

Abstract
The estrogen receptor (ER/ESR1) is expressed in a majority of breast cancers and drugs that inhibit ER signaling are the cornerstone of breast cancer pharmacotherapy. Currently, aromatase inhibitors are the frontline endocrine interventions of choice although their durability in metastatic disease is limited by activating point mutations within the ligand binding domain (LBD) of ESR1 that permit ligand independent activation of the receptor. It has been suggested that the most commonly occurring ESR1 mutations would likely compromise the clinical activity of selective estrogen receptor downregulators (SERDs) and selective estrogen receptor modulators (SERMs) when used as second-line therapies. It was unclear, however, how these mutations, which are likely coexpressed in cells with ERWT, may impact response to ER ligands in a clinically meaningful manner. To address this issue, we dissected the molecular mechanism(s) underlying ESR1 mutant pharmacology in models relevant to metastatic disease. These studies revealed that the response of ESR1 mutations to ligands was dictated primarily by the relative coexpression of ERWT in cells. Specifically, dysregulated pharmacology was only evident in cells in which the mutants were overexpressed relative to ligand-activated ERWT; a finding that highlights the role of allelism in determining ER mutant pharmacology. Importantly, we demonstrated that the antagonist activity of the SERM, lasofoxifene, was not impacted by mutant status; a finding that has led to its clinical evaluation as a treatment for patients with advanced ER-positive breast cancer whose tumors harbor ESR1 mutations.
Keywords: ESR1 mutants, endocrine resistant breast cancer, lasofoxifene, pharmacological development, personalized medicine
Introduction:
The estrogen receptor (ER/ESR1) is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors and is expressed in the majority of luminal breast cancers (1,2). Upon binding an estrogenic ligand, this transcription factor regulates the expression of genes required for cancer cell proliferation and survival. Not surprisingly, drugs that inhibit estrogen actions are the cornerstone of pharmacotherapy of breast cancers that express ER. Among the interventions most commonly used are the selective estrogen receptor modulator (SERM) tamoxifen, a drug which functions as an ER antagonist in breast cancer cells, and aromatase inhibitors (AIs) (letrozole, anastrozole, or exemestane), competitive inhibitors of CYP19 (aromatase), the enzyme that converts androgens into estrogens (3,4). Whereas both classes of drug effectively inhibit ER signaling in breast cancer, it is now standard practice to use aromatase inhibitors in the adjuvant setting as frontline endocrine therapy in postmenopausal patients or in high-risk premenopausal patients when combined with ovarian suppression (4). Tamoxifen is primarily reserved for the adjuvant treatment of premenopausal breast cancer patients at low-risk for recurrence with or without interventions to achieve ovarian suppression (3,5). These endocrine therapies have had a very significant impact on disease-free and overall survival in patients with breast cancer, although de novo and acquired resistance to either type of drug remains a significant clinical issue (6–9). However, the observation that ER remains engaged in the regulation of processes of importance in cancers that have escaped frontline endocrine interventions have led to the continued exploitation of this receptor as a therapeutic target (10).
Fulvestrant, a selective estrogen receptor downregulator (SERD), is used in patients who progress on frontline endocrine therapies and is given as monotherapy or in combination with targeted therapies (11). Drugs of this class function primarily as competitive inhibitors of agonist binding to ER, but their inhibitory activity is reinforced by a drug-induced conformational change that targets the receptor for proteasomal degradation (11,12). Currently, fulvestrant is the only clinically approved SERD. Whereas this drug is a very effective inhibitor and downregulator of ER expression in cellular and animal models of breast cancer, its clinical utility is limited by its poor pharmaceutical properties and by the need to administer it as a large bolus intramuscularly (13,14). Further, it is not clear to what extent ER within tumors is occupied by fulvestrant at the maximum doses that can be delivered to patients (15). This has driven the search for oral SERDs (or SERMs) that are as effective as fulvestrant in inhibiting ER activity but which have tissue exposure levels sufficient to saturate the receptor. From these efforts emerged the first generation oral SERDs GW5638, AZD9496 (NCT02248090, NCT03236874) and GDC-0810 (NCT01823835), all of which demonstrated efficacy in late state disease but whose development has been discontinued (16–21). Other oral SERDs, like RAD1901 (NCT02338349), are currently in clinical development (22,23).
Whereas the mechanisms underlying resistance to endocrine therapies are varied and complex, it is now clear that gain of function point mutations within the ligand binding domain (LBD) of ESR1 that permit it to exhibit constitutive transcriptional activity can confer resistance to aromatase inhibitors (24–26). Although rare in primary breast tumors, mutations in the ESR1 LBD (ERmut) occur in up to 40% of metastatic lesions, a finding that is consistent with their selection by conditions of extreme estrogen deprivation (24,25,27–31). Two of the most common mutations, Y537S, and D538G (ERY537S, and ERD538G), account for roughly 70% of all ESR1 mutations identified in patients with metastatic breast cancer (24,25,27–31). In addition to constitutively activating transcription, these mutations also exhibit distinct neomorphic activities that likely contribute to disease progression (32,33). Notwithstanding these important differences, most attention has been focused on how these disease-associated mutations reduce the ER binding affinity of some clinically important antagonists, an activity that may limit their therapeutic utility (24,25,27–31). The development of most SERDs was initiated before the prevalence of ERmuts was fully appreciated, and it is now apparent that, as with fulvestrant, the affinity of ERmuts for even the most contemporary SERDs is substantially reduced (õne order of magnitude) (18,23,34). Thus, in addition to addressing whether inhibition of ER with these drugs is a viable approach to inhibit ER-positive, endocrine therapy-refractive disease, there remains an open question as to their efficacy in cancers expressing the ERmuts (24–26). Thus, the primary goal of this study was to define the impact of ERmuts on the pharmacology of ER ligands with a view to prioritizing existing drugs for clinical evaluation in patients. Additionally, elucidation of the molecular mechanisms underlying the dysregulated pharmacology of ERmuts was also undertaken with the goal of informing the identification of the next generation of ER modulators for use in the treatment of advanced breast cancer.
Methods:
Cell lines and Reagents:
Fulvestrant (1047) and Raloxifene (2280), were purchased from Tocris. Estradiol (E8875) and 4-hydroxytamoxifen (H7904) were purchased from Sigma. Bazedoxifene (S2128) was purchased from Selleckchem. Lasofoxifene (HYA0038K), RAD1901 (HY19822A), GDC-0810 (HY12864) and AZD9496 (HY12870) were purchased from MedChem Express. SKBR3 and the MCF7 cells used to generate the MCF7I lines were purchased from ATCC which employs STR analysis. MCF7B and T47D cell lines were published previously (32,33). The McDonnell laboratory routinely completes PCR based Mycoplasma testing on all cell lines. All cell lines were used within 10–15 passages of thawing.
SKBR3 Luciferase Transcriptional Reporter Assay:
SKBR3 cells were co-transfected with the 3X- ERE-TATA luciferase reporter gene (35) and expression constructs for either wild-type or mutant receptors using Fugene transfection reagent (Promega). pCMV-β-gal was used as a control for transfection efficiency. Ligands (dose titration of antagonists in the presence of 1 nM E2) were added five hours post transfection. Cells were lysed 24 hours later and the luciferase and β-gal assays were performed as described previously (36).
MCF7 Luciferase Transcriptional Reporter Assay:
Cells were cultured in DMEM/F12 supplemented with 8% charcoal dextran-treated FBS. For siRNA transfection experiments, cells were plated over aliquoted siRNAs targeting the 3’ UTR of ER (to knockdown endogenous ER) using Lipofectamine RNAiMax (Thermo-Fisher Scientific) per the manufacturer protocol. After 48 hours, cells were co-transfected with the 7X-ERE-TATA luciferase reporter gene (35) and pCMV-β-gal. Assays were performed as described above for SKBR3, with the exception of 0.1nM E2 being used as a competitor. A detailed description of the derivation of MCF7 cells and the siRNA sequences utilized in these studies is described in Supplemental Methods.
