

Abstract
The O-glycoprotein apolipoprotein E (APOE), the strongest genetic risk factor for Alzheimer’s disease, associates with lipoproteins. Cerebrospinal fluid (CSF) APOE binds only high-density lipoproteins (HDLs), while plasma APOE attaches to lipoproteins of diverse sizes with binding fine-tuned by the C-terminal loop. To better understand the O-glycosylation on this critical molecule and differences across tissues, we analyzed the O-glycosylation on APOE isolated from the plasma and CSF of aged individuals. Detailed LC-MS/MS analyses allowed the identification of the glycosite and the attached glycan and site occupancy for all detectable glycosites on APOE and further three-dimensional modeling of physiological glycoforms of APOE. APOE is O-glycosylated at several sites: Thr8, Thr18, Thr194, Ser197, Thr289, Ser290 and Ser296. Plasma APOE held more abundant (20.5%) N-terminal (Thr8) sialylated core 1 (Neu5Acα2–3Galβ1–3GalNAcα1-) glycosylation compared to CSF APOE (0.1%). APOE was hinge domain glycosylated (Thr194 and Ser197) in both CSF (27.3%) and plasma (10.3%). CSF APOE held almost 10-fold more abundant C-terminal (Thr289, Ser290 and Ser296) glycosylation (36.8% of CSF peptide283–299 was glycosylated, 3.8% of plasma peptide283–299), with sialylated and disialylated (Neu5Acα2–3Galβ1–3(Neu5Acα2–6) GalNAcα1-) core 1 structures. Modeling suggested that C-terminal glycosylation, particularly the branched disialylated structure, could interact across domains including the receptor-binding domain. These data, although limited by sample size, suggest that there are tissue-specific APOE glycoforms. Sialylated glycans, previously shown to improve HDL binding, are more abundant on the lipid-binding domain of CSF APOE and reduced in plasma APOE. This indicates that APOE glycosylation may be implicated in lipoprotein-binding flexibility.
Keywords: APOE, Alzheimer’s disease, glycoproteomics, lipoprotein binding, tissue-specific glycosylation
Introduction
Apolipoprotein E (APOE) is the most significant genetic risk factor for late-onset Alzheimer’s disease (AD), the most common form of AD (Corder et al. 1993; Lambert et al. 2013). Compared to the most common APOE3 allele, each APOE4 allele increases risk of AD 2.5 times (Harold et al. 2009), while the APOE2 allele decreases risk (Wu and Zhao 2016). The APOE alleles encode a mature secreted protein of 299 amino acids, with single amino acid substitutions accounting for each isoform (E2: Cys112, Cys158; E3: Cys112; Arg158; E4: Arg112, Arg158) (Rall et al. 1982).
APOE is essential for lipid transport in the brain and plasma and is able to bind lipoproteins of diverse sizes and shapes to carry out its complex roles. In the brain, APOE is expressed by astrocytes, microglia and the choroid plexus (Pitas et al. 1987a; Xu et al. 2006; Achariyar et al. 2016). APOE associates with small high-density lipoproteins (HDLs) in the brain which increase in size in the cerebrospinal fluid (CSF) (Pitas et al. 1987b; LaDu et al. 1998; Koch et al. 2001). CSF APOE diffuses into the plasma via arachnoid granulations in the superior sagittal sinus (Segal 2005).
The APOE lifecycle is more complex in the periphery taking part in the HDL, exogenous and endogenous cholesterol pathways. Expressed primarily by hepatocytes as well as macrophages (Mahley 1988; Kockx et al. 2008), APOE is found on plasma HDL (Otvos 2002). The endogenous pathway begins with nascent very low-density lipoprotein (VLDL) production in the liver containing APOE and released into the plasma. Plasma VLDLs are hydrolyzed to intermediate density lipoproteins which can contain multiple APOE molecules before APOE is lost (Beisiegel 1998). The exogenous pathway involves intestinal derived chylomicrons which enter the plasma where they gain APOE from circulated HDL and reduce in size by lipoprotein lipase becoming remnant particles which are removed by the liver via APOE receptor binding (Patsch 1998; Dawson and Rudel 1999; Mahley and Ji 1999).
APOE is an O-glycoprotein that contains an N-terminal four-helix receptor-binding domain, a central flexible hinge region and a C-terminal three helix lipid-binding domain (Lalazar et al. 1988; Sakamoto et al. 2008; Nguyen et al. 2010; Chen et al. 2011). Although models differ slightly, on binding to lipoprotein, APOE undergoes a change separating the N and C terminals (Chen et al. 2011; Frieden et al. 2017). This alteration exposes the previously buried receptor-binding domain, ensuring that optimal binding to members of the LDL receptor (LDLR) family is achieved only with fully lipidated APOE (Chen et al. 2011). Given the flexibility of the APOE structure and the significance of this characteristic to its function, the position and nature of its O-glycosylation are particularly important.
Mucin-like O-glycosylation is made up of eight core structures, with cores 1–4 principally in humans, that can be extended by the addition of monosaccharides such as sialic acid (N-acetylneuraminic acid, Neu5Ac) (Brockhausen et al. 2009), often biologically significant due to its size and negative charge (Ali et al. 2014; Chaudhury et al. 2016). Biosynthesis begins with the addition of an N-acetyl galactosamine (GalNAc) to the hydroxyl group of a serine or threonine by 1 of 20 redundant UDP-GalNAC: polypeptide N-acetylgalactosaminyl-transferases (GalNAc-T) (Brockhausen et al. 2009; Bennett et al. 2012). These enzymes differ in tissue expression and substrate specificities; for example, GalNAcT1 and 2 are expressed ubiquitously while others show narrow tissue particularity (Bennett et al. 2012). The core 1 structure (Galβ1–3GalNAcα1-), a simple and very common structure, is then completed by the addition of a galactose (Gal) by core 1 β3-galactosyltransferase (Ju and Cummings 2002). Sialic acid can be added by a range of linkage and monosaccharide-dependent sialytransferases most commonly creating two to three or two to six linkages with the adjoining monosaccharide generating (Patel and Balaji 2006), for example, the linear sialylated (Neu5Acα2–3Galβ1–3GalNAcα1-) or disialylated (Neu5Acα2–3Galβ1–3(Neu5Acα2–6) GalNAcα1-) core 1 structures.
Early work first identified that Thr194, within the hinge region, could be glycosylated in plasma APOE (Wernette-Hammond et al. 1989). While there is evidence that intracellular APOE is more heavily glycosylated than secreted or plasma APOE (Zannis et al. 1986; Lee et al. 2010), there has been no study to characterize the glycans attached and site occupancy at all sites between the CSF and plasma. To better understand APOE glycosylation and its potential role on its varied functions, we undertook glycoproteomic analyses of APOE isolated from CSF and plasma. We used these glycoprofiles to model CSF and plasma glyco-APOE variants.
Results
N-terminal glycosylation
APOE was isolated by immunoprecipitation from older adult CSF (n = 4, mean age 58.25 years) and plasma (n = 4, mean age 66.75 years) of known APOE genotype (Supplementary Table SI). APOE tryptic peptides and glycopeptides were analyzed using a top 30 method using a TripleTOF© 6600 QTOF (Sciex) allowing at least 10 points on the curve. All HexHexNAc structures identified by mass spectrometry are assumed to be GalGalNAc, forming a core 1 structure of known linkage (Galβ1–3GalNAcα1-). Branched di-Neu5Ac HexHexNAc with spectra showing diagnostic ions representing sialylation of both Hex and HexNAc residues are understood to have the following Neu5Ac linkages: Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1-, forming the disialylated core 1 structure. The linear Neu5AC-GalGalNAc structure identified by mass spectrometry to have a sialylated Hex is assumed to be the Neu5Ac α2–3 linkage, creating the Neu5Acα2–3Galβ1–3GalNAcα1- structure as have all been extensively studied (Brockhausen et al. 2009).
