

Abstract
Base editing technology efficiently generates nucleotide conversions without inducing excessive double-strand breaks (DSBs), which makes it a promising approach for genetic disease therapy. In this study, we generated a novel hereditary tyrosinemia type 1 (HT1) mouse model, which contains a start codon mutation in the fumarylacetoacetate hydrolase (Fah) gene by using an adenine base editor (ABE7.10). To investigate the feasibility of base editing for recombinant adeno-associated virus (rAAV)-mediated gene therapy, an intein-split cytosine base editor (BE4max) was developed. BE4max efficiently induced C-to-T conversion and restored the start codon to ameliorate HT1 in mice, but an undesired bystander mutation abolished the effect of on-target editing. To solve this problem, an upstream sequence was targeted to generate a de novo in-frame start codon to initiate the translation of FAH. After treatment, almost all C-to-T conversions created a start codon and restored Fah expression, which efficiently ameliorated the disease without inducing off-target mutations. Our study demonstrated that base editing-mediated creation of de novo functional elements would be an applicable new strategy for genetic disease therapy.
Graphical Abstract
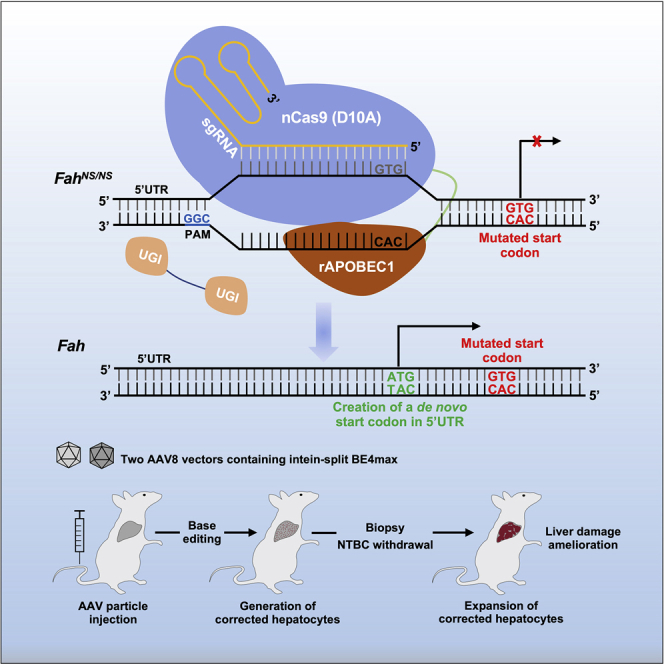
Li and colleagues have generated a start codon-mutated HT1 mouse model via ABE editing and developed two CBE-mediated therapeutic strategies through either restoration of the start codon or creation of a de novo initiation codon in the Fah gene to ameliorate inherited metabolic liver disease in this mouse model.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system is a versatile tool that has been widely used in biomedical research and exhibits prospective applications in gene therapy.1, 2, 3 With the guidance of sequence-specific single guide RNAs (sgRNAs), Cas9 protein generates very efficient and precise DNA double-strand breaks (DSBs) that evoke various intrinsic DNA damage and repair pathways, including microhomology-mediated end joining (MMEJ),4,5 classical non-homologous end joining (cNHEJ), or homology-directed repair (HDR), in the presence of donor templates.6 Although HDR is the ideal strategy to correct genetic mutations due to its high precision, this DNA repair pathway only occurs in proliferating cells, limiting its application in the majority of adult tissues.7 As one of the critical in vivo gene therapy target organs, the liver is a promising tissue for HDR-mediated gene integration therapy, as partial hepatocytes proliferate constantly. However, Cas9-mediated HDR efficiency in adult animal livers is very low,8,9 even using rAAV vectors. Thus, only a limited number of genetic diseases, such as hemophilia and hereditary tyrosinemia type 1 (HT1), have been successfully ameliorated via HDR.8,10,11 Cas9 is a versatile genetic tool that has been shown to correct various hereditary diseases through non-HDR pathways. Through the NHEJ repair pathway, premature stop codons or de novo cryptic splicing sites have been removed to ameliorate Duchenne muscular dystrophy (DMD)3,12, 13, 14 and Hutchinson-Gilford progeria syndrome (HGPS)15,16 in mice, respectively. Cas9-induced DSBs are able to generate precise therapeutic gene correction at some pathogenic microduplications through the MMEJ pathway.5 Although Cas9-mediated deletion could remove DNA fragments such as pathogenic genomic elements, it is difficult to create de novo functional DNA elements using this system.
It is reported that 58% of the known human pathogenic DNA variants are point mutations that need to be corrected through nucleotide conversions.17 The innovative base editors (BEs), including cytosine BEs (CBEs) and adenine BEs (ABEs), are ideal genetic tools to correct nucleotide conversions without inducing significant DSBs.18,19 CBEs were developed through the fusion of cytidine deaminases, such as rAPOBEC1, AID, hAPOBEC3A, and their variants, with Cas9 nickase and uracil glycosylase inhibitor (UGI) to generate efficient C⋅G-to-T⋅A conversions.17,20 ABEs are composed of a wild-type (WT) non-catalytic TadA monomer, an evolved TadA∗ monomer, and a Cas9 nickase to stimulate A⋅T-to-G⋅C base substitutions.19 These BEs have been widely used in animal models,21, 22, 23 plants,24,25 bacteria,26 and human cells.27, 28, 29 Several studies have shown the promising applications of BEs for therapy of genetic disorders, such as DMD,22 HT1,30,31 and phenylketonuria (PKU),32 but the delivery of BEs is challenging. Although adenoviral vectors30 or hydrodynamic tail vein injection (HTVI) of naked plasmid DNA31 is effective in animal models, it is not clinically applicable currently. Clinical studies have demonstrated that the rAAV vector is a safe and efficient system;33 however, its limited payload capacity (≤4.7 kb) hinders the efficient delivery of BEs of large size. To solve this problem, BEs have been split and delivered via two rAAV vectors either by trans-splicing AAV vectors (tsAAVs)22 or by the intein-split system.32,34
To examine the feasibility of rAAV-mediated SpCas9-BE4max35 (hereafter, BE4max) delivery for gene therapy, we first generated a novel tyrosinemia mouse model by converting the ATG start codon of the fumarylacetoacetate hydrolase (Fah) gene to GTG using the ABE system. Through rAAV vectors, an intein-mediated split BE4max system was delivered to either rescue the start codon or create a de novo in-frame start codon in the 5′ UTR region of the Fah gene to ameliorate the genetic disease in adult mice. Our study is a proof-of-concept work to demonstrate that novel gene therapy strategies could be developed not only via base editing-mediated in situ correction but also through creation of de novo functional genetic elements.