Cofactor Profiling:
HepG2 cells were maintained in Basal Medium Eagles containing 8% fetal bovine serum. For mammalian two-hybrid based ER cofactor assay, cells were seeded in 96-well plates and transfected with VP16-ER, 5XGal4Luc3, Gal4DBD-peptide fusion constructs (pM-peptides), and pCMV β-gal using Lipofectin as previously described (21,37–39). Detailed description of peptide sequences and generation can be found in Supplemental Methods. Cells were then treated with saturating concentrations of ligands (10 μM for ER antagonists) for 48 hours. Assays were performed as described above. The data were standardized to avoid bias due to signal strength and clustered with the Ward hierarchical clustering method using JMP Pro 13 (SAS). The hierarchical cluster dendrogram was ordered by the first principal component.
Statistics:
Two-way ANOVA was utilized, comparing the logIC50 of all three independent experiments, to determine if there were significant differences between the WT and mutant receptors. Significant differences (p-value < 0.05) were appropriately noted.
Results:
The expression of clinically relevant ER mutants (ERmuts) does not alter the pharmacology of ER ligands in cells expressing ERWT.
Prior studies that informed our current understanding of the pharmacology of ERmuts in breast cancer cells were performed in model systems in which the mutants were expressed absent the WT receptor (ERWT) (24–26,34,40). Whereas this may be an appropriate way to model the pharmacology of compounds in cells homozygous for the mutants, this approach does not take into account the heterogeneity of ERWT/ ERmut expression in advanced ER-positive breast tumors cell that results from the selective pressure of endocrine therapy (26). To address this issue, we performed a comprehensive analysis of ER ligand pharmacology in cellular models in which ERWT is expressed alone or in combination with ERmut, the latter a scenario that is likely to represent what occurs within the majority of tumor cells in patients with metastatic disease.
To enable the evaluation of ERmut pharmacology, we created MCF7 cell derivatives that express ERWT alone (MCF7B-WT) or both ERWT and individual ER mutants (MCF7B-Y537S and MCF7B-D538G) (32). The structures of the antagonists evaluated in this study are shown in Supplemental Fig. S1 and include most of the clinically relevant SERMs and SERDs that are available (16,17,22,41). Interestingly, there was no change in cellular proliferation of cells expressing ERmuts when compared to the MCF7B-WT which expresses ERWT (Fig. 1B–G). Importantly, a similar result was observed when ER transcription, as opposed to proliferation, was used to monitor ER activity (Supplemental Fig. S2). Specifically, as expected, basal (ligand-independent) ER transcriptional activity, assessed using an ERE-luciferase reporter, was higher in both MCF7B-Y537S and MCF7B-D538G cells when compared to the isogenic MCF7B-WT cells. Further, as observed in MCF7B-WT cells, treatment with 17β-estradiol (E2) increased ER-dependent transcriptional activity in both MCF7B-Y537S and MCF7B-D538G cell models (Supplemental Fig. S2A). Notably, however, no significant shift in potency or efficacy was observed for any of the ER ligands tested in this assay when comparing either MCF7B-Y537S or MCF7B-D538G with MCF7B-WT (Supplemental Fig. S2 B–I). Previous studies which demonstrated shifts in ligand potency in similar models were performed in hormone stripped media where the activity of ERWT is minimally active (32,42).
Figure 1: Cells expressing both the ERWT and ERmuts have similar pharmacological responses to antiestrogens when compared to cells only expressing ERWT.
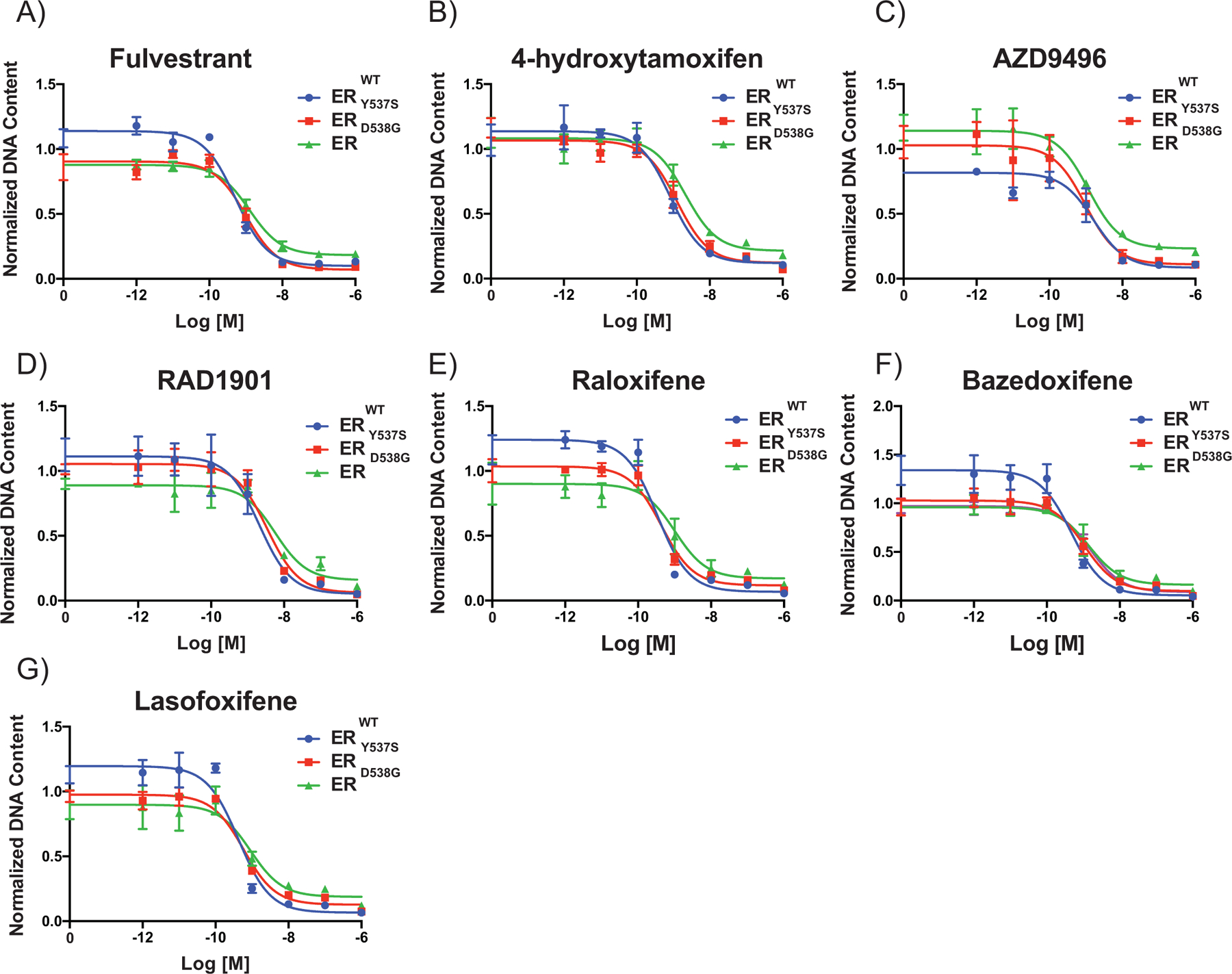
(A-G) MCF7B cells were grown in DMEM-F12 media containing 2% FBS for 7 days while being treated with ER antagonists (10−12-10−6 M). Cellular proliferation was assessed by measuring DNA content (Hoechst stain) and DNA content is normalized to vehicle. Data points are the mean of three technical replicates, and error bars are the standard deviation of these replicates. Data presented is a representative of three independent experiments. Two-way ANOVA was utilized, comparing the logIC50 of all three independent experiments, to determine if there were significant differences between the WT and mutant receptors. No significant differences (p-value < 0.05) were determined.
We, and others, have reported extensively on the role of cell context in regulating ER pharmacology, a likely consequence of differences in coregulator expression (43,44). Thus, we extended our studies to evaluate ER pharmacology in a second model, in which ERWT expressing T47D cells were engineered to express ERY537S or ERD538G in addition to endogenous ERWT(Supplemental Fig. S3 A and B) (33). Interestingly, of the five mutant clones tested, only ERY537SA displayed resistance to any of the compounds analyzed (Supplemental Fig. S3A). Further, using In-Cell Western assays in the MCF7B cells (and derivatives), it was demonstrated that the potency of SERDs as assessed by receptor turnover was not affected by mutation status (Supplemental Fig. S4). We also confirmed that the expression of ERWT and ERmut did not change over time and were maintained under the conditions of our in vitro assays (Supplemental Fig. S5) (45). Interestingly, the ERY537SA clone that displays partial resistance has a higher allelic frequency of ERY537S compared to the ERY537SB clone that does not show resistance (Supplemental Fig. S3A and Supplemental Fig. S5). However, our results appear to conflict with in vitro studies from others in which it was determined that ERY537S and ERD538G display an altered response to clinically relevant SERMs and SERDs (24–26,33,34,40).
