

Abstract
Genomic analysis of individuals or organisms is predicated on the availability of high-quality reference and genotype information. With the rapidly dropping costs of high-throughput DNA sequencing, this is becoming readily available for diverse organisms and for increasingly large populations of individuals. Despite these advances, there are still aspects of genome sequencing that remain challenging for existing sequencing methods. This includes the generation of long-range contiguity during genome assembly, identification of structural variants in both germline and somatic tissues, the phasing of haplotypes in diploid organisms and the resolution of genome sequence for organisms derived from complex samples. These types of information are valuable for understanding the role of genome sequence and genetic variation on genome function, and numerous approaches have been developed to address them. Recently, chromosome conformation capture (3C) experiments, such as the Hi-C assay, have emerged as powerful tools to aid in these challenges for genome reconstruction. We will review the current use of Hi-C as a tool for aiding in genome sequencing, addressing the applications, strengths, limitations and potential future directions for the use of 3C data in genome analysis. We argue that unique features of Hi-C experiments make this data type a powerful tool to address challenges in genome sequencing, and that future integration of Hi-C data with alternative sequencing assays will facilitate the continuing revolution in genomic analysis and genome sequencing.
Keywords: chromosome conformation capture, genome assembly, haplotype phasing, structural variants, metagenomics, 3D genome organization
Introduction
DNA sequencing technologies have undergone a revolution since the original draft sequence of the human genome was published (1). This includes rapidly plunging costs of so-called short-read sequencing, as well as dramatic improvements in the length and sequence quality of long-read technologies (2,3). In addition, novel methodological developments in sequencing library construction are allowing existing sequencing platforms to be repurposed to generate higher quality data (4–6). As a result of these technological improvements, generating high-quality sequence data for any individual or organism is now no longer a rate-limiting step in genomic analysis. One of the primary challenges now is to fully reconstruct accurate genome sequence information from an individual or organism. This problem manifests currently in several technical challenges in genomic analysis. Namely, some of the primary limitations in current genome sequencing methods involve scaffolding during genome assembly, haplotype phasing of individuals or reference genomes, identification of structural variants, and deconvolution of genomes in complex samples. Numerous methodological approaches have been developed to address these problems (2,4–6), but one of particular interest has been the use of methods originally designed for analyzing chromosome folding (7), in particular the Hi-C assay (8) and other variants of chromatin conformation capture experiments (9). In this review, we will discuss the use of chromatin conformation capture-based assays to address what we generally term ‘genome reconstruction’. This includes both the underlying basis for why 3D genome structure can inform genome reconstruction and the specific experimental and analytical approaches developed by different groups. We will also discuss important limitations and future applications of these methods.
Chromatin conformation captures critical features for genome reconstruction
Although it is typical to represent genomes as linear sequences of nucleic acids, DNA adopts higher order conformations in all kingdoms of life. Most prokaryote genomes are known to be folded through supercoiling (10) and can adopt additional, more complex configurations (11–15). DNA in eukaryotes is wrapped around histones to form nucleosomes (16) before being compressed into chromatin (17), and can adopt even more complex higher order structures (8,18–22). How 3D genome structure varies across individuals and species and what role it plays in gene regulation in development, disease and evolution have been the major topics in biology. Classical methods for studying chromatin organization in vivo have typically centered on imaging-based methods. An alternative approach focuses on the use of molecular methods, such as the chromosome conformation capture (3C) method (7) and subsequent variants of this assay (23,24), to identify sequences in close 3D space through proximity ligation. With the development of high-throughput sequencing methods, these were generalized into genome-wide assays, such as Hi-C (8) and TCC (25), or genome-wide variants that focused on regions of the genome bound by specific factors, such as ChIA-PET (26), HiChIP (27) and PLAC-seq (28). Hi-C has been a powerful tool to investigate the structure of many genomes, such as human (22), mouse (19), Drosophila (21) and bacteria (11,15), as well as generating data for structural modeling of 3D architecture (29,30).
The Hi-C assay is based on proximity ligation, and the initial steps in the assay are similar to the original 3C method. Cells are first cross-linked by formaldehyde and then digested by a restriction endonuclease. In a 3C experiment, the DNA ends are ligated together and are used as the template for PCR. In current Hi-C-based methods, digested ends are filled with nucleotides, one of which is linked to a biotin moiety, and ends are then ligated, with ligation events occurring preferentially between ends that are spatially adjacent. DNA is then purified, sheared and prepared for paired-end DNA sequencing. The biotin labeling in the end-filling step enables the concentration of fragments that contain ligations using streptavidin beads before library preparation, though sequencing can be performed on samples that have not been enriched by biotin fill in and pull down (31). Paired sequencing reads can then be aligned to the reference genome. The number of distinct reads spanning from one locus to another indicates the interaction frequency between those two loci in the 3D space. The interaction frequencies between any two loci in the genome can be visualized in a 2D genome-wide contact matrix (Figure 1A).
Figure 1.
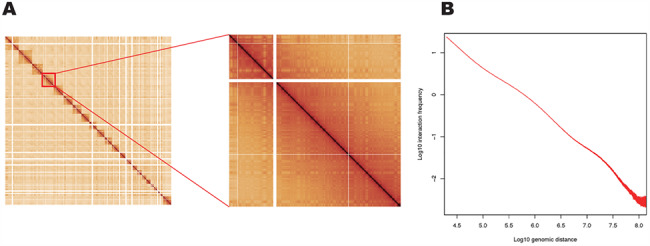
Features of Hi-C data that facilitate genome reconstruction. (A) A whole-genome Hi-C contact matrix for the GM12878 cell line (22) showing chromosome territories (squares along diagonal are individual chromosomes). Intra-chromosomal contacts are higher than inter-chromosomal contacts. (B) Average Hi-C interaction frequency as a function of genomic distance separating interacting bins at a bin size of 10 kb. The plot shows a typical distance-dependent decay curve.
Despite the fact that 3D genome organization is variable between species, cell-types (8,19,22,32), cell-cycle stages (33,34) and even within homogeneous populations (33,35), canonical patterns emerge from the data (36). First, as a result of the fact that chromosomes occupy distinct territories (37) and exhibit distinct spatial preferences in the nucleus (38,39), Hi-C interaction frequency tends to be much higher within the same chromosome. Interactions between different chromosomes, on the contrary, tend to be relatively sparse (Figure 1A). Second, based on the fact that in the in situ Hi-C protocol, genomic DNA is fixed within an intact nucleus, and cross-linking between nuclei is rare. As a result of this principle, single-cell Hi-C methods have been developed by separating nuclei after proximity ligation in bulk (33,40,41). Third, Hi-C interaction frequency displays a distance-dependent decay (Figure 1B) because loci nearby in linear sequence interact more frequently according to principles of polymer physics (42,43). These three principles underlie the utility of Hi-C for genome reconstruction. Specifically, the feature of distance-dependent decay is critical for genome assembly and structural variant analysis; the organization of chromosomes into territories is essential for haplotype phasing and structural variant identification, and the paucity of inter-cellular contacts is critical for the use of Hi-C in metagenomics. These features have thus far been observed to be fundamental features of genome organization across cell types and species and, therefore, allow Hi-C to be widely applied for genome reconstruction.
