

Abstract
Purpose:
Generation of antigen-specific T cells from cancer patients employs large numbers of peripheral blood cells and/or tumor infiltrating cells to generate antigen-presenting and effector cells commonly requiring multiple rounds of re-stimulation ex vivo. We used a novel paramagnetic, nanoparticle-based artificial antigen presenting cell (nano-aAPC) that combines anti-CD28 co-stimulatory and human MHC class I molecules that are loaded with antigenic peptides to rapidly expand tumor antigen-specific T cells from melanoma patients.
Experimental Design:
Nano-aAPC expressing HLA-A*0201 molecules and costimulatory anti-CD28 antibody and loaded with MART-1 or gp100 class I restricted peptides were used to stimulate CD8 T cells purified from the peripheral blood of treatment-naïve or PD-1 antibody-treated patients with stage IV melanoma. Expanded cells were re-stimulated with fresh peptide-pulsed nano-aAPC at day 7. Phenotype analysis and functional assays including cytokine release, cytolysis, and measurement of avidity were conducted.
Results:
MART-1-specific CD8 T cells rapidly expanded up to 1000-fold by day 14 after exposure to peptide-pulsed nano-aAPC. Expanded T cells had a predominantly stem cell memory CD45RA+/CD62L+/CD95+ phenotype, expressed ICOS, PD-1, Tim3, and LAG3 and lacked CD28. Cells from patients with melanoma were polyfunctional, highly avid, expressed IL-2, IFN-gamma, TNF-alpha and exhibited cytolytic activity against tumor cell lines. They expanded 2–3-fold after exposure to PD-1 antibody in vivo, and expressed a highly diverse TCR V beta repertoire.
Conclusions:
Peptide-pulsed nanoparticle aAPC rapidly expanded polyfunctional antigen-specific CD8 T cells with high avidity, potent lytic function and a stem-memory phenotype from melanoma patients.
Keywords: Effector cells, Cytokines, Functional avidity, MART-1, gp100, Nanoparticles
Introduction
The emerging field of adoptive T cell therapy (ACT) has established that durable, complete regressions can be achieved with cell transfer strategies in cancer patients (1). ACT commonly depends on the use of autologous antigen-presenting cells (APC) either as feeder cells or to deliver antigen to promote the expansion of CD8 effector T cells that are optimally antigen-specific, stem memory cells with long telomeres that are derived from tumor infiltrating lymphocytes (TIL) or peripheral blood and can persist in vivo. Dendritic cells (DCs) are felt to be the most potent APC to expand T cells for adoptive transfer, which have also shown some clinical success (2). Ex vivo generated dendritic cells have been employed in a number of ACT trials and have themselves been used in therapeutic trials by intranodal, intradermal and even intravenous injection with limited clinical benefit (3). However, the use of DCs as antigen-presenting cells is associated with several manufacturing procedures to expand tumor-specific T cells from naïve precursor T cells, requiring a number of weeks or months to produce the large number of tumor-specific T cells required for ACT (4–6). DC-based expansion is limited by the requirement for enough DCs which effectively limits their use (7). Additionally, DCs are cumbersome to make, are highly variable in availability and function due to pretreatment and underlying disease limiting the ability to predict or control presentation to cognate TCRs. Furthermore, DCs from tumor-bearing patients can be dysfunctional due to the presence of the tumor microenvironment, contributing to immune non-responsiveness (8). DCs do not constitute an easily renewable and reliable “off the shelf” reagent readily adaptable for large scale clinical trials.
Various artificial antigen-presenting approaches from cell-based to acellular platforms have been developed to generate T cells for ACT (9–16). K562 cell-based artificial APC and Dynabeads conjugated with anti-CD3 and anti-CD28 antibody have been used to expand T cells in clinical trials (17,18). Although these modalities promote the polyclonal expansion of T-cells they can lead to suboptimal expansion because these signals are not naturally presented by APC resulting in activation-induced cell death and diminished T-cell viability (19,20). A further pitfall in the use of non-specific expansion of T cells for ACT is the induction of effectors that can lead to expansion of self-reactive T cells, increasing the chances for off-target toxicity (21,22). Using tumor-specific T cells derived from effectors expressing the autologous naïve T cell repertoire has some advantages compared to the use of engineered TCRs that have an improved affinity for antigen, since they may exhibit reduced autoimmune off-target toxicities (23) and avoid tumor escape through antigenic down-modulation.
In order to overcome these deficiencies, we designed and developed an acellular T cell expansion platform, consisting of nanoparticle-based artificial antigen-presenting cells (nano-aAPC) which are paramagnetic iron-dextran nanoparticles, 80–100 nm in diameter, conjugated with costimulatory anti-CD28 antibody and MHC-Ig molecules that were loaded with HLA-A2 restricted peptides to promote T cell expansion (24). This technology facilitates the expansion not only of human memory T cells (24) but also tumor-specific T cells derived from the naïve repertoire from healthy individuals (25).
In the current work, we show that tumor-associated antigen (TAA) peptide-loaded human class I MHC and anti-CD28 can be covalently linked to non-cell based magnetic nanoparticles to create nano-aAPC that can rapidly stimulate and expand potent antigen-specific CD8+ T cells derived from the peripheral blood of melanoma patients. An Enrichment + Expansion (E + E) platform has been developed so that T cells binding the magnetic nano-aAPC are easily purified in a sterile manner with commercial magnets that facilitates the expansion of antigen-specific T cells more rapidly and effectively than dendritic cells or other natural antigen-presenting cells. The resulting T cells are MHC Class I restricted, highly antigen-specific, polyfunctional and have a stem-memory phenotype with long telomeres. Combined, these cellular attributes characterize T cells with anti-tumor properties that may confer potent and durable effect when adoptively transferred to patients. We also demonstrated that this approach expands T cells from post-anti-PD-1 treated melanoma patients with an even higher yield.
Materials and Methods
Patient samples
The clinical studies were performed in accordance with the Declaration of Helsinki. All Peripheral blood samples were obtained from patients with metastatic melanoma under NYU Institutional Review Board (IRB)-approved protocols (NCT01783938 (26), and NCT02983006), and written informed consent was obtained from all subjects from whom peripheral blood samples were obtained. Samples were coded with an anonymized 5-digit number and their identity was unknown to those performing the experiments. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient with 1.077 g/mL Ficoll-Paque (GE Healthcare, Chicago, IL). For cryopreservation, PBMCs were resuspended in human AB serum with 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich, St Louis, Mo), frozen at −80°C for 2–3 days and then stored in liquid nitrogen.
Preparation of nano-aAPC and reagents
Nano-aAPC were provided by Neximmune Inc (Gaithersburg, MD). Briefly, nano-aAPC were manufactured by direct conjugation of humanized HLA-Ig dimer and anti-CD28 antibody to MACS Microbeads (Miltenyi Biotec) as described previously (24).
Recombinant human IL-2 (Aldesleukin, Prometheus Laboratories, San Diego, CA), IL-7, IL-15, IL-21 and GM-CSF (R&D systems) were commercially obtained. The cytokine mix (IL-2, IL-4, IL-6, IL-1 beta and gamma interferon) was provided by Neximmune Inc. CD8+ T-cells and CD14+ monocytes were isolated through EasySep magnetic negative or positive selection kit (StemCell Technologies, Vancouver, BC, Canada).
Cell lines
Melanoma cell lines A375, Malme-3M, MeWo, SK-MEL-28 and T lymphoblast T2 cells were purchased from the American Type Culture Collection (ATCC). Acute lymphoblastic leukemia cell line 1301 was obtained from European Collection of Authenticated Cell Cultures (ECACC). Cell lines were not authenticated and used for experiments between 8 and 12 passages. A375 cells were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS and antibiotic antimycotic solution (Sigma-Aldrich). Malme-3M cells were cultured in DMEM supplemented with 20% FBS and antibiotic antimycotic solution. MeWo and SK-MEL-28 cells were cultured in EMEM (Thermo Fisher Scientific) supplemented with 10% FBS and antibiotic antimycotic solution. T2 and 1301 cells were cultured in RPMI supplemented with 10% FBS and antibiotic antimycotic solution. All cell lines were cultured in a humidified 5% CO2, 37 °C incubator.
Flow cytometry analysis
Cells were washed using phosphate-buffered saline (PBS) with 2% FBS, and were then stained with monoclonal antibodies (mAbs) specific for CD8 (RPA-T8, BD), CD45RA (HI100, BD), CD95 (DX-2, BD), CD62L (DREG-56, eBioscience, San Diego, CA), CD69 (FN50, BD), 4–1BB (4B4–1, Biolegend), ICOS (DX29, BD), CD27 (M-T271, BD), CD28 (CD28.2, BD), PD-1 (MIH4, BD), CD223 (LAG-3) (3DS223H, eBioscience), Tim-3 (F38–2E2, BD) and KLRG1 (13F12F2, eBioscience). MART-1- or gp100-specific CD8+ T cells were detected by MART-1 or gp100/HLA-A*0201 PE-labelled tetramers (MBL, Woburn, MA). Intracellular staining of Ki67 (B56, BD) was performed after fixation and permeabilization using a staining kit (eBioscience) according to the manufacturer’s instructions. Live Dead dyes (Invitrogen, Carlbad, CA) was used to assess viable cells. The cells in all experiments were analyzed with an Attune NXT flow cytometer (ThermoFisher Scientific, Waltham, MA) and analysed using FlowJo 10 software (Ashland, Oregon).
Generation of MART-1 Tet+ T-cells by nano-aAPC
As previous described (25), nano-aAPC were stored at a concentration of 10 OD/mL, and all volumes refer to particles at this concentration assessed by optical density. Ten million CD8-enriched lymphocytes at ~108 cells/ml were incubated with 20 μl of nano-aAPC for 1 hour at 4°C. Cell-particle mixtures were subsequently passed through a magnetic enrichment column (Miltenyi). Isolated positive fractions were mixed and cultured for 14 days in medium supplemented with the cytokine cocktail (4 ng/ml IL-2, 0.3 ng/ml IL-4, 0.4 ng/ml IL-6, 0.2 ng/ml IL-1β, 1 ng/ml IFNγ) mentioned above in a humidified 5% CO2, 37 °C incubator. Specificity of CTL was monitored on day 0, 7 and 14 and function of CTL was assessed on day 14. The number of antigen-specific cells was calculated by multiplying the number of total live cells by the fraction of CD8 and antigen-specific cells.
