

Abstract
Rationale:
Peripheral artery disease, common in metabolic syndrome and diabetes, responds poorly to medical interventions, and is characterized by chronic vessel immaturity leading to lower extremity amputations.
Objective:
To define the role of reversible palmitoylation at the endothelium in the maintenance of vascular maturity.
Methods and Results:
Endothelial knockout of the depalmitoylation enzyme acyl-protein thioesterase 1 (APT1) in mice impaired recovery from chronic hindlimb ischemia, a model of peripheral artery disease. Endothelial APT1 deficiency decreased fibronectin processing, disrupted adherens junctions, and inhibited in vitro lumen formation. In an unbiased palmitoylation proteomic screen of endothelial cells from genetically modified mice, R-Ras, known to promote vessel maturation, was preferentially affected by APT1 deficiency. R-Ras was validated as an APT1 substrate, and click chemistry analyses demonstrated increased R-Ras palmitoylation in cells with APT1 deficiency. APT1 enzyme activity was decreased in endothelial cells from db/db mice. Hyperglycemia decreased APT1 activity in HUVECs, due in part to altered acetylation of the APT1 protein. Click chemistry analyses demonstrated increased R-Ras palmitoylation in the setting of hyperglycemia. Altered R-Ras trafficking, increased R-Ras palmitoylation, and fibronectin retention were found in diabetes models. Loss of R-Ras depalmitoylation caused by APT1 deficiency constrained R-Ras membrane trafficking as shown by TIRF imaging. To rescue cellular phenotypes, we generated an R-Ras molecule with an inserted hydrophilic domain to circumvent membrane rigidity caused by defective palmitoylation turnover. This modification corrected R-Ras membrane trafficking, restored fibronectin processing, increased adherens junctions, and rescued defective lumen formation induced by APT1 deficiency.
Conclusions:
These results suggest that endothelial depalmitoylation is regulated by the metabolic milieu and controls plasma membrane partitioning to maintain vascular homeostasis.
Keywords: Peripheral artery disease, palmitoylation, diabetes
Subject Terms: Angiogenesis, Cell Biology/Structural Biology, Metabolism, Peripheral Vascular Disease
GRAPHICAL ABSTRACT
As many as 10% of proteins are palmitoylated, a modification involving the addition of the fatty acid palmitate to cysteine residues to promote interaction with cell membranes. More than 20 acyltransferases catalyze this reaction, and several are expressed in endothelial cells, but a small number of enzymes remove palmitate to allow for palmitoylation cycling (repeated palmitoylation followed by depalmitoylation). Here we show that the depalmitoylation enzyme APT1 is required for vascular remodeling in chronic ischemia and the activity of this enzyme is decreased in diabetes. Hyperglycemia decreased acetylation of APT1 and treatment with a class I and II HDAC inhibitor, but not a class III (sirtuin) inhibitor, restored the acetylation of APT1. R-Ras, important for vessel maturation, was identified in a palmitoylation proteomic screen of endothelial cells. In APT1 deficient endothelial cells, impaired R-Ras membrane trafficking was rescued by modifying the palmitoylated R-Ras molecule to promote dissociation from membranes. These observations identifypalmitoylation cycling as a potential therapeutic target in the treatment of vascular disease.

INTRODUCTION
Peripheral artery disease, atherosclerotic occlusive disease of the lower extremities, affects more than 200 million individuals globally and its prevalence is increasing with the aging of the population.1 The disease is common in people with the metabolic syndrome, insulin resistance, and diabetes,2–4 and it predicts major cardiovascular events, stroke, and death.5 Patients with peripheral artery disease treated with statins are less likely to die from coronary events, but medical therapy does not prevent limb loss.6–8 For people with diabetes, myocardial infarctions have declined but amputations have not.9, 10
Impaired tissue perfusion in occlusive arterial disease is associated with disturbed homeostasis in endothelial cells. In the setting of decreased perfusion and diabetes or insulin resistance, vascular remodeling is distorted,11 characterized by decreased assembly of adherens junctions and other structural defects. Persistent exposure to angiogenic signals activates endothelial cells and triggers vascular instability. Excessive endothelial activation leads to deposition of fibronectin, prompting vascular inflammation, oxidative stress, and pathological remodeling that produces immature, unstable blood vessels.11, 12 Current therapies do not correct abnormal vascular remodeling.
During development, fibronectin is required for embryogenesis and vasculogenesis,13 but in adults, fibronectin contributes to provisional matrix utilized for tissue repair.14 Derived from both local synthesis and uptake from the circulation, fibronectin engages integrin receptors to assemble extracellular fibrillar networks and organize the intracellular cytoskeleton.15, 16 Disruption of this process leads to retention of insoluble fibronectin that impairs blood vessel maturation.17 Abnormal integrin signaling hampers fibronectin remodeling,18 and the translocation and ectopic activation of β1 integrin triggers contractile forces that destabilize adherens junctions.19
Since current medical therapy is suboptimal for preventing limb loss, unconventional biomarkers like fibronectin and fatty acids might suggest novel approaches. Circulating fibronectin is increased in humans with peripheral artery disease and those with metabolic disorders like diabetes.20, 21 Dietary saturated fatty acids like palmitate are associated with peripheral artery disease and type 2 diabetes.22–24 How the lipid milieu alters fibronectin metabolism to destabilize blood vessels is unknown, but lipid modification of proteins has been implicated in infections, premature aging, cancer, and diabetes.24, 25
One such modification is palmitoylation (S-acylation), the formation of a thioester bond between palmitate and cysteine.26 Protein palmitoylation promotes association with hydrophobic domains such as those found in membranes, and it is reversible, allowing adaptive membrane affinity of proteins through de/repalmitoylation cycling. Cytosolic and membrane proteins are thought to be palmitoylated by a large family of DHHC acyltransferases, many expressed in endothelial cells.27 The family of depalmitoylation enzymes is small and includes acyl-protein thioesterases 1 and 2 (APT1 and APT2), two distinct proteins that are conserved in all vertebrates.28 APT1 is thought to be important for active depalmitoylation,29 but its physiology in vascular disease is unknown. Using mouse models, cell systems, and arterial samples from humans with end-stage peripheral artery disease, we tested the hypothesis that inadequate palmitoylation cycling promotes endothelial instability, a hallmark of chronic arterial occlusive disease.
METHODS
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Mouse protocols.
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University. An APT1 floxed mouse model was generated at Washington University. Mice were fed standard chow in a specific pathogen-free facility with a 12-h light–dark cycle.
APT1 activity.
HUVECs were trypsinized and incubated with DPP330 (1 μM) in PBS (1% FBS) for 20 min at room temperature. The emission fluorescence of the cell suspension was acquired by digital FACScan (Cytek Development) using a blue laser (488 nm) and FL1 filter (530/30). Both data collection and post-acquisition analysis were performed in Flowjo. Background signals, obtained from samples without staining, were subtracted from the data. For endothelial cells from db/db mice, lungs were digested using collagenase and single cell suspensions were stained for CD45 (PerCp/Cy5.5, BioLegend) and CD31(APC, Thermo). After washing, APT1 was assayed using DPP3 (5 μM).
Statistical analyses.
Analyses were performed using GraphPad software (La Jolla, CA). Data are represented by scatter dot plot (with mean ± SEM) or box/whisker plot, and p<0.05 was considered significant.
Additional Methods are described in the Online Supplement.
RESULTS
Endothelial APT1 deficiency impairs vascular remodeling after hindlimb ischemia.
We generated a mouse with floxed Lypla1 (encoding the APT1 protein, Online Fig. IA–C). APT1 floxed mice (homozygous at the floxed allele) were bred with Tie2-Cre mice to yield endothelial cell-specific APT1 knockout mice. Lypla1 transcript (Online Fig. ID) and APT1 protein (Fig. 1A) were decreased in primary cultures of aortic endothelial cells from Tie2-APT1 KO animals, which grew normally as compared to floxed controls (without Cre) to six months of age.