Results
Generation of an HT1 Mouse Model through ABE-Mediated Disruption of the Start Codon
HT1 is a fatal liver disease caused by FAH gene mutation, which leads to liver failure due to the accumulation of toxic metabolites in the tyrosine metabolic pathway.36 It is estimated that start codon mutations account for 2.1% of total mutations in HT1 patients.36 Administration of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), a chemical inhibitor of an upstream enzyme,37 is the most effective clinical treatment. This disease is an ideal model for gene therapy since the repaired hepatocytes containing functional Fah have a growth advantage to substitute for mutant cells and repopulate the entire liver.38 To generate a novel HT1 mouse model, we targeted the start codon of the mouse Fah gene using adenine BE ABE7.10 to mimic the 1A>G/p.M1 mutation in human patients,36 as described in our previous report21 (Figure 1A). Since this model contains a disrupted start codon of the Fah gene, we refer to this mouse strain as FahNS/NS hereafter. Western blotting, immunohistochemical staining, and qRT-PCR revealed the absence of FAH protein and Fah mRNA in FahNS/NS mouse liver tissue (Figures 1B–1D). Compared to WT mice, FahNS/NS mice lost 20% of their body weight at 3 weeks after NTBC withdrawal, whereas the mice maintained on NTBC water thrived as well as did WT mice (Figure 1E). 3 weeks after NTBC withdrawal, the serum level of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total bilirubin (TBIL) were significantly increased, but the serum albumin (ALB) was decreased in FahNS/NS mice compared with control groups (Figure 1F). Histopathological analyses of FahNS/NS mouse livers at 3 weeks after NTBC withdrawal revealed massive necrosis and inflammation (Figure 1G). These results indicated that our FahNS/NS model faithfully represented the major phenotypes observed in human HT1 patients and other HT1 animal models,8,39 suggesting the successful generation of a Fah gene start codon-mutated HT1 disease model.
Figure 1.

Phenotypic Characterization of FahNS/NS HT1 Mice
(A) Schematic view of the mutated start codon, which is replaced by the triplet GTG for valine (underlined) in the mouse Fah gene. (B) Western blot analysis of FAH expression in liver tissue from 6-week-old WT and FahNS/NS mice. (C) FAH IHC staining of liver tissue sections from 6-week-old WT and FahNS/NS mice. Scale bar, 100 μm. (D) Relative mRNA expression of Fah gene in liver tissue from 6-week-old WT and FahNS/NS mice. (E) Body weight curves of WT (left) and FahNS/NS mice with (center) or without (right) NTBC in drinking water. Body weight was normalized to the weight on day of NTBC withdrawal. Mice with more than 20% weight loss were euthanized. n = 6 mice per group. (F) Serum AST, ALT, TBIL, and ALB levels in peripheral blood from WT and FahNS/NS mice 3 weeks after NTBC withdrawal. (G) Hematoxylin and eosin (H&E) staining of liver tissue sections from WT and FahNS/NS mice, 3 weeks after NTBC withdrawal. In all graphs, values and error bars represent mean ± SD (n = 3 mice per group), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n.s., not significant.
BE4-Mediated In Situ Correction of the Fah Start Codon with Plasmid DNA Injection
Since the editing window of the BE4max is typically located 4–8 nt downstream of the 5′ end of the targeted sequence distal to the protospacer-adjacent motif (PAM) site, we designed two sgRNAs (sg1 and sg2) for BE4max to generate G-to-A conversion and restore the ATG start codon. The desired target G (G1 in Figure 2A) was in protospacer position 7 for sg1 and 8 for sg2, respectively (Figure 2A). We also noticed that there was a bystander G (G3) in protospacer position 5 and 6 corresponding to sg1 and sg2, respectively. To restore the G1TG3 codon to A1TG3, only the first G (G1) should be converted to A. If the bystander G3 is mutated, the start codon would not be restored. To investigate the base editing efficiency and the product sequence of these two sgRNAs, a HEK293T reporter cell line with stably integrated 5′ UTR and exon 1 of the FahNS/NS allele was generated through lentiviral infection. BE4max was transfected along with sg1 or sg2 into this reporter cell line. High-throughput sequencing (HTS) revealed that both G1 and G3 were spontaneously edited in the majority of the sequence reads (Figures 2B and 2C). The total conversion efficiency at G1 is 71% for sg1 and 66.9% for sg2, while their corresponding efficiency at G3 is 71.9% and 77.1%, respectively (Figure 2B). The percentage of the desired editing product (A1TG3) of the sg1 and sg2 was extremely low (0.78% and 0.74% respectively), while byproducts (A1TA3, G1TA3, and other non-A1TG3 outcomes) dominated (73.7% and 77.4%) (Figure 2C).
Figure 2.

Cytosine Base Editing-Mediated In Situ Correction of Fah Start Codon Mutation
(A) Diagram of two sgRNAs (sg1 and sg2) designed to target the mutant allele of FahNS/NS. PAM sites are indicated with blue lines. Protospacers are indicated with red lines. Blue number indicates the position of an individual base within the protospacers. The disease-causing mutation 1A>G (p.M1>V) is shown in red. The C1-to-T1 (shown in green) conversion in the non-coding strand leads to the desired G1-to-A1 (shown in green) conversion in the coding strand, which restores the start codon for methionine (shown in green). (B) Editing efficiencies of G1 and G3 targeted by sg1 or sg2 in HEK293T reporter cells. Values and error bars represent the mean ± SD of three independent biological replicates. (C) HTS reads of genomic DNA from HEK293T reporter cells. Correct reads (desired editing products) possess G-to-A conversion at G1 only. Any reads that include non-G-to-A conversions and G-to-A conversions at positions other than G1 are considered incorrect reads (byproducts). Values and error bars represent the mean ± SD of three independent biological replicates. n.s., not significant. (D) Editing efficiencies of G1 and G3 in BE4max-sg1-treated mice from the high-dose or low-dose groups 7 and 67 days after injection. Values and error bars represent mean ± SD (n = 3 mice per group). (E) HTS reads of liver genomic DNA in mice 7 and 67 days after injection. Values and error bars represent mean ± SD (n = 3 mice per group). (F) FAH IHC staining of liver tissue sections. Scale bar, 200 μm. (G) Body weight curves of mice injected with rAAVs or PBS. Body weight was normalized to the weight on pre-injection day (day 0). Liver biopsy was performed on day 7 (black arrow), after which NTBC water was withdrawn. Mice with more than 20% weight loss were euthanized.