Genome assembly
One of the fastest growing applications of Hi-C data is in de novo genome assembly. One of the major current challenges in genome assembly is resolving repetitive sequences to generate chromosome scale assemblies. Although sequence read length has increased dramatically in the last decade (2), when genomic repeats are longer than the sequenced reads, the assembly will remain fragmented in contigs due to the ambiguity introduced by repetitive sequences (Figure 2A). The process of connecting and ordering contigs, also called scaffolding, requires linked sequences uniquely assigned to different contigs. Diverse strategies to generate such information have been developed (Table 1). In ranges less than 40 kb, traditional methods such as Illumina mate-pair sequencing (44) and paired-end sequencing of fosmids (45) are being replaced by sequencing technologies with longer reads (46–49), which can close most gaps of this size. In ranges in intervals longer than 100 kb, solutions include paired-end sequencing of bacteria artificial chromosomes (BAC) (50,51), optical mapping (52) and traditional genetic mapping (53). However, these applications have limitations. BAC libraries are laborious to generate and not able to solve gaps longer than 200 kb. Optical maps work better for longer gaps, but currently, they are expensive to generate sufficient data for large genomes. Genetic maps are usually not available for genomes other than human and model organisms.
Figure 2.
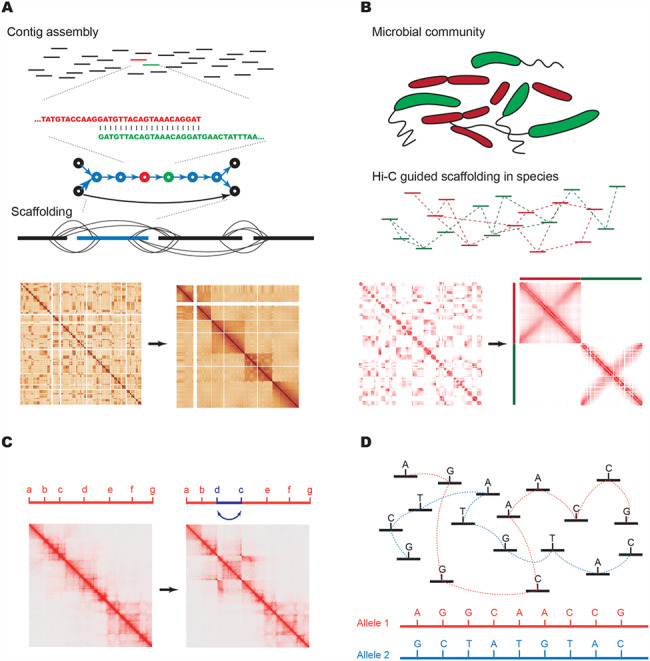
Application of Hi-C in genome reconstruction. (A) In genome assembly, sequence reads are assembled by overlapping unique sequences into contigs. Contigs end when there is no sequence present or when repetitive regions create ambiguity in terms of the order of contigs. Using Hi-C data, contigs can be scaffolded to chromosomal scale. Also shown are examples of Hi-C matrices before scaffolding (left) and after (right). (B) In metagenomics, contigs are first grouped into those derived from the same species, either using Hi-C or alternative methods such as computational binning (represented by red versus green species and contigs). After grouping by species, contigs can be scaffolded into genome scale assemblies. Also shown are Hi-C heat maps from two bacterial species with simulated, ungrouped and unordered contigs (left) and the same sequences after binning and assembly (right). (C) An example of the effect of structural variation on Hi-C heat maps. The heat map on the left is from K562 cells where the region has no Structural Variations (SVs), whereas the heat map on the right is from KBM7 cells where there is a ~1 Mb inversion of the segments from ‘c’ to ‘d’. This leaves a ‘bowtie’ or ‘butterfly’ like pattern in the heat map at the location of the breakpoints. (D) A cartoon example of how haplotypes can be phased according to Hi-C data connecting Single Nucleotide Polymorphisms (SNPs) within same allele. Unphased sequence variants can be linked together by Hi-C reads into two parental haplotypes (colored red and blue).
Table 1. Comparison of several methods used for scaffolding.
| Scaffolding methods | Spanning distance | Cost | Throughput/Contacts per run |
|---|---|---|---|
| lllumina mate pair | 2–30 kb | Low | High |
| Fosmid | 40 kb | Median | Low |
| BAC | 40–200 kb | High | Low |
| Optical mapping | 20–500 kb | High | High |
| Genetic map | Chromosomal | Impractical | Very low |
| Hi-C | 1 kb – 20 Mb | Low | High |
With the ability to profile long-range interactions across whole genomes in high-throughput, Hi-C can inform key aspects of genome assembly. Seminal papers from Kaplan et al. and Burton et al. showed that Hi-C data can be used to perform scaffolding of contigs during genome assembly. Since the publication of these pioneering studies, Hi-C has been used to aid in genome assemblies of goat (48), alligator (54), frog (55), mosquito (56,57), Schmidtea mediterranea (58), barley (59), lettuce (60), durian (61), quinoa (62), wombat (63), opossum (63), raccoon (63), band-tailed pigeon (63) and subterranean clover (64). Unlike mate-pair sequencing and clone-based paired-end sequencing, which have relatively fixed insert sizes, the distance between two ends in Hi-C can vary from 1 kb to many megabases within the same chromosome (8,65,66). This endows Hi-C with the power to theoretically cover any gap that is shorter than a human chromosome. Given the universal properties of Hi-C discussed in the previous section, contigs from the same chromosome can be easily grouped and sorted according to their Hi-C interaction frequency with each other (Figure 2A) (67,68). The cost of Hi-C experiments is generally low, and it has been suggested that de novo assembly of large genomes can cost as little as $1000 (63). In addition, Hi-C requires no specialized equipment and is capable of being performed in any molecular biology laboratory (8,22). Hi-C is also not restricted to model organisms as long as proximity ligation can be carried out in nuclei. In summary, Hi-C is a widely applicable, powerful tool for genome assembly with low cost. As a result, Hi-C is being used as a key assay for genome scaffolding by several large genome assembly consortia, including the Genome 10 k project and the DNA zoo (56,69).