Figure 1. Endothelial ablation of APT1 disrupts vascular remodeling.

(A) Western blots for APT1 in mouse aortic endothelial cells. (B) Perfusion imaging following hindlimb ischemia, and analysis in (C) (n=10 mice for both control and Tie2-APT1 KO). (D) X-ray angiographs, with collateral growth highlighted on the right. (E) Vessel area (n=6 for control and n=5 for Tie2-APT1 KO). Non-ligated (F) and ligated (G) images (left) for SMA (red), CD11b (green), and DAPI (blue), and quantitation of lumen perimeter (right). (H) Quantitation of CD31 normalized to myocytes and NG2 positive microvessels in gastrocnemius muscle (n=6 for control and n=7 for Tie2-APT1 KO). Representative images are shown in Figure S1E. (I) Western blots for APT1 in mouse aortic endothelial cells with or without 4-hydroxytamoxifen treatment (1 μM for 48 h). (J) Perfusion imaging following hindlimb ischemia, and analysis in (K) (n=3 for control and n=6 for VEC-APT1 KO). (L) X-ray angiographs, with collateral growth highlighted on the right. Quantitation of vessel area (M) is shown (n=5 for control and n=7 for VEC-APT1 KO). Non-ligated (N) and ligated (O) images (left), and quantitation of lumen perimeter (right). (P) Quantitation of CD31 normalized to myocytes and NG2 positive microvessels in gastrocnemius muscle (n=5 for control and n=9 for VEC-APT1 KO). Representative images are shown in Figure S1F. Statistical analyses performed by 2 way ANOVA (C, E, K, M), or by unpaired t test with Welch’s correction or Mann-Whitney test (F, G, H, N, O, P). Scale=50 microns.
When these mice were subjected to femoral artery ligation, a model of peripheral artery disease, the restoration of limb perfusion assessed by Doppler imaging was decreased in Tie2-APT1 KO mice (Fig. 1B,C). Ischemia-induced perfusion recovery involves remodeling of arteries (arteriogenesis) and capillaries (angiogenesis).31 Using X-ray angiography (Fig. 1D), vessel area (Fig. 1E) was reduced in Tie2-APT1 KO mice compared to controls. Quantitative histology of vessels in the adductor muscle group on the non-ligated side showed no genotype difference in lumen circumference (Fig. 1F, 110 arteries from 3 controls, 77 arteries from 3 knockouts). Measurements of the ligated side confirmed decreased arteriogenesis as lumen size of arteries in knockout animals was smaller with endothelial APT1 deficiency (Fig. 1G, 49 arteries from 5 controls, 81 arteries from 6 knockouts). For angiogenesis, examination of microvessels (Fig. 1H for quantitation and Online Fig. IE for representative images) showed decreased pericyte (NG2 positive cells) coverage of microvascular networks in Tie2-APT1 KO mice with ischemia, a manifestation of vascular immaturity. To confirm these findings, the floxed mice were crossed with animals carrying an inducible Cre, VE-cadherin-CreERT2.32 Following postnatal activation of Cre with 4-OH-tamoxifen, APT1 protein was decreased in cultured endothelial cells derived from VEC-APT1 KO (VE-cadherin-CreERT2: APT1 f/f) mice but not floxed controls without Cre (Fig. 1I). Tamoxifen-treated VEC-APT1 KO mice showed decreased hindlimb perfusion recovery after femoral artery ligation (Fig. 1J,K). Deficient recovery of perfusion was associated with reduced arterial expansion assessed by angiography (Fig. 1L,M). On the non-ligated side, there was no genotype difference in lumen circumference (Fig. 1N, 55 arteries from 3 controls, 68 arteries from 3 knockouts). Measurements on the ligated side showed decreased arteriogenesis with APT1 deficiency (Fig. 1O, 29 arteries for 3 controls, 32 arteries for 3 knockouts). Ischemia was also associated with decreased pericyte coverage of microvessels as well as decreased numbers of both capillaries and pericytes (Fig. 1P for quantitation and Online Fig. IF for representative images). These findings confirm those in Tie2-APT1 KO mice and indicate that endothelial APT1 is required for vascular remodeling and vessel maturity in chronic ischemia.
APT1 coordinates fibronectin processing by endothelial cells.
Stable endothelial cells interact with matrix enriched in collagen IV and laminin and depleted of fibronectin. Following injury, vessel repair requires efficient processing of provisional matrix proteins like fibronectin. Fibronectin exists in a soluble form consisting of dimeric proteins that self-associate to form fibrils, and an insoluble form produced by ongoing fibronectin association to yield large branched fibril complexes. Tie2-APT1 KO aortas compared to control aortas had increased soluble and insoluble fibronectin (Fig. 2A), which may promote vascular immaturity. Two weeks after femoral artery ligation, fibronectin was increased in the intima but not the media of lower extremity arteries of Tie2-APT1 KO mice as compared to controls (Fig. 2B), and increased in the intima but not the media of lower extremity arteries of VEC-APT1 KO mice (Online Fig. IIA). The finding of increased vascular fibronectin content prompted us to study fibronectin metabolism in the setting of endothelial APT1 deficiency.
Figure 2. Loss of APT1 impairs endothelial cell remodeling of fibronectin.

(A) Representative Western blots of DOC (deoxycholate)-soluble and DOC-insoluble extractions of mouse aorta (left two panels), as well as the quantitation of normalized fibronectin contents in the DOC-soluble and –insoluble fractions (right two panels) (n=8 mice for control and n=10 mice for Tie2APT1KO). (B) Representative cross-sections (left) of thigh muscle arteries immunostained for FN (green), CD31 (red), and SMA (blue), as well as the quantitation (right) of FN contents in intima and media (n=6 mice for control and n=8 mice for Tie2-APT1 KO, two weeks after surgeries). Scale=20 microns. I: intima, M: media, arrows indicate intimal fibronectin deposition. (C) Representative Western blots of exogenous biotin-labeled fibronectin [FN(biotin)] from DOC extractions of cells during pulse-chase. (D) Quantitation of conversion ratios of FN(biotin) (insoluble/soluble fractions) in pulsed cells (time=0, n=4 for control and Tie2-APT1 KO cells). (E) Retained fibronectin(biotin) contents in DOC–insoluble fractions after 2 h chase (n=3 for control and Tie2APT1KO cells). (F) Representative stitched confocal images of arteries from human peripheral artery disease patients stained for fibronectin (green), SMA (yellow), and CD31 (red). Scale=1 mm. IEL: internal elastic lamina with blue autofluorescence. (G) Pearson correlation analysis of intimal fibronectin deposition (ratios of intimal to medial FN stained fluorescence signal) with intima-media thickness, n=11 samples from 8 patients. Data are presented as mean ± SEM by scatter dot plot for A,B,D,E with statistical analyses performed by unpaired t test with Welch’s correction or Mann-Whitney test.
Assembly of fibronectin matrix was decreased in APT1-deficient cells. In aortic endothelial APT1 KO cells (Online Fig. IIB) and APT1-KD (knockdown) HUVECs (Online Fig. IIC), the ratio of insoluble/soluble fibronectin was decreased compared to controls, suggesting impaired self-association required for mature matrix. Decreased maturation of fibronectin matrix was particularly prominent in APT1-KD cells (Online Fig. IID), where fibrils were not only sparse but also more isotropic as compared to control cells (SC). Turnover of exogenous fibronectin was also decreased in APT1-deficient cells. In pulse-chase studies using biotin-labeled fibronectin, the 0 time point of the chase showed increased soluble fibronectin but decreased insoluble fibronectin in APT1 KO compared to control cells (Fig. 2C, Fig. 2D). During the chase, labeled insoluble fibronectin was mostly cleared in control cells, but retained in APT1 KO cells (Fig. 2C, Fig. 2E). Similar results were seen in APT1-KD HUVECs (Online Fig. IIE,F).
