

Abstract
Human immunodeficiency virus (HIV) is the primary infectious agent of acquired immunodeficiency syndrome (AIDS), and non-nucleoside reverse transcriptase inhibitors (NNRTIs) are the cornerstone of HIV treatment. In the last 20 years, our medicinal chemistry group has made great strides in developing several distinct novel NNRTIs, including 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT), thio-dihydro-alkoxy-benzyl-oxopyrimidine (S-DABO), diaryltriazine (DATA), diarylpyrimidine (DAPY) analogues, and their hybrid derivatives. Application of integrated modern medicinal strategies, including structure-based drug design, fragment-based optimization, scaffold/fragment hopping, molecular/fragment hybridization, and bioisosterism, led to the development of several highly potent analogues for further evaluations. In this paper, we review the development of NNRTIs in the last two decades using the above optimization strategies, including their structure–activity relationships, molecular modeling, and their binding modes with HIV-1 reverse transcriptase (RT). Future directions and perspectives on the design and associated challenges are also discussed.
Keywords: HIV-1, NNRTIs, HENTs, S-DABOs, DATAs, DAPYs, Structure-based optimization, Fragment-based drug design, Molecular hybridization, Bioisosterism
Abbreviations: AIDS, acquired immunodeficiency syndrome; DAPY, diarylpyrimidine; DATA, diaryltriazine; DLV, delavirdine; DOR, doravirine; ECD, electronic circular dichroism; ee, enantiomeric excess; EFV, efavirenz; ETR, etravirine; FDA, U.S. Food and Drug Administration; HAART, highly active antiretroviral therapy; HENT, napthyl-HEPT; HEPT, 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine; HIV, human immunodeficiency virus; INSTI, integrase inhibitor; NNIBP, NNRTI binding pocket; NNRTI, non-nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; NVP, nevirapine; PI, protease inhibitor; PK, pharmacokinetic; PROTAC, proteolysis targeting chimera; RPV, rilpivirine; RT, reverse transcriptase; SAR, structure–activity relationship; SBDD, structure-based drug design; S-DABO, thio-dihydro-alkoxy-benzyl-oxopyrimidine; SFC, supercritical fluid chromatography; SI, selectivity index; UNAIDS, the Joint United Nations Programme on HIV/AIDS
Graphical abstract
Human immunodeficiency virus (HIV) is the primary infectious agent of acquired immunodeficiency syndrome (AIDS), and non-nucleoside reverse transcriptase inhibitors (NNRTIs) are the cornerstone of HIV treatment. In this review, we summarize the development of NNRTIs in our past two decades using the multiple optimization strategies.
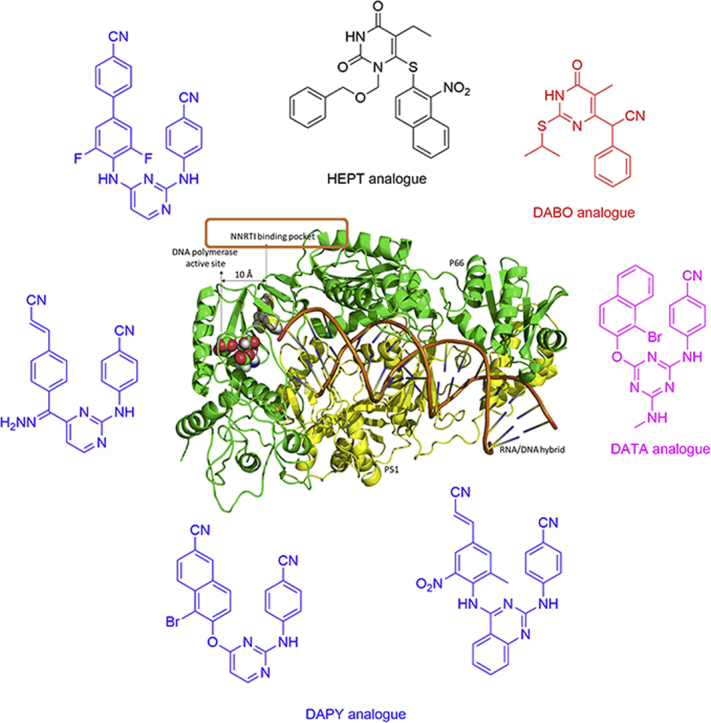
1. Introduction
Acquired immunodeficiency syndrome (AIDS) is considered as a pandemic caused by human immunodeficiency virus (HIV) infection. According to the latest data from the Joint United Nations Programme on HIV/AIDS (UNAIDS), 36.9 million people globally were living with HIV, and 21.7 million people were receiving antiretroviral therapy in 20171. Remarkable reduction in AIDS-related mortality has been made possible by the current combination therapy, termed highly active antiretroviral therapy (HAART). Typically, HAART targets multiple viral replication cycles and includes two nucleoside reverse transcriptase inhibitors (NRTIs), a non-nucleoside reverse transcriptase inhibitor (NNRTI), and a protease inhibitor (PI) or an integrase inhibitor (INSTI)2, 3, 4. The NNRTIs are essential components in the drug combination therapies due to their unique antiviral activity, high specificity, and low toxicity5, 6, 7. To date, more than 50 chemotypes have been identified as NNRTIs, including 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymines (HEPTs), dihydro-alkoxy-benzyl-oxopyrimidines (DABOs), diarylpyrimidines (DAPYs), etc.7, 8, 9. Among these, U.S. Food and Drug Administration (FDA) approved six NNRTIs for HIV-1 treatment (Fig. 1A), namely nevirapine (NVP, 1996), delavirdine (DLV, 1997), efavirenz (EFV, 1998), etravirine (ETR, 2008), rilpivirine (RPV, 2011), and doravirine (DOR, 2018)3,10. NNRTIs inhibit reverse transcriptase (RT) by binding to an allosteric site, namely NNRTI binding pocket (NNIBP), located about 10 Å away from the DNA polymerase active site (Fig. 1B). Although the NNRTIs are structurally different and unrelated, they fit within the NNIBP and their binding modes are in a similar conformation to be considered as a “butterfly”, “horseshoe”, or “U” mode with a central scaffold and two “wings” (Fig. 1A). This feature provides valuable information for the discovery of NNRTIs and lead optimizations.
Figure 1.

(A) The chemical and three-dimension structures of FDA-approved NNRTI drugs obtained from the crystal complexes with HIV-1 RT showing similar binding conformations: a “butterfly”, “horseshoe”, or “U” mode with a central scaffold and two “wings” (B) Structure of HIV-1 RT (P66/P51) in complex with RNA/DNA and a NNRTI, NVP, are highlighted in different colors (PDB entry: 4PUO).
However, a growing number of drug-resistant HIV-1 strains in NNRTI-treated cells and patients with HIV-1 are being reported11. Mutations within or near the NNIBP dramatically reduce the efficacy of NNRTIs, resulting in treatment failures. Therefore, drug resistance poses a considerable challenge in anti-HIV-1 drug development. In the last 20 years (1999–2019), our medicinal chemistry group tackled the development of novel anti-HIV-1 drugs using multiple optimization strategies involving the classical NNRTIs, including structure- and fragment-based drug design, scaffold hopping, molecular/fragment hybridization, and bioisosterism to identify novel NNRTIs with high antiviral activity against wild-type (WT) and mutant viruses. With ongoing efforts, many derivatives have been obtained; in particular, two DAPY analogues have advanced to preclinical evaluations. In this article, we summarize our efforts on structure optimizations and development strategies, structure–activity relationship (SAR) studies, and computational analysis. Future directions and perspectives on the design and development of anti-HIV agents and associated challenges are also discussed.
2. Development of NNRTIs
2.1. Naphthyl-HEPT (HENT) analogues
In 1989, the first discovered NNRTI, HEPT (Fig. 2) was reported by Miyasaka et al.12 as a novel anti-HIV-1 lead compound, showing promising antiviral activity (EC50 = 7 μmol/L) and selectivity index (selectivity index, SI = 106). Subsequent optimizations led to a more potent ethoxymethyl analogue MKC-442, also named emivirine (Coactinon), which was selected as a candidate drug for phase III clinical trials in AIDS patients (NCT00002413). However, the clinical trial by Triangle Pharmaceuticals in 2002 was terminated due to lower drug potency compared to other antiretrovirals13. Besides, the drug resistance mediated by the NNIBP mutations limited the use of HEPT analogues13.
Figure 2.

Structure optimization workflow for the HEPT analogues developed by our group. The modified parts are colored.
As shown in Fig. 3A, the MKC-442-RT co-crystal complex showed that (1) MKC-442 was constrained by a strong hydrogen-bonding interaction between the 3-NH of the pyrimidine ring with the carbonyl oxygen of K101 and water-mediated hydrogen bonds with K101 and E138; (2) the 6-benzyl ring of the inhibitor, inserting into the hydrophobic region consisting of Y181, Y188, and W229 residues, were conformationally different from those in the unliganded RT structure and formed π–π stacking interactions with these aromatic residues14. Steric bulky groups could not replace the isopropyl group located at the E138 site due to limited space in the Y181 and E138 channel. The ethoxymethyl tail was situated in the F227, Y318, and P236 region. Therefore, the hydrophilic hydroxyl group was replaced by a hydrophobic group to improve the antiviral activity15.
Figure 3.