As a general strategy for genome assembly, initial sequencing is usually performed using at least one conventional sequencing method to generate contigs assembled from massive parallel sequencing (Figure 2A). This can be in the form of short-read Illumina sequencing or long-read sequencing. Once this initial assembly is generated, Hi-C data, generated in parallel, can be aligned to the contigs and used to determine the order and orientation of contigs, so-called ‘scaffolding’. According to examples from recent genome projects and simulations, Hi-C requires an input assembly with contig N50 sizes in the range of 36–200 kb to reach a proper genome assembly with chromosomal scale scaffolding, something that is readily achievable with both short-read- and long-read-based contigs (56,57,63,67,68,70). Various computational tools have been developed for Hi-C-based genome assembly. In this review, we will only summarize the major approaches taken by different Hi-C-based assembly algorithms without going in depth into the details of each method. In our view, there are two general approaches that are taken for Hi-C-based genome scaffolding. First are the graph-based approaches. Graph-based methods consider contigs as nodes and Hi-C interactions between contigs as edges in a graph. LACHESIS (67), 3D DNA (56) and SALSA/SALSA2 (70,71) are graph-based approaches. These methods largely differ in how the graphs are constructed and how the final ‘path’ through the graph, representing the order and orientation of contigs, is determined. Graph-based methods are a logical extension of the original assembly, as most modern assemblers use de Bruijn graphs to construct contigs during assembly (72–74). The second general strategy in Hi-C-based assembly is to use probabilistic models. This includes graph-free methods, such as DNA Triangulation (68) and GRAAL (75), and hybrid approaches such as HiRise (76) that constructs and orients a contig–contig linking graph based on a likelihood model. Methods such as SALSA/SALSA2 and 3D-DNA are routinely capable of generating chromosome scale scaffolds from input short- or long-read assemblies (56,57,64,70,71). The primary remaining challenges appear to be scaffolding of short contigs, resolving highly repetitive regions and ensuring proper contig orientation (56,63,70). The recently proposed SALSA2 method actually attempts to merge graph-based Hi-C scaffolding and de Bruijn graph-based assembly by performing Hi-C scaffolding in a manner that is aware of edges in the de Bruijn graph, which in theory should greatly aid in ordering and orienting contigs during assembly (70).
One specific hurdle for Hi-C-based genome assembly, compared with other methods, is that despite the universal existence of distance-dependent decay in Hi-C contact maps, long-range Hi-C interactions also contain signals that result from biological aspects of chromatin organization. For instance, two distant loci in the same topologically associated domain (TAD) (19–21) may have higher interaction frequencies than two loci in adjacent TADs that lie closer in linear proximity along the genome. This appears as fluctuations and rebounds in the distance-dependent decay curve (41,65), which can potentially cause misestimation of the distance between them during assembly. In order to address this issue, the Chicago method was developed (76), in which high-molecular-weight (>500 kb) genomic DNA is extracted and reconstituted in vitro with purified histones and chromatin assembly factors before fixation by formaldehyde. This in vitro sample is then processed through the regular Hi-C protocol. The in vitro assembly of chromatin largely excludes confounding signals from complicated high-order structures of the genome in vivo. However, very-long range and chromosome length contact information is lost, as the procedure for isolating genomic DNA inevitably breaks the longest DNA fragments, and therefore, chromosome length scaffolds remain a challenge. In summary, the development of Hi-C-based experimental procedures and bioinformatic tools has demonstrated that Hi-C is one of the major techniques for genome assembly. Future efforts will likely continue to integrate long-read sequencing, Hi-C and optical maps. Further improvements in sequencing technology and the development of new long-range linkage assays and physical mapping technologies will continue the revolution in this area.
Metagenomics
Microbial communities are essential components of diverse ecosystems. There is growing appreciation for the role that microbial communities play in environments including ‘microbiomes’ that impact human health (77), environmental ecology (78), and biomass degradation and fuel production (79). Therefore, there is a great desire to study the species within microbial communities to better understand how they function both as individuals and as a system. The problem is that relatively few strains of bacteria can be successfully cultured in the laboratory, and specific micro-organisms may be difficult to isolate from large heterogeneous populations of thousands of species (80,81). This has led to efforts to study microbial communities en masse without isolation by examining DNA content from samples containing mixtures of species. Studying microbial genomes in bulk, so-called metagenomics, has provided an attractive way to study microbial communities while avoiding issues related to sample isolation or cultivation.
A classical method for studying microbial identity is by sequencing of the 16S ribosomal RNA genes (82). When coupled with high-throughput sequencing methods, this can facilitate the determination of the types and the abundance of species within a microbial community (83). However, 16S rRNA sequencing has limitations in the complete characterization of microbes. Specifically, 16S rRNA sequencing requires a taxonomic database for species identification, and strain-level differences will not be readily determined. Furthermore, non-chromosomal DNA sources, such as phages or plasmids, will not be resolved. To address these limitations, shotgun sequencing-based methods have been applied to metagenomic samples to fully characterize DNA sequences within a population (84–86). These samples can then be subject to de novo genome assembly. The challenge is that the regions of the genome conserved across species would be considered as ‘repetitive’, causing fragmented assemblies. As a result, it can be unclear what contigs within the assembly correspond to which species. Assigning sequences to individual species is difficult and sometimes requires assistance from comparative genomic analysis using known genome and protein sequences (87–89). However, with little knowledge of karyotypes and species composition of samples, the ability to deconvolute genomic samples from a mixture of genomes largely depends on the quality of initial assembly (87). Therefore, there is a need for methods that can help to group contigs together within species so that the nature of microbes within a community can be better understood.
Given the fact that genomes are fixed, digested and ligated within individual cells in Hi-C experiments, it is intuitive to apply this technology to metagenomic studies for the deconvolution of individual genomes. After initial studies demonstrating the ability to perform genome-wide chromatin conformation capture experiments in bacteria (11,90), Burton et al., Beitel et al. and Mourbouty et al. showed that proximity ligation can aid in clustering scaffolds into species in artificial microbial communities (31,91,92) as well as on environmental samples such as river sediment (31). Subsequent studies have applied this approach to metagenomics samples from a variety of sources, including cow rumen (93,94), mouse (12) and human (95) fecal samples and Belgian lambic style beer (96). The most common approach in these studies is to generate genome assemblies from short-read whole genome sequencing experiments, though some studies have also used long-read (PacBio) sequencing (94). The contigs assembled from these species are then clustered by species using contact frequencies from the Hi-C data. Once contigs are grouped by species, scaffolding can be performed to generate complete genome assemblies (Figure 2B) (31,92). Of note, Marbouty et al. have also shown that the parallel generation of whole genome sequencing libraries may not be necessary and that the Hi-C libraries themselves may be used for initial assembly. This may in part be affected by the specific experimental strategy applied, as the Marbouty study did not enrich for chimeric fragments as in a typical Hi-C experiment (thus the name, meta3C), and therefore, most of the sequenced reads represent typical shotgun sequencing and not chimeric reads. One potential concern for such an approach is the generation of chimeric contigs as a result of ligation events in the sample, but the frequency of such chimeras appears to be low (31).
While Hi-C can be used to group contigs by species in metagenomic sequencing studies, it is not the only method for doing so. A purely bioinformatic approach named metagenomic binning can also be applied to assign contigs to species (97–99). Binning works by clustering contigs according to metrics such as tetranucleotide frequency (100), gene covariance across populations (101), identified 16S rRNA sequence (98) or differential protein alignment results against reference databases (102). Metagenomic binning is particularly attractive as it requires no additional experimentation beyond the initial sequencing and assembly. Several studies have performed the direct comparisons of Hi-C and metagenomic binning for contig clustering that have yielded mixed results in terms of performance (93–95,103). In a head-to-head comparison on a sample from cow rumen, metagenomic binning using the MetaBAT2 tool resulted in more complete assemblies in comparison with Hi-C-based contig clustering based on the ProxiMeta algorithm (93). In a different cow rumen study using both short-read (Illumina) and long-read (PacBio) derived contigs, Hi-C-based clustering using ProxiMeta resulted in a similar number of genomes clusters compared with metagenomic binning from MetaBat using short-read-based assemblies, but Hi-C generated substantially more genome clusters than metagenomic binning when using long-read-based contigs (94). In contrast, two studies of metagenomic assembly on a human fecal sample (using the same underlying data) both demonstrated that Hi-C-based clustering using either ProxiMeta or the bin3C algorithm resulted in more genome clusters and more complete genomes than metagenomic binning using MaxBin, with evidence that bin3C outperforms ProxiMeta in terms of Hi-C-based clustering (95,103). Notably, these studies both showed that Hi-C-based clustering resulted in substantially lower levels of contamination that binning from MaxBin.