Generation of monocyte-derived dendritic cells
CD14+ cells that were purified from PBMCs using a Human CD14 Positive Selection Kit, (StemCell) were cultured with 20 ng/ml GM-CSF and 20 ng/ml IL-4 (R&D systems) in X-VIVO15 medium (ThermoFisher Scientific) with 5% human serum AB (Omega Scientific). The medium was replaced with fresh medium containing cytokines at Day 3 and 6. To generate mature dendritic cells (mDCs), 1 μg/ml LPS (Sigma-Aldrich) were added to immature DC (iDCs) cultures for 48 hours (27).
Generation of MART-1 Tet+ T-cells by DCs
Antigen-specific CD8+ T cells were induced as described previously with modifications (27,28). Mature DCs were pulsed with 10 μM HLA-A*0201 restricted MART-126–35 (ELAGIGILTV) peptide (BioSynthesis, Lewisville, TX) for two hours. Mature LPS-treated DCs were cultured with sorted CD8+ T cells generated previously in fresh medium containing 20 IU/ml IL-2, 10 ng/ml IL-7, 10 ng/ml IL-15, and 20 ng/ml IL-21 (29,30) or the cytokine mix described above.
Polyclonal T-cell expansion by CD3/CD28 Dynabeads
Isolated CD8+ T cells were mixed with CD3/CD28 Dynabeads (ThermoFisher Scientific) and cultured with 30 IU/mL IL-2 according to the manufacturer’s instructions. The medium was replaced with fresh medium containing IL-2 every third day.
Intracellular cytokine staining
Expanded CD8+ T cells were stimulated for 3–4 hours with MART-126–35 A27L peptide-loaded T2 cells or melanoma cell lines. Golgi plug (protein transport inhibitor containing brefeldin A, BD) and Golgi stop (Monensin, BD) were added 2 hours before staining. Cells were stained for cell surface markers followed by intracellular cytokines (IFNγ [4S. B3, BD], TNFα [MAb11, BD], and IL-2 [MQ1–17H12, BD]) after fixation and permeabilization. For perforin (B-D48, BioLegend) and granzyme B (GB11, BD) staining, effector cells were cocultured with T2 cell for 16 hours. To analyze cytokine production of T cells, effector cells were cocultured with T2 cells for 24 hours and supernatants were collected. Cytokines were analyzed using a Luminex system (ThermoFisher Scientific).
Functional avidity analysis and cytotoxicity assay
T2 cells labeled with CFSE (2.5 μM, Invitrogen) were incubated with mature-DC or nano aAPC-expanded CD8+ T cells for 12–16 hours, then their specific lysis was analyzed by flow cytometry. The EC50 of target cell lysis was measured by Prism version 8 software (GraphPad, San Diego, CA). The target melanoma cell lines were labeled with CFSE (5 μM, Invitrogen) and incubated with CD8+ T cells for 12–16 hours, then specific lysis of cell lines was analyzed by flow cytometry. The specific lysis of target melanoma cells was measured at different effector versus target ratios (ET ratio: 0.01, 0.1, 1 and 10). The percent specific lysis was calculated as follows: 100-[(target cell number in the presence of effector cells)/(target cell number in the absence of effector cells) ×100].
Detection of telomere length
The telomere length of cells was assessed using a Telomere PNA kit (DAKO, #K5327). Briefly, an equivalent number of CD8+ T cells and 1301 cells which have longer telomere length were mixed, then a FITC-conjugated PNA probe that bind to telomeres was added. The relative telomere length was calculated using the manufacturer’s instructions.
TCR seq analysis
Antigen-specific CD8+ T cells were sorted based on MART-1 tetramer expression using the MoFlo XDP (BD Biosciences). Total genomic DNA was isolated with the DNeasy Blood and Tissue Kit (Qiagen, Venlo, the Netherlands). The DNA was used for deep sequencing of the β-chain, using the ImmunoSEQ platform (Adaptive Biotechnologies, Seattle, WA, USA). Details of the deep sequencing assay are as follows: the TCRβ and TCRγ CD3 region was amplified and sequenced using the ImmunoSEQ assay (Adaptive Biotechnologies, Seattle, WA). Specifically, CD8+ T cells from patients were either sequenced pre-expansion or expanded for MART-1 antigen using DCs or nano-aAPC, before sorting for MART-1 tetramer positive cells, and then sequenced. Diversity metrics, clone distribution, and Vβ gene usages were examined both pre- and post-expansion for each patient. The diversity metrics that we examined were: productive entropy, which is Shannon’s Entropy ( where pi is the frequency of a given clone) for all productive rearrangements within a sample, with larger numbers representing greater diversity; and productive clonality ( where H is productive entropy and N is the number of unique CDR3 clones) which ranges from 0 to 1 with lower values representing greater diversity.
Statistical analysis
The required sample size which varied depending on the experiment was determined in G*Power using a priori analysis, set at significance level 0.05 and power 0.9. Differences between two groups were analyzed by Student’s two-tailed non-paired or paired t test using Prism version 8 software. One-way ANOVA and Tukey’s multiple comparison post-hoc test was used for group comparisons. P values < 0.05 were considered statistically significant.
Results
Frequency of MART-1 Tet+ cells expanded with nanoparticle aAPC or autologous DCs
For enrichment by nano-aAPC, CD8+ T-cells were isolated from frozen PBMC that were thawed, and incubated with MART-1 peptide-loaded nano-aAPC, then the cell-particle mixture was passed through a magnetic column, and the antigen-specific fraction was enriched by eluting the magnetic-bound fraction containing T cells. After enrichment, the magnet-bound fractions of enriched cells and nano-aAPC were cultured in vitro (Fig. 1A) with cytokines. Seven days after enrichment, additional nano-aAPC and fresh media with cytokines were supplied. The antigen-specific CD8+ T-cells were detected by tetramer at day 7 and 14. We evaluated the frequency of MART-1 Tet+ CD8+ T-cells expanded by CD3/CD28 Dynabeads, nano-aAPC or lipopolysaccharide (LPS)-matured DCs pulsed with MART-1 peptide from the same stage IV melanoma patients. In general, the frequency of MART-1 Tet+ cells prior to nano-aAPC expansion by either method ranged from undetectable levels to approximately 0.01% of total CD8+ T cells. After 14 days, the frequency of MART-1 Tet+ cells expanded by nano-aAPC increased to 53.6% of total CD8+ T-cells. In contrast, Dynabeads or mature DCs did not expand MART-1 Tet+ cells to greater than 5% of total CD8+ T-cells (Fig. 1B). The frequency of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC were significantly increased compared to those expanded by Dynabeads or DCs (Fig. 1C). These data suggest that E + E with nano-aAPC can expand MART-1 Tet+ CD8 T+ cells more effectively than mature DCs.
Figure 1. Frequency of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC or autologous DCs.
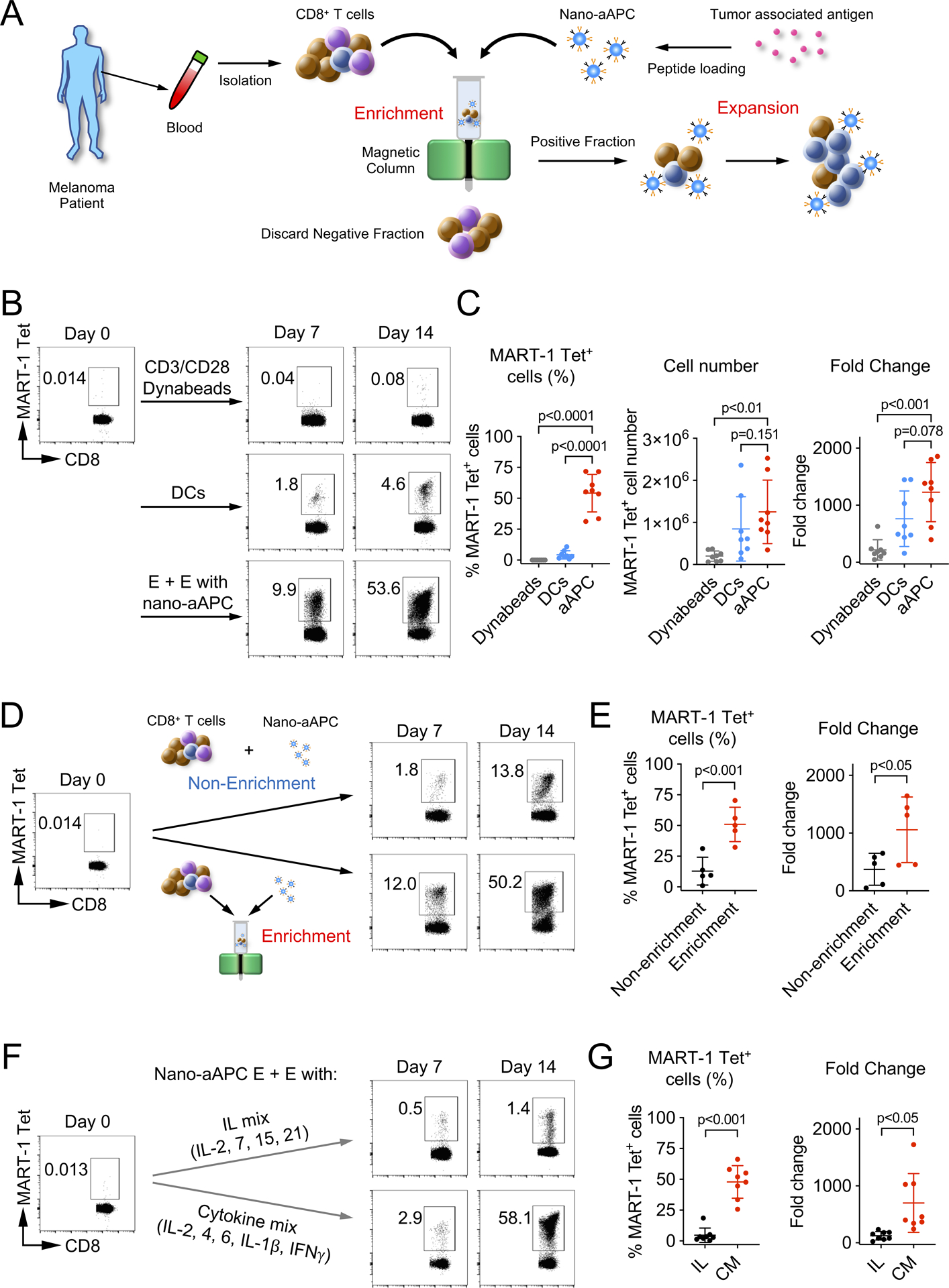
(A) Schematic diagram of Enrichment + Expansion using nano-aAPC. Isolated CD8+ T-cells were incubated with nano-aAPC loaded with MART-1 peptide, and then the cell-particle mixture was passed through a magnetic column. After enrichment, magnet-bound fractions of enriched cells and nano-aAPC were eluted and cultured in vitro. (B) Representative data of frequency of MART-1 Tet+ CD8+ T-cells expanded by CD3/CD28 Dynabeads, mature DCs or nano-aAPC at day 0 (pre-expansion), day 7 and day 14. (C) Summary of frequency, cell number and fold change of MART-1 Tet+ CD8+ T-cells from the same melanoma patients (n=8). (D) Representative data of frequency of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC with Enrichment or Non-enrichment method at day 0 (pre-expansion), day 7 and day 14. (E) Summary of frequency and fold change of MART-1 Tet+ CD8+ T-cells from the same melanoma patients (n=5). (F) Representative data of frequency of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC in the presence of IL mix (IL) or cytokine mix (CM) at day 0 (pre-expansion), day 7 and day 14. (G) Summary of frequency and fold change of MART-1 Tet+ CD8+ T-cells from the same melanoma patients (n=8). Success rates were 51/53 for E + E with nano-aAPC, 16/16 for DCs, 8/10 for Dynabeads from stage IV melanoma patients. Significance was assessed by Student’s two-tailed paired t test.
