

Summary
The fly trachea is the equivalent of the mammalian lung and is a useful model for human respiratory diseases. However, little is known about the molecular mechanisms underlying tracheal air filling during larval development. In this study, we discover that PTPMT1 has a function in tracheal air filling. PTPMT1 is a widely conserved, ubiquitously expressed mitochondrial phosphatase. To reveal PTPMT1's functions in genetically tractable invertebrates and whether those functions are tissue specific, we generate a Drosophila model of PTPMT1 depletion. We find that fly PTPMT1 mutants show impairments in tracheal air filling and subsequent activation of innate immune responses. On a cellular level, these defects are preceded by aggregation of mitochondria within the tracheal epithelial cells. Our work demonstrates a cell-type-specific role for PTPMT1 in fly tracheal epithelial cells to support air filling and to prevent immune activation. The establishment of this model will facilitate exploration of PTPMT1's physiological functions in vivo.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
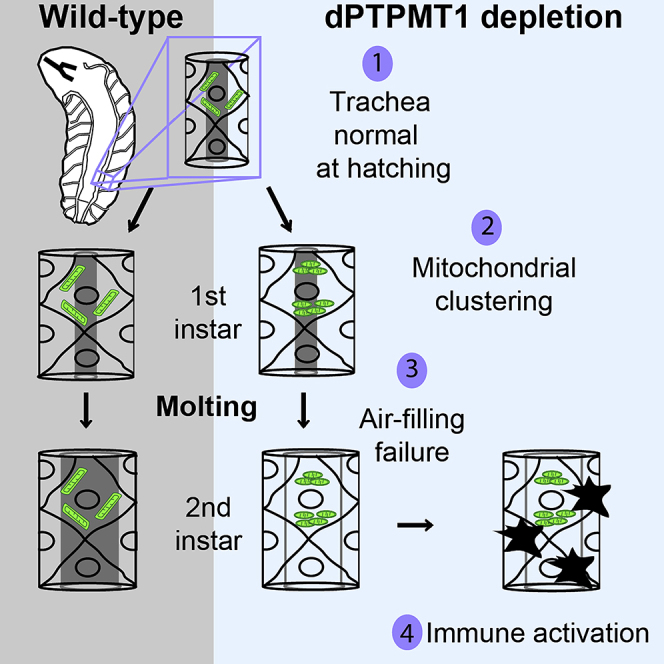
Highlights
-
•
A Drosophila model of PTPMT1 depletion is generated
-
•
PTPMT1 mutants show impairments in tracheal air filling
-
•
Mitochondria aggregate within the tracheal epithelial cells prior to air-filling failure
-
•
Depletion of the cardiolipin pathway components does not mimic PTPMT1 deficiency
Biological Sciences; Molecular Biology; Cell Biology
Introduction
The fly respiratory system (i.e., the trachea) is the equivalent of the mammalian lung with similarities in development and immunity, which make it a useful model for numerous human respiratory diseases (Bergman et al., 2017). The tracheal system consists of a network of air-filled epithelial tubes. These tubes contain a cuticular lining that provides structural support, through ridges known as taenidia. As the larva grows the trachea must also grow to match the increased oxygen demand of the tissue. During molting the old cuticular lining separates from the tracheal epithelium, and what is presumed to be molting fluid fills the space between the old and new lining. The old lining collapses and is removed from the trachea, followed immediately by filling of the trachea with air and removal of molting fluid (Park et al., 2002). However, little is known about the molecular mechanisms underlying larval tracheal air filling.
The trachea is open to the environment and thus vulnerable to infection. Tracheal epithelial cells have a number of innate defenses including the expression of antimicrobial peptides (AMPs) (Tzou et al., 2000; Wagner et al., 2009; Bergman et al., 2017) and melanization, an arthropod-specific response to wounds or infections (Tang et al., 2008). The production of melanin is catalyzed by prophenoloxidases (PPOs), zymogens that are activated by serine protease cascades (Tang et al., 2006; Dudzic et al., 2019). Outside the trachea, melanization is typically mediated by specialized immune cells known as crystal cells, which can be triggered to release PPOs into the hemolymph (Tang, 2009).
Mitochondria are extremely multifaceted organelles with roles in metabolism, apoptosis, cell signaling, and more. Reversible phosphorylation is one mechanism by which mitochondrial activities are regulated (Pagliarini and Dixon, 2006). Protein-tyrosine-phosphatase localized to the mitochondrion 1 (PTPMT1) is a phosphatase that resides on the inner mitochondrial membrane, where it regulates mitochondrial functions through its phosphatase activity (Pagliarini et al., 2004, 2005). Studies from vertebrate systems have shown that PTPMT1 controls pyruvate importation and utilization in the mitochondria through regulation of phosphatidylinositol phosphate (PIP) levels, modulates crista structure and respiratory chain function through the synthesis of cardiolipin (CL)—the hallmark phospholipid of the mitochondria (Zhang et al., 2011; Xiao et al., 2011; Teh et al., 2013), and regulates the tricarboxylic acid (TCA) cycle through succinate dehydrogenase (Nath et al., 2015). Loss of PTPMT1 in a variety of mammalian cell types results in decreased mitochondrial respiration and increased glycolysis. These metabolic changes have drastic impacts on stem cells and cancer cells, but not on other cell types, demonstrating that the functional consequences of PTPMT1 depletion are highly cell-type-specific (Pagliarini et al., 2005; Shen et al., 2011; Zhang et al., 2011; Niemi et al., 2013; Yu et al., 2013; Zheng et al., 2018). Although PTPMT1 is broadly expressed and highly conserved among species (Pagliarini et al., 2004, 2005; Shen et al., 2011; Teh et al., 2013), to date, it has only been examined in a narrow number of tissues and cell types and predominately in vertebrates, limiting our understanding of PTPMT1's distinct cell-type-specific roles in vivo. Drosophila melanogaster is an unparalleled genetic model for studying tracheal biology and gene function, with methods for the rapid generation of mutants as well as expansive collections of existing mutants and RNA interference (RNAi) lines. Here, we examine the physiological functions of PTPMT1 in Drosophila and report a role for PTPMT1 in fly tracheal epithelial cells.
Results
dPTPMT1 Is an Essential Gene in Drosophila
To study the function of Drosophila PTPMT1 (dPTPMT1), we employed CRISPR-CAS9 to create a defined deletion in dPTPMT1 that would remove most of its coding sequence (Gratz et al., 2013, 2015) (Figure S1A). We failed to isolate the intended deletion but were able to obtain three independent lines with small deletions in the regions targeted by our gRNAs: dPTPMT1mut1 (c.487delA), dPTPMT1mut2 (c.479_489delCGGATGGACTC), and dPTPMT1mut3 (c.12 + 82delT and c487delA) (Figures 1A, S1B, and S1C). All three lines were homozygous lethal and also lethal over two deficiency lines covering dPTPMT1 (Df(3R)BSC490 and Df(3R)Exel9013). The failure to complement indicates that lethality is likely caused by mutations in dPTPMT1. In all lines, both as homozygotes and transheterozygotes (over Df(3R)BSC490), third instar larvae were present, albeit at reduced frequencies, likely due to the increased early lethality (Figure 1B). None of these larval escapers were able to undergo pupariation and survive to adulthood (Figure 1C). We collected age-synchronized embryos (3 h after egg laying–AEL) and examined viability 24 h AEL. We found approximately 80% of first instar larvae of dPTPMT1mut1 that hatched died soon after (Figure 1B). The lethality rates were comparable between dPTPMT1mut1 homozygotes and transheterozygotes. These results suggest that dPTPMT1mut1represents a null allele or strong hypomorph.
Figure 1.

dPTPMT1 Is an Essential Gene in Drosophila
(A) Regions in dPTPMT1 targeted by the three RNAi lines. Thin black bars indicate regions targeted by RNAi, dark gray bar is the 5′ UTR, light gray bar is the 3′ UTR, purple bars are exons, and thick black bars are introns. Asterisk indicates the location of the deletion in dPTPMT1mut1.