When considered in comparison with purely computational methods such as metagenomic binning, it is important to ask what are the potential advantages of using Hi-C for metagenomic analysis. There are likely several key aspects where Hi-C can still provide critical information beyond that provided by metagenomic binning. First, all of the direct comparisons of Hi-C and metagenomic binning demonstrate that Hi-C derived clusters show considerably the greater representation of mobile genomic elements, such as phages and plasmids (12,94). Second, Hi-C defined clusters and metagenomic binning-defined clusters were not redundant, indicating that different species in the population may be preferentially targeted by each approach (93,94). In this regard, combining binning and Hi-C-based clustering may prove to be an attractive approach in the future. Third, metagenomic binning only groups contigs by species, whereas Hi-C can aid in scaffolding of contigs into complete genome sequences (12,31,92). This can be particularly important in bacteria, as functionally related genes can be grouped in regions of the genome, and understanding long-range gene order and organization can potentially provide insights into function. Finally, metagenomic binning relies on conserved features that differ between species, and as a result, it will struggle with strain-level identification, or any other aspect that causes a divergence from conservation. A particularly notable example of this came from a study using Hi-C for metagenomic analysis of a beer sample (96). This study actually identified a novel yeast hybrid. Identifying such hybrid species through metagenomic binning would be highly unlikely as binning uses features shared within species for contig clustering.
In the future, it will be critical to continually re-examine the use of Hi-C data for metagenomic analysis with the further development of long-read sequencing. Long-read sequencing (104) can provide additional assistance in metagenomic analysis by generating longer contigs and scaffolds. The weakness of long-read-based assembly lies in its cost and that it requires high coverage (20–30×) to correct and assemble reads with each other; therefore, covering thousands of species in a metagenomic sample with variable species abundance is extremely expensive. However, the cost of long-read sequencing continues to drop while the read lengths continues to increase, with reports of nanopore sequencing reads now exceeding 1 million base pairs (105). In this sense, long-reads will likely someday approach the length of many bacterial chromosomes. We still believe that Hi-C can provide critical information in this context in metagenomic analysis, in particular with regards to associating extra-chromosomal elements or delineating novel species with multiple chromosomes. Our belief is the approaches that combine complementary methods will ultimately be the most fruitful for future metagenomic analysis.
Structural variation
One of the more striking features in Hi-C contact maps is the impact of structural variants, including translocations, inversions, deletions and insertions. This phenomenon was noted by multiple groups in early 4C or Hi-C studies (67,106–108). Recent efforts have sought to leverage these features to identify structural variants de novo from Hi-C data in an unbiased manner (109–112).
Structural variation has a notable effect on Hi-C contact maps due to some of the basic features that define chromatin organization (Figure 2C). First, as the distance-dependent-decay of Hi-C contact frequency follows an exponential or power law distribution, the existence of a structural variant will generate contacts with orders of magnitude higher frequency relative to the genome-wide expectation in the vicinity of the breakpoint site. When this occurs between the regions of the genome that would be expected to have low contact frequency, such as long-range interactions in cis or trans contacts between different chromosomes, this leaves a signature of dramatically elevated contact frequency in the vicinity of the SV breakpoint. Second, as chromosomes are generally organized into territories, the elevated interaction frequencies typically extend long distances from the breakpoint site, including extending the entire length of the rearranged chromosome.
Several tools have been developed to identify these signatures (110–113). The common feature they share is that they search for dramatically elevated contact frequencies relative to what would be expected from the genome-wide background. We have previously developed an approach that first uses probabilistic models of Hi-C interaction frequencies to assign a probability of observing a given interaction based on genome-wide background interaction frequencies, and then uses a 2D peak finding algorithm to identify regions showing elevated interaction frequencies (112). Another approach scans the data using a local sub-matrix and models’ interaction frequencies as Z-scores to find elevated interactions (111). An alternative approach uses binary segmentation algorithms to identify significant changes in interaction frequency indicative of translocation breakpoints (110). Finally, the recently described HiNT method can also use chimeric read information to detect the precise breakpoint sites at base pair resolution (113). It is currently unclear how these methods perform relative to each other. Additionally, several tools have been developed to identify copy number changes in Hi-C data (109,110,113–115). As Hi-C is based on high throughput sequencing, regions of the genome with elevated copy number tend to show elevated raw coverage similar to Whole Genome Sequencing (WGS) data. These methods take advantage of this feature to identify gains or losses in copy. Hi-C data has an additional challenge compared with conventional WGS data that experimental biases are introduced due to the use of restriction enzymes (116). The biases introduced by restriction enzyme digestion lead to more variable patterns of coverage in Hi-C data relative to WGS data and, therefore, must be corrected (109,110,113–115). Once the corrected coverage profiles are generated, the Hi-C data can then be segmented into regions of distinct copy number using methods similar to what have been used previously for copy number estimation from array or WGS data (109,110,113–115). In our view, if used in isolation, WGS is still likely better than Hi-C at identifying copy number changes. However, these tools can allow researchers to extract copy number changes from existing Hi-C data sets.
Hi-C has certain advantages and disadvantages with regards to identifying SVs compared with alternative methods. In terms of advantages, as SVs leave elevated contact frequencies surrounding the breakpoint site for up to megabases away from the break, Hi-C can identify SVs that occur in repetitive or unmappable regions of the genome (112). Our previous work identified some SVs where the breakpoints appear to map to centromeric regions of the genome (112), something that would be nearly undetectable with existing short-read sequencing methods. Likewise, the fact that the elevated contact frequencies can extend great distances from SV breakpoints also provides insights into complex SVs. We had shown that in the case of certain complex SVs, Hi-C shows elevated contact frequencies between the regions that are not directly joined by the SVs but are instead indirectly linked by additional sequences (112). As a result, this can provide insights into which SVs may be shared on a single allele, and has the potential in the future to be useful for the SV phasing or reconstruction of complex chromosomal rearrangements. Hi-C does have certain clear disadvantages in terms of identifying SVs as well. Namely, as the feature identified by these methods is elevated contact frequency relative to background, this becomes harder to identify when the background expected interaction frequency goes up. As a result, we have seen that Hi-C struggles to identify SVs smaller than 1 Mb in the genome (112). Likewise, Hi-C has the potential to identify false positive events where long-range interactions may be dramatically elevated due to specific properties of chromosome organization. Recent studies in olfactory neurons have demonstrated that these form hubs of trans interactions that may be identified as SVs if such methods are applied (117). In our experience, however, these kinds of events are not frequent in Hi-C data, but researchers should be aware of the potential for false positives due to such uniquely strong chromatin contacts.
