

Abstract
Merkel cell carcinoma (MCC), a neuroendocrine cancer of the skin, is caused by integration of Merkel cell polyomavirus (MCV) and persistent expression of Large T antigen (LT) and Small T antigen (ST). We report that ST in complex with MYCL and the EP400 complex activates expression of LSD1 (KDM1A), RCOR2, and INSM1 to repress gene expression by the lineage transcription factor ATOH1. LSD1 inhibition reduces growth of MCC in vitro and in vivo. Through a forward-genetics CRISPR-Cas9 screen, we identified an antagonistic relationship between LSD1 and the non-canonical BAF (ncBAF) chromatin remodeling complex. Changes in gene expression and chromatin accessibility caused by LSD1 inhibition could be partially rescued by BRD9 inhibition, revealing that LSD1 and ncBAF antagonistically regulate an overlapping set of genes. Our work provides mechanistic insight into the dependence of MCC on LSD1 and a tumor suppressor role for ncBAF in cancer.
Introduction
Approximately 80% of Merkel cell carcinoma (MCC) contains integrated Merkel cell polyomavirus (MCPyV or MCV) DNA and expression of viral LT and ST1. MCV LT and ST function as oncoproteins, maintaining the viability of virus-positive MCC cell lines, promoting cellular transformation, and inducing tumor formation in mice2. MCC harboring MCV typically contains few cellular mutations, consistent with a significant role for LT and ST in MCC oncogenesis3. In contrast, virus-negative MCC contains many somatic mutations due to sunlight exposure. Both types of MCC share certain histological features with normal Merkel cells including expression of KRT20 (CK20) and chromogranin A4.
MCV ST recruits MYCL (L-Myc) and MAX to the EP400 (p400) transcriptional activator complex. This activity is required for the ST-mediated transformation of normal cells5. To identify genes regulated by the ST-MYCL-EP400 complex, ChIP-seq of ST, MAX, and EP400 and RNA-seq following depletion of EP400, MYCL, or ST was performed in MCC cell lines5. One validated target gene of the ST- MYCL-EP400 complex is MDM2 that represses wild-type p53 activity in virus-positive MCC6.
LSD1 is a histone demethylase that removes H3K4 mono- and di-methylation transcription marks7. An alternatively spliced form of LSD1 (+8a) can demethylate H3K9me8. LSD1 assembles into a core ternary transcriptional repressor complex containing RCOR1 (CoREST), RCOR2, or RCOR3 and is recruited to chromatin by the SNAG domain-containing proteins INSM1, GFI1, GFI1B, SNAI1, and SNAI2 that play key roles in development and oncogenesis9,10.
Small molecule inhibitors of LSD1 have promising activity in preclinical models of acute myeloid leukemia (AML), small cell lung cancer (SCLC), and medulloblastoma11–14. In addition to inactivating its enzymatic activity, some LSD1 inhibitors disrupt the interaction between LSD1 and SNAG domain proteins10 and interaction with chromatin11. The exact mechanism of how LSD1 inhibition interferes with cancer cell growth has yet to be determined12.
The mammalian SWI/SNF (mSWI/SNF, BRG1/BRM-Associated Factor, BAF) complexes contribute to regulation of genes involved in differentiation15. Over 20% of all human cancers harbor mutations in mSWI/SNF complex components15. The 29 gene products assemble combinatorically to produce three related mSWI/SNF complexes: canonical BAF (cBAF), polybromo-associated BAF (PBAF), and non-canonical BAF (ncBAF) complex15–17. Each complex contains SMARCA4 (BRG1) or SMARCA2 (BRM) but are distinguished by complex-specific subunits15,16. BRD9, a BET family protein that reads acetylated lysine histone marks, is unique to the ncBAF complex along with GLTSCR1 (BICRA) and GLTSCR1-like (BICRAL)16,17. Targeting the ncBAF complex confers synthetic lethality in synovial sarcoma and malignant rhabdoid tumors, and mis-splicing of BRD9 provides growth advantages in SF3B1-mutated cancers, suggesting a specific role in cancer16–19.
Results
MCV ST activates the LSD1 repressor complex
RNA-seq performed on MCC cell lines after RNAi-mediated knockdown of MYCL, EP400, or ST identified changes in the levels of many expressed genes5. Integrative analysis of RNA- and ChIP-seq revealed that reduced levels of genes were significantly associated with promoters bound by ST, MAX, and EP400 (Extended Data Fig. 1a). In contrast to genes directly activated by the ST-MYCL-EP400 complex, depletion of EP400, MYCL, or ST led to increased levels of genes in differentiation and cancer pathways (Extended Data Fig. 1d and Source Data 1). We suspected that MCV ST could transactivate a transcriptional repressor and identified several components of a histone demethylase complex, including LSD1 (KDM1A), RCOR2, and INSM1 (Fig. 1a-c, Extended Data Fig. 1b and 1e). Depletion of EP400 led to reduced levels of RCOR2 and LSD1 mRNA, as determined by RT-qPCR (Fig. 1d and Extended Data Fig. 1c). While LSD1 expression is ubiquitous, RCOR2 expression is developmentally stage-specific and INSM1 is predominantly expressed in developing neuroendocrine tissues and the nervous system20. INSM1 has been reported to be a specific immunohistochemical marker for MCC21.
Fig. 1. MCV ST transactivates components of the LSD1 complex.

a-c. Two biological replicates of MAX (MAX-1 and MAX-2), EP400 (EP400–1 and EP400–2), and MCV ST (ST-1 and ST-2) ChIP-seq reveal that MCV ST in a complex with MAX and EP400 binds to the promoters of LSD1 (KDM1A), RCOR2 and INSM1. The UCSC genomes browser was used to visualize peaks41. d. RT-qPCR assesses EP400, RCOR2, and LSD1 levels after expression of inducible EP400 or control shRNA in MKL-1. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05. e. ChIP-qPCR indicates that MAX, EP400, and MCV ST bind specifically to RCOR2, LSD1, and ATOH1 promoters in MKL-1. KRT9 promoter and the intergenic region serve as negative controls. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05; **<0.005. f. Immunoprecipitation and mass spectrometry analysis (MudPIT) shows that LSD1 and RCOR2 form a multi-protein complex in MCC cell lines. The Venn diagram shows 123 overlapping binding partners of LSD1 and RCOR2 from four independent mass spectrometry analyses (Source Data 1). g. PHL cells with MCV T antigens (+) have increased levels of RCOR2 and LSD1 mRNA. The INSM1 levels were below detection limits in PHL cells. The signals were normalized to RPLP0. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05; **<0.005. h. PHL cells with MCV T antigens (+) have increased protein levels of RCOR2 and LSD1. The experiment was performed at least three times. See Unprocessed gels Figure 1. i. RCOR2 levels increase, and LSD1 binds to RCOR2 in PHL cells transformed with MCV T antigens (+). Immunoprecipitation of LSD1, followed by western blotting, was performed three times. # non-specific band. See Unprocessed gels Figure 1. j. ChIP-qPCR of MAX, EP400, and MCV ST in PHL cells expressing T antigens indicate that T antigens enhance MAX and EP400 binding to RCOR2 promoter. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05; **<0.005.
We performed large scale immunoprecipitation (IP) with antibodies specific for endogenous RCOR2 and LSD1 in MKL-1 and WaGa MCC cell lines followed by Multidimensional Protein Identification Technology (MudPIT) to identify associated proteins22. LSD1 and RCOR2 formed a complex with INSM1 in MCC cells together with HDAC1 and other reported components of the CoREST complex (Fig. 1f and Source Data 1)23. ChIP-qPCR confirmed specific binding of ST, MAX, and EP400 to promoters of RCOR2 and LSD1 (Fig. 1e).
We previously reported that MCV T antigens could induce anchorage-independent growth of IMR90 fibroblasts that co-expressed a dominant-negative form of p53 (p53DD), hTERT, and MYCL (L) called PHL cells5,24. To determine if the expression of MCV T antigens affected levels of LSD1, RCOR2, and INSM1, RNA, and protein levels were assessed in PHL cells with or without MCV T antigens (Fig. 1g and h). T antigens increased levels of LSD1 and RCOR2 but not INSM1. Furthermore, LSD1 could specifically co-immunoprecipitate RCOR2 in PHL cells with MCV T antigens (Fig. 1i). ChIP-qPCR for ST, MAX, and EP400 was performed to assess enrichment for the promoters of LSD1 and RCOR2 (Fig. 1j). We found that MAX and EP400 enrichment increased in the presence of T antigens. These results indicate that ST-MYCL-EP400 can activate the expression of the LSD1 complex components. Of note, expression of an ST mutant unable to bind the EP400 complex in PHL cells (PH2L) increased levels of LSD1 and RCOR2, suggesting that T antigens may have additional means to activate these genes (Extended Data Fig. 1f-g).
LSD1 inhibition abrogates T antigen-driven transformation
We assessed RNA and protein levels of RCOR2, LSD1, and INSM1 in several virus-positive (MKL-1, MKL-2, MS-1, WaGa, PeTa, and BroLi), virus-negative MCC cell lines (UISO, MCC13, and MCC26), and human foreskin fibroblasts (HFF). LSD1, RCOR2, and INSM1 transcripts (Fig. 2a-c) and protein (Fig. 2d) were significantly more abundant in the virus-positive MCC cell lines than in virus-negative MCC or HFF. Although there is a controversy regarding the ontology of the virus-negative MCC cell lines, the result shows that MCC cell lines expressing T antigens express LSD1, RCOR2, and INSM1 abundantly.
Fig. 2. LSD1 inhibition abrogates MCV T antigen-dependent transformation.