The antagonist potency of SERDs and SERMs is reduced in cells expressing ERmuts alone.
To reconcile the discrepancies between our results presented here and those reported by others, we employed an overexpression model comparable to those that had been used previously to evaluate ERmut pharmacology (24–26,34,40). Vectors expressing ERWT, ERY537S or ERD538G, together with an ERE-luciferase reporter, were co-transfected into ER-negative SKBR3 breast cancer cells. Western immunoblot analysis was used to confirm that ERWT, ERY537S or ERD538G are expressed at comparable levels (Supplemental Fig. S6). Using this model system, we demonstrated, as was observed in MCF7B cells, that both ERY537S and ERD538G exhibited constitutive transcriptional activity (Fig. 2A). However, while the efficacy of fulvestrant and 4-hydroxytamoxifen were comparable for all three receptors, the antagonist potency of these two clinically important compounds in cells expressing ERY537S or ERD538G was reduced by approximately one order of magnitude when compared to ERWT (Fig. 2B and C). The acidic SERDs, AZD9496 and GDC-0810, were found to be inactive as antagonists on ERY537S, and indeed the latter compound functioned as a partial agonist in this assay (Fig. 2D and E). This is similar to our previous finding demonstrating that GW7604, a structurally distinct acidic SERD that is the 4-hydroxylated analog of GW5638, had reduced efficacy when assayed in ERY537S expressing ovarian cancer cells (46). Additionally, the potency of RAD1901, raloxifene, and bazedoxifene were reduced with subtle differences in the pharmacology noted when assayed on either ERY537S or ERD538G (Fig. 2F–H). One of the most interesting findings in this study was that lasofoxifene, a SERM originally developed for the treatment/prevention of osteoporosis, was the only compound found to be as potent an antagonist when evaluated in cells expressing ERY537S or ERD538G when compared to ERWT (Fig. 2I). This latter observation is in agreement with the findings of a recent study from our group showing that lasofoxifene was as effective an inhibitor of ERmuts as ERWT in cellular models of gynecological cancers (46). These findings have important clinical implications that could inform the optimal selection of ER antagonists for the treatment of patients with ERmuts in advanced disease.
Figure 2: ERmuts confer antiestrogen resistance when expressed alone.
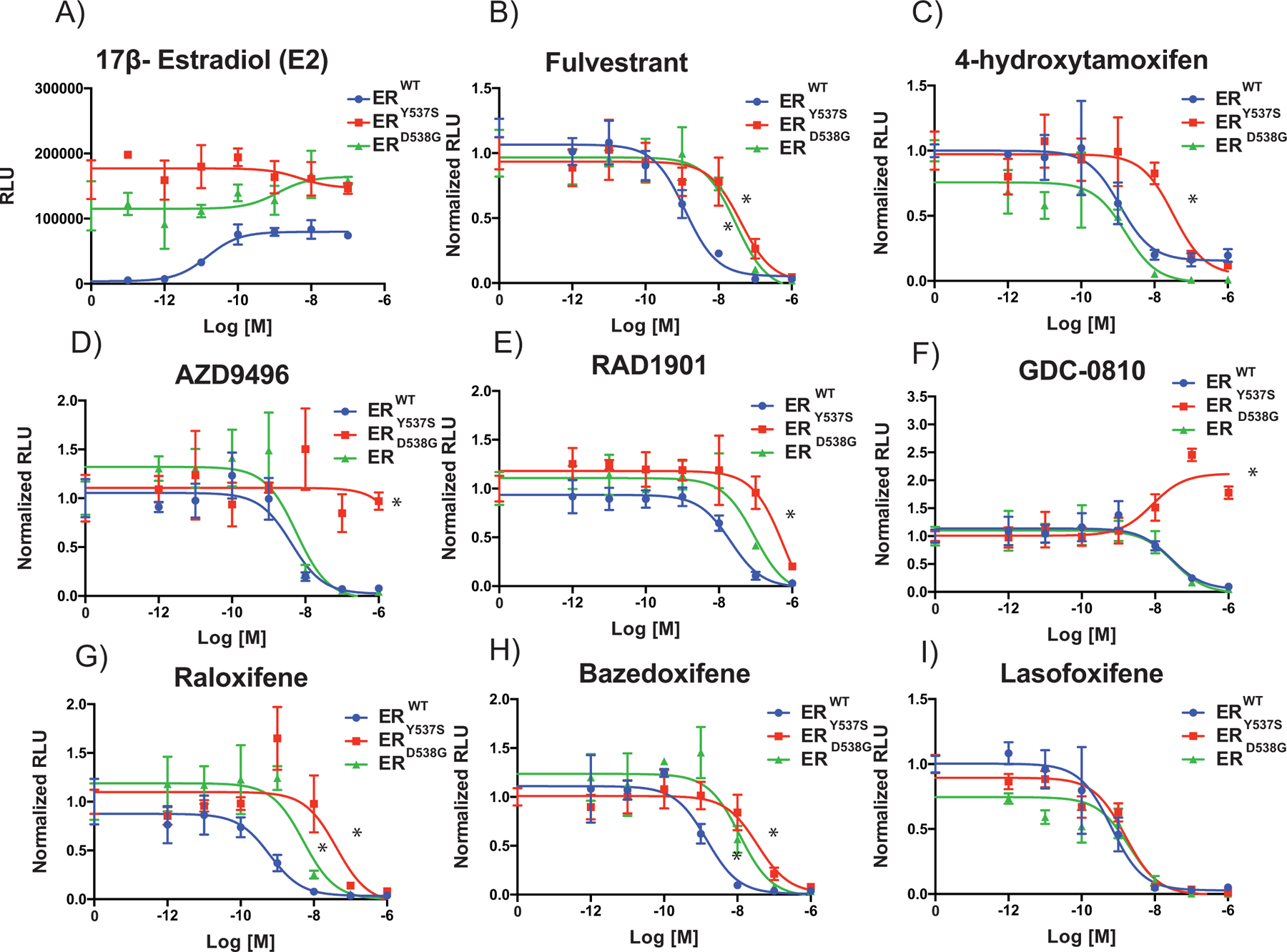
SKBR3 (ER- negative breast cancer) cells were plated in phenol red free media and transfected with an estrogen responsive reporter gene (3X-ERE-tata-Luc) in the presence of ERWT, ERY537S or ERD538G. After 5 hours, cells were treated with 17β-estradiol (1 nM) (A) and ER antagonists (10−12 M to 10−6 M) (B-I). Firefly luciferase activity was assessed and normalized to β-galactosidase transfection control (Y-Axis). Data points are the mean of three technical replicates, and error bars are the standard deviation of these replicates. Data presented is a representative of three independent experiments. Two-way ANOVA was utilized, comparing the logIC50s of all three independent experiments, to determine if there were significant differences between the WT and mutant receptors. Significant differences (p-value < 0.05) of the mutant IC50s when compared to that of the WT that were determined by this analysis are represented with a star. For GDC-0810 and AZD9496 on ERY537S the highest dose tested (10−6 M) was used as a surrogate, as the IC50 is greater than this value. The only compound that did not reach a significant difference for either mutant isoform was lasofoxifene.
ER ligands exhibit subtle differences in their ability to facilitate the interaction of ERmuts with coregulators.
We next embarked on studies to define the molecular basis of the differences in the pharmacology of ERWT, ERY537S or ERD538G. Resolution of this issue, we anticipated, would allow for the optimal use of existing endocrine therapies, and inform the development of the next generation of ER modulators for breast cancer. Receptor conformation has emerged as the primary mechanism by which information flows from a ligand through the receptor to the transcriptional machinery (44,47,48). Similarly conformed ER ligand complexes can exhibit diverse activities in different cell contexts as a consequence of the cell-selective expression and differential recruitment of functionally distinct coregulators. Further, subtle changes in ER structure, induced by structurally similar ligands, can result in different transcriptional outputs on individual target genes (37,49,50). Thus, it is possible that differences in the pharmacology of the ERmuts noted in cellular models of ER-positive (MCF7B and T47D cells) or ER-negative (SKBR3 cells) breast cancer could result from differences in cofactor expression and their differential recruitment by ERWT, ERY537S or ERD538G upon ligand activation.