Since intimal retention of fibronectin characterizes our model systems of APT1 deficiency, we quantified fibronectin immunostaining in cross sections of human arterial samples prepared from minimally diseased tissue isolated at the time of lower extremity amputations in 8 patients (age 58 ± 4, BMI 31 ± 2, 6 of 8 had type 2 diabetes, 2 of 8 were females, also see Online Table I). The ratio of intima fibronectin content to media fibronectin content was positively associated with intima-media thickness (Pearson correlation r2=0.455, p=0.023, 11 samples in 8 patients, Fig. 2F,G), a measure of the extent of luminal occlusion in arterial disease.
Fibronectin matrix assembly and turnover requires the binding of fibronectin to integrins, so APT1 deficiency would be expected to uncouple the integrin-fibronectin interaction. Consistent with this prediction, co-localization of β1 integrin-enriched fibrillar adhesions (using 9EG7 antibody in Fig. 3A–D and confirmed using MB1.2 antibody in Fig. 3E–H) and endogenous fibronectin was disrupted in APT1 KO cells. In APT1 deficiency, some regions of fibronectin staining were punctate and remote from integrin staining.
Figure 3. Uncoupling of integrin-fibronectin interaction in APT1 deficiency.
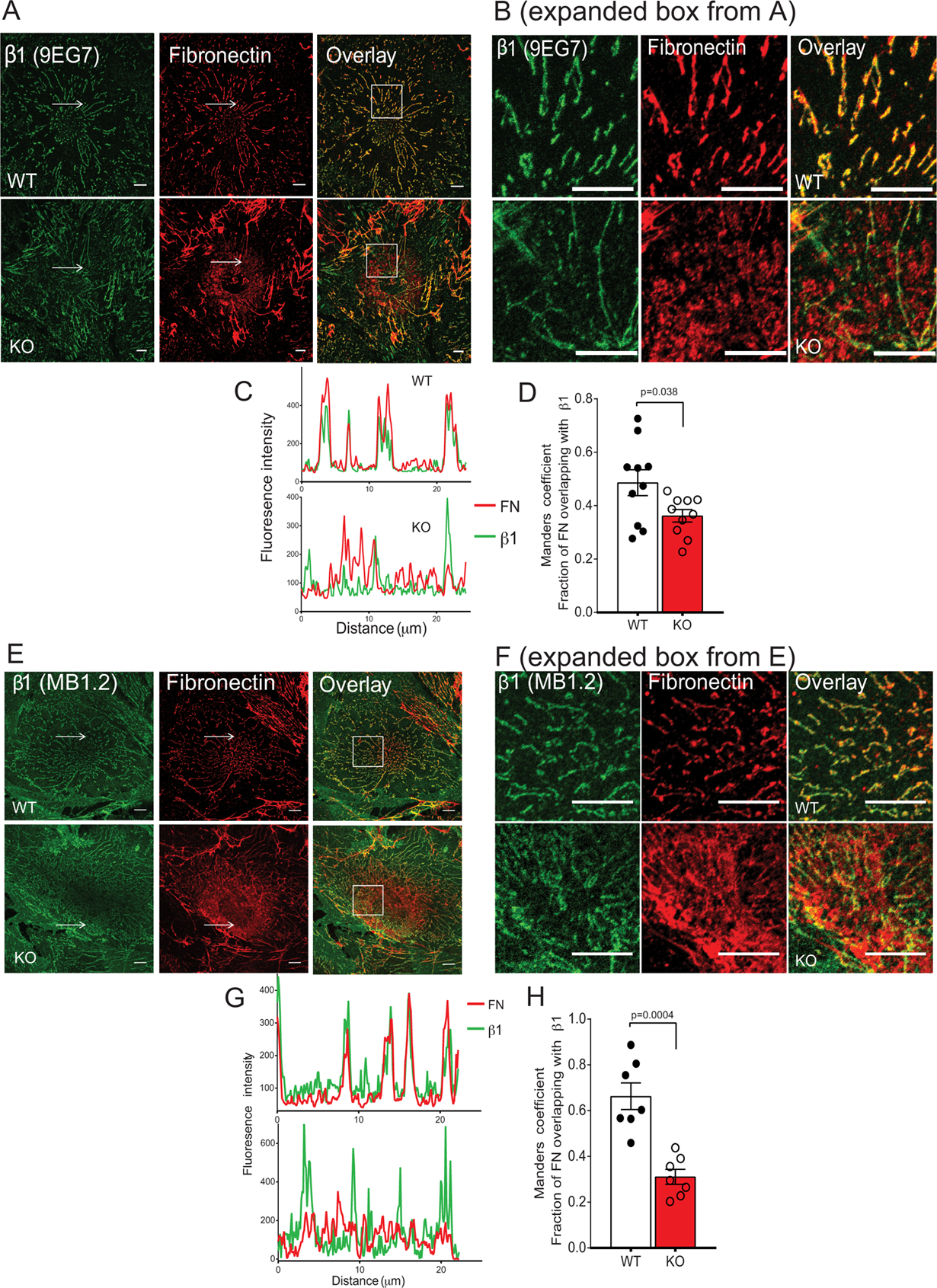
(A) Representative confocal images of cells traced by 9EG7 antibody (green) followed by immunostaining for endogenous fibronectin (red). Scale=10 microns. Boxes are expanded in (B). Arrows indicate areas of intensity profiles shown in (C), with colocalization between two channels (fractions of fibronectin colocalized with β1 integrin) expressed by Manders coefficients (D) (n=10 for both control and Tie2APT1KO cells). (E) Representative confocal images of cells traced by MB1.2 antibody (green) followed by immunostaining for endogenous fibronectin (red). Scale=10 microns. Boxes are expanded in (F). Arrows indicates areas of intensity profiles shown in (G), with colocalization between two channels (fractions of fibronectin colocalized with β1 integrin) expressed by Manders coefficients (H) (n=7 for control and Tie2APT1KO cells). Data are presented as ± SEM by scatter dot plot. Statistical analyses performed by unpaired t test with Welch’s correction (D and H).
Altered lumen formation and cell adhesion with APT1 deficiency.
In two-dimensional vitro angiogenesis assays using Matrigel (solubilized basement membrane matrix), the tubular network formed by APT1-deficient endothelial cells was not decreased (Online Fig. IIIA). In contrast, APT1-deficient cells coated on Cytodex microcarrier beads and embedded in a three-dimensional fibrin gel formed disorganized sprouts. Unlike control cells, APT1 KO endothelial cells gave rise to tree-like structures with broken branches and isolated cells (Fig. 4A). The same pattern was seen with APT1 KD HUVECs (Online Fig. IIIB). Moreover, tube diameter induced by fibroblast co-culture was decreased with APT1 deficiency compared to controls (representative image and quantitation in Fig. 4B), an observation confirmed by confocal imaging (Fig. 4C). Abnormal sprouting in a three-dimensional matrix suggests that APT1 deficiency impairs endothelial cell adaptations inherent to vessel remodeling mediated by cell-matrix and cell-cell interactions.33
Figure 4. APT1 deficiency affects cell adhesions.

