2012; Fries, 2009). It has been suggested that gamma oscillations are involved in attention (Engel et al., 2001; Fries, 2009), temporal encoding (Hopfield, 1995), sensory binding (Gray and Singer, 1989), and storage and recall of memories (Lisman and Idiart, 1995). Conversely, disruption of gamma oscillations is thought to underlie some psychiatric disorders, such as schizophrenia (Lewis et al., 2005; Uhlhaas and Singer, 2010).
Despite substantial progress, our knowledge about the mechanisms mediating brain state transitions and state-dependent neuronal activity remains incomplete. Given the global nature of brain states, mechanisms acting over longer timescales and on a brain-wide spatial scale could likely be involved. The interstitial ionic environment, which bathes neurons and glial cells alike, is capable of influencing not only individual neurons, but also large assemblies of neurons. Central to these state changes are widespread networks of neuromodulatory transmitters. While these systems are well known to have direct effects on the activation-specific receptors necessary for eliciting gamma oscillations and other neuronal rhythms, substantial evidence from peripheral tissue as well as the brain suggests that these modulators can directly impact the activity and expression of the Na+, K+-ATPase. This relationship (reviewed in Therien and Blostein, 2000), generally supports increased Na+, K+-ATPase activity following application of neuromodulators through protein kinase A-dependent activation and protein kinase C inactivation (Bertorello et al., 1991; de Lores Arnaiz and Ordieres, 2014). Though, work have shown a differential effect of norepinephrine on the activity of the Na+, K+-ATPase in neurons and glial cells; with pump activity increasing and decreasing in neurons and glial cells, respectively (Baskey et al., 2009). In the brain the interplay of norepinephrine and serotonin in regulating the Na+, K+-ATPase is well described as being dependent upon the glial Na+, K+-ATPase (Hernández-R., 1992; Peña-Rangel et al., 1999). This modulation has complex effects, with very high crosstalk between neuromodulatory systems and Na+, K+-ATPase activity: For instance, the Na+, K+-AT-Pase in the C. elegans neuromuscular junction is essential to the formation of the nicotinic acetylcholine receptor dense synapses necessary for proper development (Doi and Iwasaki, 2008). In this way, neuromodulators have complex, cell- and tissue-specific effects on homeostatic regulation of interstitial ions.
Strikingly, this effect has been known for decades, when it was demonstrated that release of neuromodulatory transmitters through direct stimulation of the mesencephalic reticular formation - an area containing several neuromodulator nuclei and intimately associated with brain state - initiates a 1.5–2 mM increase in [K+]o across all cortical layers of the cat primary visual cortex (Singer et al., 1976). A similar 0.4 mM increase is seen during the application of a cocktail of neuromodulators in the presence of tetrodotoxin (Ding et al., 2016) and is mirrored in cholinergically-induced gamma oscillations (Fisahn et al., 1998; Huchzermeyer et al., 2008; Kann et al., 2011) and serotonergically-induced locomotor rhythms (Table 1; Brocard et al., 2013). Emerging recent work has further implicated interstitial ion changes in driving the transition between different well-defined brain states, including the transition from sleep to wake (Ding et al., 2016; Rasmussen et al., 2017) and from quiet to active wakefulness (Rasmussen et al., 2017). These studies suggest that these neuromodulators have widespread facilitatory and inhibitory effects on neuronal processing beyond direct activation of neuronal activity through individual cells. To understand this role, we will discuss ion homeostasis and neuromodulation in these sleep-wake and within-wakefulness state transitions, and potential mechanisms for these shifts.
4.1. The composition of interstitial ions controls the sleep-wake cycle
If interstitial ions are indeed causally driving the transition from sleep to awake, the concentrations of such ions would have to be changing in predictable ways as a function of the sleep-wake cycle. Measuring [K+]o, [Ca2+]o, and [Mg2+]o with ion-sensitive electrodes in the cortex when mice naturally fall asleep or awaken revealed that these ion species change notably around the time of waking up: [K+]o increased by 0.4 mM, while [Ca2+]o and [Mg2+]o decreased by 0.13 and 0.11 mM, respectively (Ding et al., 2016) (Fig. 5A). These findings were confirmed by microdialysis, suggesting that the observed interstitial ion changes are indeed global in nature. Interestingly, when the authors artificially imposed the interstitial ion concentrations seen in either sleeping or waking, the LFP activity shifted toward that state: Mimicking sleep ion concentrations caused a marked increase in 1–4 Hz delta oscillations, while mimicking awake ion concentrations produced the opposite effect, regardless of the actual behavioral state of the animal (Fig. 5B). Furthermore, changing interstitial ion concentrations drove reversible transitions between periods of sleep and awake behavioral states, as determined by the absence and presence of muscle tone, respectively (Fig. 5B). These findings were later implemented as constraints for detailed theoretical modeling simulations, testing the contribution of interstitial ions for the transition from sleep-like to awake-like activity in individual neurons (Rasmussen et al., 2017). These simulations showed that resetting interstitial ion concentrations to those measured during wakefulness notably reduced the threshold for triggering the transition from sleep to wakefulness, by playing a permissive role in that transition. Collectively, these data show that the interstitial ion changes measured during the sleep-wake cycle are indeed capable of causally regulating neuronal delta oscillations and the overall behavioral state of animals, suggesting that interstitial ions play an integral part in the transition from sleep to awake.
Fig. 5. Interstitial ion concentration changes causally determine global brain states.
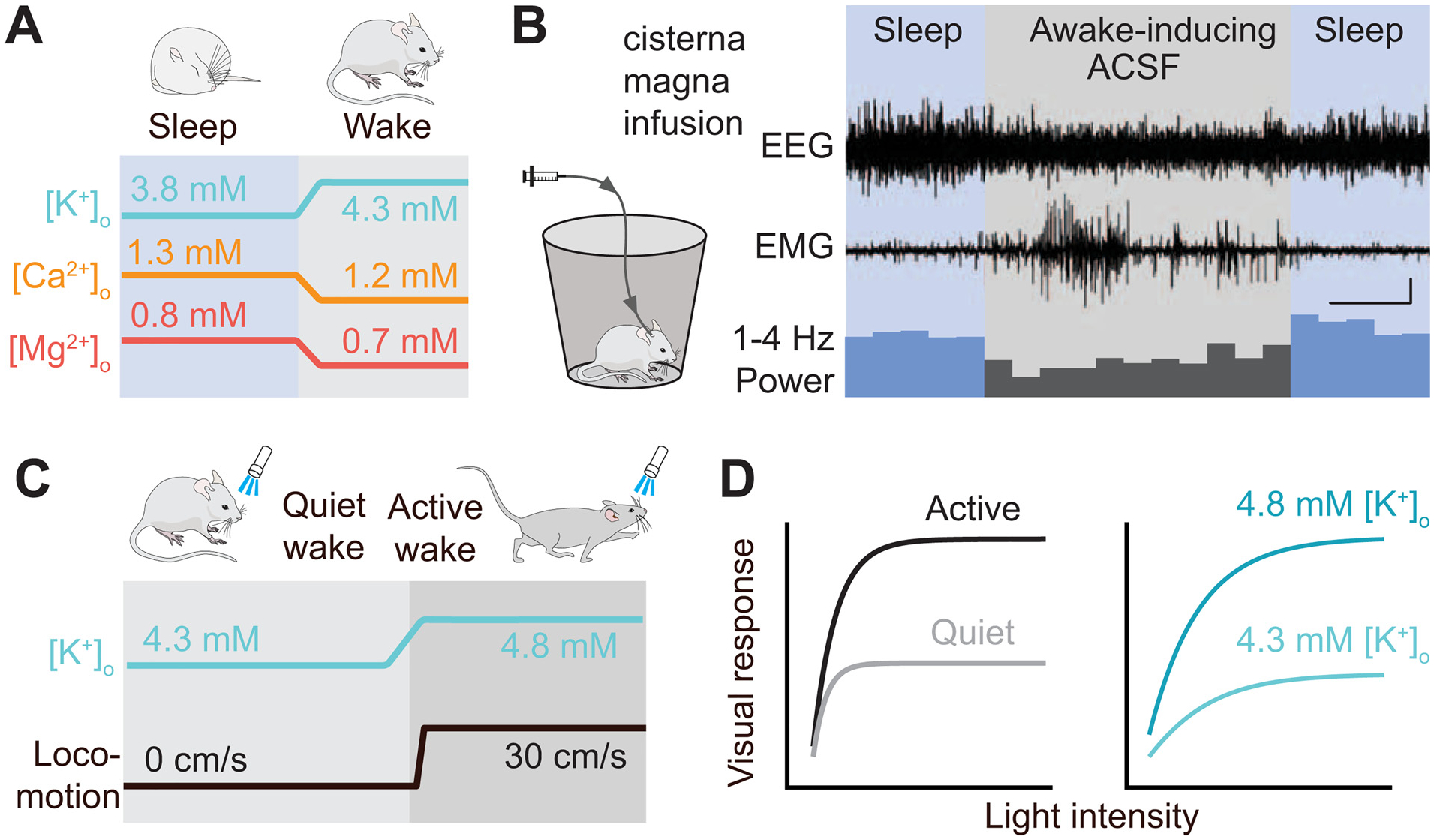
(A) Upon the transition from sleep to wakefulness [K+]o, [Ca2+]o, and [Mg2+]o show robust, concerted changes. (B) Globally changing the interstitial ion concentrations by intracisternal infusion of an “awake-inducing artificial CSF solution” (containing elevated [K+], and lowered [Ca2+] and [Mg2+]) is sufficient to transition both the electroencephalogram (EEG) and the behavioral state (electromyogram [EMG]) recordings from sleep to wakefulness patterns. Scale bar, x =30 min, y =0.5 mV. Panels (A) and (B) are reproduced based on Ding et al., 2016. (C) Preceding the behavioral state change from quiet to active wakefulness, [K+]o increases globally across the cortex. (D) During the active waking state, responses to visual stimuli are multiplicatively gained (left); this phenomenon can be fully reproduced by increasing [K+]o locally in quietly awake animals (right). Panels (C) and (D) are reproduced based on Rasmussen et al., 2019.
How do these concerted interstitial ion changes produce the stereotypical changes in neuronal delta oscillations characteristic of the sleep-wake cycle? Although this question has not yet been experimentally addressed in-vivo, we propose that the following mechanisms might be at play: Increasing [K+]o tonically depolarizes the membrane potential of neurons, thereby reducing the activation of T-type Ca2+ channels, while also reducing the driving force for Ca2+-activated K+ currents, known to be involved in sleep duration (Tatsuki et al., 2016). In this scenario, the decreasing [Ca2+]o and [Mg2+]o further depolarizes the membrane by reducing surface charge screening, while also decreasing T-type Ca2+ channel and Ca2+-activated K+ channel activations and increasing NMDA receptor activation. These combined effects are likely necessary (if not sufficient) to drive the transition from synchronous burst firing to asynchronous tonic firing of individual neurons, which in turn gives rise to the reduction in delta oscillations measured in the EEG or LFP from sleep to awake.
4.2. Changes in interstitial K+ regulate neuronal activity in awake states
As noted above, it is increasingly recognized that within the condition of wakefulness the brain transitions between at least two well-defined sub-states: Quiet and active wakefulness, and these associate with a decrease in delta and increase in gamma oscillations. Yet, the mechanisms contributing to state-dependent neuronal activity within the awake brain are incompletely understood. Work performed in hippocampal brain slices has shown that increasing [K+]o by 0.5–2 mM is sufficient for evoking short episodes of gamma oscillations (LeBeau et al., 2002), inviting the possibility that dynamic [K+]o changes could be involved in mediating the change between awake states. This work was complimented by theoretical modeling showing that concerted, subtle changes in interstitial ions could suffice to drive the transitioning of individual neurons from an activity state resembling quiet wakefulness to a state similar to that observed in active wakefulness (Rasmussen et al., 2017) (Fig. 4C, D): When increasing [K+]o by 0.5 mM and decreasing [Ca2+]o and [Mg2+]o by 0.15 and 0.1 mM, respectively, the authors documented a marked decrease in delta oscillations and a concomitant prominent increase in gamma oscillations, and this effect was primarily driven by the increase in [K+]o (Rasmussen et al., 2017). These works show that changes in interstitial ions, and especially [K+]o, are capable of evoking neuronal activity changes similar to those seen when animals transition from the quiet to the active wakefulness state, including the enhancement of gamma oscillations. It is worth noting that the work by LeBeau et al. (2002) used a 0.5–2 mM [K+]o increase to elicit gamma oscillations, while the work of Rasmussen et al. (2017) used only 0.5 mM. This slight discrepancy is likely explained, at least in part, by differences in baseline [K+]o together with different levels of [Ca2+]o and [Mg2+]o and varying levels of neuromodulator, but the different methods do not ultimately affect the proposal that increases in [K+]o could regulate awake state-dependent neuronal activity.
This theory was very recently tested experimentally by measuring [K+]o in multiple cortical areas and layers when mice spontaneously transitioned between periods of locomotion and quiescence (Rasmussen et al., 2019). This work demonstrated that [K+]o increased by 0.6–1 mM in the transition from quiet to active, and that this event occurred approximately 1 s before the behavioral state change (Fig. 5C). Similar to observations during the sleep-wake cycle, this rapid [K+]o increase was observed across cortical areas, including the motor, sensory, and visual cortices, suggesting a global nature. An increase in [K+]o of this magnitude caused a tonic membrane potential depolarization of 4 mV, i.e. decreasing from −70 to −66 mV (Rasmussen et al., 2019). This effect could as such explain the persistent 2–4 mV membrane potential depolarization and the decline of downstate episodes observed in cortical neurons during locomotion (Bennett et al., 2013; McGinley et al., 2015a; Polack et al., 2013; Reimer et al., 2014; Schiemann et al., 2015). Unfortunately, the authors of this study did not report on the relationship between the increase in [K+]o and the prevalence of gamma oscillations in their LFP recordings. This made it impossible to determine if the [K+]o increase was involved in regulating gamma oscillations during the transition from quiet to active, although the magnitude of the [K+]o increase indicates that it is possible (LeBeau et al., 2002; Rasmussen et al., 2017).