(B) Viability of dPTPMT1mut1 first instar larvae at 24 h AEL. n = 4–5 replicates, 56–285 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(C) Adult viability of dPTPMT1mut1 mutants. Genotypic frequency is calculated for adults and then normalized to expected genotypic frequency. n = 3–4 replicates; 0- to 2-day-old adults. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(D) Percentage of third instar males with ubiquitous dPTPMT1 RNAi surviving to pupation and eclosion. Larvae are on a yellow mutant background (hereafter indicated by “y”). Statistical significance is given relative to Act5C-GAL4 crossed to the respective RNAi background controls at the same developmental stage. n = 3 replicates, 25–26 for each repeat for RNAi 1-2 and controls. n = 3 replicates, 12–44 for each repeat for RNAi 3 and control.
Data are represented as mean ± SEM and ∗∗p < 0.01, ∗∗∗p < 0.001 by chi-square test. See also Figure S1.
To complement the CRISPR mutants, we obtained three UAS-dPTPMT1 RNAi lines from the Vienna Drosophila RNAi Center and the Drosophila Transgenic RNAi Project collections (Dietzl et al., 2007; Perkins et al., 2015). All three UAS-dPTPMT1 RNAi insertions are at independent genomic locations. RNAi 1 and 2 target the same region within the third exon of dPTPMT1, whereas RNAi 3 targets the fourth exon of dPTPMT1, allowing us to control for off-target effects (Figure 1A). Ubiquitous dPTPMT1 RNAi with the driver Act5C-GAL4 reduced dPTPMT1 transcripts to below 20% of wild-type levels for all three lines, as detected by quantitative real-time PCR (qPCR) (Figure S1D). dPTPMT1 RNAi 1 and 3 were pupal lethal (Figure 1D). RNAi 2 allowed adult survivors but with a significant decrease in viability compared with wild-type controls (Figure 1D). Given the conservation in protein sequence between PTPMT1 and PTEN's catalytic regions (Pagliarini et al., 2004), we also verified that dPTPMT1 RNAi did not affect fly PTEN (dPten) transcript levels by qPCR (Figure S1E).
To study the structure-function relationship of dPTPMT1 in vivo, we generated UAS-dPTPMT1 transgenic fly lines, including wild-type (WT) dPTPMT1, catalytic-dead (CD; with a C141S mutation) dPTPMT1, and dPTPMT1 with a deletion of its putative mitochondrial targeting sequence (ΔMTS; removing amino acids 1–31) (Pagliarini et al., 2005). All transgenes were inserted into the same genomic region using the PhiC31 integrase-mediated transgenesis system (Markstein et al., 2008). To test the functional conservation of PTPMT1 between flies and humans, we also generated UAS-human PTPMT1 (hPTPMT1) and UAS-hPTPMT1-CD (C132S) (Zhang et al., 2011). Because we were unable to generate an antibody or identify a commercially available antibody that recognized dPTPMT1, we tagged all transgenes with 3×FLAG. We confirmed that the expression of these transgenes driven by Act5C-GAL4 were comparable using immunoblotting against FLAG (Figures S1F and S1G).
To examine the localization of exogenously expressed dPTPMT1, we performed mitochondrial enrichments using adults or larvae with ubiquitous expression of each of our transgenes. We used the inner mitochondrial membrane protein, dMIC60, as a positive control for enrichment of mitochondria (Tsai et al., 2018). Exogenously expressed dPTPMT1-WT-3×FLAG was detected in the mitochondrial-enriched fraction, although it was also detected in the cytosolic fraction (Figure S1H). Deletion of the putative MTS decreased dPTPMT1-3×FLAG levels in the mitochondrial-enriched fraction (Figure S1I), suggesting that the N-terminal amino acids 1–31 direct dPTPMT1 import into mitochondria. These results are consistent with a previous study in yeast showing that heterologously expressed dPTPMT1 localizes to mitochondria (Teh et al., 2013). Importantly, dPTPMT1-CD-3×FLAG was also detected in the mitochondrial fraction (Figure S1J). We next assayed flies with ubiquitous expression of UAS-hPTPMT1-WT-3×FLAG and UAS-hPTPMT1-CD-3×FLAG and detected both versions of hPTPMT1 in the mitochondrial fraction (Figure S1K). Collectively, the generation of these transgenes allowed us to perform the following functional studies of PTPMT1 in flies.
To corroborate that the lethality observed in dPTPMT1 CRISPR mutants was caused by a lack of dPTPMT1, we ubiquitously expressed UAS-dPTPMT1-3×FLAG in dPTPMT1mut1 transheterozygotes. Ubiquitous expression of UAS-dPTPMT1-3×FLAG allowed mutants to survive to adulthood, eclosing at the expected genotypic ratio (Figure 1C). This result confirms that the lethality of dPTPMT1 CRISPR mutants is caused by the loss of dPTPMT1. It should be noted that we were unable to quantify viability in this rescue experiment by counting the numbers of survivors of larvae and pupae as implemented in Figure 1D, because one of the lines necessary for the identification of genotypes of third instars could not be generated. Instead, we quantified viability by calculating the genotypic ratio of surviving adults and then normalizing it to the expected genotypic frequency. Importantly, expression of ΔMTS-dPTPMT1-3×FLAG and dPTPMT1-CD-3×FLAG failed to rescue viability of CRISPR mutants (Figure 1C), indicating that mitochondrial localization and catalytic activity are required for dPTPMT1's function. Ubiquitous expression of UAS-hPTPMT1-WT-3×FLAG, but not UAS-hPTPMT1-CD-3×FLAG, was also able to rescue viability (Figure 1C), indicating that human and fly PTPMT1 are functionally conserved. Notably, expression of dPTPMT1 in specific tissues such as the trachea by btl-GAL4, neurons by elav-GAL4, or muscles by Mhc-GAL4 did not rescue the viability of dPTPMT1mut1 transheterozygotes, indicating that dPTPMT1 is required in multiple tissues in Drosophila for survival. Collectively, these data suggest that dPTPMT1 is essential for viability in the fly.
Depletion of dPTPMT1 Causes Tracheal Blackening
We noticed that ubiquitous dPTPMT1 RNAi resulted in tracheal blackening in both adults and third instar larvae (Figure 2A). All three dPTPMT1 RNAi lines showed a significantly higher frequency of tracheal blackening than controls (Figures 2B–2D). By contrast, ubiquitous GFP RNAi did not cause significant tracheal blackening, indicating that this phenotype is not an artifact of the RNAi system (Figures 2B and 2C). Using transmission electron microscopy (TEM) on a section of trachea from a third instar larva with ubiquitous dPTPMT1 RNAi, we discovered large electron-dense regions between the tracheal cuticle and the tracheal epithelial cells (Figure S2A). To test whether this tracheal blackening was due to melanization, we used RNAi to ubiquitously knockdown MP1, a serine protease reported to be involved in activating tracheal melanization (Tang et al., 2006, 2008). Using qPCR, we confirmed a significant reduction of MP1 transcripts (Figure S2B). We saw a substantial decrease in the strength and frequency of tracheal blackening in dPTPMT1 RNAi flies by MP1 RNAi but not by GFP RNAi (Figures 2B and 2C). These results indicate that tracheal blackening is at least partially a result of melanization.
Figure 2.

dPTPMT1 Depletion Results in Tracheal Blackening
(A) Ubiquitous dPTPMT1 RNAi results in tracheal blackening (arrows); 0- to 2-day-old adult males and third instar males are shown. Scale bars, 250 μm.
(B and C) Percentage of 0- to 2-day-old adult males with tracheal blackening (B) and strength of tracheal blackening (C). n = 3 replicates, 48–138 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(D) Percentage of third instars with tracheal blackening; mixed-sex, females are heterozygous for y and males are hemizygous for y (hereafter denoted as y/y+). n = 3 replicates, 25–43 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(E) Viability of tracheal-specific dPTPMT1 RNAi larvae. Statistical significance is given relative to btl-GAL4 crossed to respective RNAi background controls at the same developmental stage. n = 2–3 replicates, 50–100 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(F) Percentage of second instar, tracheal-specific dPTPMT1 RNAi larvae with tracheal blackening. n = 2–3 replicates, 38–94 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(G) A representative image of a second instar larva with tracheal-specific dPTPMT1 RNAi shows tracheal blackening (arrow). Scale bars, 250 μm.