To investigate the effect of enrichment on subsequent T cell proliferation, we compared cells that had undergone the enrichment process to those that had not been enriched (Fig. 1D). Enrichment significantly enhanced both frequency and fold change of MART-1 Tet+ CD8+ T-cells (p<0.001 for frequency, p<0.05 for fold change, Fig. 1E).
Cytokine mix versus IL-2, 7, 15, and 21 for expansion of MART-1 Tet+ CD8+ T-cells by nano-aAPC
Next, we compared a cytokine cocktail with well-established γc chain cytokines (IL mix; IL-2, 7, 15, and 21) (29) for the expansion of MART-1 Tet+ CD8+ T-cells using nano-aAPC. In a representative sample shown, cytokine mix expanded MART-1-positive cells to 58% of CD8+ T-cells, whereas IL mix expanded MART-1 Tet+ CD8+ T-cells only to 1.4% of CD8+ T-cells in the same patients (Fig. 1F). The frequency and fold change of MART-1 Tet+ CD8+ T-cells expanded by the cytokine mix were significantly higher than those expanded with IL mix (p<0.001 for frequency, p<0.05 for fold change, Fig. 1G). These results suggest that the cytokine mix promotes the expansion of MART-1 Tet+ CD8+ T-cells to a higher frequency than IL-2, 7, 15, and 21.
Comparison of nano-aAPC T cell expansion in melanoma patients and healthy donors.
We evaluated whether there was a difference between melanoma and healthy donor samples in the degree of expansion of MART-1 Tet+ CD8+ T-cells by nano-aAPC. After 14 days, the frequency of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC increased to approximately 50% of total CD8+ T cells from both healthy donors and melanoma patients (Fig. 2A and B). There was no significant difference in the frequency, cell number and fold change of MART-1 Tet+ CD8+ T-cells from 8 healthy donors and 8 melanoma patients (Fig. 2B).
Figure 2. Comparison of nano-aAPC T cell expansion from melanoma patients and healthy donors.
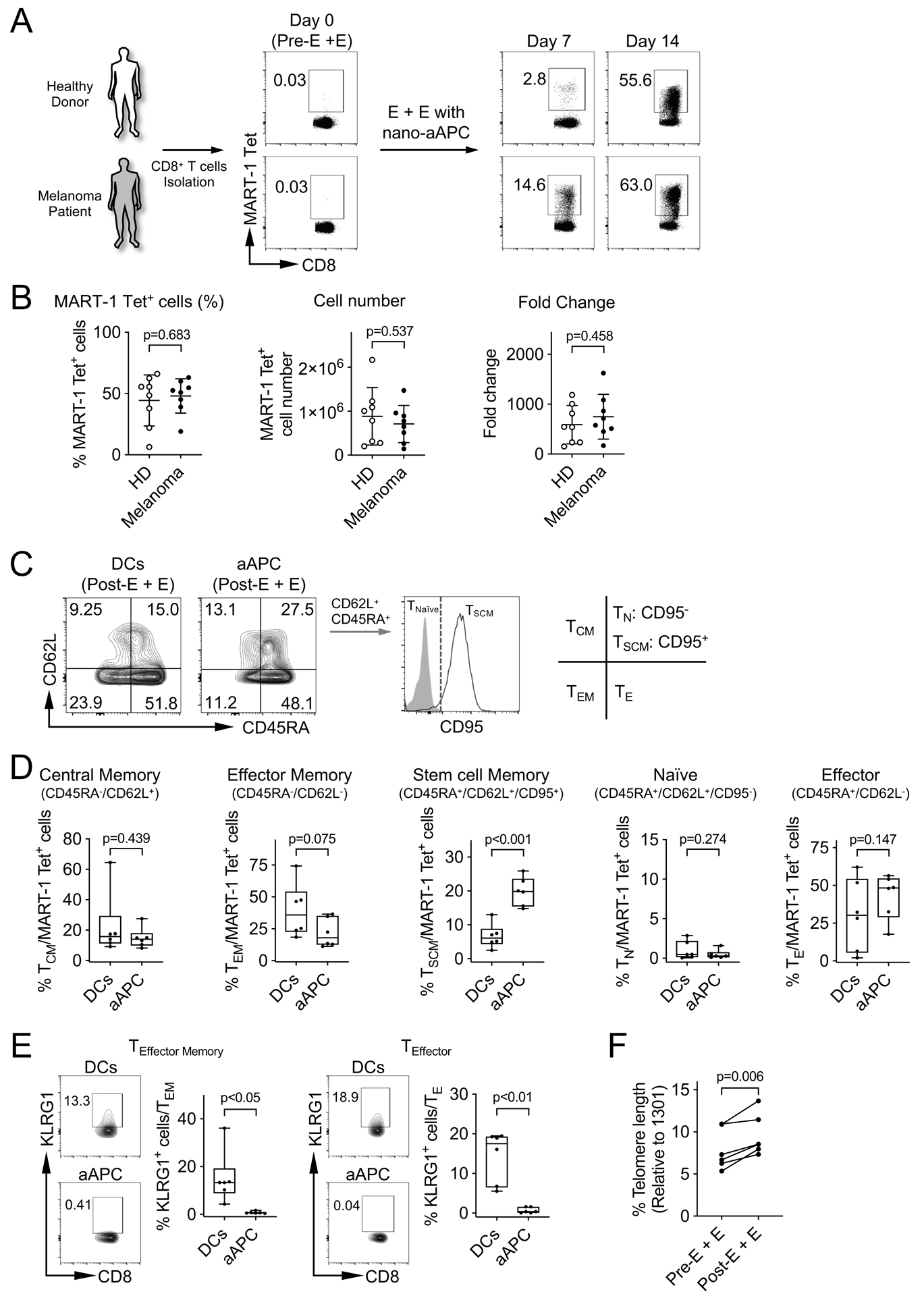
(A) The frequency of MART-1 Tet+ CD8+ T-cells by nano-aAPC expansion on day 0, 7 and 14 from healthy individuals and melanoma patients. Representative flow data of one melanoma patient and one healthy donor were shown. (B) Summary of frequency, fold change and cell number of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC in 8 healthy donors (HD) and 8 melanoma patients (Melanoma). Significance was assessed by Student’s two-tailed t test. (C) Representative staining of MART-1 Tet+ CD8+ T-cells from melanoma patients expanded by DCs or nano-aAPC for CD45RA, CD62L (L-selectin) and CD95 at day 14. The staining pattern of TSCM and TNaïve was shown. (D) Summary of phenotypes on MART-1 Tet+ CD8+ T-cells expanded by DCs or nano-aAPC from the same melanoma patients (n=6). Phenotypes of T-cells were categorized based on CD45RA, CD62L and CD95 expression into central memory CD45RA−, CD62L+, (TCM), effector memory CD45RA−, CD62L− (TEM), stem cell memory CD45RA+, CD62L+, CD95+ (TSCM), naïve CD45RA+, CD62L+, CD95− (TN), and effector CD45RA+, CD62L− (TE) populations. (E) Expression of KLRG1 on TEffector Memory and TEffector cells of MART-1 Tet+ CD8+ T-cells was analyzed (n=6). (F) Relative telomere length from 6 melanoma patients was analyzed based on those of 1301 cells which has long telomere length. Error bars indicate mean ± SD. Significance was assessed by Student’s two-tailed paired t test.
Phenotype, activation and checkpoint expression of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC or DCs.
To analyze the phenotype of MART-1 Tet+ CD8+ T-cells expanded by DCs or nano-aAPC, MART-1 Tet+ CD8+ T-cells were categorized based on CD62L (L-selectin), CD45RA and CD95 expression into central memory CD45RA−CD62L+ (TCM), effector memory CD45RA−CD62L− (TEM), stem cell memory cells CD45RA+, CD62L+, CD95+ (TSCM), naïve CD45RA+CD62L+CD95− (TN), and effector CD45RA+CD62L− (TE) populations (Fig. 2C). The use of nano-aAPC expansion increased the frequency of central memory and stem cell memory MART-1 Tet+ CD8+ T-cells (TCM: P=0.0044 and TSCM: P=0.0002), and decreased the frequency of naïve and effector memory cells (TN: P=0.0058 and TEM: P=0.002) compared with pre-expansion CD8+ T-cells (Supplementary Fig. S1A and S1B). We then compared the phenotype of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC to those expanded by DCs. The enhanced frequency of stem cell memory phenotype was observed in nano-aAPC expanded MART-1 Tet+ CD8+ T-cells rather than DCs (Fig. 2D). Adoptive cell transfer strategies require effector cell expansion and persistence in vivo after engraftment of T cells to optimize durable clinical efficacy. We investigated the expression of KLRG1, a senescence marker of T cells, on expanded effector memory and effector type CD8+ T-cells. A higher expression of KLRG1 was observed on MART-1 Tet+ CD8+ T-cells that were expanded by DCs than those expanded by nano-aAPC (p<0.05 for TEM, p<0.01 for TE, Fig. 2E). Next, we determined whether MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC E + E have a long telomere length that is associated with achieving long-term persistence after transfer. Expanded MART-1 Tet+ CD8+ T-cells from individual melanoma patients were mixed with 1301 cells which have a defined long telomere length. Fluorescence in situ hybridization was performed using a FITC-conjugated PNA probe to detect the telomere length of T cells. The relative telomere length of expanded CD8+ T-cells was significantly higher than those of pre-E + E CD8+ T-cells (P=0.006, Fig. 2F).