The active cortical state seen during periods of locomotion has a substantial functional impact, as it is accompanied by marked changes in sensory processing (Castro-Alamancos, 2004; Crochet and Petersen, 2006; Otazu et al., 2009). For example, visually-evoked responses in the primary visual cortex are multiplicatively gained when animals are locomoting (Bennett et al., 2013; Dadarlat and Stryker, 2017; Neske and McCormick, 2018; Niell and Stryker, 2010; Polack et al., 2013) (Fig. 4D), which has been functionally linked to improved visual detection (Bennett et al., 2013). Yet our knowledge about the mechanisms mediating state-dependent modulation of sensory processing is incomplete. If the state-dependent increase in [K+]o contributes to this, it would be expected that selectively increasing the [K+]o alone, in the absence of the behavioral state change, should replicate, at least in part, the sensory gain modulation observed when animals shift from quiet to active. Interestingly, when Rasmussen et al. (2019) locally imposed a [K+]o increase of ~0.6 mM in the visual cortex of stationary mice by changing the ionic content of the artificial cerebrospinal fluid bathing the cranial window, they observed a multiplicative gain modulation of visually-evoked responses that was strikingly similar to that seen during locomotion (Fig. 5D). This result suggests that the [K+]o increase observed during locomotion is able to modulate visual sensory processing in awake animals by producing gain modulation within the visual cortex. How might tonic changes in [K+]o produce multiplicative gain modulation in the awake cortex? In-vivo, there is considerable variance in the membrane potential of neurons on a timescale of seconds. Consequently, the relationship between the membrane potential and firing rate in response to a sensory input follows a power law (Anderson et al., 2000; Haider and Mccormick, 2009; Miller and Troyer, 2002; Murphy and Miller, 2003). This power law relationship means that tonic depolarization can produce multiplicative gain modulation (Murphy and Miller, 2003). Although tonic depolarization is traditionally thought to be driven by changes in excitation and inhibition, we here propose an additional mechanism whereby the reliable [K+]o increase during periods of active wakefulness can depolarize cortical neurons and thus modulate their input-output properties in a brain state-dependent manner. In future experiments, the potential causality between the tonic depolarization observed during active wakefulness and the concomitant increase in [K+]o should be tested. In addition, it will be important to determine if the awake state-dependent increase in [K+]o is causally involved in regulating the notable emergence of gamma oscillations, seen during periods of locomotion and increased arousal. Such insights could open a whole new avenue for studying and implicating dynamic interstitial ion changes in higher cognitive functions, such as attention, temporal coding, sensory binding, and memory storage. Finally, only [K+]o has hitherto been explored during different waking states; it may well be found that [Ca2+]o and [Mg2+]o also undergo dynamic changes that accompany the frequent transitions between quiet and active wakefulness in awake, behaving animals. This is a critical question, as it may point to concerted interstitial ion changes being a universal control mechanism across the spectrum of brain states, ranging from sleep to active wakefulness.
4.3. A central role of astrocytes in state-dependent control of interstitial K+
The concerted change in interstitial ion concentrations during state transitions relies on the stable regulation of homeostatic set-points for these ions. The principle mechanism for maintaining ion homeostasis in the brain is the Na+, K+-ATPase. This electrogenic transporter extrudes 3 Na+ ions for every 2 K+ brought into the cell, helping to set the membrane potential, and providing the electromotive driving force for many other ion channels. In the brain, the expression of specific subunit isoforms helps to maintain homeostasis throughout activity: Synaptic expression of the voltage-sensitive α3 subunit in neurons helps to maintain intracellular Na+ concentration during periods of sustained activity; and the [K+]o-sensitive α2 subunit, selectively expressed in astrocytes, prevents excess accumulation of [K+]o during activity (Larsen et al., 2016, 2014). During periods of increased neuronal activity, as seen for example in gamma oscillation, repeated action potentials drive high-levels of Na+, K+-ATPase activity to restore neuronal and astrocytic membrane potentials. This activity requires an enormous amount of energy (Kann et al., 2016), with neurons over time potentially consuming the majority of energy used by all cells in the brain (Attwell and Iadecola, 2002). As maintaining metabolic homeostasis and effective maintenance of interstitial ions to permit recovery within neurons is critical to brain function, the substantial metabolic demand imposed by neuronal activity is met by the combined action of neurons and astrocytes in regulating the flow of metabolites within the brain (Clasadonte et al., 2017).
Beginning with work in the 1960s (Kuffler et al., 1966; Orkand et al., 1966), astrocytes have long been recognized as the primary cell type responsible for buffering activity-dependent shifts in [K+]o (Verkhratsky and Nedergaard, 2018; Walz, 2000). This function has been attributed to high astrocytic K+ conductivity, driven by the Na+, K+-ATPase and the inward rectifying K+ channel, Kir4.1, as well as the ability of these cells to redistribute ions and solutes through connexin 30 and 43 (Cx30, Cx43)-mediated coupling with adjacent astrocytes. With widespread expression and a reversal potential near −80 mV, the Kir4.1 channel is the primary channel responsible for the highly hyperpolarized membrane potential, as well as the low input resistance, of astrocytes. This effect drives a highly-responsive, largely passive, quasinernstian membrane potential in astrocytes that mirrors slow cortical oscillations (Amzica and Massimini, 2002). While a consensus has yet to be reached for the precise role of Kir4.1 in regulating interstitial K+ - with direct blocking by Ba2+ injection showing little effect on activity-dependent [K+]o uptake (D’Ambrosio et al., 2002), and conditional knockouts showing an effect (Chever et al., 2010) - these studies also found differences in post-activity, Na+, K+-ATPase-dependent undershoot of [K+]o, and increased baseline [K+]o. This suggests that the precise permeability of astrocytes to K+ helps to govern the temporal dynamics of activity-dependent [K+]o increases and the compartmentalization of brain K+ between interstitial and intra-astrocytic space.
Coupled to their high permeability and sensitivity to [K+]o, astrocytes also exhibit high levels of interconnectivity through gap junctions. In early work, these connections were studied from the perspective of ‘spatial buffering’ of K+ during periods of high activity as gap junction formation is regulated by intracellular pH (Ransom and Sontheimer, 1992) and Ca2+ in a membrane potential-dependent manner (Enkvist and McCarthy, 2002). Networks arising from astrocytic gap junctions may provide a role in diffusive K+ coupling, as disruption of these channels impairs [K+]o clearance and volume regulation (Amzica et al., 2002; Pannasch et al., 2011); these networks may also provide potential routes for metabolic support and neuroglial coupling (Clasadonte et al., 2017) at the interface of multiple ionic pathways (reviewed in Charvériat et al., 2017; Hertz et al., 2013; Petit and Magistretti, 2016). As such, the mechanisms governing the compartmentalization of solutes in the brain is highly dependent on glial permeability and connectivity - providing instantaneous ‘passive’ responses to changes in interstitial ions, metabolites, and pH, while fluidly altering the baseline expression and activity of these responses through neuromodulator-dependent changes in the activity and expression of these same pumps and channels.
The disruption or mutation of astrocytic proteins (Kir4.1, α2-Na+, K+-ATPase, and Cx30/43) have been associated with changes in acetylcholine-dependent exploratory behavior (Dere et al., 2003; Frisch et al., 2003), depression (Ernst et al., 2011), and disrupted paired-pulse facilitation (Pannasch et al., 2011). The important question then becomes: How might astrocytes maintain a stable [K+]o of ~3.8 mM during sleep, 4.3 mM during quiet wakefulness, and 4.8 mM during locomotion (Fig. 4)? Most likely, the state-dependent astrocytic K+ buffering is controlled by arousal-related neuromodulators, including norepinephrine, serotonin, acetylcholine and orexin. Norepinephrine is perhaps the most important mediator of arousal and wakefulness: Optogenetic stimulation of locus coeruleus neurons triggers a frequency-dependent increase in cortical activity, sleep-to-wake transitions, and general locomotor arousal; these neuromodulators are also centrally implicated in the sleep-wake cycle (Lee and Dan, 2012). In light of this, we therefore propose that, in addition to their direct effects on neurons, a key role of neuromodulators is through the regulation of interstitial ion levels via astrocytes (Fig. 6). This proposal is supported by in-vitro experiments showing that applying a cocktail of neuromodulators to electrically-silenced cortical slices results in an increase in [K+]o of approximately 0.4 mM (Ding et al., 2016). Other lines of work show that G-protein coupled receptor-mediated activation of astrocytes boost K+ uptake by a Ca2+-dependent mechanism (Wang et al., 2012a). Astrocytes express an abundance of neuromodulator receptors, including Gq-linked α1-adrenergic receptors (Ding et al., 2013). Similar lines of evidence show that astrocytic intracellular Ca2+ signaling is intimately linked with arousal: Ca2+ activity in astrocytes is suppressed by anesthesia (Thrane et al., 2012) and activated during wakefulness, when compared with the conditions of non-rapid eye movement and rapid-eye movement sleep (Bojarskaite et al., 2019; Thrane et al., 2012). Furthermore, arousal and locomotion evoke cortical-wide increases in astrocytic Ca2+, but these global Ca2+ increases, as well as baseline Ca2+ activity, are blocked by the α1-adrenergic receptor antagonist prazosin (Ding et al., 2013; Paukert et al., 2014; Slezak et al., 2019; Srinivasan et al., 2015). The cortex-wide increase in astrocytic Ca2+ likely reflects the widespread release of norepinephrine from locus coeruleus projections during arousal (Bellesi et al., 2016). The global pattern of α1-adrenergic receptor-mediated astrocytic Ca2+ signaling matches the cortex-wide changes in [K+]o observed when mice transition from quiet wakefulness to arousal or locomotion (Rasmussen et al., 2019). Thus, a considerable amount of data supports the concept that astrocytes respond in a receptor-mediated Ca2+-dependent pathway to locus coeruleus activity, and in turn control the interstitial K+ levels. Whether norepinephrine alters astrocytic Kir4.1 expression and localization as seen in the heart (Duan et al., 2019), or if changes in norepinephrine driven shifts in connexin 43 localization (Nuriya et al., 2018) are state dependent, remains unknown. Nonetheless, astrocytic networks may act as a potent intermediary in translating state-dependent changes in neuromodulation to shifts in the balance of interstitial ions and consequent impacts on neuronal activity (Fig. 6). In particular, as astrocytes appear to show intracellular Ca2+ and K+ transients that mirror and precede bursts of neuronal activity (Chever et al., 2010; Poskanzer and Yuste, 2016), future studies are needed to determine if the magnitude of these shifts are altered by neuromodulatory activity, and whether this change in astrocytic ion homeostasis directly relates to the initiation of complex oscillations in neuronal microcircuits and networks. What about ions other than K+? How might they be regulated across brain states? Both [Ca2+]o and [Mg2+]o are, as discussed above, lower during wakefulness than during sleep (Ding et al., 2016). While it may be that astrocytes also control baseline [Ca2+]o and [Mg2+]o, the mechanisms responsible have not yet been described. It is possible that pathways controlling other cations, or also neuromodulators, may respond indirectly to changes in [K+]o. For example, the drosophila “blood”-brain barrier is more permeable to Mg2+ at night than during daytime (Zhang et al., 2018). Additional studies are clearly needed to establish these unknown mechanisms, which might identify new targets for the therapeutic modulation of sleep.
Fig. 6. Neuromodulator release drives state changes by altering interstitial ion concentrations through astrocytes.