The N-terminal tryptic peptide 1–15 was found to be glycosylated at Thr8, the only possible site on the peptide sequence, KVEQAVETEPEPELR (Figure 1). The first lysine residue of the mature APOE protein was not trypsinized; this was compared in each sample, and pep2–15 was orders of magnitude lower in abundance compared to pep1–15 (Supplementary Figure S1). Pep1–15 (Figure 1A) was identified at the expected parent mass of an attached sialylated core 1 structure. MS/MS of this parent (Figure 1B) gave Neu5Ac (m/z 274.09 and 292.10) and Gal-GalNAc (m/z 366.15) fragments confirming the sialylated core 1 structure. The Neu5Acα2–3Galβ1–3GalNAcα1- linear form was confirmed by the identification of the Neu5Ac-Gal peak at m/z 454.15 (Supplementary Figure S2) using an APOE plasma standard and a specific targeted MS method. No other attached glycan could be identified and confirmed by MS/MS in the samples tested, although the disialylated core 1 structure was able to be detected in the standard APOE at high concentrations using a directed method (Supplementary Figure S2).
Fig. 1.
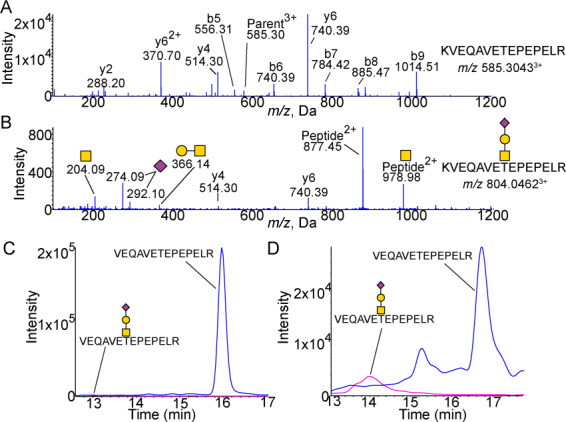
Glycosylation of the N-Terminal 1–15 peptide. (A) MS/MS spectra of unglycosylated KVEQAVETEPEPELR peptide. Expected m/z 585.3037. (B) MS/MS spectra of peptide with Neu5Acα2–3Galβ1–3GalNAcα1 attached showing peaks of Neu5Ac (m/z 274.09 and 292.10) as well as Galβ1–3GalNAc (m/z 366.14) and GalNac (m/z 204.09). Expected m/z 804.0463. XICs of unglycosylated and sialylated core 1 glycosylated 1–15 peptide from (C) CSF (n = 4) and (D) plasma (n = 4). CSF shows a higher proportion of unglycosylated peptide with the glycosylated peptide. The background is low except for the unglycosylated peptide in plasma which shows a higher background generally as well as an additional unrelated peak (confirmed to be unrelated by MS/MS) at 15.3 min. Blue is peptide. Pink is peptide with Neu5Acα2–3Galβ1–3GalNAcα1 structure attached. All masses are observed masses.
While only the Neu5Acα2–3Galβ1–3GalNAcα1- structure was identified, the site occupancy was different between CSF and plasma. Quantitation by area under the curve was determined from extracted ion chromatogram (XIC) and relative quantitation shown for each form of the peptide. CSF APOE showed very low glycosylation, with the Neu5Acα2–3Galβ1–3GalNAcα1- pep1–15 only 0.1% of the total quantified pep1–15 (Figures 1C and 4B and C). Plasma, however, showed a significantly different (P < 0.05) relative abundance of the glycosylated form with more glycosylation compared to CSF: 20.5% of pep1–15 was glycosylated with the Neu5Acα2–3Galβ1–3GalNAcα1- structure (Figures 1D and 5A).
Fig. 4.
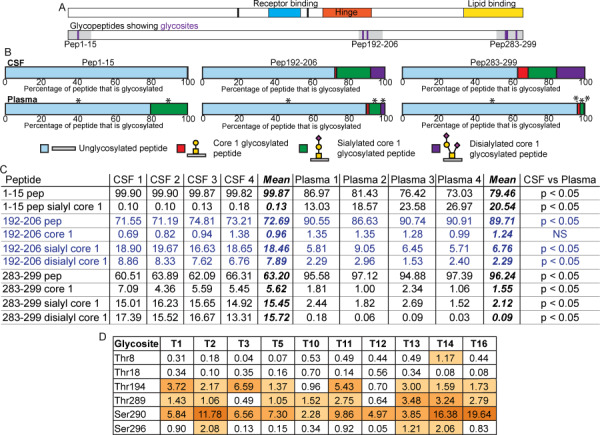
Comparison of the glycoprofiles of CSF and plasma APOE. (A) Schematic of APOE domain structures showing amino acids 112 and 158 in black, the receptor-binding domain in blue, the hinge in orange and the lipid-binding domain in yellow. The schematic below shows glycosylated tryptic peptides in grey with position of glycosites in purple. (B) Average percentage of identified peptide that was unglycosylated (pale blue), glycosylated with Galβ1–3GalNAcα1- (red), Neu5Acα2–3Galβ1–3GalNAcα1- (green) or Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1- (purple) from CSF (n = 4, top) and plasma (n = 4, bottom) samples. Asterisks (*) above the plasma bars designates peptides that showed a statistically significant difference between the CSF and plasma samples where the P value was less than 0.05 as determined by a Mann–Whitney nonparametric test. P values were FDR adjusted to correct for multiple comparisons. (C) Relative quantitative data and statistics for individual CSF and plasma samples shown in (B). All data are shown as percentages, and the mean follows the four CSF and four plasma samples. As described above, Mann–Whitney non-parametric tests were performed to compare the results obtained for each peptide from the CSF and plasma samples. P values were FDR-adjusted to correct for multiple comparisons. These P value results are shown at the end of the table with significance designated as a P value of less than 0.05. Blue text is used only to delineate peptides for ease of reading. (D) ISOGlyP results for APOE glycosites. T refers to the individual GalNAc-T. Results are shown as EVP (enhancement value product) which refers to the preference a GalNAc-T shows towards glycosylating a glycosite: greater than 1 correlates with a positive likelihood of that glycosite being able to be glycosylated by that GalNAc-T, and less than 1 suggests a negative correlation with that specific GalNAc-T. The EVP does not correlate with the probability of that glycosite being glycosylated. ISOGlyP results are GalNAc-T-specific as it only considers 10 of the 20 known enzymes; others may contribute to glycosylation at any particular glycosite.
Fig. 5.