(A) Crystal complex of MKC-442 with RT (PDB: 1RT1); and molecular docking models of RT with compounds 1 (B), 2 (C), 4 (D) and 9 (E). The figures were generated by PyMOL.
Based on the structural biology information, two parts of HEPT or MKC-442 have been optimized, and the representative structure optimization workflow is summarized in Fig. 216, 17, 18, 19, 20, 21, 22, 23, 24. (1) Replacement of the 6-benzyl ring by a naphthyl ring could enhance π-stacking interactions in the NNIBP13. (2) Introducing an aromatic ring such as a phenyl group at the ethoxymethyl tail aimed at increasing the hydrophobic interactions with the Y318/P236 channel. Consistent with the co-crystal information, our 3D-QSAR study also revealed that the 6-benzyl ring of MKC-442 could be replaced by the α-naphthyl ring (abbreviated as HENT)21,23. The resulting compound 1 exhibited an EC50 of 17 nmol/L against HIV-1 WT strain in MT-4 cells (HEPT, EC50 = 7 μmol/L; MKC-442, EC50 = 20 nmol/L)13 and relatively low cytotoxicity (SI = 2229). Compounds 2 and 3 with phenyl and benzyl groups at the ethoxymethyl tail had similar potency and cytotoxicity as compound 122. Molecular docking studies showed that the naphthyl group located at the hydrophobic region strengthened the π–π stacking interactions with Y181, Y188, and W229 (Fig. 3B and C), and the phenyl ring brought an additional hydrophobic interaction with the Y318 (Fig. 3C).
Carbon to sulfur variation is observed commonly in drug development using a bioisosterism strategy. A new series of naphthylthio HEPT (S-HENT) was synthesized based on the original HEPT and bioisosteric replacement17. Compound 4 with α-naphthyl had an EC50 of 48 nmol/L while the analogue with β-naphthyl (compound 5) decreased the anti-HIV-1 activity by 13.5-fold, indicating limited space or flexibility for the naphthyl group at the hydrophobic pocket (Fig. 3D). Enhanced π–π stacking interactions by introducing an α-nitro group in compound 6 were probably due to the reduced electron density. Consequently, the antiviral activity of compound 6 improved by 10-fold with an EC50 of 65 nmol/L. However, the amino substitution (7) with a different electronic effect dramatically decreased the interaction, with an EC50 of 3650 nmol/L. A carbonyl group between the naphthyl and central pyrimidine ring was tolerated at the binding pocket (Fig. 3E)18. However, the antiviral activity decreased by 6–8 fold for compounds 8 and 9, potentially due to lack of π–π interaction with Y181 by the flipped naphthyl ring. Further SAR studies illustrated that the ester group presenting on the phenyl side chain played a critical role in the antiviral activity. The replacement of the β-oxygen of the N-1 side chain with a carbonyl group (e.g., compound 10) significantly suppressed the anti-HIV-1 activity20.
2.2. DABO analogues
The DABO analogues were first reported in 1992 as novel NNRTIs25. Chemically, the DABOs bearing the 4-pyrimidinone scaffold, are structurally similar to HEPTs with the N-1 substitution changed to the C-2 position (Fig. 4), thus leading to reasonably high anti-HIV activity26,27. The molecular docking study (Fig. 5) showed that (1) the central pyrimidinone scaffold was located at the E138, K101 and K103 region forming two hydrogen bonds with K101; (2) at the C-5 position, the hydrophobic cavity of V179 and K103 residues had limited space, suggesting use of small chemical groups such as methyl, ethyl and isopropyl group at this site; (3) optimization of the side chain at the C-2 position could be attempted because it was pointing to a hydrophobic channel surrounded by V106, F227, and P236; (3) the oxygen could be replaced by sulfur using a bioisosterism strategy and a hydrogen-bond acceptor (e.g., –OCH3) could be introduced to improve the activity.
Figure 4.

Structure optimization workflow for the DABO analogues developed by our group. The modified parts are colored.
Figure 5.

Molecular docking models of RT with compounds 11 (A), 13 (B), 18 (C), 17-S (D), 17-R (E) and the superposition mode of 17-S and 17-R (F). The figures were generated by PyMOL.
Thus, the structure-based drug design (SBDD) was conducted to obtain several potent analogues. Compounds 11 and 12 showed great potency in the nanomolar range and high SI values compared with the micromolar-range DABOs (Fig. 4)25,28. Similar to HEPT, the dimethylbenzyl ring was inserted into the hydrophobic tunnel of Y181, Y188, F227, and W229 residues (Fig. 5A). A naphthyl group was introduced to obtain compounds 13 and 14 with an EC50 of 30 and 45 nmol/L, respectively (Fig. 5B)29, 30, 31.
Subsequently, the linkers between the central pyrimidinone and side chains were modified. First, we focused on the left part of the molecule. The carbonyl group of the C-2 alkylthio chain was changed to a thioether (compound 15, EC50 = 120 nmol/L), and the amide was generated by introducing a 4-chlorinated aniline (compound 19 with a decreased EC50 of 210 nmol/L), proving the importance of the carbonyl group of the C-2 alkylthio chain32,33. Nevertheless, the carbonyl group did not show any hydrogen-bonding interaction with the surrounding residues (Fig. 5A and B). In contrast, the linker on the molecule's right part was also changed. Reports34,35 show suitable modifications of the methylene bridge between the aryl moiety and pyrimidine might favor the π-stacking interactions between the electron-deficient pyrimidine ring and the electron-rich phenyl ring of RT Tyr188 or Tyr181. Mai et al.34, encouraged by the conformational restriction of a methyl linker, replaced the methylene by a carbonyl group to obtain compound 16, which exhibited an EC50 of 290 nmol/L in HIV-1 infected cells35. However, it was less potent than the methyl analogue probably due to an unfavorable orientation of the 3-F-benzyl moiety and weaker hydrophobic interactions with V106, P225, and F227.
Next, we changed the methyl to a cyano group, commonly considered as a bioisostere of carbonyl, hydroxyl, carboxyl, and halogen groups36. The newly obtained compounds 17 and 20 had better potency (EC50 ∼90 nmol/L). Meanwhile, a chiral center was introduced at the CH–CN position. Based on molecular docking analysis, the two isomers of compound 17 (Fig. 5D and E) showed similar superpositioned binding modes (Fig. 5F) as the cyano group was acceptable in the hydrophobic tunnels of V179/E138 and V106/F22737. Finally, we undertook structural simplifications of the cyano analogues38. When the p-methoxylbenzyl was removed, the alkyl groups, e.g., isopropyl, were accepted in the V106/F227 tunnel (Fig. 5C). Compound 18 had the best EC50 (2.0 ± 0.2 nmol/L) and SI value (4600) in this series. However, higher cytotoxicity (CC50 = 0.8 μmol/L) was observed compared to benzylthio compounds.
2.3. DATA analogues
The DATA, bearing a triazine amine scaffold, is an important family of HIV-1 NNRTIs (Fig. 6)39,40. The co-crystal complex of compound 21 (R129385) with RT (PDB: 1S9E) provided detailed insight for structure-based optimization41. As shown in Fig. 7A, compound 21 interacted with RT in a typical “butterfly” conformation. The 4-cyanophenyl was inserted into the hydrophobic pocket containing F227/P236 residues. The central triazine amine was located at the entrance channel of NNRTI binding pocket, a largely open solvent-exposed region in front of K101, E138, and V179 residues. The amides and N-3 formed hydrogen bonds with residues E138 and K101. The 2,6-dichlorophenoxy group was inserted into the aromatic-rich W229 subpocket.
Figure 6.

Structure optimization workflow for the DATA analogues developed by our group. The modified parts are colored.
Figure 7.