Given the primacy of receptor structure in determining pharmacological output on ER, we evaluated the impact of SERMs and SERDs on the conformation of different receptor-ligand complexes using a cofactor peptide binding assay, the utility of which we have described previously (21,37–39). In this assay, short peptides identified using combinatorial peptide phage display, and peptides derived from the receptor interaction domains of validated coactivators (CoA) and corepressors (CoR), are expressed as GAL4-DBD peptide fusions (Fig. 3A). Additionally, a control peptide that interacts with ER in the presence of any ligand (αII) was also utilized. Sequences and detailed information on all of the peptides used are included in the Supplemental Methods. ERWT, ERY537S or ERD538G were modified to contain a VP-16 acidic activation domain at their amino termini (Fig. 3A). The interaction of the VP-16-ER proteins with the GAL4-peptide fusions in the presence of each ligand was assessed by measuring transcriptional activity on a GAL4-responsive luciferase reporter.
Figure 3: Differential cofactor recruitment reveals modest changes in overall receptor conformation between the WT and mutant receptors:
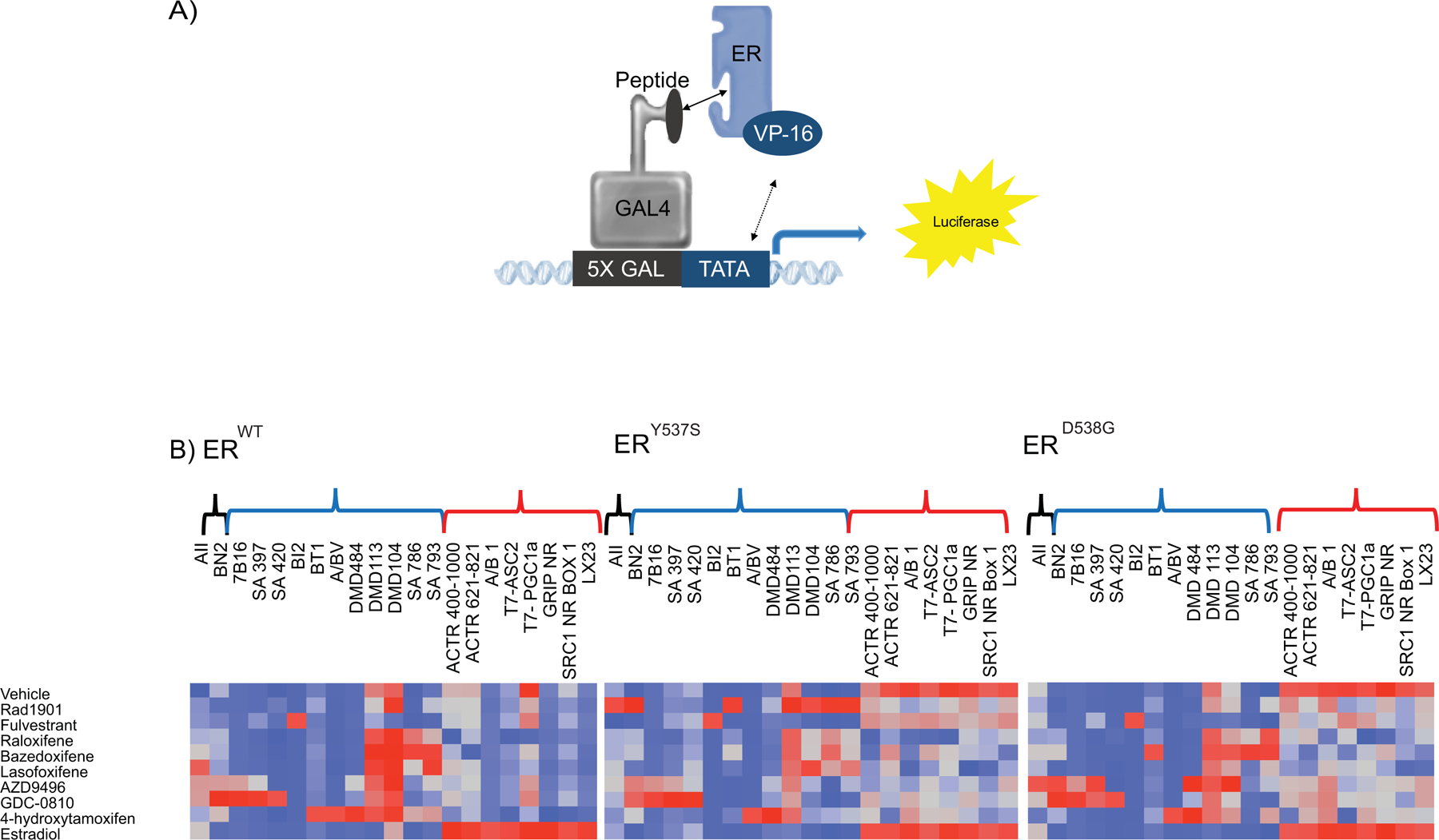
(A) A mammalian two-hybrid assay was used to evaluate ligand-dependent recruitment of peptides that mimic ER coregulators. (B) Hep-G2 cells were co-transfected with VP-16 tagged WT or mutant ER, Gal4DBD tagged peptides and a Gal4-responsive reporter gene and pCMV β-gal. 24 hours later cells were treated with saturating concentrations of ligands (10 μM) and incubated for 48 hours. Normalized response, which was obtained by normalizing luciferase activity to β-galactosidase activity, was used as input for Ward hierarchical clustering. Heat maps of mutant ERs are re-ordered to match the WT receptor. Results demonstrated a change in receptor conformation in response to ER activating mutations. At the top of the graph, there are three classes of peptides: ligand indiscriminate (black), peptides associated with receptor inhibition (blue) and receptor activation (red). Data presented is a representative of two independent experiments.
As expected given their constitutive activity, both mutant receptors interact in a ligand-independent manner, albeit to different degrees, with CoA-like peptides (designated with red brackets), and these interactions are further elevated upon the addition of E2 (Fig. 3B). The CoA interaction profiles of E2-activated ERWT, ERY537S or ERD538G are surprisingly indistinguishable. Importantly, the constitutive interaction of the mutants with CoA peptides is only partially attenuated upon the addition of SERMs/SERDs (Fig. 3B). Notably, raloxifene and lasofoxifene appear to be the two most effective inhibitors of CoA-peptide binding to ERY537S or ERD538G. Subtle quantitative differences in the binding of CoR-peptides (blue brackets) to the receptors in the presence of different ligands were also noted but there were no obvious differences in peptide binding preferences. One exception is the robust interaction of the RAD1901/ERY537S complex with a subset of the CoR peptides. We infer this to mean that this particular ligand-receptor complex may have an increased ability to recruit corepressors to ERmut. However, when taken together, it appears that the ERWT can adopt different conformational states upon binding different ligands and these interactions are substantially similar in each of the mutant receptors. Taking into account the limitations of this study (i.e. surfaces on ER not probed with our current technology), we concluded that it is unlikely that the differences in the pharmacology of the ERmuts observed in different cells can be attributed to differential coregulator binding alone.
The altered pharmacology of ERmuts is only evident when their expression in cells exceeds that of the WT receptor.