One additional advantage of Hi-C data for SV calling that we believe represents an exciting area for future exploration is the use of Hi-C to identify SVs in formaldehyde fixed paraffin embedded (FFPE) samples (111). FFPE is the standard method for archival of clinical specimens for histological analysis. There is great interest in using such archived samples for a number of molecular and biochemical assays. One challenge in applying sequencing-based assays to such samples is that FFPE can degrade DNA, rendering it no longer suitable for long-read-based sequencing. As Hi-C normally starts with a formaldehyde fixation step, it would appear that this storage method may be amenable for Hi-C experiments. A recent report has demonstrated the first use of Hi-C on FFPE samples and was able to identify numerous known translocations in such samples (111). This would represent an exciting opportunity to profile SVs in rare or hard to obtain samples or in cases where the annotation of samples in FFPE blocks using traditional histological methods may have already been applied. This represents a potential unique space for Hi-C to excel as an SV finding method, as using FFPE for many genomic assays has proved challenging.
Haplotype phasing
Another application for Hi-C in genome reconstruction is for haplotype phasing, either on a chromosome wide scale (66) or over focal regions of the genome (118,119). Humans like all diploid organisms inherit half of their genetic material from each parent. As a result, we also inherit half of our genetic variation from each parent. The problem of haplotype phasing is to resolve which sets of variants along a chromosome were inherited from each parent for a given individual. The ability to genotype variants has become cost effective with the development of SNP microarrays and whole genome sequencing. The challenge of haplotype phasing is to then determine which genotypes were co-inherited from each parent. Phasing haplotypes in the genome remains a strong need in genetic research. Studying haplotypes of human individuals provides insights into population structure and evolutionary history (5,120,121). Haplotype-solved cancer genomes can reveal the mechanisms of haplotype-specific activation of oncogenes (122).
Haplotype phasing is typically accomplished using one of two approaches: statistical phasing or experimental phasing. Statistical phasing relies on determining which local haplotypes occur frequently in the population due to linkage disequilibrium. Statistical phasing, therefore, does not require any additional experiments beyond genotype information, but it does require large sets of population genetic information. Statistical phasing is highly accurate locally, but the classical limitation of statistical phasing is that it will inevitably contain numerous errors (‘switch errors’) often at recombination hotspots due to the fact that linkage disequilibrium will only account for co-inheritance of alleles that are proximal along the chromosome. Despite this, improving methods for statistical phasing is generating longer and longer haplotypes, in particular when phasing large numbers of individuals from closely related populations (123).
The alternative general approach for phasing is experimental phasing. This relies on the presence of multiple variants within single or paired/linked sequence reads. The reason why this problem is non-trivial is that the average density of heterozygous polymorphisms ranges from 1 variant per 1000–1500 base pairs depending on the population (124), while the typical fragment sequenced in short-read Illumina sequencing is around 500 bp. As a result, using short-read data, few variants can be linked together. One solution to this problem has involved using long fragment sequencing, including the use of mate-pair sequencing, long-read sequencing (PacBio) or linked reads (5,6). These methods can begin to achieve longer local haplotypes but inevitably fail to extend to generate haplotype N50 s longer than several megabases in length or to phase across centromeres (125). Hi-C has the potential to overcome some of these limitations, as read pairs captured in Hi-C experiments can in theory span the entire length of a chromosome. Importantly, as was demonstrated in our initial description of Hi-C-based phasing, read pairs that align to the same homologous chromosome are more likely to also arise from the same haplotype (Figure 2D) (66). This effect is dependent on the distance between the pairs of reads, such that as reads are further separated along a chromosome, the likelihood of the reads occurring on the same haplotype approaches the likelihood of reads aligning to separate homologous chromosomes in trans.
Several computational algorithms have thus far been described for using Hi-C data to phase haplotypes. Our original description relied on the use of HapCUT (126), an algorithm that uses max-cuts in a graph-based representation of haplotypes and sequence reads to identify an optimal pair of haplotypes. This has been recently modified as HapCUT2 (127), a maximum likelihood-based approach for improved performance and explicit handling of Hi-C specific errors. Alternative methods also include the SpectralPhasing algorithm, which uses Hi-C data to extend partial haplotypes from short-read sequencing by embedding Hi-C data in 3D Euclidean space (128). In addition, a recent method using a 1D spin model to perform two-tiered phasing using linked-read and Hi-C data has been recently described (129).
Our original description of Hi-C-based haplotype phasing was able to generate chromosome length haplotypes for all somatic chromosomes. The limitation was that these were sparse haplotypes, containing only ~20% of heterozygous variants along a given chromosome. Many of the missing variants could be ‘imputed’ based on the seed with high-accuracy, but this is only able to rescue a subset of variants and is biased towards common variants. Current experimental efforts to move beyond these initial sparse haplotypes have focused on combining alternative data types with Hi-C-based haplotypes. For example, recent reports integrating Hi-C data with linked-read sequencing-based approaches can improve the fraction of phased variants to upwards of 98.9% (127,129). An alternative approach to integrating Hi-C with alternative sequencing methods is to use Hi-C data to construct a phased, diploid genome assembly (130). Such an approach has recently been described by integrating Hi-C and PacBio sequencing data using the ‘FALCON-phase’ method. PacBio long-reads are first used to generate a de novo assembly. PacBio-based assembly can generate phased contigs (termed ‘haplotigs’) that are interrupted by unphased contigs due to the regions of low heterozygosity. By integrating Hi-C data, a phased diploid assembly can be resolved by stitching haplotigs together into complete phased genomes.
Hi-C is one of relatively few experimental methods which allows for chromosome length haplotype phasing. As statistical phasing and phasing based on alternative methods such as linked-reads, long-reads or fosmid-based sequencing can all achieve high local accuracy, one important question is what is the utility of obtaining the long haplotypes generate by Hi-C compared with alternative approaches? There are likely several intriguing potential applications. One potential application is with regards to non-invasive prenatal genome sequencing. The initial reports of using cell-free DNA to sequence the genome of a fetus in utero relied on phased haplotypes of the parents in order to accurately genotype which variants were inherited by the fetus (131–133). The authors note that longer haplotypes yield greater accuracy of fetal genotypes (132). Indeed, one report that generated chromosome length haplotypes using trio-based phasing-by-transmission indicated that with chromosome length phasing in the parents, highly accurate fetal genotypes can be inferred using only shallow sequencing of cfDNA (134). Trio-based approaches required the whole genome sequencing of both parents and all four grandparents of the fetus. Therefore, an Hi-C-based chromosome length haplotype may obviate the need to sequencing an entire pedigree. This application is practically limited by the fact that genome-wide Hi-C likely fails to phase many rare variants due to the sparsity of the experimentally derived haplotypes, so sequencing to identify rare Mendelian disease associated variants may have limited power. This limitation has been solved, however, using targeted approaches (135), indicating that 3C-based methods may be most readily applied when targeting specific rare variants.