Co-crystal complex of compound 21 with RT (PDB: 1S9E) (A); and molecular docking models of RT with compounds 22 (B), 23 (C), and 24 (D). The figures were generated by PyMOL.
Our 3D-QSAR study42 and the subsequently disclosed co-crystal complex41 suggested that increasing the volume of the substituent(s) on the C-6 of triazine ring might favor strengthening the π–π stacking interactions between the ligand and the highly conserved hydrophobic region (W229)43. In addition, the entrance channel was another suitable site for ligand optimization44. The newly synthesized compound 22 with a β-naphthyl and N-allyl group showed promising anti-HIV-1 activity (EC50 = 34 ± 1 nmol/L), low cytotoxicity (CC50 = 220 μmol/L), and a good SI value (6475). As expected, the molecular docking analysis showed that the naphthyl group strengthened the π–π stacking interactions with Y181, Y188, and W229, which could be improved dramatically by the electron-withdrawing bromo group. The N-allyl group occupied the entrance channel and maintained the hydrogen bond with residue E138 (Fig. 7B). However, the interaction was not critical to the activity (Fig. 7C) as replacement of allyl by a methyl (compound 23) group improved activity with an EC50 of 9 ± 3 nmol/L and higher SI value (15,385)45. Finally, the effect of the α/β-naphthyl was determined. Compound 24 with the α-naphthyl group had an EC50 value of 93 ± 40 nmol/L, which was 10-fold lower than that of the β-naphthyl compound 23. Similar to DABOs, the π-stacking interactions weakened as α-naphthyl was flipped, placing one of the phenyl rings outside the hydrophobic pocket (Fig. 7D). In addition, a series of fluorine-containing DATA analogues (structure not shown) was obtained with no significantly improved antiviral activity46. Further optimization by introducing biphenyl to replace the naphthyl ring enhanced the π-stacking interactions with W229 (see details in section 2.4.). Compound 25 with a pyrrolidinol significantly improved the activity against not only WT but also mutant strains except F227L + V106A double mutants. However, its high cytotoxicity (CC50 = 4.5 ± 2.6 nmol/L) made it not suitable for further in vivo evaluation47.
2.4. DAPY analogues
The DAPY, first reported in 2001, is a novel class of NNRTIs with highly potent antiviral activity, and ultimately yielded ETR and RPV approved by the FDA48. They form a flexible “butterfly” conformation, minimizing the loss of binding stability41,48. However, a growing number of drug-resistant mutants, especially E138K, are recognized from the failure of RPV or ETR treatments in the clinic49,50. Besides, the absolute oral bioavailability of ETR in humans is still unknown, due to poor solubility and lack of suitable intravenous formulation51,52. The high cytotoxicity (CC50 ∼5 μmol/L) of ETR and RPV cannot be ignored. Thus, we focused our efforts to find novel NNRTIs using ETR or RPV as the lead compound, aiming at overcoming drug resistance, and improving safety and pharmacokinetic (PK) profiles (Figure 8, Figure 9, Figure 10).
Figure 8.

Structure (left “wing”) optimization workflow for the DAPY analogues developed by our group. The modified parts are colored.
Figure 9.

Molecular docking models of RT (PDB: 3MEC) with compounds 26 (A), 27 (B), 28 (C) and 33 (F); RT (PDB: 2ZD1) with 29 (D), and 30 (E). The figures were generated by PyMOL.
Figure 10.

Linker optimization workflow for the DAPY analogues developed by our group. The modified parts are colored.
Similar to previous HENT, DABO, and DATA scaffolds, the naphthyl group was also introduced into the left “wing” on the DAPY C4-position based on the co-crystal complex of ETR with RT (Fig. 8)53. Compound 26 with α-bromo-β-naphthyl showed an IC50 value of 2.4 nmol/L against HIV-1 infected MT-4 cells and low cytotoxicity (CC50 = 154 μmol/L) with a high SI of 65,591. However, the activity against the double mutants (K103N + Y181C) was weak (EC50 = 6570 nmol/L). Molecular docking analysis showed that: (1) the central pyrimidine and the NH bridge of the cyanophenyl wing formed two hydrogen-bonding interactions with K101 (Fig. 9A); (2) the naphthyl and cyanophenyl groups inserted into the hydrophobic pockets formed strong π–π interactions (Fig. 9B). Further optimizations of the naphthyl group by introducing a cyano group at C6-position (e.g., compound 27) exhibited higher potency with an extremely high SI value (181,247) against WT HIV-1 but also against the K103N + Y181C double mutant than for compound 26. The calculated results showed enhanced π-stacking interaction between the inhibitor and W229 with an interaction energy (Eint) of −5.55 kcal/mol, much higher than the non-cyano analogue (Eint = −1.76 kcal/mol)54. The SAR study showed that the dual-substituted compounds at C1- and C3-positions of the naphthyl ring exhibited more potent activity against the mutant viruses. The di-methoxy group (28) was introduced to constrain the conformation and improve the π-stacking interaction (Fig. 9C), and effectively inhibited WT HIV-1 (EC50 = 5 nmol/L) and K103N + Y181C double mutant (EC50 = 160 nmol/L), showing low cytotoxicity (CC50 > 118 μmol/L) and high SI (>25,000).
To extend the length of the π–π stacking conjugated system in the left “wing”, the biphenyl ring was utilized for fragment-based drug design of biphenyl-DAPYs55,56. The Alexandre group and our QSAR study57,58 both demonstrated that interaction of a polar cyano group with W229 was particularly beneficial to improve mutant sensitivity to drugs. The resulting compound 29 with a 4′-cyano group on the biphenyl ring, displayed EC50 values of 1.0, 1.3, 0.8, 1.5, 11.0, 2.0, and 10.0 nmol/L against WT HIV-1, L100I, K103N, Y181C, Y188L, E138K, and K103N + Y181C mutants, respectively55. The docking modes showed that the biphenyl ring markedly strengthened the π–π stacking effect by inserting deeply into the W229 pocket as proposed (Fig. 9D). However, 29 exhibited a high cytotoxicity value CC50 of 2.1 μmol/L and a relative low SI of 2059. The aqueous solubility and liver metabolic stability were still unsatisfactory.
The thiophene [3,2-d]pyrimidine scaffold first reported by Liu and Zhan's groups59, exhibited excellent safety, low toxicity, and high SI values. We further introduced a thiophene ring into our biphenyl-DAPYs by a scaffold-hopping strategy aiming at overcoming the issue of high cytotoxicity of biphenyl DAPYs (Figure 8, Figure 9E). In this series, compound 30 with the di-fluoro group on the biphenyl ring had much lower cytotoxicity (CC50 = 216.9 μmol/L) and higher SI of 16,094 than compound 29, maintained marked inhibitory activity (EC50 = 13.5, 9.4, 17.0, 52.0, and 58.2 nmol/L) against WT, K103N, E138K, L100I, and Y181C mutants, respectively, and moderate activity towards double mutants56. To further explore the conformationally constrained effects, substituents at different positions of the biphenyl ring have been introduced60. As a result, the 2′-methyl group, attributed to enlarge the dihedral angle of biphenyl rings and compound 31 with dimethyl groups, exhibited potent biological activity against WT HIV-1-infected MT-4 cells (EC50 = 14 nmol/L) and adequate activity against mutant variants with moderate cytotoxicity (CC50 = 66 μmol/L). Due to high cytotoxicity and metabolic potential of the dimethyl phenyl analogues, a series of non-dimethylphenyl-diarylpyrimidines were also developed. Compound 32 with di-fluoro biphenyl moiety showed good antiviral activity against not only WT (EC50 = 1.3 nmol/L) but also mutant strains (EC50 = 10.8 nmol/L, L100I; 2.6 nmol/L, K103N; 6.1 nmol/L, Y181C; 130 nmol/L, Y188L; 1.9 nmol/L, E138K), and RT enzyme (EC50 = 4.7 nmol/L). The hydrochlorate form of compound 32 exhibited improved water solubility of 5.6 μg/mL much higher than ETR (<<1 μg/mL) and 29, better liver metabolic stability, and a great oral bioavailability of 44% as a drug candidate. In addition, no apparent toxicity was observed in an acute toxicity assay (2 g/kg) and HE staining did not show any damage to the mouse organs.
In contrast, the saturated rings, e.g., cycloalkyl rings, were developed based on bioisosterism principle61. The left “wing” of DAPYs and three linkers (O, NH or S) between the cycloalkyl and the central pyrimidine were maintained (Fig. 8). The SAR demonstrated that the antiviral activity generally improved in series O < NH < S. The bulky cycloalkyl groups affected the activity as follows: cyclopropyl < cyclopentyl < cyclohexyl, indicating the importance of the left “wing”. Compound 33 with a cyclohexyl group had moderate anti-HIV-1 activity (EC50 = 1160 nmol/L) and no significant improvement was observed with nitrogen replacement (compound 34, EC50 = 960 nmol/L). Compound 35, with a sulfur linker, showed a marked increase in antiviral activity against WT HIV-1 (EC50 = 55 nmol/L, SI = 7290) in cells. The activity in cells might be attributed to the effect on the physicochemical properties but not by strengthening the π–π interaction with W229 pocket (Fig. 9F).
Encouraged by the series of compounds 33–35, the linker variations between the left “wing” and the central pyrimidine moiety are other important factors for optimization. Additionally, the conformational flexibility and positional adaptability of the linker, are vitally important in the design of NNRTIs62. The hydroxyimino, hydrazinyl, alkylamino, hydroxyl, halogen, and cyano groups were introduced on the methylene linker (Fig. 10).
The best compound 36 in the hydroxyiminate series inhibited HIV-1 in infected cells with an IC50 of 6 nmol/L but lacked activity against double mutants63. Predicted binding modes explained the activity (Fig. 11). The molecule was in the “horseshoe” conformation, and a new hydrogen-bonding interaction between the hydroxyimino and E138 was observed. However, the compound did not increase the interaction with the W229 in the hydrophobic region (Fig. 11A). Similarly, hydrazinyl linker (37) was suitable to DAPYs with an EC50 of 9 nmol/L and SI value of 39,59964. With the introduction of a cyanovinyl group, compound 38 increased the antiviral activity (EC50) to 1.8 nmol/L and SI to 106,367, probably due to enhanced interactions of the cyanovinyl group with Y181, Y188, F227, and W229 residues (Fig. 11B)65. The linker, when replaced by different alkylamino groups led to lower potency and activity in the series isopropylamino > n-propylamino > methylamino > ethylamino ≈ cyclopropylamino66. The hydrogen–bonding interaction of hydrazinyl with E138 disappeared compared with 38, the cyclopropyl group inserted into the Y181/E138 channel, and the di-fluoro groups limited the rotational freedom of the phenyl ring (Fig. 11C), collectively leading to compound 39 with an EC50 of 99 nmol/L.
Figure 11.