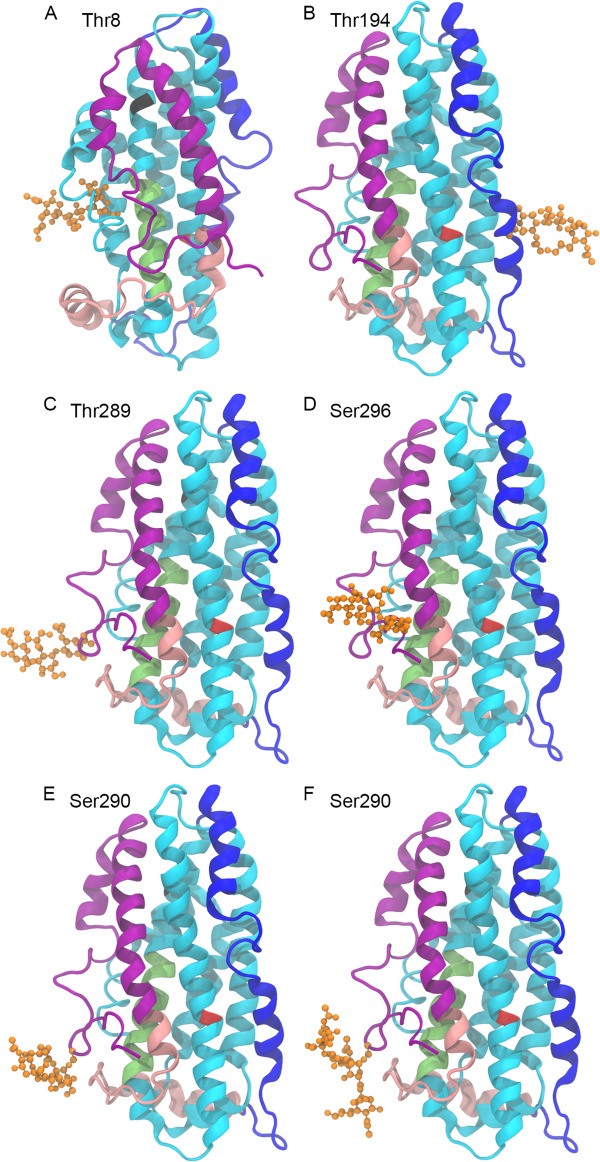
Structures of CSF and plasma glycovariants. All APOE structures are APOE3 NMR crystal structure 2L7B with the following glycosylation modeled at the indicated glycosites. (A) Thr8 glycosylated with Neu5Acα2–3Galβ1–3GalNAcα1-, most abundant in plasma APOE. (B) Thr194 glycosylated with Neu5Acα2–3Galβ1–3GalNAcα1-, identified in plasma and CSF APOE. (C–F) C-terminal glycosites, most abundant in CSF APOE. (C) Thr289 glycosylated with Neu5Acα2–3Galβ1–3GalNAcα1. (D) Ser296 glycosylated with Neu5Acα2–3Galβ1–3GalNAcα1. (E) Ser290 glycosylated with Neu5Acα2–3Galβ1–3GalNAcα1-. (F) Ser290 glycosylated with Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1-. The protein backbone is displayed in NewCartoon. The N-terminal region is cyan with the LDL receptor-binding domain in lime. The hinge region is in blue. The C-terminal is in pink with the lipid-binding region in purple. Amino acid 112 is red, amino acid 158 is black. The glycan is in orange displayed in CPK.
An additional low occupancy N-terminal glycopeptide, pep16–25, the next tryptic peptide, was also identified, but only when a high amount of standard APOE was analyzed. The peptide with mono and disialylated core 1 structures attached was identified, with the disialylated the more abundant (MS/MS Supplementary Figure S3).
Hinge glycosylation
The flexible hinge region was identified to contain a single glycopeptide, pep192–206, AATVGSLAGQPLQER. Although the glycopeptide shows two possible glycosites, there have been multiple studies identifying Thr194 as glycosylated in APOE (Wernette-Hammond et al. 1989; Halim et al. 2013). Our data suggests that, although two glycopeptides are not chromatographically separated, both Thr194 and Ser197 appear to be glycosylated giving a mixed glycopeptide spectra. This is apparent by the loss of H2O peak previously shown to indicate the deglycosylated amino acid attachment site (Greis and Hart 1998; Ko and Brodbelt 2015). Both the b3+ (m/z 244.13) and deglycosylated b3+ ion (m/z 226.12, Thr194) as well as the b6+ (m/z 487.25) and deglycosylated b6+ ion (m/z 469.24, Ser197) were observed in the same spectra (Figure 2). A considerably higher intensity of the deglycosylated b3+ ion suggests Thr194 is more heavily glycosylated than Ser197, as fits with previous findings of the identification of Thr194 over Ser197 despite their close proximity and the limited and/or imperfect site specificity of O-glycosylation. Pep192–206 was identified to hold three glycosylation types: core 1 (Figure 2A), sialylated core 1 (Figure 2B) and disialylated core 1 structures (Figure 2C). The distinction between the sialylated and disialylated core 1 glycosylated peptides is apparent by the difference in parent mass as well as relative intensity of sialic acid peaks (m/z 274.09 and 292.10) and the y-series ions. The MS/MS of the unglycosylated pep192–206 is shown in the Supplementary Figure S4. The linear Neu5Acα2–3Galβ1–3GalNAcα1- structure was again confirmed by the very low abundance m/z 454.15 fragment (Figure 2B). The Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1- structure was confirmed by the presence of the Neu5Acα2–3GalNAc fragment observed at m/z 495.18 (Figure 2C).
Fig. 2.
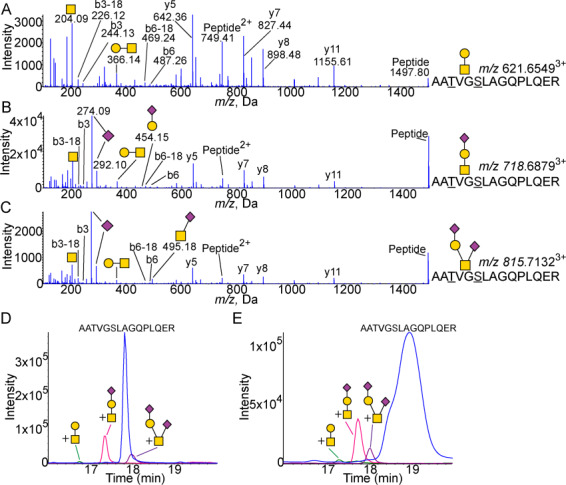
Glycosylation of the hinge domain 192–206 peptide. (A) MS/MS spectra of core 1 glycosylated AATVGSLAGQPLQER pep192–206 showing peaks Galβ1–3GalNAc (m/z 366.14) and GalNac (m/z 204.09). Expected m/z 621.6496. (B) MS/MS spectra of the peptide with Neu5Acα2–3Galβ1–3GalNAcα1 attached showing peaks of Neu5Ac (m/z 274.09 and 292.10) as well as Galβ1–3GalNAc (m/z 366.14) and GalNac (m/z 204.09). The linear structure is confirmed by the m/z 454.15 Neu5Acα2–3Gal fragment. Expected m/z 718.6813. (C) MS/MS spectra of peptide with Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1 attached showing peaks of Neu5Ac (m/z 274.09 and 292.10) as well as Galβ1–3GalNAc (m/z 366.14), Neu5Acα2–6GalNAc (m/z 495.18) and GalNac (m/z 204.09). Expected m/z 815.7131. XICs of unglycosylated and glycosylated 192–206 peptide from (D) CSF (n = 4) and (E) plasma (n = 4). Blue is peptide. Green is peptide with core 1 structure attached. Pink is peptide with sialylated core 1 structure attached. Purple is peptide with disialylated core 1 structure attached. All masses are observed masses.
The relative proportions of the three glycans were similar between the CSF (Figure 2D) and plasma (Figure 2E) APOE, with the highest proportion of Neu5Acα2–3Galβ1–3GalNAcα1- glycosylated pep192–206 followed by the Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1- structure and, finally, a very small proportion of the Galβ1–3GalNAcα1- structure. The site occupancy, however, differs between the source of APOE (Figure 4B and C, P < 0.05 for all peptides except the unmodified core 1 glycosylated peptide): CSF APOE held an overall 27.3% of glycosylated pep192–206 (disialylated 7.9%, sialylated 18.5%, unmodified 1.0%) compared to only 10.3% in plasma APOE (disialylated 2.3%, sialylated 6.8%, unmodified 1.2%).
Lipid-binding domain
One peptide within the lipid-binding domain was identified as O-glycosylated: pep283–299, the C-terminal peptide, VQAAVGTSAAPVPSDNH. The peptide was identified to hold only core 1 structures including the unmodified, monosialylated and disialylated forms (MS/MS in Supplementary Figure S5). The peptide contains three possible glycosites, Thr289, Ser290 and Ser296. All three sites were found to be glycosylated in CSF APOE (Figure 3). However, only a single site was found to be glycosylated on any given peptide, resulting in three MS/MS-confirmed chromatographic peaks for each of the Neu5Acα2–3Galβ1–3GalNAcα1- (Figure 3A) and Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1- forms (Figure 3B). Glycosites were determined for the monosialylated glycoform using loss of H2O peaks (Supplementary Figure S6) and disialylated glycoforms by relative retention times. The plasma APOE glycosylation was less intense, and a single chromatographic peak was identified for each glycoform (Supplementary Figure S7). Standard plasma APOE showed more glycoforms. As with the CSF, the plasma Thr289 and Ser290 Neu5Acα2–3Galβ1–3GalNAcα1- glycoforms were most intense, although difficult to separate (Supplementary Figure S7).