a-c. RT-qPCR of six virus-positive (MKL-1, MKL-2, MS-1, WaGa, PeTa, and BroLi), virus-negative MCC lines (UISO, MCC13, and MCC26), and primary human cells (HFF) were performed in triplicate. The signals were normalized to the geomean of three loading controls (18s rRNA, RPLP0, and β-actin (ACTB)). Data are shown as mean of n=3 ± SD; two-sided t-test, **<0.005; ****<0.00005. d. MCV-positive MCC cell lines express abundant levels of INSM1 and RCOR2. The experiment was performed at least three times. See Unprocessed gels Figure 2. e. Virus-positive (MCV+), but not virus-negative f. (MCV-), MCC cell lines are sensitive to LSD1 inhibition. Cells were treated with various concentrations of LSD1 inhibitor GSK2879552 for 12 days. The CellTiter-Glo assay measured relative viability. Data are shown as mean and reflect three biological replicates. g. Anchorage-independent growth of IMR90 PHL cells expressing MCV T antigens and MKL-1 MCC cells with an LSD1 inhibitor (GSK-LSD1, 0.1 μM) shows that colony formation of normal cells by MCV T antigens and MCC maintenance was dependent on LSD1 activity. h. Quantification of the number of soft agar colonies (g) is shown. Data are shown as mean of n=4 ± SD; two-sided t-test, *P<0.05; **<0.005. i. MKL-1 cells were treated with GSK2879552 for eight days. The finding was observed at least five times.
To determine if MCC cell lines were sensitive to LSD1 inhibition, we treated the same panel of MCC cell lines with LSD1 inhibitors GSK2879552, CPI-242, and GSK-LSD112. Cell viability was assessed by CellTiter-Glo assay after 12 days of treatment (Fig. 2e and f and Extended Data Fig. 2). All virus-positive MCC cell lines tested were sensitive to nanomolar concentrations of LSD1 inhibitors, whereas the virus-negative cell lines were highly resistant.
To determine if the transformation of normal cells by MCV T antigens required LSD1 activity, we performed an anchorage-independent growth assay using MKL-1 and PHL cells with MCV T antigens24 (Fig. 2g and h). The addition of the GSK-LSD1 inhibitor reduced the transformed phenotype of PHL cells with MCV T antigens, as shown by the reduced number of colonies formed when cultured in soft agar. Similarly, virus-positive MKL-1 MCC cells were able to grow in soft agar, but treatment with the GSK-LSD1 inhibitor completely blocked colony formation.
Integrated LSD1 target analysis reveals critical gene expression changes during LSD1 inhibition
The virus-positive MCC cells grow as loose clusters in suspension, formed tight floating spheroids in the presence of LSD1 inhibitors (Fig. 2i). To assess for changes in gene expression, RNA-seq was performed on six virus-positive and the virus-negative UISO MCC cell lines treated with various LSD1 inhibitors for one or three days (Fig. 3a-c). Despite differences in treatment parameters, similar gene expression changes were observed in response to LSD1 inhibition in all six virus-positive cell lines but not in UISO cells when assessed by RNA-seq and RT-qPCR (Fig. 3b and d). Principal Component Analysis (PCA) of the RNA-seq data showed similar global shift changes in gene expression in all six virus-positive MCC cells with LSD1 inhibition (Fig. 3c). Levels of many genes including FAM5B (BRINP2), ID1, CDH11, CALB2, PROM1, GFI1, SMARCA1, SMAD9, ID2, and HES1 were significantly increased by LSD1 inhibition in virus-positive cells but not in virus-negative cells (Fig. 3a, 3b, 3d, and Extended Data Fig. 3a, 3b, and 3d). These changes were accompanied by increased levels of H3K4me1 but not H3K4me2, suggesting that LSD1 inhibition specifically targets mono-methylation of H3K4 in MCC cells (Fig. 3e).
Fig. 3. RNA-seq reveals critical gene expression changes during LSD1 inhibition in MCC.

a. MKL-1 cells were treated with GSK-LSD1 (0.1 μM) for three days and processed for RNA-seq. NS= Not Significant; Log 2 FC= Fold change cutoff 1.5; P=P-value cutoff 10e-6. The Wald test was performed using the DEseq2 R package42 with the p-values adjusted by Benjamini-Hochberg. n=3. b. RNA-seq of six virus-positive MCC and virus-negative UISO cell lines treated with LSD1 inhibitors (GSK-LSD1 for three days or CPI-670242 for one day). n=3. c. The PCA analysis displays global gene expression changes caused by LSD1 inhibition. d. MKL-1, WaGa, and UISO cell lines were treated with GSK-LSD1 (0.05 μM) for three days. The signals were normalized to untreated samples and RPLP0 in each sample. Data are shown as mean of n=3 ± SD; two-sided t-test; * P<0.05, **<0.005, ***<0.0005 and ****<0.00005. e. LSD1 inhibition increases global levels of H3K4me1 and LSD1 target genes. MKL-1 and WaGa cells were treated with GSK-LSD1 (0.05 μM) for six days, and the whole-cell lysates and histone extracts were prepared. LT= MCV LT; ST=MCV ST; VINC=Vinculin. The experiment was performed at least three times. See Unprocessed gels Figure 3. f. Cells were treated with LSD inhibitor GSK-LSD1 (0.05 μM) for three or six days. LSD1 inhibition activates the BMP pathway as assessed by increased levels of phosphorylated SMAD1/5/9 (P-SMAD1/5/9). The experiment was performed at least three times. See Unprocessed gels Figure 3. g. Cells were transduced with either control or LSD1 targeting shRNA for six days and harvested for western blotting. The experiment was performed at least three times. See Unprocessed gels Figure 3. h. LSD1 inhibition perturbs gene expression in the virus-positive MCC cell lines but not in the virus-negative MCC cell lines. Cells were treated with GSK-LSD1 (0.05 μM) for three days. The experiment was performed at least three times. See Unprocessed gels Figure 3.
The LSD1 target gene analysis predicted that BMP (Bone morphogenetic protein) signaling factors, including Inhibitors of DNA binding 1 (ID1), ID2, ID3, and SMAD9 were targets (Fig. 3 and Extended Data Fig. 3b-d). The BMP pathway regulates embryonic patterning and neuroectodermal development25. Soluble BMP proteins bind to their receptors to promote phosphorylation of SMAD1, SMAD5, and SMAD9 that, in turn, transactivates specific target genes, including IDs25. To determine if LSD1 inhibition activates BMP, we performed western blotting and observed increased phosphorylation of SMAD1, SMAD5, and SMAD9 as well as levels of ID1 RNA and protein (Fig. 3e-h and Extended Data Fig. 3c).
To test the specificity of the LSD1 inhibitors, we depleted LSD1 using an shRNA in MKL-1 and WaGa cells and assessed levels of LSD1 target genes by western blotting (Fig. 3g). We found that knockdown of LSD1 led to increased levels of PROM1, ID1, and SMARCA1, similar to their response to LSD1 inhibitors. LSD1 inhibitor did not affect protein levels of ID1, PROM1, and SMARCA1 in the virus-negative MCC cell lines (Fig. 3h).
To identify genes directly regulated by the LSD1 repressor complex, we performed ChIP-seq of LSD1 and RCOR2 in MKL-1 cells (Fig. 4a and Extended Data Fig. 4). We observed that LSD1 and RCOR2 peaks were enriched in the genes whose expression was perturbed by LSD1 inhibition, such as SMAD9, HES1, and ID1 (Fig. 4b). RNA-seq was correlated with LSD1 ChIP-seq26 to identify LSD1 targets (1567 genes) in MCC cell lines (Source Data 3). Remarkably, target genes directly repressed by LSD1 in MCC were enriched in similar pathways as those genes upregulated by EP400 depletion (Extended Data Fig. 1d, 3e-f, and Source Data 4). These pathways included neuron development as well as BMP and TGFβ signaling.
Fig. 4. LSD1 and RCOR2 bind to the ATOH1 transcription factor binding motif close to transcription start sites of genes involved in differentiation pathways.

a. Tag heatmaps of LSD1, RCOR2, and ATOH1 ChIP-seq and the LSD1-RCOR2-ATOH1 overlapping peaks show the factors’ shared occupancy in the transcription start site regions (+/− 4,000bp). Peaks from two independent replicates of ChIP-seq were combined, and the shared peaks were determined using the BEDtools43 (Source Data 4). b. Two biological replicates of RCOR2 (_1 and _2), LSD1, and ATOH1 ChIP-seq peaks in the SMAD9, HES1, and ID1 genes are shown. The peaks were visualized with the IGV genome browser44. c. The SeqPos motif tool27 was used to determine the LSD1 binding motifs in MKL-1. The binding motifs of ATOH1, as well as OLIG2 and ASCL2, were enriched in the LSD1 (LSD1_1) ChIP-seq. d-e. Peak frequency in the transcription start site regions (TSSs) reveals that LSD1, RCOR2, and ATOH1 bind close to TSSs. The global +/− 2,000bp TSSs (d; promoters) and +/−10,000bp TSSs (e; promoters and proximal enhancers) are shown. f. ChIP-seq annotations indicate that LSD1, RCOR2, and ATOH1 bind to shared genomic regions. g. ChIP-qPCR of LSD1 in MKL-1 MCC cells shows that LSD1 enrichment decreases following LSD1 inhibition. MKL-1 cells were treated with GSK-LSD1 for three days. Levels of LSD1 binding decreased after treatment with LSD1 inhibitor. Data are shown as mean of n=4 ± SD; two-sided t-test, *P<0.05; **<0.005. h. ChIP-qPCR shows ATOH1 binds to LSD1 target genes, ID1 and SMAD9. MKL-1 cells were treated with GSK-LSD1 for three days and harvested for ChIP with ATOH1. Data are shown as mean of n=3 ± SD; two-sided t-test; * P<0.05.
To gain insight into the gene expression program regulated by LSD1 in MCC, we performed motif identification analysis of LSD1 and RCOR2 ChIP-seq in the MKL-1 cell line. The SeqPos motif tool revealed that the binding motifs of the basic helix-loop-helix transcription factors ATOH1, OLIG2, and ASCL2 were most significantly enriched (Fig. 4c, and Extended Data Fig. 4d)27. ATOH1 is a master transcription factor required for normal Merkel cell lineage28. Moreover, ATOH1 was significantly more abundant in virus-positive MCC cells compared to virus-negative cells, while ST, MAX, and EP400 bound specifically to the ATOH1 promoter (Fig. 1e and Extended Data Fig. 3i).
ChIP-seq of ATOH1 in MKL-1 cells revealed that LSD1, RCOR2, and ATOH1 bind to an overlapping set of genes (Fig. 4a, b, and Extended Data Fig. 4a-c). ATOH1 binding appeared to be centered on transcription start sites (TSSs) of target genes, while LSD1 and RCOR2 peaks were within 500 bp of the TSSs (Fig. 4d and e). LSD1, RCOR2, and ATOH1 binding was enriched in similar proportions to proximal promoters (<=1 kbp) (15 to 17%), intragenic (~45%), and distal intergenic regions (35%) (Fig. 4f and Source Data 4). Pathway analysis of the of LSD1, RCOR2, and ATOH1 peaks revealed that they bind to genes enriched for axon guidance and stem cell pluripotency (Extended Data Fig. 4e).
We next sought to determine the effect of LSD1 inhibitors on the DNA occupancy of LSD1, RCOR2, and ATOH1. ChIP-qPCR in untreated MKL-1 cells detected significant enrichment of LSD1, RCOR2, and ATOH1 at the ID1 and SMAD9 promoters. Following LSD1 inhibition, enrichment of LSD1 at these target genes, as well as at ZNF781, HES1, and DLL1, was markedly decreased (Fig. 4g). In contrast, LSD1 inhibition led to increased ATOH1 enrichment at ID1 and SMAD9 (Fig. 4h). This result indicates that LSD1-RCOR2 and ATOH1 compete for binding to a common set of genes.
LSD1 inhibitors have been shown to perturb the neuroendocrine transcription program in SCLC by disrupting LSD1 interactions with chromatin, mediated by the SNAG domain protein INSM110. In MCC, we determined that LSD1 associates with INSM1 (Fig. 1f and Source Data 1). We performed IP-mass spectrometry (MudPIT) with an INSM1 antibody using MKL-1 cells treated with the LSD1 inhibitor (GSK-LSD1) and found that the INSM1 interaction with LSD1 decreased with inhibitor treatment (Source Data 4). IP-western blotting experiments confirmed that INSM1 has reduced binding to LSD1 after the inhibitor treatment (Extended Data Fig. 3g and h).
LSD1 inhibition reduces the growth of MCC cells in mice and perturbs neuronal gene expression in human and mouse tissues
To identify changes at the protein level with LSD1 inhibition, we performed a multiplexed isobaric tag-based profiling29 of the MKL-1 virus-positive MCC cell line treated with GSK-LSD1 inhibitor for eight days. LSD1 inhibition led to major perturbations in protein expression (Fig. 5a). Proteins upregulated by LSD1 inhibition were enriched for pathways in cell adhesion, axonogenesis, and neuron differentiation (Extended Data Fig. 5a and b). About 30% of proteins significantly increased in response to LSD1 inhibition were also identified as LSD1 targets by the integrated RNA-seq and ChIP-seq analysis, including CALB2, PROM1, FAM5B, DLL1, and GFI1 (Extended Data Fig. 3e and Source Data 5).
Fig. 5. Multiplexed Isobaric Tag-Based Profiling of MKL-1 cell line and MCC-derived xenografts reveals that LSD1 inhibition perturbs neuronal gene expression in human and mouse tissues.