We analyzed whether the nano-aAPC based expansion affected expression of activation (CD69, 4–1BB, ICOS and OX-40, Fig. 3A), co-stimulatory (CD27 and CD28, Fig. 3B), checkpoint (PD-1, Tim3 and LAG3, Fig. 3C), proliferation (Ki67, Fig. 3D), and senescence markers (KLRG1, Fig. 3E) on MART-1 Tet+ CD8+ T-cells. Expansion with nano-aAPC significantly increased the frequency of CD69, 4–1BB, ICOS, CD27, OX-40, Ki-67, PD-1, Tim3 and LAG3 expression on MART-1 Tet+ CD8+ T-cells compared with CD8+ T-cells before expansion. In contrast, expansion with nano-aAPC decreased CD28 expression by MART-1 Tet+ CD8+ T-cells (Supplementary Fig. S1C). MART-1 Tet+ CD8+ T-cells expanded by both DCs and nano-aAPC showed similar expression levels of each molecules except for ICOS, Tim3, Ki67 and KLRG1 (Fig. 3). Expansion with nano-aAPC significantly increased the frequency of ICOS (p<0.05), Tim3 (p<0.01) and Ki67 (p<0.05) expression on MART-1 Tet+ CD8+ T-cells. In contrast, DCs induced expression of KLRG1 (p<0.01).
Figure 3. Analysis of activation, maturation, checkpoint, proliferation and senescence markers on MART-1 Tet+ CD8+ T-cells expanded by DCs or nano-aAPC.
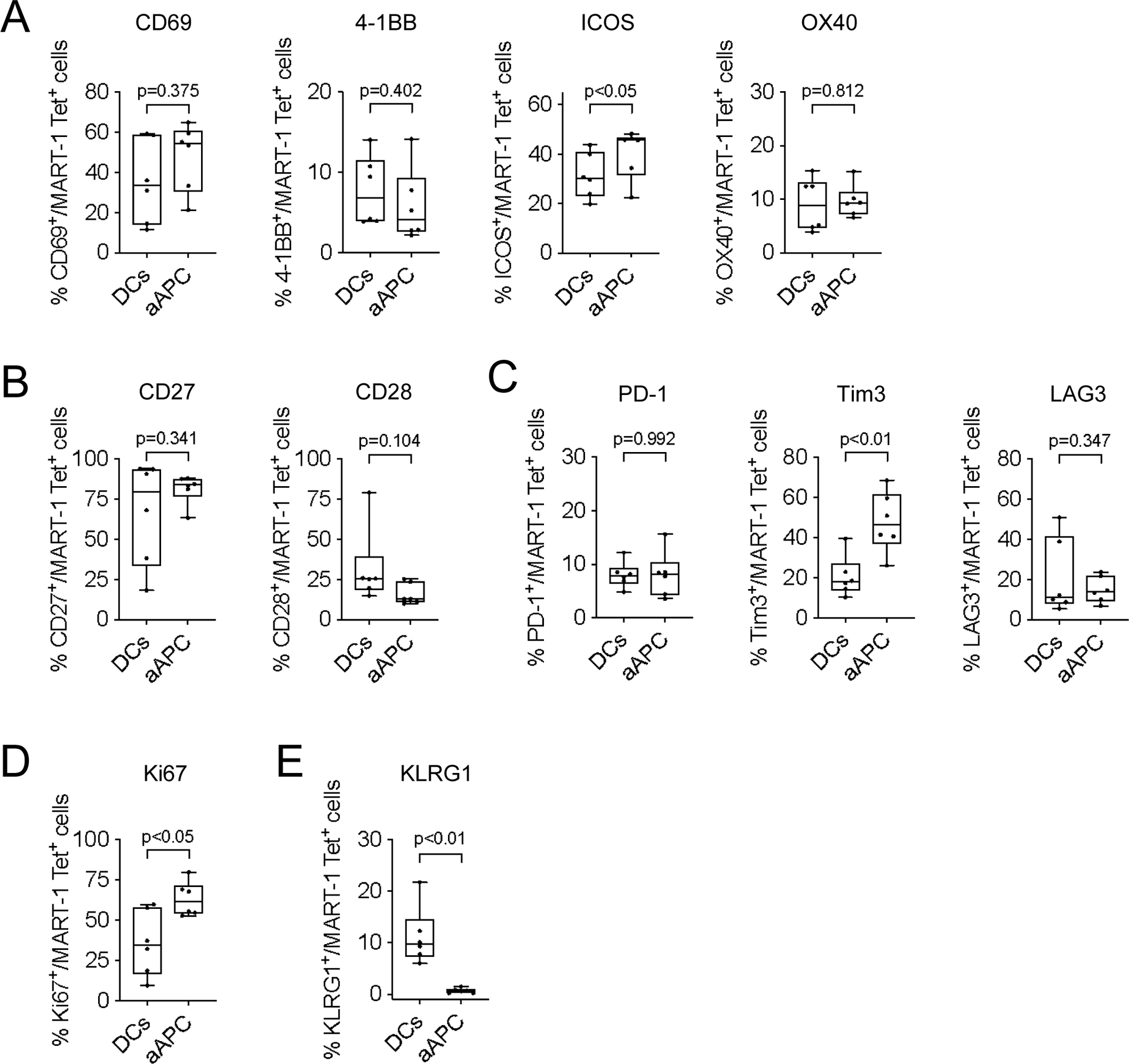
(A-E) Expression of activation (A, CD69, 4–1BB, ICOS and OX-40), maturity (B, CD27 and CD28), checkpoint (C, PD-1, Tim3 and LAG3), proliferation (D, Ki67) and senescence molecules (E, KLRG1) on MART-1 Tet+ CD8+ T-cells expanded by DCs or nano-aAPC from the same melanoma patients (n=6). Significance was assessed by Student’s two-tailed paired t test.
CD8+ T-cells expanded with nano-aAPC are polyfunctional
To assess the function of CD8+ T cells which were expanded with nano-aAPC from melanoma patients, we performed intra-cellular cytokine analysis of MART-1 Tet+ CD8+ T-cells. Expanded cells were co-cultured with MART-126–35 A27L peptide-pulsed T2 cells. Expression of CD107a, and production of TNFα, IFNγ and IL-2 were observed by MART-1 Tet+ CD8+ T-cells co-cultured with MART-1 peptide-loaded T2 cells but not with control T2 cells (Fig. 4A and B). In contrast, MART-1 Tet− CD8+ T-cells produced minimal levels of cytokines when co-cultured with MART-1 peptide-loaded or control T2 cells (Supplementary Fig. S2A and S2B). Expanded MART-1 Tet+ CD8+ T-cells also expressed perforin and granzyme B after incubation with target cells (Fig. 4C and Supplementary Fig. S2C and S2D). We analyzed cytokine release from nano-aAPC expanded MART-1 Tet+ CD8+ T-cells following exposure to peptide-pulsed or control T2 cells by Luminex. Secreted cytokines granzyme B, IFNγ, TNFα, IL-2, IL-4, IL-5, IL-13 were detected in response to peptide-pulsed T2 cells but not to control T2 cells (Supplementary Fig. S2E). Polyfunctionality analysis of MART-1 Tet+ CD8+ T-cells (Fig. 4D), showed that about 20% of cells produced all three cytokines measured (TNFα, IFNγ and IL-2) and were CD107a positive, and approximately 75% of cells exhibited multiple functional markers compared to those expressing only one (p=0.001, Fig. 4E). To determine whether expansion methods affected the functionality of expanded T cells, we analyzed the expression profile of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC or peptide-pulsed mature DCs using the same patient samples. MART-1 Tet+ CD8+ T-cells expanded by peptide-pulsed DCs exhibited multifunctionality but the frequency of polyfunctional effectors expressing all four effector functions was lower than that achieved with T cells expanded with nano-aAPC (Fig. 4F and Supplementary Fig. S3A). T cells expanded with mature-peptide-pulsed DC had a lower frequency of IL-2 and CD107a expression (p<0.01 for CD107a, p=0.01 for IL-2, Supplementary Fig. S3B and S3C), indicating that MART-1 Tet+ CD8+ T-cells expanded with nano-aAPC possess highly specific polyfunctionality against target cells.
Figure 4. Nano-aAPC expanded MART-1 Tet+ CD8+ T-cells are polyfunctional.