Another potential application of Hi-C-based haplotype phasing is in allele-resolved epigenomic analysis. Studying the differential epigenetic and transcriptomic states between alleles can yield important insights into phenomena such as genetic imprinting (136), X-chromosome inactivation (137) and the effects of regulatory variants acting in cis. This has historically been accomplished using crosses between inbred mouse strains or by the use of samples with known haplotypes from trio-based phasing. However, the ability to generate chromosome length haplotype phasing can allow this type of analysis to be performed in any setting. For example, this approach has been applied to phase human ES cell lines and their differentiated progeny to study allele specific gene regulation as well as phasing haplotypes from healthy patient samples to study such effects in tissue samples (32,138). One challenge of such approaches is that Hi-C-based haplotype phasing typically requires high sequencing coverage in order to obtain complete haplotype phasing. Therefore, in the future to make more widespread use of these approaches, there is a need to develop methods that can generate chromosome length haplotype phasing using limited or shallow sequencing.
Conclusions
The development and application of Hi-C and its derivative methods has provided insights into aspects of genome biology even beyond its original purpose for studying 3D genome architecture. Hi-C has been applied to study the order of linear sequences, genome rearrangements, and heterogeneous composition of genomes in different (metagenomics) or the same (haplotype phasing) nucleus. Future improvements of Hi-C for this purpose may include developments that bring higher resolution and less background, higher efficiency of ligation and also new computational tools that better integrate data from Hi-C with other genomic assays. Due to the largely invariant basic features that define Hi-C data, we believe that this is an assay that is likely here to stay as a tool for genome reconstruction.
Key points
3C assays can aid in genome sequencing, including contributing to genome scaffolding, metagenomic assembly, haplotype phasing and structural variant identification.
These methods have been applied to a wide variety or organisms and individuals, and have the potential to be applied to unique sample types, such as clinical FFPE samples.
Some limitations to Hi-C as a tool for genome reconstruction are apparent, including orientation errors in scaffolding and the lack of sensitivity for detecting small rearrangements in structural variant identification.
Integrating Hi-C data with alternative data types, such as long-read or linked-read sequencing, are likely to increase the power of this approach in the future.
Zhichao Xu is a post-doctoral fellow in the Dixon Lab at the Salk Institute for Biological Studies. He is currently focusing on finding the connections between 3D chromatin architecture, somatic mutations, and gene regulation in cancer.
Jesse R. Dixon is a fellow in the Salk Helmsley Fellows program at the Salk Institute for Biological Studies. He is interested in 3D genome organization, and how germline and somatic sequence variants impact genome architecture and gene regulation.
Zhichao Xu is a post-doctoral fellow in the Dixon Lab at the Salk Institute for Biological Studies. He is currently focusing on finding the connections between 3D chromatin architecture, somatic mutations and gene regulation in cancer.
Funding
This work was supported by an NIH Director’s Early Independence Award (DP5OD023071) and the Leona M. and Harry B. Helmsley Charitable Trust grant #2017-PG-MED001.
References
- 1. Shendure J, Lieberman Aiden E. The expanding scope of DNA sequencing. Nat Biotechnol 2012;30:1084–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eid J, Fehr A, Gray J, et al. . Real-time DNA sequencing from single polymerase molecules. Science 2009;323:133–138. [DOI] [PubMed] [Google Scholar]
- 3. Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 2015;12:733–735. [DOI] [PubMed] [Google Scholar]
- 4. Amini S, Pushkarev D, Christiansen L, et al. . Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat Genet 2014;46:1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kitzman JO, Mackenzie AP, Adey A, et al. . Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol 2011;29:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng GX, Lau BT, Schnall-Levin M, et al. . Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 2016;34:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science 2002;295:1306–1311. [DOI] [PubMed] [Google Scholar]
- 8. Lieberman-Aiden E, Berkum NL, Williams L, et al. . Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sajan SA, Hawkins RD. Methods for identifying higher-order chromatin structure. Annu Rev Genomics Hum Genet 2012;13:59–82. [DOI] [PubMed] [Google Scholar]
- 10. Sinden RR, Pettijohn DE. Chromosomes in living Escherichia coli cells are segregated into domains of supercoiling. Proc Natl Acad Sci U S A 1981;78:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le TB, Imakaev MV, Mirny LA, Laub MT. High-resolution mapping of the spatial organization of a bacterial chromosome. Science 2013;342:731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marbouty M, Baudry L, Cournac A, Koszul R. Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci Adv 2017;3:e1602105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marbouty M, Le Gall A, Cattoni DI, et al. . Condensin- and replication-mediated bacterial chromosome folding and origin condensation revealed by Hi-C and super-resolution imaging. Mol Cell 2015;59:588–602. [DOI] [PubMed] [Google Scholar]
- 14. Wang X, Le TB, Lajoie BR, et al. . Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev 2015;29:1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trussart M, Yus E, Martinez S, et al. . Defined chromosome structure in the genome-reduced bacterium Mycoplasma pneumoniae. Nat Commun 2017;8: 14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olins AL, Olins DE. Spheroid chromatin units (v bodies). Science 1974;183:330–332. [DOI] [PubMed] [Google Scholar]
- 17. Woodcock CL. A milestone in the odyssey of higher-order chromatin structure. Nat Struct Mol Biol 2005;12:639–640. [DOI] [PubMed] [Google Scholar]
- 18. Duan Z, Andronescu M, Schutz K, et al. . A three-dimensional model of the yeast genome. Nature 2010;465:363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dixon JR, Selvaraj S, Yue F, et al. . Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nora EP, Lajoie BR, Schulz EG, et al. . Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012;485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sexton T, Yaffe E, Kenigsberg E, et al. . Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012;148:458–472. [DOI] [PubMed] [Google Scholar]
- 22. Rao SS, Huntley MH, Durand NC, et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simonis M, Klous P, Splinter E, et al. . Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 2006;38:1348–1354. [DOI] [PubMed] [Google Scholar]
- 24. Dostie J, Richmond TA, Arnaout RA, et al. . Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 2006;16:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalhor R, Tjong H, Jayathilaka N, et al. . Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat Biotechnol 2011;30:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fullwood MJ, Liu MH, Pan YF, et al. . An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009;462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mumbach MR, Rubin AJ, Flynn RA, et al. . HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 2016;13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fang R, Yu M, Li G, et al. . Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 2016;26:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stevens TJ, Lando D, Basu S, et al. . 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017;544:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lesne A, Riposo J, Roger P, et al. . 3D genome reconstruction from chromosomal contacts. Nat Methods 2014;11:1141–1143. [DOI] [PubMed] [Google Scholar]