(H) Tracheal blackening (arrows) in dPTPMT1mut1 third instar larvae. Scale bars, 250 μm.
(I) Percentage of dPTPMT1mut1 third instars with tracheal blackening. n = 2–4 replicates, 16–110 for each repeat.
Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test. See also Figures S2 and S3.
To determine whether this blackening phenotype was cell autonomous, we performed a tracheal-specific RNAi knockdown of dPTPMT1, using btl-GAL4 (Kim et al., 2018). Both dPTPMT1 RNAi 1 and 3 resulted in early lethality (Figure 2E). We found that over half of dPTPMT1 RNAi larvae were dead by 72 h AEL (Figure 2E). It should be noted that, in this particular experiment at 72 h AEL, the majority of larvae (control and RNAi) were second instars (Figure S2C), due to the developmental delay caused by sorting first instars at room temperature (22°C) at hatching (see Methods). Many of these larvae were found dead on the side of the vial, suggesting that they died of anoxia as second instar larvae (Wagner et al., 2009). Importantly, first instar larvae with tracheal dPTPMT1 RNAi showed no sign of tracheal blackening (btl-GAL4—Control for RNAi 1: 0% ± 0%, btl-GAL4>UAS-dPTPMT1 RNAi 1: 0% ± 0%; btl-GAL4—Control for RNAi 3: 0% ± 0%, btl-GAL4>UAS-dPTPMT1 RNAi 3: 1% ± 1%; n = 2–3 replicates, 37–97 for each repeat). However, nearly all second instar larvae had blackened trachea (Figures 2F and 2G). TEM on a section of trachea from a third instar escaper with tracheal-specific dPTPMT1 RNAi also revealed large electron-dense regions between the tracheal cuticle and the tracheal epithelial cells (Figure S2D), like ubiquitous dPTPMT1 RNAi (Figure S2A). Therefore, tracheal blackening occurs during the transition from first to second instar in tracheal-specific dPTPMT1 RNAi larvae.
Melanization can be activated by the trachea (Tang et al., 2008). However, it can also be mediated by phenol oxidases, released by crystal cells, in the hemolymph (Tang, 2009). Knockdown of MP1 specifically in the trachea has been shown to block tracheal-specific melanization (Tang et al., 2008). To determine whether tracheal blackening in dPTPMT1 RNAi larvae was a result of tracheal-specific melanization being activated, we knocked down MP1 in the trachea by btl-GAL4. Tracheal-specific MP1 RNAi did not reduce tracheal blackening; however, it did partially rescue viability (Figures S2E and S2F), suggesting that MP1 mediates tracheal blackening through other tissues in dPTPMT1 RNAi.
Data from Flybase (www.flybase.org) show that dPTPMT1 is ubiquitously expressed. To determine whether dPTPMT1 is required in non-tracheal tissues to prevent tracheal blackening, we knocked down dPTPMT1 using drivers specific to the nervous system (multiple elav-GAL4 drivers and ey-GAL4), the muscles (Mhc-GAL4 and C57-GAL4), and the fat bodies/hemocytes (Pxn-GAL4, Cg-GAL4). dPTPMT1 RNAi by these drivers did not result in any apparent lethality, blackening phenotypes, or other visible phenotypes (Figure S2G), suggesting that tracheal epithelial cells are highly sensitive to loss of dPTPMT1.
Next, we examined dPTPMT1 CRISPR mutants and found that third instar escapers from all three CRISPR lines showed substantial tracheal blackening as homozygotes and transheterozygotes (Figures 2H, 2I, S3A, and S3B). Ubiquitous expression of dPTPMT1 in dPTPMT1mut1 transheterozygotes resulted in no tracheal blackening in rescued adults (n = 4 replicates, 47–58 adults for each repeat). Interestingly, although ubiquitous expression of hPTPMT1-3×FLAG rescued viability in dPTPMT1mut1 transheterozygotes (Figure 1C), 20% of these adults showed tracheal blackening (n = 3 replicates, 29–48 adults for each repeat). Importantly, ubiquitous expression of hPTPMT1 but not hPTPMT1-CD also caused tracheal blackening in a wild-type background. Twenty percent of adults with hPTPMT1 expression showed tracheal blackening compared with 0% of adults with hPTPMT1-CD expression (n = 3 replicates, 10 adults for each repeat), suggesting that hPTPMT1 acts as a weak dominant negative for tracheal blackening in Drosophila. We were also able to rescue tracheal blackening in dPTPMT1mut1 transheterozygotes with tracheal-specific expression of wild-type dPTPMT1-3×FLAG but not CD or ΔMTS-dPTPMT1-3×FLAG (Figure 2I). Collectively, these data demonstrate that dPTPMT1 is essential to prevent tracheal blackening in a cell-autonomous manner and this function is dependent upon dPTPMT1's mitochondrial localization and phosphatase activity.
dPTPMT1 Depletion Results in Drosomycin Expression in the Trachea
Because tracheal melanization has been shown to induce the expression of one of the AMPs, Drosomycin (Drs) (Tang et al., 2008), we assayed dPTPMT1 RNAi and CRISPR mutant larvae for expression of Drs. qPCR revealed that Drs was significantly upregulated in ubiquitous dPTPMT1 RNAi adults and in dPTPMT1mut1 homozygous third instar escapers (Figures 3A and 3B). Ubiquitous dPTPMT1 RNAi third instars also showed increased Drs expression by qPCR, although this increase was not statistically significant (Figure S3C). We next examined localization of Drs-GFP expression in adults and third instar larvae with ubiquitous dPTPMT1 RNAi, using Drs-GFP reporter flies (Ferrandon et al., 1998). We saw an increased percentage of both adults and larvae with Drs-GFP expression in their trachea (Figures 3C–3F). We also examined Drs-GFP in white RNAi trachea, which have a knockdown of the transporter involved in pigmentation of the eye (Mount, 1987). We did not see a significant increase in the percentage of white RNAi adults with Drs-GFP in the trachea (Figures 3C and 3D), indicating that the lack of dPTPMT1, rather than the RNAi system itself, is responsible for this phenotype.
Figure 3.

dPTPMT1 Depletion Results in Drs Expression in the Trachea
(A and B) (A) qPCR analysis of Drs expression using whole-fly lysates. n = 3–6 replicates of 15–20 0- to 2-day-old adult males (A) or third instars (B). Data are represented as mean ± SEM and ∗∗p < .01, ∗∗∗p < 0.001, by one-tailed Student's t test.
(C) Drs-GFP expression in dPTPMT1 RNAi adult females, 0–2 days old. Arrow points to signal in the leg trachea. Females are heterozygous for y mutation (y/y+). Scale bars, 250 μm.
(D) Percentage of adult females with Drs-GFP in the trachea. Females are heterozygous for y mutation (y/y+). n = 3 replicates, 17–57 for each repeat, 0–2 days old. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(E) Drs-GFP expression in dPTPMT1 RNAi third instar males. Arrow points to Drs-GFP expression in the dorsal trunk of the trachea and posterior spiracles. Dashed box shows a higher magnification image of the affected area. Scale bars, 250 μm.
(F) Percentage of third instar males with Drs-GFP expression in the trachea. n = 4–7 replicates, 12–53 for each repeat.
Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test. See also Figure S3.