a. Multiplexed Isobaric Tag-Based Profiling of MKL-1 virus-positive MCC cell line treated with GSK-LSD1 for eight days displays global changes in the proteome. Changes in the differentially expressed proteins are shown (Source Data 5). Tukey’s Honest Significance Test was performed posthoc on-peptide level linear models for each protein and the p-values were adjusted by Benjamini-Hochberg. The levels of proteins identified with the arrows significantly changed in fold change. n=5. Blue: Log2 Fold Change (FC) > 1.5; Light Blue: Log2 FC > 1.5 and FDR < 0.05; Pink: Not significant. b. MKL-1 and WaGa MCC cell lines grown as xenografts in NSG mice have reduced growth rates with LSD1 inhibitors. # Day 22 data not plotted as only 3 of 8 mice were alive. Data are shown as mean of n=8 two-sided t-test; *P<0.05, **<0.005, and ***0.0005 (Supplementary Tables 1–2). c-g. Tumors from MKL-1 or WaGa model (n=2/group/model) were harvested six hours after the last dose for the TMT-10 plex isobaric tag-labeling quantitative mass spectrometry experiment. c. Heatmap shows the relative abundance of 30 most upregulated and 30 most downregulated human (tumor) proteins after the treatment (refer to Methods). d-g. Selected GOTERM biological processes (BP) and cell compartments (CC) –log10 of Bonferroni adjusted p-values for the pathways enriched with the top 100 most differentially expressed proteins in the following categories are shown. The clustering test was implemented using kappa statistics45. d. Upregulated human proteins (Source Data 5). e. Downregulated human proteins. f. Upregulated mouse proteins. g. Downregulated mouse proteins.
We tested the efficacy of targeting LSD1 in vivo using MKL-1 and WaGa virus-positive MCC cells grown as xenografts in NOD SCID gamma (NSG) mice. When the subcutaneous tumors reached 150 mm3, mice were treated with the CPI-242 LSD1 inhibitor dosed once weekly by oral gavage. LSD1 inhibition significantly decreased the rate of tumor growth in both MCC xenograft models (Fig. 5b and Supplementary Tables 1–2).
To explore the tumor response to LSD1 inhibition in vivo, we assessed the proteome of the treated MKL-1 and WaGa xenografts by performing a similar multiplexed isobaric tag-based profiling experiment (Fig. 5c-g, Extended Data Fig. 5c, 6a-d, and Source Data 5)29. We observed that proteins involved in neuron differentiation, including CNTN2 (Contactin 2), NEFL (neurofilament light), NEFM (neurofilament medium), and SYT4 (synaptotagmin 4) were significantly increased in both MKL-1 and WaGa xenografts treated with the LSD1 inhibitor (Fig. 5c). Detection of CNTN2, SYT4, NEFL, and NEFM is typically restricted to the brain and some neuronal lineage tissues30. Gene ontology analysis of the proteins upregulated during LSD1 inhibition was enriched for cytoskeleton organization in neuron projections (Fig. 5d). Interestingly, mouse neuronal proteins including Rph3a (Rabphilin 3A), Nefl (neurofilament light), and Dclk1 (doublecortin-like kinase 1) also increased during LSD1 inhibition, indicating that LSD1 inhibition affected neuronal differentiation in mouse tissues in the xenograft experiment (Fig. 5f and Extended Data Fig. 5c).
In contrast to the factors up-regulated by LSD1 inhibition, proteins with decreased levels were involved in epithelial growth signaling and extracellular organization including MUC1 (mucin 1), CNPY1 (Canopy FGF Signaling Regulator 1), SPRR1A (Small Proline Rich Protein 1A), CDC6 (Cell Division Cycle 6), and PCOLCE2 (Procollagen C-Endopeptidase Enhancer 2) (Fig. 5e). Mouse proteins decreased during LSD1 inhibitor treatment of xenografts were enriched for wound healing and inflammatory response pathways, consistent with the observed shrinkage of tumors (Fig. 5g and Extended Data Fig. 5c).
Knockout of ncBAF complex components confers resistance to LSD1 inhibition in MCC
To gain insight into the mechanism of LSD1 inhibition-mediated cell growth inhibition in MCC, we performed a genome-wide CRISPR-Cas9 screen31. MKL-1 cells were transduced with human CRISPR-Cas9 knockout pooled lentivirus libraries followed by treatment with 1.5 nM (IC30) of GSK-LSD1 or vehicle for 20 days. Transduced cells were collected on days 0 and 20 of treatment and sequenced for short guide RNAs (Fig. 6a and b). Reads from day 20 samples (control and treated) were normalized to those from day 0 sample to identify differentially selected genes (Fig. 6c-e and Source Data 6). Knockout of the positively selected genes (Group A: 3624 genes) promoted proliferation during LSD1 inhibition, whereas knockout of the negatively selected genes (Group B: 2207 genes) augmented LSD1 inhibition in blocking cell growth (Fig. 6d). KMT2C (Lysine Methyltransferase 2C, MLL3) was positively selected during LSD1 inhibition (Fig. 6e and f). KMT2C is the primary mammalian H3K4 mono- and di-methyltransferase involved in the activation of cell-type-specific gene expression during differentiation32. The positive selection of KMT2C in the resistance screen suggests that it may be a writer of the active histone marks erased by LSD1.
Fig. 6. CRISPR-Cas9 genome-wide screen for resistance to LSD1 inhibitors creates positive pressure for loss of ncBAF complex components.