(A) Sprouting images of primary cultured mouse endothelial cells coated on microbeads embedded in fibrin gel. Arrows indicate migrated cells disconnected from major branches. (B) Images of lumenized sprouts (red arrows) from HUVEC cells coated on fibrin-embedded microbeads with quantitation of tube diameters (n=77 for SC control and n=68 for KD sprouts). (C) Confocal images of sprouts from HUVEC cells stained for fibronectin (red) and nuclei (blue), including longitudinal projections (x-y view) and transverse sections (y-z view, through the white lines crossing the lumens). (D) F-actin staining of primary cultured mouse endothelial cells with quantitation of stress fiber-containing cells (n=3 independent experiments). (E) Images of spreading cells stained for 9EG7 (green), F-actin (red), and paxillin (blue) with (F) quantitation of spreading areas and size distributions (n=137 for control and n=146 for APT1 KO). (G) VE-cadherin staining of primary cultured mouse endothelial cells (green for VEC and blue for DAPI) with quantitation of junctional signals (n=9 random regions for both control and KO). (H) Confocal images of cell-cell contacts in confluent HUVEC cells immunostained for VE-cadherin (green), F-actin (red), and fibronectin (FN, grey). SC and KD: scrambled or APT1 knockdown. Scale=50 microns for A-B, D, G; scale=20 microns for C,E; scale=10 microns for H. Data are presented as mean ± SEM by scatter dot plot (D,G), or box/whisker plot (B and F). Statistical analyses performed by unpaired t test with Welch’s correction or Mann-Whitney test (B,D,F-G).
Endothelial APT1 deficiency is associated with increased retention of fibronectin (Fig. 2), which enhances integrin signaling, increases cell spreading, and reorganizes cytoskeletal stress fibers.34 F-actin stress fibers were increased in APT1-deficient endothelial cells from mice (Fig. 4D). These cells also had increased cell-matrix adhesions reflected by more integrin β1-enriched fibrils (clustered on the ventral surface of the cell body) that co-localized with F-actin and paxillin-containing adhesion complexes (Fig. 4E). Cell spreading was increased in APT1 KO compared to control cells (Fig. 4F). Similar findings were made using APT1 KD HUVECs (Online Fig. IIIC–G).
Cell-cell interactions required for angiogenesis are mediated in part by adherens junctions, characterized by the protein VE-cadherin. Adherens junctions were reduced in confluent APT1 KO endothelial cells (Fig. 4G) as well as APT1 KD HUVECs (Online Fig. III H) compared to controls. VE-cadherin appeared to accumulate intracellularly in APT1-deficient ECs (Online Fig. III I). Total VE-cadherin protein content was not decreased in APT1-deficient HUVECs or aortic endothelial cells (Online Fig. III J,K), suggesting that defective palmitoylation is associated with appropriate production of VE-cadherin but impaired assembly of adherens junctions. One reading of these findings is that loss of palmitoylation cycling increases fibronectin, leading to endothelial cell activation and increased intracellular tension that disrupts adherens junctions. Consistent with this interpretation, imaging showed increased F-actin distribution centrally, decreased F-actin distribution at the cell cortex, and decreased VE-cadherin with APT1 deficiency (Fig. 4H).
APT1 depalmitoylates R-Ras protein.
We used proteomics to characterize APT1 substrates (Online Fig. IVA). Palmitoylated protein-enriched samples from primary cultured mouse endothelial cells were individually labeled with tandem mass tags for multiplexed quantitation. Three matched groups of control and knockout cell preps were analyzed and normalized fold changes with p values were generated for individual proteins (Online Fig. VI B–C). Among >3,200 identified proteins, 74 palmitoylated proteins were significantly increased or decreased between genotypes (moderated p values ≤0.05 and ≥2 peptides, Online Table II). Fifty-two were candidate APT1 substrates (increased palmitoylation in knockouts without differences in protein abundance for that candidate), including several known APT1 substrates (Online Fig. IVD). Increased palmitoylation was confirmed for H-Ras and Flot1 (Online Fig. IVE). Potential APT1 substrates were analyzed by functional annotation clustering (DAVID, Online Table III). Consistent with our observation that APT1 deficiency altered cell-matrix interactions, the highest-scoring significant pathway (Fig. 5A) was focal adhesion (Gene Ontology Term: 0005925, p=0.014 by Benjamini). Of the seven proteins in this cluster (Fig. 5B), R-Ras has been implicated in vessel maturation, integrin trafficking, fibronectin biology, and adherens junction integrity.35–39
Figure 5. Palmitoylation proteomic identification of R-Ras as an endothelial substrate of APT1.

(A) Functional annotation analysis of candidate proteins. A cluster of focal adhesion is highlighted in red, which consists of 7 proteins as shown in (B). (C) Western blot analysis of palmitoylated R-Ras and Caveolin-1 by acyl-RAC assays in HUVEC cells, primary cultured mouse endothelial cells from aorta (D), APT1 KO mouse muscle (E), and APT2 KO mouse muscle (F). (G) Click chemistry analysis of palmitoylated R-Ras (fluorescent azide in green, R-Ras protein in red) in cells pulsed with palmitic acid alkyne, or (H) pulse-chased for indicated times. +APT1: APT1 overexpression. (I) GFP-R-Ras was expressed in control or APT1 Kd HUVECs, palmitic acid alkyne was pulsed, then GRP-R-Ras was immunoprecipitated, labeled with biotin-azide, and detected by blotting. (J) Quantitation of click chemistry analysis of palmitoylated R-Ras normalized to GFP-R-Ras from (I). Results in I,J were replicated in at least 3 experiments.
R-Ras palmitoylation was increased in APT1-deficient HUVECs, APT1-deficient mouse endothelial cells, and in skeletal muscle (an endothelial-rich tissue) from mice with APT1 deficiency (Fig. 5C–E). R-Ras palmitoylation was not increased in skeletal muscle tissues of APT2 knockout mice (Fig. 5F), suggesting specificity of the effect of APT1 on R-Ras. R-Ras can affect Akt signaling,39 but Akt phosphorylation was not altered in APT-deficient endothelial cells (Online Fig. VA).
Transfection of tagged APT1 (levels of expressed and endogenous APT1 are shown in Online Fig. VB) decreased the association of a palmitic acid analog with R-Ras in a click chemistry-based palmitoylation assay (Fig. 5G) and promoted its removal from R-Ras in chase experiments (Fig. 5H). GFP-R-Ras expression in control and APT1-deficient HUVECs followed by palmitic acid alkyne labeling and time dependent detection of R-Ras showed increased palmitoylated R-Ras with APT1 deficiency (Fig. 5I,J), consistent with a role for APT1 in R-Ras palmitoylation dynamics in endothelial cells.
Increased R-Ras palmitoylation and decreased APT1 enzyme activity in diabetes models.
Peripheral artery disease is associated with diabetes and metabolic syndrome. APT1 mRNA and protein were not reduced in cells cultured in high glucose or aortic extracts from db/db mice, a model of type 2 diabetes characterized by striking hyperglycemia (Online Fig. VIA,B). However, when GFP-R-Ras was expressed in HUVECs cultured under normal glucose and high glucose conditions, click chemistry palmitic acid alkyne labeling followed by time dependent detection of R-Ras demonstrated increased palmitoylated R-Ras in the high glucose condition (Fig. 6A,B), suggesting decreased APT1 enzyme activity with hyperglycemia. APT1 enzyme activity (determined using DPP3, a molecule with a thioester bond that fluoresces after cleavage by depalmitoylases30) was decreased in endothelial cells isolated from the lungs of control and db/db mice (Fig. 6C). Consistent with decreased ATP1 enzyme activity, R-Ras palmitoylation was increased in muscle tissue from db/db mice (Fig. 6D), and there was increased retention of insoluble fibronectin in aortic tissue from db/db mice (Online Fig. VIC). R-Ras palmitoylation was also increased in HUVECs cultured in high glucose (Fig. 6E). APT1 enzyme activity determined using the fluorescent substrate DPP3 was decreased in HUVECs cultured in high glucose (Fig. 6F).
Figure 6. R-Ras palmitoylation is increased and APT1 enzyme activity is decreased in diabetes models.