Lastly, we asked whether tracheal blackening and Drs elevation were specific to the loss of dPTPMT1, or a general by-product of mitochondrial dysfunction. To answer this question, we examined larvae lacking dMIC60, a key mitochondrial player for maintaining mitochondrial structure and function (Tsai et al., 2017, 2018). We found that only 2.3% of third instars (n = 86) had tracheal blackening as compared with 46%–100% in dPTPMT1 mutants and only 7% had tracheal Drs-GFP expression (±4% versus w1118 control: 0% ± 0%, n = 2–3 replicates, 10–28 for each repeat) as compared with 59%–90% in dPTPMT1 mutants. This result suggests that tracheal blackening and Drs elevation are unlikely non-specific responses to general mitochondrial dysfunction. Taken together, dPTPMT1 is required to prevent both tracheal blackening and Drs upregulation.
dPTPMT1 Depletion Causes Tracheal Air-Filling Defects
Because we found that tracheal blackening was absent in first instar larvae with tracheal-specific dPTPMT1 RNAi, but highly penetrant in second instars, we performed an in-depth examination of these larvae during the first-second instar molt. We inspected the trachea of dPTPMT1 RNAi first instars and discovered that these mutants failed to fill their trachea with air upon completion of molting (Figures 4A and 4B). Failure of tracheal air filling was only observed in dPTPMT1 RNAi but not in GFP RNAi larvae (Figures 4A and 4B). First instars with tracheal-specific dPTPMT1 RNAi examined at hatching (24 h AEL, n = 5, btl-GAL4 > UAS-dPTPMT1 RNAi 1, UAS-mito-GFP) and just prior to molting (48 h AEL, n = 5, btl-GAL4 > UAS-dPTPMT1 RNAi 1, UAS-mito-GFP) showed no air-filling defects in the trachea, suggesting that these defects occur during or immediately following molting. We examined dPTPMT1mut1 homozygous and transheterozygous third instar escapers and found that they also showed defects in tracheal air filling, although this phenotype was less penetrant, possibly due to genetic backgrounds and the resistance of escapers to stressors (Figure 4C). In addition, TEM on the trachea of a third instar larva with ubiquitous dPTPMT1 RNAi and on a larva with tracheal-specific dPTPMT1 RNAi revealed that the tracheal lumina of these larvae were filled with unidentifiable membranous materials as well as what we presumed to be molting fluid (Figures S2A and S2D). Together, these data show that dPTPMT1 mutants fail to fill their trachea with air and fail to clear fluid from the tracheal lumen.
Figure 4.

dPTPMT1 Depletion Results in Air Filling and Mitochondrial Defects in the Trachea
(A) Air-filling defects in second instars with tracheal-specific dPTPMT1 RNAi. Red arrows indicate trachea. In dPTPMT1 RNAi a portion of the posterior dorsal trunk shows air filling, but the remainder of the dorsal trunk is unfilled. Trachea filled with air appears darker than the adjacent tissues owing to differences in the refractive indices of liquid and air. Scale bars, 100 μm.
(B) Percentage of second instars with air-filling defects with tracheal-specific dPTPMT1 RNAi. n = 3 replicates, 6–12 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(C) Percentage of dPTPMT1mut1 third instars showing tracheal air-filling defects. n = 3–6 replicates, 14–33 for each repeat. Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test.
(D) Taenidial folds (arrows) in dissected trachea captured by light microscopy show no defects in tracheal-specific dPTPMT1 RNAi second instars. Note that these mutant tracheae are transparent because of the lack of air. The same results were seen in 8–22 larvae from two independent crosses. Scale bars, 100 μm.
(E) Timeline of events in tracheal-specific dPTPMT1 RNAi larvae undergoing first to second instar molt.
(F) Live imaging of mito-GFP within the dorsal trunk of the trachea of first instars at hatching, just prior to molting (“pre-molting”), and newly molted second instars (“post-molting”). The same results were seen in ten larvae from two independent crosses. Scale bars, 5 μm.
(G) Quantification of fluorescence ratios of mito-roGFP2-Grx1 expressed in trachea of second instars. n = 32–38 from three independent crosses. Boxes show 25th/75th percentiles, whiskers are the minimum and maximum values, and x is the median marker and ∗∗∗p < 0.001 by one-tailed Student's t test.
(H) Relative ATP levels, normalized to total protein. n = 7 independent experiments. Five-day-old Pink11RV (control) and Pink1B9 adult males (Park et al., 2006) were used as a positive control. Ubiquitous dPTPMT1 RNAi were adult males (0–2 days old) and dPTPMT1mut1 were third instars. Statistical significance is given relative to respective controls (black bar). Data are represented as mean ± SEM and ∗p < 0.05, ∗∗∗p < 0.001 by one-tailed Student's t test.
(I) Viability of adults with tracheal-specific RNAi of enzymes involved in CL biosynthesis raised at 25°C, 0- to 2-day-old adults. For simplicity only one background control is shown. Genotypic frequency is calculated for the genotype of interest and then normalized to its expected genotypic frequency. Statistical significance is given relative to respective controls. dPGS1 RNAi viability is significantly increased relative to its control. Data are represented as mean ± SEM and ∗p < 0.05, ∗∗∗p < 0.001 by chi-square test.
(J) Tracheal blackening and Drs-GFP expression in adult females (0–2 days old) with ubiquitous RNAi of CL biosynthesis enzymes. Females are heterozygous for y mutation (y/y+). Statistical significance is given relative to Act5C-GAL4>white RNAi. n = 3 replicates, 17–83 for each repeat. Data for Drs-GFP for controls and dPTPMT1 RNAi are the same as in Figure 3D.
Data are represented as mean ± SEM and ∗∗∗p < 0.001 by chi-square test. See also Figures S3 and S4.
To exclude the possibility that structural defects were responsible for failed fluid clearance and air filling, we examined the taenidia of second instars and saw no obvious defects in the structure of taenidia in either of our two independent RNAi lines, despite the obvious lack of air in their trachea (Figure 4D). TEM on third instar larvae with ubiquitous and tracheal-specific dPTPMT1 RNAi did show some abnormally formed taenidia (Figures S2A and S2D). This result suggests that these tracheal phenotypes are not caused by gross cuticular defects; however, subtler defects in taenidial structure may play a role.
To determine the temporal relationship between failure of air filling and tracheal blackening, we performed a time course analysis. We found that failure of air filling was followed by blackening of the trachea, within 30–90 min of molting completion (n = 15–17, 2 independent crosses) (Figure 4E). Importantly, although we observed nearly complete penetrance of both tracheal blackening and tracheal air filling defects in tracheal-specific dPTPMT1 RNAi larvae (Figures 2F and 4B), we found that dPTPMT1mut1 homozygous and transheterozygous third instar escapers showed a higher frequency of tracheal blackening than air-filling defects (Figures 2I and 4C). These results suggest that, although the failure of tracheal air filling precedes tracheal blackening in tracheal-specific dPTPMT1 RNAi, the air-filling failure is not absolutely required for blackening. It is likely both phenotypes are a consequence of an initial defect in the trachea due to a lack of dPTPMT1.
dPTPMT1 Depletion Leads to Mitochondrial Aggregates Prior to Failure of Air Filling
We set out to search for any defect that occurred earlier than tracheal air-filling failure and blackening in our mutants. Because dPTPMT1 is a mitochondrially localized phosphatase, we hypothesized that dPTPMT1 deficiency disrupted mitochondrial function in tracheal epithelial cells, which consequently interfered with air filling during molting. We examined mitochondrial function in first and second instars with tracheal-specific dPTPMT1 RNAi by btl-GAL4, because they showed a strong, penetrant phenotype with a precise onset.