One of the key differences between MCF7B (and T47D cells) and SKBR3 models is that in the latter cell line ERY537S or ERD538G are expressed in the absence of ERWT. We considered it possible that in the MCF7B (and T47D) cell background, ERWT pharmacology dominates and normalizes the transcriptional activity of the mutants. We considered it likely, therefore, that by overexpressing the mutants relative to ERWT in MCF7 cells, that the altered mutant pharmacology apparent in SKBR3 cells would emerge. To test this hypothesis, we generated MCF7 cells in which ERWT, ERY537S or ERD538G expression was regulated in a doxycycline-inducible manner, allowing titratable expression of these proteins (MCF7I) over endogenous ERWT. Western immunoblot analysis confirmed that the increasing protein expression levels with increasing doses of doxycycline were comparable in each cell line (Supplemental Fig. S7). Considering the pharmacology noted in SKBR3 cells, we selected fulvestrant (potency shift observed with both mutants), AZD9496 (loss of efficacy as an inhibitor of ERY537S) and lasofoxifene (potency and efficacy unaffected by mutation status) for analysis in these model systems. The transcriptional activity and pharmacology of receptor combinations were assessed using a transfected ERE-luciferase reporter gene (Fig. 4). Relative to its activity on ERWT, it was noted that increased expression of ERD538G resulted in a reduction in fulvestrant potency, and a trend towards reduced potency was also noted with ERY537S (Fig. 4A). Likewise, the potency of AZD9496 on ERY537S was considerably reduced upon its overexpression in MCF7I cells (Fig. 4B). The pharmacology of lasofoxifene was unaffected by receptor expression levels (Fig. 4C). No changes were noted in the activity of any ligand in cells overexpressing ERWT alone (Supplemental Fig. S8). These data suggest that the altered pharmacology of ERmuts may only be manifest when they are expressed at a higher level than ERWT.
Figure 4: The altered pharmacology of ERmuts can be manipulated by their expression level.
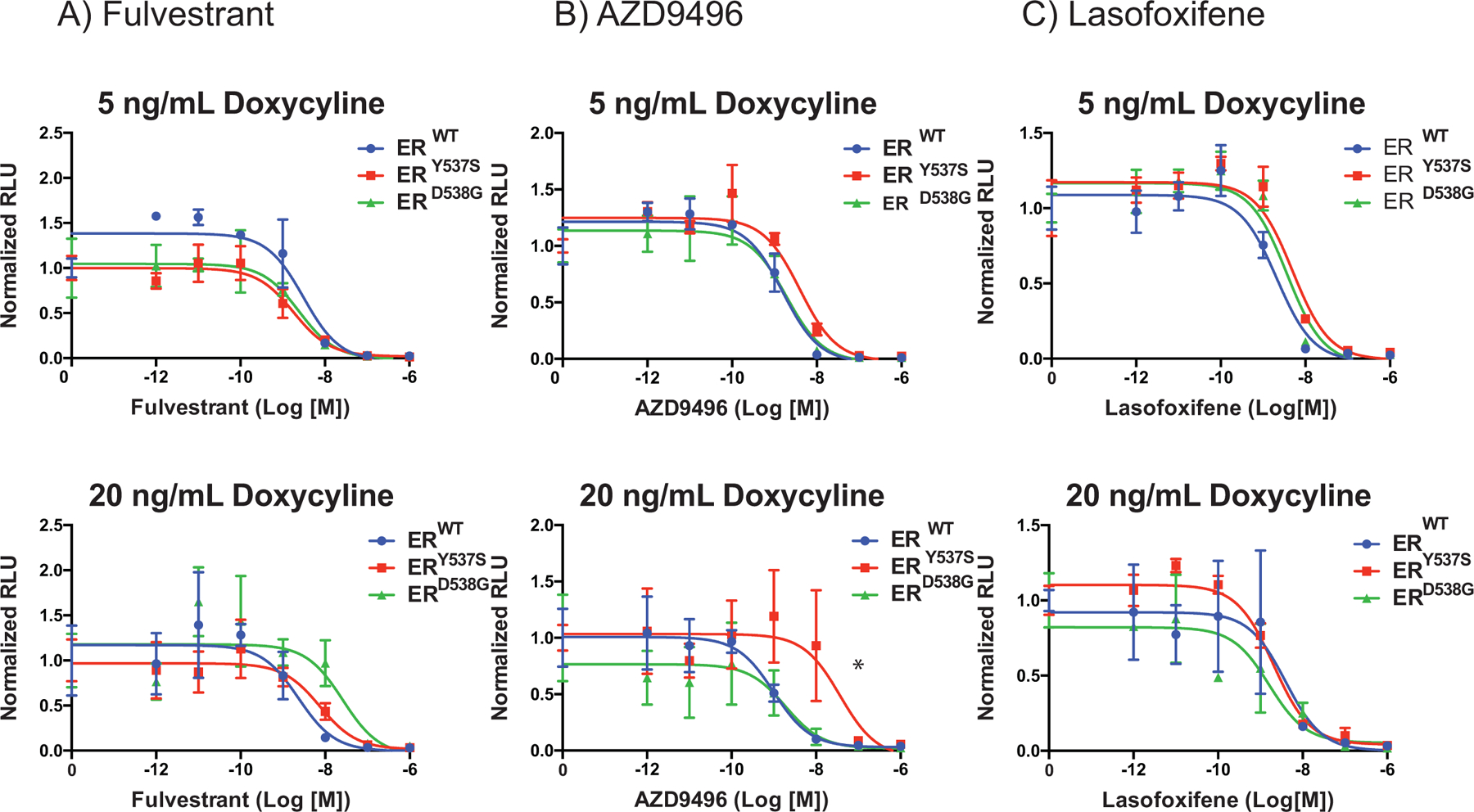
(A-C) MCF7 (ER-positive breast cancer) cells were engineered to express the WT or mutant receptors in a dose-dependent manner in response to doxycycline treatment over the endogenous ERWT. Cells were plated in phenol red-free media for 48 hours with doxycycline 5 or 20 ng/ml as indicated and then transfected with an estrogen responsive reporter gene (7X-ERE-tata-Luc). After 5 hours, cells were treated with 17β-estradiol (0.1 nM) and ER antagonists (10−12 M to 10−6 M). Firefly luciferase activity was assessed and normalized to β-galactosidase transfection control (Y-Axis). Data points are the mean of three technical replicates, and error bars are the standard deviation of these replicates. Data presented is a representative of three independent experiments. Two-way ANOVA was utilized, comparing the logIC50 of all three independent experiments, to determine if there were significant differences between the WT and mutant receptors. Significant differences (p-value < 0.05) of the mutant IC50s when compared to that of the WT that were determined by this analysis are represented with a star.
A series of experiments were designed to examine whether the altered pharmacology of select ERmuts was solely an artifact of their overexpression or if overexpression was required to outcompete a normalizing effect of ERWT. To this end, the impact of altering the expression of ERWT relative to ERmuts was evaluated initially in SKBR3 cells. Western blots were used to confirm that the desired changes in receptor/mutant expression was accomplished (Supplemental Fig. S9). Consistent with the results presented in Figure 2, the potency of fulvestrant and AZD9496 was reduced in SKBR3 cells expressing ERY537S or ERD538G alone (Fig. 5A and B). However, as the expression of the ERWT was increased to comparable levels with ERmut, the pharmacology of fulvestrant and AZD9496 was normalized to that which mirrored their activity on ERWT (Fig. 5 A–B). Lasofoxifene antagonist efficacy remained unchanged as the expression levels of ERWT and ERmut were altered (Fig. 5C). It is important to note that in the absence of ligand, the constitutive activity of the mutant receptors is observed even when ERWT is present (Supplemental Fig. S10). Thus, in the absence of hormone, the mutant is functionally in excess indicating that ERWT activity, and not its expression alone, is required to achieve the normalization of ER pharmacology noted. This finding supports previous data in the literature that demonstrate that under conditions of extreme hormone deprivation, the resistance of ERmuts to ER ligands is not affected by the presence of ERWT(32,42). To support these findings, we performed an analogous experiment in MCF7I cells (Fig. 5D–F). In this context, we expressed ERmuts in cells expressing endogenous ERWT and consistent with our prior observations, the pharmacology of cells expressing ERWT or ERmuts were found to be indistinguishable. However, when the expression of the endogenous receptor was reduced using an siRNA directed against the 3’ UTR of the ER mRNA, the mutant pharmacology emerged. Doxycycline-induced expression of ERWT and ERmuts and the effectiveness of the siRNA mediated knockdown of endogenous ER protein levels were confirmed by In-Cell Western (Supplemental Fig. S11). As observed in SKBR3 cells, the constitutive activity of the mutant receptors was not diminished by coexpression of ERWT (Supplemental Fig. S12). Importantly, the impact of ERWT/ERmut status on ligand pharmacology, established using a synthetic reporter assay, was also seen when the activity of ligands were assessed using endogenous target gene transcription on PgR and GREB1 (Supplemental Fig. S13). Together, these results indicate that activated ERWT can normalize the pharmacology of ERY537S and ERD538G, and that tumor response to ER ligands following aromatase inhibitor therapy will depend on the relative co-expression of ERmuts and ERWT in breast cancer cells. For reasons yet to be determined the pharmacology of the SERM lasofoxifene is not affected by mutant status.