a. Experimental design for CRISPR-Cas9 screening. b. MKL-1 cells display sensitivity to an LSD1 inhibitor (GSK-LSD1) in a dose-dependent manner. IC30 (1.5 nM) and IC50 (4 nM) are shown. For the CRISPR-Cas9 screen, IC30 was used. Data are shown as mean of n=3. c. MAGeCKFlute normalization results in a normal distribution of beta scores (selection) for the control and treated samples (LSD1i, 1.5 nM for 20 days)46. d. Genes positively (Group A) or negatively selected (Group B) during the CRISPR screen were determined based on the beta scores of the control (DMSO 20 days) and treated samples (LSD1i 20 days; IC30) (Source Data 6). e. Gene ranks based on the beta scores show positively and negatively selected genes. The top 20 genes in each group are shown (Source Data 6). f. Top 10 most significantly positively selected genes in the LSD1i screen are shown. The p-values were estimated using the MAGeCK-MLE model and adjusted by FDR47. g. Two independent BRD9 MudPIT in MKL-1 cells identified ncBAF complex components, including GLTSCR1 and GLTSCR1L1, not the cBAF or PBAF complex specific components. dNSAF values shown (refer to Methods and Source Data 6).
In addition to KMT2C, the loss of several components of the ncBAF complex, including BRD9 and GLTSCR1 (BICRA), were among the most significant, positively selected genes in the screen (Fig. 6e, 6f)16–18. Other significant, positively selected, ncBAF components included SMARCD1, SMARCC1, and SMARCA2 (Extended Data Fig. 7a). To determine if BRD9 formed the ncBAF complex in MKL-1 cells, we performed large scale immunoprecipitation of the endogenous BRD9, followed by MudPIT to identify associated proteins (Fig. 6g and Source Data 6). We observed that BRD9 co-precipitated GLTSCR1 and SMARCA4, and several additional components of the ncBAF complex. This finding was confirmed by western blot in MCC cells (Extended Data Fig. 7b). In contrast, BRD9 did not associate with components restricted to the BAF and PBAF complexes such as ARID1A, ARID2, PBRM1, and BRD7.
LSD1 and BRD9 regulate an overlapping set of genes in MCC
To assess the role of the ncBAF complex in MCC, we used a bifunctional inhibitor, dBRD9, that selectively degrades BRD9 by bridging the bromodomain and the cereblon (CRBN) E3 ubiquitin ligase complex (Extended Data Fig. 7d)33. To determine if depletion of BRD9 confers resistance to LSD1 inhibition, we treated MKL-1 cells with GSK-LSD1 and dBRD9 or a related BRD9 inhibitor (BRD9i, BI-7273) without the linker to CRBN for six days and measured viability by the XTT assay (Fig. 7a). The addition of dBRD9 or BRD9i partially rescued the decreased levels of cell viability caused by LSD1 inhibition in MKL-1 cells (Fig. 7a, b, and Extended Data Fig. 7c).
Fig. 7. LSD1 and BRD9 regulate an overlapping set of genes in MCC.
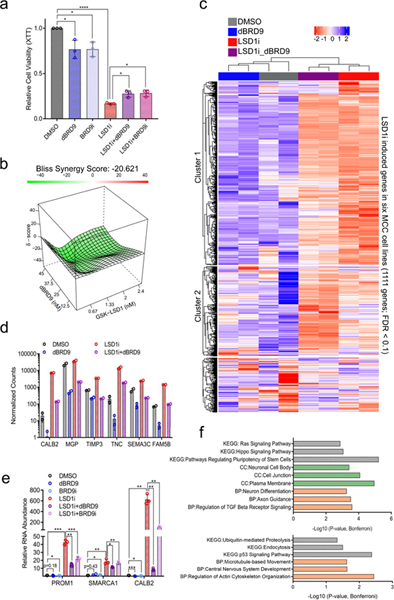
a. BRD9 degradation or inhibition partially rescues the reduced cell viability caused by LSD1 inhibition in MCC. MKL-1 cells were treated with DMSO, GSK-LSD1 (LSD1i, 0.1 μM), dBRD9 (1 μM), BRD9i (BI-7273, 1 μM), or combinations of LSD1i with dBRD9 or BRD9i for six days. The XTT assay was used to measure relative cell viability. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05, ****<0.00005. b. MKL-1 cells were treated with varying doses of GSK-LSD1 or dBRD9 for six days. SynergyFinder48 was used to calculate negative synergy (rescue) scores. c. RNA-seq was performed with two replicates of MKL-1 cells treated with DMSO, GSK-LSD1 (LSD1i, 0.1 μM), dBRD9 (0.1 μM), or both GSK-LSD1 and dBRD9 for six days. n=2. The heatmap shows expression changes in 1,111 genes induced by LSD1i in six virus-positive MCC cell lines (Fig. 3a-c and Source Data 7). d. DESeq2 counts of selected LSD1 target genes are shown. Multiple t-test was performed and p-valued were adjusted by FDR (See Supplementary Table 3). Data are shown as mean of n=2 ± SD. e. Selected GOTERM biological processes (BP), cellular components (CC), or KEGG -log10 of p-values for the pathways enriched with the Cluster 1 and 2 genes (Source Data 7). The clustering test was implemented using kappa statistics45. n=2. f. MKL-1 cells were treated with DMSO, LSD1i (GSK-LSD1, 0.05 μM), dBRD9 (1μM), BRD9i (BI-7273, 1 μM), LSD1i with dBRD9, or LSD1i with BRD9i for three days. The signals were normalized to the DMSO treated sample and RPLP0. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05; **<0.005; ***<0.0005.
Since LSD1 inhibition led to the de-repression of specific genes, we suspected that this de-repression required the ncBAF complex. We tested if simultaneous inhibition of LSD1 and BRD9 could dampen expression of the LSD1 target genes. We performed RNA-seq with MKL-1 cells treated with GSK-LSD1 (LSD1i), dBRD9, or both for six days (Fig. 7c-e). PCA of the RNA-seq data revealed that inhibition of BRD9 or LSD1 caused global changes in gene expression, while the dual inhibition led to changes distinct from those of either single treatment alone (Extended Data Fig. 7e). The addition of BRD9 degrader significantly dampened the induction of more than half of LSD1 target genes (Cluster 1: 622/1111 genes) (Fig. 7c, d, and Extended Data Fig. 8 and Source Data 7).
We validated by RT-qPCR that levels of PROM1, SMARCA1, and CALB2 transcripts increased with LSD1 inhibition, and the addition of dBRD9 or BI-7273 dampened this response (Fig. 7e). Also, the shRNA-mediated knockdown of BRD9 led to decreased levels of PROM1, SMARCA1, and ID1 that were increased by LSD1 depletion (Extended Data Fig. 7f). Genes induced by LSD1 inhibition and repressed by dBRD9 were enriched for pathways involved in neuron differentiation and cell morphology changes (Fig. 7f and Source Data 7).
BRD9 is required to de-repress the expression of LSD1 target genes during LSD1 inhibition.
We hypothesized that BRD9 is required for increasing levels of LSD1 target genes during LSD1 inhibition. To test this possibility, we performed ATAC-seq to determine the chromatin state of MKL-1 MCC cells treated with LSD1 and BRD9 inhibitors. The PCA analysis of the ATAC-seq peaks revealed distinct patterns of global changes in chromatin states following LSD1 inhibition, BRD9 degradation, or both (Fig. 8a). Remarkably, the combination treatment (LSD1i_dBRD9) had a similar shifting pattern to the control, suggesting that BRD9 degradation restored structural changes in chromatin caused by LSD1 inhibition. We observed that ATAC-seq peaks reside close to the TSSs in control cells, while LSD1 inhibition increased, and dBRD9 decreased the open chromatin peaks close to the TSSs (Fig. 8b-c and Extended Data Fig. 9a). By comparing the ATAC-seq peaks, we determined that the peaks gained by adding dBRD9 to LSD1i (Combo_gained) reside close to the TSSs whose gene products are involved in pathways such as axon guidance and cell-to-cell communications crucial for differentiation and cancer progression (Fig. 8d, 8e, Extended Data Fig. 9b-d and Source Data 8). A close examination of the ATAC-seq peaks in the promoters of LSD1 target genes such as MGP, TNC, ID1, SYT4, and CALB2 whose expression was dampened by addition of dBRD9 revealed that LSD1 inhibition increased levels of open chromatin in the promoters and addition of dBRD9 dampened this increase (Fig. 8f-I, and Extended Data Fig. 9e). These results suggest that BRD9 was required to promote the open chromatin states following LSD1 inhibition.
Fig. 8. BRD9 is required to de-repress the expression of LSD1 target genes.