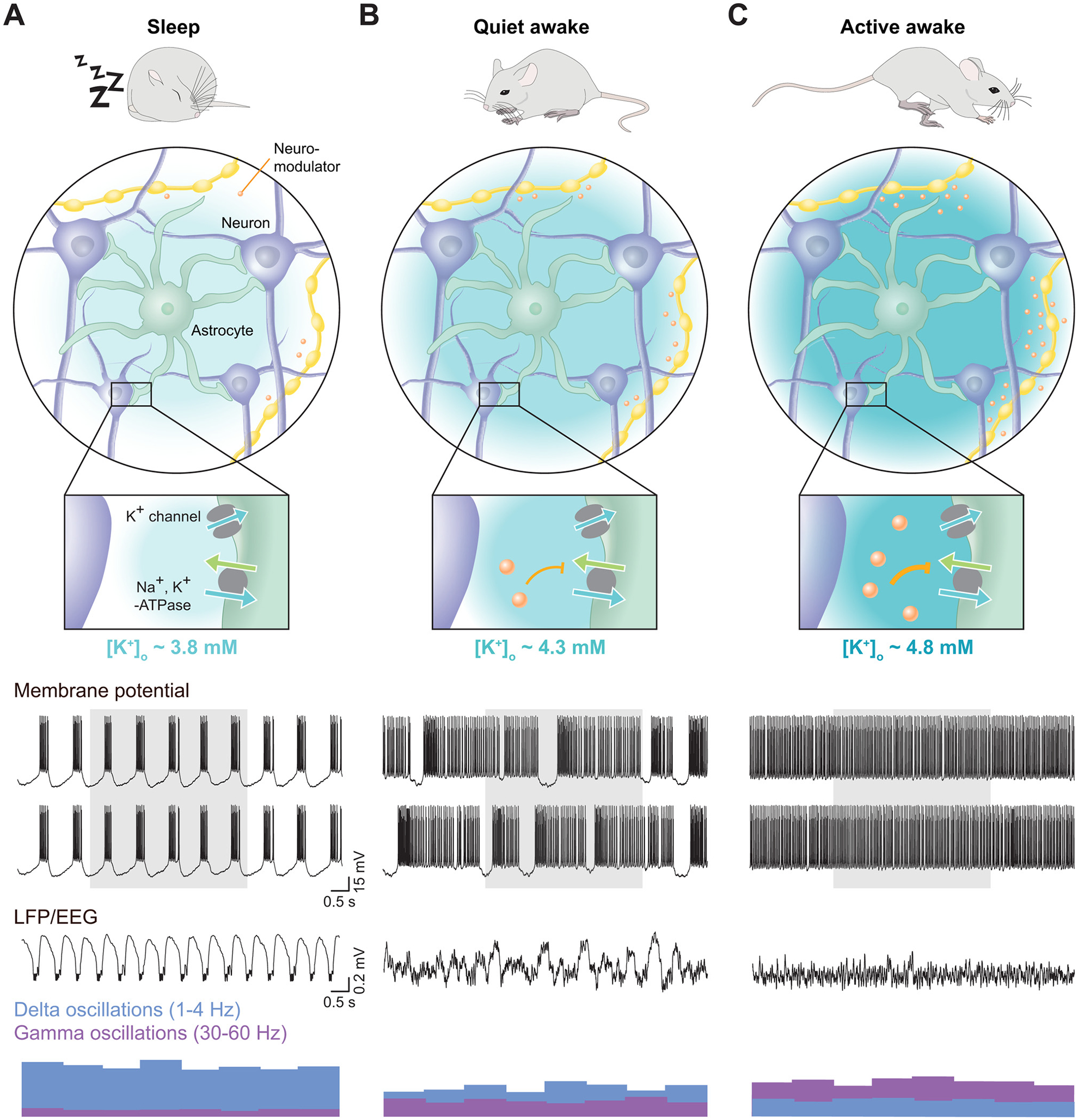
(A) Top panel: Diagram of one astrocyte surrounded by several local excitatory neurons and varicosities from long-projection, branching neuromodulatory neurons. Inset: During sleep, low-level release of neuromodulators permits high activity of the astrocytic Na+, K+-ATPase (blue arrow = K+, green = Na+). This is coupled with weak enhancement of inward rectifying K+ currents (Kir), leading to an [K+]o of ~3.8 mM. Membrane Potential: Two sample, simulated neurons are shown. In the sleep state, low [K+]o is coupled to high [Ca2+]o and [Mg2+]o and an expanded (~21%) interstitial space volume. This results in high network synchrony as seen in the concurrent bursts of activity in both neurons during up states. Gray shading highlights the neuronal synchrony. EEG: The simulated EEG of neurons in sleep is shown. Under the interstitial ionic conditions seen in sleep, high synchrony, phasic 1–4 Hz delta oscillations predominate, with minimal higher frequency activity, as seem in the relative delta versus gamma oscillation power at the bottom. (B) In quite wakefulness neuromodulator release is increased. This results in inhibition of the astrocytic Na+, K+-ATPase, and increased K+ conductance through Kir channels. The net result of this activity is a [K+]o of ~4.3 mM, decreased interstitial space, [Ca2+]o and [Mg2+]o. This results in increased activity of simulated neurons, and asynchrony between nearby neurons. This decreases slower frequency activity within the EEG and enhances more complex, faster frequency oscillations, as shown with decreased delta and increased gamma power. (C) In active awake states, such as during locomotion, neuromodulator release within the brain peaks. This results in maximal inhibition of the astrocytic Na+, K+-ATPase, and activation of Kir channel currents. These alterations lead to high [K+]o of ~4.8 mM, and minimal [Ca2+]o, [Mg2+]o, and interstitial space volume, resulting in tonic depolarization and tonic firing of individual neurons. This in turn results in high levels of gamma frequency oscillations and minimal delta frequency activity. It should be noted that neuromodulators can also have direct effects on neurons that would contribute to [K+]o regulation, such as for example metabotropic receptor-mediated opening or closing of K+-conducting ion channels.
5. Interstitial ions and their role in brain diseases
As highlighted above, interstitial ions are integral to both the normal functioning and modulation of neural activity in the brain. While the neuronal Na+, K+-ATPase is known to consume a substantial amount of energy (Attwell and Iadecola, 2002), mutations in astrocytic proteins necessary for ion homeostasis suggest at least an equivalent role for astrocytic ion buffering in disrupted neuromodulator-dependent changes in interstitial ions. Three basic mechanisms exist for astrocytic regulation of interstitial ions: 1) spatial buffering, as seen in the transfer of K+ through the glial syncytium by gap junctions; 2) ionotropic channel-mediated passive transport, including inward-rectifying K+ channels, and voltage-, ligand-, and calcium-gated transport; and 3) carrier-mediated transport through ATP-dependent pumps or facilitated exchange diffusion and cotransport along with biomolecules. Disruption of ion homeostasis causes significant clinical impairment in several CNS pathologies. For example, mutations of the astrocyte-specific α2 subunit of the Na+, K+-ATPase is implicated in familial hemiplegic migraine (Staehr et al., 2019), whereas there are widespread mutations in connexins and Kir4.1 in epilepsy (Köhling and Wolfart, 2016), Huntington’s disease (Tong et al., 2014), and EAST syndrome (presence of epilepsy, ataxia, sensorineural deafness, and tubulopathy) (Bockenhauer et al., 2009; Nwaobi et al., 2016), whereas Kir2.1 mutations are noted in Andersen-Tawil syndrome and the short QT3, autism-epilepsy phenotype (Ambrosini et al., 2014). Extending beyond these rare conditions, a range of K+ channels have been shown to play roles in aspects of fragile X syndrome, Alzheimer’s disease, and epilepsy (Noh et al., 2019). While these channels, receptors, and transporters must necessarily play fundamental roles in both interstitial and intracellular ion homeostasis, few studies to date have generated data documenting the importance of disturbed interstitial ions in these disease states. While the range of proteins implicated across different brain diseases is striking, recent evidence suggests that disrupted K+ homeostasis may represent part of a shared pathology, with striking down-regulation of diverse types of K+ channels (Windrem et al., 2017). To understand how disruption in [K+]o could drive neuropathology, we here consider the historical study of [K+]o in epilepsy, as well as new studies demonstrating a more direct role for disrupted K+ homeostasis in the pathologies of Huntington’s disease and schizophrenia.
Beginning with the discovery that small cortical infusions of KCl could elicit seizures in cats, the K+ hypothesis of epilepsy emerged (Feldberg and Sherwood, 1957; Fertziger and Ranck, 1970). Under the framework of this hypothesis, elevated [K+]o resulting from neuronal activity, altered buffering, or both would lead to neuronal depolarization, increased firing, and ultimately synchronous burst-firing of neurons (Traynelis and Dingledine, 1988). However, several decades of evidence in intact systems has demonstrated that, while artificially elevated [K+]o can indeed drive population spikes similar to those seen in ictal events, elevated [K+]o alone is insufficient to reliably elicit seizures without support from an additional factor (Somjen, 2004). Why might an acute drastic increase in [K+]o fail to drive seizures, while pathology of channels involved in the maintenance of [K+]o gradients can manifestly promote epilepsy? First, epileptic activity is highly variable in its manifestation, ranging from focal to whole-brain seizure activity. The specific features of seizure activity thereby naturally arise from the cellular and subcellular expression of local neuronal populations and, critically, the capacity of both neurons and glia to buffer local ion changes. This heterogeneity of response to changes in [K+]o seen in different neuronal populations also reflects the different spatial architecture and channel expression profiles present in different brain regions (He et al., 2019). Among the relevant factors here are long-term maladaptive changes in AMPA receptors (Maneepark et al., 2012), slow-conductance K+ channels (Oliveira et al., 2010), and hyperpolarization-activated nucleotide-gated channels (Arnold et al., 2019). However, this diversity in regional channel expression, which is often invoked to explain differences in regional seizure susceptibility, belies the broad differences in the handling of interstitial K+. Finally, we note that the story of how [K+]o has been implicated in epilepsy etiology appears to be mirrored in the search for an explanation of spreading depression (Grafstein, 1956); here too the experimental data have proved unsatisfactory for causally relating [K+]o increases to the propagation of spreading depression (Somjen, 2001). Thus, the view that epilepsy and spreading depression result solely from an increase in [K+]o has been abandoned: They result from interlocking cascades of signaling pathways wherein [K+]o changes are an integral part.
Considering [K+]o as a potential mediator of excitotoxic injury, recent work on Huntington’s disease has found that dysfunctional astrocytic Kir4.1-mediated K+ buffering leads to elevated [K+]o. In this work, disrupted Kir4.1 currents were found to precede atrophy and gliosis, resulting in a significant (6 mV) depolarization of medium spiny neurons in the striatum, thus providing a potential pathogenic mechanism within the R6/2 mouse model of Huntington’s disease (Tong et al., 2014). Supporting this K+-hypothesis, studies using induced pluripotent stem cells from human volunteers, engrafted into R6/2 mice to form astrocytes, found that healthy astrocytes partially restored normal [K+]o buffering, while engrafting human cells carrying mutant huntingtin protein into disease-free mice was sufficient to drive [K+]o increases (Benraiss et al., 2016). Strikingly, an analogous study using induced pluripotent stem cells from volunteers with schizophrenia engrafted into mice resulted in both behavioral changes and broad changes in the expressions of various K+ channels (Windrem et al., 2017). These studies suggest that changes in [K+]o following disrupted expression of K+ channels may constitute an emergent phenotype in seemingly unrelated brain pathologies. Surprisingly, behavioral differences noted in the schizophrenia model included reduced socialization and increased anxiety, which are among the core symptoms of the human disease. Taken together, these studies suggest that astrocytes play an essential role in the buffering of interstitial K+ to maintain normal local neural activity as well as to provide the stable background for the emergence of coherent network activity. Coupled with the finding that local disruption in [K+]o can alter neuronal membrane potential dynamics, these intriguing studies demonstrate the need for a multifaceted approach to better understanding of the high inter-regional variability in interstitial ion dynamics, including the use of broad behavioral measures.
The need to maintain [K+]o within normal levels likely plays a role in regulating feedback changes in pre- and post-synaptic protein expression that, in turn, alter the properties of local neuronal membranes. However, as the preceding studies have shown, while local neuronal expression and connection patterns are key to understanding highly stereotyped diseases, disrupted interstitial ion homeostasis may act as a generalized stressor with wide-ranging effects. In addition to the surprising findings of widespread changes in K+-channels in schizophrenia, another recent study suggests that the Na+, K+-ATPase and glial K+ buffering may accompany neuronal activity in regulating infraslow brain-wide neural oscillation (Krishnan et al., 2018). While the importance of Na+, K+-ATPase here is far from proven, these oscillations are thought to underlie the systems that maintain established brain connectivity patterns - with the potential to strongly influence behavior. As such, astrocytes appear to have a necessary role in the buffering of interstitial K+ to maintain normal local neural activity, as well as to provide the stable background for oscillatory network activity. Coupled to the finding that local disruption in [K+]o can alter neuronal membrane potential dynamics; this demonstrates the need for a broad approach to understanding interstitial ion dynamics because high inter-regional variability and diaschisis necessitate the use of both local mechanistic analysis and broad behavioral measures.
6. Past and future perspectives
Most of the fundamental knowledge regarding interstitial ions was established in the 1970s, when the technology for fabrication of ion-selective microelectrodes had advanced sufficiently to match the broad interest in electrical excitability. However, with the acceptance in the late 1970s of glutamate as the major excitatory transmitter in the CNS, the focus of neuroscience research shifted sharply to the study of how glutamate and other neurotransmitters coordinated electrical activity in the brain. This booming interest in glutamatergic transmission in the 1980s effectively put to a halt the ongoing studies of interstitial ions. Publications on this topic became sparse and disconnected. As the early interest in ionic balance faded, the dominating view held that interstitial ion concentrations were of little interest, as they were epiphenomena of synaptic activity. In that scenario, intense neuronal firing may transiently increase [K+]o, but homeostatic mechanisms will quickly return [K+]o to some baseline. The predominance of this rather simplistic view seems somewhat surprising, since electrophysiologists had for decades been deliberately controlling neuronal excitability in slice preparations by manipulating the concentrations of K+, Ca2+, or Mg2+ in the bath solution.
Recently it was noticed that the CNS employs the same trick to control excitability in different states of brain activity: Interstitial ion concentrations follow the sleep-wake cycle, and application of a cocktail of neuromodulators triggers ion changes in cortical slices in presence of tetrodotoxin (Ding et al., 2016). In this way, neuromodulators may act in part by controlling excitability by altering interstitial ion concentrations. The relatively long timescale of the action of neuromodulators (seconds) mirrors the timescale of changes in interstitial ions. Similar cortex-wide changes in interstitial [K+]o have been noted when mice transition from quiet wakefulness into active running (Rasmussen et al., 2019). As discussed above, astrocytes respond with cortex-wide Ca2+ increases upon state transition (awakening, running, startle), which will transiently boost their capacity for K+ buffering (Kjaerby et al., 2017; Wang et al., 2012a). Thus, an emerging line of work suggests that arousal states are at least in part controlled by concerted changes in interstitial ions. Conversely, we speculate that dysregulation of interstitial ion concentrations may contribute to a broad range of both acute and chronic disturbances of cognitive dysfunctions. For example, the postanesthetic state is characterized by a quick normalization of [K+]o, while [Ca2+]o and [Mg2+]o remain abnormally high for more than 30 min (Ding et al., 2016). The high [Mg2+]o is expected to block NMDA receptors and negatively impact cognitive function, which in turn may contribute to postanesthetic delirium. Another area that clearly needs to be viewed from the perspective of interstitial ions are channelopathies, which are inherited diseases caused by defects in ion channels or associated proteins. While a great deal of effort has been devoted to studying the impact of channel mutations on the electrical properties of the neuronal membrane, very few studies have explored the possible consequences on interstitial ion concentrations. Very recent evidence links a range of disorders spanning from Huntington’s disease to schizophrenia to a general decline in K+ channel expression and a resultant increase in interstitial [K+]o. Restoration of Kir4.1 channel expression or replacement of defective astrocytes were both sufficient to restore [K+]o and neural function in animal models (Benraiss et al., 2016; Liu et al., 2019; Tong et al., 2014; Windrem et al., 2017). Such findings may have considerable importance for the understanding of cognitive disturbances in neurodegenerative diseases and psychiatric disorders.