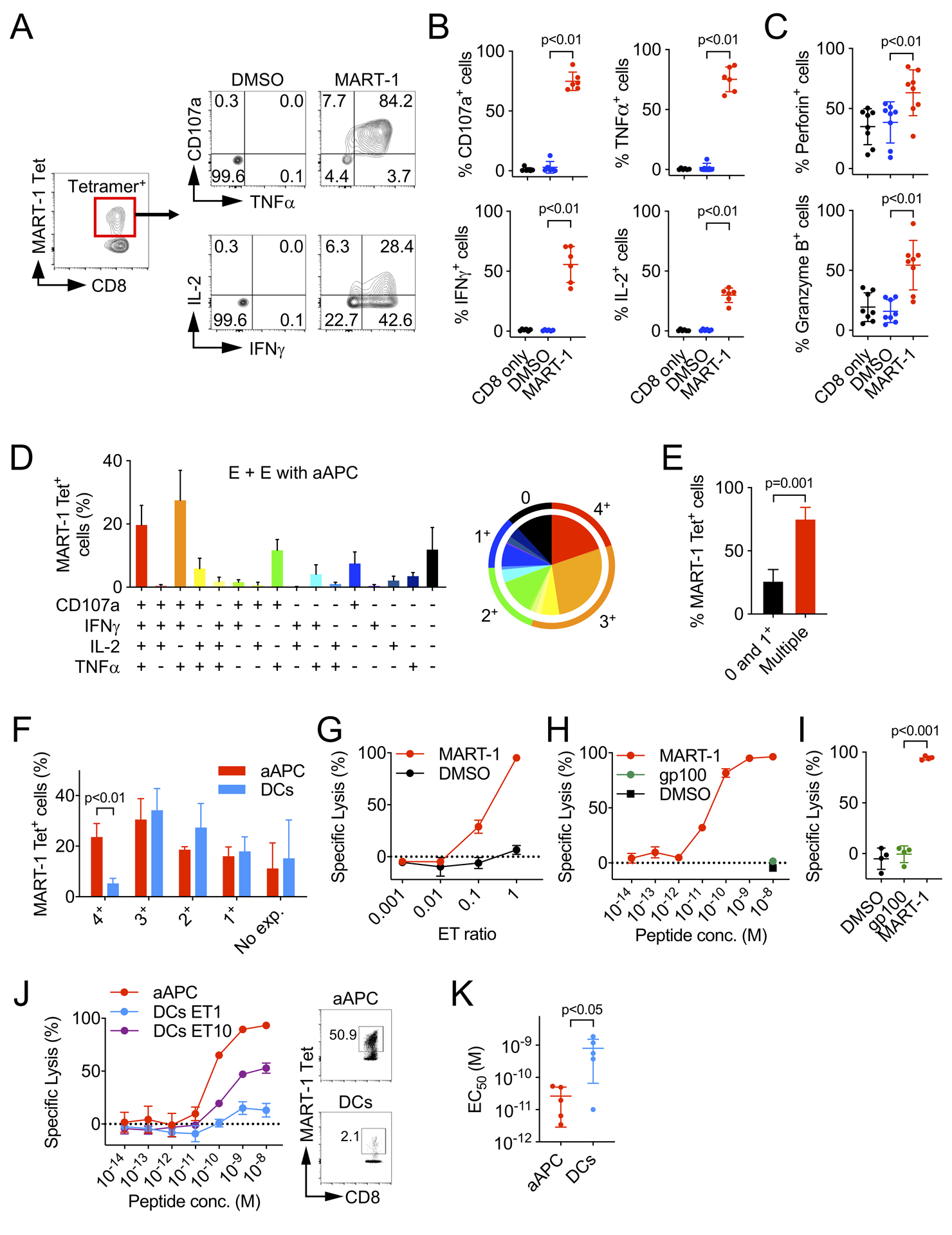
(A) Representative staining pattern of MART-1 Tet+ CD8+ T-cells for CD107a and indicated cytokines. Expanded CD8+ T-cells were co-cultured with T2 cells pulsed with the MART-126–35 A27L peptide. (B) Summary of expression of CD107a, intracellular TNFα, IFNγ, IL-2 from 6 melanoma patients. (C) Summary of perforin and granzyme B expression from 8 melanoma patients. (D) Polyfunctional analysis of MART-1 Tet+ CD8+ T-cells from 6 melanoma patients. All combinations of responses were shown. The data were summarized in a pie chart. (E) MART-1 Tet+ CD8+ T-cells expressing 2 or more functions were compared with those expressing 0 and 1 function. (F) The frequency of MART-1 Tet+ CD8+ T-cells that expressed each cytokine (from 1+ to 4+) and no cytokines (No exp.). (G) Cytotoxic activity of MART-1 Tet+ CD8+ T-cells at various E:T ratios (n=3). (H and I) Cytotoxic activity of MART-1 Tet+ CD8+ T-cells from 4 melanoma patients. The percent specific lysis against T2 cells which were pulsed with MART-1 or gp100 peptide (10−8 (M) respectively). (J) Specific lysis of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC (ET=1) or DCs (ET=1 and 10) from the same melanoma patient. (K) The functional avidity of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC or DCs from same patients (n=5). Error bars indicate mean ± SD. Significance was assessed by Student’s two-tailed t test.
Additionally, we determined whether MART-1 Tet+ CD8+ T-cells generated by E + E with nano-aAPC possessed killing activity against antigen-expressing target cells. Expanded MART-1 Tet+ CD8+ T-cells were incubated with CFSE-labeled T2 cells which were pulsed with MART-126–35 A27L peptide for 12–16 hours at various ET ratio. Specific cytotoxicity against MART-1 peptide-loaded T2 cells was observed (Fig 4G). Peptide concentration-dependent cytotoxicity was also observed at ET ratio 1:1 (Fig. 4H). MART-1 Tet+ CD8+ T-cells derived from 4 melanoma patients uniformly exhibited specific and high levels of lytic activity against antigen-expressing target cells (Fig. 4I). To determine whether APC expansion methods affected the cytotoxic activity of expanded effector cells, we co-cultured peptide-pulsed or control T2 cells and MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC or peptide-pulsed mature DCs from the same melanoma patients (Fig. 4J). CD8+ T-cells expanded by DCs exhibited a lesser degree of lytic activity due to their lower purity of MART-1 Tet+ CD8+ T-cells (Fig. 4J and Supplementary Fig. S3D). Moreover, MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC exhibited higher functional avidity compared to MART-1 Tet+ CD8+ T-cells expanded by autologous DCs (Fig. 4K), indicating that CD8+ T-cells generated by nano-aAPC possessed higher tumor-specific lytic activity than those expanded by mature peptide-pulsed DCs.
We used the MART-126–35 A27L peptide to assess the functionality of MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC, so to validate whether those expanded effector cells recognized native peptide, we tested whether nano-aAPC expanded T cells showed lytic activity against T2 cells loaded with naturally processed peptides, MART-126–35 EAAGIGILTV and MART-127–35 AAGIGILTV. As shown in a T2 stabilization assay which indicated affinity of peptides (Supplementary Fig. S4A), similar lytic activity was observed against MART-126–35 A27L or naturally processed MART-127–35 peptide-loaded T2 cells, although less against MART-126–35 peptides-loaded targets (Supplementary Fig. S4B).
To evaluate whether aAPC-stimulated T cells exhibited cytotoxic activity against naturally processed, antigen-expressing melanoma cell lines, T cells were incubated with target melanoma cell lines which were labeled with 5 μM of CFSE for 12–16 hours. Expanded CD8+ T-cells which retained functionality against cell lines (Supplementary Fig. S4C and S4D) exhibited cytotoxic activity against MeWo and Malme-3M (HLA-A*0201+, MART-1+) but not to SK-MEL-28 (HLA-A*0201−, MART-1+) or A375 (HLA-A*0201+, MART-1−) melanoma cell lines (Fig. 5A and Supplementary Fig. S4E). A high level of specific lysis by MART-1 Tet+ CD8+ T-cells expanded from 5 separate melanoma patients against MeWo was observed suggesting that antigen-specific CD8+ T-cells with potent functionality against naturally processed MART-1 antigen were efficiently expanded by nano-aAPC (Fig. 5B).
Figure 5. Cytotoxic activity of nano-aAPC expanded CD8+ T-cells against melanoma cell lines.
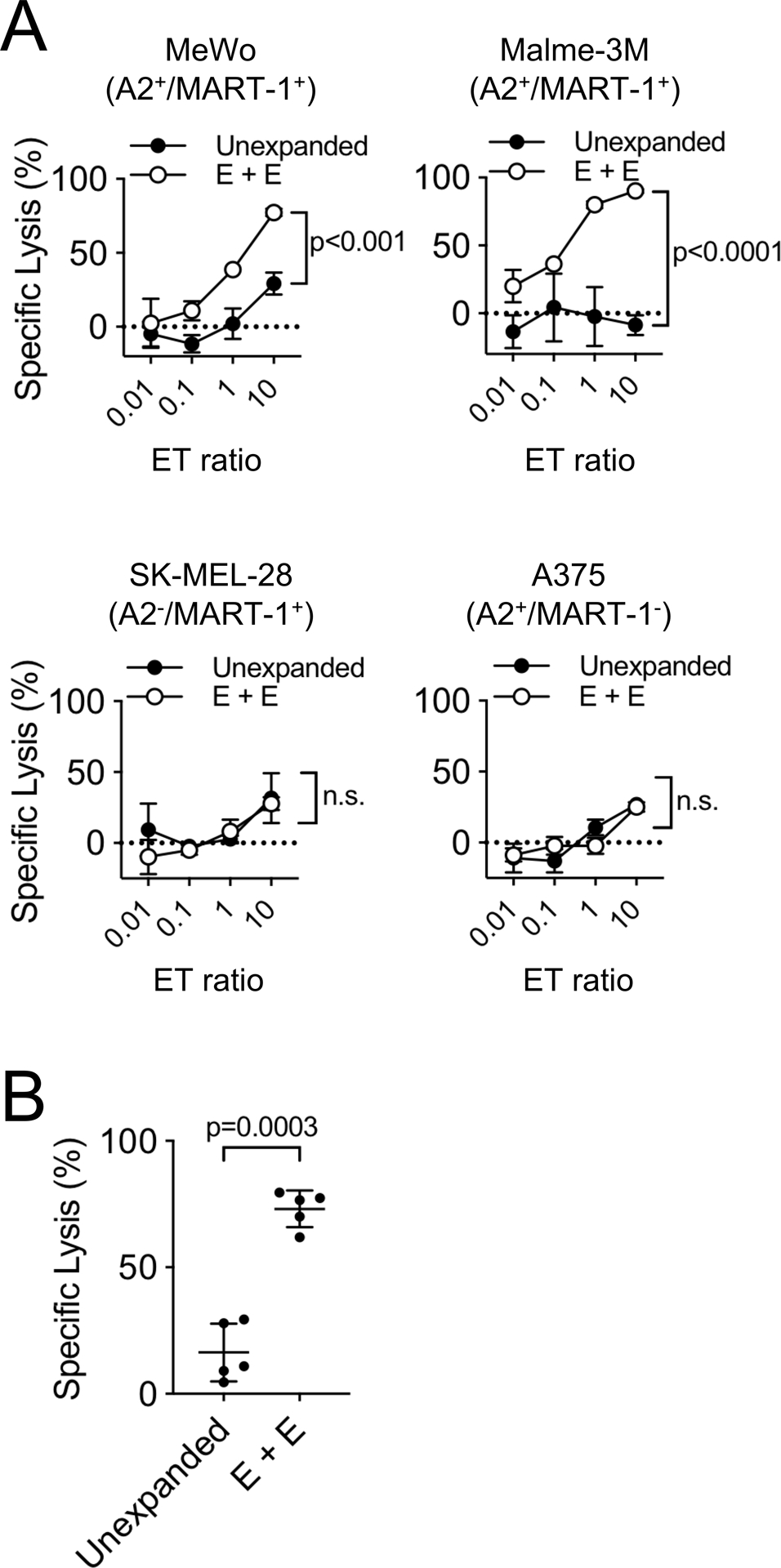
(A) Specific lysis of CD8+ T-cells were analyzed. CD8+ T-cells were co-cultured with melanoma cell lines SK-MEL-28 (HLA-A*0201−, MART-1+), A375 (HLA-A*0201+, MART-1−), MeWo (HLA-A*0201+, MART-1+), Malme-3M (HLA-A*0201+, MART-1+) at various ET ratios. Significance was assessed by Student’s two-tailed t test at ET=10. (B) Specific lysis of nano-aAPC expanded CD8+ T-cells against MeWo at ET=10 comparing with unexpanded CD8+ T-cells from 5 melanoma patients. Error bars indicate mean ± SD. Significance was assessed by Student’s two-tailed paired t test.