a-i. Inhibition of LSD1 and BRD9 alters chromatin accessibility of an overlapping set of genes in an antagonistic manner (Source Data 8). Two biological replicates of ATAC-seq with MKL-1 cells treated with DMSO (Control), GSK-LSD1 (LSD1i, 0.1 μM), dBRD9 (0.1 μM) or both LSD1i and dBRD9 (LSD1i+dBRD9) for three days were performed. a. The PCA plot displays global changes in chromatin accessibility following the mentioned treatments. b. Peak count frequencies in the transcription start sites (TSSs, +/− 2,000 bp) for the mentioned conditions are shown. The shaded areas display 95% confidence intervals. c. Peak count frequencies of ATAC-seq with the mentioned conditions are shown (TSSs +/− 500). d. Frequencies of the differentially enriched peaks between LSD1i and LSD1i+dBRD9 are shown. ‘Combo_gained’ displays frequencies of peaks gained in the LSD1i+dBRD9 combination compared to LSD1i, whereas ‘Combo_lost’ shows frequencies of peaks lost in the combination. e. GOTERM biological process pathways enriched with the genes annotated with Combo_gained (n=358) and Combo_lost (n=431) peaks are shown. The enrichment test was performed on hypergeometric distribution using the clusterProfiler R package49 and the p-values were adjusted by FDR49. f-g. DESeq2 normalized counts of MGP (f) and TNC (g) from the RNA-seq of MKL-1 cells treated with DMSO (DMSO), GSK-LSD1 (LSD1i, 0.1 μM), dBRD9 (0.1 μM) or both LSD1i and dBRD9 (LSD1i+dBRD9) are shown (Fig. 7c-e). Differentially expressed genes were found by comparing each condition with DESeq242, and p-values were adjusted by Benjamini-Hochberg. h-i. ATAC-seq peaks in the promoters of MGP (h) and TNC (i) in the mentioned conditions with two biological replicates (_1 and _2) are shown. j-k. ChIP-qPCR reveals that LSD1 binding decreases (j), whereas BRD9 binding increases during LSD1 inhibition (k). MKL-1 cells were treated with GSK-LSD1 for three days. Data are shown as mean ± SD; * P<0.05, ** P<0.005, and ***P<0.0005. Data are shown as mean of n=3 ± SD; two-sided t-test, *P<0.05; **<0.005; ***<0.0005.
ChIP-qPCR was performed to assess for LSD1 and BRD9 enrichment on LSD1 target genes in MKL-1 cells (Fig. 8j and k). LSD1 binding to the ID1, ID2, SMAD9, and ZNF781 genes decreased after three days of LSD1 inhibition, whereas BRD9 enrichment increased significantly. The observation indicates that as LSD1 left from chromatin, BRD9, presumably in the ncBAF complex, was recruited to these sites. Together with the ATAC-seq results, these findings indicate that BRD9 and the ncBAF complex is required for de-repressing expression of a subset of LSD1 target genes during LSD1 inhibition.
Discussion
Here, we report that MCV ST generates an essential dependence on LSD1 activity through transactivation of LSD1 complex components, leading to repression of multiple LSD1-RCOR2-INSM1 target genes that were in turn activated by the ncBAF complex (Extended Data Fig. 9f). Through a comprehensive targetome analysis integrating RNA-seq, ChIP-seq, ATAC-seq, and quantitative mass spectrometry, we establish that LSD1 inhibition leads to increased expression of genes involved in neuron differentiation. Our findings support a specific role for LSD1 in repressing BMP signaling, previously linked to fate specification of neurons and skin cells34. Our results are consistent with the finding that Keratin 14-driven overexpression of BMP in skin dampens Merkel cell development35. LSD1 repression of BMP signaling may also inhibit MCC differentiation that, in turn, regulates neuronal gene expression.
It is important to note that MYCL and the EP400 complex mediate MCV ST oncogenic transformation and LSD1 expression. In this context, the ST-MYCL-EP400 complex represses developmentally related genes by transactivation of the LSD1-RCOR2-INSM1 complex. The MYC and MYCN paralogs of MYCL have been reported to repress specific gene targets, by binding directly to a repressor such as MIZ-1 (ZBTB17) or G9a (EHMT2) or by transactivation of a repressor such as EZH2 that in turn represses specific gene targets36,37. Here we find that MYCL functions to indirectly repress a specific set of developmental genes through the LSD1-RCOR2-INSM1 complex. This activity does not exclude the possibility that MYCL may have additional associated repressor activities mediated by MIZ-1, G9a, and EZH2.
We observed that DNA binding of LSD1 and ATOH1 co-localize in the proximity of LSD1 target genes. ATOH1 is the master transcription factor for Merkel cell specification28. In this role, ATOH1 may act as a pioneer factor to make active transcription sites accessible for other transcription factors, while LSD1 serves to repress specific genes in these sites based on cellular needs.
We found that all virus-positive, but not virus-negative, MCC cell lines tested responded to LSD1 inhibition. To understand the mechanism of LSD1 inhibition, we determined that the ncBAF chromatin remodeling complex was required to de-repress LSD1 target genes. Inhibition of BRD9, a core component of ncBAF, rescued loss of cell viability caused by LSD1 inhibition. Loss of ncBAF or its downstream target genes may serve as a mechanism of resistance for targeting LSD1 in neuroendocrine cancers.
The role of ncBAF in cancer is largely unknown. The perturbation of canonical BAF complexes (i.e., the SMARCB1 subunit) generates a dependency on BRD9 in synovial sarcoma18,38. On the other hand, in cancers with SF3B1 mutations, loss of BRD9 by mi-splicing promotes proliferation, suggesting a tumor suppressor role for BRD919.
One promising LSD1 combination therapy may involve immunotherapy. A recent report indicated that LSD1 depletion enhanced PD-L1 (CD274) checkpoint blockade in mouse melanoma by increasing type 1 interferon response and anti-tumor T cell infiltration39. Historically, therapeutic options for MCC have been limited to surgery, radiation, and cytotoxic chemotherapy, although several recent reports demonstrated response rates as high as 62% to checkpoint blockade therapy with PD-1 and PD-L1 inhibitors40. A strategy combining checkpoint blockade therapy and LSD1 inhibition may prove to be a useful therapeutic strategy for MCC and other cancers.
Methods
Plasmids, Cell, and Cell culture
Expression vectors include pLIX402 inducible empty or pLenti CMV Blast empty vector (w263–1), gifts from David Root (41394; Addgene) and Eric Campeau (17486; Addgene), respectively. Lentiviral packaging plasmid psPAX2 and envelope plasmid pMD2.G were gifts from Didier Trono (12260, 12259; Addgene). The shRNA clone (#43; CAGGAGAAGCTGCTGGTATCA) targeting KDM1A was obtained from Cellecta and was carried in the Cellecta 3rd generation lentiviral plasmid, pRSI-U6wt-sh-cPPT-UbiC-zsG-2A-Puro. The generation of the shRNA clones for EP400 was described in Cheng et al5. The shRNA clones targeting RCOR2 (HsSH00209621 and HsSH00209662) and BRD9 (HsSH00206761 and HsSH00206889) were obtained from the Harvard plasmid database.
In generating cell lines stably expressing these constructs, IMR90 human lung fibroblast and MKL-1 MCC cells were transduced using a three-vector lentivirus transduction System5. MCC cell lines were gifts from Masa Shuda (University of Pittsburgh, PA), Jürgen Becker (Medical University Graz, Austria), and Roland Houben (University of Wuerzburg, Germany). 293T and IMR90 cells were obtained from ATCC. Generation of MKL-1 MCC cell lines with inducible expression of shRNAs and IMR90 transformation using p53DD, MYCL, and hTERT constructs were described previously5,24. IMR90 and primary human foreskin fibroblast cells (HFF) were cultured in DMEM supplemented with 15% FBS, antibiotics and non-essential amino acids and MCC cell lines were grown in RPMI supplemented with 10% FBS and antibiotics.
Cell viability assay and anchorage-independent growth assay
GSK-LSD1 (SML1072; Sigma Aldrich), GSK2879552 (A-1385; Active Biochem), CPI-242, and other LSD1 inhibitors (Constellation Pharmaceuticals), dBRD9 (a gift from Jun Qi, DFCI), and BI-7273 (20311; Cayman Chemical) were reconstituted in DMSO and added directly to culture media. CPI-242 is an orally available, small molecule, covalent styrenylcyclopropane inhibitor of LSD1. The structure and activity of this molecule will be covered in “Design and synthesis of styrenylcyclopropylamine LSD1 inhibitors”, By Gehlling, V. et al. (Manuscript in preparation).
CPI-670242 and other LSD1 inhibitors were arrayed in a 10-point dilution series on the plates (10 μM maximum concentration), prior to the addition of cells. Cultures were split every fourth day, and an aliquot was removed for viable cell counts and the remainder was re-plated at a ~1:4 dilution into compound-arrayed plates. Cell viability assays were performed using Cell Proliferation Kit II (XTT) (Roche) or Cell Titer-Glo luminescent cell viability assay (Promega) according to the manufacturers’ instructions. GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA USA) was used for curve fitting and determination of GI50 values. Synergy testing was performed using SynergyFinder48. Anchorage-independent growth was performed as described24 using 6-well dishes with SeaPlaque Agarose (Lonza) at concentrations of 0.3% top and 0.6% bottom layers. Agarose was diluted with 2X MEM (Gibco) supplemented with 2X Gluta-max (Gibco), 2X pen-strep (Gibco), and 30% FBS. IMR90 cells were seeded in triplicate in the top agarose layer. Wells were fed with top agarose twice per week. After four weeks, cells were stained with 0.005% crystal violet (Sigma) in PBS and colonies were counted in a blind manner.
Immunoprecipitation and immunoblotting
Confluent cultures of cells were washed with ice-cold phosphate-buffered saline (PBS) and resuspended in EBC lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.5% NP-40, 1:10,000 β-mercaptoethanol, 0.5 mM EDTA) for 10 min on ice and then centrifuged. Clarified lysates were quantified and boiled in Laemmli sample buffer for western blotting or incubated overnight with antibodies and magnetic protein A/G beads (PureProteome magnetic beads, EMD Millipore) for immunoprecipitation. The beads were washed with high-salt EBC buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 0.5% NP-40, 0.5 mM EDTA) 5 times and boiled in Laemmli sample buffer before SDS polyacrylamide gel electrophoresis. The Roeder 2 step lysis protocol was modified to prepare histone extracts50. Briefly, cytoplasmic fractions were separated in Buffer A (10mM Tris, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, Benzonase) and the nuclear pellets were resuspended in Buffer C (20 mM Tris, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.42 M NaCl, 0.5 mM DTT, Benzonase). After clarifying the nuclear fraction, the insoluble nuclear material was sonicated in Buffer B (20 mM Tris, pH 7.9, 1.5 mM MgCl2, 1M NaCl), and the clarified extract was combined with the soluble nuclear fraction.
Immunoprecipitation and western blotting were performed with antibodies to LSD1 (2139; Cell signaling technology; 1:3000), INSM1 Mouse (SC-271408; Santa Cruz Biotechnology; 1:250), RCOR2 (23969–1-AP; Proteintech Group; 1:2000), ID1 (SC-488; Santa Cruz Biotechnology; 1:250), MYCL (14584–1-AP; Proteintech Group; 1:1000), PROM1 (CD133, 18470–1-AP; Proteintech Group; 1:3000), SMAD9 (LS-C161303–400; Lifespan Biosciences; 1:1000), Phospho-Smad1 (Ser463/465)/Smad5(Ser463/465)/ Smad9(Ser465/467) (13820T; Cell Signaling Technology; 1:3000), SMARCA1 (9450S; Cell Signaling Technology; 1:3000), BRD9 (61537; Active Motif Rabbit; 1:3000), BICRA (GLTSCR1; SC-515086; Santa Cruz Biotechnology; 1:250), Cleaved Notch1 (Val1744) (4147T; Cell Signaling Technology; 1:3000), HES1 (11988S; Cell Signaling Technology; 1:3000), H3 (9715; Cell Signaling Technology; 1:3000), di-methyl-histone H3 (Lys4) (C64G9) (9725; Cell Signaling Technology; 1:3000), mono-methyl-histone H3 (Lys4) (61634; Active Motif; 1:2000) and normal Rabbit IgG (2729; Cell signaling technology; 1:3000). Mouse monoclonal antibodies Ab3 and Ab5 (1mg/ml; 1:1000 dilution was used for blotting) against MCV T antigens were generated against MCV large T antigen residues 1 to 260 and produced as a glutathione S-transferase (GST) fusion protein in bacteria51.
MudPIT
MudPIT was performed with MKL-1 and WaGa suspension cells harvested in EBC lysis buffer. Clarified cell extract (100–300 mg) was incubated overnight at 4°C with 20 μg antibodies cross-linked to 30 mg protein G agarose beads by dimethyl pimelimidate (DMP). Beads were washed with high salt wash buffer five times, then eluted with 0.2 M glycine pH 3 and neutralized with 1 M Tris pH 8.0. Proteins were precipitated with 1/5 TCA overnight at 4°C and washed with cold acetone twice and analyzed by MudPIT as described22. The endoproteinase Lys-C and trypsin digested peptides were loaded on a split-triple-phase fused-silica micro-capillary column and placed in-line with a linear ion trap mass spectrometer (LTQ, Thermo Scientific), coupled with a Quaternary Agilent 1100 Series HPLC system. A fully automated 10-step chromatography run was carried out. Each full MS scan (400–1600 m/z) was followed by five data-dependent MS/MS scans.
The MS/MS data set was searched using ProLuCID (v. 1.3.3)52 against a database consisting of 36,628 non-redundant Homo sapiens proteins (downloaded from NCBI RefSeq 2016–06-10), 193 common contaminants, and, to estimate false discovery rates (FDRs), 36,821 randomized amino acid sequences derived from each NR protein entry. To account for alkylation by CAM, 57 Da were added statically to the cysteine residues. To account for the oxidation of methionine to methionine sulfoxide, 16 Da was added as a differential modification to the methionine residue. Peptide/spectrum matches were sorted and selected to an FDR less than 5% at the peptide and protein levels, using DTASelect in combination with swallow, an in-house software. Original mass spectrometry data can be accessed from the ProteomeXchange Consortium with ProteomeXChange accession: PXD012516 http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD012516
ChIP-qPCR and RT-qPCR
The ChIP method was modified from protocols described in Schmidt et al.53. MKL-1 and IMR90 cells were cross-linked using dual cross-linking with disuccinimidyl glutarate (DSG) and formaldehyde. After cross-linking, cells were lysed using SimpleChIP buffer A and B (Cell signaling), and DNA was processed with the Micrococcal nuclease (New England Biolabs Inc.) for 30 minutes at 37°C followed by sonicating for 20s pulses 5 times at 4°C. For RT-qPCR, total RNA was purified using RNeasy Plus Mini Kit (Qiagen), and cDNA was synthesized using the High-Capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative PCR was performed using Brilliant III Ultra-Fast SYBR Green qPCR Master Mix (Agilent Technologies). The primer information can be found in Supplementary Table 4.
Genome-wide CRISPR screening
CRISPR lentiviral libraries H1 and H2 each contain 92,817 pooled sgRNAs targeting 18,493 human genes. CRISPR screen was performed by following a previous protocol54. Briefly, 2×108 MKL-1 cells were transduced with H1 and H2 CRISPR libraries separately at MOI 0.3 to ensure single sgRNA incorporation per cell. After 6 days of 1 μg/ml puromycin selection, surviving cells from each sgRNA library transduction were split into three groups, 3×107 cells were saved as initial state controls, the remaining two-thirds were divided into two groups and they were treated with either DMSO or IC30 of GSK-LSD1 (1.4 nM; Sigma Aldrich). These two groups were cultured for 20 days, with at least 3×107 cells maintained and used as final state samples. Genomic DNA was extracted, and 200 μg from each sample were used to PCR amplify integrated sgRNAs and to generate four libraries for next-generation sequencing. 50 million reads were obtained for each sequencing library. Copy numbers of every 50-kb segment of the MKL-1 genome were called from the input of ChIP-seq experiments using QDNAseq software5. Segmented copy numbers were converted to copy numbers per gene based on gene coordinates. MAGeCK and MAGeCKFlute pipelines were used to assess data quality, correct copy number variation effect and identify statistically significant targets46,54.
Omni-ATAC-seq
Two independent replicates of 70,000 MKL-1 MCC cells were harvested for ATAC-seq after treatment with GSK-LSD1 0.1μM, dBRD9 0.1μM, or both for three days. Cells were washed with PBS twice to achieve >85% viable cells in pellets. ATAC-seq protocol was modified from Corces et al55. The transpose reaction was performed using TDE1 Tn5 transposase (15027865; Illumina) and TD buffer (15027866; Illumina). The reactions were cleaned up using MinElute PCR Purification Kit (28004; Qiagen) and amplified using NEBNext High-Fidelity 2X PCR Master Mix (M0541S; New England Biolabs Inc.) with a total of 7 cycles. The libraries were cleaned up using MinElute PCR Purification Kit and additionally with AMPure XP beads (Beckman Coulter) to remove primer dimers and large fragments.
To sequence the libraries, NextSeq 550 systems (Illumina) was used at the Center for Cancer Computational Biology (CCCB) and Molecular Biology Core Facilities (MBCF-DFCI) following the manufacturer’s paired-end sequencing protocol. For analysis, the adapters were removed using NGmerge56 and the reads were mapped to hg38 reference using Bowtie257 and sorted by SAMtools58. ATAC-seq peaks were merged, annotated, and visualized using the BEDtools43 and the ChIPseeker59, ChIPpeakAnno27, and DiffBind60 R packages and Integrative genome viewer (IGV; Broad Institute)44.
Pathway analysis of gene expression
RNA-seq of MKL-1 and WaGa MCC cell lines expressing three independent shRNAs targeting EP400, an shRNA and a microRNA targeting MYCL, or an shRNA targeting MCV ST was reported in Cheng et al5. Differentially induced genes in each condition were selected with the fold change cutoff of 1.5 and FDR cutoff of 0.1. The pathway analysis was performed using the clusterProfiler R package49, and the differentially enriched GOTERM biological process pathway terms were called using the FDR cutoff of 0.05 (refer to Source Data 1).
ChIP-seq analysis
For ChIP-seq, 30 ng of DNA from ChIP experiments or input DNA were prepared for sequencing with NEBNext ChIP-seq Library Prep Reagent Set for Illumina (New England BioLabs). Amplified libraries were cleaned up using AMPure XP beads (Beckman Coulter) and checked on a Bioanalyzer (Agilent) to confirm a narrow distribution with a peak size of around 275 bp. Diluted libraries were used for 50 cycles single-end sequencing on HiSeq 2000 or NextSeq 550 systems (Illumina) at the Center for Cancer Computational Biology (CCCB) and Molecular Biology Core Facilities (MBCF-DFCI) following the manufacturer’s protocol. ChIP-seq mapping was performed using Bowtie2 against human genome version hg19, allowing only uniquely mapping reads57. Peak calling was done using MACS2 on either single replicate mapped files or replicates merged as mapped bam files61. ChIP-seq peaks were merged, annotated, and visualized using the BEDtools43 and the ChIPseeker59, ChIPpeakAnno27, and DiffBind60 R packages and Integrative genome viewer (IGV; Broad Institute)44.
RNA-seq analysis and Target prediction
Merkel cell lines in the exponential growth phase were treated in triplicate with 1 μM CPI-242 (MKL-2, WaGa, BroLi, PeTa, and UISO) or GSK-LSD1 (MKL-1 and MS-1) for 24 or 72 hours. Total RNA was purified using RNeasy Plus Mini Kit (Qiagen). mRNA was isolated with NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs). Sequencing libraries were prepared with Illumina TruSeq RNA Sample Preparation Kit (v2; Illumina) or NEBNext mRNA Library Prep Master Mix Set for Illumina (New England BioLabs) and passed Qubit, Bioanalyzer, and qPCR QC analyses. 50 cycles single-end sequencing was performed on HiSeq 2000 or NextSeq 550 systems. Reads were mapped to the Hg19 genome by STAR62. HTSeq was used to create a count file containing gene names63. The R package DESeq2 was used to normalize counts, determine differential gene expression, and visualize the results42. Gene Ontology (GO) term enrichment was performed using DAVID Bioinformatics Resources (NIH)45, and the terms were summarized and visualized using REVIGO64. Heatmaps depict the average standardized expression profiles and were created using the ‘Heatmap’ function from the R package ’ComplexHeatmap’65, and the volcano plot showing global gene expression changes was made with the R package ‘Enhancedvolcano’66. ChIP-seq data were integrated with individual differential expression data from RNA-seq using Binding and Expression Target Analysis (BETA) software package, which infers activating or repressive function of transcription factors and predict the target genes based on the rank product of binding potential and differential expression26.
Xenograft Efficacy Study
Female NSG mice, seven weeks old (The Jackson Laboratory, ME), were implanted with 10 × 106 MKL-1 or WaGa MCC cells with 50% Matrigel subcutaneously. When the tumors reached the size of 150 mm3, a group of 8 mice for each cell line model were randomized and treated once weekly with vehicle or CPI-242 (Constellation Pharmaceuticals, 40 mg/kg, 0.5% methylcellulose (400 cP) in 50 mM phosphate buffer, pH 6.8) by oral gavage. The study was terminated when the tumor volume exceeded the maximum permissible size of 2000 mm3 in the vehicle control group. The DFCI Institutional Animal Care and Use Committee (IACUC) approved this study. The study is compliant with all relevant ethical regulations regarding animal research.
Multiplexed isobaric tag-based profiling of proteins
Quantitative multiplexed proteomics was performed as described previously67. Cells were lysed in 8M Urea (Sigma Aldrich) containing a cocktail of phosphatase and protease inhibitors (Roche) buffered with HEPES (Sigma Aldrich) to pH 8.5. Xenograft tissues were placed in the same urea lysis buffer and homogenized with a handheld tissue drill. All lysates were homogenized by passing the lysate through a 22-gauge needle before passage through a 25-gauge needle followed by sedimentation by centrifugation at 21,000 x g for 15 minutes. The supernatant was transferred to a new tube, and protein concentration was determined by a bicinchoninic acid (BCA) assay (ThermoFisher Scientific). The proteins were then reduced and alkylated to block reactive cysteine groups, and chloroform-methanol precipitated. Proteins were resuspended in 200mM EPPS pH 8.5 and digested with Lys-C (Wako) overnight at room temperature and subsequently digested with sequencing grade trypsin (Promega) for 6 hours at 37°C. Digests were then labeled with a TMT10 reagent for 90mins at room temperature and then quenched with hydroxylamine. The quenched reaction was then combined at a 1:1:1:1:1:1:1:1:1:1 ratio, flash frozen and dried down in a vacuum centrifuge. The sample was resuspended in 1% formic acid, desalted using C18 solid-phase extraction (SPE) (Sep-Pak, Waters), and dried down in a vacuum centrifuge before resuspension in 10mM ammonium bicarbonate and 5% acetonitrile for off-line basic pH reversed-phase (BPRP) fractionation. Off-line BPRP HPLC was performed on an Agilent 1260 pump. A gradient of 13%−37% acetonitrile in 10mM ammonium bicarbonate was used over 50 minutes. Whole proteome fractions were collected in a 96-well plate and combined into 24 fractions, of which only non-adjacent samples were analyzed, as described previously67. The pooled TMT-labelled sample and the phosphopeptide enriched sample were each separated into 96 fractions by the instrument. For each fractionation experiment, fractions were collected in a 96-well plate and combined into 24 fractions as previously described. The 24 fractions were acidified to 1% formic acid and dried down in a vacuum centrifuge. Dried down fractions were resuspended in 5% acetonitrile and 5% formic acid for LC-MS/MS analysis.
Data for all Quantitative TMT LC-MS/MS experiments were collected on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with LC separation performed on an attached Proxeon EASY-nLC 1200 liquid chromatography (LC) pump (Thermo Fisher Scientific). LC-MS/MS method was modified from a previous study A 100μm inner diameter microcapillary column packed with 35cm of Accucore C18 resin (2.6 μm, 150Å, Thermo Fisher Scientific) was used to separate peptides. Approximately 2 μg of the peptide was loaded onto the column for analysis. A 150 min gradient of 6% to 25% acetonitrile in 0.125% formic acid was used at a flow rate of ~450 nL/min to separate peptides from the pooled TMT-labelled samples: MS1 spectra (Orbitrap resolution 120,000; mass range: 350–1,400 m/z; automatic gain control (AGC) target: 5×105; maximum injection time: 100ms). We then used a Top10 method to select precursors for further downstream analysis. MS2 spectrum were collected after collision-induced dissociation (CID) (AGC target: 2×104; Normalized collision Energy (NCE): 35%; maximum injection time: 120 ms; and isolation window of 0.7 Th). MS2 analysis was performed in the ion trap. We performed an MS3 analysis for each MS2 scan acquired by isolating multiple MS2 fragment ions that were used as precursors for the MS3 analysis with a multi-notch isolation waveform. We detected the MS3 analysis in the Orbitrap (resolution 50,000) after high energy collision-induced dissociation (HCD) (NCE: 65% with instrument parameters: AGC target: 2.5×105; maximum injection time: 150 ms; and isolation window of 1.3 Th).
TMT LC-MS/MS Data Analysis
Spectra acquired from LC-MS/MS experiments for the TMT experiments were processed using a Sequest-based software pipeline. First, a modified version of ReAdW.exe converted spectra to the mzXML format. These files were then searched against a database that contained the human proteome (Uniprot Database Organism ID: 9606, downloaded May 26, 2018) and Merkel Cell Polyoma Virus small and large T antigens concatenated to a database of all protein sequences reversed. For xenograft studies, a mouse proteome was appended to the database (Uniprot Database Organism ID: 10090, downloaded May 26, 2018) before reversal and concatenation. Mouse and human proteins are distinguished by their peptide identifications. The Sequest search engine takes care of assignments of peptides to mass spectra68. A precursor ion tolerance of 50ppm and a production tolerance of 0.9Da were used as search parameters. Static modifications for TMT tags (+229.163Da) on lysine residues and the peptide’s N termini and carbamidomethylation (+57.021 Da) on cysteine residues were used in conjunction with a variable modification for oxidation (+15.995 Da) on methionine. Peptide-spectrum matches (PSMs) were then filtered using the linear discriminant analysis to a false discovery rate (FDR) of 1% as described previously69. XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy were used as parameters for the LDA. The false discovery rate was estimated by using the target-decoy method. Peptides were identified and collapsed using principles of parsimony to a final protein-level FDR of 1%. For quantitation, we extracted the signal-to-noise (S:N) ratio of the closest matching centroid to the expected mass of the TMT reporter ion for each TMT channel from MS3 scans triggered by MS2 scans. MS3 spectra were filtered for a minimum TMT reporter ion sum S:N of 200 and an isolation specificity of at least 0.5. Protein level fold changes were calculated by comparing the sum signal to noise ratios for all observed peptides of a given protein. Proteins were ranked by nominal p-values obtained from Tukey’s Honest Significance Test performed posthoc on-peptide level linear models for each protein. These p-values were adjusted for multiple testing by employing Benjamini-Hochberg at a condition pairwise level to adjust false discovery rates. Pattern matching was performed by converting a desired protein expression pattern into a vector of 8 values, corresponding to the TMT channels used in the experiment, with each value taking a number between 0 (no expression) and 100 (full expression). This vector was compared to a vector of the scaled TMT values for each protein by calculating a cosine similarity value with values closer to 1 indicating more similarity.
Statistics and Reproducibility
To ensure reproducibility, all the western blotting experiments were performed at least three times, and only the representative data with similar results are reported in the study. Statistical analysis details can be found in figure legends and methods.
Data and Code availability
ATAC-seq, ChIP-seq, and RNA–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE124856, GSE124857, GSE124861, GSE124864, and GSE140505. The mass spectrometry data are available at the ProteomeXChange under accession code PXD012516. All other data supporting the findings and computer codes implemented in this study are available from the corresponding author on reasonable request.
Extended Data
Extended Data Fig. 1|.