We expressed UAS-mito-GFP in tracheal-specific dPTPMT1 RNAi larvae and live-imaged mito-GFP within the trachea. We found that, although mito-GFP signal appeared normal at hatching, enlarged mito-GFP puncta started to form just prior to molting in first instars and persisted in recently molted second instars (Figure 4F). At both developmental stages, these puncta were present throughout the dorsal trunk and tracheal branches. Notably, there was a greater number of large mito-GFP puncta in late first instars than in early second instars (Figure 4F). Importantly, this mitochondrial phenotype preceded the air-filling phenotype (Figure 4E). To exclude the possibility that these mito-GFP puncta were an artifact of our RNAi system, we examined mito-GFP in the trachea of larvae with tracheal-specific knockdown of white. In these larvae, mito-GFP appeared normal, as in controls (Figure 4F). Large mito-GFP puncta were also present in dPTPMT1 RNAi trachea using two additional independent mitochondrially localizing GFP lines (Figure S3D). These large, irregular mito-GFP puncta may represent clusters of mitochondria. Indeed, under TEM we saw individual mitochondria with intact crista structure aggregated in a tracheal epithelial cell of a third instar escaper with tracheal-specific dPTPMT1 RNAi (Figure S3E). To eliminate the possibility that the air-filling failure we observed in dPTPMT1 mutants was a result of tracheal epithelial cell death, we examined tracheal cells using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. We saw no significant differences in the number of TUNEL-positive cells between mutants and controls (Figure S3F), suggesting that dPTPMT1-depleted tracheal epithelial cells are not apoptotic. Collectively, these results suggest that a defect in mitochondria could be the earliest phenotype in dPTPMT1 mutant trachea (Figure 4E).
dPTPMT1 Depletion Compromises Mitochondrial ROS Production in the Trachea and ATP Levels
Defective tracheal air filling causes anoxia, which consequently impacts mitochondrial aerobic respiration. We examined mitochondrial reactive oxidative species (ROS), which are a key by-product of mitochondrial oxidative phosphorylation, in tracheal epithelial cells. We used two genetically encoded mitochondrial ROS sensors: UAS-mito-roGFP2-Grx1, a reporter of the glutathione redox potential, which indicates general ROS and reactive nitrogenous species (RNS) (Lushchak, 2012), and UAS-mito-roGFP2-Orp1, a hydrogen peroxide (H2O2) sensor (Albrecht et al., 2011; Barata and Dick, 2013). roGFPs are redox-sensitive GFPs that react to oxidants and reductants, resulting in ratiometric changes in fluorescence emitted at 500–530 nm when the probe is excited at 405 and 488 nm (Dooley et al., 2004; Hanson et al., 2004). An increase in the 405/488 nm ratio indicates increased oxidant concentration. Coupling roGFP to Grx1 and Orp1 enhances its specificity for glutathione and H2O2, respectively (Gutscher et al., 2008, 2009). We did not observe significant differences in the fluorescence ratio (405/488 nm) of the two ROS reporters between controls and tracheal-specific RNAi of dPTPMT1 by btl-GAL4 at 24 h AEL (first instars) (Figures S3G and S3H). However, we did see a significant decrease in the fluorescence ratio of mito-roGFP2-Grx1 in second instars of dPTPMT1 RNAi (Figure 4G). We also detected a decrease in H2O2 levels as measured by mito-roGFP2-Orp1 in second instars of dPTPMT1 RNAi, although this difference was not statistically significant (Figure S3H). These results indicate reduced ROS levels in dPTPMT1 RNAi. The timing of the occurrence of ROS reduction coincided with the failure of tracheal air filling in dPTPMT1 RNAi. Both phenotypes were not present in first instars but were present after molting in second instars. It is likely that the lack of air within the trachea reduced mitochondrial oxidative phosphorylation and consequently decreased ROS generation. Consistent with this hypothesis, ubiquitous dPTPMT1 RNAi adults, dPTPMT1mut1 homozygous, and transheterozygous third instar escapers all showed lowered ATP levels (Figure 4H). We used Pink1B9 adult males, which have been reported to show decreased ATP levels, to validate our ATP assay (Park et al., 2006). Taken together, our findings demonstrate that the lack of dPTPMT1 compromises mitochondrial ROS production in the trachea and reduces ATP levels.
Depletion of the CL Pathway Components Does Not Mimic dPTPMT1 Deficiency
Next, we set out to explore the underlying molecular mechanism by which dPTPMT1 deficiency causes tracheal air-filling failure and immune activation. Because we have demonstrated that the catalytic activity of dPTPMT1 is required to prevent tracheal blackening in dPTPMT1 mutants, we reasoned that dPTPMT1 might act through its known substrates. PTPMT1 has been implicated in CL synthesis in mammals (Figure S4A), acting as a phosphatidylglycerophosphate (PGP) phosphatase (Zhang et al., 2011). It has been found that dPTPMT1 dephosphorylates PGP to phosphatidylglycerol (PG) in vitro (Teh et al., 2013). Furthermore, expression of dPTPMT1 in yeast deficient for the PGP phosphatase, GEP4, rescues decreased viability (Teh et al., 2013), suggesting that PTPMT1's role as a PGP phosphatase may be conserved in the fly.
To determine whether loss of dPTPMT1 causes tracheal phenotypes via the CL pathway, we examined tracheal phenotypes in mutants lacking additional enzymes involved in CL synthesis. We identified a Drosophila homolog (CG7718: dPGS1) of mammalian phosphatidylglycerophosphate synthase 1 (PGS1), which acts upstream of PTPMT1 in CL synthesis (Figure S4A) (Kawasaki et al., 1999). dPGS1 shares 43% identity to the amino acid sequence of human PGS1. We also tested CL synthase (CG4774; dCLS) and tafazzin (CG8766; dTaz), which act downstream of PTPMT1 in CL synthesis (Figure S4A) (Xu et al., 2006; Acehan et al., 2011; Dudek, 2017). Using qPCR, we confirmed significant knockdown of dPGS1, dCLS, and dTaz by RNAi (Figure S4B). None of these RNAi lines were lethal when driven by the tracheal-specific driver, btl-GAL4, at 25°C or 30°C (Figures 4I and S4C). Furthermore, none of the surviving adults showed tracheal blackening. We also examined adults with ubiquitous RNAi of dCLS and dTaz driven by Act5C-GAL4 and did not see tracheal blackening or expression of Drs-GFP in the trachea (Figures 4J and S4D). Ubiquitous dPGS1 RNAi was pupal lethal, so we examined third instars and again saw no expression of Drs-GFP in the trachea, no tracheal blackening, and no tracheal air-filling defects (n = 51–56 third instar female larvae). We further examined third instars of dCLS mutants (dCLSe01021/dCLSC01874) (Thibault et al., 2004) and saw no evidence of tracheal blackening (w1118 control: 1% ± 0% versus dCLS mutant 0% ± 0%; n = 2–3 replicates, 22–102 third instar larvae for each repeat), consistent with our findings with dCLS RNAi. Together, these data suggest that the CL biosynthesis pathway is not responsible for the defects observed in the tracheal system with dPTPMT1 deficiency.
Discussion
In this paper, we have revealed a physiological role of dPTPMT1 in the Drosophila tracheal system. Loss of dPTPMT1 causes a sequence of defects in the trachea. In first instar larvae, with tracheal-specific dPTPMT1 RNAi, we have observed mitochondrial clustering just prior to molting. These defects are followed by a failure to fill the trachea with air upon completion of molting and subsequent blackening of the trachea. We have provided evidence suggesting the hypoxic impacts of tracheal air-filling defects on mitochondria, including compromised mitochondrial ROS production and decreased ATP levels. Immune responses are also activated in these mutants, as demonstrated by MP1-dependent melanization and Drs upregulation. Our results show that the lack of dPTPMT1 in tracheal epithelial cells interferes with the ability of epithelial cells to perform air filling and activates immune responses.
To date, the mechanisms of tracheal air filling are poorly defined. Our current understanding suggests that this process involves the removal of liquid from the lumen through active and passive ion transport across epithelial cell membranes and the production of gas in the lumen through the dehydration of bicarbonate ions (Förster and Woods, 2013). Mitochondria play a key role in maintaining homeostasis of ions and metabolites and generating ATP to power ion exchanges. Mitochondrial metabolism and oxidative phosphorylation require a series of biochemical reactions within the mitochondria. One possibility is that dPTPMT1 depletion causes a defect in a specific biochemical reaction in mitochondria, which subsequently impedes the ability of tracheal epithelial cells to conduct air filling. Interestingly, ablation of the carnitine acylcarnitine translocase, COLT, results in failure of air filling at the time of hatching, likely due to an energy deficiency from impaired fatty-acid oxidation (Oey et al., 2005; Hartenstein et al., 1997). It has been shown that loss of PTPMT1 in mammalian cell models causes decreased mitochondrial respiration, which in some cell types is accompanied by decreased ATP levels (Shen et al., 2011; Zhang et al., 2011; Yu et al., 2013; Zheng et al., 2018). In our fly models, we have observed reduced mitochondrial ROS production and decreased ATP levels (Figures 4G and 4H). The impairments in second instars appear to be a consequence of failed tracheal air filling; however, the lack of dPTPMT1 may also affect specific metabolic reactions that cause the mitochondrial aggregation observed in first instars prior to molting. It should be noted that general mitochondrial impairments are unlikely to be responsible for the sensitivity of tracheal epithelial cells to the loss of dPTPMT1, as we have not seen substantial tracheal blackening and Drs-GFP expression in dMIC60 mutants (Tsai et al., 2017, 2018).