Molecular docking models of RT (PDB: 3MEC) with compound 36 (A), 38 (B), 39 (C), 40-R (D), 40-S (E) and 43 (F). The figures were generated by PyMOL.
In 2011, we also designed and synthesized a series of DAPYs bearing a hydroxyl on the methylene linker67. They possessed excellent activities against WT HIV-1 at nanomolar concentrations and moderate activities against the double mutant K103N + Y181C. The racemic compound 40 showed an EC50 of 9 nmol/L and SI of 4115. The chiral center on the hydroxymethyl linker was taken into consideration, and supercritical fluid chromatography (SFC) afforded both compound (+)-40 and (−)-40 with enantiomeric excess (ee) value > 99% and purity >99%68. Their absolute configurations were assigned as R and S, respectively, based on the experimental electronic circular dichroism (ECD) calculations. They exhibited different antiviral activity: the (+)-(R)-40 had an EC50 of 5 nmol/L, which was 12-fold more potent than the (−)-(S)-40 (EC50 = 65 nmol/L). Docking studies showed that they targeted a similar RT conformation as other DAPYs. A hydrogen–bonding interaction between the hydroxyl group and Y188 was observed in the (+)-(R)-40-RT model (Fig. 11D) while not seen in the (−)-(S)-40-RT model (Fig. 11E), thus interpreting the biological result. Further alkyl modification of the linker produced new CR2(OH)-DAPYs and the best compound 41 in this series had an EC50 of only 67 nmol/L69. The halogen substitution was optimized to obtain compound 42 with an EC50 of 5 nmol/L70. For the positive influence of the cyano substitution on the phenyl ring, the chloride was replaced by a cyano group to obtain compound 43 with superior antiviral activity (EC50 = 2 nmol/L) and high SI value (>118,595)71. As shown in Fig. 11F, the R/S enantiomers of compound 43 displayed similar binding conformations with the cyano group inserted in the Y181/E138 tunnel, which might be a key feature for the activity. However, compound 43 was inactive towards the K103N + Y181C double mutant.
2.5. Molecular hybrids
Molecular hybridization or fragment-based optimization is a highly efficient strategy in drug discovery72. This method is also applied widely in the design of next-generation NNRTIs to overcome drug resistance, improve safety, and/or physicochemical properties (Fig. 12)7,72.
Figure 12.

Molecular hybridization workflow for the DAPY analogues developed by our group. The modified parts are colored.
In 2015, a series of hybrids were designed by hopping the thioacetanilide moiety onto the right “wing” of DAPY, based on the superposition of lower-energy binding conformations of ETR and VRX-480,77373. Compound 44 displayed moderate inhibitory activity against WT HIV-1 RT with an EC50 value of 240 nmol/L and bound to the NNRTI binding pocket in a “U” mode (Fig. 13A). The SAR study showed an encouraging activity of the electron-withdrawing substituents on the right “wing”. The antiviral activity was lost when the amide group was removed, indicating its importance74. The sulfide of compound 44 was oxidized to the sulfone, and the strong electron-withdrawing nitro group introduced to obtain compound 45, had 180-fold better antiviral activity and a much higher SI value of 45,83075. Molecular docking showed that the compound bound well to RT (Fig. 13B) and super-positioned with compound 44 (Fig. 13C); two hydrogen-bonding interactions between the sulfone and K101 and the nitro, and F227 (Fig. 13B) were observed. Inspired by the promising activity of the above hybrids, a family of piperidin-4-yl-aminopyrimidine derivatives was obtained by combining ETR-VRX-480,773 hybrids and piperidine-linked aminopyrimidines, another family of NNRTIs76. Compounds 46 and 47 displayed highly potent activity against WT HIV-1 with an EC50 of 7.0 and 1.9 nmol/L. However, compound 47 showed high toxicity (CC50 = 1.6 μmol/L), low SI value (845) and lacked potency against the double mutant strains K103N + Y181C, thus needing further optimization.
Figure 13.

Molecular docking models of RT (PDB: 3MEC) with compounds 44 (A), 45 (B), 44 and 45 (C), 48 (D), 50 (E) and 52 (F). The figures were generated by PyMOL.
Then, we focused on fusing pharmacophores to the left “wing” of DAPYs. The diaryl ethers were reported as a class of NNRTIs with excellent oral bioavailability and anti-HIV potency77. Compound 48 combining the important pharmacophores of DAPYs and diaryl ethers, displayed good activity against WT HIV-1 with an EC50 of 13 nmol/L78. The newly introduced 2-fluorophenylamide group was located in the W229 hydrophobic pocket (Fig. 13D). Similarly, compound 49 combined the structural features of DAPY and GW678248 (structure shown in Fig. 15) led to a reduced anti-HIV activity (EC50 = 790 nmol/L)79. We attempted to synthesize hybrid DAPY with the integrase antiviral inhibitor, the first quinolone-based anti-HIV drug, elvitegravir80. The newly designed diarlyprimidine-quinolone hybrid compound 50 displayed an EC50 value of 280 nmol/L against WT HIV-1 infected cells. An enzymatic assay with the compound showed an EC50 of only 7.2 ± 0.5 μmol/L, indicating the highly polar quinolone 3-carboxylic acid moiety was not suitable for binding to the hydrophobic pocket (Fig. 13E). The follow-up work showed that the length of the N-substituted alkyl group and the introduction of an iodine atom on the quinolone backbone affected the activities. Compound 51 displayed a significant EC50 value of 9.6 nmol/L against WT HIV-1 and of 0.98 μmol/L against K103N + Y181C81. This strategy of integrating pharmacophores of different types of inhibitors might lead to compounds with dual targeting effects, needing further characterization.
Figure 15.

Other antiviral inhibitors developed by our group. The modified parts are colored.
Finally, we focused our attention on the entrance channel by fusing a phenyl ring (similar as compound 30) based on the superposition model of DPC083 or EFV, and ETR in the binding pocket of HIV-1 RT82,83. For instance, compound 52, with the two “wings” of ETR unchanged, had impressive activity against WT HIV-1 with an EC50 value of 1.8 nmol/L, and with an SI value of 111,954. It was also highly active in the nanomolar range against a broad range of viral mutants including L100I (EC50 = 18 nmol/L), K103N (EC50 < 3.6 nmol/L), E138K (EC50 = 11 nmol/L), Y181L (EC50 = 31 nmol/L), Y188L (EC50 = 36 nmol/L), F227L + V106A (EC50 = 107 nmol/L), and K103N + Y181C (EC50 = 60 nmol/L)82. As proposed, the fused phenyl ring was located at the entrance channel of E138/K103/K101 residues (Fig. 13F), probably maintaining the conformational flexibility to compensate for the resistant mutants’ effect.
Very recently, we developed a series of novel diarylbenzopyrimidine analogues by hybridizing FDA-approved drugs ETR and EFV83. Introduction of the important fragment of RPV, cyanovinyl group and substituent modifications on the left “wing” resulted in the development of new derivatives with the combined strength of the two drugs. Compound 53 showed promising activity towards the EFV-resistant K103N mutant (EC50 = 10.2 nmol/L) but was not better against E138K (EC50 = 17.7 nmol/L) than ETR. The PK profile was acceptable with a rat oral bioavailability of 15.5%. Substitution optimizations on the fused phenyl ring led to compound 54 with much higher activity than ETR and EFV against the WT, E138K, and K103N variants (EC50 = 3.4, 4.3, and 3.6 nmol/L, respectively) and a rat oral bioavailability of 16.5%, which should be better than that of ETR. Molecular docking (Fig. 14) analysis showed that three hydrogen bonds stabilized the “butterfly” conformation and the fused phenyl ring occupied the E138/K101 channel. In the mutants, the orientation of the new K138 was altered due to the existence of the adjacent K101, making the space more accessible to the fused ring. For the N103 mutant, the 2-methyl-6-nitro group restricted the conformation, and it was oriented towards new N103, potentially resulting in a water-mediated hydrogen bond with the NH of amide. Collectively, the binding features explain the improved antiviral activity and guide further DAPY optimization to improve the activity against WT and the mutants.
Figure 14.