Merkel small T antigen transactivates LSD1 complex components. a, Integrated ChIP- and RNA-seq analysis of the MCV ST target genes predicts that MCV ST forms an activator complex. b, RNA-seq results show that EP400 depletion in MKL-1 leads to a reduction in mRNA levels of RCOR2, LSD1, INSM1, and additional components of the LSD1 complex. DESeq2 normalized counts were plotted. Differentially expressed genes were found by comparing each condition with DESeq2 and p-values were adjusted by Benjamini-Hochberg. c, Two independent shRNAs against EP400 decrease levels of EP400 and RCOR2 levels but increase levels of DLL1, HES1, LGR6, KRT17, KRT9, and CDH1 in MKL-1 cells. The RT-qPCR signals were normalized to each uninduced sample and the geomean of 36B (RPLP0) and 18 s rRNA. d, Depletion of EP400, MYCL, or MCV ST by shRNA leads to increased levels of genes involved in critical cancer and differentiation pathways in MKL-1 and WaGa MCC cell lines (n = 3 independent biological replicates used in each condition). The enrichment test was performed on hypergeometric distribution and the p-values were adjusted by FDR (Supplementary Tables 1–3). e, RNA-seq of MKL-1 cells expressing three independent shRNAs targeting EP400 or ST in MKL-1 and WaGa cells was performed. Mean expression was plotted against log fold change. f, The LSD1 and RCOR2 levels were significantly higher in PHEL ( + MCV LT and wild-type ST) and PH2L ( + MCV LT and the ST mutant E86S-E87S unable to bind the EP400) than in PHL (-MCV T antigens). The fold changes in the western blot signals from four replicate experiments were averaged. Data are shown as mean of n = 4 ± SD; two-sided t-test, *P < 0.05; **<0.005. g, A representative blot for Extended Data Fig 1g is shown. The experiment was performed four times. See Unprocessed Gels Extended Data Fig. 1. Source data
Extended Data Fig. 2|.