Although we speculate that the fluid detected within the lumen of affected tracheal cells is molting fluid, it is also possible that it is hemolymph that has infiltrated into the trachea as a result of disruptions in the integrity of the tracheal epithelium. Abnormal tracheal cell-cell junctions trigger tracheal air-filling failure owing to loss of the tracheal paracellular barrier (Ile et al., 2012). Additionally, deformations in the tracheal cuticle and taenidia have been connected with defects in tracheal air filling (Skouloudaki et al., 2019; Scholl et al., 2019; Jaspers et al., 2014; Yue et al., 2019; Rosa et al., 2018; Swanson et al., 2009). Light microscopy reveals no gross morphological defects in the taenidia of dPTPMT1-depleted trachea (Figure 4D), whereas TEM images show some abnormally formed taenidia (Figures S2A and S2D). However, it is difficult to assess the nature of these abnormalities owing to obliquities in sectioning, and this remains a possible explanation for air-filling failure in dPTPMT1 depletion.
Intriguingly, in mammals, one of PTPMT1's enzymatic products, PG, is the second most abundant phospholipid within lung surfactant—a mixture of lipids and proteins that reduces surface tension in the lung and prevents alveolar collapse (Agassandian and Mallampalli, 2013). PG is produced in the mitochondria and endoplasmic reticulum (Schlame et al., 1986; Agassandian and Mallampalli, 2013) and has critical functions in immunity in the lung surfactant (Kuronuma et al., 2009; Numata et al., 2010, 2012; Kandasamy et al., 2011). PTPMT1 may act in these organelles to generate PG for surfactant (Teh et al., 2013). Although there is currently no direct evidence that flies produce surfactant in the tracheal lumen (Förster and Woods, 2013), impairments in surfactant production could explain both the air-filling defects and immune activation demonstrated in dPTPMT1 mutants. However, in our study, RNAi knockdown of dPGS1, which acts upstream of dPTPMT1 in the synthesis of PG, does not phenocopy dPTPMT1 RNAi, suggesting that reduction of PG synthesis is not the reason for the activation of immune responses in mutant flies.
In addition to air-filling defects, we have observed blackening of the trachea following dPTPMT1 RNAi. Tracheal blackening in ubiquitous dPTPMT1 RNAi adults is partially blocked by knockdown of MP1, a known activator of PPO, which catalyzes melanization (Tang et al., 2006, 2008). Interestingly, tracheal-specific knockdown of MP1 does not block tracheal blackening in dPTPMT1 RNAi larvae, suggesting that melanization of the trachea is not cell autonomous but instead likely the result of melanization being activated by the hemolymph. Alternatively, this result could indicate that MP1 plays a dispensable role in the activation of tracheal-specific melanization, consistent with recent reports of its more redundant role in systemic immunity (Dudzic et al., 2019).
Notably, tracheal blackening occurs after the failure of gas filling in tracheal-specific dPTPMT1 RNAi second instars. Tracheal blackening following failure of tracheal air filling has been reported elsewhere, although the mechanistic basis for this effect has not be elucidated (Fisk and Thummel, 1998; Xu et al., 2019). Although in our study tracheal epithelial cells show no evidence of apoptosis (Figure S3F), the possibility remains that these cells are undergoing necrosis, either prior to the occurrence of air-filling defects or following these defects as a result of hypoxia. Importantly, necrotic cells have been shown to activate innate immune pathways, including melanization and AMP expression, likely through damage-associated molecular patterns (DAMPs) (Chew et al., 2004; Link et al., 2007; Obata et al., 2014; Shaukat et al., 2015). Hypoxia may also directly activate immune responses (Abdelsadik, 2012; Bandarra et al., 2014). Flies grown in hypoxic conditions have been reported to activate AMP expression in the trachea, including Drs (Abdelsadik, 2012). Additionally, mitochondrial stress has been shown to lead to the release of mitochondrial DAMPs and the activation of innate immune responses (Nakahira et al., 2011; Zhou et al., 2011; Rongvaux et al., 2014; White et al., 2014; West et al., 2015). These are a few explanations for the melanization and Drs elevation observed in the trachea, although our current study does not rule out a role for pathogens in the induction of these immune responses.
Our work using a Drosophila model of dPTPMT1 depletion adds to our understanding of the importance of dPTPMT1 in sustaining cellular and organismal functions. Mitochondrial activities mediated by dPTPMT1 likely determine the ability of tracheal epithelial cells to support air filling, which in turn enables mitochondrial oxidative phosphorylation and all essential cellular activities. Our dPTPMT1 mutants and transgenes generated here will provide a useful toolkit to dissect the tissue-specific functions of dPTPMT1 and may help us elucidate additional roles for this phosphatase in mitochondrial biology.
Limitations of the Study
As mentioned in the text, tracheal sectioning for TEM was performed transversely, and thus obliquity in the sections limits our ability to make conclusions about defects in the taenidial ultrastructure. Although we described a number of tracheal phenotypes resulting from dPTPMT1 deficiency, the molecular mechanisms responsible for their manifestations remain undefined.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Xinnan Wang at xinnanw@stanford.edu.
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate/analyze datasets/code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the Department of Defense (PR150380, X.W.), Stanford Gabilan and McCormick Fellowship (X.W.), the Shurl and Kay Curci Foundation (X.W.), the Graduate Research Fellowship Program of the National Science Foundation (A.M.P.), and in part National Center for Research Resources (NCRR) (ARRA, 1S10RR026780-01). The contents of this work do not necessarily represent the official views of the NCRR or the National Institutes of Health. We thank Drs. Bingwei Lu, Liqun Luo, and Clarissa Cheney for providing flies; Drs. Melissa Harrison, Kate O'Connor-Giles, and Jill Wildonger for providing CRISPR plasmids; and Dr. John Perrino and the Stanford Cell Science Imaging EM Facility for support with TEM. We also thank Drs. Margaret Fuller, Richard Reimer, William Talbot, Vafa Bayat, Karen Mruk, Atossa Shaltouki, Pei-I Tsai, Chung-Han Hsieh, Roeland Vanhauwaert, Vinita Bharat, Li Li, Arnaldo Carreira-Rosario, Oguz Kanca, Catherine Baker, Jesse Isaacman-Beck, James Purzner, Susanna Brantley, and Cameron Berry, as well as Todd Galitz, Benjamin Bolival, Xue Yang, Teni Anbarchian, Julia Wucherpfennig, and Ashley Gonzalez for their advice and support.
Author Contributions
A.M.P. designed and performed the experiments, analyzed the results, and wrote the manuscript. M.J.K. assisted with the CRISPR screen. X.W. supervised the project, designed the experiments, and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101285.