A major bottleneck for future studies is that the current technology - largely ion-selective electrodes - has not kept pace with research requirements. These electrodes remain difficult to produce and are unstable and not always selective. Furthermore, the electrode tips measure ions in a very restricted space of less than 10 μm and therefore only detect interstitial ions in very localized regions. On the other hand, one advantage of these ion-selective electrodes is their ability to measure absolute ion concentrations, something not possible with any other method. While chemical intracellular Ca2+ indicators and more recent genetic tools have revolutionized our understanding of intracellular Ca2+ signaling, none can be used for imaging interstitial ion concentrations, and nor are they able to measure absolute ion concentrations. Genetic K+ indicators have recently been developed (Bischof et al., 2017; Shen et al., 2019), but have not yet been modified for measuring interstitial ions. Advances in the development of high sensitivity (mM), genetically encoded ion sensors for the most common interstitial ions, particularly K+, could fast-track progress.
Hopefully, in the future, the research community will become more open to the idea that the brain is sophisticated enough to use both neurotransmitters and ion signaling in executing its complex functions.
Acknowledgements
The authors thank Hajime Hirase, Paul Cumming and Sara Oakeley for critical comments on the manuscript, and Dan Xue for expert graphical design.
Funding
R.R. was supported by a Lundbeck Foundation PhD Scholarship (R230-2016-2326). J.O. was supported by the National Institute of Neurological Disorders and Stroke (U.S. National Institutes of Health: F32NS105365). M.N. was supported by The Novo Nordisk, Lundbeck Foundation, National Institute of Neurological Disorders and Stroke and the National Institute on Aging (U.S. National Institutes of Health: R01NS100366; RF1AG057575), and the U.S. Army Research Office (grant no. MURI W911NF1910280). The views and conclusions contained in this manuscript are solely those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the National Institutes of Health, Army Research Office, or the U.S. government. The U.S. government is authorized to reproduce and distribute reprints for government purposes notwithstanding any copyright notation herein.
Footnotes
Appendix A. The Peer Review Overview and Supplementary data
The Peer Review Overview and Supplementary data associated with this article can be found in the online version, at doi:https://doi.org/10.1016/j.pneurobio.2020.101802.
References
- Aitken PG, Somjen GG, 1986. The sources of extracellular potassium accumulation in the CA1 region of hippocampal slices. Brain Res. 369, 163–167. 10.1016/0006-8993(86)90524-X. [DOI] [PubMed] [Google Scholar]
- Ambrosini E, Sicca F, Brignone MS, D’Adamo MC, Napolitano C, Servettini I, Moro F, Ruan Y, Guglielmi L, Pieroni S, Servillo G, Lanciotti A, Valvo G, Catacuzzeno L, Franciolini F, Molinari P, Marchese M, Grottesi A, Guerrini R, Santorelli FM, Priori S, Pessia M, 2014. Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum. Mol. Genet 23, 4875–4886. 10.1093/hmg/ddu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames A, Sakanoue M, Endo S, 1964. Na, K, Ca, Mg, and Cl concentrations in choroid plexus fluid and cisternal fluid compared with plasma ultrafiltrate. J. Neurophysiol 27, 672–681. 10.1152/jn.1964.27.4.672. [DOI] [PubMed] [Google Scholar]
- Amzica F, Massimini M, 2002. Glial and Neuronal Interactions during Slow Wave and Paroxysmal Activities in the Neocortex. Cereb. Cortex 12, 1101–1113. 10.1093/cercor/12.10.1101. [DOI] [PubMed] [Google Scholar]
- Amzica F, Massimini M, Manfridi A, 2002. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J. Neurosci 22, 1042–1053 doi:22/3/1042 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amzica F, Steriade M, 2000. Neuronal and glial membrane potentials during sleep and paroxysmal oscillations in the neocortex. J. Neurosci 20, 6648–6665 doi:20/17/6648 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiou CA, Koch C, 2015. Ephaptic coupling to endogenous electric field activity: why bother? Curr. Opin. Neurobiol 31, 95–103. 10.1016/j.conb.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Anastassiou CA, Perin R, Markram H, Koch C, 2011. Ephaptic coupling of cortical neurons. Nat. Neurosci 14, 217–223. 10.1038/nn.2727. [DOI] [PubMed] [Google Scholar]
- Anderson JS, Lampl I, Gillespie DC, Ferster D, 2000. The contribution of noise to contrast invariance of orientation tuning in cat visual cortex. Science (80-.) 290, 1968–1972. 10.1126/SCIENCE.290.5498.1968. [DOI] [PubMed] [Google Scholar]
- Arnold EC, McMurray C, Gray R, Johnston D, 2019. Epilepsy-Induced Reduction in HCN Channel Expression Contributes to an Increased Excitability in Dorsal, But Not Ventral, Hippocampal CA1 Neurons. eneuro 6. ENEURO.0036–19.2019. 10.1523/ENEURO.0036-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Iadecola C, 2002. The neural basis of functional brain imaging signals. Trends Neurosci. 25, 621–625. 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Aitken PG, Somjen GG, 1986. The effects of moderate changes of extracellular K+ and Ca2+ on synaptic and neural function in the CA1 region of the hippocampal slice. Brain Res. 377, 229–239. 10.1016/0006-8993(86)90863-2. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Somjen GG, 1988. Concentration of carbon dioxide, interstitial pH and synaptic transmission in hippocampal formation of the rat. J. Physiol 396, 247–266. 10.1113/jphysiol.1988.sp016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P, 2007. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci 8, 45–56. 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Baskey G, Singh A, Sharma R, Mallick BN, 2009. REM sleep deprivation-induced noradrenaline stimulates neuronal and inhibits glial Na-K ATPase in rat brain: in vivo and in vitro studies. Neurochem. Int 54, 65–71. 10.1016/j.neuint.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Baylor DA, Nicholls JG, 1969. Changes in extracellular potassium concentration produced by neuronal activity in the central nervous system of the leech. J. Physiol 203, 555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ, 2004. Potassium Model for Slow (2–3 Hz) In Vivo Neocortical Paroxysmal Oscillations. J. Neurophysiol 92, 1116–1132. 10.1152/jn.00529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, 2007. The action potential in mammalian central neurons. Nat. Rev. Neurosci 8, 451–465. 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Bellesi M, Tononi G, Cirelli C, Serra PA, 2016. Region-Specific Dissociation between Cortical Noradrenaline Levels and the Sleep/Wake Cycle. Sleep 39, 143–154. 10.5665/sleep.5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C, Arroyo S, Hestrin S, 2013. Subthreshold mechanisms underlying state-dependent modulation of visual responses. Neuron 80, 350–357. 10.1016/j.neuron.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benraiss A, Wang S, Herrlinger S, Li X, Chandler-Militello D, Mauceri J, Burm HB, Toner M, Osipovitch M, Jim Xu Q, Ding F, Wang F, Kang N, Kang J, Curtin PC, Brunner D, Windrem MS, Munoz-Sanjuan I, Nedergaard M, Goldman SA, 2016. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun 7, 11758. 10.1038/ncomms11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger H, 1931. Über das Elektrenkephalogramm des Menschen. Arch. Psychiatr. Nervenkr 94, 16–60. 10.1007/BF01835097. [DOI] [Google Scholar]
- Bertorello AM, Aperia A, Walaas SI, Nairn AC, Greengard P, 1991. Phosphorylation of the catalytic subunit of Na+,K(+)-ATPase inhibits the activity of the enzyme. Proc. Natl. Acad. Sci. U. S. A 88, 11359–11362. 10.1073/pnas.88.24.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevensee MO, Weed RA, Boron WF, 1997. Intracellular pH regulation in cultured astrocytes from rat hippocampus. I. Role Of HCO3-. J. Gen. Physiol 110, 453–465. 10.1085/jgp.110.4.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijak M, 1989. Daily and seasonal variations in Na+, K+, Ca2+ and Mg2+ contents in the cingulate cortex of the mouse brain. Folia Biol 37, 3–11. [PubMed] [Google Scholar]
- Bischof H, Rehberg M, Stryeck S, Artinger K, Eroglu E, Waldeck-Weiermair M, Gottschalk B, Rost R, Deak AT, Niedrist T, Vujic N, Lindermuth H, Prassl R, Pelzmann B, Groschner K, Kratky D, Eller K, Rosenkranz AR, Madl T, Plesnila N, Graier WF, Malli R, 2017. Novel genetically encoded fluorescent probes enable real-time detection of potassium in vitro and in vivo. Nat. Commun 8, 1422. 10.1038/s41467-017-01615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow G, Nilsson B, Siesjö BK, 1985. Influence of Reduced Oxygen Availability on Cerebral Metabolic Changes during Bicuculline-Induced Seizures in Rats. J. Cereb. Blood Flow Metab 5, 439–445. 10.1038/jcbfm.1985.59. [DOI] [PubMed] [Google Scholar]
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R, 2009. Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy, and KCNJ10 Mutations. N. Engl. J. Med 360, 1960–1970. 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarskaite L, Bjørnstad DM, Pettersen KH, Cunen C, Hermansen GH, Åbjørsbråten KS, Sprengel R, Vervaeke K, Tang W, Enger R, Nagelhus EA, 2019. Ca2+ signaling in astrocytes is sleep-wake state specific and modulates sleep. bioRxiv 750281. 10.1101/750281. [DOI] [Google Scholar]
- Borst JGG, 2010. The low synaptic release probability in vivo. Trends Neurosci. 33, 259–266. 10.1016/J.TINS.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Borst JGG, Sakmann B, 1999. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J. Physiol 521, 123–133. 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury MW, Leeman CR, Bagdoyanh, Berberian A, 1968. The calcium and magnesium content of skeletal muscle, brain, and cerebrospinal fluid as determined by atomic bsorption flame photometry. J. Lab. Clin. Med 71, 884–892. [PubMed] [Google Scholar]
- Branco T, Häusser M, 2011. Synaptic Integration Gradients in Single Cortical Pyramidal Cell Dendrites. Neuron 69, 885–892. 10.1016/j.neuron.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branston NM, Strong AJ, Symon L, 1977. Extracellular potassium activity, evoked potential and tissue blood flow: Relationships during progressive ischaemia in baboon cerebral cortex. J. Neurol. Sci 32, 305–321. 10.1016/0022-510X(77)90014-4. [DOI] [PubMed] [Google Scholar]
- Brocard F, Shevtsova NA, Bouhadfane M, Tazerart S, Heinemann U, Rybak IA, Vinay L, 2013. Activity-Dependent Changes in Extracellular Ca2+ and K+ Reveal Pacemakers in the Spinal Locomotor-Related Network. Neuron 77, 1047–1054. 10.1016/j.neuron.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SH, Kang J, Akk G, Chiang LW, Schulman H, Huguenard JR, Prince DA, 2001. Kinetic and Pharmacological Properties of GABAA Receptors in Single Thalamic Neurons and GABA A Subunit Expression. J. Neurophysiol 86, 2312–2322. 10.1152/jn.2001.86.5.2312. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Anastassiou CA, Koch C, 2012. The origin of extracellular fields and currents-EEG, ECoG, LFP and spikes. Nat. Rev. Neurosci 13, 407–420. 10.1038/nrn3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Draguhn A, 2004. Neuronal Oscillations in Cortical Networks. Science (80-.) 304, 1926–1929. 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Wang X-J, 2012. Mechanisms of Gamma Oscillations. Annu. Rev. Neurosci 35, 203–225. 10.1146/annurev-neuro-062111-150444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G, 1992. Long-term Potentiation in the Striatum is Unmasked by Removing the Voltage-dependent Magnesium Block of NMDA Receptor Channels. Eur. J. Neurosci 4, 929–935. 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- Cao L, Händel B, 2019. Walking enhances peripheral visual processing in humans. PLoS Biol. 17, e3000511. 10.1371/journal.pbio.3000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, 2004. Absence of Rapid Sensory Adaptation in Neocortex during Information Processing States. Neuron 41, 455–464. 10.1016/S0896-6273(03)00853-5. [DOI] [PubMed] [Google Scholar]
- Charvériat M, Naus CC, Leybaert L, Sáez JC, Giaume C, 2017. Connexin-Dependent Neuroglial Networking as a New Therapeutic Target. Front. Cell. Neurosci 11, 174. 10.3389/fncel.2017.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, 2003. Regulation and modulation of pH in the brain. Physiol. Rev 83, 1183–1221. 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kraig RP, 1987. Intracellular pH of astrocytes increases rapidly with cortical stimulation. Am. J. Physiol 253, R666–70. 10.1152/ajpregu.1987.253.4.R666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chever O, Djukic B, McCarthy KD, Amzica F, 2010. Implication of Kir4.1 Channel in Excess Potassium Clearance: An In Vivo Study on Anesthetized Glial-Conditional Kir4.1 Knock-Out Mice. J. Neurosci 30, 15769–15777. 10.1523/JNEUROSCI.2078-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Shivacharan RS, Wei X, Gonzalez-Reyes LE, Durand DM, 2019. Slow periodic activity in the longitudinal hippocampal slice can self-propagate non--synaptically by a mechanism consistent with ephaptic coupling. J. Physiol 597, 249–269. 10.1113/JP276904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasadonte J, Scemes E, Wang Z, Boison D, Haydon PG, 2017. Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron 95 10.1016/j.neuron.2017.08.022. 1365–1380.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JE, Fields RD, 2004. Extracellular Calcium Depletion in Synaptic Transmission. Neuroscientist. 10.1177/1073858403259440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg P, Patterson L, Purves MJ, 1977. The pH of brain extracellular fluid in the cat. J. Physiol 272, 137–166. 10.1113/jphysiol.1977.sp012038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Petersen CCH, 2006. Correlating whisker behavior with membrane potential in barrel cortex of awake mice. Nat. Neurosci 9, 608–610. 10.1038/nn1690. [DOI] [PubMed] [Google Scholar]