LSD1 inhibition reduces the growth of virus-positive MCC cell lines. a-g, Virus-positive (a-d), but not virus-negative (e-g), MCC cell lines are sensitive to two independent LSD1 inhibitors (GSK2879552 and CPI-670242) in a dose-dependent manner. Relative viability was measured at 12 days of treatment by the CellTiter-Glo assay. Data are shown as mean ± SD and reflect three biological replicates.
Extended Data Fig. 3|.

Integrative ChIP-seq and RNA-seq LSD1 targetome analysis reveal that LSD1 regulates neuronal differentiation pathways in MCC. a, Differential gene expression analysis was performed using DESeq2 and p-values were adjusted by Benjamini-Hochberg. b, c, RT-qPCR (b) and western blot (c) assessment of LSD1 target genes in MKL-1 cells of LSD1 inhibition (GSK-LSD1, 1 μM) in triplicate. For b, the signals were normalized to untreated samples and RPLP0 in each sample. See Unprocessed Gels Extended Data Fig. 3. d, Cells were treated with GSK-LSD1 (0.1 μM) for three days. Data are shown as mean of n = 3 ± SD; two-sided t-test, *P
Extended Data Fig. 4|.

ChIP-seq of LSD1, RCOR2, and ATOH1 indicates shared DNA occupancy. a, Integrated ChIP- and RNA-seq analysis35 reveals that genes perturbed by LSD1 inhibition are direct LSD1 targets. b, Tag heat maps of two replicates of LSD1 (LSD1_1 and LSD1_2), RCOR2 (RCOR2_1 and RCOR2_2), and ATOH1 (ATOH1_1 and ATOH1_2) ChIP-seq show that the factors bind to common regions close to the transcription start sites (TSSs + /− 4,000 bp). c, The heatmap displays correlations among LSD1, RCOR2, and ATOH1 ChIP-seq peaks. d, The SeqPos motif tool36 was used to determine the LSD1 binding motifs in MKL-1. The binding motifs of ATOH1, as well as OLIG2 and ASCL2, were enriched in the RCOR2 (RCOR2_1) ChIP-seq. e, GOTERM biological process pathway analysis of the two independent replicates of LSD1, RCOR2, and ATOH1 ChIP-seq revealed that the factors regulate genes involved in neuronal functions and developmental signaling. The enrichment test was performed on hypergeometric distribution and the p-values were adjusted by FDR.
Extended Data Fig. 5|.

Multiplexed Isobaric Tag-Based Profiling of MCC-derived xenografts reveals that LSD1 inhibition perturbs neuronal gene expression in human and mouse tissues. a-b, The multiplexed Isobaric Tag-Based Profiling of the MKL-1 virus-positive MCC cell line treated with GSKLSD1 for eight days identified genes that are differentially expressed during LSD1 inhibition. n = 5 independent biological replicates were used for analysis. Selected GOTERM biological process and cellular compartment terms are shown for the upregulated (a) and downregulated (b) proteins. c, Eight mice were injected with MKL-1 or WaGa MCC cells, and when the tumor size reached 150 mm3, two of each four mice for each MCC model were treated with CPI-670242 (40 mg/kg) orally once a week for 22 days. The tumors were harvested for the TMT-10 plex isobaric tag-labeling quantitative mass spectrometry experiment. c, The heatmap shows the relative abundance of 30 most upregulated and 30 most downregulated mouse proteins after the treatment.
Extended Data Fig. 6|.

Multiplexed Isobaric Tag-Based Profiling of MCC-derived xenografts profiles global changes in tumor proteomes. The volcano plots display global protein expression changes of MKL-1 human proteins (a), WaGa human proteins (b), MKL-1 mouse proteins (c), and WaGa mouse proteins (d) with or without the LSD1 inhibitor. Tukey’s Honest Significance Test was performed post-hoc on-peptide level linear models for each protein and the p-values were adjusted by Benjamini-Hochberg. Each protein is plotted based on its log2 fold change against -log10 of adjusted p-values. The five most upregulated and five most downregulated proteins in each plot are labeled.
Extended Data Fig. 7|.

CRISPR screen shows that LSD1 inhibition creates positive pressure for deleting ncBAF complex components. a, Gene ranks based on the differences of the beta (selection) scores between the treatment (GSK-LSD1, 20 days, IC30–1.5 nM) and control (DMSO 20 days) screens show the positively and negatively selected mSWI/SNF components genes including the ncBAF complex components BRD9, GLTSCR1, SMARCA2, SMARCD1, and SMARCC1 (Supplementary Tables 18–20). The rank list contains all the previously reported mSWI/SNF components. b, BRD9 binds to SMARCA4 (BRG1) and GLTSCR1 (BICRA) in MKL-1. IP using a BRD9 antibody followed by western blotting was performed to determine interactions among BRD9, GLTSCR1, and SMARCA4. The experiment was performed at least three times. See Unprocessed Gels Extended Data Fig. 7. c, BRD9 degradation restores the loss of cell viability caused by LSD1 inhibition in MCC. MKL-1 cells were treated with varying doses of the dBRD9 and LSD1 inhibitor (GSK-LSD1) for six days. The XTT assay measured relative cell viability. d, dBRD9 degrades BRD9 efficiently. MKL-1 cells were treated with GSK-LSD1 (0.1 μM), dBRD9 (0.1 μM), or both for three days and harvested for western blotting. The experiment was performed at least three times. See Unprocessed Gels Extended Data Fig. 7. e, The PCA plot shows that the degradation of BRD9 by dBRD9 partially rescues the global gene expression changes caused by LSD1 inhibition. n = 2 independent biological replicates were used for analysis. f, BRD9 depletion by shRNAs rescues gene expression changes caused by LSD1 depletion. MKL-1 cells were transduced with an LSD1-targeting shRNA either with a control shRNA or two distinct BRD9-targeting shRNAs for six days and harvested for western blotting. The experiment was performed at least three times. See Unprocessed Gels Extended Data Fig. 7.
Extended Data Fig. 8|.

Heatmaps of gene expression changes following LSD1 and BRD9 inhibition indicate that LSD1 and BRD9 regulate an overlapping set of genes in an antagonistic manner. a, b, RNA-seq was performed with n = 2 biologically independent replicates of MKL-1 cells treated with DMSO, GSK-LSD1 (LSD1i, 0.1 μM), dBRD9 (0.1 μM), or both GSK-LSD1 and dBRD9 for six days. a, Top 50 most differentially expressed genes between LSD1i and LSD1i + dBRD9 are shown. Differential gene expression analysis was performed using DESeq2 and p-values were adjusted by Benjamini-Hochberg. b, All differentially expressed genes (3392 genes; FDR
Extended Data Fig. 9|.