Supplemental Information
References
- Abdelsadik A.M.K. Christian-Albrechts Universität Kiel; 2012. Hypoxia Induces Processes Related to Inflammation and Remodelling in the Airways of the Fruit Fly Drosophila melanogaster. [Google Scholar]
- Acehan D., Malhotra A., Xu Y., Ren M., Stokes D.L., Schlame M. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 2011;100:2184–2192. doi: 10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agassandian M., Mallampalli R.K. Surfactant phospholipid metabolism. Biochim. Biophys. Acta. 2013;1831:612–625. doi: 10.1016/j.bbalip.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht S.C., Barata A.G., Großhans J., Teleman A.A., Dick T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011;14:819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Bandarra D., Biddlestone J., Mudie S., Muller H.A., Rocha S. Hypoxia activates IKK–NF-κB and the immune response in Drosophila melanogaster. Biosci. Rep. 2014;34:e00127. doi: 10.1042/BSR20140095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barata A.G., Dick T.P. In vivo imaging of H2O2 production in Drosophila. Methods Enzymol. 2013;526:61–82. doi: 10.1016/B978-0-12-405883-5.00004-1. Elsevier. [DOI] [PubMed] [Google Scholar]
- Bergman P., Esfahani S.S., Engström Y. Drosophila as a model for human diseases—focus on innate immunity in barrier epithelia. Curr. Top. Dev. Biol. 2017;121:29–81. doi: 10.1016/bs.ctdb.2016.07.002. Elsevier. [DOI] [PubMed] [Google Scholar]
- Chew S.K., Akdemir F., Chen P., Lu W.-J., Mills K., Daish T., Kumar S., Rodriguez A., Abrams J.M. The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev. Cell. 2004;7:897–907. doi: 10.1016/j.devcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Dietzl G., Chen D., Schnorrer F., Su K.-C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Dooley C.T., Dore T.M., Hanson G.T., Jackson W.C., Remington S.J., Tsien R.Y. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 2017;5:90. doi: 10.3389/fcell.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudzic J.P., Hanson M.A., Iatsenko I., Kondo S., Lemaitre B. More than black or white: melanization and Toll share regulatory serine proteases in Drosophila. Cell Rep. 2019;27:1050–1061.e3. doi: 10.1016/j.celrep.2019.03.101. [DOI] [PubMed] [Google Scholar]
- Ferrandon D., Jung A., Criqui M.C., Lemaitre B., Uttenweiler-Joseph S., Michaut L., Reichhart J.M., Hoffmann J. A drosomycin–GFP reporter transgene reveals a local immune response in Drosophila that is not dependent on the Toll pathway. EMBO J. 1998;17:1217–1227. doi: 10.1093/emboj/17.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk G.J., Thummel C.S. The DHR78 nuclear receptor is required for ecdysteroid signaling during the onset of Drosophila metamorphosis. Cell. 1998;93:543–555. doi: 10.1016/s0092-8674(00)81184-8. [DOI] [PubMed] [Google Scholar]
- Förster T.D., Woods H.A. Mechanisms of tracheal filling in insects. Biol. Rev. 2013;88:1–14. doi: 10.1111/j.1469-185X.2012.00233.x. [DOI] [PubMed] [Google Scholar]
- Gratz S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., O’Connor-Giles K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz S.J., Rubinstein C.D., Harrison M.M., Wildonger J., O'Connor-Giles K.M. CRISPR-Cas9 genome editing in Drosophila. Curr. Protoc. Mol. Biol. 2015;111:31.32.1–31.2.20. doi: 10.1002/0471142727.mb3102s111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutscher M., Pauleau A.-L., Marty L., Brach T., Wabnitz G.H., Samstag Y., Meyer A.J., Dick T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods. 2008;5:553. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- Gutscher M., Sobotta M.C., Wabnitz G.H., Ballikaya S., Meyer A.J., Samstag Y., Dick T.P. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 2009;284:31532–31540. doi: 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson G.T., Aggeler R., Oglesbee D., Cannon M., Capaldi R.A., Tsien R.Y., Remington S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Hartenstein K., Sinha P., Mishra A., Schenkel H., Török I., Mechler B.M. The congested-like tracheae gene of Drosophila melanogaster encodes a member of the mitochondrial carrier family required for gas-filling of the tracheal system and expansion of the wings after eclosion. Genetics. 1997;147:1755–1768. doi: 10.1093/genetics/147.4.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ile K.E., Tripathy R., Goldfinger V., Renault A.D. Wunen, a Drosophila lipid phosphate phosphatase, is required for septate junction-mediated barrier function. Development. 2012;139:2535–2546. doi: 10.1242/dev.077289. [DOI] [PubMed] [Google Scholar]
- Jaspers M.H., Pflanz R., Riedel D., Kawelke S., Feussner I., Schuh R. The fatty acyl-CoA reductase Waterproof mediates airway clearance in Drosophila. Dev. Biol. 2014;385:23–31. doi: 10.1016/j.ydbio.2013.10.022. [DOI] [PubMed] [Google Scholar]
- Kandasamy P., Zarini S., Chan E.D., Leslie C.C., Murphy R.C., Voelker D.R. Pulmonary surfactant phosphatidylglycerol inhibits Mycoplasma pneumoniae-stimulated eicosanoid production from human and mouse macrophages. J. Biol. Chem. 2011;286:7841–7853. doi: 10.1074/jbc.M110.170241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K., Kuge O., Chang S.C., Heacock P.N., Rho M., Suzuki K., Nishijima M., Dowhan W. Isolation of a chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. The Journal of biological chemistry. 1999;274:1828–1834. doi: 10.1074/jbc.274.3.1828. [DOI] [PubMed] [Google Scholar]
- Kim D.-H., Kim Y.-J., Adams M.E. Endocrine regulation of airway clearance in Drosophila. Proc. Natl. Acad. Sci. U S A. 2018;115:1535–1540. doi: 10.1073/pnas.1717257115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuronuma K., Mitsuzawa H., Takeda K., Nishitani C., Chan E.D., Kuroki Y., Nakamura M., Voelker D.R. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J. Biol. Chem. 2009;284:25488–25500. doi: 10.1074/jbc.M109.040832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link N., Chen P., Lu W.-J., Pogue K., Chuong A., Mata M., Checketts J., Abrams J.M. A collective form of cell death requires homeodomain interacting protein kinase. J. Cell Biol. 2007;178:567–574. doi: 10.1083/jcb.200702125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak V.I. Glutathione homeostasis and functions: potential targets for medical interventions. J. Amino Acids. 2012;2012:736837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M., Pitsouli C., Villalta C., Celniker S.E., Perrimon N. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 2008;40:476. doi: 10.1038/ng.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount S.M. Sequence similarity. Nature. 1987;325:487. doi: 10.1038/325487c0. [DOI] [PubMed] [Google Scholar]
- Nakahira K., Haspel J.A., Rathinam V.A., Lee S.-J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12:222. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A.K., Ryu J.H., Jin Y.N., Roberts L.D., Dejam A., Gerszten R.E., Peterson R.T. PTPMT1 inhibition lowers glucose through succinate dehydrogenase phosphorylation. Cell Rep. 2015;10:694–701. doi: 10.1016/j.celrep.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi N.M., Lanning N.J., Westrate L.M., Mackeigan J.P. Downregulation of the mitochondrial phosphatase PTPMT1 is sufficient to promote cancer cell death. PLoS One. 2013;8:e53803. doi: 10.1371/journal.pone.0053803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata M., Chu H.W., Dakhama A., Voelker D.R. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus–induced inflammation and infection. Proc. Natl. Acad. Sci. U S A. 2010;107:320–325. doi: 10.1073/pnas.0909361107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata M., Kandasamy P., Nagashima Y., Posey J., Hartshorn K., Woodland D., Voelker D.R. Phosphatidylglycerol suppresses influenza A virus infection. Am. J. Respir. Cell Mol. Biol. 2012;46:479–487. doi: 10.1165/rcmb.2011-0194OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata F., Kuranaga E., Tomioka K., Ming M., Takeishi A., Chen C.-H., Soga T., Miura M. Necrosis-driven systemic immune response alters SAM metabolism through the FOXO-GNMT axis. Cell Rep. 2014;7:821–833. doi: 10.1016/j.celrep.2014.03.046. [DOI] [PubMed] [Google Scholar]