- 31. Marbouty M, Cournac A, Flot JF, et al. . Metagenomic chromosome conformation capture (meta3C) unveils the diversity of chromosome organization in microorganisms. elife 2014;3:e03318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dixon JR, Jung I, Selvaraj S, et al. . Chromatin architecture reorganization during stem cell differentiation. Nature 2015;518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagano T, Lubling Y, Varnai C, et al. . Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017;547:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gibcus JH, Samejima K, Goloborodko A, et al. . A pathway for mitotic chromosome formation. Science 2018;359 pii: eaao6135. doi: 10.1126/science.aao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bintu B, Mateo LJ, Su JH, et al. . Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018;362 pii: eaau1783. doi: 10.1126/science.aau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oddes S, Zelig A, Kaplan N. Three invariant Hi-C interaction patterns: applications to genome assembly. Methods 2018;142:89–99. [DOI] [PubMed] [Google Scholar]
- 37. Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol 2010;2:a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun HB, Shen J, Yokota H. Size-dependent positioning of human chromosomes in interphase nuclei. Biophys J 2000;79:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szczepinska T, Rusek AM, Plewczynski D. Intermingling of chromosome territories. Genes Chromosomes Cancer 2019;58(7):500–506. [DOI] [PubMed] [Google Scholar]
- 40. Ramani V, Deng X, Qiu R, et al. . Massively multiplex single-cell Hi-C. Nat Methods 2017;14:263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flyamer IM, Gassler J, Imakaev M, et al. . Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017;544:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fudenberg G, Mirny LA. Higher-order chromatin structure: bridging physics and biology. Curr Opin Genet Dev 2012;22:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rippe K. Making contacts on a nucleic acid polymer. Trends Biochem Sci 2001;26:733–740. [DOI] [PubMed] [Google Scholar]
- 44. Potato Genome Sequencing Consortium, Xu X, Pan S, et al. . Genome sequence and analysis of the tuber crop potato. Nature 2011;475:189–195. [DOI] [PubMed] [Google Scholar]
- 45. Williams LJ, Tabbaa DG, Li N, et al. . Paired-end sequencing of Fosmid libraries by Illumina. Genome Res 2012;22:2241–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Michael TP, Jupe F, Bemm F, et al. . High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat Commun 2018;9:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vaser R, Sovic I, Nagarajan N, Sikic M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 2017;27:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bickhart DM, Rosen BD, Koren S, et al. . Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet 2017;49:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma ZS, Li L, Ye C, et al. . Hybrid assembly of ultra-long Nanopore reads augmented with 10x-genomics contigs: demonstrated with a human genome. Genomics 2019;111(6):1896–1901. [DOI] [PubMed] [Google Scholar]
- 50. Wei X, Xu Z, Wang G, et al. . pBACode: a random-barcode-based high-throughput approach for BAC paired-end sequencing and physical clone mapping. Nucleic Acids Res 2017;45:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gordon D, Huddleston J, Chaisson MJ, et al. . Long-read sequence assembly of the gorilla genome. Science 2016;352:aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stankova H, Hastie AR, Chan S, et al. . BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotechnol J 2016;14:1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature 2004;431:931–945. [DOI] [PubMed] [Google Scholar]
- 54. Rice ES, Kohno S, John JS, et al. . Improved genome assembly of American alligator genome reveals conserved architecture of estrogen signaling. Genome Res 2017;27:686–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Session AM, Uno Y, Kwon T, et al. . Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016;538:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dudchenko O, Batra SS, Omer AD, et al. . De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017;356:92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Matthews BJ, Dudchenko O, Kingan SB, et al. . Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 2018;563:501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grohme MA, Schloissnig S, Rozanski A, et al. . The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms. Nature 2018;554:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mascher M, Gundlach H, Himmelbach A, et al. . A chromosome conformation capture ordered sequence of the barley genome. Nature 2017;544:427–433. [DOI] [PubMed] [Google Scholar]
- 60. Reyes-Chin-Wo S, Wang Z, Yang X, et al. . Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat Commun 2017;8: 14953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Teh BT, Lim K, Yong CH, et al. . The draft genome of tropical fruit durian (Durio zibethinus). Nat Genet 2017;49:1633–1641. [DOI] [PubMed] [Google Scholar]
- 62. Jarvis DE, Ho YS, Lightfoot DJ, et al. . The genome of Chenopodium quinoa. Nature 2017;542:307–312. [DOI] [PubMed] [Google Scholar]
- 63. Dudchenko O, Shamim MS, Batra SS, et al. . The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Biorxiv 2018. doi: 10.1101/254797preprint: not peer reviewed. [DOI] [Google Scholar]
- 64. Dudchenko O, Pham M, Lui C, et al. . Hi-C yields chromosome-length scaffolds for a legume genome, Trifolium subterraneum. bioRxiv 2018. doi: 10.1101/473553. [DOI] [Google Scholar]
- 65. Lajoie BR, Dekker J, Kaplan N. The Hitchhiker's guide to Hi-C analysis: practical guidelines. Methods 2015;72:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Selvaraj S, Dixon JR, Bansal V, Ren B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol 2013;31:1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Burton JN, Adey A, Patwardhan RP, et al. . Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 2013;31:1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kaplan N, Dekker J. High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat Biotechnol 2013;31:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Genome 10K Community of Scientists Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered 2009;100:659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ghurye J, Rhie A, Walenz BP, et al. . Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol 2019;15:e1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ghurye J, Pop M, Koren S, et al. . Scaffolding of long read assemblies using long range contact information. BMC Genomics 2017;18:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gnerre S, Maccallum I, Przybylski D, et al. . High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A 2011;108:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008;18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li D, Liu CM, Luo R, et al. . MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31:1674–1676. [DOI] [PubMed] [Google Scholar]
- 75. Marie-Nelly H, Marbouty M, Cournac A, et al. . High-quality genome (re) assembly using chromosomal contact data. Nat Commun 2014;5:5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Putnam NH, O'Connell BL, Stites JC, et al. . Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res 2016;26:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lloyd-Price J, Mahurkar A, Rahnavard G, et al. . Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017;550:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Annu Rev Mar Sci 2011;3:347–371. [DOI] [PubMed] [Google Scholar]
- 79. Hess M, Sczyrba A, Egan R, et al. . Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011;331:463–467. [DOI] [PubMed] [Google Scholar]
- 80. Hug LA. Sizing up the uncultured microbial majority. mSystems 2018;3 pii: e00185–18. doi: 10.1128/mSystems.00185-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rappe MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol 2003;57:369–394. [DOI] [PubMed] [Google Scholar]
- 82. Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 1991;173:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Davidson RM, Epperson LE. Microbiome sequencing methods for studying human diseases. Methods Mol Biol 2018;1706:77–90. [DOI] [PubMed] [Google Scholar]
- 84. Venter JC, Remington K, Heidelberg JF, et al. . Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004;304:66–74. [DOI] [PubMed] [Google Scholar]
- 85. DeLong EF, Preston CM, Mincer T, et al. . Community genomics among stratified microbial assemblages in the ocean's interior. Science 2006;311:496–503. [DOI] [PubMed] [Google Scholar]
- 86. Rusch DB, Halpern AL, Sutton G, et al. . The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 2007;5:e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Qin J, Li R, Raes J, et al. . A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yooseph S, Nealson KH, Rusch DB, et al. . Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature 2010;468:60–66. [DOI] [PubMed] [Google Scholar]