Nano-aAPC E + E of CD8+ T cells specific for gp100
We assessed whether Enrichment + Expansion with nano-aAPC was effective using other target antigens. E + E was performed using nano-aAPC bearing the gp100209–217 tumor antigen peptide. The cell number and purity of gp100 Tet+ CD8+ T-cells were increased at day 14 (Supplementary Fig. S5A) indicating that E + E platform using nano-aAPC is applicable using other TAA for the expansion of CTL. Expanded gp100 Tet+ CD8+ T-cells also exhibited a more stem cell memory phenotype compared with pre-E + E CD8+ T-cells (Supplementary Fig. S5B). These results were consistent with those observed with MART-1 Tet+ CD8+ T-cells (Supplementary Fig. S1B).
Expansion of MART-1 Tet+ cells pre- and post-PD-1 treatment
To evaluate whether anti-PD-1 treatment influenced the expansion and TCR repertoire of MART-1 Tet+ CD8+ T-cells expanded with nano-aAPC, we analyzed 6 patients who had paired peripheral blood samples obtained prior to, and after, anti-PD-1 treatment. These patients included 3 responders and 3 non-responders to treatment with PD-1 blockade. Nano-aAPC increased the MART-1 Tet+ CD8+ T-cells from 0.01–0.1% at Day 0 to approximately 20–90% of total CD8+ T cells at Day 14 (Supplementary Fig. S6A and S6B). In five of six post-PD-1 treatment samples, anti-PD-1 treatment induced a higher frequency of MART-1 Tet+ CD8+ T-cells cells compared with the baseline pre-PD-1 (Supplementary Fig. S6C). Anti-PD-1 treatment increased the cell number in all three responders, and in two of three non-responders (Supplementary Fig. S6C). We also evaluated whether anti-PD-1 treatment affected the function of expanded MART-1 Tet+ CD8+ T-cells from anti-PD-1 treated patients, and they retained multifunctionality against antigen-expressing target cells (Supplementary Fig. S7).
TCR repertoire analysis of nano-aAPC expanded MART-1 Tet+ CD8+ T-cells
To analyze the impact of nano-aAPC expansion on the TCR repertoire, MART-1 Tet+ CD8+ T-cells were isolated after E + E using nano-aAPC, and a TCR repertoire analysis was performed by DNA sequencing. Results were compared with pre-expansion CD8+ T-cells. The TCR repertoire of expanded CD8+ T-cells specific for the MART-1 antigen using nano-aAPC was compared to the repertoire of cells expanded with peptide-pulsed DCs. Pre-expansion CD8+ T cells had similar distributions of clonotypes and Vβ gene usage (Fig. 6A and B). Clonotype distribution varied depending on the expansion method (Supplementary Fig. S8A and S8B). Vβ gene usage was also considerably different both between patients and depended on the expansion methods. For instance, T-cells expanded by DCs from patient 3 (P3) primarily used Vβ19, whereas nano-aAPC expanded cells from the same patient used Vβ06. Similarly, while T-cells expanded by DCs from patient 7 (P7) used primarily Vβ28, nano-aAPC expanded T cells used both Vβ28 but also Vβ13. We then compared the MART-1 repertoires of healthy donors and melanoma patients after nano-aAPC expansion and found that they were similarly diverse (Supplementary Fig. S8C and S8D). Moreover, they had similar clonotype distributions (Fig. 6C) and used a similar dominant set of Vβs (Vβ06, Vβ04, and Vβ28) (Fig. 6D). Based on an analysis of productive entropy and clonality, the different expansion methods both resulted in a less diverse T cell receptor repertoire (Fig. 6E and F).
Figure 6. MART-1 TCR Repertoires from healthy donors and melanoma patients post expansion with nano-aAPCs.
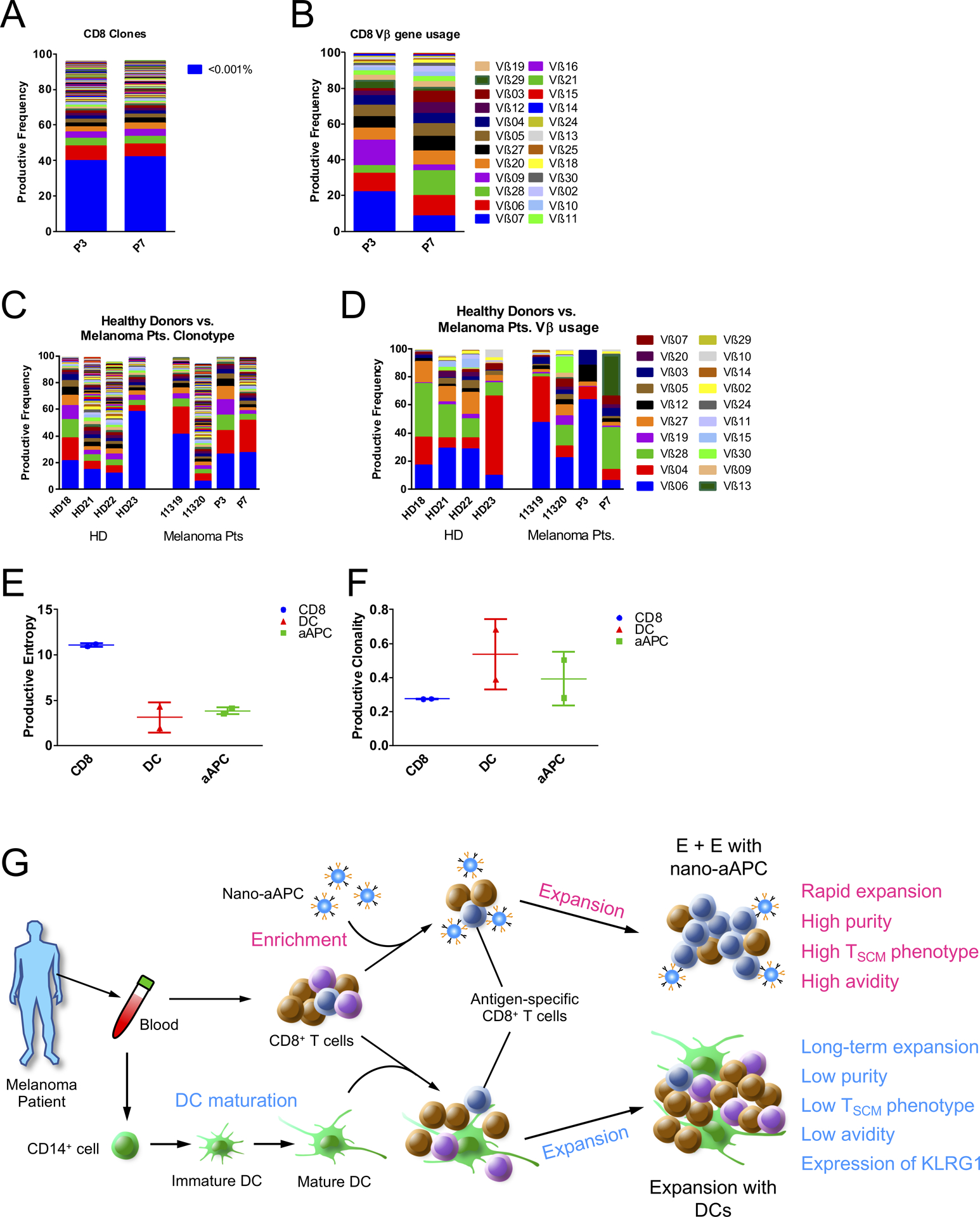
(A) Productive frequency of clones in CD8+ T-cell samples of two patients, with blue representing the sum of all clones with frequencies lower than 0.001%. (B) Vβ gene usage of CD8 samples in two patients prior to DCs or nano-aAPC expansion. (C) Frequency of individual MART-1-specific T cell clones ordered from highest to lowest frequency for healthy donor (HD) vs. melanoma patients. (D) Frequency of each Vβ for MART-1-expanded samples from healthy donors and melanoma patients. (E) Productive Entropy of CD8 samples from two patients compared to their corresponding MART-1 Tet+ CD8+ T-cells expanded by DCs or nano-aAPC. (F) Productive clonality of CD8 samples from two samples compared to their corresponding MART-1 Tet+ CD8+ T-cells expanded by DCs or nano-aAPC. (G) Summary of expansion by nano-aAPC or DCs.
Discussion
In this work, we employed nanoparticle aAPCs offering an “off the shelf” platform to rapidly expand large numbers of highly functional, antigen-specific T cells derived from patients with metastatic melanoma. MART-1 specific CD8+ T cells were rapidly expanded up to 1000-fold in a 14-day exposure to peptide-loaded nano-aAPC. The E + E system with peptide loaded nano-aAPC effectively expanded tumor antigen-specific T cells in terms of number and purity of cells, suggesting that this technology could potentially be employed to expand cells for ACT.
We demonstrated that E + E with nano-aAPC generated highly enriched MART-1 specific CD8+ T cells from both healthy donors and metastatic melanoma patients. Effective expansion of MART-1 responding cells has been reported by several groups (12,31–35). Although their protocols have shown the generation of a high precursor frequency against MART-1, they require extensive culture time or repetitive stimulation to expand T cells or an additional enrichment process for T cells (36). Our nano-aAPC E + E platform generated large numbers and a high proportion of MART-1 Tet+ CD8+ T-cells with only 1 re-stimulation within 14 days. An autologous DC-based ACE-CD8 platform has been shown to be a promising method that produces a large number of CD8+ T-cells (7), however this approach, along with other DC-based stimulations, is clinically difficult if more than one round of DC-based stimulation is required since production of autologous DC will become limiting, requiring additional leukaphereses. In addition, DCs from patients with cancer are often dysfunctional (37,38), or are often immunosuppressive (39,40). In contrast, the use of nano-aAPC omits the need for autologous cellular APC or to use feeder cells.