To test whether CBE-mediated restoration of the ATG start codon would have therapeutic effects, we chose sg1 for in vivo studies since the editing activity was similar between sg1 and sg2 (Figures 2B and 2C). First, the convenient HTVI method was employed to deliver the plasmid DNA encoding BE4max and sg1. 7 days after injection, NTBC was withdrawn to allow the proliferation of corrected hepatocytes.8 Mice injected with plasmids started to gain weight 21 days after NTBC withdrawal, whereas the FahNS/NS control mice lost weight 14 days after NTBC withdrawal until they were euthanized due to a huge weight decrease (Figure S1A). Ten weeks after NTBC withdrawal, mice were euthanized, and the livers were collected for analysis. Sanger sequencing indicated both G1 and G3 were partially converted to A, and a higher G1-to-A1 conversion rate was observed than for G3-to-A3 conversion (Figure S1B). Moreover, immunohistochemistry (IHC) staining also confirmed that FAH-positive hepatocytes occupied most of the tissue due to the expansion of corrected hepatocytes, which was further confirmed by western blotting with an anti-FAH antibody (Figures S1C and S1D). These data demonstrated that this strategy was able to restore the expression of Fah. However, HTVI is not a clinically acceptable method to deliver genetic vectors. Thus, we next examined rAAV vectors.
Split-BE4max Delivered through rAAV Corrected the Mutation in FahNS/NS Mice
To circumvent the payload limitations of rAAV, an intein-mediated split-BE4max strategy was leveraged.40 Initially, BE4max was split into two parts based on the previously reported split site of SpCas941 and fused to a corresponding split-intein moiety from cyanobacteria Nostoc punctiforme (Npu),40 resulting in two constructs named BE4max-N and BE4max-C (Figure S2A). To our surprise, the split constructs exhibited very low editing efficiency compared to the full-length version (Figure S2B). We speculated that the fusion of UGI in the C terminus of BE4max-C might affect the reconstitution of full-length BE4max, since a parallel experiment to split ABEmax was successful (data not shown). Previous study has shown that co-expression of free UGI not only suppresses the formation of indels and non-C-to-T conversions but it also increases C-to-T editing efficiency.42 Thus, we replaced the linker between nCas9 and UGI with a P2A self-cleaving peptide (referred to as BE4max-C-P2A) (Figure S2A). Co-transfecting BE4max-N and BE4max-C-P2A showed comparable editing efficiency with full-length BE4max (Figure S2B). Thus, we successfully developed an intein-mediated split-BE4max system.
To increase the tissue specificity of genome editing, the cytomegalovirus (CMV) promoter of the split-BE4max was replaced by a strong liver-specific promoter Lp1 and packaged into AAV8 serotype vectors (referred to as rAAV8-BE4max-N and rAAV8-BE4max-C-P2A-sg1). 6-week-old FahNS/NS mice were tail vein injected with two rAAV vectors (rAAV8-BE4max-N and rAAV8-BE4max-C-P2A-sg1 at a 1:1 ratio of vector genomes (vg); 1 × 1012 vg per rAAV per mouse in the high-dose group or 1 × 1010 vg per rAAV per mouse in the low-dose group). Injected mice were supplied with NTBC water for 7 days to allow base editing but to prevent the expansion of the corrected cells. On day 7, liver biopsy was performed. HTS showed that the editing efficiency was 2.7% (2.1%–3.3%) and 2.4% (2.1%–3.1%) for G1 and G3, respectively, in the high-dose group, but very low in the low-dose group (Figures 2D and S3). The efficiency of the desired correction to restore ATG was 0.7% (0.5%–0.85%) in the high-dose group since simultaneous G1/G3 conversion occurred in most of the alleles (Figures 2E and S3). IHC showed FAH-positive hepatocytes scattered in the livers of the high-dose group, which was consistent with HTS data (Figure 2F). To investigate the inflammatory response in the high-dose group, qRT-PCR was performed to examine the mRNA level of some pro-inflammatory cytokines, such as interleukin 6 (IL-6), IL-10, interferon β1 (IFN-β1), and tumor necrosis factor α (TNF-α), whose expression was elevated at day 7 post-injection, suggesting a mild inflammatory response (Figure S4). The body weight of treated mice increased gradually (Figure 2G). All mice were sacrificed 2 months (day 67) after NTBC withdrawal. HTS of the liver genomic DNA demonstrated that the percentage of correctly edited alleles increased dramatically due to the expansion of FAH-positive hepatocytes (Figures 2D, 2E, and S3). Specifically, 16.7% (15.2%–18.5%) and 15.6% (13.5%–18.8%) of the HTS reads were the desired sequence with restored ATG start codons from mice receiving high-dose and low-dose injections, respectively (Figures 2E and S3). However, the ratio of the sequence containing mutations without start codon restoration also increased, suggesting that some of the expanded cells contained both functional and non-functional mutations. IHC studies demonstrated that FAH-positive hepatocytes expanded in clones and occupied the majority of the liver tissue after 2 months of treatment (Figure 2F).
Creation of a De Novo Start Codon by BE4max in the Fah Gene to Ameliorate HT1
Although BE4max is efficient, it could not selectively catalyze distinct cytidines in our FahNS/NS model within an optimized editing window. The above data showed that a bystander mutation counteracted the desired G1-to-A1 conversion and greatly compromised the efficiency of restorative editing. We hypothesized that if a de novo start codon was created in the 5′ UTR of the FahNS/NS allele, in-frame with the coding sequence, it could restore Fah expression and function. Theoretically, conversion of the triplet code ATA to ATG could be catalyzed by an ABE targeting the coding strand; ACG and GTG could be converted to ATG by CBEs targeting the coding strand and non-coding strand, respectively. We identified a G−12TG−10 triplet 12 nt upstream of the start codon of the Fah gene as a possible target and designed a sgRNA (sg3) in which the desired G−12 nucleotide was located on protospacer position 4 within the editing window (Figure 3A). This placed the bystander nucleotide G−10 at protospacer position 2, which usually is not within the CBE editing window (Figure 3A). After testing in HEK293T reporter cells, we found that 71.1% of the HST reads contained A−12TG−10 while G−10 was almost untouched (Figures 3B and 3C). Although the G−13 (position 5) was also converted, it would not affect the de novo-created start codon (A−12TG−10) in the 5′ UTR (Figures 3A and 3B).
Figure 3.