- Cuddapah VA, Zhang SL, Sehgal A, 2019. Regulation of the Blood-Brain Barrier by Circadian Rhythms and Sleep. Trends Neurosci. 42, 500–510. 10.1016/j.tins.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, Ma S, Sang K, Tang S, Li Y, Shen Y, Berry H, Wu S, Hu H, 2018. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554, 323–327. 10.1038/nature25752. [DOI] [PubMed] [Google Scholar]
- Czeh G, Somjen GG, 1989. Changes in extracellular calcium and magnesium and synaptic transmission in isolated mouse spinal cord. Brain Res 486, 274–285. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Gordon DS, Winn HR, 2002. Differential Role of KIR Channel and Na +/K+ Pump in the Regulation of Extracellular K+ in Rat Hippocampus. J. Neurophysiol 87, 87–102. 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Dadarlat MC, Stryker MP, 2017. Locomotion Enhances Neural Encoding of Visual Stimuli in Mouse V1. J. Neurosci 37, 3764–3775. 10.1523/JNEUROSCI.2728-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby MA, 1969. Epilepsy and 3 per second spike and wave rhythms. A clinical, electroencephalographic and prognostic analysis of 346 patients. Acta Neurol. Scand Suppl 40:3+. [PubMed] [Google Scholar]
- de Baaij JHF, Hoenderop JGJ, Bindels RJM, 2015. Magnesium in Man: Implications for Health and Disease. Physiol. Rev 95, 1–46. 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- de Lores Arnaiz GR, Ordieres MGL, 2014. Brain Na(+), K(+)-ATPase Activity In Aging and Disease. Int. J. Biomed. Sci 10, 85–102. [PMC free article] [PubMed] [Google Scholar]
- Dere E, De Souza-Silva MA, Frisch C, Teubner B, Sohl G, Willecke K, Huston JP, 2003. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur. J. Neurosci 18, 629–638. 10.1046/j.1460-9568.2003.02784.x. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M, 1999. Spatiotemporal analysis of local field potentials and unit discharges in cat cerebral cortex during natural wake and sleep states. J. Neurosci 19, 4595–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH, 2001. Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron 32, 1107–1117. 10.1016/s0896-6273(01)00535-9. [DOI] [PubMed] [Google Scholar]
- Diekelmann S, Born J, 2010. The memory function of sleep. Nat. Rev. Neurosci 11, 114–126. 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Dietz AG, Goldman SA, Nedergaard M, 2019. Glial cells in schizophrenia: a unified hypothesis. The Lancet Psychiatry. 10.1016/S2215-0366(19)30302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD, 1980. Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp. Brain Res 40, 432–439. 10.1007/BF00236151. [DOI] [PubMed] [Google Scholar]
- Ding F, O’Donnell J, Thrane AS, Zeppenfeld D, Kang H, Xie L, Wang F, Nedergaard M, 2013. α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 54, 387–394. 10.1016/j.ceca.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, O’Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M, 2016. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352, 550–555. 10.1126/science.aad4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Somjen G, 1981. Calcium dependence of synaptic transmission in the hippocampal slice. Brain Res 207, 218–222. [DOI] [PubMed] [Google Scholar]
- Doi M, Iwasaki K, 2008. Na+/K+ ATPase regulates the expression and localization of acetylcholine receptors in a pump activity-independent manner. Mol. Cell. Neurosci 38, 548–558. 10.1016/j.mcn.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domich L, Oakson G, Steriade M, 1986. Thalamic burst patterns in the naturally sleeping cat: a comparison between cortically projecting and reticularis neurones. J. Physiol 379, 429–449. 10.1113/jphysiol.1986.sp016262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X-P, Gu L, Xiao Y, Gao Z-X, Wu P, Zhang Y-H, Meng X-X, Wang J-L, Zhang D-D, Lin D-H, Wang W-H, Gu R, 2019. Norepinephrine-Induced Stimulation of Kir4.1/Kir5.1 Is Required for the Activation of NaCl Transporter in Distal Convoluted Tubule. Hypertension 73, 112–120. 10.1161/HYPERTENSIONAHA.118.11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Lynch G, 1979. The relationship between extracellular calcium concentrations and the induction of hippocampal long-term potentiation. Brain Res. 169, 103–110. [DOI] [PubMed] [Google Scholar]
- Egelman DM, Montague PR, 1999. Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys. J 76, 1856–1867. 10.1016/S0006-3495(99)77345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P, 2012. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci 13, 7–21. 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AK, Fries P, Singer W, 2001. Dynamic predictions: Oscillations and synchrony in top-down processing. Nat. Rev. Neurosci 2, 704–716. 10.1038/35094565. [DOI] [PubMed] [Google Scholar]
- Enkvist KMO, McCarthy KD, 2002. Astroglial Gap Junction Communication Is Increased by Treatment with Either Glutamate or High K+ Concentration. J. Neurochem 62, 489–495. 10.1046/j.1471-4159.1994.62020489.x. [DOI] [PubMed] [Google Scholar]
- Ernst C, Nagy C, Kim S, Yang JP, Deng X, Hellstrom IC, Choi KH, Gershenfeld H, Meaney MJ, Turecki G, 2011. Dysfunction of Astrocyte Connexins 30 and 43 in Dorsal Lateral Prefrontal Cortex of Suicide Completers. Biol. Psychiatry 70, 312–319. 10.1016/j.biopsych.2011.03.038. [DOI] [PubMed] [Google Scholar]
- Evarts EV, 1964. TEMPORAL PATTERNS OF DISCHARGE OF PYRAMIDAL TRACT NEURONS DURING SLEEP AND WAKING IN THE MONKEY. J. Neurophysiol 27, 152–171. 10.1152/jn.1964.27.2.152. [DOI] [PubMed] [Google Scholar]
- Feeney KA, Hansen LL, Putker M, Olivares-Yañez C, Day J, Eades LJ, Larrondo LF, Hoyle NP, O’Neill JS, Van Ooijen G, 2016. Daily magnesium fluxes regulate cellular timekeeping and energy balance. Nature 532, 375–379. 10.1038/nature17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldberg W, Sherwood SL, 1957. Effects of calcium and potassium injected into the cerebral ventricles of the cat. J. Physiol 139, 408–416. 10.1113/jphysiol.1957.sp005901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertziger AP, Ranck JB, 1970. Potassium accumulation in interstitial space during epileptiform seizures. Exp. Neurol 26, 571–585. 10.1016/0014-4886(70)90150-0. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Pike FG, Buhl EH, Paulsen O, 1998. Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature 394, 186–189. 10.1038/28179. [DOI] [PubMed] [Google Scholar]
- Formenti A, De Simoni A, Arrigoni E, Martina M, 2001. Changes in extracellular Ca2+can affect the pattern of discharge in rat thalamic neurons. J. Physiol 535, 33–45. 10.1111/j.1469-7793.2001.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frace AM, Maruoka F, Noma A, 1992. External K+ increases Na+ conductance of the hyperpolarization-activated current in rabbit cardiac pacemaker cells. Pflugers Arch 421, 97–99. 10.1007/BF00374739. [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL, 1957. The action of calcium on the electrical properties of squid axons. J. Physiol 137, 218–244. 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries P, 2009. Neuronal Gamma-Band Synchronization as a Fundamental Process in Cortical Computation. Annu. Rev. Neurosci 32, 209–224. 10.1146/annurev.neuro.051508.135603. [DOI] [PubMed] [Google Scholar]
- Frisch C, Theis M, De Souza Silva MA, Dere E, Sohl G, Teubner B, Namestkova K, Willecke Ki, Huston JP, 2003. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur. J. Neurosci 18, 2313–2318. 10.1046/j.1460-9568.2003.02971.x. [DOI] [PubMed] [Google Scholar]
- Fröhlich F, Bazhenov M, Iragui-Madoz V, Sejnowski TJ, 2008. Potassium Dynamics in the Epileptic Cortex: New Insights on an Old Topic. Neurosci. 14, 422–433. 10.1177/1073858408317955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich F, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ, 2006. Slow State Transitions of Sustained Neural Oscillations by Activity-Dependent Modulation of Intrinsic Excitability. J. Neurosci 26, 6153–6162. 10.1523/JNEUROSCI.5509-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, Kasai N, Torimitsu K, 2009. Effect of Mg2+ on neural activity of rat cortical and hippocampal neurons in vitro. Magnes. Res 22, 174S–181S. 10.1684/mrh.2009.0179. [DOI] [PubMed] [Google Scholar]
- Gambino F, Pagès S, Kehayas V, Baptista D, Tatti R, Carleton A, Holtmaat A, 2014. Sensory-evoked LTP driven by dendritic plateau potentials in vivo. Nature 515, 116–119. 10.1038/nature13664. [DOI] [PubMed] [Google Scholar]
- Gervasoni D, Lin S-C, Ribeiro S, Soares ES, Pantoja J, Nicolelis MAL, 2004. Global Forebrain Dynamics Predict Rat Behavioral States and Their Transitions. J. Neurosci 24, 11137–11147. 10.1523/JNEUROSCI.3524-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goaillard JM, Taylor AL, Schulz DJ, Marder E, 2009. Functional consequences of animal-to-animal variation in circuit parameters. Nat. Neurosci 12, 1424–1430. 10.1038/nn.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb D, Shedmi A, Curtu R, Ermentrout GB, 2006. Persistent Synchronized Bursting Activity in Cortical Tissues With Low Magnesium Concentration: A Modeling Study. J. Neurophysiol 95, 1049–1067. 10.1152/jn.00932.2005. [DOI] [PubMed] [Google Scholar]
- Grafstein B, 1956. Mechanism of cortical spreadin depression. J. Neurophysiol 19, 154–171. 10.1152/jn.1956.19.2.154. [DOI] [PubMed] [Google Scholar]
- Gray CM, Singer W, 1989. Stimulus-specific neuronal oscillations in orientation columns of cat visual cortex. Proc. Natl. Acad. Sci 86, 1698–1702. 10.1073/pnas.86.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoriou GG, Gotts SJ, Zhou H, Desimone R, 2009. High-frequency, long-range coupling between prefrontal and visual cortex during attention. Science 324, 1207–1210. 10.1126/science.1171402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoriou GG, Paneri S, Sapountzis P, 2015. Oscillatory synchrony as a mechanism of attentional processing. Brain Res. 1626, 165–182. 10.1016/j.brainres.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Gregoriou GG, Rossi AF, Ungerleider LG, Desimone R, 2014. Lesions of prefrontal cortex reduce attentional modulation of neuronal responses and synchrony in V4. Nat. Neurosci 17, 1003–1011. 10.1038/nn.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grichtchenko II, Chesler M, 1994a. Depolarization-induced acid secretion in gliotic hippocampal slices. Neuroscience 62, 1057–1070. 10.1016/0306-4522(94)90343-3. [DOI] [PubMed] [Google Scholar]
- Grichtchenko II, Chesler M, 1994b. Depolarization-induced alkalinization of astrocytes in gliotic hippocampal slices. Neuroscience 62, 1071–1078. 10.1016/0306-4522(94)90344-1. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ, Lundervold A, 1981. Hippocampal excitability and changes in extracellular potassium. Exp. Neurol 71, 410–420. 10.1016/0014-4886(81)90099-6. [DOI] [PubMed] [Google Scholar]
- Haider B, Mccormick DA, 2009. Review Rapid Neocortical Dynamics : Cellular and Network Mechanisms. Neuron 62, 171–189. 10.1016/j.neuron.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K-S, Guo C, Chen CH, Witter L, Osorno T, Regehr WG, 2018. Ephaptic Coupling Promotes Synchronous Firing of Cerebellar Purkinje Cells. Neuron 100 10.1016/j.neuron.2018.09.018. 564–578.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ, 1985. Effect of anoxia on ion distribution in the brain. Physiol. Rev 65, 101–148. 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Harris KD, Thiele A, 2011. Cortical state and attention. Nat Rev Neurosci 12, 509–523. 10.1038/nrn3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RJ, Symon L, Branston NM, Bayhan M, 1981. Changes in extracellular calcium activity in cerebral ischaemia. J. Cereb. Blood Flow Metab 1, 203–209. 10.1038/jcbfm.1981.21. [DOI] [PubMed] [Google Scholar]
- He LS, Rue MCP, Morozova EO, Powell DJ, James EJ, Kar M, Marder E, 2019. Rapid adaptation to elevated extracellular potassium in the pyloric circuit of the crab, Cancer borealis. bioRxiv 636910. 10.1101/636910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zorumski CF, Mennerick S, 2002. Contribution of Presynaptic Na+ Channel Inactivation to Paired-Pulse Synaptic Depression in Cultured Hippocampal Neurons. J. Neurophysiol 87, 925–936. 10.1152/jn.00225.2001. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Konnerth A, Pumain R, Wadman WJ, 1986. Extracellular calcium and potassium concentration changes in chronic epileptic brain tissue. Adv. Neurol 44, 641–661. [PubMed] [Google Scholar]