Therapies that require an infusion of a large population of purely antigen-specific T cells using DC-based approaches require multiple rounds of re-stimulation ex vivo, which can render effectors hyporesponsive to re-stimulation (41). The use of non-specific stimulation with anti-CD3/anti-CD28 Dynabead leads to a decrease in the percent and number of antigen-specific T cells, possibly due to expansion of bystander cells (9,31). A system that enables both enrichment and expansion as in the current work offers a proliferative advantage to rare antigen-specific T cells (25), thus reducing the amount of re-stimulation as well as shortening the culture time to achieve a high number and frequency of antigen-specific T cells, and allowing for a less exhausted, more stem-like cellular product.
There are a variety of technologies for isolating antigen-specific T cells, such as flow sorting of T cells using tetramers, 4–1BB positivity, or OX-40 positivity (1), IFN-γ capture systems (42), or anti-PE/APC magnetic beads post-tetramer staining (43). There are also a variety of “off-the-shelf” technologies for expansion of antigen-specific T cells, such as cellular aAPCs, based on the K562 human leukemic cell line (9,13,44), or acellular platforms such as liposomes (45), exosomes (46), polymeric beads (47–49), or T cell stimulating matrices (50). However, the technology described in this paper is unique in its ability to both isolate and simultaneously expand rare antigen-specific T cells.
We also assessed the telomere length of nano-aAPC expanded CD8+ T-cells, a factor which has been shown to be associated with T cell persistence in vivo and durability of ACT responses (51). CD8+ T-cells expanded with peptide-loaded nano-aAPC exhibited a longer telomere length than unexpanded cells. Compared to autologous DCs, the nano-aAPC E + E platform expanded more T cells with stem cell memory phenotype (52), associated with durable responses and long-term survival with the use of adoptive cell transfer. Moreover, MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC expressed very low levels of the senescence molecule KLRG1, consistent with continuous proliferative activity of T-cells.
We investigated expression of activation, proliferation, senescence and checkpoint molecules on MART-1 Tet+ CD8+ T-cells expanded by nano-aAPC. Our E + E platform enhanced expression of CD69, 4–1BB, ICOS, OX-40 and Ki67 indicating the expansion of highly activated and proliferative T cells. The fact that expression of the checkpoint molecules PD-1, Tim3 and LAG3 was also upregulated, suggests that combination of adoptively transferred nano-aAPC expanded T cells with immune checkpoint inhibitors could be useful for future immunotherapy approaches.
Although PD-1 blocking antibodies alone or in combination with ipilimumab are associated with objective response rates of 40 to 55% in patients with metastatic melanoma (53,54), almost half of patients ultimately do not obtain benefit. Our work demonstrated that the nano-aAPC E + E platform induced a higher frequency of MART-1 Tet+ CD8+ T-cells in post-nivolumab treatment samples, suggesting that antigen-specific cells can be expanded after PD-1 blockade, even in non-responder patients. Expanded MART-1 Tet+ CD8+ T-cells from post treatment patients also showed polyfunctionality against target cells (Supplementary Fig. S7) indicating that T-cells produced by nano-aAPC after anti-PD-1 treatment are functional and might be useful for ACT.
T-cell activation by CD3/CD28 Dynabeads with exogenous interleukin-2 (IL-2) supplementation has been used for non-antigen specific expansion of T cells in clinical trials (18). However, these cultures may produce functionally limited or dysregulated T cells (55,56). We demonstrated that MART-1 specific CD8+ T-cells generated by E + E with nano-aAPC were highly polyfunctional. The majority of MART-1 Tet+ CD8+ T-cells showed multifunctionality, with over 20% of cells producing TNFα, IFNγ and IL-2, which was decreased to 5.2% in cells expanded by autologous DCs (Fig. 4F and Supplementary Fig. S3). These results are consistent with previous work where mature DC-generated T cells are less functional (57). We also demonstrated that the high purity of target T cells induced by the nano-aAPC E + E platform is associated with efficient target killing activity (Fig. 4J and Supplementary Fig. S3D). Moreover, the functional avidity of MART-1 Tet+ CD8+ T-cells generated by nano-aAPC was higher than those of cells expanded by mature peptide-pulsed DCs (Fig. 4J and K), indicating that expansion of antigen-specific T cells with nano-aAPC can produce highly functional T-cells with high avidity and long telomeres, which might contribute to, and are thought to be critical for, in vivo anti-tumor killing activity (58).
The expansion of large numbers of rare, neoantigen specific T-cells can be important for future ACT but is challenging due to a low precursor frequency of such cells in the periphery. Although neoantigen peptide-pulsed autologous mature DCs could be useful for expanding neoantigen-specific T cells, the need to culture DCs with multiple rounds of re-stimulation increases the cost and resources needed to adequately expand T cells (59). The nano-aAPC E + E platform induces large numbers of high-purity tumor-specific T cells within 2 weeks. Moreover, nano-aAPC are highly adaptable and could be loaded with different target peptides for multi-antigen specific T cell expansion, not only TAA such as gp100 (Supplementary Fig. S5) but also neoantigens. Although some of the samples expanded with nano-aAPC resulted in a low yield due to the low frequency of antigen-specific T-cells pre-expansion (MART-1; 0.0037% to 6.5%, gp100; 0.005% to 7.1%), suggesting that certain minimal levels of CD8+ T-cells could be needed for a less dominant antigen, a single leukapheresis could yield enough cells to overcome that hurdle. Because of the adaptable approach of the aAPC design, additional HLA allele molecules, co-stimulatory molecules (eg, 4–1BB, OX-40) and/or cytokines such as IL-2 can also be added (60). Additionally, the nano-aAPC platform could be used to generate multiple antitumor T-cells simultaneously using a nano-aAPC cocktail targeting multiple target peptides (25). The nano-aAPC can also be used for direct therapy, since in animal models they have been injected safely intravenously and mediated tumor regression (24).
In summary, peptide-loaded nano-aAPC can rapidly expand polyfunctional tumor antigen-specific CD8+ T-cells that have high avidity, potent cytotoxic activity, long telomere length and a stem cell memory phenotype (Fig. 6G) from melanoma patients. This generates a T cell product with measurable in vivo characteristics critical for potent and durable in vivo anti-tumor activity. These expanded tumor specific T cells could be a powerful tool for adoptive immunotherapy of cancer.
Supplementary Material
Statement of translational relevance :
Adoptive cell therapies employ large numbers of T cells requiring multiple re-stimulations ex vivo to successfully expand cells necessary for effective treatment. Although dendritic cells (DCs) are potent antigen-presenting cells (APC) to expand T cells for adoptive transfer, which have shown some clinical success, expansion with DCs requires a number of weeks or months to produce large numbers of tumor-specific T cells. Herein we have developed nanoparticle artificial APC “off the shelf” to expand large numbers of antigen-specific T cells from melanoma patients within 14 days. The Enrichment + Expansion platform produces high purity and highly functional antigen-specific T cells with high avidity and long telomeres. This technology could be applicable to adoptive cell transfer immunotherapy for cancer.
Acknowledgement:
We acknowledge the Cytometry and Cell Sorting Laboratory at NYU Langone Health for their assistance. This work was supported by grant funding from the National Cancer Institute NCI R01CA175732 (J.S.W.). A.Y.I. is supported by the National Science Foundation Graduate Research Fellowship. This work was funded by support from the National Institutes of Health R33-CA229042 (J.P.S. and J.S.W.), P41-EB028239 (J.P.S.) and a gift from the Wojcicki-Troper foundation (J.P.S.).
Financial Support:
National Cancer Institute: NCI R01CA175732 (J.S.W.)
National Institutes of Health: R33-CA229042 (J.P.S. and J.S.W.), P41-EB028239 (J.P.S.)
Wojcicki-Troper foundation (J.P.S.)
National Science Foundation Graduate Research Fellowship (A.Y.I.)
Footnotes
A conflict of interest disclosures statement:
Junya Ichikawa: Employee of Daiichi Sankyo Co., Ltd.
Tatsuya Yoshida: None.
Ariel Y. Isser: None.
Andressa S. Laino: None.
Melinda Vassallo: None.
David M. Woods: Stock ownership in BMS.
Sojung Kim: Employee of NexImmune Inc.
Mathias Oelke: Employee of NexImmune Inc.
Kristi Jones: Employee of NexImmune Inc.
Jonathan P. Schneck: Under a licensing agreement between NexImmune and The Johns Hopkins University, Jonathan Schneck is entitled to a share of royalty received by the university on sales of products described in this article. He is also a founder of NexImmune and owns equity in the company and he serves as a member of NexImmune’s Scientific Advisory Board. The terms of these arrangements have been reviewed and approved by The Johns Hopkins University in accordance with its conflict of interest policies.
Jeffrey S. Weber: Honoraria and travel from BMS, Merck, GSK, Genentech, Astra Zeneca, Pfizer, CytoMx, EMD Serono, Incyte. Stock in Biond, Altor, Protean, CytoMx, Celldex, Sellas. Research funding from NextCure. All other clinical research funding to my institution, not me. Named on a patent for a PD-1 biomarker by Biodesix not used in this work. Named on a CTLA-4 biomarker patent by Moffitt Cancer Center not used in this work. Named on a patent for the use of 4–1BB antibody for tumor infiltrating lymphocyte growth by Moffitt Cancer Center not used in this work.