Cytosine Base Editing of the 5′ UTR of FahNS/NS Allele to Create a De Novo Start Codon
(A) Diagram of sg3 designed to target the 5′ UTR of the FahNS/NS allele. PAM site is indicated with a blue line. Protospacer is indicated with a red line. The position of individual base within the protospacer is indicated with a blue number. The disease-causing mutation 1A>G (p.M1>V) is shown in red. C−12-to-T−12 (shown in green) conversion in the non-coding strand leads to the desired G−12-to-A−12 (shown in green) conversion in the coding strand, which creates a de novo start codon (A−12TG−10). Thus, four amino acids (methionine, leucine, valine, and serine, all shown in green) will be added to the N terminus of FAH protein. (B) Editing efficiencies of G−13, G−12, and G−10 targeted by sg3 in HEK293T reporter cells. Values and error bars represent the mean ± SD of three independent biological replicates. (C) HTS reads of genomic DNA from HEK293T reporter cells. Correct reads entail G-to-A conversion that creates a start codon (A−12TG−10) in the 5′ UTR (conversion at G−12, with or without conversion at G−13, but excluding conversion at G−10). Any reads that involve base substitutions unable to create an ATG start codon in the 5′ UTR are defined as incorrect reads. Values and error bars represent the mean ± SD of three independent biological replicates. (D) Editing efficiencies of G−13, G−12, and G-10 in BE4max-sg3-treated mice from high-dose or low-dose group 7 and 67 days after injection. Values and error bars represent mean ± SD (n = 3 mice per group). (E) HTS reads of liver genomic DNA in mice 7 and 67 days after injection. Values and error bars represent mean ± SD (n = 3 mice per group). (F) FAH IHC staining of liver tissue sections. Scale bar, 200 μm. (G) Body weight curves of mice injected with rAAVs or PBS. Body weight was normalized to the weight on pre-injection day (day 0). Liver biopsy was performed on day 7 (black arrow), after which NTBC water was withdrawn. Mice with more than 20% weight loss were euthanized. (H) The percentage of reads containing indels as well as an in-frame start codon in total reads with indels.
Mice were injected with rAAV particles expressing BEmax-N and BE4max-C-P2A-sg3, and the initial editing rate was determined 7 days after injection. In the high-dose group, HTS showed that the editing efficiency was 3.8% (2.2%–5.6%) for the targeted base G−12 (Figures 3D and S5), and almost all of the edited alleles (average 3.7% of the reads) created the de novo start codon (Figures 3E and S5). Moreover, consistent with the results from the HEK293T reporter cells, no significant editing occurred on G−10 (Figures 3D and S5). Surprisingly, no FAH-positive hepatocytes were distinguished by IHC at day 7 but they were expanded in clones after 2 months of treatment (Figure 3F). The body weight of BE4max-sg3-treated mice decreased slightly after NTBC withdrawal and then increased dramatically (Figure 3G). The group receiving low-dose rAAVs also recovered, although they required a longer time (Figure 3G). HTS of the liver genomic DNA from treated mice 2 months after NTBC withdrawal revealed that G-to-A conversion rates increased (Figures 3D and S5), and almost all of them bore the desired conversion (31% [29.1%–33.4%] and 9.1% [7.5%–10.6%] of total sequencing reads in the high-dose group and the low-dose group, respectively) to generate the new start codon (Figures 3E and S5). Intriguingly, the indel rates also dramatically increased, which was not observed in BE4max-sg1-treated mice (Figure 3E). Further analysis of the HTS reads with indels revealed that nearly all of them contained an in-frame ATG start codon (Figures 3H, S6, and S7), indicating that these indels were functional to restore Fah gene expression. We reasoned that since CBEs might yield low but detectable indels,18,43 a portion of indels generated ATG start codon to restore Fah expression in some cells that were also heathy for repopulation.
We noticed that in IHC studies, the FAH-positive signal density varied in different clusters of hepatocytes in BE4max-sg3-treated mice, while in BE4max-sg1-treated liver tissue FAH expression was homogeneous in all converted cells (Figure 4A), suggesting that the FAH protein level varied in distinct clusters of hepatocytes in BE4max-sg3-treated mice. Western blotting study revealed that the protein level of FAH was much lower in BE4max-sg3-treated liver tissues (Figure 4B). The lower FAH protein level was not due to the correction rate, since the start codon restoration efficiency, as well as the mRNA level, was higher in BE4max-sg3-treated mice (Figures 4C and 4D). In BE4max-sg3-treated mice, 4 aa were extended to the N terminus of FAH protein, and several genotypes of the start codon-restored alleles were detected (Figures 3A and S5–S7), which might induce variant protein level and result in lower FAH protein in the liver (Figures 4A and 4B). The extent of liver damage in BE4max-sg3-treated mice was significantly ameliorated compared to that in control mice (Figures 4E and 4F), suggesting that this 4-aa-extended FAH protein is functional and that BE4-mediated creation of a de novo start codon is feasible to ameliorate HT1 in our mouse model.
Figure 4.

In Vivo Cytosine Base Editing Rescued the Liver Damage Phenotype in FahNS/NS Mice
(A) FAH IHC staining of liver sections from BE4max-sg1- and BE4max-sg3-treated mice from the high-dose group 67 days after injection. Scale bar, 500 μm. (B) Western blot analysis of FAH expression in liver tissue from treated mice 67 days after injection. (C) The percentage of HTS reads with an in-frame ATG start codon in the Fah gene. These reads include correct reads (as defined in Figures 2 and 3) and reads with indels as well as an in-frame start codon. (D) Relative Fah mRNA expression in liver tissue 67 days after injection. (E) Serum AST, ALT, TBIL, and ALB levels in peripheral blood from treated mice 67 days after injection. Peripheral blood from PBS-injected mice was collected the day when mice lost 20% of their original weight. (F) H&E staining of liver tissue sections from treated mice. Liver tissue from PBS-injected mice was collected the day when mice lost 20% of their original weight. Scale bar, 200 μm. In all graphs, values and error bars represent mean ± SD (n = 3 mice per group). ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n.s., not significant.