Figure 5: The altered pharmacology of ERmuts is only evident when expressed at a level higher than the WT receptor:
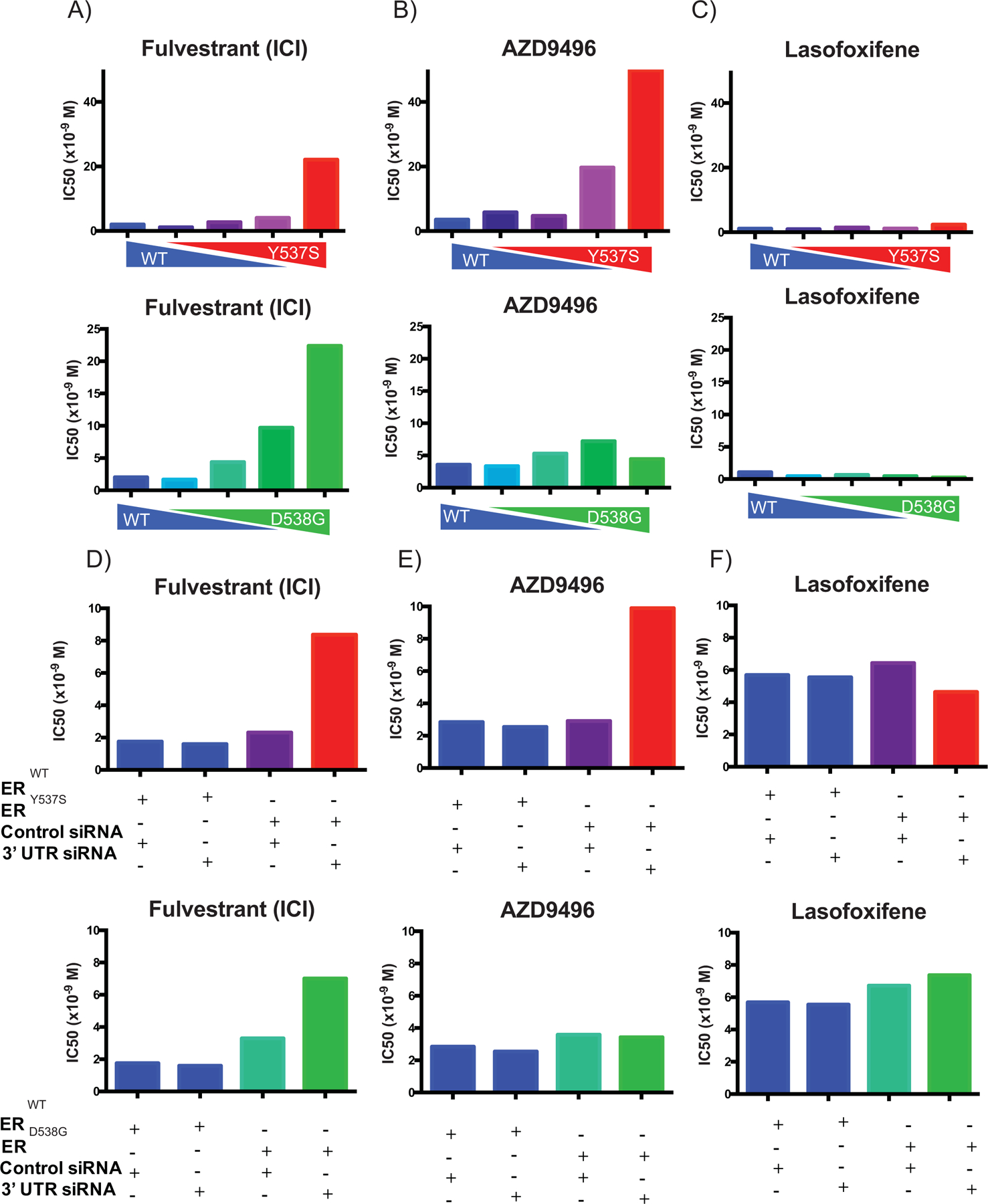
(A-C) SKBR3 (ER-negative breast cancer) cells plated in phenol red-free media supplemented with charcoal stripped serum and transfected with an estrogen responsive reporter gene (3X-ERE-tata-Luc) in the presence of different WT to mutant ER (Y537S or D538G) construct ratios. After 5 hours, cells were treated with 17β-estradiol (1 nM) and ER antagonists (10−12 M to 10−6 M). Firefly luciferase activity was assessed and normalized to β-galactosidase transfection control. The IC50s of each dose-response curve are plotted. Two-way ANOVA was utilized, comparing the logIC50s of all three independent experiments, to determine if there were significant differences between the WT and mutant receptors. Significant differences (p-value < 0.05) of the mutant IC50s when compared to that of the WT that were determined by this analysis are represented with a star. (D-F) MCF7 (ER-positive breast cancer) cells were engineered to express the WT or mutant receptors in response to doxycycline treatment. Cells were plated in phenol red-free media for 48 hours with doxycycline and siRNA (control or targeting 3’ UTR to knockdown the endogenous WT receptor) and then transfected with an estrogen responsive reporter gene (7X-ERE-tata-Luc). After 5 hours, cells were treated with 17β- estradiol (0.1 nM) and ER antagonists (10−12 M to 10−6 M). Firefly luciferase activity was assessed and normalized to β-galactosidase transfection control. The IC50s of each dose response curve are plotted. Two-way ANOVA was utilized, comparing the logIC50s of all three independent experiments, to determine if there were significant differences between the WT and mutant receptors. Significant differences (p-value < 0.05) of the mutant IC50s when compared to that of the WT that were determined by this analysis are represented with a star. Data presented is a representative of three independent experiments.
Discussion:
The goal of this study was to define the molecular basis for the altered pharmacology exhibited by the most clinically relevant ERmuts, information we anticipate could inform the selection of existing drugs for use in patients with advanced ER-positive breast cancer whose tumors harbor these mutations. In cell-based models of breast cancer, we made the important observation that when compared to ERWT, the pharmacology, most notably antagonist potency, of these mutants was significantly impacted by the relative co-expression of ERWT and ERmut. Previously, we and others have observed that the potency of existing ER antagonists was reduced in cells expressing either of the two most frequently occurring ERmuts (ERD538G and ERY537S) (24,25,34,40,46). In this study, we have demonstrated that such differences are dependent on the relative expression level of both the ERWT and ERmuts and are only apparent under conditions where ERmuts are substantially overexpressed relative to ERWT. Given the previously reported neomorphic activities of the ERmuts, it is possible that the differences in response to ER ligands may only be manifest on select endogenous target genes (32,33). However, in our system, the activity of the mutants in the transcriptional reporter assays mirror their activities, in the presence of various ligands, when cell proliferation or endogenous target gene transcription is used as the readout. These findings are significant as prior studies that have informed our current understanding of the importance of ERmuts in the pharmacotherapy of breast cancer were performed in cells expressing only ERmuts in the absence of the ERWT (24–26,34,40).
Whereas we have been able to confirm using several experimental models that ERWT normalizes the activity of coexpressed ERmuts, the mechanism(s) by which this activity occurs is elusive. One possibility is that ERWT preferentially dimerizes with ERmuts and simply outcompetes ERmut homodimers. However, such a simple mechanism would require that the ERmuts would exhibit reduced homodimerization/heterodimerization activity. It is more likely that in cells where ERWT and ERmut are present, and assuming no differences in dimerization ability, that the majority of the receptor (75%) would exist in an ERWT/ERWT or ERWT/ERmut complex and that the presence of the WT receptor normalizes the response (potency) to ligands. The recent cryo-EM structure of the ER coregulator complex is informative as to how ERWT may normalize the activity of the mutant (51). Specifically, it was observed that the establishment of a productive transcription complex requires each monomer in an ER dimer to engage a p160 coregulator (i.e. SRC-3) to establish a platform upon which p300 can be recruited. Thus, in an ERWT/ERWT or ERWT/ERmut complex, the conformational change(s) induced in ERWT by antagonists would result in the expulsion of one or two SRC proteins from the complex and a productive transcriptional complex could not form. Using peptide binding/cofactor binding studies, we have demonstrated that the interaction of ERmuts with coactivators is substantially inhibited upon the addition of saturating concentrations of most antagonists, explaining why the efficacy of existing inhibitors is not affected by the most commonly occurring mutations.