Fig. 3.
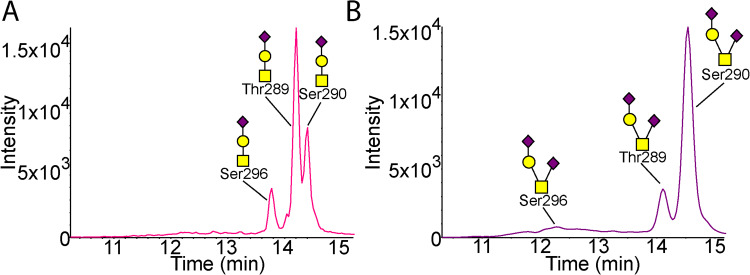
Glycosylation of the lipid-binding domain 283–299 peptide in CSF. (A) XIC of Neu5Acα2–3Galβ1–3GalNAcα1 glycosylated 283–299 peptide from CSF (n = 4). (B) XIC of Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1 glycosylated 283–299 peptide from CSF (n = 4). Each chromatogram shows three glycoforms of the 283–299 peptide for the two glycan structures as confirmed by MS/MS. MS/MS are shown in Fig. S6. Pink is peptide with sialylated core 1 structure attached. Purple is peptide with disialylated core 1 structure attached.
The APOE isolated from CSF held approximately 10 times more total pep283–299 glycosylation (36.8%) compared to plasma pep283–299 (3.8%, Figure 4B and C). The glycan proportions were also statistically significantly different (P < 0.05 for all pep283–299 peptides); plasma pep283–299 had a lower proportion of the core 1 disialylated structure (disialylated 0.1%, sialylated 2.1%, unmodified 1.6%) compared to CSF pep283–299 (disialylated 15.7%, sialylated 15.5%, unmodified 5.6%).
Comparison of glycosylation and GalNAc-T preference between CSF and plasma APOE
Relative quantitation of each glycovariant of the analyzed glycopeptides is shown as a percentage for each of the total of all variants identified for that peptide from CSF and plasma samples (individual sample data, Figure 4C). APOE from the two samples, CSF and plasma, show considerable glycosylation differences (Figure 4B and C). Plasma APOE has more abundant N-terminal glycosylation, and CSF APOE has more abundant C-terminal, lipid-binding domain, glycosylation. Both CSF and plasma APOE have hinge region glycosylation, although to a greater extent on CSF APOE (Figure 4A, B and C).
The suitability of each glycosite to be a substrate for specific GalNAc-Ts, GalNAc-T1–3, 5, 10–14 and 16, was also analyzed using ISOGlyP (Kong et al. 2015). ISOGlyP site preference results (Figure 4D) are given as EVP (enhancement value product): the likelihood the given GalNAc-T contributed to glycosylating that glycosite, where below 1 the GalNac-T is unlikely to have contributed and above 1 the likelihood is enhanced (Kong et al. 2015). The N-terminal sites Thr8 and Thr18 were all below 1 for the available enzymes except for Thr8 and GalNAc-T14, with an EVP 1.17. Thr-194 was shown to prefer the ubiquitous GalNAc-T1 (EVP 3.72) as well as T3 (EVP 6.59) and T11 (EVP 5.43). The C-terminal glycosites instead had a preference for less common GalNAc-Ts. Thr289 showed a preference for T13, T14 and T16 (EVP 3.48, 3.24, 2.79), and Thr290 showed a marked preference for T16 (EVP 19.64), followed by T14 (EVP 16.38) and T2 (EVP 11.78). Thr296 showed less GalNAc-T specificity overall though favored T2, T14 and T13 (EVP 2.08, 2.06, 1.21). These data suggest that the common hinge glycosite Thr194 and the CSF dominant C-terminal glycosites (Thr289, Ser290 and Ser296) may be preferential substrates for a different subset of GalNAc-Ts.
Modeling of CSF and plasma glyco-APOE
To better understand APOE glycoforms, low energy conformations of the glycans identified by mass spectrometry and linkages described above were generated at GLYCAM-Web (www.glycam.org) (Woods-Group 2005–2019) and attached to the glycosites we identified. The full-length APOE3 NMR structure (PDBID: 2L7B) (Chen et al. 2011) was used for all structures, the only isoform available as a full-length structure. In the case of Thr8, it was necessary to adjust the side chain of the amino acid to remove steric clashes between the glycan and protein. Models were generated using the most abundant biologically relevant CSF and plasma APOE glycoforms (Figure 5A). N-terminal Thr8 is situated at the posterior side of helix N1 (AA 6–9) with a side chain that has relatively low accessibility (31% (Hubbard and Thornton 1993)) restricting the possible orientation of the GalNAc of the glycan at this site (Figure 5A). The linear glycan extends out from the space between helix N1 and helix 1 (AA 26–40); a more detailed view of the glycosylated Thr8 is shown in Supplementary Figure S8.
The hinge region Thr194 residue is widely open and can hold the core 1, sialylated and disialylated structures. The sialylated core 1 Thr194 glycoform, the most abundant Thr194 glycoform in both the CSF and plasma, is shown in Figure 5B.
The lipid-binding domain pep283–299 contains glycosites Thr289, Ser290 and Ser296. However, we found that any given peptide held only a single occupied site in adult CSF and plasma. Thus, individual 3D models were generated for each site with the same Neu5Acα2–3Galβ1–3GalNAcα1- structure attached (Figure 5C, D and E). In contrast to Thr8, each site had good accessibility (Thr289: 47%, Ser290: 51% and Ser296: 51% (Hubbard and Thornton 1993)). In each of these three structures, the large linear Neu5Acα2–3Galβ1–3GalNAcα1- is projected away from the protein backbone and into the solution. The addition of the α2–6 Neu5Ac residue to the GalNAc changes the overall topology of the glycan structure (compare Figure 5E and F). The additional negatively charged Neu5Ac residue is proximal to the protein backbone, as shown in more detail in Figure 6. This α2–6-linked Neu5Ac of the Ser290 glycan could potentially form interactions with the C-terminal domain (pink) via Val232 and Asp230, as well as the N-terminal domain (cyan) via Glu132 and Glu131, which are adjacent to the LDLR-binding domain (lime green).
Fig. 6.
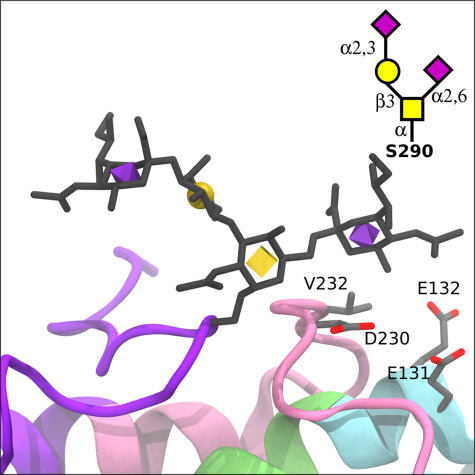
Structural changes due to sialylated glycovariants in the lipid-binding domain. The APOE3 crystal structure with a disialylated core 1 glycan (Neu5Acα2–3Galβ1–4[Neu5Acα2–6]GalNAcα) modeled at Ser290 (displayed as licorice with 3D-SNFG icons), which is part of the C-terminal lipid-binding region (purple). Relative to the monosialylated core 1 structure, the α2–6 linked Neu5Ac could potentially form interactions with the C-terminal domain (pink) via V232 and D230, as well as the LDL receptor-binding domain (lime green) via E132 and E131.
Discussion
Our study has shown that APOE glycosylation varies between the CSF and plasma, with this difference apparent despite the small sample size. CSF APOE is more heavily glycosylated and holds significantly more sialylated and disialylated core 1 structures within the C-terminal lipid-binding domain. In contrast, plasma APOE holds the sialylated core 1 O-glycan on the N-terminal domain. Both APOE from the central nervous system (CNS) and the periphery hold glycosylation at the hinge with similar glycan distributions, though the CSF APOE to a greater extent. This work also demonstrates our ability to monitor glyco-APOE from small sample aliquots and as CSF is cleared into the bloodstream; if with further investigation these data are consistent across broader closely matched sample sets, it opens up the possibility of identifying brain-derived APOE from plasma samples.