Postponing Withdrawal of NTBC Revealed a Much Higher Initial Correction Rate
Although we had shown that both of the two base-editing strategies were feasible to ameliorate the disease phenotype of the FahNS/NS mouse model, the initial editing efficiency was quite low in vivo compared with the experiments in HEK293T reporter cells. We speculated that 7 days was too short for the viral infection, protein expression, and BE functioning since rAAV8-mediated ectopic protein expression reached its highest level 56 days after infection and maintained this for several months.44 To evaluate the real in vivo editing efficiency of BE4max-sg3, three mice were treated with a high dose of rAAV8-BE4max-sg3 particles and maintained on NTBC water for 21 days. As expected, plenty of FAH-positive hepatocytes were detected by IHC (Figure 5A), and these cells did not form clustered clones, suggesting that these FAH-positive cells did not expand from a few corrected cells. On day 21, the editing efficiency of G−12 and G−13 was increased by 2.7- and 3-fold, respectively, and the desired correct editing rate was increased by 2.7-fold, compared with day 7 (Figures 5B and 5C). These data suggest that rAAV-mediated delivery of intein-split BE4max is very efficient in vivo. To determine the off-target effects of the two sites targeted by BE4max, 10 top predicted off-targeted sites of each site were analyzed. Deep sequencing of these off-targeted sites in liver DNA from treated mice (high-dose group) 67 days after injection showed no evidence of editing (Figure S8).
Figure 5.

Postponing Withdrawal of NTBC Revealed a Much Higher Initial Correction Rate
(A) FAH IHC staining of liver tissue sections from BE4max-sg3-treated mice maintained on NTBC water for 21 days before euthanasia. Scale bars, 200 μm for top panel and 100 μm for bottom panel. (B) Editing efficiencies of G−13, G−12, and G−10 in BE4max-sg3-treated mice 7 or 21 days after injection without NTBC withdrawal. (C) The percentage of HTS reads with correct editing (G-to-A conversion that creates an ATG start codon [A−12TG−10] in the 5′ UTR) in liver genomic DNA from BE4max-sg3-treated mice. In all graphs, values and error bars represent mean ± SD (n = 3 mice per group). ∗p < 0.05, ∗∗p < 0.005.
Discussion
Base editing is an innovative technology to generate targeted base conversion through nCas9/sgRNA-guided direct nucleotide deamination without inducing excessive DSBs. In this study, we generated a novel HT1 mouse model with a mutant start codon through ABE7.10. We then used an intein-split system to deliver the BE4max/sgRNA via rAAV vectors in vivo. BE4max-mediated restoration of the original start codon or creation of a de novo start codon in the 5′ UTR of the Fah gene ameliorated HT1 disease symptoms in FahNS/NS mice, suggesting that split-BE4max is efficient and feasible to correct genetic disease through rAAV-based delivery systems.
Several studies have shown promising potential applications of BEs for in vivo editing or gene therapy. Various methods have been employed to deliver CBE components for gene editing or disease therapy, such as ribonucleoprotein (RNP) transfection in post-mitotic mouse inner ear cells,45 adenoviral vector targeting liver tissues in both adults and fetuses to treat metabolic diseases,30,46 and HTVI of ABE plasmid DNA to treat tyrosinemia.31 Although the above delivery systems are efficient, none of them is a clinically applicable approach for in vivo gene therapy to date. We successfully used the intein-split strategy for the BE4max system, which is generally the most efficient CBE variant with a longer coding sequence, to ameliorate an inherited liver metabolic disease. As BEs are able to catalyze base conversions in non-dividing cells, their broader applications would be achieved for gene therapy in other adult tissues, such as in brain, retina, heart, and skeletal muscle, as demonstrated in a recent report.34 We also found that direct fusion of two copies of UGI in an intein-split system greatly reduced BE4max activity, but the P2A fusion constructs enabled efficient editing in vitro and in vivo. Interestingly, one or two UGIs directly fused to intein-split SpCas9 at another split site exhibited efficient editing,34 suggesting that the split site of SpCas9 would affect the reconstitution of full-length BE4max in the intein-split CBE system.
Bystander mutations created by BEs are major obstacles for using precise editing to treat genetic diseases. To solve this problem, researchers have developed BE3 variants with narrowed editing windows (such as YEE-BE3 with the editing window narrowed to 2 nt [positions 5–6]), which dramatically increased editing precision.47 Through rational design and screening of human APOBEC3A variant mutations, eA3A-BE3 was developed to preferentially catalyze C-to-T conversions in specific TC motifs (with a TCR>TCY>VCN hierarchy) with greatly reduced bystander editing.48 These CBEs substantially increased the editing precision. However, due to limitations of the PAM sequence and editing window, the targeting scope of these editors is still restricted. Most recently, Liu et al.49 developed an innovative prime editing method that directly introduces genetic modifications (targeted insertions, deletions, and all 12 types of nucleotide conversions) into a specified DNA site without requiring DSBs and donor DNA templates. It is a promising technology, but still in its infancy, and the editing efficiency is lower than base editing.
A set of ingenious strategies using CRISPR variants to treat specific genetic diseases has been used to achieve considerable therapeutic effect in vivo. CRISPR-dCas9-mediated transcriptional upregulation of Lama1 was used to compensate for the lack of Lama2 in treating muscular dystrophy type 1A (MDC1A) in mice.50 Perturbation of the GATA1 binding site at the +58 BCL11A erythroid enhancer or genome editing of the HBG1 promoter that mimics the 13-nt HPFH (hereditary persistence of HbF) deletion observed in patients with SCD (sickle cell disease) was used to treat β-hemoglobinopathies.2,51,52 Furthermore, deletion of an upstream gene in the tyrosine synthesis metabolic pathway served to treat HT153 or primary hyperoxaluria type 1 (PH1),54 and genome editing-mediated allelic exchange treated a disease caused by compound heterozygous mutations.55 In our current study, we developed a method to target the sequence upstream of the mutation to create a de novo functional element (start codon) for the therapy of HT1 in mice. The extension of 4 aa at the N terminus of FAH protein might influence the protein level as indicated by variant intensity of IHC signals and lower FAH protein level in BE4max-sg3-treated liver tissue (Figures 4A and 4B), but it is functional to rescue the lethal phenotype, suggesting that the additional 4 aa might slightly affect protein translation or stability. This provides a novel strategy for genetic modification to treat hereditary diseases, especially for mutations that affect the start or stop codon as well as other non-coding region-related dysfunction, but whether it works well in other diseases needs to be further investigated.
Although no off-target editing has been identified after analysis of 10 top predicted sites of each target, target-independent DNA56,57 and RNA58,59 off-targeting effects induced by deaminases should be seriously considered, since rAAV-mediated CBE expression would be maintained for months. However, cytosine deaminase-induced off-target editing is very difficult to determine. Through introducing variant mutations in rAPOBEC1 and human APOBEC3A, several engineered CBEs have been developed to dramatically reduce the off-target editing on RNA58, 59, 60 or DNA61 substrates. These improved CBE variants might be suitable for gene therapy in clinical trials in the future.