- Heinemann U, Lux H, Gutnick MJ, 1977. Extracellular free calcium and potassium during paroxsmal activity in the cerebral cortex of the cat. Exp. Brain Res 27, 237–243. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD, 1977. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. 120, 231–249. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Schaible HG, Schmidt RF, 1990. Changes in extracellular potassium concentration in cat spinal cord in response to innocuous and noxious stimulation of legs with healthy and inflamed knee joints. Exp. brain Res 79, 283–292. [DOI] [PubMed] [Google Scholar]
- Hernández-R J, 1992. Na+/K+-ATPase regulation by neurotransmitters. Neurochem. Int 20, 1–10. 10.1016/0197-0186(92)90119-C. [DOI] [PubMed] [Google Scholar]
- Hertz L, 1965. Possible Role of Neuroglia: A Potassium-Mediated Neuronal - Neuroglial - Neuronal Impulse Transmission System. Nature 206, 1091–1094. 10.1038/2061091a0. [DOI] [PubMed] [Google Scholar]
- Hertz L, Xu J, Song D, Yan E, Gu L, Peng L, 2013. Astrocytic and neuronal accumulation of elevated extracellular K+ with a 2/3 K+/Na+ flux ratio—consequences for energy metabolism, osmolarity and higher brain function. Front. Comput. Neurosci 7, 114. 10.3389/fncom.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, 1978. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J 22, 283–294. 10.1016/S0006-3495(78)85489-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, Woodhull AM, Shapiro BI, 1975. Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions, and pH. Philos. Trans. R. Soc. Lond. B. Biol. Sci 270, 301–318. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Fourment A, Marc ME, 1983. Sleep-related variations of membrane potential in the lateral geniculate body relay neurons of the cat. Brain Res. 259, 308–312. 10.1016/0006-8993(83)91264-7. [DOI] [PubMed] [Google Scholar]
- Holmes MD, Dewaraja AS, Vanhatalo S, 2004. Does Hyperventilation Elicit Epileptic Seizures? Epilepsia 45, 618–620. 10.1111/j.0013-9580.2004.63803.x. [DOI] [PubMed] [Google Scholar]
- Hopfield JJ, 1995. Pattern recognition computation using action potential timing for stimulus representation. Nature 376, 33–36. 10.1038/376033a0. [DOI] [PubMed] [Google Scholar]
- Hsiao C-F, Wu N, Levine MS, Chandler SH, 2002. Development and serotonergic modulation of NMDA bursting in rat trigeminal motoneurons. J. Neurophysiol 87, 1318–1328. 10.1152/Jn00469.2001. [DOI] [PubMed] [Google Scholar]
- Hu H, Jonas P, 2014. A supercritical density of Na+ channels ensures fast signaling in GABAergic interneuron axons. Nat. Neurosci 17, 686–693. 10.1038/nn.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huchzermeyer C, Albus K, Gabriel H-J, Otahal J, Taubenberger N, Heinemann U, Kovacs R, Kann O, 2008. Gamma Oscillations and Spontaneous Network Activity in the Hippocampus Are Highly Sensitive to Decreases in pO2 and Concomitant Changes in Mitochondrial Redox State. J. Neurosci 28, 1153–1162. 10.1523/JNEUROSCI.4105-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg B. a., Peng W, Gundersen G. a., Benveniste H, Vates GE, Deane R, Goldman S. a., Nagelhus E. a., Nedergaard M, 2012. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid. Sci. Transl. Med 4, 147ra111. 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R, 1984. Voltage-dependent burst-to-tonic switching of thalamic cell activity: an in vitro study. Arch. Ital. Biol 122, 73–82. [PubMed] [Google Scholar]
- Javaheri S, De Hemptinne A, Vanheel B, Leusen I, 1983. Changes in brain ECF pH during metabolic acidosis and alkalosis: a microelectrode study. J. Appl. Physiol 55, 1849–1853. 10.1152/jappl.1983.55.6.1849. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y, 1994. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J. Neurophysiol 71, 831–839. [DOI] [PubMed] [Google Scholar]
- Jung MW, Larson J, Lynch G, 1990. Long-term potentiation of monosynaptic EPSPs in rat piriform cortex in vitro. Synapse 6, 279–283. 10.1002/syn.890060307. [DOI] [PubMed] [Google Scholar]
- Kadala A, Verdier D, Morquette P, Kolta A, 2015. Ion Homeostasis in Rhythmogenesis: The Interplay Between Neurons and Astroglia. Physiology 30, 371–388. 10.1152/physiol.00023.2014. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS, 1994. Residual Ca2 +and short-term synaptic plasticity. Nature 371, 603–606. 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- Kann O, Hollnagel J-O, Elzoheiry S, Schneider J, 2016. Energy and Potassium Ion Homeostasis during Gamma Oscillations. Front. Mol. Neurosci 9, 47. 10.3389/FNMOL.2016.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Huchzermeyer C, Kovács R, Wirtz S, Schuelke M, 2011. Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain 134, 345–358. 10.1093/brain/awq333. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Frambach D.a, Proenza LM, 1985. Laminar profile of resistivity in frog retina. J. Neurophysiol 54, 1607–1619. 10.1152/jn.1985.54.6.1607. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Ben Haim L, Schirmer L, Tyzack GE, Tolman M, Miller JG, Tsai HH, Chang SM, Molofsky AV, Yang Y, Patani R, Lakatos A, Ullian EM, Rowitch DH, 2018. Kir4.1-Dependent Astrocyte-Fast Motor Neuron Interactions Are Required for Peak Strength. Neuron 98, 306–319. 10.1016/j.neuron.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JS, Krnjevic K, Somjen G, 1969. Divalent cations and electrical properties of cortical cells. J. Neurobiol 1, 197–208. 10.1002/neu.480010207. [DOI] [PubMed] [Google Scholar]
- Kirkland AE, Sarlo GL, Holton KF, 2018. The Role of Magnesium in Neurological Disorders. Nutrients 10. 10.3390/NU10060730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaerby C, Rasmussen R, Andersen M, Nedergaard M, 2017. Does Global Astrocytic Calcium Signaling Participate in Awake Brain State Transitions and Neuronal Circuit Function? Neurochem. Res 10.1007/s11064-017-2195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, Wolfart J, 2016. Potassium Channels in Epilepsy. Cold Spring Harb. Perspect. Med 6, a022871. 10.1101/cshperspect.a022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R, 1987. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J. Neurophysiol 57, 325–340. 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Ferreira-Filho CR, Nicholson C, 1983. Alkaline and acid transients in cerebellar microenvironment. J. Neurophysiol 49, 831–850. 10.1152/jn.1983.49.3.831. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Nicholson C, 1978. Extracellular ionic variations during spreading depression. Neuroscience 3, 1045–1059. 10.1016/0306-4522(78)90122-7. [DOI] [PubMed] [Google Scholar]
- Krishnan GP, González OC, Bazhenov M, 2018. Origin of slow spontaneous resting-state neuronal fluctuations in brain networks. Proc. Natl. Acad. Sci 115, 6858–6863. 10.1073/pnas.1715841115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjević K, Morris ME, Reiffenstein RJ, 1982. Stimulation-evoked changes in extracellular K+ and Ca2+ in pyramidal layers of the rat’s hippocampus. Can. J. Physiol. Pharmacol 60, 1643–1657. 10.1139/y82-243. [DOI] [PubMed] [Google Scholar]
- Kryger MH, Roth T. (Tom), Dement WC, 2017. Principles and practice of sleep medicine, 6th ed.. [Google Scholar]
- Kuffler SW, Nicholls JG, Orkand RK, 1966. Physiological properties of glial cells in the central nervous system of amphibia. J. Neurophysiol 29, 768–787. 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, Macaulay N, 2014. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62, 608–622. 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen BR, Stoica A, MacAulay N, 2016. Managing Brain Extracellular K(+) during Neuronal Activity: The Physiological Role of the Na(+)/K(+)-ATPase Subunit Isoforms. Front. Physiol 7, 141. 10.3389/fphys.2016.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavzin M, Rapoport S, Polsky A, Garion L, Schiller J, 2012. Nonlinear dendritic processing determines angular tuning of barrel cortex neurons in vivo. Nature 490, 397–401. 10.1038/nature11451. [DOI] [PubMed] [Google Scholar]
- LeBeau FEN, Towers SK, Traub RD, Whittington MA, Buhl EH, 2002. Fast network oscillations induced by potassium transients in the rat hippocampus in vitro. J. Physiol 542, 167–179. 10.1113/jphysiol.2002.015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-HH, Dan Y, 2012. Neuromodulation of Brain States. Neuron 76, 109–222. 10.1016/j.neuron.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW, 2005. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci 6, 312–324. 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lewis DV, Schuette WH, 1975. Temperature dependence of potassium clearance in the central nervous system. Brain Res. 99, 175–178. 10.1016/0006-8993(75)90623-X. [DOI] [PubMed] [Google Scholar]
- Lisman J, Idiart M, 1995. Storage of 7 +/− 2 short-term memories in oscillatory sub-cycles. Science (80-.) 267, 1512–1515. 10.1126/science.7878473. [DOI] [PubMed] [Google Scholar]
- Liu Z, Osipovitch M, Benraiss A, Huynh NPT, Foti R, Bates J, Chandler-Militello D, Findling RL, Tesar PJ, Nedergaard M, Windrem MS, Goldman SA, 2019. Dysregulated Glial Differentiation in Schizophrenia May Be Relieved by Suppression of SMAD4- and REST-Dependent Signaling. Cell Rep. 27, 3832–3843. 10.1016/j.celrep.2019.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone MS, Hubel DH, 1981. Effects of sleep and arousal on the processing of visual information in the cat. Nature 291, 554–561. 10.1038/291554a0. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Magee JC, 2006. Integrative Properties of Radial Oblique Dendrites in Hippocampal CA1 Pyramidal Neurons. Neuron 50, 291–307. 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Lux HD, 1974. Fast recording ion specific microelectrodes: Their use in pharmacoeogical studies in the CNS. Neuropharmacology 13, 509–517. 10.1016/0028-3908(74)90140-3. [DOI] [PubMed] [Google Scholar]
- Lux HD, Neher E, 1973. The equilibration time course of [K+]o in cat cortex. Exp. brain Res 17, 190–205. [DOI] [PubMed] [Google Scholar]
- Madeja M, 2000. Extracellular Surface Charges in Voltage-Gated Ion Channels. News Physiol. Sci 15, 15–19. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Lancaster B, Zucker RS, 1992. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron 9, 121–128. 10.1016/0896-6273(92)90227-5. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA, 1993. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 16, 521–527. [DOI] [PubMed] [Google Scholar]
- Maneepark M, Srikiatkhachorn A, Bongsebandhu-phubhakdi S, 2012. Involvement of AMPA Receptors in CSD-Induced Impairment of LTP in the Hippocampus. Headache J. Head Face Pain 52, 1535–1545. 10.1111/j.1526-4610.2012.02229.x. [DOI] [PubMed] [Google Scholar]
- Maquet P, 2001. The Role of Sleep in Learning and Memory. Science (80-.) 294, 1048–1052. 10.1126/science.1062856. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM, 2006. Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Markram H, Muller E, Ramaswamy S, Reimann MW, Abdellah M, Sanchez CA, Ailamaki A, Alonso-Nanclares L, Antille N, Arsever S, Kahou GAA, Berger TK, Bilgili A, Buncic N, Chalimourda A, Chindemi G, Courcol J-D, Delalondre F, Delattre V, Druckmann S, Dumusc R, Dynes J, Eilemann S, Gal E, Gevaert ME, Ghobril J-P, Gidon A, Graham JW, Gupta A, Haenel V, Hay E, Heinis T, Hernando JB, Hines M, Kanari L, Keller D, Kenyon J, Khazen G, Kim Y, King JG, Kisvarday Z, Kumbhar P, Lasserre S, Le Bé J-V, Magalhães BRC, Merchán-Pérez A, Meystre J, Morrice BR, Muller J, Muñoz-Céspedes A, Muralidhar S, Muthurasa K, Nachbaur D, Newton TH, Nolte M, Ovcharenko A, Palacios J, Pastor L, Perin R, Ranjan R, Riachi I, Rodríguez J-R, Riquelme JL, Rössert C, Sfyrakis K, Shi Y, Shillcock JC, Silberberg G, Silva R, Tauheed F, Telefont M, Toledo-Rodriguez M, Tränkler T, Van Geit W, Díaz JV, Walker R, Wang Y, Zaninetta SM, DeFelipe J, Hill SL, Segev I, Schürmann F, 2015. Reconstruction and Simulation of Neocortical Microcircuitry. Cell 163, 456–492. 10.1016/j.cell.2015.09.029. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadóttir H, Mölle M, Born J, 2006. Boosting slow oscillations during sleep potentiates memory. Nature 444, 610–613. 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Massimini M, Amzica F, 2001. Extracellular calcium fluctuations and intracellular potentials in the cortex during the slow sleep oscillation. J. Neurophysiol 85, 1346–1350. [DOI] [PubMed] [Google Scholar]
- Massimini M, Ferrarelli F, Huber R, Esser SK, Singh H, Tononi G, 2005. Breakdown of cortical effective connectivity during sleep. Science 309, 2228–2232. 10.1126/science.1117256. [DOI] [PubMed] [Google Scholar]
- Matyushkin DP, Krivoi II, Drabkina TM, 1995. Synaptic feed-backs mediated by potassium ions. Gen. Physiol. Biophys [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB, 1984. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263. 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA, 1985. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J. Neurophysiol 54, 782–806. 10.1152/jn.1985.54.4.782. [DOI] [PubMed] [Google Scholar]
- McGinley MJ, David SV, McCormick DA, 2015a. Cortical Membrane Potential Signature of Optimal States for Sensory Signal Detection. Neuron 87, 179–192. 10.1016/j.neuron.2015.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinley MJ, Vinck M, Reimer J, Batista-Brito R, Zagha E, Cadwell CR, Tolias AS, Cardin JA, McCormick DA, 2015b. Waking State: Rapid Variations Modulate Neural and Behavioral Responses. Neuron 87, 1143–1161. 10.1016/j.neuron.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S, 2004. Selective Effects of Potassium Elevations on Glutamate Signaling and Action Potential Conduction in Hippocampus. J. Neurosci 24, 197–206. 10.1523/JNEUROSCI.4845-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JS, Gotoh F, 1960. Metabolic and electroencephalographic effects of hyperventilation. Experimental studies of brain oxygen and carbon dioxide tension, pH, EEG and blood flow during hyperventilation. Arch. Neurol 3, 539–552. 10.1001/archneur.1960.00450050059007. [DOI] [PubMed] [Google Scholar]