(A) GFP-R-Ras was expressed in HUVECs cultured in 5 mM or 25 mM glucose, palmitic acid alkyne was pulsed, then GFP-R-Ras was immunoprecipitated, labeled with biotin-azide, and detected by blotting. (B) Quantitation of click chemistry analysis of palmitoylated R-Ras normalized to GFP-R-Ras from (A). Results in A,B were replicated in at least 3 experiments. (C) Mouse lungs were subjected to collagenase digestion. APT1 activity of gated endothelial cells (red boxes in left panel) stained by DPP3 was detected (middle panel), and quantified (right panel, unpaired t test). (D) Acyl-RAC palmitoylation assays of muscle tissue from db/m (control) and db/db mice. (E) Acyl-RAC palmitoylation assays in HUVEC cells treated with normal (5 mM) or high glucose (25 mM) media. (F) Flow cytometry detection of APT1 activity in HUVECs treated with normal (5 mM) or high glucose (25 mM) media or high glucose media in the presence of 5 μM TSA (representative signals left panel, quantitation right panel with comparisons by ANOVA). Data presented in C and F as mean ± SEM by scatter dot plot.
APT1 is known to be subject to acetylation,40 a common modification linked to altered metabolism,41 and pull down assays showed decreased APT1 acetylation in HUVECs cultured in high glucose (Online Fig. VID). Histone deacetylase (HDAC, also known as lysine deacetylase because targets are not limited to histones) inhibition may improve experimental diabetes complications. Treatment with the class I and II HDAC inhibitor trichostatin A (TSA), but not the class III (sirtuin) inhibitor nicotinamide, increased acetylation of tagged APT1 (Online Fig. VIE), and TSA treatment caused a time-dependent increase in APT1 enzyme activity in HUVECs (Online Fig. VIF). In HUVECs, TSA partially reversed the inhibition of APT1 enzyme activity by high glucose (Fig. 6F).
To further implicate acetylation as a potential molecular mechanism in the alteration of ATP1 activity by hyperglycemia, we examined specific APT1 residues. K224 of APT1 is exposed on the C-terminal α-helix.28 K224R and K224A mutations but not a K224Q mutation (mimicking acetylated lysine) decreased the activation potential of APT1 in an activity-based in vitro pull-down assay (Online Fig. VIIA). Moreover, purified recombinant K224Q mutant APT1 protein had increased enzyme activity compared to the recombinant wild type enzyme (Online Fig. VIIB–E). Increased acetylation of wildtype APT1 protein with TSA treatment was blunted in APT1 protein bearing the K224R mutation (Online Fig. VIIF), consistent with a role for lysine residue 224 of APT1 in the posttranslational control of APT1 activity.
Hyperglycemia also decreased VE-cadherin junctional signals in HUVECs (Online Fig. VIIIA), and impaired exogenous fibronectin turnover as reflected by a decreased ratio of insoluble/soluble label at time 0 and increased insoluble retention after a 2 h chase (Online Fig. VIIIB–D), mirroring fibronectin processing defects found in endothelial cells with genetic inactivation of APT1 (Online Fig. IIB,C,E). Hyperglycemia thus produces functional APT1 deficiency that mimics genetic APT1 deficiency in terms of increased R-Ras palmitoylation, altered cell contacts, and fibronectin metabolism.
APT1 and membrane trafficking.
Since palmitoylation promotes association with hydrophobic domains, we examined membrane trafficking in the presence and absence of APT1. Crude membrane fractionation revealed no APT1-dependent effects on the global membrane localization of R-Ras (Fig. 7A). However, cell fractionation by density gradient centrifugation in the absence of detergents showed that APT1 deficiency caused R-Ras to preferentially localize in bottom fractions (Fig. 6B, with the shift to non-raft fractions indicated by a red box). This shift to non-raft fractions was not seen with Flot1, suggesting that APT1 deficiency has relatively specific effects on R-Ras.
Figure 7. Depalmitoylation maintains R-Ras membrane trafficking of R-Ras.

(A) Western blots of R-Ras and APT1 proteins in crude membrane fractions and cytoplasmic fractions of HUVEC cells. (B) Western blots of R-Ras and Flot-1 proteins in fractions from sucrose gradient ultracentrifugation of HUVECs following cell lysis by high pH sodium bicarbonate. Light fractions (rafts) are denoted by the top 1–6 fractions. APT1 deficiency-induced redistribution of signal into non-raft fractions is indicated by a red box. (C) Representative still TIRF images of GFP-R-Ras in live primary endothelial cells, as well as (D-E) quantitation of turnover rates (slopes of intensity changes of adhesion-like structures). Disassembly rates are calculated as pooled adhesions (n=331 for control and 564 for KO) from primary endothelial cells. Assembly rates are calculated as pooled adhesions (n=110 for control and 213 for KO) from primary endothelial cells. (F) Representative still TIRF images of GFP-R-Ras with the overlay of particle tracking results as maximal speed, as well as quantitation of global membrane dynamics in primary cells as mean speed (G), mean displacement (H), and mean duration (I) of tracks (n=28 for control or APT1 KO cells). Sc and Kd: scrambled or APT1 knockdown. Scale=10 microns for C, and scale=1 micron for F. Data are presented as box/whisker plots. Statistical analyses performed by unpaired t test with Welch’s correction or Mann-Whitney test (D-E and G-I). Please see Online Video 1.
In HUVECs, high glucose (which decreases APT1 enzyme activity) also caused R-Ras but not Flot1 to shift to non-raft heavy fractions (Online Fig. IXA, with the R-Ras shift denoted by a red box). To begin to address the nature of this heavy fraction compartment, fractionation was performed in control HUVECs after palmitic acid alkyne labeling. After a one hour pulse of the palmitic acid alkyne in wild type cells, palmitoylated R-Ras was preferentially detected in the heavy fraction compartment (Online Fig. IXB). As compared to control HUVECs, APT1 deficient HUVECs showed increased palmitic acid alkyne labeling of heavy fractions at 30 min.
We imaged R-Ras tagged with N-terminal monomeric GFP (mEGFP-R-Ras). Like native R-Ras, GFP-tagged R-Ras shifted to include heavy (non-raft) fractions in APT1-knockdown cells (Online Fig. XA, with the shift to non-raft fractions indicated by a red box). Cells coated on gelatin showed little R-Ras at the plasma membrane, but cells cultured on fibronectin (Online Fig. XB) contained tagged R-Ras at cell junctions. With APT1 deficiency, R-Ras was localized away from cell junctions (and structures such as lipid rafts) toward the cell center/perinuclear region (Online Fig. XC), consistent with cell fractionation data showing a shift of R-Ras toward a non-raft compartment. Using confocal microscopy, R-Ras was identified at the cell surface in control cells (SC) and at intracellular vesicles in APT1 KD cells (KD, Online Fig. XD). Using TIRF imaging of live primary endothelial cells, R-Ras formed matrix-adhesion-like structures close to the cell surface in control cells, and these were expanded in APT1 KO cells (Fig. 7C), suggesting that the loss of depalmitoylation disrupted membrane trafficking involving focal adhesions. Time lapse TIRF imaging of mEGFP-R-Ras revealed reduced turnover of R-Ras-positive structures (less assembly and less disassembly) in knockout as compared to control cells (Fig. 7D–E). Particle tracking analyses (Fig. 7F) showed that R-Ras proteins had decreased speed and displacement distance in KO as compared to control cells (Fig. 7G–H), and decreased turnover (as reflected by increased track duration, Fig. 7I and Online Video I) in KO as compared to control cells.
An R-Ras mutant rescues APT1 deficiency in vitro.
To provide proof of principle that the adverse effects of APT1 deficiency are mediated by delayed R-Ras trafficking, we generated mutations at the R-Ras C-terminus (CPCVLL, including the palmitoylation cysteine at residue 213 and prenylation cysteine at residue 215, Fig. 8A) with a goal of decreasing C-terminal hydrophobicity. The non-polar proline residue between the two lipidated cysteines was converted to a positively charged Histidine to generate an H mutant with the modestly altered terminus CHCVLL, and the short hydrophilic sequence RSGRSG was Inserted to generate an I mutant with the more hydrophilic terminus CRSGRSGPCVLL (Fig. 8A). Wild type, H mutant and I mutant R-Ras were expressed under conditions allowing similar confocal detection of each tagged protein at the cell surface in HUVECs (Online Fig. XIA). Live cell imaging by TIRF microscopy in APT1 KD cells showed increased migration speed for the I mutant but not the H mutant (Fig. 8B,C, Online Video II), suggesting that decreasing hydrophobic affinity can correct the effect of constitutive palmitoylation on R-Ras trafficking. Akt phosphorylation was the same in cells expressing wild type R-Ras and the H and I R-Ras mutants (Online Fig. XIB).