There are several immediate clinical implications of this work. It is clear that selection for ERmuts by estrogen deprivation (aromatase inhibitor) manifests as resistance. However, given that most mutants would be expected to be co-expressed in breast cancer cells with ERWT, it was unclear how they would impact the pharmacology of fulvestrant and other clinically important SERDs and SERMs. Our findings suggest that in ERWT expressing cells the presence of a mutant receptor is unlikely to have any significant impact on response to existing antagonists unless its expression vastly exceeds that of ERWT (or in cells in which it is solely expressed). Some clinical data supports that assertion (29). Specifically, baseline and on treatment evaluation of ERmuts in circulating tumor DNA was evaluated and correlated to fulvestrant response (alone or in combination with PI3K inhibition) as a part of the Phase II FERGI study (NCT01437566). The findings of this study demonstrated that median ESR1 allele frequency was low at 0.45% and as such progression-free survival was not different in patients with ERmuts compared with ERWT patients (29). Conversely, in the PALOMA- 3 study (NCT01942135), there were observed differences in fulvestrant progression-free survival in response to mutation status (52,53). Interestingly, these studies report a higher whole tumor allele frequency of ERmuts, with a reported expression fraction of 0.10 (or 10%). This study also suggested that ERmut containing clones were a small fraction of the whole tumor and as such the low allele frequency estimate was not representative of each individual cell. It is clear that cells expressing ERY537S emerged in the fulvestrant only arm of the PALOMA-3 arm and this has been taken as definitive evidence that this mutation reduces the potency of fulvestrant. We propose the alternative hypothesis that fulvestrant exposure is not sufficient to efficiently occupy ERWT, or the ERmuts, and that cells expressing the constitutively active mutants have a fitness advantage.
In our study, dysregulated ERmut pharmacology is only manifest when the expression of the mutant receptor(s) exceeds that of its ERWT counterpart. It is not clear how often this occurs in individual tumor cells and further research is needed to adequately assess allelism at the cellular level. Mutations in ESR1 can be detected in clinical tumor samples and circulating tumor DNA using next-generation sequencing and ddPCR (24,25,27–31,52,53). However, these assays are not designed to establish allelic frequency (homozygous versus heterozygous ESR1 alleles) on a single cell basis. The likely importance of ERmut allelism was suggested in a recent study that revealed a propensity for a loss of heterozygosity of ERWT when an ERmut is also present in the tumors of patients on endocrine therapies (54). Specifically, in breast cancer patients that harbored ESR1 mutants, LOH of the WT allele drove 78% of ESR1 mutant specific allele balance, while background loss of alleles for non-mutant containing tumors also on endocrine therapy was only 30%. These data suggest that the ERWT is important in determining ERmut response to therapy and that tumors having a lower expression ERWT have a survival advantage. The inability to assess ERWT/ERmut allelism in a facile manner reinforces the need to understand the relationship between ER expression level and ligand potency/efficacy as a means to select/develop pharmaceuticals for use in the treatment of patients whose mutants harbor ERmuts.
It is likely that even in situations where the ERmuts is expressed at a higher level compared to ERWT, it is only of significance when potency is a limiting property of a drug (i.e. fulvestrant). However, our work suggests that most of the liabilities of the mutants can be mitigated by increasing the dose (assuming dose-proportional exposure and tolerable side-effect profile) of individual drugs as antagonist efficacy is not compromised by the expression of the most commonly occurring ERmuts. This highlights the importance of drug exposure when considering new/existing drugs for use in the treatments of patients with mutant receptors. One approach that has been developed to address the reduced affinity of the mutants is to develop Selective Estrogen Receptor Covalent Antagonists (SERCAs) (55,56). The first of this new class of drugs, currently in clinical development, essentially converts tamoxifen into a covalent ER binder, thus mitigating the impact of the mutation on binding affinity. One SERCA is currently in clinical trials for metastatic breast cancer patients progressing on endocrine therapy (NCT03250676) (56). However, it is likely that locking the receptor in a “tamoxifen-induced conformation” is going to result in the selection of cancer cells which support the partial agonist activity of tamoxifen, an activity that is associated with acquired resistance (21,57,58).
The SERM lasofoxifene appears to have attributes that would make it particularly useful in patients where there is concern as to the contribution of ERmuts to drug response. In this study, and an earlier study in gynecological cancers, we demonstrated that this drug is an efficient antagonist whose actions are not influenced by mutant status (46). Lasofoxifene was initially developed for the treatment of climacteric symptoms and osteoporosis associated with menopause. It is currently under evaluation in the ELAINE trial (NCT03781063) to assess its efficacy compared to fulvestrant, post aromatase, and CDK4/6 inhibitor therapy, as a treatment for patients whose tumors harbor ERmuts. We also noted that the acidic SERDs (represented by GDC-0810, AZD9496 and previously GW7604 (46)) are ineffective inhibitors of ERY537S. Thus, it is likely that the efficacy of this class of drugs will be diminished as the allelic frequency of ERY537S increases in patients. Currently, there are numerous new ER modulators, including LSZ102 (NCT202734615), AZD9833 (NCT03616586), GDC-9545 (NCT03332797), SAR429859 (NCT03284957), G1T48 (NCT03455270) and Zn-C5 (NCT03560531) under evaluation in the clinic (59). Notwithstanding the potential impact of ERY537S on the response to acidic SERDs, it appears as if the mutant status of tumors may not be a significant issue for drugs that achieve significant exposure to offset the decreased potency noted (for all but lasofoxifene). We believe the studies presented herein should emphasize approaches to achieve maximal drug exposure in tumors as opposed to developing new molecules that demonstrate increased affinity for the mutant receptors.
Supplementary Material
Acknowledgements:
Supporting funding provided by the NIDDK R01 DK048807 (D.P. McDonnell), NIGMS T32 GM 007105 and NCI F31 CA220978 (K.J. Andreano).
DPM receives research funding from Novartis. SS receives research funding from Astra Zeneca and Eli Lilly.
Footnotes
Conflict of interest statement:
KJA, CYC and DPM are inventors on a Duke patent for the use of lasofoxifene as a treatment for breast tumors harboring ESR1 mutations. This has been licensed to Sermonix Pharmaceuticals and these authors could receive royalties through Duke.
SEW and DPM are inventors on Duke patents for the use of RAD1901 in breast cancer. This has been licensed to Radius Health and these authors could receive royalties through Duke.
The other authors declare no potential conflicts of interest.