Molecular docking models of compounds 53 and 54 with the HIV-1 WT and E138K/K103N mutant RT (A) WT with 53 (B) E138K mutant RT with 54 (C) K103N mutant RT with 53 (D) WT with 54 (E) E138K mutant RT with 54 (F) K103N mutant RT with 54. The figures were generated by PyMOL.
2.6. Others
Alternatively, we attempted to hybridize the pharmacophore of DAPY into early generation NNRTIs84,85. For instance, compound 55 was developed by introducing the 4-cyanophenyl group into EFV, with an EC50 value of 0.84 nmol/L against the WT HIV-1 strain, and 3.5 and 66.0 nmol/L against E138K and K103N + Y181C mutants. However, the cytotoxicity was high (CC50 = 1.9 μmol/L)84. The B-ring of GW678248 was replaced by naphthyl ring as NNRTIs85. However, they showed only moderate activity against WT HIV-1 and mutated viruses. Compound 56, the best one in this series, showed an EC50 of 4.8 nmol/L against WT HIV-1 and 2.1 μmol/L against the HIV-1 double mutant K103N + Y181C, exhibiting much less potency than the parent GW678248.
3. Conclusions and future perspectives
In the past 20 years, several commonly used medicinal chemistry strategies, including structure-based drug design, fragment-based optimization, scaffold/fragment hopping, molecular/fragment hybridization and bioisosterism, have been utilized in our NNRTI drug development. Progress in structural optimizations of HEPT, DABO, DATA, and DAPY scaffolds have been made, leading to highly potent derivatives against not only WT HIV-1 but also single/double mutant variants. However, the growing incidence of resistance to nearly all current drugs is investigated continually. Resistance-associated mutations occur within or near the NNIBP, resulting in decreased efficacy of NNRTIs86, 87, 88. Thus, more novel compounds to treat HIV infections are urgently needed. Nevertheless, novel and high-quality lead compounds, especially towards mutants remains a significant challenge in the current anti-HIV drug discovery. There are some perspectives and directions to be considered for the development of next-generation NNRTIs.
-
(1)
The number of rotatable bonds is essential for antiviral activity. Except for DLV, the FDA approved drugs having more freedom of rotation in the binding site had better activity against RT mutants3. For instance, RPV showed significantly improved activity in various mutants with the introduction of the cyanovinyl group. In addition, the cyanovinyl group inserted into the W229 hydrophobic tunnel strengthened the π–π stacking interactions. Our compounds 53 and 54 with cyanovinyl showed good anti-HIV-1 WT, E138K and K103N activities. The biphenyl derivative compounds 29 and 30, and the methylene linker optimized compounds (36–43) with more rotatable bonds showed much higher potency and antiviral profiles.
-
(2)
Forming additional hydrogen-bonding interactions with the targeted protein is a common strategy to enhance the binding affinity. In the NNRTI development, this method has been used widely to improve the activity against WT HIV-1 or prevent efficacy reduction towards mutants89. For instance, the sulfone of compound 45 formed additional hydrogen-bonding interactions with K101, possessing 180-fold better antiviral activity than the corresponding sulfide compound 4475.
-
(3)
The W229 hydrophobic region has to be occupied for improving π–π stacking interactions with surrounding aromatic residues, e.g., the introduction of naphthyl, biphenyl, cyanovinyl group, or others. The halogen interaction90, methyl effect91, and cyano group36 could be applied for further optimizations.
-
(4)
The optimization of the PK profiles will be addressed in the future. For instance, ETR has very poor water solubility and lacks an intravenous formulation leading to unknown absolute oral bioavailability51,52,83. High plasma protein binding (>95%) of DLV, EFV, ETR, and RPV was observed, and >60% NVP binding with a long half-life, should be considered during clinical treatments3. Introducing halogen, especially F, CF3 group, sulfur, phosphor, silicium, and others might increase the antiviral activity and PK profiles. Introducing solubilizing groups at the entrance channel and other suitable sites, and design of prodrugs are other strategies to improve the physicochemical properties. Compound 31 with fluoro substitutions exhibited an improved PK profile as well as reduced cytotoxicity. Liu and Zhan et al.52,92, 93, 94, 95, 96 succeeded in enhancing the PK, water solubility, and oral bioavailability by structural optimizations of multiple NNRTIs.
-
(5)
Clinically, the use of NNRTIs is always in conjunction with two NRTIs as HHART3. The efficacy, especially the safety of several drug regimens, is of great importance in the clinical evaluation. From the medicinal chemistry view, it is possible to make a NNRTI and NRTI conjugate act as a bifunctional inhibitor due to the proximity (∼10 Å, Fig. 1B) of the NRTI and NNRTI binding sites in RT. In 2013, the bivalent NNRTI and NRTI inhibitors had been developed by Anderson et al.97, and the representative compound (the triphosphate of d4T-4PEG-DAPY analogue conjugate, structure not shown here) inhibited RT polymerase at low nanomolar concentrations, which was more potent than the two parent drugs. Besides, a prodrug design of these agents or other modern strategies, such as proteolysis targeting chimeras (PROTACs) to degrade HIV-1 RT, are prospected. Recently, a class of small molecule antivirals were developed to induce proteasomal degradation of HCV viral proteins to overcome resistant viral variants. This work highlights reference to HIV targeted protein degradation and a new paradigm for the development of molecules with superior resistance profiles98.
The above-discussed directions will undoubtedly attract further interest in the coming years and predict that novel NNRTI-based drugs will be discovered using modern drug discovery strategies and new chemical sources. Phenotypic screening, fragment-based drug design, structure/ligand-based virtual screening, DNA-encoded library generation and screening, conjugation chemistry, prodrugs, PROTACs, etc. will be applied widely for the rapid development of future antiviral drugs against HIV infection.
Acknowledgments
This work was funded by grants from the National Natural Science Foundation of China (81872791 and 21372050); the Young Elite Scientists Sponsorship Program by the China Association for Science and Technology (2017QNRC061); National Key R&D Program of China (2017YFA0506000) and the Key Research and Development Program of Ningxia (2019BFG02017 and 2018BFH02001, China). We thank Ningxia Medical University for providing the sources of molecular modeling.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.11.010.
Author contributions
Chunlin Zhuang and Fener Chen generated the manuscript draft. Christophe Pannecouquec and Erik De Clercq edited and revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.UNAIDS global HIV & AIDS statistics—2019 fact sheet. http://www.unaids.org/en/resources/fact-sheet Available from:
- 2.Brechtl J.R., Breitbart W., Galietta M., Krivo S., Rosenfeld B. The use of highly active antiretroviral therapy (HAART) in patients with advanced HIV infection: impact on medical, palliative care, and quality of life outcomes. J Pain Symptom Manag. 2001;21:41–51. doi: 10.1016/s0885-3924(00)00245-1. [DOI] [PubMed] [Google Scholar]
- 3.Namasivayam V., Vanangamudi M., Kramer V.G., Kurup S., Zhan P., Liu X. The journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J Med Chem. 2019;62:4851–4883. doi: 10.1021/acs.jmedchem.8b00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shafer R.W., Vuitton D.A. Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomed Pharmacother. 1999;53:73–86. doi: 10.1016/s0753-3322(99)80063-8. [DOI] [PubMed] [Google Scholar]
- 5.de Clercq E. The design of drugs for HIV and HCV. Nat Rev Drug Discov. 2007;6:1001–1018. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 6.Martins S., Ramos M.J., Fernandes P.A. The current status of the NNRTI family of antiretrovirals used in the HAART regime against HIV infection. Curr Med Chem. 2008;15:1083–1095. doi: 10.2174/092986708784221467. [DOI] [PubMed] [Google Scholar]
- 7.Zhan P., Chen X., Li D., Fang Z., de Clercq E., Liu X. HIV-1 NNRTIs: structural diversity, pharmacophore similarity, and implications for drug design. Med Res Rev. 2013;33(Suppl 1):E1–E72. doi: 10.1002/med.20241. [DOI] [PubMed] [Google Scholar]