- 89. Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, et al. . Uncovering Earth's virome. Nature 2016;536:425–430. [DOI] [PubMed] [Google Scholar]
- 90. Umbarger MA, Toro E, Wright MA, et al. . The three-dimensional architecture of a bacterial genome and its alteration by genetic perturbation. Mol Cell 2011;44:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Beitel CW, Froenicke L, Lang JM, et al. . Strain- and plasmid-level deconvolution of a synthetic metagenome by sequencing proximity ligation products. PeerJ 2014;2:e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Burton JN, Liachko I, Dunham MJ, Shendure J. Species-level deconvolution of metagenome assemblies with Hi-C-based contact probability maps. G3 (Bethesda) 2014;4:1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Stewart RD, Auffret MD, Warr A, et al. . Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat Commun 2018;9:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bickhart DM, Watson M, Koren S, et al. . Assignment of virus and antimicrobial resistance genes to microbial hosts in a complex microbial community by combined long-read assembly and proximity ligation. Genome Biol 2019;20:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Press MO, Wiser AH, Kronenberg ZN, et al. . Hi-C deconvolution of a human gut microbiome yields high-quality draft genomes and reveals plasmid-genome interactions. biorxiv 2017; 198713. doi: 10.1101/198713, preprint: not peer reviewed. [DOI] [Google Scholar]
- 96. Smukowski Heil C, Burton JN, Liachko I, et al. . Identification of a novel interspecific hybrid yeast from a metagenomic spontaneously inoculated beer sample using Hi-C. Yeast 2018;35:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Albertsen M, Hugenholtz P, Skarshewski A, et al. . Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 2013;31:533–538. [DOI] [PubMed] [Google Scholar]
- 98. Tyson GW, Chapman J, Hugenholtz P, et al. . Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004;428:37–43. [DOI] [PubMed] [Google Scholar]
- 99. Flot JF, Marie-Nelly H, Koszul R. Contact genomics: scaffolding and phasing (meta) genomes using chromosome 3D physical signatures. FEBS Lett 2015;589:2966–2974. [DOI] [PubMed] [Google Scholar]
- 100. Teeling H, Waldmann J, Lombardot T, et al. . TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics 2004;5:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Carr R, Shen-Orr SS, Borenstein E. Reconstructing the genomic content of microbiome taxa through shotgun metagenomic deconvolution. PLoS Comput Biol 2013;9:e1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Huson DH, Beier S, Flade I, et al. . MEGAN Community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 2016;12:e1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. DeMaere MZ, Darling AE. bin3C: exploiting Hi-C sequencing data to accurately resolve metagenome-assembled genomes. Genome Biol 2019;20:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tsai YC, Conlan S, Deming C, et al. . Resolving the complexity of human skin metagenomes using single-molecule sequencing. MBio 2016;7:e01948–e01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Payne A, Holmes N, Rakyan V, Loose M. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 2018;35(13):2193–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Engreitz JM, Agarwala V, Mirny LA. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS One 2012;7:e44196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Naumova N, Imakaev M, Fudenberg G, et al. . Organization of the mitotic chromosome. Science 2013;342:948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Simonis M, Klous P, Homminga I, et al. . High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nat Methods 2009;6:837–842. [DOI] [PubMed] [Google Scholar]
- 109. Harewood L, Kishore K, Eldridge MD, et al. . Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol 2017;18:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chakraborty A, Ay F. Identification of copy number variations and translocations in cancer cells from Hi-C data. Bioinformatics 2017;34(2):338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Troll CJ, Putnam NH, Hartley PD, et al. . Structural variation detection by proximity ligation from formalin-fixed, paraffin-embedded tumor tissue. J Mol Diagn 2019;21:375–383. [DOI] [PubMed] [Google Scholar]
- 112. Dixon JR, Xu J, Dileep V, et al. . Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 2018;50:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wang S, Lee S, Chu C, et al. . HiNT: a computational method for detecting copy number variations and translocations from Hi-C data. biorxiv 2019; 657080. doi: 10.1101/657080, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Servant N, Varoquaux N, Heard E, et al. . Effective normalization for copy number variation in Hi-C data. BMC Bioinformatics 2018;19:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wu HJ, Michor F. A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 2016;32:3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat Genet 2011;43:1059–1065. [DOI] [PubMed] [Google Scholar]
- 117. Monahan K, Horta A, Lomvardas S. LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 2019;565:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Selvaraj S, Schmitt AD, Dixon JR, Ren B. Complete haplotype phasing of the MHC and KIR loci with targeted HaploSeq. BMC Genomics 2015;16:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Vree PJ, Wit E, Yilmaz M, et al. . Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol 2014;32:1019–25. [DOI] [PubMed] [Google Scholar]
- 120. Meyer M, Kircher M, Gansauge MT, et al. . A high-coverage genome sequence from an archaic Denisovan individual. Science 2012;338:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. International HapMap Consortium, Frazer KA, Ballinger DG, et al. . A second generation human haplotype map of over 3.1 million SNPs. Nature 2007;449:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Adey A, Burton JN, Kitzman JO, et al. . The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 2013;500:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Loh PR, Palamara PF, Price AL. Fast and accurate long-range phasing in a UK biobank cohort. Nat Genet 2016;48:811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. 1000 Genomes Project Consortium, Abecasis GR, Auton A, et al. . An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Choi Y, Chan AP, Kirkness E, et al. . Comparison of phasing strategies for whole human genomes. PLoS Genet 2018;14:e1007308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics 2008;24:i153–i159. [DOI] [PubMed] [Google Scholar]
- 127. Edge P, Bafna V, Bansal V. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 2017;27:801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ben-Elazar S, Chor B, Yakhini Z. Extending partial haplotypes to full genome haplotypes using chromosome conformation capture data. Bioinformatics 2016;32:i559–i566. [DOI] [PubMed] [Google Scholar]
- 129. Tourdot RW, Zhang C-Z. Whole chromosome haplotype phasing from long-range sequencing. biorxiv 2019; 629337. doi: 10.1101/629337, preprint: not peer reviewed. [DOI] [Google Scholar]
- 130. Kronenberg ZN, Rhie A, Koren S, et al. . Extended haplotype phasing of de novo genome assemblies with FALCON-Phase. biorxiv 2019; 327064. [Google Scholar]
- 131. Lo YM, Chan KC, Sun H, et al. . Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2:61ra91. [DOI] [PubMed] [Google Scholar]
- 132. Kitzman JO, Snyder MW, Ventura M, et al. . Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012;4:137ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Fan HC, Gu W, Wang J, et al. . Non-invasive prenatal measurement of the fetal genome. Nature 2012;487:320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chen S, Ge H, Wang X, et al. . Haplotype-assisted accurate non-invasive fetal whole genome recovery through maternal plasma sequencing. Genome Med 2013;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Vermeulen C, Geeven G, Wit E, et al. . Sensitive monogenic noninvasive prenatal diagnosis by targeted haplotyping. Am J Hum Genet 2017;101:326–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xie W, Barr CL, Kim A, et al. . Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012;148:816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zylicz JJ, Bousard A, Zumer K, et al. . The implication of early chromatin changes in X chromosome inactivation. Cell 2019;176:182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Leung D, Jung I, Rajagopal N, et al. . Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 2015;518:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]