Figure 8. R-Ras mutant restores trafficking and rescues APT1 deficiency.

(A) Diagrams of wild type and mutant R-Ras proteins. (B) Representative still TIRF images of GFP-R-Ras (wild type, H, or I mutant) with overlay of particle tracking results as maximal speed, as well as quantitation of global membrane dynamics in HUVEC cells as mean speed (C) of the tracks (n=22 cells for SC control and n=14–23 cells for APT1 KD groups in each mutant). (D) Representative Western blots of exogenous biotin-labeled fibronectin during pulse-chase of cells expressing wild type, H, or I mutant R-Ras in SC control or APT1 KD cells with (E) quantitation (n=4/condition). (F) VE-cadherin staining of HUVECs (red for VEC and green for GFP-R-Ras), as well as (G) quantitation of junctional signals (n=8 random regions). (H) Images of lumenized sprouts (marked by red asterisks and arrows at low and high magnification, respectively) from HUVEC cells expressing various R-Ras constructs coated on fibrin-embedded microbeads. (I) Quantitation of tube diameters (n=53–80 sprouts for SC control and KD groups in each mutant). Scale=1 micron in B, 20 microns in F, and in H 50/20 microns for low/high magnification, respectively. SC and KD: scrambled or APT1 knockdown. Data are presented as mean ± SEM by box/whisker plot (C,I) or scatter dot plot (E,G). Statistical analyses performed by one way ANOVA with Tukey’s multiple comparisons test. Please see Online Video 2.
In addition to disturbing R-Ras trafficking, APT1 deficiency impairs fibronectin metabolism (Fig. 2), disrupts the formation of adherens junctions (Fig. 4G,H), and inhibits lumen formation (Fig. 4A–C). Processing of labeled fibronectin was decreased in APT1-deficient cells expressing wild type R-Ras (KD-W) as compared to control cells expressing wild type R-Ras (SC-W), but fibronectin metabolism was restored in APT1-deficient cells expressing the I mutant R-Ras (Fig. 8D,E). Adherens junctions were decreased in APT1-deficient cells expressing wild type R-Ras (KD-W) as compared to control cells expressing wild type R-Ras (SC-W), but increased in APT1-deficient cells expressing the I mutant R-Ras (Fig. 8F,G). Endothelial cell sprouting assays using fibrin gel beads showed that expression of the I mutant, but not the H mutant, rescued the decreased lumen diameter in APT1 knockdown cells (Fig. 8H,I). Thus, in the setting of APT1 deficiency, reducing the hydrophobicity of R-Ras improves R-Ras trafficking, fibronectin metabolism, adherens junction assembly, and lumen formation.
DISCUSSION
Our findings implicate deficient endothelial palmitoylation cycling (the inability to remove palmitate from proteins to allow dynamic re-palmitoylation and trafficking) in peripheral artery disease. As depicted in Online Fig. XII, palmitoylation cycling is required for normal R-Ras trafficking that coordinates fibronectin turnover and cell contacts. Decreased APT1 activity results in constitutively palmitoylated R-Ras with decreased membrane motility, which alters the cytoskeleton and impairs vascular stability.
Endothelial APT1 deficiency impaired angiogenesis, disrupted fibronectin remodeling, and produced features of vascular activation. An unbiased approach identified increased palmitoylation of the GTPase R-Ras in APT1 deficiency. Hyperglycemia decreased APT1 enzyme activity, increased R-Ras palmitoylation, and altered R-Ras partitioning. Diabetes models showed increased palmitoylated R-Ras, consistent with decreased APT1 activity, and vascular accumulation of fibronectin. These results suggest that attenuated endothelial depalmitoylation can be caused by hyperglycemia to promote vascular immaturity related to functional deficiency of proteins like R-Ras.
APT1 deficiency and hyperglycemia caused increased palmitoylation associated with a redistribution of R-Ras away from light fractions and toward heavy fractions that do not contain rafts. Palmitoylation promotes the association of proteins with raft domains.42 Raft domains are thought to be comprised of membrane lipids in constant transition between ordered and disordered phases, as shown in Online Fig. XII. Our results suggest that palmitoylation cycling provides re-palmitoylated proteins with renewed access to raft domains, driving appropriate signaling in the setting of fluctuating membrane states. When depalmitoylation is impaired, R-Ras can be diverted to heavy fractions that may represent submembrane compartments with slow recycling potential leading to disrupted trafficking.
Other proteins in addition to R-Ras that play signaling roles in the vascular wall are depalmitoylated by APT1, and defective palmitoylation cycling for proteins such as the Src family tyrosine kinase Lyn43 likely contributes to vascular defects in APT1 deficiency. We focused molecular studies on R-Ras because this protein appeared in the highest-scoring statistically significant pathway in cluster analysis of palmitoylation proteomics data using endothelial cells from APT deficient and control mice. Moreover, R-Ras is expressed at high levels in adult endothelial cells and known to promote vessel maturity35–39 through processes we found to be disrupted in APT1 deficiency. Decreasing the hydrophobicity of palmitoylated R-Ras in the setting of APT1 deficiency rescued intracellular trafficking, fibronectin processing, adherens junctions, and defective lumen formation in vitro. These results provide proof of principle that interfering with the predicted effect of defective palmitoylation cycling in endothelial cells, namely the inability of APT1 substrate proteins to dissociate from the plasma membrane through depalmitoylation, restores cell physiology.
eNOS is known to be a substrate for APT144 but eNOS did not appear in our unbiased screen and functional deficiency of eNOS is unlikely to be involved in the endothelial-specific APT1 knockout phenotype. Adherens junctions and cortical actin are stabilized by defects in eNOS,45 while adherens junctions and cortical actin are disrupted by defects in palmitoylation cycling that occur with APT1 deficiency.
Abnormal metabolism of fibronectin occurred with APT1 deficiency. Endothelial α5β1 interacts with soluble dimeric fibronectin to generate insoluble multimeric forms through the incompletely understood process of fibrillogenesis that contributes to matrix formation.14 Extracellular matrix accumulates in vascular diseases, especially those associated with diabetes, through multiple mechanisms.46 Our results showing that remodeling of exogenous fibronectin is defective in APT1-deficient endothelial cells, and that mice and humans accumulate intimal fibronectin suggest that the endothelium promotes vessel immaturity in peripheral artery disease. These findings are consistent with recent work demonstrating that circulating fibronectin contributes to expansion of the matrix-rich mesangium to impair kidney function in mouse diabetes,47 and that subendothelial fibronectin promotes inflammation.48
Impaired fibronectin remodeling, loss of adherens junctions, and increased R-Ras palmitoylation caused by genetic inactivation of APT1 also occur with diabetes models, consistent with the acquisition of functional deficits in APT1. Hyperglycemia decreases APT1 enzyme activity in association with decreased APT1 acetylation, and kinetic analysis of mutated APT1 mimicking acetylation shows increased enzyme activity. These observations suggest that APT1-dependent palmitoylation cycling is regulated by acetylation, another dynamic form of lipidation known to affect non-nuclear proteins involved in metabolism.49, 50 Phosphorylation increases APT1 activity in cancer cells,51 and our findings raise the possibility that another posttranslational modification, acetylation, could be targeted pharmacologically to treat a disorder that is often intractable, peripheral artery disease.
Compartmentalized depalmitoylation contributes to the selective subcellular retention of proteins in neurons.52 Similar processes appear to be operative in endothelial cells, and their disruption by abnormal systemic metabolism represents a previously unrecognized mechanism underlying chronic vascular disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE
What Is Known?