Brain and liver tissue, both of which express the APOE analyzed in this study, show differences in GalNAc-T expression (Bennett et al. 2012). The nervous system expresses all 20 known GalNAc-Ts including several that are exclusive (GalNAc-T9), highly restricted (GalNAc-T20) or enriched (GalNAc-T19) in the brain or nervous system (Bennett et al. 2012; Raman et al. 2012). Hepatocytes show a reduced profile with primary GalNAc-T expression from ubiquitous GalNAc-T1 and 2; to a lesser extent, GalNAc-T4, 10, 11, 15 and 18 (Schjoldager et al. 2015); and possibly additional low expression (Bennett et al. 2012; Raman et al. 2012). ISOGlyP analyzes the substrate preferences of 10 specific GalNAc-Ts based on their activity on 195 peptide substrates (Kong et al. 2015). Interestingly, we found using ISOGlyP that CNS and hepatocyte common GalNAc-T1 and T11 are likely to use Thr194 as a substrate, as does the CNS GalNAc-T3, consistent with the commonality of Thr194 glycosylation in CSF and plasma and higher abundance in CSF. ISOGlyP strongly predicted the brain-expressed GalNAc-T16, T14 and 13 (Bennett et al. 2012) to glycosylate the C-terminal glycosites, particularly Ser290. The ubiquitous GalNAc-Ts are also suggested to be able to utilize these sites as a substrate, however, to a much lesser extent, again supporting the increased glycosylation of these sites in the CNS compared to systemic APOE. The N-terminal sites were more difficult to explain with no GalNAc-T showing a strong inclination towards these sites as substrates. This observation could indicate that other GalNAc-Ts not included in ISOGlyP are involved; also, the unusual buried nature of these sites may make GalNAc-T prediction more difficult. Although ISOGlyP does not include several GalNAc-Ts that would be of particular interest for APOE glycosylation including GalNAc-T18 (Schjoldager et al. 2015) (expressed in liver) and GalNac-T9, 20 and 19 (Bennett et al. 2012; Raman et al. 2012) (expressed strongly in the brain), the available GalNAc-T data indicate that APOE glycosites show differential transferase preferences that may account for the tissue glycosylation specificity.
Previous studies have identified glycosylation sites on APOE. Early work identified Thr194 as a glycosite (Wernette-Hammond et al. 1989), with a later study identifying Ser290 and suggesting that sialylated core 1 structures were attached (Lee et al. 2010). More recent work has been able to progressively identify the N-terminal (Thr8 and Thr18) and C-terminal (Thr289, Ser290 and Ser296) glycosites using a combination of cancer cell lines and large volumes of CSF (Nilsson et al. 2009; Steentoft et al. 2011; Halim et al. 2013). Identification of the attached glycan is a technical challenge, and although it has been shown that APOE can hold core 1 structures in CSF, the glycopeptide isolation method used destroyed the attached glycan, making its complete identification impossible (Nilsson et al. 2009; Halim et al. 2013). Here, we have made a comprehensive analysis of all detectable identified glycosites of glycosylated APOE from the plasma and CSF, including the attached glycans and site occupancy, allowing a greater fundamental understanding of the APOE molecule across tissues. We achieved this using 25 μL of CSF compared to previous work with CSF starting volumes in the order of milliliters while still isolating glycopeptides (Halim et al. 2013). The site occupancy for most sites may appear relatively low; however, two things should be taken into consideration. First, it has been suggested that APOE is more heavily glycosylated when in the cell compared to secreted forms of the protein (Zannis et al. 1986; Lee et al. 2010) which would include both the CSF and plasma samples studied here. Second, relative quantitation is hampered by the suppression of sialic acid-holding peptides in positive ion mode (Stavenhagen et al. 2013). This suppression is reduced with the use of nano-electrospray ionization (Stavenhagen et al. 2013) as used in this study; however, the sialylated structures may be underestimated. Suppression did not hinder the detailed identification of the attached glycan structures observed in this study.
The hinge glycosylation of pep192–206, previously shown to be glycosite Thr194 (Wernette-Hammond et al. 1989; Halim et al. 2013) and suggested here to also be glycosylated at Ser197, is most similar between the CSF and plasma samples. The hinge flexibility is essential for unfolding of APOE and as the glycosite is situated on the Hinge H2 helix (Chen et al. 2011), rather than on the intervening loop regions, suggesting it would not affect unfolding. Thr8 N-terminal Neu5Acα2–3Galβ1–3GalNAcα1- glycosylation, however, was more abundant on plasma APOE compared to CSF APOE, and the Thr18 glycosite was below detection in the samples tested (though identified in a similarly processed APOE standard). The Thr18 site has been identified previously, also using more sample than used in this study and required unique sample preparation conditions for identification (Halim et al. 2013). We identified that both glycosites are buried: Thr8 is located at the interior side of the N1 helix and Thr18 at the interior side of the second turn of the N2 helix (Chen et al. 2011). This space restriction further confirms the linear monosialylated structure assignment. Accessibility of the more abundant Thr8 glycosite is not improved by the first step of APOE unfolding which is fast and reversible (Garai et al. 2010; Chen et al. 2011). It is exposed by the second step of unfolding; however, this is slow and, although reversible, requires lipid or heparin binding (Garai et al. 2010; Chen et al. 2011) making it unlikely to occur for the addition of O-glycosylation. The close proximity of Thr8 to the N-terminal suggests that there may be inherent flexibility allowing GalNAc-T accessibility to the glycosite. This flexibility of the N-terminal has also been suggested by hydrogen-deuterium exchange experiments, although further along the N-terminal, and may present a larger difference between the APOE3 and APOE4 protein molecules (Frieden et al. 2017).
The C-terminal glycosylation is situated within the lipid-binding region and thus is of great import for APOE function. It also shows the greatest difference between CSF and plasma, with CSF APOE holding much greater glycosylation. The lipoprotein-binding profiles of CSF and plasma APOE also differ markedly, as plasma APOE must bind to lipoproteins with large ranges of size, shape and composition, while APOE in the CNS interacts only with HDL. HDL from the brain is elliptical and small (8–15 nm) while in the CSF (Pitas et al. 1987b; LaDu et al. 1998; Koch et al. 2001) it is larger (12–20 nm, with a small population up to 30 nm) and spherical. APOE in the plasma, however, has a much larger range of binding particles including the very large chylomicrons (75–1200 nm) which reduce in size to remnant particles (30–80 nm) (Patsch 1998; Dawson and Rudel 1999; Mahley and Ji 1999). Plasma APOE also binds large polyhedral VLDL (30–100 nm) (Yu et al. 2016), which change in size and shape to the IDL (25–35 nm) stage (Beisiegel 1998), and small plasma HDL particles (7–14 nm) (Otvos 2002). In order for one protein to be able to bind all of these varied structures, even as they change in size, is exceptionally accommodating. The reduced C-terminal glycosylation of plasma APOE provides a less encumbered lipid-binding domain that may be important for this structural flexibility.
The removal from APOE of amino acid 244 onwards (from middle of helix C2) eliminates all lipoprotein binding (Westerlund and Weisgraber 1993); however, the binding preferences of this region are lipoprotein-specific. The C-terminal region (AA 273–299), made up of the helix C3 (AA 271–276) and loop C (AA 277–299) (Chen et al. 2011), has been shown to mediate the binding to HDL (Sakamoto et al. 2008). This binding preference is APOE genotype-dependent, with the APOE4 protein more dependent on the C-terminal 273–299 region than APOE3 (Sakamoto et al. 2008; Nguyen et al. 2010). The upstream portion of the lipid-binding region is more necessary for VLDL and LDL binding (Westerlund and Weisgraber 1993). Self-association is also completely terminated by the loss of the AA273–299 region for APOE4 but not for APOE3 (Sakamoto et al. 2008). Self-association may be important for the construction of large complexes with HDL particles able to hold at least two APOE proteins (Chen et al. 2011); APOE4-positive genotypes produce smaller lipoprotein particles in older adult CSF compared to APOE4-negative genotypes (Heinsinger et al. 2016). Therefore, changes in the C-terminal region 273–299 may affect HDL binding more dramatically than other types of lipoproteins as well as self-association and possibly affect the APOE4 protein to a higher degree than the APOE3 protein.