- Miller KD, Troyer TW, 2002. Neural Noise Can Explain Expansive, Power-Law Nonlinearities in Neural Response Functions. J. Neurophysiol 87, 653–659. 10.1152/jn.00425.2001. [DOI] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U, 1987. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J. Neurophysiol 57, 869–888. 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- Monai H, Wang X, Yahagi K, Lou N, Mestre H, Xu Q, Abe Y, Yasui M, Iwai Y, Nedergaard M, Hirase H, 2019. Adrenergic receptor antagonism induces neuroprotection and facilitates recovery from acute ischemic stroke. Proc. Natl. Acad. Sci 116, 11010–11019. 10.1073/pnas.1817347116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BK, Miller KD, 2003. Multiplicative Gain Changes Are Induced by Excitation or Inhibition Alone. J. Neurosci 23, 10040–10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray Sherman S, 2001. Tonic and burst firing: Dual modes of thalamocortical relay. Trends Neurosci. 24, 122–126. 10.1016/S0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Mutch WAC, Hansen AJ, 1984. Extracellular pH Changes during Spreading Depression and Cerebral Ischemia: Mechanisms of Brain pH Regulation. J. Cereb. Blood Flow Metab 4, 17–27. 10.1038/jcbfm.1984.3. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP, 2013. Physiological roles of Aquaporin-4 in brain. Physiol. Rev 93, 1543–1562. 10.1152/physrev.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neske GT, McCormick DA, 2018. Distinct waking states for strong evoked responses in primary visual cortex and optimal visual detection performance. bioRxiv 437681. 10.1101/437681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton TH, Abdellah M, Chevtchenko G, Muller EB, Markram H, 2019. Voltage-sensitive dye imaging reveals inhibitory modulation of ongoing cortical activity. bioRxiv 812008. 10.1101/812008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, 1979. Brain cell microenvironment as a communication channel. In: Schmitt FO, Worden FG (Eds.), The Neurosciences: Fourth Study Program. MIT Press, pp. 457–476. [Google Scholar]
- Nicholson C, Bruggencate GT, Steinberg R, Stöckle H, 1977. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc. Natl. Acad. Sci. U. S. A 74, 1287–1290. 10.1073/pnas.74.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Phillips JM, 1981. Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J. Physiol 321, 225–257. 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, ten Bruggencate G, Stöckle H, Steinberg R, 1978. Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J. Neurophysiol 41, 1026–1039. 10.1152/jn.1978.41.4.1026. [DOI] [PubMed] [Google Scholar]
- Niell CM, Stryker MP, 2010. Modulation of Visual Responses by Behavioral State in Mouse Visual Cortex. Neuron 65, 472–479. 10.1016/j.neuron.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen FH, Johnson LK, 2010. Magnesium (Mg) supplementation improves magnesium status and decreases elevated C-reactive protein in adults older than 51 years with poor quality sleep. Faseb J. 24, 158–168. 10.1684/mrh.2010.0220. [DOI] [PubMed] [Google Scholar]
- Noh W, Pak S, Choi G, Yang Sungchil, Yang Sunggu, 2019. Transient Potassium Channels: Therapeutic Targets for Brain Disorders. Front. Cell. Neurosci 13, 265. 10.3389/fncel.2019.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte M, Reimann MW, King JG, Markram H, Muller EB, 2019. Cortical reliability amid noise and chaos. Nat. Commun 10, 3792. 10.1038/s41467-019-11633-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A, 1984. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462–465. 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Nuriya M, Morita A, Shinotsuka T, Yamada T, Yasui M, 2018. Norepinephrine induces rapid and long-lasting phosphorylation and redistribution of connexin 43 in cortical astrocytes. Biochem. Biophys. Res. Commun 504, 690–697. 10.1016/J.BBRC.2018.09.021. [DOI] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML, 2016. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 132, 1–21. 10.1007/s00401-016-1553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Gangwani MR, Allam SL, Tran D, Huang S, Hoang-Trong TM, Golshani P, Rumbell TH, Kozloski JR, Khakh BS, 2019. Transient, Consequential Increases in Extracellular Potassium Ions Accompany Channelrhodopsin2 Excitation. Cell Rep 27 10.1016/j.celrep.2019.04.078. 2249–2261.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana O, Sakmann B, 1998. Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. J. Physiol 513, 135–148. 10.1111/j.1469-7793.1998.135by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MS, Skinner F, Arshadmansab MF, Garcia I, Mello CF, Knaus H-G, Ermolinsky BS, Otalora LFP, Garrido-Sanabria ER, 2010. Altered expression and function of small-conductance (SK) Ca(2+)-activated K+ channels in pilocarpine-treated epileptic rats. Brain Res. 1348, 187–199. 10.1016/j.brainres.2010.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW, 1966. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol 29, 788–806. [DOI] [PubMed] [Google Scholar]
- Oster A, Faure P, Gutkin BS, 2015. Mechanisms for multiple activity modes of VTA dopamine neurons. Front. Comput. Neurosci 9, 95. 10.3389/fncom.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otazu GH, Tai L-H, Yang Y, Zador AM, 2009. Engaging in an auditory task suppresses responses in auditory cortex. Nat. Neurosci 12, 646–654. 10.1038/nn.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Shai AS, Reeve JE, Anderson HL, Paulsen O, Larkum ME, 2014. NMDA spikes enhance action potential generation during sensory input. Nat. Neurosci 17, 383–390. 10.1038/nn.3646. [DOI] [PubMed] [Google Scholar]
- Pan E, Stringer JL, 1997. Role of Potassium and Calcium in the Generation of Cellular Bursts in the Dentate Gyrus. J. Neurophysiol 77, 2293–2299. 10.1152/jn.1997.77.5.2293. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Vargová L, Reingruber J, Ezan P, Holcman D, Giaume C, Syková E, Rouach N, 2011. Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. U. S. A 108, 8467–8472. 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack M, Smirnov S, Kaila K, 1996. Proton modulation of functionally distinct GABA(A) receptors in acutely isolated pyramidal neurons of rat hippocampus. Neuropharmacology 35, 1279–1288. 10.1016/S0028-3908(96)00075-5. [DOI] [PubMed] [Google Scholar]
- Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, Bergles DE, 2014. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82, 1263–1270. 10.1016/j.neuron.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Rangel MT, Mercado-C R, Hernández-Rodríguez J, 1999. Regulation of Glial Na +/K+-ATPase by Serotonin: Identification of Participating Receptors. Neurochem. Res 24, 643–649. 10.1023/A:1021048308232. [DOI] [PubMed] [Google Scholar]
- Penn Y, Segal M, Moses E, 2016. Network synchronization in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A 113, 3341–3346. 10.1073/pnas.1515105113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CCH, 2019. Sensorimotor processing in the rodent barrel cortex. Nat. Rev. Neurosci 20, 533–546. 10.1038/s41583-019-0200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen AV, Jensen CS, Crépel V, Falkerslev M, Perrier J-F, 2017. Serotonin Regulates the Firing of Principal Cells of the Subiculum by Inhibiting a T-type Ca2+ Current. Front. Cell. Neurosci 11, 60. 10.3389/fncel.2017.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit J-M, Magistretti PJ, 2016. Regulation of neuron-astrocyte metabolic coupling across the sleep-wake cycle. Neuroscience 323, 135–156. 10.1016/j.neuroscience.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Polack PO, Friedman J, Golshani P, 2013. Cellular mechanisms of brain state-dependent gain modulation in visual cortex. Nat. Neurosci 16, 1331–1339. 10.1038/nn.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Kocsis JD, 1990. Dendritic action potentials activated by NMDA receptor-mediated EPSPs in CA1 hippocampal pyramidal cells. Brain Res. 524, 342–346. 10.1016/0006-8993(90)90714-M. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Mauk MD, Kocsis JD, 1987. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol 58, 404–416. [DOI] [PubMed] [Google Scholar]
- Poskanzer KE, Yuste R, 2016. Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sci. U. S. A 2016, 1–10. 10.1073/pnas.1520759113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner JB, Plum F, 1967. Spinal-fluid pH and neurologic symptoms in systemic acidosis. N. Engl. J. Med 277, 605–613. 10.1056/NEJM196709212771201. [DOI] [PubMed] [Google Scholar]
- Poulet JFA, Crochet S, 2019. The Cortical States of Wakefulness. Front. Syst. Neurosci 12, 64. 10.3389/fnsys.2018.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet JFA, Petersen CCH, 2008. Internal brain state regulates membrane potential synchrony in barrel cortex of behaving mice. Nature 454, 881–885. 10.1038/nature07150. [DOI] [PubMed] [Google Scholar]
- Pumain R, Kurcewicz I, Louvel J, 1983. Fast extracellular calcium transients: involvement in epileptic processes. Science (80-.) 222, 177–179. 10.1126/science.6623068. [DOI] [PubMed] [Google Scholar]
- Raimondo JV, Tomes H, Irkle A, Kay L, Kellaway L, Markram H, Millar RP, Akerman CJ, 2016. Tight Coupling of Astrocyte pH Dynamics to Epileptiform Activity Revealed by Genetically Encoded pH Sensors. J. Neurosci 36, 7002–7013. 10.1523/JNEUROSCI.0664-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo JV, Burman RJ, Katz AA, Akerman CJ, 2015. Ion dynamics during seizures. Front. Cell. Neurosci 9, 419. 10.3389/fncel.2015.00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom B, Yamate C, Connors B, 1985. Activity-dependent shrinkage of extracellular space in rat optic nerve: a developmental study. J. Neurosci 5, 532–535. 10.1523/JNEUROSCI.05-02-00532.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom BR, Sontheimer H, 1992. The Neurophysiology of Glial Cells. J. Clin. Neurophysiol 9, 224–252. 10.1097/00004691-199204010-00005. [DOI] [PubMed] [Google Scholar]
- Rasmussen R, Jensen MH, Heltberg ML, 2017. Chaotic Dynamics Mediate Brain State Transitions, Driven by Changes in Extracellular Ion Concentrations. Cell Syst. 5, 591–603. 10.1016/j.cels.2017.11.011. [DOI] [PubMed] [Google Scholar]
- Rasmussen R, Nicholas E, Petersen NC, Dietz AG, Xu Q, Sun Q, Nedergaard M, 2019. Cortex-wide Changes in Extracellular Potassium Ions Parallel Brain State Transitions in Awake Behaving Mice. Cell Rep. 28, 1182–1194. 10.1016/j.celrep.2019.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausche G, Igelmund P, Heinemann U, 1990. Effects of changes in extracellular potassium, magnesium and calcium concentration on synaptic transmission in area CA1 and the dentate gyrus of rat hippocampal slices. Pflugers Arch. 415, 588–593. [DOI] [PubMed] [Google Scholar]
- Reimer J, Froudarakis E, Cadwell CR, Yatsenko D, Denfield GH, Tolias AS, 2014. Pupil Fluctuations Track Fast Switching of Cortical States during Quiet Wakefulness. Neuron 84, 355–362. 10.1016/j.neuron.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robello M, Baldelli P, Cupello A, 1994. Modulation by extracellular pH of the activity of GABAA receptors on rat cerebellum granule cells. Neuroscience 61, 833–837. 10.1016/0306-4522(94)90406-5. [DOI] [PubMed] [Google Scholar]
- Robinson HP, Kawahara M, Jimbo Y, Torimitsu K, Kuroda Y, Kawana a, 1993. Periodic synchronized bursting and intracellular calcium transients elicited by low magnesium in cultured cortical neurons. J. Neurophysiol 70, 1606–1616. 10.1152/jn.1993.70.4.1606. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR, 1997. Gap junctions equalize intracellular Na+ concentration in astrocytes. Glia 20, 299–307. . [DOI] [PubMed] [Google Scholar]
- Ruminot I, Gutierrez R, Pena-Munzenmayer G, Anazco C, Sotelo-Hitschfeld T, Lerchundi R, Niemeyer MI, Shull GE, Barros LF, 2011. NBCe1 Mediates the Acute Stimulation of Astrocytic Glycolysis by Extracellular K+. J. Neurosci 31, 14264–14271. 10.1523/JNEUROSCI.2310-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Fine A, 2003. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron 37, 287–297. 10.1016/S0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak IA, Shevtsova NA, St-John WM, Paton JFR, Pierrefiche O, 2003. Endogenous rhythm generation in the pre-Botzinger complex and ionic currents: Modelling and in vitro studies. Eur. J. Neurosci 18, 239–257. 10.1046/j.1460-9568.2003.02739.x. [DOI] [PubMed] [Google Scholar]