In summary, we generated a novel HT1 mouse model by mutating the start codon and developed an intein-split method to deliver BE4max through rAAV vectors. Through two base editing-based strategies to either restore the start codon or create a de novo initiation codon, an inherited metabolic liver disease was ameliorated. Our study demonstrated that a BE is able to be delivered through the rAAV system for clinical purposes and that the de novo creation of functional genomic elements could be an important strategy for the treatment of genetic diseases.
Materials and Methods
Plasmids and Cloning
The plasmid encoding BE4max (pCMV_BE4max_3xHA) was a gift from David Liu (Addgene plasmid; catalog no. 112096). N- and C-terminal intein sequences were codon-optimized and synthesized by Genewiz (Suzhou, China). Schematic views of BE4max-N, BE4max-C, and BE4max-C-P2A are shown in Figure S2A. Corresponding amino acid sequences are listed in Table S1. Lentiviral vector plasmid for generating the reporter HEK293T cell line was constructed by inserting the 5′ UTR and exon 1 of the FahNS/NS allele and a DsRed cassette driven by the EF-1α promoter into the lentiCRISPR v2 (a gift from Feng Zhang, Addgene plasmid; catalog no. 52961) backbone. Plasmids used for targeting the FahNS/NS allele in the reporter HEK293T cell line and the endogenous HEK293T genomic loci contain sgRNAs driven by the U6 promoter and EGFP driven by a CMV promoter.
Cell Culture, Transfection, and Genomic DNA Preparation
HEK293T (ATCC CRL-3216) cells were maintained in DMEM (Gibco) supplemented with 10% FBS (Gibco), 100 U/mL penicillin, and 100 mg/mL streptomycin (basal media) at 37°C and 5% CO2. Cells (∼2 × 105 cells per well) were seeded onto 24-well plates (Corning Life Sciences) and transfected at 70% confluence using polyethylenimine (PEI) (Polysciences) following the manufacturer’s recommended protocol. For full-length BE4max transfection, 400 ng of pCMV_BE4max_3xHA, 200 ng of sgRNA expression plasmid, and 400 ng of pCDNA3.1 were combined in the transfection master mix. For split BE4max transfection, 400 ng of BE4max-N encoding plasmid, 400 ng of BE4max-C (or BE4max-C-P2A) encoding plasmid, and 200 ng of sgRNA expression plasmid were combined. Each well was transfected with a total of 1 μg of plasmids. 72 h after transfection, GFP-positive cells were sorted and genomic DNA was extracted using a TIANamp blood DNA kit (Tiangen) according to the manufacturer’s instructions.
Lentiviral Vector Production
HEK293T cells were seeded on a 15-cm dish (Corning Life Sciences) and transfected at 80% confluence using PEI (Polysciences) following the manufacturer’s recommended protocol. 10 μg of psPAX2, 10 μg of pMD2.G, and 10 μg of lentiviral vector plasmid were co-transfected. 8 h after transfection, the medium was replaced. The supernatant containing lentiviral particles was harvested and filtered using a 0.2-μm syringe filter (Pall) at 48 h after transfection.
Lentiviral Transduction
HEK293T cells were seeded on a 15-cm dish (Corning Life Sciences). 24 h later, cells were transduced with viral supernatants. Two days after transduction, cells were selected in puromycin (2 μg/mL), and stably transduced cells were enriched for 7 days.
rAAV Vector Production
HEK293T cells were seeded on 15-cm dishes before transfection. 10 μg of AAV8 capsid plasmid, 10 μg of AAV helper plasmid, and 10 μg of AAV expression vector were co-transfected into HEK293T cells at 80% confluence using PEI following the manufacturer’s recommended protocol. 8 h after transfection, the medium was replaced. 60–72 h after transfection, cells were re-suspended by pipetting and harvested by centrifugation (3,500 rpm). The cell pellet was re-suspended in lysis buffer (150 mM NaCl, 20 mM Tris [pH 8.0]) and lysed through three consecutive freeze-thaw cycles between a dry ice/ethanol bath and 37°C water bath. MgCl2 (1 mM final concentration) and Benzonase (25 U/mL final concentration) (Merck) were added into the cell lysate followed by incubation at 37°C for 30 min. The supernatant was harvested and centrifuged at 5,500 rpm, 4°C for 20 min to remove the cell debris. The virus was further purified by iodixanol gradient ultracentrifugation (60,000 rpm for 90 min at 16°C in a Beckman Ti70 rotator) and concentrated to about 1 mL by ultrafiltration. Finally, the virus titer was determined by quantitative real-time PCR.
Animal Studies
C57BL/6 strain mice purchased from Shanghai Laboratory Animal Center were housed in standard cages in a specific pathogen-free facility on a 12-h light/12-h dark cycle with ad libitum access to food and water. All animal experiments conformed to the regulations drafted by the Association for Assessment and Accreditation of Laboratory Animal Care in Shanghai and were approved by the East China Normal University Center for Animal Research. The FahNS/NS mouse was obtained as previously described21 and maintained on NTBC water at a concentration of 10 mg/L. For hydrodynamic tail vein plasmid injection, 200 μg of BE4max encoding plasmid and 100 μg of sgRNA expressing plasmid were diluted into 2 mL of saline and injected via the tail vein in 5–7 s into 8-week-old mice. For systematic rAAV delivery, 6-week-old mice were injected with 1 × 1012 (high-dose group) or 1 × 1010 (low-dose group) viral vector genomes (for each rAAV) per mouse. Mice were humanely euthanized by carbon dioxide asphyxiation if over 20% of body weight was lost after NTBC withdrawal.
Western blot, IHC, and Histology
Mice were humanely sacrificed by carbon dioxide asphyxiation. Mouse liver tissue was lysed in radioimmunoprecipitation assay (RIPA) buffer with proteinase and phosphatase inhibitors. Total protein was quantified using a Pierce protein bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific). Proteins were resolved on SDS-PAGE, transferred to nitrocellulose membranes, and detected by anti-FAH antibody (AbboMax, 1:2,000) and anti-actin antibody (Sigma, 1:5,000). For IHC and hematoxylin and eosin (H&E) staining, mouse livers were fixed with 4% paraformaldehyde (PFA), embedded in paraffin, and sectioned at 5 μm. Anti-FAH antibody (AbboMax, 1:2,000) was used for IHC to detect FAH-positive hepatocytes.
qRT-PCR
Total RNA was extracted from mouse liver using RNAiso Plus (Takara) and reverse transcribed using the HiScript II Q RT SuperMix (Vazyme) kit. qRT-PCR reactions were performed on QuantStudio 3 (Applied Biosystems) using Hieff qPCR SYBR Green master mix (Yeasen Biotechnology). Data were normalized to β-actin. Primers used for qRT-PCR are listed in Table S2.