APOE glycosylation and, in particular, sialylation, has previously been shown to affect the lipoprotein-binding preference of APOE. In a study of APOE glycosylation on lipoprotein binding, APOE was de-sialylated with a neuraminidase that preferentially removes α2–3-linked Neu5Ac but also α2–6-linked Neu5Ac (Marmillot et al. 1999) (as we have shown here, the most common sialic acid residues on APOE). A comparison in the binding of sialylated and de-sialylated APOE revealed that HDL bound five times more effectively to sialylated APOE, while VLDL bound only two times more effectively to sialylated APOE (Marmillot et al. 1999). HDL-binding strength of de-sialylated APOE was rescued by the re-addition of sialic acid using sialyltransferases from rat liver golgi (Marmillot et al. 1999) confirming the significance of glycosylation with the charged Neu5Ac to HDL binding. De-sialylation would remove Neu5Ac from all APOE glycosites, and thus, we cannot rule out the importance of the N-terminal or hinge glycosites. However, the low abundance of N-terminal glycosylation in the CSF, compared to the high abundance in the plasma APOE, does suggest it is not essential for HDL binding. Also, the glycosites within the lipid-binding region are the most Neu5Ac-rich, the only APOE region which shows equivalent amounts of the Neu5Acα2–3Galβ1–3GalNAcα1- and Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1- structures, while all others show a marked preference for the Neu5Acα2–3Galβ1–3GalNAcα1- structure. This is particularly relevant as sialyltransferases decrease with age (Zahn et al. 2006; Itakura et al. 2016) and in AD (Maguire et al. 1994; Maguire and Breen 1995) suggesting a possible explanation for APOE HDL-binding deficiencies over time and in AD.
The addition of O-glycosylation can affect the structure and function of a protein (Grinstead et al. 2002), and the location of glycosylation can have wide-reaching effects (Chaffey et al. 2017). Based on our modeling, the C-terminal lipid-binding domain glycosylation has the potential to interact across APOE domains. This was apparent by the proximity of the α2–6-linked Neu5Ac of the disialylated core 1 glycosylated Ser290 to the N-terminal domain, adjacent to the LDLR-binding domain. This proximity is of particular interest given that the Ser290 site shows a higher abundance of glycosylation with the disialylated core 1 glycan compared to the monosialylated version, unlike the other C-terminal glycosites, suggesting that this domain interaction may be favorable. A glycosylation-dependent relationship between the N and C terminals could have implications for APOE protein dynamics given that protein unfolding at the hinge, separating the two domains, is necessary for LDLR binding (Chen et al. 2011) and therefore healthy metabolism of APOE. This N and C-terminal interaction has been shown to be more significant to APOE and its function than initially understood (Chen et al. 2011; Frieden et al. 2017).
In conclusion, we have identified that the C-terminal loop structure of APOE, important for tailoring lipoprotein binding preference, remains almost exclusively unglycosylated within the plasma, potentially leaving it open for the wide range of lipoproteins that bind there as is required of the APOE in plasma. In the CNS, however, higher abundance glycosylation and sialylation may tailor HDL binding by the addition of negatively charged sialylated glycans previously shown to improve HDL binding. Although this current study is limited by sample size, this observation is worthy of further investigation given the critical importance of APOE to AD and the current lack of understanding of APOE as a glycosylated molecule.
Materials and methods
Experimental design and statistical rationale
Glycoproteomics analysis was undertaken on APOE from CSF and plasma from older individuals. Purchased APOE from plasma was used as a standard during these analyses. Glycopeptide relative quantification was carried out to allow for comparison and to determine the most appropriate structures to use for glyco-APOE modeling. Glycopeptide relative quantification was compared between CSF and plasma samples using non-parametric tests (Mann–Whitney test) due to the low sample numbers and P values adjusted for multiple comparisons.
Sample information
Human lumbar puncture CSF samples (n = 4) were from Washington University in St Louis, collected as control samples as part of a study on CSF biomarkers in AD. As previously described, CSF (20–30 mL) was collected by routine lumbar puncture at 8 a.m. following overnight fasting (Fagan et al. 2006; Sutphen et al. 2015). CSF was immediately aliquoted (500 μL) and stored at −80°C (Sutphen et al. 2015). All samples and clinical information were anonymized, all individuals gave written informed consent and the study was approved by the Human Research Protection Office at Washington University. Amyloid status was confirmed, where possible, by amyloid Pittsburgh compound B positron emission tomography.
Human plasma samples (n = 4) were collected in EDTA. Samples were from the Georgetown Brain Bank tissue and biofluid repository. Blood samples were taken and processed into plasma with all samples processed within 1 h of sampling. Blood was centrifuged at 3000 rpm for 15 min at 4°C, and plasma was then aliquoted (1 mL) and immediately stored at −80°C in the clinic. All samples and clinical information were anonymized, all individuals gave written informed consent and the study was approved by the Institutional Review Board at Georgetown University Medical Center. All individuals underwent a mini-mental state exam at the time of sampling to confirm cognitive state. Sample information is found in Supplementary Table SI. Standard APOE was from fresh human plasma (rPeptide).
Immunoprecipitation
Samples were precleared with Protein A Sepharose® beads (Sigma) for 1 h at 4°C with rotation. An excess of beads was used to preclear the higher immunoglobulin content of plasma. Protease (Pierce Mini Tablets with EDTA, 88665) and phosphatase (Pierce phosphatase inhibitor mini tablets 88667) inhibitors were added to the IP buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 1% NP40). The amount of antibody used for IP was optimized for each sample type. Precleared samples were incubated with fresh beads and goat polyclonal anti-APOE (K74190G, Meridian Life Science) in IP buffer for 16 h at 4°C with rotation. Beads were washed five times with IP buffer and the sample removed from beads with NuPAGE® LDS Sample Buffer (Thermo Fisher Scientific).
Glycoproteomics sample preparation
IP samples were separated on 4–12% NuPAGE® gels (Thermo Fisher Scientific) using MOPS SDS running buffer (50 mM MOPS, 50 mM Tris, 0.1% SDS, 1 mM EDTA) followed by reduction and alkylation using 50 mM DTT for 1 h at 65°C and 125 mM iodoacetamide 30 min, RT in the dark, and the reaction stopped with 125 mM DTT. Standard plasma APOE (rPeptide) was included on gels.
All solvents were of MS quality. APOE bands were excised including gel to account for low-abundance modifications. Bands were washed until destained (40 min at 37°C, 100 mM ammonium bicarbonate twice followed by 50% acetonitrile in 100 mM ammonium bicarbonate twice until destained) and dried before trypsinization with 500 ng of Trypsin Gold, MS grade trypsin (Promega) for 16 h, 37°C, to ensure adequate trypsinization of APOE. Peptides and glycopeptides were extracted with water followed by 50% acetonitrile/0.1% trifluoroacetic acid and samples dried ready for MS analysis.