- Schiemann J, Puggioni P, Dacre J, Pelko M, Domanski A, van Rossum MCW, Duguid I, 2015. Cellular mechanisms underlying behavioral state-dependent bidirectional modulation of motor cortex output. Cell Rep. 11, 1319–1330. 10.1016/j.celrep.2015.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y, 2000. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature 404, 285–289. 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Schurr A, Miller JJ, Payne RS, Rigor BM, 1999. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J. Neurosci 19, 34–39. 10.1523/jneurosci.19-01-00034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneviratne U, Cook MJ, D’Souza WJ, 2017. Electroencephalography in the Diagnosis of Genetic Generalized Epilepsy Syndromes. Front. Neurol 8, 499. 10.3389/fneur.2017.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Wu S-Y, Rancic V, Aggarwal A, Qian Y, Miyashita S-I, Ballanyi K, Campbell RE, Dong M, 2019. Genetically encoded fluorescent indicators for imaging intracellular potassium ion concentration. Commun. Biol 2. 10.1038/s42003-018-0269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PY, Savtchenko LP, Kamasawa N, Dembitskaya Y, McHugh TJ, Rusakov DA, Shigemoto R, Semyanov A, 2013. Retrograde Synaptic Signaling Mediated by K+ Efflux through Postsynaptic NMDA Receptors. Cell Rep. 5, 941–951. 10.1016/j.celrep.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Shin DS, Carlen PL, 2008. Enhanced Ih Depresses Rat Entopeduncular Nucleus Neuronal Activity From High-Frequency Stimulation or Raised Ke+. J. Neurophysiol 99, 2203–2219. 10.1152/jn.01065.2007. [DOI] [PubMed] [Google Scholar]
- Siemkowicz E, Hansen AJ, 1981. Brain extracellular ion composition and EEG activity following 10 minutes ischemia in normo- and hyperglycemic rats. Stroke 12, 236–240. 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- Singer W, Tretter F, Cynader M, 1976. The effect of reticular stimulation on spontaneous and evoked activity in the cat visual cortex. Brain Res. 102, 71–90. [DOI] [PubMed] [Google Scholar]
- Slais K, Vorisek I, Zoremba N, Homola A, Dmytrenko L, Sykova E, 2008. Brain metabolism and diffusion in the rat cerebral cortex during pilocarpine-induced status epilepticus. Exp. Neurol 209, 145–154. 10.1016/j.expneurol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Slezak M, Kandler S, Van Veldhoven PP, Van den Haute C, Bonin V, Holt MG, 2019. Distinct Mechanisms for Visual and Motor-Related Astrocyte Responses in Mouse Visual Cortex. Curr. Biol 29, 3120–3127. 10.1016/J.CUB.2019.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky I, Abumaria N, Wu LJ, Huang C, Zhang L, Li B, Zhao X, Govindarajan A, Zhao MG, Zhuo M, Tonegawa S, Liu G, 2010. Enhancement of Learning and Memory by Elevating Brain Magnesium. Neuron 65, 165–177. 10.1016/j.neuron.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Smith DAS, Connick JH, Stone TW, 1989. Effect of changing extracellular levels of magnesium on spontaneous activity and glutamate release in the mouse neocortical slice. Br. J. Pharmacol 97, 475–482. 10.1111/j.1476-5381.1989.tb11975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SL, Smith IT, Branco T, Häusser M, 2013. Dendritic spikes enhance stimulus selectivity in cortical neurons in vivo. Nature 503, 115–120. 10.1038/nature12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Bergsman JB, Harata NC, Scheller RH, Tsien RW, 2004. Recordings from Single Neocortical Nerve Terminals Reveal a Nonselective Cation Channel Activated by Decreases in Extracellular Calcium. Neuron 41, 243–256. 10.1016/S0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- Somjen G, Tombaugh G, 1998. pH modulation of neuronal excitability and central nervous system functions. In: Kaila K, Ransom B (Eds.), PH and Brain Function. John Wiley & Sons, pp. 373–393. [Google Scholar]
- Somjen GG, 2004. Ions in the Brain: Normal Function, Seizures, and Stroke. Oxford University press. [Google Scholar]
- Somjen GG, 2002. Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8, 254–267. [DOI] [PubMed] [Google Scholar]
- Somjen GG, 2001. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev 81, 1065–1096. [DOI] [PubMed] [Google Scholar]
- Somjen GG, 1980. Stimulus-evoked and seizure-related responses of extracellular calcium activity in spinal cord compared to those in cerebral cortex. J. Neurophysiol 44, 617–632. 10.1152/jn.1980.44.4.617. [DOI] [PubMed] [Google Scholar]
- Somjen GG, Müller M, 2000. Potassium-induced enhancement of persistent inward current in hippocampal neurons in isolation and in tissue slices. Brain Res. 885, 102–110. 10.1016/S0006-8993(00)02948-6. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS, 2015. Ca2+ signaling in astrocytes from Ip3r2−/− mice in brain slices and during startle responses in vivo. Nat. Neurosci 10.1038/nn.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, Rothhammer V, Chan A, Gold R, Berthele A, Bennett JL, Korn T, Hemmer B, 2012. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N. Engl. J. Med 367, 115–123. 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehr C, Rajanathan R, Matchkov VV, 2019. Involvement of the Na+,K+-ATPase isoforms in control of cerebral perfusion. Exp. Physiol 104, 1023–1028. 10.1113/EP087519. [DOI] [PubMed] [Google Scholar]
- Steriade M, Amzica F, Nuñez A, 1993a. Cholinergic and noradrenergic modulation of the slow (approximately 0.3 Hz) oscillation in neocortical cells. J. Neurophysiol 70, 1385–1400. [DOI] [PubMed] [Google Scholar]
- Steriade M, Domich L, Oakson G, 1986. Reticularis thalami neurons revisited: activity changes during shifts in states of vigilance. J. Neurosci 6, 68–81. 10.1523/JNEUROSCI.06-01-00068.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ, 1993b. Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685. 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F, 2001. Natural waking and sleep states: a view from inside neocortical neurons. J. Neurophysiol 85, 1969–1985. 10.1016/j.neuroimage.2009.03.074. [DOI] [PubMed] [Google Scholar]
- Stickgold R, 2005. Sleep-dependent memory consolidation. Nature 437, 1272–1278. 10.1038/nature04286. [DOI] [PubMed] [Google Scholar]
- Sturm P, Wimmers S, Schwarz JR, Bauer CK, 2005. Extracellular potassium effects are conserved within the rat erg K+ channel family. J. Physiol 564, 329–345. 10.1113/jphysiol.2004.078840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, 2013. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Kosugi Y, Kawakami E, Piao Y-S, Hashimoto T, Oyanagi K, 2009. Magnesium concentration in the cerebrospinal fluid of mice and its response to changes in serum magnesium concentration. Magnes. Res 22, 266–272. 10.1684/mrh.2009.0186. [DOI] [PubMed] [Google Scholar]
- Suomalainen P, 1938. Production of Artificial Hibernation. Nature 142 10.1038/1421157a0. 1157–1157. [DOI] [Google Scholar]
- Syková E, 2003. Diffusion Parameters of the Extracellular Space. Isr. J. Chem 43, 55–69. 10.1560/11GA-DQD7-DC41-Q1U7. [DOI] [Google Scholar]
- Syková E, 1992. Ionic and Volume Changes in the Microenvironment of Nerve and Receptor Cells, Progress in Sensory Physiology. Springer, Berlin Heidelberg, Berlin, Heidelberg. 10.1007/978-3-642-76937-5. [DOI] [Google Scholar]
- Syková E, Nicholson C, 2008. Diffusion in brain extracellular space. Physiol. Rev 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syková E, Rothenberg S, Krekule I, 1974. Changes of extracellular potassium concentration during spontaneous activity in the mesencephalic reticular formation of the rat. Brain Res. 79, 333–337. 10.1016/0006-8993(74)90428-4. [DOI] [PubMed] [Google Scholar]
- Syková E, Svoboda J, Polák J, Chvátal A, 1994. Extracellular Volume Fraction and Diffusion Characteristics during Progressive Ischemia and Terminal Anoxia in the Spinal Cord of the Rat. J. Cereb. Blood Flow Metab 14, 301–311. 10.1038/jcbfm.1994.37. [DOI] [PubMed] [Google Scholar]
- Taira T, Smirnov S, Voipio J, Kaila K, 1993. Intrinsic proton modulation of excitatory transmission in rat hippocampal slices. Neuroreport 4, 93–96. 10.1097/00001756-199301000-00024. [DOI] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TNT, Kim TTK, Jarsky T, Yao Z, Levi BB, Gray LT, Sorensen SA, Dolbeare T, Bertagnolli D, Goldy J, Shapovalova N, Parry S, Lee CC, Smith K, Bernard A, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H, Yao Z, Lee CC, Shapovalova N, Parry S, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H, 2016. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 10.1038/nn.4216. advance on, 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuki F, Sunagawa GA, Shi S, Susaki EA, Yukinaga H, Perrin D, Sumiyama K, Ukai-Tadenuma M, Fujishima H, Ohno R, Tone D, Ode KL, Matsumoto K, Ueda HR, 2016. Involvement of Ca2+-Dependent Hyperpolarization in Sleep Duration in Mammals. Neuron 90, 70–85. 10.1016/j.neuron.2016.02.032. [DOI] [PubMed] [Google Scholar]
- Taylor C, Dudek F, 1982. Synchronous neural afterdischarges in rat hippocampal slices without active chemical synapses. Science (80-.) 218, 810–812. 10.1126/science.7134978. [DOI] [PubMed] [Google Scholar]
- Terlau H, Ludwig J, Steffan R, Pongs O, Stühmer W, Heinemann SH, 1996. Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflügers Arch. - Eur. J. Physiol 432, 301–312. 10.1007/s004240050137. [DOI] [PubMed] [Google Scholar]
- Therien AG, Blostein R, 2000. Mechanisms of sodium pump regulation. Am. J. Physiol. Physiol 279, C541–C566. 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- Thrane AS, Rangroo Thrane V, Zeppenfeld D, Lou N, Xu Q, Nagelhus EA, Nedergaard M, 2012. General anesthesia selectively disrupts astrocyte calcium signaling in the awake mouse cortex. Proc. Natl. Acad. Sci. U. S. A 109, 18974–18979. 10.1073/pnas.1209448109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Somjen GG, 1996. Effects of extracellular pH on voltage-gated Na+, K + and Ca2+ currents in isolated rat CA1 neurons. J. Physiol 493, 719–732. 10.1113/jphysiol.1996.sp021417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson M.a, Mody I, Olsen ML, Sofroniew MV, Khakh BS, 2014. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci 17, 694–703. 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG, 1990. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345, 347–350. 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R, 1988. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J. Neurophysiol 59, 259–276. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W, 2010. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci 11, 100–113. 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- Utzschneider D, Kocsis J, Devor M, 1992. Mutual excitation among dorsal root ganglion neurons in the rat. Neurosci. Lett 146, 53–56. 10.1016/0304-3940(92)90170-C. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM, 1997. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys. J 72, 2103–2116. 10.1016/S0006-3495(97)78853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, 2018. Physiology of Astroglia. Physiol. Rev 98, 239–389. 10.1152/physrev.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinck M, Batista-Brito R, Knoblich U, Cardin JA, 2015. Arousal and Locomotion Make Distinct Contributions to Cortical Activity Patterns and Visual Encoding. Neuron 86, 740–754. 10.1016/j.neuron.2015.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voipio J, Kaila K, 1993. Interstitial PCO2 and pH in rat hippocampal slices measured by means of a novel fast CO2/H(+)-sensitive microelectrode based on a PVC-gelled membrane. Pflugers Arch. 423, 193–201. 10.1007/bf00374394. [DOI] [PubMed] [Google Scholar]
- Walz W, 2000. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int 36, 291–300. [DOI] [PubMed] [Google Scholar]
- Wang F, Smith N. a., Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M, 2012a. Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci. Signal 5, ra26. 10.1126/scisignal.2002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Xu Q, Wang W, Takano T, Nedergaard M, 2012b. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc. Natl. Acad. Sci. U. S. A 109, 7911–7916. 10.1073/pnas.1120380109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger Combremont A-L, Bayer L, Dupré A, Mühlethaler M, Serafin M, 2016. Slow Bursting Neurons of Mouse Cortical Layer 6b Are Depolarized by Hypocretin/Orexin and Major Transmitters of Arousal. Front. Neurol 7, 88. 10.3389/fneur.2016.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Osipovitch M, Liu Z, Bates J, Chandler-Militello D, Zou L, Munir J, Schanz S, McCoy K, Miller RH, Wang S, Nedergaard M, Findling RL, Tesar PJ, Goldman SA, 2017. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 21 10.1016/j.stem.2017.06.012. 195–208.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M, 2013. Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z-G, Lu W-Y, MacDonald JF, 1997. Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc. Natl. Acad. Sci 94, 7012–7017. 10.1073/pnas.94.13.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto L, Heinemann U, 1983. Aspartate and glutamate induced reductions in extracellular free calcium and sodium concentration in area CA1 of “in vitro” hippocampal slices of rats. Neurosci. Lett 35, 79–84. 10.1016/0304-3940(83)90530-X. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Yue Z, Arnold DM, Artiushin G, Sehgal A, 2018. A Circadian Clock in the Blood-Brain Barrier Regulates Xenobiotic Efflux. Cell 173 10.1016/j.cell.2018.02.017. 130–139.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Nguyen KT, Barrett EF, David G, 2010. Vesicular ATPase Inserted into the Plasma Membrane of Motor Terminals by Exocytosis Alkalinizes Cytosolic pH and Facilitates Endocytosis. Neuron 68, 1097–1108. 10.1016/j.neuron.2010.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]