Serum Biochemical Analysis
Mouse blood was collected using retro-orbital puncture and centrifuged at 12,000 rpm for 10 min at 4°C for serum collecting. For control groups, mouse blood was collected when weight loss was more than 20%. Mouse serum was kept at −80°C before testing. AST, ALT, TBIL, and ALB levels were measured by Adicon Clinical Laboratory.
Targeted Deep Sequencing and Off-Target Analysis
HEK293T or mouse liver genomic DNA was isolated using the TIANamp blood DNA kit (Tiangen) according to the manufacturer’s instructions. PCR products for targeted deep sequencing were prepared as described in Hi-TOM kits (Novogene). Mixed samples were sequenced on the Illumina HiSeq platform as previously described.62 Off-target sites were predicted using CRISPOR.63 Predicted potential off-target sites are listed in Table S3. Next-generation sequencing (NGS) data were analyzed using BE-Analyzer.64 All primers used for Hi-TOM deep sequencing are listed in Table S4.
Data Availability
The deep-sequencing data from this study have been deposited in the NCBI Sequence Read Archive (SRA) with BioProject: PRJNA593170 and PRJNA593901.
Author Contributions
D.L., L.Y., L.W., and Mi.L. designed the experiments. L.Y., L.W., Y. Huo, X.C., S.Y., Y. Hu, X.Z, R.Z., H.G, H.H., X.M., Me.L., H.L., W.Y. and J.W. performed the experiments and analyzed the data. D.L., L.Y., L.W., Mi.L., and Y. Huo wrote the manuscript. D.L. supervised the research.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Dr. Stefan Siwko for proofreading this manuscript and the support of the ECNU Public Platform for Innovation (011). This work was partially supported by grants from the National Key R&D Program of China (2019YFA0110802, 2019YFA0802802); the National Natural Science Foundation of China (81670470, 81873685); the Innovation Program of the Shanghai Municipal Education Commission (2019-01-07-00-05-E00054); the Shanghai Municipal Commission for Science and Technology (18411953500); and by the Fundamental Research Funds for the Central Universities.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.05.001.
Supplemental Information
References
- 1.Matharu N., Rattanasopha S., Tamura S., Maliskova L., Wang Y., Bernard A., Hardin A., Eckalbar W.L., Vaisse C., Ahituv N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science. 2019;363 doi: 10.1126/science.aau0629. eaau0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakade S., Tsubota T., Sakane Y., Kume S., Sakamoto N., Obara M., Daimon T., Sezutsu H., Yamamoto T., Sakuma T., Suzuki K.T. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014;5:5560. doi: 10.1038/ncomms6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer S., Suresh S., Guo D., Daman K., Chen J.C.J., Liu P., Zieger M., Luk K., Roscoe B.P., Mueller C. Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nature. 2019;568:561–565. doi: 10.1038/s41586-019-1076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 7.Cox D.B.T., Platt R.J., Zhang F. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao Y.J., Wang L.R., Guo N.N., Wang S.F., Yang L., Li Y.J., Wang M., Yin S., Han H., Zeng L. Cas9-nickase-mediated genome editing corrects hereditary tyrosinemia in rats. J. Biol. Chem. 2018;293:6883–6892. doi: 10.1074/jbc.RA117.000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin H., Song C.Q., Dorkin J.R., Zhu L.J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anguela X.M., Sharma R., Doyon Y., Miller J.C., Li H., Haurigot V., Rohde M.E., Wong S.Y., Davidson R.J., Zhou S. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood. 2013;122:3283–3287. doi: 10.1182/blood-2013-04-497354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guan Y., Ma Y., Li Q., Sun Z., Ma L., Wu L., Wang L., Zeng L., Shao Y., Chen Y. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016;8:477–488. doi: 10.15252/emmm.201506039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu L., Park K.H., Zhao L., Xu J., El Refaey M., Gao Y., Zhu H., Ma J., Han R. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther. 2016;24:564–569. doi: 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beyret E., Liao H.K., Yamamoto M., Hernandez-Benitez R., Fu Y., Erikson G., Reddy P., Izpisua Belmonte J.C. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019;25:419–422. doi: 10.1038/s41591-019-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santiago-Fernández O., Osorio F.G., Quesada V., Rodríguez F., Basso S., Maeso D., Rolas L., Barkaway A., Nourshargh S., Folgueras A.R. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat. Med. 2019;25:423–426. doi: 10.1038/s41591-018-0338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X., Li J., Wang Y., Yang B., Wei J., Wu J., Wang R., Huang X., Chen J., Yang L. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat. Biotechnol. 2018;36:946–949. doi: 10.1038/nbt.4198. [DOI] [PubMed] [Google Scholar]
- 21.Yang L., Zhang X., Wang L., Yin S., Zhu B., Xie L., Duan Q., Hu H., Zheng R., Wei Y. Increasing targeting scope of adenosine base editors in mouse and rat embryos through fusion of TadA deaminase with Cas9 variants. Protein Cell. 2018;9:814–819. doi: 10.1007/s13238-018-0568-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 23.Kim K., Ryu S.M., Kim S.T., Baek G., Kim D., Lim K., Chung E., Kim S., Kim J.S. Highly efficient RNA-guided base editing in mouse embryos. Nat. Biotechnol. 2017;35:435–437. doi: 10.1038/nbt.3816. [DOI] [PubMed] [Google Scholar]
- 24.Zhang R., Liu J., Chai Z., Chen S., Bai Y., Zong Y., Chen K., Li J., Jiang L., Gao C. Generation of herbicide tolerance traits and a new selectable marker in wheat using base editing. Nat. Plants. 2019;5:480–485. doi: 10.1038/s41477-019-0405-0. [DOI] [PubMed] [Google Scholar]
- 25.Zong Y., Wang Y., Li C., Zhang R., Chen K., Ran Y., Qiu J.L., Wang D., Gao C. Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat. Biotechnol. 2017;35:438–440. doi: 10.1038/nbt.3811. [DOI] [PubMed] [Google Scholar]