ATAC-seq of MKL-1 cells treated with LSD1 and BRD9 inhibitors suggests that BRD9 is required to de-repress a subset of LSD1 target genes. a, Tag heat maps of ATAC-seq peaks show open chromatin regions localize close to the transcription start sites (TSSs, +/− 2000 bp). Combined peaks from two replicates of ATAC-seq of MKL-1 cells treated with DMSO (Control), dBRD9 (0.1 μM), GSK-LSD1 (LSD1i, 0.1 μM), or both dBRD9 and LSD1i (LSD1i_dBRD9) are shown. b–d, The differentially enriched ATAC-seq peaks between Control vs. LSD1i (b), Control vs. dBRD9 (c), and LSD1i vs. LSD1i + dBRD9 (d) are shown with the peak abundance (log concentration) and log fold change in peak scores. The differentially enriched peaks were called using the Diffbind R package with the Wald test and FDR p-value correction. e, ATAC-seq peaks in the promoters of LSD1 target genes ID1, SYT4, and CALB2 with the mentioned conditions are shown. f, Model: Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis.
Supplementary Material
Acknowledgments
We are grateful to Cigall Kadoch (Dana-Farber Cancer Institute, DFCI) and members of the Kadoch laboratory for helpful discussions relating to mSWI/SNF and specifically ncBAF complex biology and experimental approaches; to Yang Shi (Boston Children’s Hospital) for providing valuable insights on LSD1; to X. Shirley Liu (DFCI) for consultation about CRISPR-Cas9 screen analysis; and to Stephen Gygi (Harvard Medical School), Jun Qi (DFCI), and Myles Brown (DFCI) for sharing reagents and equipment.
This work was supported in part by U.S. Public Health Service grants F31CA213464 to D.E.P., R01GM132129 to J.A.P., and R35CA232128, R01CA63113, R01CA173023, and P01CA203655 to J.A.D. J.A.D. received research funding from Constellation Pharmaceuticals. S.K.S., L.F., and M.P.W. were supported by the Stowers Institute for Medical Research.
Footnotes
Declaration of Interests
JPM and PT are employees of Constellation Pharmaceuticals. JAD received research funding from Constellation Pharmaceuticals. JAD has served as a consultant to Merck & Co., Inc. and EMD Serono, Inc.
References
- 1.Shuda M et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proceedings of the National Academy of Sciences of the United States of America 105, 16272–16277, doi: 10.1073/pnas.0806526105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shuda M, Kwun HJ, Feng H, Chang Y & Moore PS Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. The Journal of clinical investigation 121, 3623–3634, doi: 10.1172/JCI46323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harms PW et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer research 75, 3720–3727, doi: 10.1158/0008-5472.CAN-15-0702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moll I, Kuhn C & Moll R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. The Journal of investigative dermatology 104, 910–915 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Cheng J et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS pathogens 13, doi: 10.1371/journal.ppat.1006668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park DE et al. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proceedings of the National Academy of Sciences of the United States of America, doi: 10.1073/pnas.1818798116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953, doi: 10.1016/j.cell.2004.12.012 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Laurent B et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Molecular cell 57, 957–970, doi: 10.1016/j.molcel.2015.01.010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saleque S, Kim J, Rooke HM & Orkin SH Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Molecular cell 27, 562–572, doi: 10.1016/j.molcel.2007.06.039 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Takagi S et al. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer research 77, 4652–4662, doi: 10.1158/0008-5472.CAN-16-3502 (2017). [DOI] [PubMed] [Google Scholar]
- 11.McGrath JP et al. Pharmacological Inhibition of the Histone Lysine Demethylase KDM1A Suppresses the Growth of Multiple Acute Myeloid Leukemia Subtypes. Cancer research 76, 1975–1988, doi: 10.1158/0008-5472.CAN-15-2333 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Mohammad HP et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer cell 28, 57–69, doi: 10.1016/j.ccell.2015.06.002 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Sugino N et al. A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 31, 2303–2314, doi: 10.1038/leu.2017.59 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Lee C et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nature communications 10, 332, doi: 10.1038/s41467-018-08269-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadoch C et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics 45, 592–601, doi: 10.1038/ng.2628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mashtalir N et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1744830464, doi: 10.1016/j.cell.2018.09.032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alpsoy A & Dykhuizen EC Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. The Journal of biological chemistry 293, 3892–3903, doi: 10.1074/jbc.RA117.001065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michel BC et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nature cell biology 20, 1410–1420, doi: 10.1038/s41556-018-0221-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue D et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature, doi: 10.1038/s41586-019-1646-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang P et al. RCOR2 is a subunit of the LSD1 complex that regulates ESC property and substitutes for SOX2 in reprogramming somatic cells to pluripotency. Stem cells (Dayton, Ohio) 29, 791–801, doi: 10.1002/stem.634 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Rush PS et al. Insulinoma-associated 1: A novel nuclear marker in Merkel cell carcinoma (cutaneous neuroendocrine carcinoma). Journal of cutaneous pathology 45, 129–135, doi: 10.1111/cup.13079 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Florens L & Washburn MP Proteomic analysis by multidimensional protein identification technology. Methods in molecular biology (Clifton, N.J.) 328, 159–175, doi: 10.1385/1-59745-026-X:159 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Paoletti AC et al. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proceedings of the National Academy of Sciences of the United States of America 103, 18928–18933, doi: 10.1073/pnas.0606379103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berrios C et al. Merkel Cell Polyomavirus Small T Antigen Promotes Pro-Glycolytic Metabolic Perturbations Required for Transformation. PLoS pathogens 12, doi: 10.1371/journal.ppat.1006020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma R et al. Bmp signaling maintains a mesoderm progenitor cell state in the mouse tailbud. Development (Cambridge, England), doi: 10.1242/dev.149955 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Wang S et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nature protocols 8, 2502–2515, doi: 10.1038/nprot.2013.150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu LJ et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC bioinformatics 11, 237, doi: 10.1186/1471-2105-11-237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright MC et al. Unipotent, Atoh1+ progenitors maintain the Merkel cell population in embryonic and adult mice. The Journal of cell biology 208, 367–379, doi: 10.1083/jcb.201407101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulo JA & Gygi SP Isobaric Tag-Based Protein Profiling of a Nicotine-Treated Alpha7 Nicotinic Receptor-Null Human Haploid Cell Line. Proteomics 18, doi: 10.1002/pmic.201700475 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fagerberg L et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & cellular proteomics : MCP 13, 397–406, doi: 10.1074/mcp.M113.035600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhanjal JK, Radhakrishnan N & Sundar D. Identifying synthetic lethal targets using CRISPR/Cas9 system. Methods (San Diego, Calif.), doi: 10.1016/j.ymeth.2017.07.007 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Lee J-EE et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2, doi: 10.7554/eLife.01503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Remillard D et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angewandte Chemie (International ed. in English) 56, 5738–5743, doi: 10.1002/anie.201611281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huelsken J, Vogel R, Erdmann B, Cotsarelis G & Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 105, 533–545 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Guha U et al. Target-derived BMP signaling limits sensory neuron number and the extent of peripheral innervation in vivo. Development (Cambridge, England) 131, 1175–1186, doi: 10.1242/dev.01013 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Tu WB et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer cell 34, 579, doi: 10.1016/j.ccell.2018.09.001 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Varlakhanova N, Cotterman R, Bradnam K, Korf I & Knoepfler PS Myc and Miz-1 have coordinate genomic functions including targeting Hox genes in human embryonic stem cells. Epigenetics & chromatin 4, 20, doi: 10.1186/1756-8935-4-20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brien GL et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 7, doi: 10.7554/eLife.41305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng W et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–1228931072, doi: 10.1016/j.cell.2018.05.052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nghiem PT et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. The New England journal of medicine 374, 2542–2552, doi: 10.1056/NEJMoa1603702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kent WJ et al. The human genome browser at UCSC. Genome research 12, 996–1006, doi: 10.1101/gr.229102 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love MI, Huber W & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson JT et al. Integrative genomics viewer. Nature biotechnology 29, 24–26, doi: 10.1038/nbt.1754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang DW a. W., Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57, doi: 10.1038/nprot.2008.211 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Wubing Zhang FW, and Binbin Wang. MAGeCKFlute: Integrative analysis pipeline for pooled CRISPR functional genetic screens. R package version 1.1.8. version 1.1.8. (2018). [Google Scholar]
- 47.Wubing Zhang FW, and Binbin Wang. MAGeCKFlute: Integrative analysis pipeline for pooled CRISPR functional genetic screens. R package version 118 version 1.1.8. (2018). [Google Scholar]
- 48.Ianevski A, He L, Aittokallio T & Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics (Oxford, England) 33, 2413–2415, doi: 10.1093/bioinformatics/btx162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu G, Wang L-GG, Han Y & He Q-YY clusterProfiler: an R package for comparing biological themes among gene clusters. Omics : a journal of integrative biology 16, 284–287, doi: 10.1089/omi.2011.0118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carey MF, Peterson CL & Smale ST Dignam and Roeder nuclear extract preparation. Cold Spring Harbor protocols 2009, doi: 10.1101/pdb.prot5330 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Cheng J, Rozenblatt-Rosen O, Paulson KG, Nghiem P & DeCaprio JA Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. Journal of virology 87, 6118–6126, doi: 10.1128/JVI.00385-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu T et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. Journal of proteomics 129, 16–24, doi: 10.1016/j.jprot.2015.07.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt D et al. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods (San Diego, Calif.) 48, 240–248, doi: 10.1016/j.ymeth.2009.03.001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mertins P et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat Protoc 13, 1632–1661, doi: 10.1038/s41596-018-0006-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature methods 14, 959–962, doi: 10.1038/nmeth.4396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaspar JM NGmerge: merging paired-end reads via novel empirically-derived models of sequencing errors. BMC bioinformatics 19, 536, doi: 10.1186/s12859-018-2579-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079, doi: 10.1093/bioinformatics/btp352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu G, Wang L-GG & He Q-YY ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics (Oxford, England) 31, 2382–2383, doi: 10.1093/bioinformatics/btv145 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Ross-Innes CS et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393, doi: 10.1038/nature10730 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome biology 9, doi: 10.1186/gb-2008-9-9-r137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, Pyl PT & Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Supek F, Bošnjak M, Škunca N & Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one 6, doi: 10.1371/journal.pone.0021800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu Z, Eils R & Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics (Oxford, England) 32, 2847–2849, doi: 10.1093/bioinformatics/btw313 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Blighe. Enhancedvolcano: Publication-ready volcano plots with enhanced colouring and labeling, 2018). [Google Scholar]
- 67.Navarrete-Perea J, Yu Q, Gygi SP & Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of proteome research 17, 2226–2236, doi: 10.1021/acs.jproteome.8b00217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ting L, Rad R, Gygi SP & Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature methods 8, 937–940, doi: 10.1038/nmeth.1714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods 4, 207–214, doi: 10.1038/nmeth1019 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ATAC-seq, ChIP-seq, and RNA–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE124856, GSE124857, GSE124861, GSE124864, and GSE140505. The mass spectrometry data are available at the ProteomeXChange under accession code PXD012516. All other data supporting the findings and computer codes implemented in this study are available from the corresponding author on reasonable request.