Mass spectrometric method
MS analyses were carried out on a TripleTOF© 6600 QTOF (quadrupole time of flight, Sciex), used in positive ion mode. A nanoACQUITY UPLC (Ultra Performance Liquid Chromatography, Waters) with an analytical ACQUITY UPLC M-Class peptide BEH C18 column (300 Å, 1.7 μm, 75 μm × 15 cm, Waters) and a nanoACQUITY UPLC symmetry C18 trap column (100 Å, 5 μm, 180 μm × 20 mm, Waters) was used. Mobile phases, solvent A (aqueous 2% acetonitrile, 0.1% formic acid) and solvent B (acetonitrile, 0.1% formic acid) were used for a 60 min gradient with a trapping flow rate of 15 μL/min and analytical flow rate of 400 nL/min. The gradient began with 1 min at 99% solvent A, and an increase in solvent B from 5 to 50% in 35 min increased to 99% solvent B in 2 min held for 3 min before returning to 99% solvent A for 20 min. Declustering potential was set to 80 and ionspray voltage 2300. A top 30 data-dependent acquisition method was used. The TOF MS accumulation time was 250 ms, 400–1250 Da. The TOF MSMS accumulation time was 50 ms, 100–1500 Da, with an intensity threshold of 100 based on background and exclusion after 2 MS/MS of 5 s based on peak width. The method allows for at least 10 points on the curve of the narrowest peak under analysis. Independent data acquisition (IDA) collision energy parameters were set as follows: written as charge, slope and intercept; unknown, 0.049 and −1; 1, 0.05, 5; 2, 0.049, −1; 3, 0.048, −2; and 4 and higher, 0.05, −2.
A blank was run after every sample to stop any unexpected carryover interference, and a standard (trypsinized β-galactosidase) was run following every fourth sample. Sample order was randomized. Standard APOE from plasma (rPeptide) was also run to ensure glycopeptide detection and expected separation.
All APOE glycopeptides in each sample were confirmed manually by parent m/z, MSMS and relative retention time. APOE protein identification information is shown in Supplementary Table SII. All glycopeptide spectra contained strong glycan (oxonium) ions indicating structure. Software was not able to identify all low abundance glycopeptides; this was possible by manual search. A large range of glycan structures were searched for the possible glycopeptides of APOE: core 1 structures, including the sialylated and disialylated forms, as well as extended forms with and without Neu5Ac residue/s; truncated core 1 structures, including Tn antigen (GalNAc) and sialyl Tn; a range of core 2 structures, including sialylated forms; extended core 2 structures with and without Neu5Ac residue/s. Peptides with more than one possible glycosylation site were searched as containing multiple glycans, focusing on glycans that may have been identified previously on that peptide.
All hexosamine residues are assumed to be GalNac and hexose residues Gal, forming a core 1 structure of known linkage (Galβ1–3GalNAcα1-). Branched disialylated core 1 structures with spectra showing diagnostic ions representing sialylation of both Gal and GalNAc residues are understood to have the following Neu5Ac linkages Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1-. The associated linear Gal-sialylated core 1 structure has the Neu5Ac α2–3 linkage, creating the Neu5Acα2–3Galβ1–3GalNAcα1- structure as has been extensively studied (Brockhausen et al. 2009). All MS/MS spectra shown in paper or supplementary for all glycoforms and structure accurately represent the level of annotation as detailed in associated text including details of diagnostic ions. All MS/MS spectra show observed masses.
MultiQuant™ software version 2.1.1 (Sciex, Framingham, MA) was used for quantitation. Quantitation was based on parent mass, and mass allowance was +/− 0.05 Da. Two peptides, LGPLVEQGR (amino acid 181–189, hinge region) and LQAEAFQAR (amino acid 252–260, C-terminal) which cannot hold glycosylation, were monitored for total APOE quantitation, chosen based on previous data showing them to be the most consistently intense peptides in all sample types. Data is shown as relative quantitation where the area under the curve was determined for non-glycosylated and all glycoforms of each given peptide and a total determined. The relative percentage of each peptide was then determined and is shown in Figure 4C for each sample and graphically in Figure 4B. The data supporting the findings of the study are available within the paper and its supplementary information files. Any further data generated during this study are available from the corresponding author on request. Quantification data from the CSF and plasma samples were compared using the Mann–Whitney nonparametric test due to the low sample number. GraphPad Prism version 8.2.1 (GraphPad Software) was used for statistical analyses. P values were FDR-adjusted to account for the multiple comparisons using R.
Glycoprotein modeling
Glycoprotein modeling was performed using a prototype of the new GLYCAM-Web glycoprotein builder (www.glycam.org/gp) (Woods-Group 2005-2019), which uses the GLYCAM06 force field (Kirschner et al. 2008) to generate 3D structures of carbohydrates. The full-length APOE3 NMR structure (Chen et al. 2011) was downloaded from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (Berman et al. 2000) (PDBID: 2L7B). Each 3D glycan structure was superimposed onto the appropriate Ser/Thr side chain of APOE3. Any atomic overlaps between the glycan and the protein were relieved by adjusting the protein side chain and glycosidic linkage dihedral angles. Accessibility was determined using NACCESS software (Hubbard and Thornton 1993). The glycosylated APOE3 structures were visualized using Visual Molecular Dynamics software (VMD 1.9.3) (Humphrey et al. 1996).
Funding
Georgetown-Howard Universities Center for Clinical and Translational Science and National Institutes of Health (R01 NS100714 to S.A.F. and G.W.R.); National Institutes of Health (U01 CA207824 and P41 GM103390 to R.J.W.); National Institutes of Health (R56 AG062305 to S.A.F.).
Supplementary Material
Acknowledgements
The authors acknowledge the good work and collaborative spirit of the Alzheimer’s Disease Research Center, Washington University, in St. Louis who supplied CSF samples (P50AG005681, P01AG003991 and PO1AG026276). They would also like to thank the Georgetown Brain Bank and Memory Disorders Program, Georgetown University Medical Center, who supplied plasma samples.
Abbreviations
AA, amino acid
AD, Alzheimer’s disease
Arg, arginine
APOE, apolipoprotein E
CNS, central nervous system
CSF, cerebrospinal fluid
Cys, cysteine
Gal, galactose
GalNAc, N-acetyl galactosamine
GalNAc-T, UDP-GalNAC: polypeptide N-acetylgalactosaminyl-transferases
HDL, high-density lipoprotein
LDL, low-density lipoprotein
LDLR, low-density lipoprotein receptor
MS, mass spectrometry
MS/MS, tandem mass spectrometry
Neu5Ac, N-acetylneuraminic acid
QTOF, quadrupole time of flight
Ser, serine
Thr, threonine
UPLC, ultra performance liquid chromatography
VLDL, very low-density lipoprotein
Conflict of interest statement
None declared.