- 8.Gu S.X., Lu H.H., Liu G.Y., Ju X.L., Zhu Y.Y. Advances in diarylpyrimidines and related analogues as HIV-1 nonnucleoside reverse transcriptase inhibitors. Eur J Med Chem. 2018;158:371–392. doi: 10.1016/j.ejmech.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Yang S., Chen F.E., de Clercq E. Dihydro-alkoxyl-benzyl-oxopyrimidine derivatives (DABOs) as non-nucleoside reverse transcriptase inhibitors: an update review (2001–2011) Curr Med Chem. 2012;19:152–162. doi: 10.2174/092986712803414169. [DOI] [PubMed] [Google Scholar]
- 10.de Clercq E. Fifty years in search of selective antiviral drugs. J Med Chem. 2019;62:7322–7339. doi: 10.1021/acs.jmedchem.9b00175. [DOI] [PubMed] [Google Scholar]
- 11.Wensing A.M., Calvez V., Gunthard H.F., Johnson V.A., Paredes R., Pillay D. 2017 Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2016;24:132–133. [PMC free article] [PubMed] [Google Scholar]
- 12.Miyasaka T., Tanaka H., Baba M., Hayakawa H., Walker R.T., Balzarini J. A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J Med Chem. 1989;32:2507–2509. doi: 10.1021/jm00132a002. [DOI] [PubMed] [Google Scholar]
- 13.El-Brollosy N.R., Jorgensen P.T., Dahan B., Boel A.M., Pedersen E.B., Nielsen C. Synthesis of novel N-1 (allyloxymethyl) analogues of 6-benzyl-1-(ethoxymethyl)-5-isopropyluracil (MKC-442, emivirine) with improved activity against HIV-1 and its mutants. J Med Chem. 2002;45:5721–5726. doi: 10.1021/jm020949r. [DOI] [PubMed] [Google Scholar]
- 14.Rodgers D.W., Gamblin S.J., Harris B.A., Ray S., Culp J.S., Hellmig B. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1995;92:1222–1226. doi: 10.1073/pnas.92.4.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka H., Takashima H., Ubasawa M., Sekiya K., Nitta I., Baba M. Synthesis and antiviral activity of deoxy analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) as potent and selective anti-HIV-1 agents. J Med Chem. 1992;35:4713–4719. doi: 10.1021/jm00103a009. [DOI] [PubMed] [Google Scholar]
- 16.Meng G., Chen F.E., de Clercq E., Balzarini J., Pannecouque C. Nonnucleoside HIV-1 reverse transcriptase inhibitors: part I. Synthesis and structure–activity relationship of 1-alkoxymethyl-5-alkyl-6-naphthylmethyl uracils as HEPT analogues. Chem Pharm Bull (Tokyo) 2003;51:779–789. doi: 10.1248/cpb.51.779. [DOI] [PubMed] [Google Scholar]
- 17.Sun G.F., Chen X.X., Chen F.E., Wang Y.P., de Clercq E., Balzarini J. Nonnucleoside HIV-1 reverse-transcriptase inhibitors, part 5. Synthesis and anti-HIV-1 activity of novel 6-naphthylthio HEPT analogues. Chem Pharm Bull (Tokyo) 2005;53:886–892. doi: 10.1248/cpb.53.886. [DOI] [PubMed] [Google Scholar]
- 18.Ji L., Chen F.E., Feng X.Q., de Clercq E., Balzarini J., Pannecouque C. Non-nucleoside HIV-1 reverse transcriptase inhibitors, part 7. Synthesis, antiviral activity, and 3D-QSAR investigations of novel 6-(1-naphthoyl) HEPT analogues. Chem Pharm Bull (Tokyo) 2006;54:1248–1253. doi: 10.1248/cpb.54.1248. [DOI] [PubMed] [Google Scholar]
- 19.Ji L., Chen F.E., Xie B., de Clercq E., Balzarini J., Pannecouque C. Synthesis and anti-HIV activity evaluation of 1-[(alkenyl or alkynyl or alkyloxy)methyl]-5-alkyl-6-(1-naphthoyl)-2,4-pyrimidinediones as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2007;42:198–204. doi: 10.1016/j.ejmech.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 20.He Y., Kuang Y., Chen F., Wang S., Ji L., de Clercq E. Non-nucleoside HIV-1 reverse transcriptase inhibitors, part 4[1]. Synthesis and anti-HIV activity of N-1-β-carbonyl-6-naphthyl-methyl analogues of HEPT. Monatshefte für Chemie-Chemical Monthly. 2005;136:1233–1245. [Google Scholar]
- 21.Meng G., He Y., Chen F.E. Three dimensional quantitative structure–activity relationship of HEPT analogues as HIV-1 reverse transcriptaseinhibitors. Chem J Chin Univ. 2002;23:1304–1308. [Google Scholar]
- 22.Meng G., Chen F.E., de Clercq E., Dai H.F. Interactive Study between two types of 1-[2-(hydroxyethoxy)methyl]-6-naphthylmethylthymines and HIV-1 reverse transcriptase. Chin J Process Eng. 2003;3:24–28. [Google Scholar]
- 23.Meng G., Chen F.E. Nonnucleoside HIV-1 reverse transcriptase inhibitors II 3D-QSAR study on 6-(1-naphthylmethyl)substituted thymines. Chin J Med Chem. 2003;13:254–259. [Google Scholar]
- 24.He Y., Hu H., Xu L., Meng G., Fan K., Chen F.E. Structure–activity relationship studies on 6-naphthylmethyl substituted HEPT derivatives as non-nucleoside reverse transcriptase inhibitors based on molecular docking. Chem J Chin Univ. 2005;26:254–258. [Google Scholar]
- 25.Botta M., Artico M., Massa S., Gambacorta A., Marongiu M.E., Pani A. Synthesis, antimicrobial and antiviral activities of isotrimethoprim and some related derivatives. Eur J Med Chem. 1992;27:251–257. [Google Scholar]
- 26.Wu Y., Tang C., Rui R., Yang L., Ding W., Wang J. Synthesis and biological evaluation of a series of 2-(((5-akly/aryl-1H-pyrazol-3-yl)methyl)thio)-5-alkyl-6-(cyclohexylmethyl)-pyrimidin-4(3H)-ones as potential HIV-1 inhibitors. Acta Pharm Sin B. 2020;10:512–528. doi: 10.1016/j.apsb.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nawrozkij M.B., Forgione M., Yablokov A.S., Lucidi A., Tomaselli D., Patsilinakos A. Effect of alpha-methoxy substitution on the anti-HIV activity of dihydropyrimidin-4(3H)-ones. J Med Chem. 2019;62:604–621. doi: 10.1021/acs.jmedchem.8b01238. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y.P., Chen F.E., de Clercq E., Balzarini J., Pannecouque C. Synthesis and biological evaluation of novel 6-substituted 5-alkyl-2-(arylcarbonylmethylthio)pyrimidin-4(3H)-ones as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Med Chem. 2008;16:3887–3894. doi: 10.1016/j.bmc.2008.01.039. [DOI] [PubMed] [Google Scholar]
- 29.He Y., Chen F., Sun G., Wang Y., de Clercq E., Balzarini J. 5-Alkyl-2-[(aryl and alkyloxylcarbonylmethyl)thio]-6-(1-naphthylmethyl) pyrimidin-4(3H)-ones as an unique HIV reverse transcriptase inhibitors of S-DABO series. Bioorg Med Chem Lett. 2004;14:3173–3176. doi: 10.1016/j.bmcl.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 30.He Y., Chen F., Yu X., Wang Y., de Clercq E., Balzarini J. Nonnucleoside HIV-1 reverse transcriptase inhibitors; part 3. Synthesis and antiviral activity of 5-alkyl-2-[(aryl and alkyloxyl-carbonylmethyl)thio]-6-(1-naphthylmethyl) pyrimidin-4(3H)-ones. Bioorg Chem. 2004;32:536–548. doi: 10.1016/j.bioorg.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Sun G.F., Kuang Y.Y., Chen F.E., de Clercq E., Balzarini J., Pannecouque C. Non-nucleoside HIV reverse transcriptase inhibitors, Part 61: synthesis and anti-HIV activity of novel 2-[(arylcarbonylmethyl)thio]-6-arylthio DABO analogues. Arch Pharm (Weinheim) 2005;338:457–461. doi: 10.1002/ardp.200400961. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y., Chen F.E., Balzarini J., de Clercq E., Pannecouque C. Non-nucleoside HIV-1 reverse-transcriptase inhibitors. Part 10. Synthesis and anti-HIV activity of 5-alkyl-6-(1-naphthylmethyl)pyrimidin-4(3H)-ones with a mono- or disubstituted 2-amino function as novel ‘dihydro-alkoxy-benzyl-oxopyrimidine’ (DABO) analogues. Chem Biodivers. 2008;5:168–176. doi: 10.1002/cbdv.200890008. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y.P., Chen F.E., de Clercq E., Balzarini J., Pannecouque C. Synthesis and in vitro anti-HIV evaluation of a new series of 6-arylmethyl-substituted S-DABOs as potential non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2009;44:1016–1023. doi: 10.1016/j.ejmech.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 34.Mai A., Artico M., Sbardella G., Massa S., Novellino E., Greco G. 5-Alkyl-2-(alkylthio)-6-(2,6-dihalophenylmethyl)-3,4-dihydropyrimidin-4(3H)-ones: novel potent and selective dihydro-alkoxy-benzyl-oxopyrimidine derivatives. J Med Chem. 1999;42:619–627. doi: 10.1021/jm980260f. [DOI] [PubMed] [Google Scholar]
- 35.Wu H.Q., Yan Z.H., Chen W.X., He Q.Q., Chen F.E., de Clercq E. Towards new C6-rigid S-DABO HIV-1 reverse transcriptase inhibitors: synthesis, biological investigation and molecular modeling studies. Bioorg Med Chem. 2013;21:6477–6483. doi: 10.1016/j.bmc.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y., Du Y., Huang N. A survey of the role of nitrile groups in protein–ligand interactions. Future Med Chem. 2018;10:2713–2728. doi: 10.4155/fmc-2018-0252. [DOI] [PubMed] [Google Scholar]
- 37.Wu H.Q., Pannecouque C., Yan Z.H., Chen W.X., He Q.Q., Chen F.E. Synthesis and biological evaluation of new conformationally restricted S-DABO hybrids as non-nucleoside inhibitors of HIV-1 reverse transcriptase. MedChemComm. 2014;5:468. [Google Scholar]