References:
- 1.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348(6230):62–8 doi 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 2008;358(25):2698–703 doi 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348(6236):803–8 doi 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res 2011;17(16):5343–52 doi 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen FT, Thisted RA, Rowley DA, Schreiber H. A systematic analysis of experimental immunotherapies on tumors differing in size and duration of growth. Oncoimmunology 2012;1(2):172–8 doi 10.4161/onci.1.2.18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res 2010;16(9):2646–55 doi 10.1158/1078-0432.CCR-10-0041. [DOI] [PubMed] [Google Scholar]
- 7.Wolfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc 2014;9(4):950–66 doi 10.1038/nprot.2014.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol 2017;45:43–51 doi 10.1016/j.coi.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4–1BB. Nat Biotechnol 2002;20(2):143–8 doi 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 10.Mandal S, Hammink R, Tel J, Eksteen-Akeroyd ZH, Rowan AE, Blank K, et al. Polymer-based synthetic dendritic cells for tailoring robust and multifunctional T cell responses. ACS Chem Biol 2015;10(2):485–92 doi 10.1021/cb500455g. [DOI] [PubMed] [Google Scholar]
- 11.Hirano N, Butler MO, Xia Z, Ansen S, von Bergwelt-Baildon MS, Neuberg D, et al. Engagement of CD83 ligand induces prolonged expansion of CD8+ T cells and preferential enrichment for antigen specificity. Blood 2006;107(4):1528–36 doi 10.1182/blood-2005-05-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirano N, Butler MO, Xia Z, Berezovskaya A, Murray AP, Ansen S, et al. Efficient presentation of naturally processed HLA class I peptides by artificial antigen-presenting cells for the generation of effective antitumor responses. Clin Cancer Res 2006;12(10):2967–75 doi 10.1158/1078-0432.CCR-05-2791. [DOI] [PubMed] [Google Scholar]
- 13.Butler MO, Ansen S, Tanaka M, Imataki O, Berezovskaya A, Mooney MM, et al. A panel of human cell-based artificial APC enables the expansion of long-lived antigen-specific CD4+ T cells restricted by prevalent HLA-DR alleles. Int Immunol 2010;22(11):863–73 doi 10.1093/intimm/dxq440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol 2015;33(1):64–72 doi 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung AS, Zhang DKY, Koshy ST, Mooney DJ. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat Biotechnol 2018;36(2):160–9 doi 10.1038/nbt.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiden J, Voerman D, Dolen Y, Das RK, van Duffelen A, Hammink R, et al. Injectable Biomimetic Hydrogels as Tools for Efficient T Cell Expansion and Delivery. Front Immunol 2018;9:2798 doi 10.3389/fimmu.2018.02798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest 2016;126(9):3363–76 doi 10.1172/JCI86721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother 2009;32(2):169–80 doi 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zappasodi R, Di Nicola M, Carlo-Stella C, Mortarini R, Molla A, Vegetti C, et al. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. Haematologica 2008;93(10):1523–34 doi 10.3324/haematol.12521. [DOI] [PubMed] [Google Scholar]
- 20.Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D, et al. A carbon nanotube-polymer composite for T-cell therapy. Nat Nanotechnol 2014;9(8):639–47 doi 10.1038/nnano.2014.154. [DOI] [PubMed] [Google Scholar]
- 21.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013;36(2):133–51 doi 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013;122(6):863–71 doi 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yee C The use of endogenous T cells for adoptive transfer. Immunol Rev 2014;257(1):250–63 doi 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- 24.Perica K, De Leon Medero A, Durai M, Chiu YL, Bieler JG, Sibener L, et al. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomedicine 2014;10(1):119–29 doi 10.1016/j.nano.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perica K, Bieler JG, Schutz C, Varela JC, Douglass J, Skora A, et al. Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano 2015;9(7):6861–71 doi 10.1021/acsnano.5b02829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL Jr., Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol 2016;17(7):943–55 doi 10.1016/S1470-2045(16)30126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, et al. Detection of self-reactive CD8(+) T cells with an anergic phenotype in healthy individuals. Science 2014;346(6216):1536–40 doi 10.1126/science.aaa1292. [DOI] [PubMed] [Google Scholar]
- 28.Lin Y, Gallardo HF, Ku GY, Li H, Manukian G, Rasalan TS, et al. Optimization and validation of a robust human T-cell culture method for monitoring phenotypic and polyfunctional antigen-specific CD4 and CD8 T-cell responses. Cytotherapy 2009;11(7):912–22 doi 10.3109/14653240903136987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med 2013;5(174):174ra27 doi 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapuis AG, Lee SM, Thompson JA, Roberts IM, Margolin KA, Bhatia S, et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J Exp Med 2016;213(7):1133–9 doi 10.1084/jem.20152021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med 2003;9(5):619–24 doi 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- 32.Xu S, Koski GK, Faries M, Bedrosian I, Mick R, Maeurer M, et al. Rapid high efficiency sensitization of CD8+ T cells to tumor antigens by dendritic cells leads to enhanced functional avidity and direct tumor recognition through an IL-12-dependent mechanism. J Immunol 2003;171(5):2251–61 doi 10.4049/jimmunol.171.5.2251. [DOI] [PubMed] [Google Scholar]
- 33.Dauer M, Schad K, Herten J, Junkmann J, Bauer C, Kiefl R, et al. FastDC derived from human monocytes within 48 h effectively prime tumor antigen-specific cytotoxic T cells. J Immunol Methods 2005;302(1–2):145–55 doi 10.1016/j.jim.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, et al. Potent costimulation of human CD8 T cells by anti-4–1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol Immunother 2008;57(2):175–83 doi 10.1007/s00262-007-0360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chauvin JM, Larrieu P, Sarrabayrouse G, Prevost-Blondel A, Lengagne R, Desfrancois J, et al. HLA anchor optimization of the melan-A-HLA-A2 epitope within a long peptide is required for efficient cross-priming of human tumor-reactive T cells. J Immunol 2012;188(5):2102–10 doi 10.4049/jimmunol.1101807. [DOI] [PubMed] [Google Scholar]
- 36.Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol 1999;162(4):2227–34. [PubMed] [Google Scholar]
- 37.Satthaporn S, Robins A, Vassanasiri W, El-Sheemy M, Jibril JA, Clark D, et al. Dendritic cells are dysfunctional in patients with operable breast cancer. Cancer Immunol Immunother 2004;53(6):510–8 doi 10.1007/s00262-003-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gigante M, Blasi A, Loverre A, Mancini V, Battaglia M, Selvaggi FP, et al. Dysfunctional DC subsets in RCC patients: ex vivo correction to yield an effective anti-cancer vaccine. Mol Immunol 2009;46(5):893–901 doi 10.1016/j.molimm.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurwitz AA, Watkins SK. Immune suppression in the tumor microenvironment: a role for dendritic cell-mediated tolerization of T cells. Cancer Immunol Immunother 2012;61(2):289–93 doi 10.1007/s00262-011-1181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y, Shurin GV, Gutkin DW, Shurin MR. Tumor associated regulatory dendritic cells. Semin Cancer Biol 2012;22(4):298–306 doi 10.1016/j.semcancer.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Liu S, Hernandez J, Vence L, Hwu P, Radvanyi L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J Immunol 2010;184(1):452–65 doi 10.4049/jimmunol.0901101. [DOI] [PubMed] [Google Scholar]
- 42.Becker C, Pohla H, Frankenberger B, Schuler T, Assenmacher M, Schendel DJ, et al. Adoptive tumor therapy with T lymphocytes enriched through an IFN-gamma capture assay. Nat Med 2001;7(10):1159–62 doi 10.1038/nm1001-1159. [DOI] [PubMed] [Google Scholar]
- 43.Campbell JD, Foerster A, Lasmanowicz V, Niemoller M, Scheffold A, Fahrendorff M, et al. Rapid detection, enrichment and propagation of specific T cell subsets based on cytokine secretion. Clin Exp Immunol 2011;163(1):1–10 doi 10.1111/j.1365-2249.2010.04261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butler MO, Lee JS, Ansen S, Neuberg D, Hodi FS, Murray AP, et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin Cancer Res 2007;13(6):1857–67 doi 10.1158/1078-0432.CCR-06-1905. [DOI] [PubMed] [Google Scholar]
- 45.Prakken B, Wauben M, Genini D, Samodal R, Barnett J, Mendivil A, et al. Artificial antigen-presenting cells as a tool to exploit the immune ‘synapse’. Nat Med 2000;6(12):1406–10 doi 10.1038/82231. [DOI] [PubMed] [Google Scholar]
- 46.Hsu DH, Paz P, Villaflor G, Rivas A, Mehta-Damani A, Angevin E, et al. Exosomes as a tumor vaccine: enhancing potency through direct loading of antigenic peptides. J Immunother 2003;26(5):440–50 doi 10.1097/00002371-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, et al. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small 2015;11(13):1519–25 doi 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caserta S, Alessi P, Guarnerio J, Basso V, Mondino A. Synthetic CD4+ T cell-targeted antigen-presenting cells elicit protective antitumor responses. Cancer Res 2008;68(8):3010–8 doi 10.1158/0008-5472.CAN-07-5796. [DOI] [PubMed] [Google Scholar]
- 49.Kosmides AK, Meyer RA, Hickey JW, Aje K, Cheung KN, Green JJ, et al. Biomimetic biodegradable artificial antigen presenting cells synergize with PD-1 blockade to treat melanoma. Biomaterials 2017;118:16–26 doi 10.1016/j.biomaterials.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hickey JW, Dong Y, Chung JW, Salathe SF, Pruitt HC, Li X, et al. Engineering an Artificial T-Cell Stimulating Matrix for Immunotherapy. Adv Mater 2019;31(23):e1807359 doi 10.1002/adma.201807359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol 2009;21(2):233–40 doi 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med 2017;23(1):18–27 doi 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372(4):320–30 doi 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 54.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34 doi 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Kurlander RJ. Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: differing impact on CD8 T cell phenotype and responsiveness to restimulation. J Transl Med 2010;8:104 doi 10.1186/1479-5876-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin C, Yu D, Hillerdal V, Wallgren A, Karlsson-Parra A, Essand M. Allogeneic lymphocyte-licensed DCs expand T cells with improved antitumor activity and resistance to oxidative stress and immunosuppressive factors. Mol Ther Methods Clin Dev 2014;1:14001 doi 10.1038/mtm.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ndhlovu ZM, Oelke M, Schneck JP, Griffin DE. Dynamic regulation of functionally distinct virus-specific T cells. Proc Natl Acad Sci U S A 2010;107(8):3669–74 doi 10.1073/pnas.0915168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang B, Baybutt TR, Berman-Booty L, Magee MS, Waldman SA, Alexeev VY, et al. Prime-Boost Immunization Eliminates Metastatic Colorectal Cancer by Producing High-Avidity Effector CD8(+) T Cells. J Immunol 2017;198(9):3507–14 doi 10.4049/jimmunol.1502672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 2016;22(4):433–8 doi 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem 2011;286(40):34883–92 doi 10.1074/jbc.M111.276329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.