References
- Achariyar TM, Li B, Peng W, Verghese PB, Shi Y, McConnell E, Benraiss A, Kasper T, Song W, Takana T et al. . 2016. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol Neurodegener. 11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali L, Flowers SA, Jin C, Bennet EP, Ekwall AK, Karlsson NG. 2014. The O-glycomap of lubricin, a novel mucin responsible for joint lubrication, identified by site-specific glycopeptide analysis. Mol Cell Proteomics. 13:3396–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisiegel U. 1998. Lipoprotein metabolism. Eur Heart Journal. 19(Suppl A):A20–A23. [PubMed] [Google Scholar]
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. 2012. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 22:736–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhausen I, Schachter H, Stanley P. 2009. O-GalNAc glycans In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of glycobiology. Cold Spring Harbor (NY). [PubMed] [Google Scholar]
- Chaffey PK, Guan X, Chen C, Ruan Y, Wang X, Tran AH, Koelsch TN, Cui Q, Feng Y, Tan Z. 2017. Structural insight into the stabilizing effect of O-glycosylation. Biochemistry. [DOI] [PubMed] [Google Scholar]
- Chaudhury NM, Proctor GB, Karlsson NG, Carpenter GH, Flowers SA. 2016. Reduced mucin-7 (Muc7) sialylation and altered saliva rheology in Sjogren’s syndrome associated oral dryness. Mol Cell Proteomics. 15:1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Q, Wang J. 2011. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci U S A. 108:14813–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 261:921–923. [DOI] [PubMed] [Google Scholar]
- Dawson PA, Rudel LL. 1999. Intestinal cholesterol absorption. Curr Opin Lipidol. 10:315–320. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA et al. . 2006. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 59:512–519. [DOI] [PubMed] [Google Scholar]
- Frieden C, Wang H, Ho CMW. 2017. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc Natl Acad Sci U S A. 114:6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai K, Mustafi SM, Baban B, Frieden C. 2010. Structural differences between apolipoprotein E3 and E4 as measured by (19) F NMR. Protein Sci. 19:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greis KD, Hart GW. 1998. Analytical methods for the study of O-GlcNAc glycoproteins and glycopeptides In: Hounsell EF, editor. Methods in molecular biology, glycoanalysis protocols. Totowa (NJ): Humana Press; p. 19–35. [DOI] [PubMed] [Google Scholar]
- Grinstead JS, Koganty RR, Krantz MJ, Longenecker BM, Campbell AP. 2002. Effect of glycosylation on MUC1 humoral immune recognition: NMR studies of MUC1 glycopeptide-antibody interactions. Biochemistry. 41:9946–9961. [DOI] [PubMed] [Google Scholar]
- Halim A, Ruetschi U, Larson G, Nilsson J. 2013. LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res. 12:573–584. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A et al. . 2009. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinsinger NM, Gachechiladze MA, Rebeck GW. 2016. Apolipoprotein E genotype affects size of ApoE complexes in cerebrospinal fluid. J Neuropathol Exp Neurol. 75:918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. 1993. ‘NACCESS’, computer program,. Hubbard, S.J. and Thornton, J.M. University College London: Department of Biochemistry and Molecular Biology. [Google Scholar]
- Humphrey W, Dalke A, Schulten K. 1996. VMD: Visual molecular dynamics. J Mol Graph. 14(33–38):27–38. [DOI] [PubMed] [Google Scholar]
- Itakura Y, Sasaki N, Kami D, Gojo S, Umezawa A, Toyoda M. 2016. N- and O-glycan cell surface protein modifications associated with cellular senescence and human aging. Cell Biosci. 6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju T, Cummings RD. 2002. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A. 99:16613–16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. 2008. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 29:622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko BJ, Brodbelt JS. 2015. Comparison of glycopeptide fragmentation by collision induced dissociation and ultraviolet photodissociation. Int J Mass Spectrom. 377:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg HJ, Buhmann C, Beisiegel U. 2001. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 42:1143–1151. [PubMed] [Google Scholar]
- Kockx M, Jessup W, Kritharides L. 2008. Regulation of endogenous apolipoprotein E secretion by macrophages. Arterioscler Thromb Vasc Biol. 28:1060–1067. [DOI] [PubMed] [Google Scholar]
- Kong Y, Joshi HJ, Schjoldager KT, Madsen TD, Gerken TA, Vester-Christensen MB, Wandall HH, Bennett EP, Levery SB, Vakhrushev SY et al. . 2015. Probing polypeptide GalNAc-transferase isoform substrate specificities by in vitro analysis. Glycobiology. 25:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, Holtzman DM. 1998. Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem. 70:2070–2081. [DOI] [PubMed] [Google Scholar]
- Lalazar A, Weisgraber KH, Rall SC Jr, Giladi H, Innerarity TL, Levanon AZ, Boyles JK, Amit B, Gorecki M, Mahley RW et al. . 1988. Site-specific mutagenesis of human apolipoprotein E. receptor binding activity of variants with single amino acid substitutions. J Biol Chem. 263:3542–3545. [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B et al. . 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kockx M, Raftery MJ, Jessup W, Griffith R, Kritharides L. 2010. Glycosylation and sialylation of macrophage-derived human apolipoprotein E analyzed by SDS-PAGE and mass spectrometry: Evidence for a novel site of glycosylation on Ser290. Mol Cell Proteomics. 9:1968–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire TM, Breen KC. 1995. A decrease in neural sialyltransferase activity in Alzheimer’s disease. Dementia. 6:185–190. [DOI] [PubMed] [Google Scholar]
- Maguire TM, Gillian AM, O'Mahony D, Coughlan CM, Dennihan A, Breen KC. 1994. A decrease in serum sialyltransferase levels in Alzheimer’s disease. Neurobiol Aging. 15:99–102. [DOI] [PubMed] [Google Scholar]
- Mahley RW. 1988. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science. 240:622–630. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Ji ZS. 1999. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 40:1–16. [PubMed] [Google Scholar]
- Marmillot P, Rao MN, Liu QH, Lakshman MR. 1999. Desialylation of human apolipoprotein E decreases its binding to human high-density lipoprotein and its ability to deliver esterified cholesterol to the liver. Metabolism. 48:1184–1192. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Dhanasekaran P, Nickel M, Nakatani R, Saito H, Phillips MC, Lund-Katz S. 2010. Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry. 49:10881–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J, Ruetschi U, Halim A, Hesse C, Carlsohn E, Brinkmalm G, Larson G. 2009. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat Methods. 6:809–811. [DOI] [PubMed] [Google Scholar]
- Otvos JD. 2002. Measurement of lipoprotein subclass profiles by nuclear magnetic resonance spectroscopy. Clin Lab. 48:171–180. [PubMed] [Google Scholar]
- Patel RY, Balaji PV. 2006. Identification of linkage-specific sequence motifs in sialyltransferases. Glycobiology. 16:108–116. [DOI] [PubMed] [Google Scholar]
- Patsch J. 1998. Influence of lipolysis on chylomicron clearance and HDL cholesterol levels. Eur Heart J. 19(Suppl H):H2–H6. [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. 1987a. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 917:148–161. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. 1987b. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E (LDL) receptors in the brain. J Biol Chem. 262:14352–14360. [PubMed] [Google Scholar]
- Rall SC Jr, Weisgraber KH, Mahley RW. 1982. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 257:4171–4178. [PubMed] [Google Scholar]
- Raman J, Guan Y, Perrine CL, Gerken TA, Tabak LA. 2012. UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferases: Completion of the family tree. Glycobiology. 22:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T, Tanaka M, Vedhachalam C, Nickel M, Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H. 2008. Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry. 47:2968–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjoldager KT, Joshi HJ, Kong Y, Goth CK, King SL, Wandall HH, Bennett EP, Vakhrushev SY, Clausen H. 2015. Deconstruction of O-glycosylation--GalNAc-T isoforms direct distinct subsets of the O-glycoproteome. EMBO Rep. 16:1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal MB. 2005. Fluid compartments of the central nervous system In: Zheng W, Chodobski A, editors. The blood-cerebrospinal fluid barrier. Boca Raton (FL): Taylor and Francis. [Google Scholar]
- Stavenhagen K, Hinneburg H, Thaysen-Andersen M, Hartmann L, Varon Silva D, Fuchser J, Kaspar S, Rapp E, Seeberger PH, Kolarich D. 2013. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: An evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J Mass Spectrom. 48:627–639. [DOI] [PubMed] [Google Scholar]
- Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H. 2011. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 8:977–982. [DOI] [PubMed] [Google Scholar]
- Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, Benzinger TLS, Stoops EEJ, Vanderstichele HMJ, Brix B et al. . 2015. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 72:1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, Rall SC Jr. 1989. Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. J Biol Chem. 264:9094–9101. [PubMed] [Google Scholar]
- Westerlund JA, Weisgraber KH. 1993. Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J Biol Chem. 268:15745–15750. [PubMed] [Google Scholar]
- Woods-Group 2005-2019. GLYCAM-Web glycoprotein builder. Woods Group: Complex Carbohydrate Research Center, University of Georgia, Athens, GA. [Google Scholar]
- Wu L, Zhao L. 2016. ApoE2 and Alzheimer’s disease: Time to take a closer look. Neural Regen Res. 11:412–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. 2006. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 26:4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Kuang YL, Lei D, Zhai X, Zhang M, Krauss RM, Ren G. 2016. Polyhedral 3D structure of human plasma very low density lipoproteins by individual particle cryo-electron tomography1. J Lipid Res. 57:1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK. 2006. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannis VI, vanderSpek J, Silverman D. 1986. Intracellular modifications of human apolipoprotein E. J Biol Chem. 261:13415–13421. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.