- 26.Zheng K., Wang Y., Li N., Jiang F.F., Wu C.X., Liu F., Chen H.C., Liu Z.F. Highly efficient base editing in bacteria using a Cas9-cytidine deaminase fusion. Commun. Biol. 2018;1:32. doi: 10.1038/s42003-018-0035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang P., Ding C., Sun H., Xie X., Xu Y., Zhang X., Sun Y., Xiong Y., Ma W., Liu Y. Correction of β-thalassemia mutant by base editor in human embryos. Protein Cell. 2017;8:811–822. doi: 10.1007/s13238-017-0475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G., Liu Y., Zeng Y., Li J., Wang L., Yang G., Chen D., Shang X., Chen J., Huang X., Liu J. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein Cell. 2017;8:776–779. doi: 10.1007/s13238-017-0458-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou C., Zhang M., Wei Y., Sun Y., Sun Y., Pan H., Yao N., Zhong W., Li Y., Li W. Highly efficient base editing in human tripronuclear zygotes. Protein Cell. 2017;8:772–775. doi: 10.1007/s13238-017-0459-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossidis A.C., Stratigis J.D., Chadwick A.C., Hartman H.A., Ahn N.J., Li H., Singh K., Coons B.E., Li L., Lv W. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018;24:1513–1518. doi: 10.1038/s41591-018-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song C.Q., Jiang T., Richter M., Rhym L.H., Koblan L.W., Zafra M.P., Schatoff E.M., Doman J.L., Cao Y., Dow L.E. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020;4:125–130. doi: 10.1038/s41551-019-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villiger L., Grisch-Chan H.M., Lindsay H., Ringnalda F., Pogliano C.B., Allegri G., Fingerhut R., Häberle J., Matos J., Robinson M.D. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 33.Basner-Tschakarjan E., Mingozzi F. Cell-mediated immunity to AAV vectors, evolving concepts and potential solutions. Front. Immunol. 2014;5:350. doi: 10.3389/fimmu.2014.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy J.M., Yeh W.H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrow G., Angileri F., Tanguay R.M. Molecular aspects of the FAH mutations involved in HT1 disease. Adv. Exp. Med. Biol. 2017;959:25–48. doi: 10.1007/978-3-319-55780-9_3. [DOI] [PubMed] [Google Scholar]
- 37.Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin. Liver Dis. 2001;21:563–571. doi: 10.1055/s-2001-19035. [DOI] [PubMed] [Google Scholar]
- 38.Paulk N.K., Wursthorn K., Wang Z., Finegold M.J., Kay M.A., Grompe M. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 2010;51:1200–1208. doi: 10.1002/hep.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L., Shao Y., Li L., Tian F., Cen J., Chen X., Hu D., Zhou Y., Xie W., Zheng Y. Efficient liver repopulation of transplanted hepatocyte prevents cirrhosis in a rat model of hereditary tyrosinemia type I. Sci. Rep. 2016;6:31460. doi: 10.1038/srep31460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Truong D.J., Kühner K., Kühn R., Werfel S., Engelhardt S., Wurst W., Ortiz O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chew W.L., Tabebordbar M., Cheng J.K., Mali P., Wu E.Y., Ng A.H., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Xue W., Yan L., Li X., Wei J., Chen M., Wu J., Yang B., Yang L., Chen J. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res. 2017;27:1289–1292. doi: 10.1038/cr.2017.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komor A.C., Zhao K.T., Packer M.S., Gaudelli N.M., Waterbury A.L., Koblan L.W., Kim Y.B., Badran A.H., Liu D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017;3 doi: 10.1126/sciadv.aao4774. eaao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 45.Yeh W.H., Chiang H., Rees H.A., Edge A.S.B., Liu D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018;9:2184. doi: 10.1038/s41467-018-04580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chadwick A.C., Wang X., Musunuru K. In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin type 9) as a therapeutic alternative to genome editing. Arterioscler. Thromb. Vasc. Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gehrke J.M., Cervantes O., Clement M.K., Wu Y., Zeng J., Bauer D.E., Pinello L., Joung J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018;36:977–982. doi: 10.1038/nbt.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kemaladewi D.U., Bassi P.S., Erwood S., Al-Basha D., Gawlik K.I., Lindsay K., Hyatt E., Kember R., Place K.M., Marks R.M. A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature. 2019;572:125–130. doi: 10.1038/s41586-019-1430-x. [DOI] [PubMed] [Google Scholar]
- 51.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw3768. eaaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L., Li L., Ma Y., Hu H., Li Q., Yang Y., Liu W., Yin S., Li W., Fu B. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Res. 2020;30:276–278. doi: 10.1038/s41422-019-0267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pankowicz F.P., Barzi M., Legras X., Hubert L., Mi T., Tomolonis J.A., Ravishankar M., Sun Q., Yang D., Borowiak M. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 2016;7:12642. doi: 10.1038/ncomms12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zabaleta N., Barberia M., Martin-Higueras C., Zapata-Linares N., Betancor I., Rodriguez S., Martinez-Turrillas R., Torella L., Vales A., Olagüe C. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat. Commun. 2018;9:5454. doi: 10.1038/s41467-018-07827-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D., Li J., Song C.Q., Tran K., Mou H., Wu P.H., Tai P.W.L., Mendonca C.A., Ren L., Wang B.Y. Cas9-mediated allelic exchange repairs compound heterozygous recessive mutations in mice. Nat. Biotechnol. 2018;36:839–842. doi: 10.1038/nbt.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364:289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin S., Zong Y., Gao Q., Zhu Z., Wang Y., Qin P., Liang C., Wang D., Qiu J.L., Zhang F., Gao C. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science. 2019;364:292–295. doi: 10.1126/science.aaw7166. [DOI] [PubMed] [Google Scholar]
- 58.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 60.Grünewald J., Zhou R., Iyer S., Lareau C.A., Garcia S.P., Aryee M.J., Joung J.K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019;37:1041–1048. doi: 10.1038/s41587-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doman J.L., Raguram A., Newby G.A., Liu D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0414-6. Published online February 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Q., Wang C., Jiao X., Zhang H., Song L., Li Y., Gao C., Wang K. Hi-TOM: a platform for high-throughput tracking of mutations induced by CRISPR/Cas systems. Sci. China Life Sci. 2019;62:1–7. doi: 10.1007/s11427-018-9402-9. [DOI] [PubMed] [Google Scholar]
- 63.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E., Kim S.T., Eils R., Kim J.S., Bae S. Web-based design and analysis tools for CRISPR base editing. BMC Bioinformatics. 2018;19:542. doi: 10.1186/s12859-018-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The deep-sequencing data from this study have been deposited in the NCBI Sequence Read Archive (SRA) with BioProject: PRJNA593170 and PRJNA593901.