- 38.Ji L., Chen F.E., de Clercq E., Balzarini J., Pannecouque C. Synthesis and anti-HIV-1 activity evaluation of 5-alkyl-2-alkylthio-6-(arylcarbonyl or alpha-cyanoarylmethyl)-3,4-dihydropyrimidin-4(3H)-ones as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem. 2007;50:1778–1786. doi: 10.1021/jm061167r. [DOI] [PubMed] [Google Scholar]
- 39.Ludovici D.W., Kavash R.W., Kukla M.J., Ho C.Y., Ye H., de Corte B.L. Evolution of anti-HIV drug candidates. Part 2: diaryltriazine (DATA) analogues. Bioorg Med Chem Lett. 2001;11:2229–2234. doi: 10.1016/s0960-894x(01)00411-5. [DOI] [PubMed] [Google Scholar]
- 40.Ludovici D.W., Kukla M.J., Grous P.G., Krishnan S., Andries K., de Bethune M.P. Evolution of anti-HIV drug candidates. Part 1: from alpha-anilinophenylacetamide (alpha-APA) to imidoyl thiourea (ITU) Bioorg Med Chem Lett. 2001;11:2225–2228. doi: 10.1016/s0960-894x(01)00410-3. [DOI] [PubMed] [Google Scholar]
- 41.Das K., Clark A.D., Jr., Lewi P.J., Heeres J., de Jonge M.R., Koymans L.M. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 42.Xiong Y.Z., Chen F.E., Feng X.Q. Three fimensional quantitative structure–activity relationship of DATA analogues as HIV-1 reverse transcriptase inhibitors. Acta Chim Sin. 2006;64:1627–1630. [Google Scholar]
- 43.Xiong Y.Z., Chen F.E., Balzarini J., de Clercq E., Pannecouque C. Non-nucleoside HIV-1 reverse transcriptase inhibitors. Part 11: structural modulations of diaryltriazines with potent anti-HIV activity. Eur J Med Chem. 2008;43:1230–1236. doi: 10.1016/j.ejmech.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Ekkati A.R., Bollini M., Domaoal R.A., Spasov K.A., Anderson K.S., Jorgensen W.L. Discovery of dimeric inhibitors by extension into the entrance channel of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2012;22:1565–1568. doi: 10.1016/j.bmcl.2011.12.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiong Y.Z., Hu H.R., Chen F.E., Balzarini J., Pannecouque C., de Clercq E. Non-nucleoside reverse transcriptase inhibitors (Part 18): synthesis and anti-HIV activity of 4-allylamino or 4-azido substituted diaryltriazines. Yao Xue Xue Bao. 2009;44:145–149. [PubMed] [Google Scholar]
- 46.Xiong Y.Z., Chen F.E., Balzarini J., de Clercq E., Pannecouque C. Non-nucleoside HIV-1 reverse transcriptase inhibitors. Part 13: synthesis of fluorine-containing diaryltriazine derivatives for in vitro anti-HIV evaluation against wild-type strain. Chem Biodivers. 2009;6:561–568. doi: 10.1002/cbdv.200800163. [DOI] [PubMed] [Google Scholar]
- 47.Jin K., Liu M., Zhuang C., de Clercq E., Pannecouque C., Meng G. Improving the positional adaptability: structure-based design of biphenyl-substituted diaryltriazines as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Acta Pharm Sin B. 2020;10:344–357. doi: 10.1016/j.apsb.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludovici D.W., de Corte B.L., Kukla M.J., Ye H., Ho C.Y., Lichtenstein M.A. Evolution of anti-HIV drug candidates. Part 3: diarylpyrimidine (DAPY) analogues. Bioorg Med Chem Lett. 2001;11:2235–2239. doi: 10.1016/s0960-894x(01)00412-7. [DOI] [PubMed] [Google Scholar]
- 49.Xu H.T., Colby-Germinario S.P., Asahchop E.L., Oliveira M., McCallum M., Schader S.M. Effect of mutations at position E138 in HIV-1 reverse transcriptase and their interactions with the M184I mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine. Antimicrob Agents Chemother. 2013;57:3100–3109. doi: 10.1128/AAC.00348-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang D., Feng D., Ginex T., Zou J., Wei F., Zhao T. Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene3,2-d.pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm Sin B. 2020;10:878–894. doi: 10.1016/j.apsb.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Intelence: European public assessment report—product information. https://www.ema.europa.eu/en/medicines/human/EPAR/intelence Available from:
- 52.Huang B., Chen W., Zhao T., Li Z., Jiang X., Ginex T. Exploiting the tolerant region I of the non-nucleoside reverse transcriptase inhibitor (NNRTI) binding pocket: discovery of potent diarylpyrimidine-typed HIV-1 NNRTIs against wild-type and E138K mutant virus with significantly improved water solubility and favorable safety profiles. J Med Chem. 2019;62:2083–2098. doi: 10.1021/acs.jmedchem.8b01729. [DOI] [PubMed] [Google Scholar]
- 53.Feng X.Q., Liang Y.H., Zeng Z.S., Chen F.E., Balzarini J., Pannecouque C. Structural modifications of DAPY analogues with potent anti-HIV-1 activity. ChemMedChem. 2009;4:219–224. doi: 10.1002/cmdc.200800334. [DOI] [PubMed] [Google Scholar]
- 54.Liang Y.H., Feng X.Q., Zeng Z.S., Chen F.E., Balzarini J., Pannecouque C. Design, synthesis, and SAR of naphthyl-substituted diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. ChemMedChem. 2009;4:1537–1545. doi: 10.1002/cmdc.200900212. [DOI] [PubMed] [Google Scholar]
- 55.Jin K., Yin H., de Clercq E., Pannecouque C., Meng G., Chen F. Discovery of biphenyl-substituted diarylpyrimidines as non-nucleoside reverse transcriptase inhibitors with high potency against wild-type and mutant HIV-1. Eur J Med Chem. 2018;145:726–734. doi: 10.1016/j.ejmech.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Sang Y., Han S., Han S., Pannecouque C., de Clercq E., Zhuang C. Follow on-based optimization of the biphenyl-DAPYs as HIV-1 nonnucleoside reverse transcriptase inhibitors against the wild-type and mutant strains. Bioorg Chem. 2019;89:102974. doi: 10.1016/j.bioorg.2019.102974. [DOI] [PubMed] [Google Scholar]
- 57.Dousson C., Alexandre F.R., Amador A., Bonaric S., Bot S., Caillet C. Discovery of the aryl-phospho-indole IDX899, a highly potent anti-HIV non-nucleoside reverse transcriptase inhibitor. J Med Chem. 2016;59:1891–1898. doi: 10.1021/acs.jmedchem.5b01430. [DOI] [PubMed] [Google Scholar]
- 58.Liang Y.H., Chen F.E. QSAR studies for diarylpyrimidines against HIV-1 reverse transcriptase wild-type and mutant strains. Eur J Med Chem. 2009;44:625–631. doi: 10.1016/j.ejmech.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 59.Kang D., Fang Z., Li Z., Huang B., Zhang H., Lu X. Design, dynthesis, and rvaluation of thiophene[3,2-d]pyrimidine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors with significantly improved drug resistance profiles. J Med Chem. 2016;59:7991–8007. doi: 10.1021/acs.jmedchem.6b00738. [DOI] [PubMed] [Google Scholar]
- 60.Sang Y., Han S., Pannecouque C., de Clercq E., Zhuang C., Chen F. Conformational restriction design of thiophene-biphenyl-DAPY HIV-1 non-nucleoside reverse transcriptase inhibitors. Eur J Med Chem. 2019;182:111603. doi: 10.1016/j.ejmech.2019.111603. [DOI] [PubMed] [Google Scholar]
- 61.Gu S.X., Yang S.Q., He Q.Q., Ma X.D., Chen F.E., Dai H.F. Design, synthesis and biological evaluation of cycloalkyl arylpyrimidines (CAPYs) as HIV-1 NNRTIs. Bioorg Med Chem. 2011;19:7093–7099. doi: 10.1016/j.bmc.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 62.Das K., Lewi P.J., Hughes S.H., Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Feng X.Q., Zeng Z.S., Liang Y.H., Chen F.E., Pannecouque C., Balzarini J. Synthesis and biological evaluation of 4-(hydroxyimino)arylmethyl diarylpyrimidine analogues as potential non-nucleoside reverse transcriptase inhibitors against HIV. Bioorg Med Chem. 2010;18:2370–2374. doi: 10.1016/j.bmc.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 64.Ma X.D., Yang S.Q., Gu S.X., He Q.Q., Chen F.E., de Clercq E. Synthesis and anti-HIV activity of aryl-2-[(4-cyanophenyl)amino]-4-pyrimidinone hydrazones as potent non-nucleoside reverse transcriptase inhibitors. ChemMedChem. 2011;6:2225–2232. doi: 10.1002/cmdc.201100334. [DOI] [PubMed] [Google Scholar]
- 65.Meng G., Liu Y., Zheng A., Chen F., Chen W., de Clercq E. Design and synthesis of a new series of modified CH-diarylpyrimidines as drug-resistant HIV non-nucleoside reverse transcriptase inhibitors. Eur J Med Chem. 2014;82:600–611. doi: 10.1016/j.ejmech.2014.05.059. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y., Meng G., Zheng A., Chen F., Chen W., de Clercq E. Design and synthesis of a new series of cyclopropylamino-linking diarylpyrimidines as HIV non-nucleoside reverse transcriptase inhibitors. Eur J Pharm Sci. 2014;62:334–341. doi: 10.1016/j.ejps.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 67.Gu S.X., He Q.Q., Yang S.Q., Ma X.D., Chen F.E., de Clercq E. Synthesis and structure–activity relationship of novel diarylpyrimidines with hydromethyl linker (CH(OH)-DAPYs) as HIV-1 NNRTIs. Bioorg Med Chem. 2011;19:5117–5124. doi: 10.1016/j.bmc.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 68.Gu S.X., Li Z.M., Ma X.D., Yang S.Q., He Q.Q., Chen F.E. Chiral resolution, absolute configuration assignment and biological activity of racemic diarylpyrimidine CH(OH)-DAPY as potent nonnucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2012;53:229–234. doi: 10.1016/j.ejmech.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 69.Yan Z.H., Huang X.Y., Wu H.Q., Chen W.X., He Q.Q., Chen F.E. Structural modifications of CH(OH)-DAPYs as new HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem. 2014;22:2535–2541. doi: 10.1016/j.bmc.2014.02.030. [DOI] [PubMed] [Google Scholar]