References:
- 1.Ignatiadis M, Sotiriou C. Luminal breast cancer: from biology to treatment. Nat Rev Clin Oncol 2013;10:494–506 [DOI] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maximov PY, Lee TM, Jordan VC. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol 2013;8:135–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rydén L, Heibert Arnlind M, Vitols S, Höistad M, Ahlgren J. Aromatase inhibitors alone or sequentially combined with tamoxifen in postmenopausal early breast cancer compared with tamoxifen or placebo - Meta-analyses on efficacy and adverse events based on randomized clinical trials. Breast 2016;26:106–14 [DOI] [PubMed] [Google Scholar]
- 5.Kim HA, Lee JW, Nam SJ, Park BW, Im SA, Lee ES, et al. Adding Ovarian Suppression to Tamoxifen for Premenopausal Breast Cancer: A Randomized Phase III Trial. J Clin Oncol 2019:JCO1900126. [DOI] [PubMed] [Google Scholar]
- 6.Brodie A Aromatase inhibitors in breast cancer. Trends Endocrinol Metab 2002;13:61–5 [DOI] [PubMed] [Google Scholar]
- 7.Dowsett M, Howell A. Breast cancer: aromatase inhibitors take on tamoxifen. Nat Med 2002;8:1341–4 [DOI] [PubMed] [Google Scholar]
- 8.Mokbel K The evolving role of aromatase inhibitors in breast cancer. Int J Clin Oncol 2002;7:279–83 [DOI] [PubMed] [Google Scholar]
- 9.Palmieri C, Patten DK, Januszewski A, Zucchini G, Howell SJ. Breast cancer: current and future endocrine therapies. Mol Cell Endocrinol 2014;382:695–723 [DOI] [PubMed] [Google Scholar]
- 10.Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol 2010;28:4594–600 [DOI] [PubMed] [Google Scholar]
- 11.Nathan MR, Schmid P. A Review of Fulvestrant in Breast Cancer. Oncol Ther 2017;5:17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonnell DP, Wardell SE, Norris JD. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J Med Chem 2015;58:4883–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howell A, Sapunar F. Fulvestrant revisited: efficacy and safety of the 500-mg dose. Clin Breast Cancer 2011;11:204–10 [DOI] [PubMed] [Google Scholar]
- 14.Robertson JF, Erikstein B, Osborne KC, Pippen J, Come SE, Parker LM, et al. Pharmacokinetic profile of intramuscular fulvestrant in advanced breast cancer. Clin Pharmacokinet 2004;43:529–38 [DOI] [PubMed] [Google Scholar]
- 15.van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov 2015;5:72–81 [DOI] [PubMed] [Google Scholar]
- 16.De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, et al. Optimization of a Novel Binding Motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Receptor Downregulator and Antagonist. J Med Chem 2015;58:8128–40 [DOI] [PubMed] [Google Scholar]
- 17.Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, et al. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J Med Chem 2015;58:4888–904 [DOI] [PubMed] [Google Scholar]
- 18.Joseph JD, Darimont B, Zhou W, Arrazate A, Young A, Ingalla E, et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, et al. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer Res 2016;76:3307–18 [DOI] [PubMed] [Google Scholar]
- 20.Hamilton EP, Patel MR, Armstrong AC, Baird RD, Jhaveri K, Hoch M, et al. A First-in-Human Study of the New Oral Selective Estrogen Receptor Degrader AZD9496 for ER. Clin Cancer Res 2018;24:3510–8 [DOI] [PubMed] [Google Scholar]
- 21.Connor CE, Norris JD, Broadwater G, Willson TM, Gottardis MM, Dewhirst MW, et al. Circumventing tamoxifen resistance in breast cancers using antiestrogens that induce unique conformational changes in the estrogen receptor. Cancer Res 2001;61:2917–22 [PubMed] [Google Scholar]
- 22.Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer 2015;22:713–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, et al. Elacestrant (RAD1901), a Selective Estrogen Receptor Degrader (SERD), Has Antitumor Activity in Multiple ER+ Breast Cancer Patient-derived Xenograft Models. . Clin Cancer Res 2017;23:4793–804 [DOI] [PubMed] [Google Scholar]
- 24.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 2014;20:1757–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 2015;7:313ra182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol 2016;34:2961–8 [DOI] [PubMed] [Google Scholar]
- 29.Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 2016;7:11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018;34:427–38.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leal MF, Haynes BP, Schuster EF, Yeo B, Afentakis M, Zabaglo L, et al. Early enrichment of ESR1 mutations and the impact on gene expression in pre-surgical primary breast cancer treated with aromatase inhibitors. Clin Cancer Res 2019 [DOI] [PubMed] [Google Scholar]
- 32.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018;33:173–86.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res 2017;19:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 Mutations Differentially Impact the Efficacy of ER Antagonists. Cancer Discov 2017;7:277–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagel SC, Hagelbarger JL, McDonnell DP. Development of an ER action indicator mouse for the study of estrogens, selective ER modulators (SERMs), and Xenobiotics. Endocrinology 2001;142:4721–8 [DOI] [PubMed] [Google Scholar]
- 36.Norris J, Fan D, Aleman C, Marks JR, Futreal PA, Wiseman RW, et al. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J Biol Chem 1995;270:22777–82 [DOI] [PubMed] [Google Scholar]
- 37.Norris JD, Paige LA, Christensen DJ, Chang CY, Huacani MR, Fan D, et al. Peptide antagonists of the human estrogen receptor. Science 1999;285:744–6 [DOI] [PubMed] [Google Scholar]
- 38.Huang HJ, Norris JD, McDonnell DP. Identification of a negative regulatory surface within estrogen receptor alpha provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol 2002;16:1778–92 [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi S, Stice JP, Kazmin D, Wittmann BM, Kimbrel EA, Edwards DP, et al. Mechanisms of progesterone receptor inhibition of inflammatory responses in cellular models of breast cancer. Mol Endocrinol 2010;24:2292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wardell SE, Ellis MJ, Alley HM, Eisele K, VanArsdale T, Dann SG, et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clin Cancer Res 2015;21:5121–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosati RL, Da Silva Jardine P, Cameron KO, Thompson DD, Ke HZ, Toler SM, et al. Discovery and preclinical pharmacology of a novel, potent, nonsteroidal estrogen receptor agonist/antagonist, CP-336156, a diaryltetrahydronaphthalene. J Med Chem 1998;41:2928–31 [DOI] [PubMed] [Google Scholar]
- 42.Kuang Y, Siddiqui B, Hu J, Pun M, Cornwell M, Buchwalter G, et al. Unraveling the clinicopathological features driving the emergence of ESR1 mutations in metastatic breast cancer NPJ Breast Cancer 2018;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol 1997;11:657–66 [DOI] [PubMed] [Google Scholar]
- 44.Wijayaratne AL, Nagel SC, Paige LA, Christensen DJ, Norris JD, Fowlkes DM, et al. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology 1999;140:5828–40 [DOI] [PubMed] [Google Scholar]
- 45.Wang T, Liu JH, Zhang J, Wang L, Chen C, Dai PG. A multiplex allele-specific real-time PCR assay for screening of ESR1 mutations in metastatic breast cancer. Exp Mol Pathol 2015;98:152–7 [DOI] [PubMed] [Google Scholar]
- 46.Gaillard SL, Andreano KJ, Gay LM, Steiner M, Jorgensen MS, Davidson BA, et al. Constitutively active ESR1 mutations in gynecologic malignancies and clinical response to estrogen-receptor directed therapies. Gynecol Oncol 2019;154:199–206 [DOI] [PubMed] [Google Scholar]
- 47.Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell 2005;18:413–24 [DOI] [PubMed] [Google Scholar]
- 48.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997;389:753–8 [DOI] [PubMed] [Google Scholar]
- 49.Norris JD, Joseph JD, Sherk AB, Juzumiene D, Turnbull PS, Rafferty SW, et al. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem Biol 2009;16:452–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gee AC, Carlson KE, Martini PG, Katzenellenbogen BS, Katzenellenbogen JA. Coactivator peptides have a differential stabilizing effect on the binding of estrogens and antiestrogens with the estrogen receptor. Mol Endocrinol 1999;13:1912–23 [DOI] [PubMed] [Google Scholar]
- 51.Yi P, Wang Z, Feng Q, Pintilie GD, Foulds CE, Lanz RB, et al. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell 2015;57:1047–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov 2018;8:1390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Leary B, Hrebien S, Morden JP, Beaney M, Fribbens C, Huang X, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun 2018;9:896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bielski CM, Donoghue MTA, Gadiya M, Hanrahan AJ, Won HH, Chang MT, et al. Widespread Selection for Oncogenic Mutant Allele Imbalance in Cancer. Cancer Cell 2018;34:852–62.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puyang X, Furman C, Zheng GZ, Wu ZJ, Banka D, Aithal K, et al. Discovery of Selective Estrogen Receptor Covalent Antagonists for the Treatment of ERα. Cancer Discov 2018;8:1176–93 [DOI] [PubMed] [Google Scholar]
- 56.Rioux N, Smith S, Korpal M, O’Shea M, Prajapati S, Zheng GZ, et al. Nonclinical pharmacokinetics and in vitro metabolism of H3B-6545, a novel selective ERα covalent antagonist (SERCA). Cancer Chemother Pharmacol 2019;83:151–60 [DOI] [PubMed] [Google Scholar]
- 57.Cocce KJ, Jasper JS, Desautels TK, Everett L, Wardell S, Westerling T, et al. The Lineage Determining Factor GRHL2 Collaborates with FOXA1 to Establish a Targetable Pathway in Endocrine Therapy-Resistant Breast Cancer. Cell Rep 2019;29:889–903.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003;95:353–61 [DOI] [PubMed] [Google Scholar]
- 59.Tria GS, Abrams T, Baird J, Burks HE, Firestone B, Gaither LA, et al. Discovery of LSZ102, a Potent, Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) for the Treatment of Estrogen Receptor Positive Breast Cancer. J Med Chem 2018;61:2837–64 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.