- Oey N.A., Ijlst L., van Roermund C.W., Wijburg F.A., Wanders R.J. dif-1 and colt, both implicated in early embryonic development, encode carnitine acylcarnitine translocase. Mol. Genet. Metab. 2005;85:121–124. doi: 10.1016/j.ymgme.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Pagliarini D.J., Dixon J.E. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem. Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Pagliarini D.J., Wiley S.E., Kimple M.E., Dixon J.R., Kelly P., Worby C.A., Casey P.J., Dixon J.E. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic β cells. Mol. Cell. 2005;19:197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Pagliarini D.J., Worby C.A., Dixon J.E. A PTEN-like phosphatase with a novel substrate specificity. J. Biol. Chem. 2004;279:38590–38596. doi: 10.1074/jbc.M404959200. [DOI] [PubMed] [Google Scholar]
- Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.-M. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Park Y., Filippov V., Gill S.S., Adams M.E. Deletion of the ecdysis-triggering hormone gene leads to lethal ecdysis deficiency. Development. 2002;129:493–503. doi: 10.1242/dev.129.2.493. [DOI] [PubMed] [Google Scholar]
- Perkins L.A., Holderbaum L., Tao R., Hu Y., Sopko R., McCall K., Yang-Zhou D., Flockhart I., Binari R., Shim H.-S. The transgenic RNAi project at Harvard Medical School: resources and validation. Genetics. 2015;201:843–852. doi: 10.1534/genetics.115.180208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A., Jackson R., Harman C.C., Li T., West A.P., de Zoete M.R., Wu Y., Yordy B., Lakhani S.A., Kuan C.-Y. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa J.B., Metzstein M.M., Ghabrial A.S. An Ichor-dependent apical extracellular matrix regulates seamless tube shape and integrity. PLoS Genet. 2018;14:e1007146. doi: 10.1371/journal.pgen.1007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M., Rüstow B., Kunze D., Rabe H., Reichmann G. Phosphatidylglycerol of rat lung Intracellular sites of formation de novo and acyl species pattern in mitochondria, microsomes and surfactant. Biochem. J. 1986;240:247–252. doi: 10.1042/bj2400247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl A., O'Brien M.J., Chandran R.R., Jiang L. The novel gene apnoia regulates Drosophila tracheal tube size. Dev. Dyn. 2019;248:477–487. doi: 10.1002/dvdy.29. [DOI] [PubMed] [Google Scholar]
- Shaukat Z., Liu D., Gregory S. Sterile inflammation in Drosophila. Mediators Inflamm. 2015;2015:369286. doi: 10.1155/2015/369286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J., Liu X., Yu W.-M., Liu J., Nibbelink M.G., Guo C., Finkel T., Qu C.-K. A critical role of mitochondrial phosphatase Ptpmt1 in embryogenesis reveals a mitochondrial metabolic stress-induced differentiation checkpoint in embryonic stem cells. Mol. Cell. Biol. 2011;31:4902–4916. doi: 10.1128/MCB.05629-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouloudaki K., Papadopoulos D.K., Tomancak P., Knust E. The apical protein Apnoia interacts with Crumbs to regulate tracheal growth and inflation. PLoS Genet. 2019;15:e1007852. doi: 10.1371/journal.pgen.1007852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson L.E., Yu M., Nelson K.S., Laprise P., Tepass U., Beitel G.J. Drosophila convoluted/dALS is an essential gene required for tracheal tube morphogenesis and apical matrix organization. Genetics. 2009;181:1281–1290. doi: 10.1534/genetics.108.099531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. Regulation and function of the melanization reaction in Drosophila. Fly. 2009;3:105–111. doi: 10.4161/fly.3.1.7747. [DOI] [PubMed] [Google Scholar]
- Tang H., Kambris Z., Lemaitre B., Hashimoto C. Two proteases defining a melanization cascade in the immune system of Drosophila. J. Biol. Chem. 2006;281:28097–28104. doi: 10.1074/jbc.M601642200. [DOI] [PubMed] [Google Scholar]
- Tang H., Kambris Z., Lemaitre B., Hashimoto C. A serpin that regulates immune melanization in the respiratory system of Drosophila. Dev. Cell. 2008;15:617–626. doi: 10.1016/j.devcel.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh P.G., Chen M.J., Engel J.L., Worby C.A., Manning G., Dixon J.E., Zhang J. Identification of a mammalian-type phosphatidylglycerophosphate phosphatase in the Eubacterium Rhodopirellula baltica. J. Biol. Chem. 2013;288:5176–5185. doi: 10.1074/jbc.M112.413617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault S.T., Singer M.A., Miyazaki W.Y., Milash B., Dompe N.A., Singh C.M., Buchholz R., Demsky M., Fawcett R., Francis-Lang H.L. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat. Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- Tsai P.-I., Lin C.-H., Hsieh C.-H., Papakyrikos A.M., Kim M.J., Napolioni V., Schoor C., Couthouis J., Wu R.-M., Wszolek Z.K. PINK1 phosphorylates MIC60/Mitofilin to control structural plasticity of mitochondrial crista junctions. Mol. Cell. 2018;69:744–756.e6. doi: 10.1016/j.molcel.2018.01.026. [DOI] [PubMed] [Google Scholar]
- Tsai P.-I., Papakyrikos A.M., Hsieh C.-H., Wang X. Drosophila MIC60/mitofilin conducts dual roles in mitochondrial motility and crista structure. Mol. Biol. Cell. 2017;28:3471–3479. doi: 10.1091/mbc.E17-03-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzou P., Ohresser S., Ferrandon D., Capovilla M., Reichhart J.-M., Lemaitre B., Hoffmann J.A., Imler J.-L. Tissue-specific inducible expression of antimicrobial peptide genes in Drosophila surface epithelia. Immunity. 2000;13:737–748. doi: 10.1016/s1074-7613(00)00072-8. [DOI] [PubMed] [Google Scholar]
- Wagner C., Isermann K., Roeder T. Infection induces a survival program and local remodeling in the airway epithelium of the fly. FASEB J. 2009;23:2045–2054. doi: 10.1096/fj.08-114223. [DOI] [PubMed] [Google Scholar]
- West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., Bestwick M., Duguay B.A., Raimundo N., MacDuff D.A. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M.J., McArthur K., Metcalf D., Lane R.M., Cambier J.C., Herold M.J., van Delft M.F., Bedoui S., Lessene G., Ritchie M.E. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J., Engel J.L., Zhang J., Chen M.J., Manning G., Dixon J.E. Structural and functional analysis of PTPMT1, a phosphatase required for cardiolipin synthesis. Proc. Natl. Acad. Sci. U S A. 2011;108:11860–11865. doi: 10.1073/pnas.1109290108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q.-Y., Deng P., Mu L.-L., Fu K.-Y., Guo W.-C., Li G.-Q. Silencing Taiman impairs larval development in Leptinotarsa decemlineata. Pestic. Biochem. Physiol. 2019;160:30–39. doi: 10.1016/j.pestbp.2019.06.013. [DOI] [PubMed] [Google Scholar]
- Xu Y., Condell M., Plesken H., Edelman-Novemsky I., Ma J., Ren M., Schlame M. A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U S A. 2006;103:11584–11588. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.-M., Liu X., Shen J., Jovanovic O., Pohl E.E., Gerson S.L., Finkel T., Broxmeyer H.E., Qu C.-K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12:62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X.Z., Li D., Lv J., Liu K., Chen J., Zhang W.Q. Involvement of mind the gap in the organization of the tracheal apical extracellular matrix in Drosophila and Nilaparvata lugens. Insect Sci. 2019;27:756–770. doi: 10.1111/1744-7917.12699. [DOI] [PubMed] [Google Scholar]
- Zhang J., Guan Z., Murphy A.N., Wiley S.E., Perkins G.A., Worby C.A., Engel J.L., Heacock P., Nguyen O.K., Wang J.H. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Yu W.-M., Shen J., Kang S., Hambardzumyan D., Li J.Y., Shen Y., Kenney A.M., Chen J., Qu C.-K. Mitochondrial oxidation of the carbohydrate fuel is required for neural precursor/stem cell function and postnatal cerebellar development. Sci. Adv. 2018;4:eaat2681. doi: 10.1126/sciadv.aat2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.