- 70.Yan Z.H., Wu H.Q., Chen W.X., Wu Y., Piao H.R., He Q.Q. Synthesis and biological evaluation of CHX-DAPYs as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem. 2014;22:3220–3226. doi: 10.1016/j.bmc.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 71.Zeng Z.S., Liang Y.H., Feng X.Q., Chen F.E., Pannecouque C., Balzarini J. Lead optimization of diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. ChemMedChem. 2010;5:837–840. doi: 10.1002/cmdc.201000045. [DOI] [PubMed] [Google Scholar]
- 72.Zhang S., Zhang J., Gao P., Sun L., Song Y., Kang D. Efficient drug discovery by rational lead hybridization based on crystallographic overlay. Drug Discov Today. 2019;24:805–813. doi: 10.1016/j.drudis.2018.11.021. [DOI] [PubMed] [Google Scholar]
- 73.Wan Z.Y., Tao Y., Wang Y.F., Mao T.Q., Yin H., Chen F.E. Hybrid chemistry. Part 4: discovery of etravirine-VRX-480773 hybrids as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem. 2015;23:4248–4255. doi: 10.1016/j.bmc.2015.06.048. [DOI] [PubMed] [Google Scholar]
- 74.Wan Z.Y., Chen F.E., de Clercq E., Daelemans D., Pannecouque C. Design, synthesis and biological evaluation of S-DAPY derivatives as novel HIV-1 inhibitors. Scientia Sinica Chimica. 2015;45:961–967. (in Chinese) [Google Scholar]
- 75.Wan Z.Y., Yao J., Mao T.Q., Wang X.L., Wang H.F., Chen W.X. Pyrimidine sulfonylacetanilides with improved potency against key mutant viruses of HIV-1 by specific targeting of a highly conserved residue. Eur J Med Chem. 2015;102:215–222. doi: 10.1016/j.ejmech.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 76.Wan Z.Y., Yao J., Tao Y., Mao T.Q., Wang X.L., Lu Y.P. Discovery of piperidin-4-yl-aminopyrimidine derivatives as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2015;97:1–9. doi: 10.1016/j.ejmech.2015.04.050. [DOI] [PubMed] [Google Scholar]
- 77.Tucker T.J., Saggar S., Sisko J.T., Tynebor R.M., Williams T.M., Felock P.J. The design and synthesis of diaryl ether second generation HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) with enhanced potency versus key clinical mutations. Bioorg Med Chem Lett. 2008;18:2959–2966. doi: 10.1016/j.bmcl.2008.03.064. [DOI] [PubMed] [Google Scholar]
- 78.Wu H.Q., Yao J., He Q.Q., Chen W.X., Chen F.E., Pannecouque C. Synthesis and biological evaluation of DAPY-DPEs hybrids as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem. 2015;23:624–631. doi: 10.1016/j.bmc.2014.11.032. [DOI] [PubMed] [Google Scholar]
- 79.Yang S., Pannecouque C., Daelemans D., Ma X.D., Liu Y., Chen F.E. Molecular design, synthesis and biological evaluation of BP-O-DAPY and O-DAPY derivatives as non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2013;65:134–143. doi: 10.1016/j.ejmech.2013.04.052. [DOI] [PubMed] [Google Scholar]
- 80.Mao T.Q., He Q.Q., Wan Z.Y., Chen W.X., Chen F.E., Tang G.F. Anti-HIV diarylpyrimidine-quinolone hybrids and their mode of action. Bioorg Med Chem. 2015;23:3860–3868. doi: 10.1016/j.bmc.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 81.Mao T.Q., He Q.Q., Chen W.X., Tang G.F., Chen F.E., de Clercq E. Structural modifications of diarylpyrimidine-quinolone hybrids as potent HIV-1 NNRTIs with an improved drug resistance profile. Curr Pharmaceut Des. 2016;22:6982–6987. doi: 10.2174/1381612823666161122125657. [DOI] [PubMed] [Google Scholar]
- 82.Zeng Z.S., He Q.Q., Liang Y.H., Feng X.Q., Chen F.E., de Clercq E. Hybrid diarylbenzopyrimidine non-nucleoside reverse transcriptase inhibitors as promising new leads for improved anti-HIV-1 chemotherapy. Bioorg Med Chem. 2010;18:5039–5047. doi: 10.1016/j.bmc.2010.05.081. [DOI] [PubMed] [Google Scholar]
- 83.Han S., Sang Y., Wu Y., Tao Y., Pannecouque C., de Clercq E. Molecular hybridization-inspired optimization of diarylbenzopyrimidines as HIV-1 nonnucleoside reverse transcriptase inhibitors with improved activity against K103N and E138K mutants and pharmacokinetic profiles. ACS Infect Dis. 2020;6:787–801. doi: 10.1021/acsinfecdis.9b00229. [DOI] [PubMed] [Google Scholar]
- 84.Jin K., Sang Y., Han S., de Clercq E., Pannecouque C., Meng G. Synthesis and biological evaluation of dihydroquinazoline-2-amines as potent non-nucleoside reverse transcriptase inhibitors of wild-type and mutant HIV-1 strains. Eur J Med Chem. 2019;176:11–20. doi: 10.1016/j.ejmech.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 85.Ma X.D., Zhang X., Dai H.F., Yang S.Q., Yang L.M., Gu S.X. Synthesis and biological activity of naphthyl-substituted (B-ring) benzophenone derivatives as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Med Chem. 2011;19:4601–4607. doi: 10.1016/j.bmc.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 86.Shafer R.W., Schapiro J.M. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 2008;10:67–84. [PMC free article] [PubMed] [Google Scholar]
- 87.Das K., Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr Opin Virol. 2013;3:111–118. doi: 10.1016/j.coviro.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Das K., Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr Opin Virol. 2013;3:119–128. doi: 10.1016/j.coviro.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhan P., Liu X., Li Z., Pannecouque C., de Clercq E. Design strategies of novel NNRTIs to overcome drug resistance. Curr Med Chem. 2009;16:3903–3917. doi: 10.2174/092986709789178019. [DOI] [PubMed] [Google Scholar]
- 90.Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G. The aalogen bond. Chem Rev. 2016;116:2478–2601. doi: 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leung C.S., Leung S.S., Tirado-Rives J., Jorgensen W.L. Methyl effects on protein–ligand binding. J Med Chem. 2012;55:4489–4500. doi: 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang D., Zhang H., Wang Z., Zhao T., Ginex T., Luque F.J. Identification of dihydrofuro[3,4-d]pyrimidine derivatives as novel HIV-1 non-nucleoside reverse transcriptase inhibitors with promising antiviral activities and desirable physicochemical properties. J Med Chem. 2019;62:1484–1501. doi: 10.1021/acs.jmedchem.8b01656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kang D., Fang Z., Huang B., Lu X., Zhang H., Xu H. Structure-based optimization of thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with improved potency against resistance-associated variants. J Med Chem. 2017;60:4424–4443. doi: 10.1021/acs.jmedchem.7b00332. [DOI] [PubMed] [Google Scholar]
- 94.Kang D., Wang Z., Zhang H., Wu G., Zhao T., Zhou Z. Further exploring solvent-exposed tolerant regions of allosteric binding pocket for novel HIV-1 NNRTIs discovery. ACS Med Chem Lett. 2018;9:370–375. doi: 10.1021/acsmedchemlett.8b00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huo Z., Zhang H., Kang D., Zhou Z., Wu G., Desta S. Discovery of novel diarylpyrimidine derivatives as potent HIV-1 NNRTIs targeting the “NNRTI adjacent” binding site. ACS Med Chem Lett. 2018;9:334–338. doi: 10.1021/acsmedchemlett.7b00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang X., Yu J., Zhou Z., Kongsted J., Song Y., Pannecouque C. Molecular design opportunities presented by solvent-exposed regions of target proteins. Med Res Rev. 2019;39:2194–2238. doi: 10.1002/med.21581. [DOI] [PubMed] [Google Scholar]
- 97.Bailey C.M., Sullivan T.J., Iyidogan P., Tirado-Rives J., Chung R., Ruiz-Caro J. Bifunctional inhibition of human immunodeficiency virus type 1 reverse transcriptase: mechanism and proof-of-concept as a novel therapeutic design strategy. J Med Chem. 2013;56:3959–3968. doi: 10.1021/jm400160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Wispelaere M., Du G., Donovan K.A., Zhang T., Eleuteri N.A. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat Commun. 2019;10:3468. doi: 10.1038/s41467-019-11429-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.