Peripheral artery disease is common, responds poorly to medical therapies, and often leads to lower extremity amputation, especially in people with diabetes.
Lower extremity vascular disease is characterized by endothelial activation, fibronectin deposition, and abnormal vascular remodeling.
The spectrum of potential therapeutic targets for peripheral artery disease is meager.
What New Information Does This Article Contribute?
We inactivated endothelial acyl-protein thioesterase 1 (APT1), which removes the fatty acid palmitate from proteins, in mice to generate a model of peripheral artery disease that retains features of human peripheral artery disease.
Deficient APT1 disrupts vascular homeostasis in part by altering the intracellular trafficking of the small GTPase R-Ras.
Hyperglycemia modifies acetylation of the APT1 protein and decreases APT1 enzyme activity in endothelial cells, an effect that can be reversed by pharmacologic inhibition of histone deacetylase (HDAC).
ACKNOWLEDGMENTS
We thank Jacqueline Mudd and J. Michael White for mouse engineering; Fanxin Long for reagents; Petra Erdmann Gilmore, Yiling Mi, and Reid Townsend for proteomics assistance; Dennis Oakley, Michael Chien-cheng Shih, and James Fitzpatrick for imaging advice; and Amanda Penrose for specimen processing.
SOURCES OF FUNDING
NIH grants R21 AG051900 (X.W.), K08 HL132060 (M.Z.), DK101392, DK020579, DK056341, UL1 TR000448, NCI P30 CA091842, R35 GM119840 (B.C.D.), the trans-NIH Knock-Out Mouse Project (KOMP), and the Washington University Centene Personalized Medicine Initiative.
Nonstandard Abbreviation and Acronyms:
- APT1
acyl-protein thioesterase 1
- DPP3
depalmitoylation probe 3
- FN
fibronectin
- DOC
deoxycholate
- TIRF
total internal reflection fluorescence
- HUVECs
human umbilical vein endothelial cells
- KD
knockdown
- SC
scrambled
Footnotes
DISCLOSURES
None.
SUPPLEMENTAL MATERIALS
Expanded Materials & Methods
Online Table I
Online Figures I – XII
References 53–75
Online Tables II – III (Excel files of proteomic data)
Online Videos I - II
Major Resources Table
REFERENCES
- 1.Criqui MH and Aboyans V. Epidemiology of peripheral artery disease. Circ Res. 2015;116:1509–26. [DOI] [PubMed] [Google Scholar]
- 2.Olijhoek JK, van der Graaf Y, Banga JD, Algra A, Rabelink TJ, Visseren FL and Group SS. The metabolic syndrome is associated with advanced vascular damage in patients with coronary heart disease, stroke, peripheral arterial disease or abdominal aortic aneurysm. Eur Heart J. 2004;25:342–8. [DOI] [PubMed] [Google Scholar]
- 3.Britton KA, Mukamal KJ, Ix JH, Siscovick DS, Newman AB, de Boer IH, Thacker EL, Biggs ML, Gaziano JM and Djousse L. Insulin resistance and incident peripheral artery disease in the Cardiovascular Health Study. Vasc Med. 2012;17:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Althouse AD, Abbott JD, Forker AD, Bertolet M, Barinas-Mitchell E, Thurston RC, Mulukutla S, Aboyans V, Brooks MM and Group BDS. Risk factors for incident peripheral arterial disease in type 2 diabetes: results from the Bypass Angioplasty Revascularization Investigation in type 2 Diabetes (BARI 2D) Trial. Diabetes Care. 2014;37:1346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohammedi K, Woodward M, Zoungas S, Li Q, Harrap S, Patel A, Marre M, Chalmers J and Group AC. Absence of peripheral pulses and risk of major vascular outcomes in patients with type 2 diabetes. Diabetes Care. 2016;39:2270–2277. [DOI] [PubMed] [Google Scholar]
- 6.Aung PP, Maxwell HG, Jepson RG, Price JF and Leng GC. Lipid-lowering for peripheral arterial disease of the lower limb. Cochrane Database Syst Rev. 2007:CD000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger JS and Hiatt WR. Medical therapy in peripheral artery disease. Circulation. 2012;126:491–500. [DOI] [PubMed] [Google Scholar]
- 8.Kumbhani DJ, Steg PG, Cannon CP, Eagle KA, Smith SC Jr., Goto S, Ohman EM, Elbez Y, Sritara P, Baumgartner I, Banerjee S, Creager MA, Bhatt DL and Investigators RR. Statin therapy and long-term adverse limb outcomes in patients with peripheral artery disease: insights from the REACH registry. Eur Heart J. 2014;35:2864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, Williams DE and Geiss L. Changes in diabetes-related complications in the United States, 1990–2010. N Engl J Med. 2014;370:1514–23. [DOI] [PubMed] [Google Scholar]
- 10.Geiss LS, Li Y, Hora I, Albright A, Rolka D and Gregg EW. Resurgence of diabetes-related nontraumatic lower-extremity amputation in the young and middle-aged adult U.S. population. Diabetes Care. 2019;42:50–54. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P and Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz MA, Vestweber D and Simons M. A unifying concept in vascular health and disease. Science. 2018;360:270–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Astrof S and Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis. 2009;12:165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarzbauer JE and DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb Perspect Biol. 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen M, Artym VV, Green JA and Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18:463–71. [DOI] [PubMed] [Google Scholar]
- 16.Caswell PT, Vadrevu S and Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10:843–53. [DOI] [PubMed] [Google Scholar]
- 17.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. [DOI] [PubMed] [Google Scholar]
- 18.Faurobert E, Rome C, Lisowska J, Manet-Dupe S, Boulday G, Malbouyres M, Balland M, Bouin AP, Keramidas M, Bouvard D, Coll JL, Ruggiero F, Tournier-Lasserve E and Albiges-Rizo C. CCM1-ICAP-1 complex controls beta1 integrin-dependent endothelial contractility and fibronectin remodeling. J Cell Biol. 2013;202:545–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, Eklund L, Ivaska J, Alitalo K and Saharinen P. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat Commun. 2015;6:5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ejim OS, Fonseca V, Coumar A, Mathur S, Bell JL and Dandona P. Fibronectin concentrations in plasma in peripheral vascular disease. Clin Chem. 1988;34:2426–9. [PubMed] [Google Scholar]
- 21.Kanters SD, Banga JD, Algra A, Frijns RC, Beutler JJ and Fijnheer R. Plasma levels of cellular fibronectin in diabetes. Diabetes Care. 2001;24:323–7. [DOI] [PubMed] [Google Scholar]
- 22.Katsouyanni K, Skalkidis Y, Petridou E, Polychronopoulou-Trichopoulou A, Willett W and Trichopoulos D. Diet and peripheral arterial occlusive disease: the role of poly-, mono-, and saturated fatty acids. Am J Epidemiol. 1991;133:24–31. [DOI] [PubMed] [Google Scholar]
- 23.Naqvi AZ, Davis RB and Mukamal KJ. Dietary fatty acids and peripheral artery disease in adults. Atherosclerosis. 2012;222:545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palomer X, Pizarro-Delgado J, Barroso E and Vazquez-Carrera M. Palmitic and oleic acid: the yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol Metab. 2018;29:178–190. [DOI] [PubMed] [Google Scholar]
- 25.Resh MD. Targeting protein lipidation in disease. Trends Mol Med. 2012;18:206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang MM and Hang HC. Protein S-palmitoylation in cellular differentiation. Biochem Soc Trans. 2017;45:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beard RS Jr., Yang X, Meegan JE, Overstreet JW, Yang CG, Elliott JA, Reynolds JJ, Cha BJ, Pivetti CD, Mitchell DA, Wu MH, Deschenes RJ and Yuan SY. Palmitoyl acyltransferase DHHC21 mediates endothelial dysfunction in systemic inflammatory response syndrome. Nat Commun. 2016;7:12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Won SJ, Davda D, Labby KJ, Hwang SY, Pricer R, Majmudar JD, Armacost KA, Rodriguez LA, Rodriguez CL, Chong FS, Torossian KA, Palakurthi J, Hur ES, Meagher JL, Brooks CL 3rd, Stuckey JA and Martin BR. Molecular mechanism for isoform-selective inhibition of acyl protein thioesterases 1 and 2 (APT1 and APT2). ACS Chem Biol. 2016;11:3374–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smotrys JE and Linder ME. Palmitoylation of intracellular signaling proteins: regulation and function. Annu Rev Biochem. 2004;73:559–87. [DOI] [PubMed] [Google Scholar]
- 30.Kathayat RS, Elvira PD and Dickinson BC. A fluorescent probe for cysteine depalmitoylation reveals dynamic APT signaling. Nat Chem Biol. 2017;13:150–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silvestre JS, Smadja DM and Levy BI. Postischemic revascularization: from cellular and molecular mechanisms to clinical applications. Physiol Rev. 2013;93:1743–802. [DOI] [PubMed] [Google Scholar]
- 32.Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M and Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–35. [DOI] [PubMed] [Google Scholar]
- 33.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, Westrom S, Claesson-Welsh L, Vestweber D and Gerhardt H. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16:309–21. [DOI] [PubMed] [Google Scholar]
- 34.Yurdagul A Jr., Finney AC, Woolard MD and Orr AW. The arterial microenvironment: the where and why of atherosclerosis. Biochem J. 2016;473:1281–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komatsu M and Ruoslahti E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat Med. 2005;11:1346–50. [DOI] [PubMed] [Google Scholar]
- 36.Sawada J, Urakami T, Li F, Urakami A, Zhu W, Fukuda M, Li DY, Ruoslahti E and Komatsu M. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell. 2012;22:235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conklin MW, Ada-Nguema A, Parsons M, Riching KM and Keely PJ. R-Ras regulates beta1-integrin trafficking via effects on membrane ruffling and endocytosis. BMC Cell Biol. 2010;11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wurtzel JG, Lee S, Singhal SS, Awasthi S, Ginsberg MH and Goldfinger LE. RLIP76 regulates Arf6-dependent cell spreading and migration by linking ARNO with activated R-Ras at recycling endosomes. Biochem Biophys Res Commun. 2015;467:785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li F, Sawada J and Komatsu M. R-Ras-Akt axis induces endothelial lumenogenesis and regulates the patency of regenerating vasculature. Nat Commun. 2017;8:1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E and Gibson BW. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A. 2013;110:6601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iyer A, Fairlie DP and Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunol Cell Biol. 2012;90:39–46. [DOI] [PubMed] [Google Scholar]
- 42.Levental I, Lingwood D, Grzybek M, Coskun U and Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci U S A. 2010;107:22050–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, Lowell CA, Du X, Cheresh DA, Malik AB and Li Z. A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood. 2013;122:4140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh DC, Duncan JA, Yamashita S and Michel T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. J Biol Chem. 1999;274:33148–54. [DOI] [PubMed] [Google Scholar]
- 45.Di Lorenzo A, Lin MI, Murata T, Landskroner-Eiger S, Schleicher M, Kothiya M, Iwakiri Y, Yu J, Huang PL and Sessa WC. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J Cell Sci. 2013;126:5541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J and Shi GP. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta. 2014;1842:2106–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klemis V, Ghura H, Federico G, Wurfel C, Bentmann A, Gretz N, Miyazaki T, Grone HJ and Nakchbandi IA. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int. 2017;91:1374–1385. [DOI] [PubMed] [Google Scholar]
- 48.Hahn C, Orr AW, Sanders JM, Jhaveri KA and Schwartz MA. The subendothelial extracellular matrix modulates JNK activation by flow. Circ Res. 2009;104:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guan KL and Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011;36:108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choudhary C, Weinert BT, Nishida Y, Verdin E and Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–50. [DOI] [PubMed] [Google Scholar]
- 51.Sadeghi RS, Kulej K, Kathayat RS, Garcia BA, Dickinson BC, Brady DC and Witze ES. Wnt5a signaling induced phosphorylation increases APT1 activity and promotes melanoma metastatic behavior. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tortosa E and Hoogenraad CC. Polarized trafficking: the palmitoylation cycle distributes cytoplasmic proteins to distinct neuronal compartments. Curr Opin Cell Biol. 2018;50:64–71. [DOI] [PubMed] [Google Scholar]
- 53.Long F, Zhang XM, Karp S, Yang Y and McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–108. [DOI] [PubMed] [Google Scholar]
- 54.Wierzbicka-Patynowski I, Mao Y and Schwarzbauer JE. Analysis of fibronectin matrix assembly. Curr Protoc Cell Biol. 2004;Chapter 10:Unit 10 12. [DOI] [PubMed] [Google Scholar]
- 55.Wei X, Schneider JG, Shenouda SM, Lee A, Towler DA, Chakravarthy MV, Vita JA and Semenkovich CF. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J Biol Chem. 2011;286:2933–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zayed MA, Wei X, Park KM, Belaygorod L, Naim U, Harvey J, Yin L, Blumer K and Semenkovich CF. N-Acetylcysteine accelerates amputation stump healing in the setting of diabetes. FASEB J. 2017;31:2686–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolte S and Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. [DOI] [PubMed] [Google Scholar]
- 59.Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS and Casey PJ. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res. 2011;52:393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin BR, Wang C, Adibekian A, Tully SE and Cravatt BF. Global profiling of dynamic protein palmitoylation. Nat Methods. 2011;9:84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erde J, Loo RR and Loo JA. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J Proteome Res. 2014;13:1885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wisniewski JR, Zougman A, Nagaraj N and Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62. [DOI] [PubMed] [Google Scholar]
- 63.Chen ZW, Fuchs K, Sieghart W, Townsend RR and Evers AS. Deep amino acid sequencing of native brain GABAA receptors using high-resolution mass spectrometry. Mol Cell Proteomics. 2012;11:M111 011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner T, Sweetman G, Savitski MF, Mathieson T, Bantscheff M and Savitski MM. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal Chem. 2014;86:3594–601. [DOI] [PubMed] [Google Scholar]
- 65.Perkins DN, Pappin DJ, Creasy DM and Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67. [DOI] [PubMed] [Google Scholar]
- 66.Koskinen VR, Emery PA, Creasy DM and Cottrell JS. Hierarchical clustering of shotgun proteomics data. Mol Cell Proteomics. 2011;10:M110 003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang da W, Sherman BT and Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arnaoutova I and Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5:628–35. [DOI] [PubMed] [Google Scholar]
- 69.Nakatsu MN, Davis J and Hughes CC. Optimized fibrin gel bead assay for the study of angiogenesis. J Vis Exp. 2007:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Preibisch S, Saalfeld S and Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hulce JJ, Joslyn C, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P and Rosen H. An in vivo active carbamate-based dual inhibitor of lysophospholipase 1 (LYPLA1) and lysophospholipase 2 (LYPLA2) Probe Reports from the NIH Molecular Libraries Program Bethesda (MD); 2010. [PubMed] [Google Scholar]
- 72.Berginski ME and Gomez SM. The Focal Adhesion Analysis Server: a web tool for analyzing focal adhesion dynamics. F1000Res. 2013;2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL and Eliceiri KW. TrackMate: An open and extensible platform for single-particle tracking. Methods. 2017;115:80–90. [DOI] [PubMed] [Google Scholar]
- 74.Fiordalisi JJ, Johnson RL, 2nd, Ulku AS, Der CJ and Cox AD. Mammalian expression vectors for Ras family proteins: generation and use of expression constructs to analyze Ras family function. Methods Enzymol. 2001;332:3–36. [DOI] [PubMed] [Google Scholar]
- 75.Liu H and Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.